Submitted:
06 August 2023
Posted:
08 August 2023
You are already at the latest version
Abstract
The relaxation processes induced by an exposure of the Ar matrices doped with CH4 (0.1 – 10%) to an electron beam were studied with a focus on the dynamics of radiolysis products – H atoms, H2 molecules, CH radicals, and energy transfer processes. Three channels of energy transfer to dopant and radiolysis products were discussed: by free charge carriers, free excitons and photons from the “intrinsic source” provided by emission of the self-trapped excitons. Radiolysis products along with the total yield of desorbing particles were monitored in a correlated manner. Analysis of methane transformation reactions induced by free excitons showed that the CH radical can be considered as a marker of the CH3 species. The competition between exciton self-trapping and energy transfer to the dopant and radiolysis products has been demonstrated. A nonlinear concentration behavior of the H atoms in doped Ar matrices has been established. Real-time correlated monitoring of optical emissions (H atom and CH3 radicals), particle ejection, and temperature revealed a nonmonotonic behavior of optical yields with a strong luminescence flash after almost an hour of exposure, which correlated with explosive pulse of particle ejection and temperature. The connection of this phenomenon with the processes of energy transfer and radical-radical recombination is discussed.
Keywords:
solid methane
; astrophysical ices
; matrix isolation
; electron irradiation
; radiolysis
; energy transfer
; particle ejection
; relaxation processes
; luminescence
1. Introduction
Radiation effects in solid methane and methane-containing frozen films, so-called ices, attract much attention in diverse fields of science and technology. Being widely present in the Universe [1,2,3,4,5,6] methane plays important role in astrophysics and astrochemistry [7]. An overview of laboratory experiments on the relationship of methane to prebiotic chemistry is presented in [8]. Properties of methane, their transformation caused by the continuous radiation of outer space, and physico-chemical processes became the topic of a large body of research. Numerous efforts have been made to simulate radiation-induced processes in the laboratory using a variety of techniques surveyed in Rev. [9]. Various types of ionizing radiation were used to study radiation behavior of methane and methane-rich ices – ions [10,11,12,13,14,15,16,17,18,19], electrons [20,21,22,23,24,25,26,27,28,29] and photons [26,30,31,32,33,34,35].
Interaction of solid methane with neutrons is closely connected to the technology of cryogenic moderators [36,37,38,39,40,41,42]. Solid methane is widely used for moderating hot neutrons to cold and ultra-cold ones because its efficiency is approximately 3.5 times that of the commonly used liquid hydrogen-based moderator [36]. Despite the attractive neutronic properties of solid methane its use in cryogenic moderators faces a problem especially for the high intensity neutron flaxes. After long exposure to neutron radiation, as it was found by Carpenter [36,37], solid methane moderator experienced sudden, violent, warm-ups and pressure rise in its vessel in some cases with followed vessel destruction. This phenomenon, called by „burp“ effect, significantly constrains the current use of solid methane moderators. The reason of this phenomenon according [36,37] is a large spontaneous release of energy. The moderator material stores a part of energy absorbed from radiation in view of frozen-in products of radiolysis. At high density of reactive species their recombination and the expansion of hydrogen, which builds up in the solid methane during irradiation by neutrons [36] result in fast energy release which destroys a moderator vessel. The pressing character of this problem stimulated the thorough study the interaction of neutron radiation with solid methane [37,38,40,41,42]. According to Carpenter’s model [37] all recombination processes in solid methane moderators proceed in one stage and are controlled by the same activation energy for defect diffusion. However, commissioning tests of the ISIS TS2 solid methane moderator, in which a „burp“-like effect was observed [40], and the study of relaxation phenomena in solid methane pre-irradiated with an electron beam [28] led to the conclusion that the radiolysis defect recombination process happens in two stages, at different temperatures, and is therefore controlled by two different activation energies. Modifying Carpenter’s approach Kirichek and coauthors implemented two different types of radiation defects (H atoms and CH3 radicals), with different recombination rates and thermal activation energies [40]. The model suggested satisfactory described results of the commissioning tests the ISIS TS2 solid methane moderator. The possible role of radiolysis product recombination processes in cryo-volcanism on comets was discussed in [37,40].
Strong explosive-like delayed ejection of particles was observed in solid methane exposed to an electron beam of the subthreshold energy [29,43] that is, under conditions when the impact mechanisms of defect formation and desorption are excluded. The effect was observed at low temperature upon reaching a critical irradiation dose of 100 eV per methane molecule and was accompanied by the sample temperature rise and a flash of the luminescence. The outburst of particles was preceded by oscillations of particles ejection with increasing amplitude. The period of these oscillations depended on the intensity of the irradiation, it decreases with an increase in the beam current density. In [44] a model has been proposed that qualitatively describes the appearance of two types of self-oscillations upon electronic excitation of solid CH4, their periods and delay time of burst. Two types of self-oscillations found are a periodic change of temperature and concentration with time of CH4 decay products – H atoms and CH3 radicals upon irradiation. The found patterns of self-oscillations was shown to determine the temporal dynamics of the delayed explosive particle ejection upon irradiation with the subthreshold electrons. The discovered effect is a new manifestation of self-oscillations similar to those considered in [45]. To the best of our knowledge, there were only a few studies in which a delayed in time burst of particles, the so-called “delayed desorption”, from solid methane exposed to other types of ionizing radiation, was observed [46,47]. In [46] solid CD4 was irradiated with MeV He + and H +. When the beam was turned on, weak desorption of D2 first appeared. Upon reaching the threshold fluence density, the yield of D2 rapidly increased by more than an order of magnitude, and then gradually decreased. The threshold fluence values for irradiation with 1.5 MeV He+ ions and 1.5 MeV H+ were 3 1014 ions cm-2 and 9 1015 ions cm-2, respectively. In [47], solid CH4 and CD4 were irradiated with 9.0 MeV α particles and 7.3 MeV protons. Doses up to 145 eV caused pressure shocks rising the chamber pressure by several orders of magnitude. This process released up to 90% of the molecules of the solid target into the gas phase. The authors concluded that the H and CH3 radicals play a major role in this phenomenon, and the observed explosion resembles a nonequilibrium process. However, in these experiments no regular oscillations were observed. The detection of complex organic molecules (COMs) in the gas phase of cold molecular clouds at such low temperatures of the cryogenic environment that they would have to condense on the surfaces raised the question of the reasons of this phenomenon. Several hypotheses have been proposed [48,49,50,51,52] and radical–radical induced explosive desorption of ice-coated interstellar nanoparticles among them [52]. In this article, the authors present the direct observation by Fourier transform infrared (FTIR) spectroscopy of rapid radical–radical reactions of formyl (HCO•) and methyl (•CH3) radicals and the reaction-induced explosive desorption during the exposure of methane and carbon monoxide ices to superthreshold 5 keV electrons. However, the formation and recombination of the H atom remained beyond the reach of this experimental approach.
To get more insight into the phenomenon the matrix isolation method was introduced in combination with a set of emission spectroscopy methods [53,54,55]. It should be noted that in contrast to pure methane and mixtures of methane with other molecular gases of astrophysical interest there are only a few studies performed in matrices of rare gases [56,57,58]. Photolysis of methane in Kr matrix was studied using synchrotron radiation [56]. Strong VUV absorption and emission features were attributed to atomic carbon C and the CH radical. Neither CH2 nor CH3 were observed in Kr matrix. In contrast to that photolysis of methane in Ne matrix resulted in appearance of wide range of hydrocarbons including CH3 and carbon clusters Cn with number of atoms up to n=20 [58].
Below, we briefly summarize the results of our previous studies of radiation effects induced by an electron beam in Ar matrices doped with CH4 [53,54,55,59]. In the cathodoluminescence (CL) spectra, the following products of radiation-induced methane transformation were recorded: H, CH and C. It has been shown that because of the small penetration depth of electrons the bulk of the matrix is excited preferentially by photons of the matrix with an energy of 9.8 eV (the most intense emission band of the self-trapped excitons), in other words “internal photolysis” occurs. Analysis of an efficiency of different channels of methane transformation under these photons and neutralization reactions indicated that the CH radical can be considered as a signature of the CH3 radicals [55]. Monitoring of the so-called nonstationary luminescence (NsL), viz. luminescence under nonstationary conditions (upon external heating under beam) in combination with the detection of thermally stimulated exoelectron emission (TSEE) and optical emission spectroscopy made it possible to reveal the contribution of the reactions of charged and neutral species. The dynamics of the main products of methane fragmentation were traced and it was found that behavior of CH radicals (therefore, CH3 radicals) and hydrogen atoms is different and depends on the concentration of methane in the matrix. This means that the assumption about the same concentration of CH3 radicals and H atoms based only on the reaction: CH4 + ΔE → CH3 + H, accepted in the delayed explosive desorption models [37,40,44], is not entirely correct. Long-term and short-term oscillations of the total particle yields were found and interpreted as thermo-concentration self-oscillations similar to those considered in [44] for the case of pure methane. It has been shown that they can be initiated either by the spontaneous release of stored energy upon reaching a critical concentration of reactants, or by external heating of the doped matrix to stimulate diffusion and release the stored chemical energy in subsequent recombination reactions [59]. Preliminary measurements of the dose dependence of the optical emission of the H atom, carried out on a lightly doped Ar matrix (C = 1%) at LHe temperature, found a puzzling fact – an increase in this emission with a pressure burst [54]. The dynamics of optical emissions from highly doped matrices upon low temperature irradiation remained unexplored.
Here we present an outgrowth of this research with a focus on H atom and CH3 radical behavior at different concentrations of CH4 in Ar matrix and processes induced by energy transfer from the matrix to dopant and products of radiolysis. Three channels of energy transfer are considered – charge transfer (by holes), transfer by free excitons, and self-trapped excitons. Special attention is paid to the recombination of H atoms and its monitoring in the matrix. This reaction proceeds nonradiatively with energy release of 218 kJ mole-1. A hydrogen molecule can be monitored by being transferred to an excited state. The thresholds of excitation of the lowest singlet (B1 Σu+) and triplet (a3 Σg+) excited states [60] are below the bands of free excitons of Ar – 12.06 eV for Γ(3/2) and 12.24 eV for Γ(1/2) [61]. So, the transfer of energy by free excitons to the hydrogen molecule can lead to its transition to the lowest excited states B1 Σu+ and a3 Σg+. Electric dipole transitions from the coupled triplet state a3 Σg+ into an unstable lower state b3 Σu+ lead to the appearance of a wide emission continuum [62] followed by dissociation of H2 molecule. The detection of H atoms proceeds under conditions of competition between exciton self-trapping and energy transfer to the dopant and radiolysis products. A nonlinear concentration behavior of the optical emission of H atoms in doped Ar matrices has been found. Real-time correlated monitoring of optical emissions (H atom and CH3 radicals), particle ejection, and temperature revealed a nonmonotonic behavior of optical yields with a strong luminescence flash after almost an hour of exposure, which correlated with surge of temperature and particle ejection. The connection of this phenomenon with the processes of energy transfer and radical-radical recombination is discussed.
2. Experimental
2.1. Choice of matrix
The electronically excited states of CH4 are of a dissociative nature, and any kind of irradiation will lead to the appearance of radiolysis products in the spectra. The Ar matrix ensures effective excitation of the dopant, since free and self-trapped Ar excitons fall into the region of the absorption band of the CH4 molecule [63], as shown in Figure 1.
The Ar matrix is transparent to the radiation of self-trapped excitons (the most intense radiation of the matrix), since these states lie below the intrinsic absorption threshold of the matrix, which makes it possible to effectively excite the methane molecule throughout the volume, stimulating its dissociation through photon processes. It is worth noting that the energies of free and self-trapped excitons in solid Ar (12.06 and 9.8 eV, respectively) are close to the photon energy of the most intense line in the VUV solar spectrum (Lyman- ɑ), and the study of excitation induced processes in an Ar matrix is of interest for planetary astrochemistry. The second reason for choosing solid Ar as a matrix is the high ionization potential I of Ar (I=15.76 eV) [64], which exceeds the ionization potential of methane itself and its radiation-induced transformation products, as shown in Table 1.
This choice ensures charge transfer from the matrix to the dopant as well as radiolysis products and prevents the reverse process of charge transfer from CH4+ and other cations to the matrix. The third reason for the choice is related to geometric parameters. The methane molecule with the kinetic diameter of 0.380 nm [65] can occupy substitutional site in Ar matrix which is about 0.376 nm [66]. When a methane concentration increases to 3%, the Ar lattice in the vicinity of the dopant begins to modify and at higher concentration of about 20% a two-phase system is obtained [67].
2.2. Sample preparation and control
The experimental procedures have been described in detail in [53]. Here we give a brief account of the procedures. We used Ar gas (99.998%) and CH4 gas (99.97%) without further purification. Mixture of Ar and CH4 of defined concentration was performed in the gas-handling system which was heated and degassed before each experiment. In these experiments, we worked in the range of methane concentrations from 0.1 to 10%. Films of solid Ar doped with methane were grown by deposition of a certain amount of premixed gas of room temperature onto a cooled to LHe temperature oxygen-free Cu substrate mounted in a high-vacuum chamber with a base pressure of <10-7 torr. Pumping out was carried out by LHe cryogenic pump and magnetic discharge pump. All measurements, both while the beam is on, and after its shutdown and subsequent heating, were performed in the dynamic pumping mode. The pressure in the experimental chamber was monitored throughout the entire experiment. Films of the same thickness (25 µm) grown in the same mode were used for all concentrations. They were fairly transparent which indicated their good structural properties and did not require annealing. The presence of impurities was controlled spectroscopically. The sample temperature was monitored during the entire experiment with a Si sensor mounted on the substrate.
To obtain some information about the structural features of the grown and irradiated samples (and the temperature ranges in which the neutralization processes will occur,) we measured the thermally stimulated exoelectronic emission (TSEE) from all preliminarily irradiated in identical way doped matrices as well as pure Ar and methane. Due to the high mobility of electrons in solid Ar and methane [68], TSEE monitoring makes it possible to obtain information on relaxation processes not only near the surface, but also in the bulk of the matrix. The irradiation was performed with a subcritical dose. In these experiments we used heating with a constant rate of 5 K min-1. Measurements of TSEE were started after decaying of the “afteremission” current to zero. Electrons released from shallow traps (defects of structure) upon heating can either neutralize cations or escape from the film yielding TSEE current. Stimulated currents were detected with an electrode kept at a small positive potential VF=+9 V and connected to the current amplifier. The TSEE curves obtained reflect the distribution of defects in the sample and provide an information on the defects of growth and radiation induced ones as well as charge accumulation. Measurements of the TSEE yields were carried out in the temperature range of 5-50 K.
Figure 2 illustrates TSEE yields from pure Ar, doped Ar matrices and pure CH4.
The low temperature peak at 8 K in the doped samples coincides with that recorded in the pure Ar matrix, but has a much lower intensity. The decrease in its intensity in doped matrices may be connected with a change of trap distribution in favor of deep ones due to the attachment of an electron to some radiolysis products with a positive electron affinity (see Table 1). The next wide feature at about 16 K represents a multipeak curve with unresolved structure incorporating components related to different defects of structure. The shape of these curves is similar for all doped samples and similar to that of the pure methane. The intensity of this feature increases with a grow of methane concentration. This suggests that this feature is related to defects induced by incorporation of methane molecules and radiolysis products into the Ar matrix and possible methane clusters formation.
2.3. Irradiation mode and optical emission measurements
The reason for choosing electrons as projectiles is that electrons are common secondary particles under any kind of irradiation and the results obtained in this way can be applied to the wider field of radiation physics. Irradiation was carried out by electrons with subthreshold energy (Ee<1.7 keV) to exclude the formation of defects and sputtering of samples via the impact mechanism, while the mechanisms of electronically stimulated defect formation and desorption, however, operate. In these experiments, the electron beam energy Ee was set to 1.5 keV with the current density of 2.5 mA cm-2. The electron gun was operating in dc regime under controlled conditions. The beam covered the icy film surface with an area of 1 cm2. The sample heating when turning on the electron beam did not exceed 0.4 K. All measurements of spectral features were carried out at 5 K. The open surface of the samples enabled us to measure cathodoluminescence (CL) spectra not only in visible but also in the vacuum ultraviolet (VUV) range as well as to measure the TSEE yields as described before. The dose deposited by irradiation was determined from the exposure time for a constant beam intensity. The dose dependencies of spectrally resolved features were measured simultaneously with a constant monitoring of desorption processes by the chamber pressure recording with an ionization detector (a Bayard-Alpert gauge). Note, that all measurements were performed in the dynamic pumping out mode.
3. Results and Discussion
As already mentioned, the electronic states of the methane molecule are dissociative, so its luminescence spectrum is determined by the products of its radiolysis. Main channels of CH4 degradation upon photon flax with the threshold wavelengths [69] in the solid Ar emission range are:
CH4 + hν → CH3 + H
CH4 + hν → CH2 + H2
CH4 + hν → CH2 + 2H
CH4 + hν → CH + H + H2
CH4 + hν → C + 2H2
In accordance with this we detected following emissive products of methane degradation: H, CH, C and H2, as can be seen from the CL spectrum shown in Figure 3.
The spectrum contains both the emission bands of the matrix and the emission bands of the radiolysis products. Emissions are observed both from the solid phase and from the gaseous due to the electronically induced desorption. The most intense feature of this spectrum at 127 nm is the well-known emission of the molecular-type self-trapped excitons (STE) – Ar2*, corresponding to the bound-free transitions from 1,3Σu+ states to a repulsive part of the ground state 1Σg+ [61]. At the short-wavelength edge of the Ar2* band, recorded in the second order, the Lyman-ɑ line, which belongs to the desorbed into the gas phase excited H atoms, was detected. Mechanisms of the excited H atoms formation and their desorption from Ar matrices, doped with CH4, were discussed recently [71]. Four scenarios for the desorption of excited H atoms were proposed, including energy transfer by holes and excitons of the Ar matrix. Own atoms of the matrix and molecules also desorb in excited states emitting corresponding lines and band (the feature with unresolved structure in Figure 3). Their desorption is driven by the exciton mechanism [72]. A necessary condition of the electronically stimulated desorption is the localization of electronic excitation on the atoms or molecules in the near-surface region with the release of energy exceeding the binding energy per atom εb (for atom of Ar matrix εb=88.8 meV [61]). For the case of Ar matrix desorption of excited particles (including H* atoms) is facilitated by negative electron affinity of Ar χ = -0.4 eV [61], in other words, excited particles are ejected under the repulsive forces between an excited atom and atoms of surrounding lattice, so-called “cavity-ejection” mechanism [72]. Interestingly, the desorption of excited argon molecules in the states 1,3Σu+(v’) was also detected as a result of self-trapped hole (Ar2+) neutralization reaction [73]. The so-called “third continuum” of Ar at 200 nm, assigned to the radiative transitions from the excited (Ar2+)* state [74], was quenched by admixture of methane. Note that the Ar2*-band recorded in the second order overlaps with a wide emission continuum of molecular hydrogen associated with transitions from the bound triplet state a3Σg+ to the unstable lower state b3Σu+, which makes it difficult to monitor the H2 molecule by this transition. According to [75], the maximum of the spectral distribution of the radiative transition probability from the level v'=0 of the term a3 Σg+ is about 260 nm. A weak band at 146 nm observed in the CL spectrum was tentatively assigned to the band 0-6 of the Lyman series B1Σu+ → X1Σg+ of molecular hydrogen. However, it was not possible to use this band for the H2 monitoring because of its low intensity. In the visible range we detected emission bands of other radiolysis products – CH radical and C atom. CH radicals were registered by the emission bands at 432 nm (the A2∆ → X2∏ transition) and 387 nm (the B2Σ- → X2∏ transition). C atoms were recorded by the emission lines at 470 nm (the 1S → 3P transition) and 295 nm (the 5S0 → 3P transition). The spectrum also contains a broad band at 184 nm unidentified at present and impurity bands of CO (the Cameron system a 3Π→X1 Σ+).
The positions and half-widths of the observed bands remain practically unchanged over the entire concentration range studied. Main changes in the spectra relate to changes in relative intensity of bands. Upon doping of Ar matrices, a strong quenching of the emission band of self-trapped excitons (that is the M-band) was observed as shown in Figure 4.
This demonstrates quite efficient energy transfer by free excitons to dopant and radiolysis products, since the diffusion length Ldif of thermalized free excitons in solid Ar (Ldif ~100 nm [76,77]) exceeds the distance between the dopants already at C=0.1%.
H atoms in the matrices were traced by a CL band centered at 166 nm, which belongs to the excimer Ar2H* [70]. Formation of Ar2H* occurs with the participation of the matrix hole Ar+ at the expense of the large affinity of Ar atom to a proton (proton affinity PA – 369.2 kJ mol-1 [64]). The quantum chemical calculations [78] on the RgnH+ species showed that the configuration with two solvent atoms of the D∞h configuration with a proton between them is the energetically favorable configuration of the proton solvated in all Rare Gas matrices. The recombination of Ar2H+ cations with electrons results in appearance of the excimers Ar2H*. It is interesting to follow the change in the relative intensity of this excimer band with the dopant concentration. The intensity of the Ar2H* emission band, hereinafter referred to as H, measured with respect to the Ar2 band of self-trapped excitons, denoted M, is shown in Figure 5.
Attention is drawn to the nonlinear behavior of the relative intensity of the H band which reflects concentration of H atoms. As can be seen from the channels (1) – (5) of CH4 fragmentation, H atoms are formed in reactions (1), (3) and (4) with the branching ratios BR=0.5: 0.2 and 0.1 respectively for a photon energy of 9.8 eV and BR=0.25; 0.5 and 0.2 for a photon energy of 12.1 eV according to [79], in which an analysis of energy-dependent branching ratios, the so-called breakdown curves, was presented. Let us analyze the reactions of secondary products of methane transformation CH3 and CH2 based on the data [79]:
and
CH3 + hν → CH2 + H
CH3 + hν → CH + 2H
CH3+ hν → CH + H2
CH3 + hν → C+H2+H
CH2 + hν → CH + H
CH2 + hν → C + 2H
CH2 + hν → C + H2
The emission of self-trapped excitons (9.8 eV) can produce H atoms from CH3 via channels (6), (7) and (9) with BR=0.28, 0.5 and 0.25 correspondingly. An excitation of CH2 by these photons induces its dissociation via channels (10) and (11) with BR=0.45 for both channels. In this case 2H atoms are produced in correlated way in channel (11). When excited by free Ar excitons (12.06 eV), the main contribution to the formation of the H atom is made by channels (6), (7) and (9) with BR=0.2, 0.8 and 0.3, respectively, moreover, reaction (7) produces 2H atoms simultaneously. Formation of H atoms from CH2 occurs most effectively via channel (11) with BR=1. Channel (10) does not operate (BR=0) at this photon energy. It should be noted that H atoms which are formed in these reactions have an excess of kinetic energy, e.g. in channel (1) the experimental mean kinetic energy of the neutral lighter fragment H appeared to be 3.1 eV [79]. In most other channels this energy exceeds 1 eV and such “fast” H atoms may diffuse for quite a long distance facilitating H + H recombination after thermalization. The mobility of H atoms in rare Gas solids and the stability of trapping sites have been the subject of numerous studies, incl. [80,81,82,83,84,85]. The case of an Ar matrix was considered in [81,84]. In Ar matrices the hydrogen atom can occupy two types of sites – interstitial Oh and substitutional ones. According [81] the EPR signals due to interstitially trapped hydrogen atoms in octahedral sites disappear near 16 K in solid Ar. The atoms trapped in the substitutional sites remain trapped [84]. However, their excitation via energy transfer leads to the excimers Ar2H* formation and the resumption of the diffusion process. As a result, the processes of H atom production and their recombination compete each other upon irradiation.
Our study of the dose dependences of the emission of H atoms and CH radicals performed at low temperature showed a sharply nonmonotonic behavior of these emissions. As follows from the analysis of BR reactions (4), (7), (8), and (10), CH3 radicals remain the main source of CH radical formation upon excitation by free excitons of the matrix; so the conclusion that CH is a CH3 marker [55] remains valid. Figure 6 shows the dose dependencies of the H and CH emission measured simultaneously with the pressure in the experimental chamber.
The dose behavior of both optical emissions was quite nonmonotonic. We first observed an increase in both optical emissions and then an exit to saturation after 350 seconds of exposure to an electron beam, followed by a very gradual decrease. Weak bursts on the radiation yield curves were detected – two on the curve for the CH band and one on the curve for the H band. These peaks correlated with small peaks on the temperature curve. The main change of yields occurred after 3250 s of irradiation. This phenomenon is quite similar to the observed in pure solid methane [29,43] interpreted as the result of thermo-concentration self-oscillations [44]. Details of the emission yields together with change of temperature and pressure are shown in Figure 7a,b.
After prolonged irradiation at about 3250 s, the temperature and intensity of the H band, which is proportional to the number of H atoms in the matrix, begin to increase, then the intensity of the H band decreases, while the temperature and pressure sharply increase. It should be noted that since the temperature sensor (Si diode) was mounted on the back of a substrate cooled by LHe, it was not possible to determine the actual surface temperature of the Ar film. However, its significant increase during explosive emission of particles is indicated by a sharp drop in the TSEE yield, measured after explosive emission and redistribution of the TSEE yield with respect to the TSEE measured after irradiation of the sample with a subcritical dose. The beginning sublimation of the sample allows one to estimate only the lower boundary of the heating pulse — 30 K. In reality, the temperature rise in the pulse exceeds the triple point temperature of Ar – 87.78 K [64]. The temperature pulse has three maxima and its duration was 100 s. After the first burst of pressure, which lasted about 20 s another maximum of pressure rise was observed but longer and less intense. During this period the change in the intensity of the H band followed the temperature course. Such an unexpected, at first glance, behavior of the H band can be understood if we consider more carefully the processes of energy transfer to the products of ongoing reactions. A high temperature of the sample during the burst is a “fingerprint” of the occurrence of exothermic reactions, in particular, the reaction of H atom recombination. Transfer of energy by free excitons of Ar matrix can populate low electronic states of H2 molecule: the singlet state B1 Σu+ and the triplet state a3 Σg+ as can be seen from Figure 8.
Transitions from the bound triplet state a3 Σg+ to the repulsion curve of the lower state b3 Σu+ result in the dissociation of the hydrogen molecule and the appearance of two “hot” H atoms, which, after thermalization, form centers responsible for the emission of the H band. Thus, the processes of recombination of H atoms with the formation of H2 molecules proceed in competition with the photon-induced dissociation of H2, which affects the dose dependence of the H band. An increase in the H band intensity during a burst of pressure was also observed at a methane concentration C=1% in the Ar matrix [54]. However, this fact was misinterpreted as the absence of a contribution from the recombination of H atoms to the delayed desorption, since long before the pressure burst in this experiment, we observed a sharp drop in the intensity of the H band, which was attributed to the recombination of H atoms.
Note that the dose dependences of the H band intensity measured on lightly (C=1%) and highly (C=10%) doped matrices are different. While in the case of a highly doped Ar matrix, the burst was observed after the stage of a slow decrease in the intensity of the H band (see Figure 6), in the case of a lightly doped matrix, the burst followed its growth as shown in Figure 9. The decrease in the burst delay time in this experiment is associated with the use of higher current density.
At a methane concentration of C=5%, the dose dependence of the H band is similar to that recorded for 1%. It should be noted that the concentration of the radiolysis products is significantly lower than that of the dopant since many different primary dissociation channels operate simultaneously. Due to this, the conditions for excitation of H2 by free excitons change with concentration: with increasing C, the efficiency of excitation of H2 by excitons rises and therefore the contribution of the recombination reaction of H atoms to the H band increases. Accordingly, the flash effect accompanying the ejection of particles appeared to be much more pronounced at 10%, as can be seen from the comparison of Figure 7a and Figure 9.
It is interesting to compare the dose dependence of the H band with that detected for the CH band in highly doped matrix (see Figure 6 and Figure 7b). An intensity of the CH band, which is a marker of the CH3 radical, follows the temperature course with an exception of one local event at about 3340 s when the temperature and intensity of the CH band change in the opposite way. Such a behavior became clear if consider the balance and interplay between C2H6 and CH3. As follows from Refs. [63,88,89] the photodissociation (absorption) cross section of ethane is close to that of methane and both free and self-trapped excitons fall into the range of ethane absorption. There are a number of primary dissociation channels which include reactions: C2H6 + hν → CH3 + CH3 and C2H6 + hν → CH3* + CH3 [88]. Note that the enthalpy ΔrH of the second reaction with the excited CH3* radical formation ΔrH = 9.54 eV [88], is close to the photon energy of self-trapped excitons in Ar (9.8 eV). The enthalpy of the first reaction is essentially lower – ΔrH = 3.81 eV [88], and both of them can proceed. Unfortunately, at present, to the best of our knowledge, there is no quantitative information on branching ratios of these reactions, however, as it was shown [88] the reaction with C-C bond fission yielding two CH3 radicals refers to active channels of the C2H6 photodissociation. Based on this conclusion, we can expect similar behavior of the CH and H bands during the burst. The results obtained (see Fig. 7 a and b) suggest that the luminescence flashes detected in these bands can be assign as indicators of the recombination of H atoms and CH3 radicals with the release of thermal energy, which ensures the explosive ejection of particles.
5. Conclusions
The influence of environment on the radiation-induced processes in Ar matrices doped with methane (C=0.1 – 10%) was studied by means of emission spectroscopy methods. Electrons of subthreshold energy (1.5 keV) were used to eliminate the knock-on defect formation and desorption. Four products of methane transformation were detected – H and C atoms, H2 molecules and CH radicals. Analysis of methane transformation reactions induced by free and self-trapped excitons showed that the CH radical can be considered as a marker of the CH3 species. Three channels of energy transfer to dopant and radiolysis products were considered: by free holes, free excitons and photons from the “intrinsic source” provided by emission of the self-trapped excitons. Special attention was paid to the dynamics of radiolysis products – H atoms, H2 molecules and CH3 radicals (via CH species). Dose dependencies of the radiolysis products along with the total yield of desorbing particles were monitored in a correlated manner. The competition between exciton self-trapping and energy transfer to the dopant and radiolysis products has been analyzed. A nonlinear concentration behavior of the H atoms in doped Ar matrices has been established. The study revealed a nonmonotonic behavior of the optical yields of H atoms and CH radicals with a strong luminescence flash (after nearly an hour irradiation) which correlated with the temperature surge and explosive pulse of particle ejection. An analysis of the energy transfer and trapping processes made it possible to explain the seemingly contradictory observation of a surge in the optical emission of H atoms and CH3 radicals as an indicator of their recombination.
Author Contributions
E. Savchenko: conceptualization, writing - original draft, review and editing, supervision. I. Khyzhniy: investigation, software, validation. S. Uyutnov: investigation, methodology M. Bludov: investigation, resources.
Acknowledgments
The authors cordially thank colleagues Vladimir Sugakov, Oleg Kirichek, Robert Kołos, Claudine Crepin-Gilbert, Hermann Rothard, Anatoli Popov and Giovanni Strazzulla, for stimulating discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Clark, R.N.; Carlson, R.; Grundy, W.; Noll , K. Observed Ices in the Solar System. In The Science of Solar System Ices; Gudipati, M.S., Castillo-Rogez, J., Eds.; Astrophysics and Space Science Library: Springer: New York, 2013; Volume 356, pp. 3–46. [Google Scholar] [CrossRef]
- Boogert, A.C.A.; Gerakines, P.A.; Whittet, D.C.B. Observation of the Icy Universe. Annual Review of Astronomy and Astrophysics 2015, 53, 541–581. [Google Scholar] [CrossRef]
- Guzmán-Marmolejo, A.; Segura, A. Methane in the Solar System. Boletín de la Sociedad Geológica Mexicana 2015, 67, 377–385. [Google Scholar] [CrossRef]
- Lacy, J.H.; Carr, J.S.; J, I.E.N.; Baas, F.; Achtermann, J.M.; Arens, J.F. Discovery of interstellar methane - Observations of gaseous and solid CH4 absorption toward young stars in molecular clouds. Astrophys. J. 1991, 376, 556–560. [Google Scholar] [CrossRef]
- Cruikshank, D.P.; Brown, R.H.; Calvin, W.M.; Roush, T.L.; Bartholomew, M.J. Ices on the satellites of Jupiter, Saturn, and Uranus. In Solar System Ices; Schmitt, B., de Bergh, C., Festou, M., Eds.; Kluwer Academic: Netherlands, 1998; pp. 579–606. [Google Scholar]
- Gibb, E.; Mumma, M.; Russo, N.D.; DiSanti, M.; Magee-Sauer, K. Methane in Oort cloud comets. Icarus 2003, 165, 391–406. [Google Scholar] [CrossRef]
- berg, K.I. Photochemistry and astrochemistry: Photochemical pathways to interstellar complex organic molecules. Chem. Rev. 2016, 116, 9631–9663. [Google Scholar]
- Kobayashi, K.; Geppert, W.D.; Carrasco, N.; Holm, N.G.; Mousis, O.; Palumbo, M.E.; Waite, J.H.; Watanabe, N.; Ziurys, L.M.; Bera, P.P.; et al. Laboratory Studies of Methane and Its Relationship to Prebiotic Chemistry. Astrobiology 2017, 17, 786–812. [Google Scholar] [CrossRef]
- Allodi, M.A.; Baragiola, R.A.; Baratta, G.A.; Barucci, M.A.; Blake, G.A.; Boduch, P.; Brucato, J.R.; Contreras, C.; Cuylle, S.H.; Fulvio, D.; et al. Complementary and Emerging Techniques for Astrophysical Ices Processed in the Laboratory. Space Sci. Rev. 2013, 180, 101–175. [Google Scholar] [CrossRef]
- Foti, G.; Calcagno, L.; Sheng, K.L.; Strazzulla, G. Micrometre-sized polymer layers synthesized by MeV ions impinging on frozen methane. Nature 1984, 310, 126–128. [Google Scholar] [CrossRef]
- Kaiser, R.I.; Roessler, K. Theoretical and laboratory study on the interaction of cosmic-ray particles with interstellar ices. III. Suprathermal chemistry-induced formation of hydrocarbon molecules in solid methane (CH4), ethylene (C2H4) and acetylene (C2H2). Astrophys. J. 1998, 503, 959–975. [Google Scholar] [CrossRef]
- Brunetto, R.; Barucci, M.A.; Dotto, E.; Strazzulla, G. Ion Irradiation of Frozen Methanol, Methane, and Benzene: Linking to the Colors of Centaurs and Trans-Neptunian Objects. Astrophys. J. 2006, 644, 646–650. [Google Scholar] [CrossRef]
- E Palumbo, M.; A Baratta, G.; Fulvio, D.; Garozzo, M.; Gomis, O.; Leto, G.; Spinella, F.; Strazzulla, G. Ion irradiation of astrophysical ices. J. Physics: Conf. Ser. 2008, 101, 012002. [Google Scholar] [CrossRef]
- Ennis, C.; Yuan, H.; Sibener, S.J.; Kaiser, R.I. On the chemical processing of hydrocarbon surfaces by fast oxygen ions. Phys. Chem. Chem. Phys. 2011, 13, 17870–17884. [Google Scholar] [CrossRef] [PubMed]
- de Barros, A.L.F.; Bordalo, V.; Duarte, E.S.; da Silveira, E.F.; Domaracka, A.; Rothard, H.; Boduch, P. Cosmic ray impact on astrophysical ices: laboratory studies on heavy ion irradiation of methane. Astron. Astrophys. 2011, 531, A160. [Google Scholar] [CrossRef]
- Mejίa, C.F.; de Barros, A.L.F.; Bordalo, V.; da Silveira, E.F.; Boduch, P.; Domaracka, A.; Rothard, H. Cosmic ray–ice interaction studied by radiolysis of 15 K methane ice with MeV O, Fe and Zn ions. Monthly Notices of the Royal Astronomical Society 2013, 433, 2368–2379. [Google Scholar] [CrossRef]
- Boduch, P.; Dartois, E.; de Barros, A.L.F.; da Silveira, E.F.; Domaracka, A.; Lv, X.-Y.; Palumbo, M.E.; Pilling, S.; Rothard, H.; Duarte, E.S.; et al. Radiation effects in astrophysical ices. J. Physics: Conf. Ser. 2015, 629. [Google Scholar] [CrossRef]
- Vasconcelos, F.A.; Pilling, S.; Rocha, W.R.M.; Rothard, H.; Boduch, P.; Ding, J.J. Ion irradiation of pure and amorphous CH4 ice relevant for astrophysical environments. Phys. Chem. Chem. Phys. 2017, 19, 12845–12856. [Google Scholar] [CrossRef]
- Mejía, C.; de Barros, A.L.F.; Rothard, H.; Boduch, P.; da Silveira, E.F. Radiolysis of Ices by Cosmic-Rays: CH4 and H2O Ices Mixtures Irradiated by 40 MeV 58Ni11+ Ions. Astrophys. J. 2020, 894, 132. [Google Scholar] [CrossRef]
- Huang, T.; Hamill, W.H. Characteristic energy loss, luminescence and luminescence excitation spectra of methane and other alkane solids under low-energy electron impact. J. Phys. Chem. 1974, 78, 2077–2080. [Google Scholar] [CrossRef]
- Bennett, C.J.; Jamieson, C.S.; Osamura, Y.; Kaiser, R.I. Laboratory Studies on the Irradiation of Methane in Interstellar, Cometary, and Solar System Ices. Astrophys. J. 2006, 653, 792–811. [Google Scholar] [CrossRef]
- Barberio, M.; Vasta, R.; Barone, P.; Manicò, G.; Xu, F. Experimental and Theoretical Study on the Ethane and Acetylene Formation from Electron Irradiation of Methane Ices. World J. Condens. Matter Phys. 2013, 03, 14–20. [Google Scholar] [CrossRef]
- Huels, M.; Parenteau, L.; Bass, A.; Sanche, L. Small steps on the slippery road to life: Molecular synthesis in astrophysical ices initiated by low energy electron impact. International Journal of Mass Spectrometry 2008, 277, 256–261. [Google Scholar] [CrossRef]
- Jones, B.M.; Kaiser, R.I. Application of Reflectron Time-of-Flight Mass Spectroscopy in the Analysis of Astrophysically Relevant Ices Exposed to Ionization Radiation: Methane (CH4) and D4-Methane (CD4) as a Case Study. J. Phys. Chem. Lett. 2013, 4, 1965–1971. [Google Scholar] [CrossRef]
- Materese, Ch.K.; Cruikshank, D.P.; Sandford, S.A.; Imanaka, H. and Nuevo, M. Ice chemistry on outer Solar System bodies: Electron radiolysis of N2-, CH4-, and CO-containing ices. Astrophys. J. 2015, 812, 150. [Google Scholar] [CrossRef]
- Abplanalp, M.J.; Jones, B.M.; Kaiser, R.I. Untangling the methane chemistry in interstellar and solar system ices toward ionizing radiation: a combined infrared and reflectron time-of-flight analysis. Phys. Chem. Chem. Phys. 2017, 20, 5435–5468. [Google Scholar] [CrossRef]
- Savchenko, E.; Khyzhniy, I.; Uyutnov, S.; Bludov, M.; Barabashov, A.; Gumenchuk, G.; Bondybey, V. Radiation effects in nitrogen and methane “ices”. Nucl. Instruments Methods Phys. Res. Sect. B: Beam Interactions Mater. Atoms 2018, 435, 38–42. [Google Scholar] [CrossRef]
- Savchenko, E.; Kirichek, O.; Lawson, C.; Khyzhniy, I.; Uyutnov, S.; Bludov, M. Relaxation processes in solid methane pre-irradiated with an electron beam. Nucl. Instrum. Meth. B, 2018; 433, 23–27. [Google Scholar] [CrossRef]
- Savchenko, E.; Khyzhniy, I.; Uyutnov, S.; Bludov, M.; Gumenchuk, G.; Bondybey, V. Effects induced by electron beam in methane ices. Nucl. Instrum. Meth. B , 2019; 460, 244–258. [Google Scholar] [CrossRef]
- Hodyss, R.; Johnson, P.V.; Stern, J.V.; Goguen, J.D.; Kanik, I. Photochemistry of methane-water ices. Icarus 2009, 200, 338–342. [Google Scholar] [CrossRef]
- Hodyss, R.; Howard, H.R.; Johnson, P.V.; Goguen, J.D.; Kanik, I. Formation of radical species in photolyzed CH4:N2 ices. Icarus 2011, 214, 748–753. [Google Scholar] [CrossRef]
- Wu, Y.-J.; Wu, C.Y.R.; Chou, S.-L.; Lin, M.-Y.; Lu, H.-C.; Lo, J.-I.; Cheng, B.-M. SPECTRA AND PHOTOLYSIS OF PURE NITROGEN AND METHANE DISPERSED IN SOLID NITROGEN WITH VACUUM-ULTRAVIOLET LIGHT. Astrophys. J. 2012, 746, 175. [Google Scholar] [CrossRef]
- Bossa, J.-B.; Paardekooper, D.M.; Isokoski, K.; Linnartz, H. Methane ice photochemistry and kinetic study using laser desorption time-of-flight mass spectrometry at 20 K. Phys. Chem. Chem. Phys. 2015, 17, 17346–17354. [Google Scholar] [CrossRef]
- Dupuy, R.; Bertin, M.; Féraud, G.; Michaut, X.; Jeseck, P.; Doronin, M.; Philippe, L.; Romanzin, C.; Fillion, J.-H. Spectrally-resolved UV photodesorption of CH4in pure and layered ices. Astron. Astrophys. 2017, 603, A61. [Google Scholar] [CrossRef]
- Krim, L.; Jonusas, M. VUV Photolysis of CH4–H2O mixture in methane-rich ices: Formation of large complex organic molecules in astronomical environments. Low Temp. Phys. 2019, 45, 606–614. [Google Scholar] [CrossRef]
- Scott, T.L.; Carpenter, J.M.; Miller, M.E. The development of solid methane neutron moderators at the intense pulsed neutron source facility of Argonne National Laboratory Preprint ANL/IPNS/CP-98533 (1999).
- Carpenter, J.M. Thermally activated release of stored chemical energy in cryogenic media. Nature 1987, 330, 358–360. [Google Scholar] [CrossRef]
- Kulagin, E.; Kulikov, S.; Melikhov, V.; Shabalin, E. Radiation effects in cold moderator materials: Experimental study of accumulation and release of chemical energy. Nucl. Instruments Methods Phys. Res. Sect. B: Beam Interactions Mater. Atoms 2004, 215, 181–186. [Google Scholar] [CrossRef]
- Feng, Q.; Kawai, T.; Zhong, B.; Wei, J.; Loong, C.-K.; Liang, T. The Solution of Cold Neutron Source using Solid Methane Moderator for the CPHS. Phys. Procedia 2012, 26, 49–54. [Google Scholar] [CrossRef]
- Kirichek, O.; Lawson, C.R.; Jenkins, D.M.; Ridley, C.J.T.; Haynes, D.J. Solid methane in neutron radiation: Cryogenic moderators and cometary cryovolcanism. Cryogenics 2017, 88, 101–105. [Google Scholar] [CrossRef]
- Kirichek, O.; Savchenko, E.; Lawson, C.; Khyzhniy, I.; Jenkins, D.; Uyutnov, S.; Bludov, M.; Haynes, D. Recombination of radiation defects in solid methane: neutron sources and cryo-volcanism on celestial bodies. 2018, 969, 012006. Journal of Physics: Conference Series 2018, 969, 012006. [Google Scholar] [CrossRef]
- Kirichek, O.; Lawson, C.; Draper, G.; Jenkins, D.; Haynes, D.; Lilley, S. Solid methane moderators: Thermodynamics and chemistry. J. Neutron Res. 2020, 22, 281–286. [Google Scholar] [CrossRef]
- Khizhny, I.V.; Uyutnov, S.A.; Bludov, M.A.; Savchenko, E.V. Explosive desorption of solid methane particles induced by an electron beam. Low Temp. Phys. 2018, 44, 1223–1225. [Google Scholar] [CrossRef]
- Bludov, M.; Khyzhniy, I.; Savchenko, E.; Sugakov, V.; Uyutnov, S. Self-oscillations in solid methane irradiated by electrons. Nucl. Phys. At. Energy 2020, 21, 312–322. [Google Scholar] [CrossRef]
- Sugakov, V.I. Lectures in Synergetics, World Scientific Series on Nonlinear Science; World Scientific: Singapore, 1998; Volume 33. [Google Scholar]
- Brown, W.; Foti, G.; Lanzerotti, L.; Bower, J.; Johnson, R. Delayed emission of hydrogen from ion bombardment of solid methane. Nucl. Instruments Methods Phys. Res. Sect. B: Beam Interactions Mater. Atoms 1987, 19-20, 899–902. [Google Scholar] [CrossRef]
- Kaiser, R.I.; Eich, G.; Gabrysch, A.; Roessler, K. Theoretical and laboratory studies on the interaction of cosmic-ray particles with interstellar ices. II. Formation of atomic and molecular hydrogen in frozen organic molecules. Astrophys. J. 1997, 484, 487–498. [Google Scholar] [CrossRef]
- d’Hendecourt, L.; Allamandola, L.; Baas, F.; Greenberg, J. Interstellar grain explosions - Molecule cycling between gas and dust. Astronomy & Astrophysics 1982, 109, L12–L14. [Google Scholar]
- Roberts, J.F.; Rawlings, J.M.C.; Viti, S.; Williams, D.A. Desorption from interstellar ices. Mon. Not. R. Astron. Soc. 2007, 382, 733–742. [Google Scholar] [CrossRef]
- Vasyunin, A.I.; Herbst, E. Reactive desorption and radiative association as possible drivers of complex molecule formation in the cold interstellar medium. Astrophys. J. 2013, 769. [Google Scholar] [CrossRef]
- Wakelam, V.; Dartois, E.; Chabot, M.; Spezzano, S.; Navarro-Almaida, D.; Loison, J.-C.; Fuente, A. Efficiency of non-thermal desorptions in cold-core conditions: Testing the sputtering of grain mantles induced by cosmic rays. Astronomy & Astrophysics 2021, 652, A63. [Google Scholar]
- Zhu, C.; Bergantini, A.; Singh, S.K.; Abplanalp, M.J.; Kaiser, R.I. Rapid Radical–Radical Induced Explosive Desorption of Ice-coated Interstellar Nanoparticles. Astrophys. J. 2021, 920, 73. [Google Scholar] [CrossRef]
- Khyzhniy, I.V.; Uyutnov, S.A.; Bludov, M.A.; Savchenko, E.V.; Bondybey, V.E. Electron-induced delayed desorption of solid argon doped with methane. Low Temp. Phys. 2019, 45, 721–726. [Google Scholar] [CrossRef]
- Savchenko, E.; Khyzhniy, I.; Uyutnov, S.; Bludov, M.; Bondybey, V. Explosive desorption from solid methane and methane-containing argon matrices. Nucl. Instruments Methods Phys. Res. Sect. B: Beam Interactions Mater. Atoms 2020, 469, 37–41. [Google Scholar] [CrossRef]
- Savchenko, E.; Khyzhniy, I.; Uyutnov, S.; Bludov, M.; Bondybey, V. Nonstationary processes in matrix-isolated methane probed by optical and current emission spectroscopy. J. Mol. Struct. 2020, 1221, 128803. [Google Scholar] [CrossRef]
- Eberlein, J.; Creuzburg, M. Dissociation of CH4 in a krypton matrix: VUV spectra of atomic C and of the radical CH. Mol. Phys. 1999, 96, 451–456. [Google Scholar] [CrossRef]
- Wu, Y.-J.; Chen, H.-F.; Camacho, C.; Witek, H.A.; Hsu, S.-C.; Lin, M.-Y.; Chou, S.-L.; Ogilvie, J.F.; Cheng, B.-M. Formation and identification of interstellar molecule linear C5H from photolysis of methane dispersed in solid neon. Astrophys. J. 2009, 701, 8–11. [Google Scholar] [CrossRef]
- Lin, M.-Y.; Lo, J.-I.; Lu, H.-C.; Chou, S.-L.; Peng, Y.-C.; Cheng, B.-M.; Ogilvie, J.F. Vacuum-Ultraviolet Photolysis of Methane at 3 K: Synthesis of Carbon Clusters up to C20. J. Phys. Chem. A 2014, 118, 3438–3449. [Google Scholar] [CrossRef] [PubMed]
- Savchenko, E.; Khyzhniy, I.; Uyutnov, S.; Bludov, M.; Bondybey, V. Explosive desorption induced by radical–radical interaction in methane-doped Ar matrices. Nucl. Instruments Methods Phys. Res. Sect. B: Beam Interactions Mater. Atoms 2023, 536, 113–118. [Google Scholar] [CrossRef]
- Sharp, T. Potential-energy curves for molecular hydrogen and its ions. At. Data Nucl. Data Tables 1970, 2, 119–169. [Google Scholar] [CrossRef]
- Song, K.S.; Williams, R.T. Self-Trapped Excitons; Springer-Verlag: Berlin, Germany, 1996. [Google Scholar]
- Smirnov, B.M.; Yatsenko, A.S. Properties of dimers. Phys. Usp 1996, 39, 211–230. [Google Scholar] [CrossRef]
- Heays, A.N.; Bosman, A.D.; van Dishoeck, E.F. Photodissociation and photoionisation of atoms and molecules of astrophysical interest. Astron. Astrophys. 2017, 602, A105. [Google Scholar] [CrossRef]
- Linstrom, P.J.; Mallard, W.G. NIST Chemistry WebBook, NIST Standard Reference Database Number 69; National Institute of Standards and Testing (NIST): Gaithersburg, MD, USA, 2013. [Google Scholar] [CrossRef]
- Ismail, A.F.; Khulbe, K.C.; Matsuura, T. Gas Separation Membranes: Polymeric and Inorganic; Springer, 2015. [Google Scholar]
- Hallam, H.E. Vibrational Spectroscopy of Trapped Species; John Wiley & Sons: London, UK, 1973. [Google Scholar]
- Chamberland, A.; Belzile, R.; Cabana, A. Infrared spectra and structure of methane – noble gas mixed crystals: the influence of temperature and methane concentration on the v3 vibration band of methane. Can. J. Chem. 1970, 48, 1129–1139. [Google Scholar] [CrossRef]
- Ferradini, C.; Jay-Gerin, J.-P. Excess Electrons in Dielectric Media; CRC Press: Boca Raton, Ann Arbor, Boston, London, 1991. [Google Scholar]
- Lo, J.-I.; Lin, M.-Y.; Peng, Y.-C.; Chou, S.-L.; Lu, H.-C.; Cheng, B.-M.; Ogilvie, J.F. Far-ultraviolet photolysis of solid methane. Mon. Not. R. Astron. Soc. 2015, 451, 159–166. [Google Scholar] [CrossRef]
- Kraas, M.; Gürtler, P. Emission and excitation spectra of rare gas hydride trimers in rare gas matrices. Chem. Phys. Lett. 1990, 174, 396–400. [Google Scholar] [CrossRef]
- Doronin, Y.S.; Vakula, V.L.; Kamarchuk, G.V.; Tkachenko, A.A.; Khyzhniy, I.V.; Uyutnov, S.A.; Bludov, M.A.; Savchenko, E.V. Desorption of excited H* atoms from free clusters Ar/CH4 and solid Ar doped with CH4. Low Temp. Phys. 2021, 47, 1058–1064. [Google Scholar] [CrossRef]
- Zimmerer, G. Electronic sputtering from rare-gas solids. Nucl. Instruments Methods Phys. Res. Sect. B: Beam Interactions Mater. Atoms 1994, 91, 601–613. [Google Scholar] [CrossRef]
- Gumenchuk, G.B.; Khyzhniy, I.V.; Bludov, M.A.; Uyutnov, S.A.; Belov, A.G.; Savchenko, E.V.; Ponomaryov, A.N.; Bondybey, V.E. Optically stimulated desorption of “hot” excimers from pre-irradiated Ar solids. Low Temp. Phys. 2008, 34, 241–244. [Google Scholar] [CrossRef]
- Ogurtsov, A.; Savchenko, E.; Becker, J.; Runne, M.; Zimmerer, G. Radiative relaxation of optically generated intrinsic charged centers in solid Ar. J. Lumin- 1998, 76-77, 478–481. [Google Scholar] [CrossRef]
- Lavrov, B.P.; Melnikov, A.S.; Käning, M.; Röpcke, J. uv continuum emission and diagnostics of hydrogen-containing nonequilibrium plasmas. Phys. Rev. E 1999, 59, 3526–3543. [Google Scholar] [CrossRef]
- Coletti, F.; Debever, J.; Zimmerer, G. Electron stimulated desorption of solid argon via exciton creation. J. de Phys. Lettres 1984, 45, 467–473. [Google Scholar] [CrossRef]
- Fugol', I. Free and self-trapped excitons in cryocrystals: kinetics and relaxation processes. Adv. Phys. 1988, 37, 1–35. [Google Scholar] [CrossRef]
- Beyer, M.; Lammers, A.; Savchenko, E.V.; Niedner-Schatteburg, G.; Bondybey, V.E. Proton solvated by noble-gas atoms: simplest case of a solvated ion. Phys. Chem. Chem. Phys. 1999, 1, 2213–2221. [Google Scholar] [CrossRef]
- IdBarkach, T.; Chabot, M.; Béroff, K.; Della Negra, S.; Lesrel, J.; Geslin, F.; Le Padellec, A.; Mahajan, T.; Díaz-Tendero, S. Breakdown curves of CH2(+), CH3(+), and CH4(+) molecules I. Construction and application to electron collisions and UV photodissociation. Astron. Astrophys. 2019, 628, A75. [Google Scholar] [CrossRef]
- Eberlein, J.; Creuzburg, M. Mobility of atomic hydrogen in solid krypton and xenon. J. Chem. Phys. 1997, 106, 2188–2194. [Google Scholar] [CrossRef]
- Vaskonen, K.; Eloranta, J.; Kiljunen, T.; Kunttu, H. Thermal mobility of atomic hydrogen in solid argon and krypton matrices. J. Chem. Phys. 1999, 110, 2122–2128. [Google Scholar] [CrossRef]
- Kiljunen, T.; Eloranta, J.; Kunttu, H. Ab initio and molecular-dynamics studies on rare gas hydrides: Potential-energy curves, isotropic hyperfine properties, and matrix cage trapping of atomic hydrogen. J. Chem. Phys. 1999, 110, 11814–11822. [Google Scholar] [CrossRef]
- Feldman, V.I.; Sukhov, F.F.; Orlov, A.Y. Hydrogen atoms in solid xenon: Trapping site structure, distribution, and stability as revealed by EPR studies in monoisotopic and isotopically enriched xenon matrices. J. Chem. Phys. 2008, 128, 214511. [Google Scholar] [CrossRef] [PubMed]
- Ozerov, G.K.; Bezrukov, D.S.; Buchachenko, A.A. Computational study of the stable atomic trapping sites in Ar lattice. Low Temp. Phys. 2019, 45, 301–309. [Google Scholar] [CrossRef]
- Sheludiakov, S.; Ahokas, J.; Järvinen, J.; Lehtonen, L.; Vasiliev, S.; Dmitriev, Y.A.; Lee, D.M.; Khmelenko, V.V. Electron spin resonance study of atomic hydrogen stabilized in solid neon below 1 K. Phys. Rev. B 2018, 97, 104108. [Google Scholar] [CrossRef]
- Sawada, K.; Fujimoto, T. Effective ionization and dissociation rate coefficients of molecular hydrogen in plasma. J. Appl. Phys. 1995, 78, 2913–2924. [Google Scholar] [CrossRef]
- Massey, H. Negative Ion. In Negative Ions; Cambridge University Press: Cambridge, 1976. [Google Scholar]
- Chang, Y.; Yang, J.; Chen, Z.; Zhang, Z.; Yu, Y.; Li, Q.; He, Z.; Zhang, W.; Wu, G.; Ingle, R.A.; et al. Ultraviolet photochemistry of ethane: implications for the atmospheric chemistry of the gas giants. Chem. Sci. 2020, 11, 5089–5097. [Google Scholar] [CrossRef]
- Gao, H. Molecular photodissociation in the vacuum ultraviolet region: implications for astrochemistry and planetary atmospheric chemistry. Mol. Phys. 2020, 119. [Google Scholar] [CrossRef]
Figure 1.
Comparison of the most intense emission band of Ar matrix (curve 2) with the absorption cross-section of gaseous CH4 (curve 1) adopted from [63].
Figure 1.
Comparison of the most intense emission band of Ar matrix (curve 2) with the absorption cross-section of gaseous CH4 (curve 1) adopted from [63].
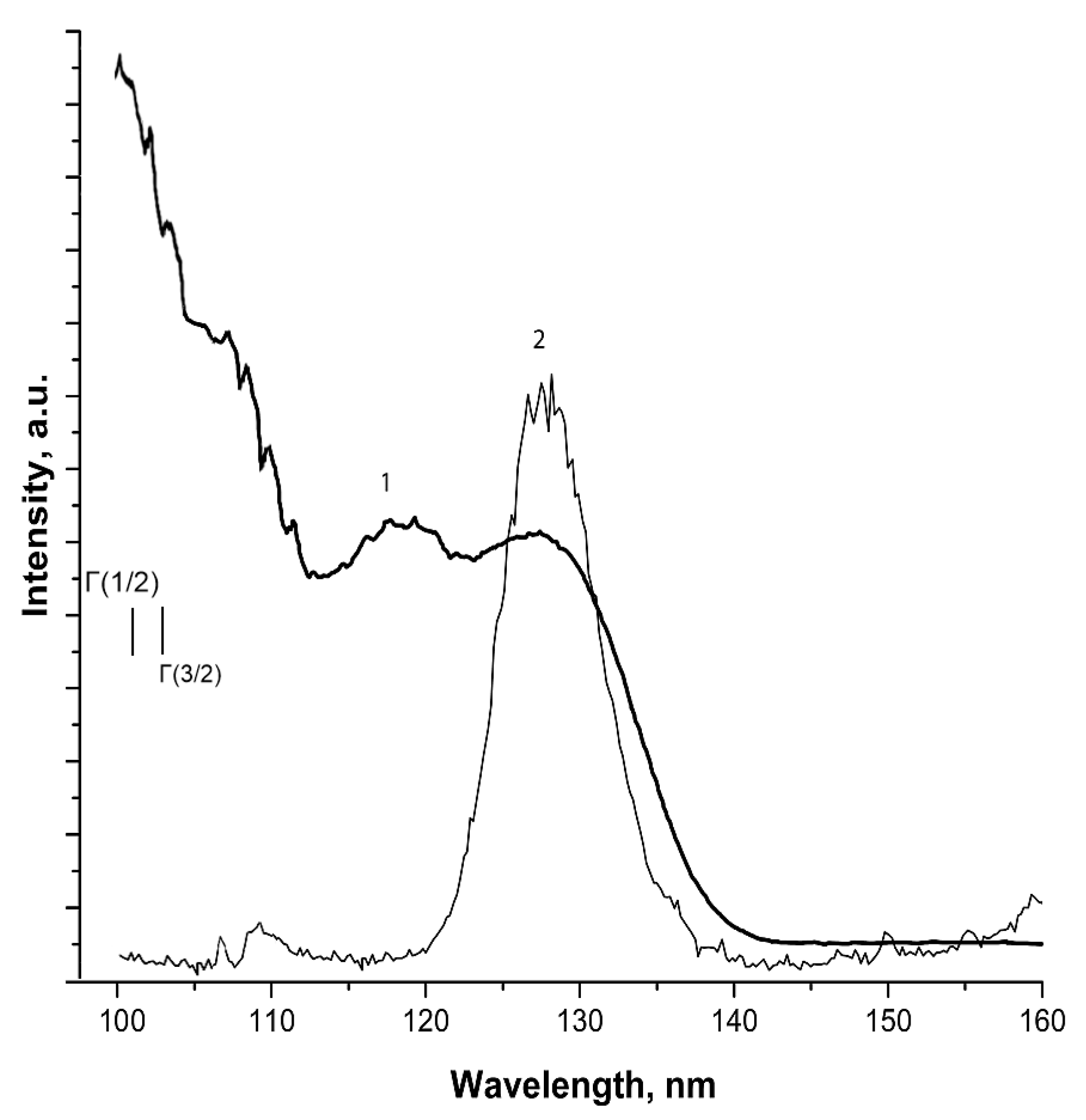
Figure 2.
TSEE yields recorded from pre-irradiated CH4, CH4-doped Ar matrices and pure Ar.
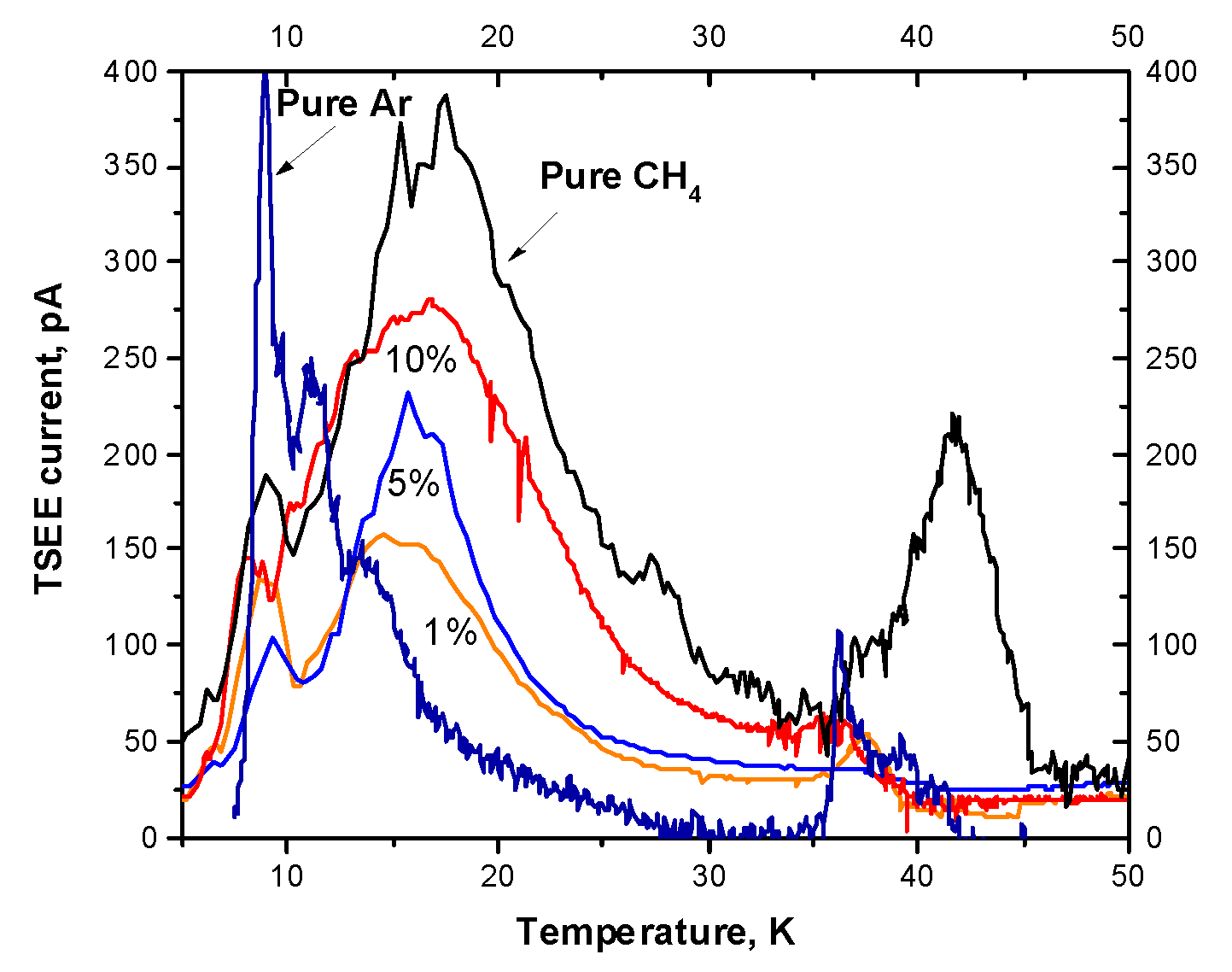
Figure 3.
CL spectrum of solid Ar doped with 1% CH4 taken upon irradiation at 5 K.
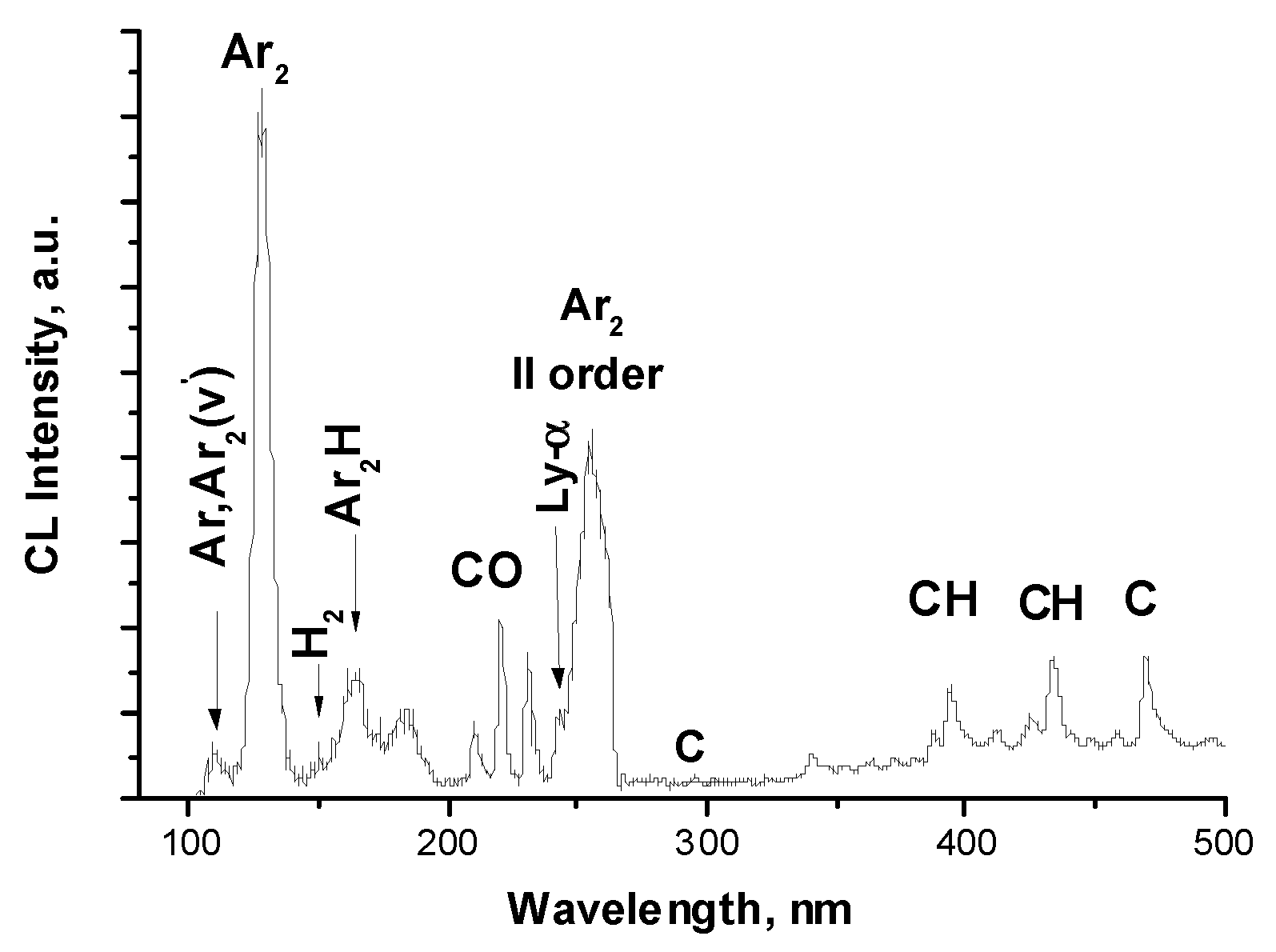
Figure 4.
Intensity of the M-band in pure Ar (first black column) and in CH4-doped Ar matrices.
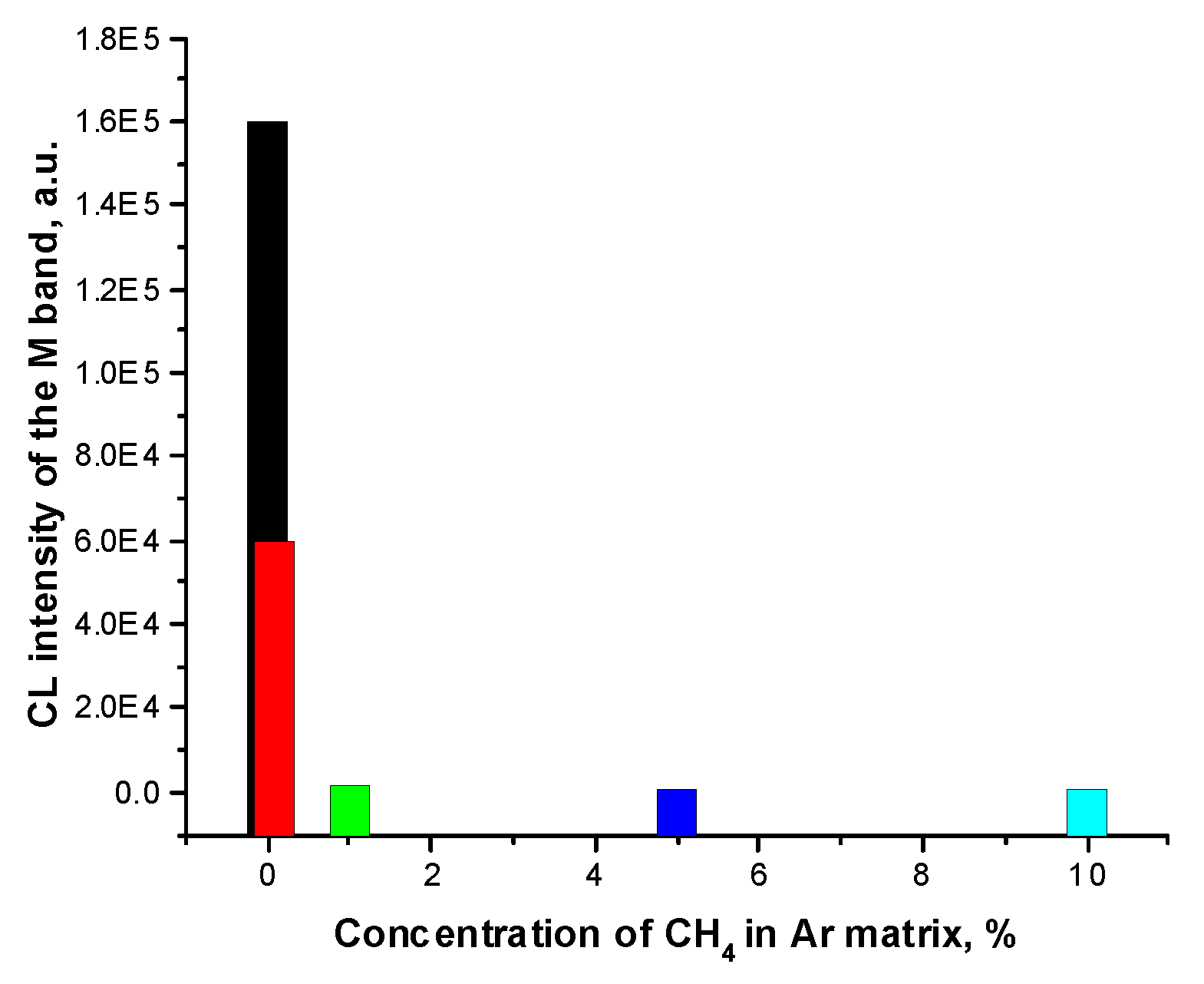
Figure 5.
The relative intensity of the Ar2H* (H) emission band.
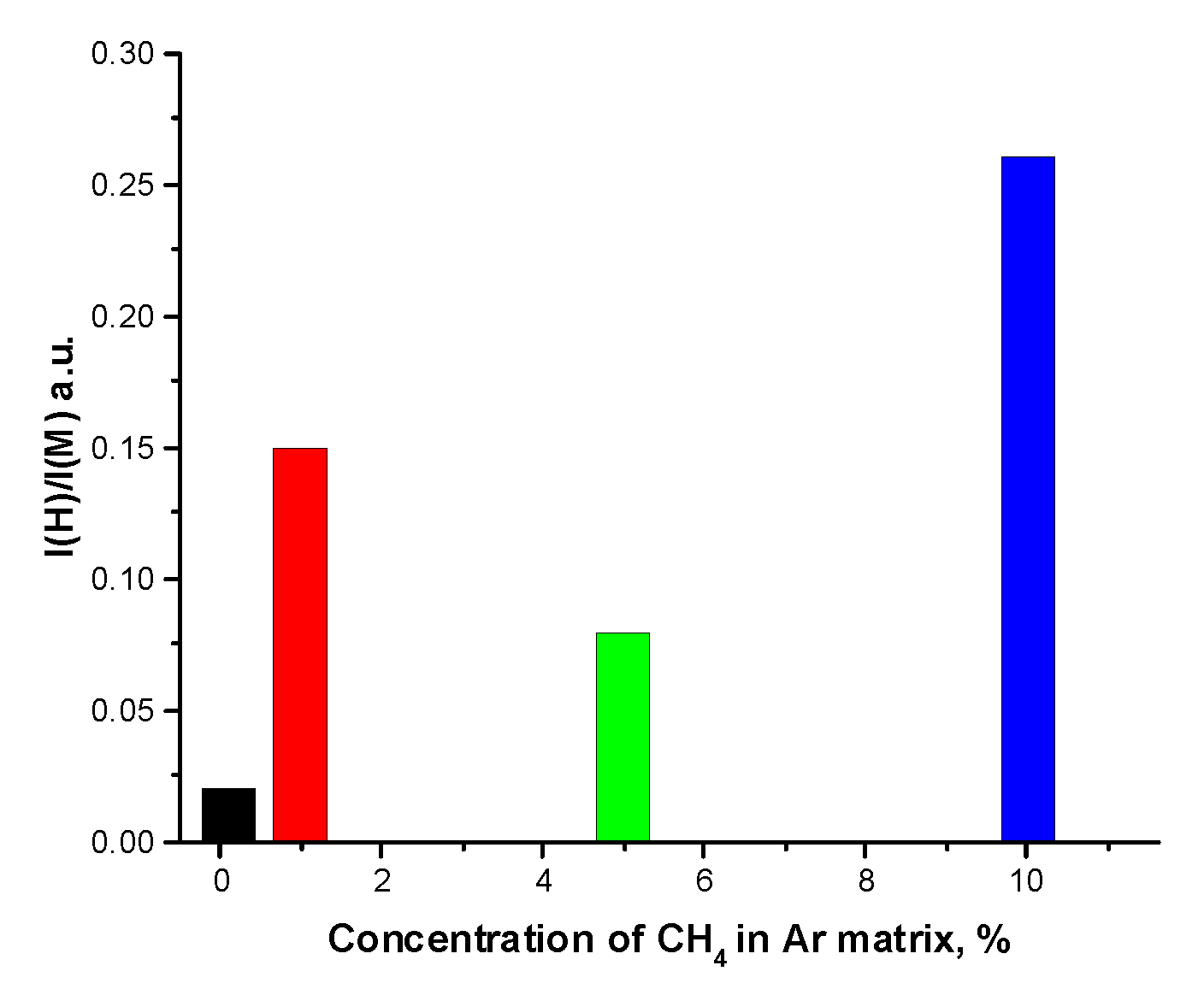
Figure 6.
Dose dependencies of optical emission of H atoms and CH radicals taken from highly doped Ar matrix (C of CH4=10%) upon irradiation with an electron beam. The Figure also shows the dose dependences of the pressure and temperature of the sample measured in a correlated way with optical emissions.
Figure 6.
Dose dependencies of optical emission of H atoms and CH radicals taken from highly doped Ar matrix (C of CH4=10%) upon irradiation with an electron beam. The Figure also shows the dose dependences of the pressure and temperature of the sample measured in a correlated way with optical emissions.
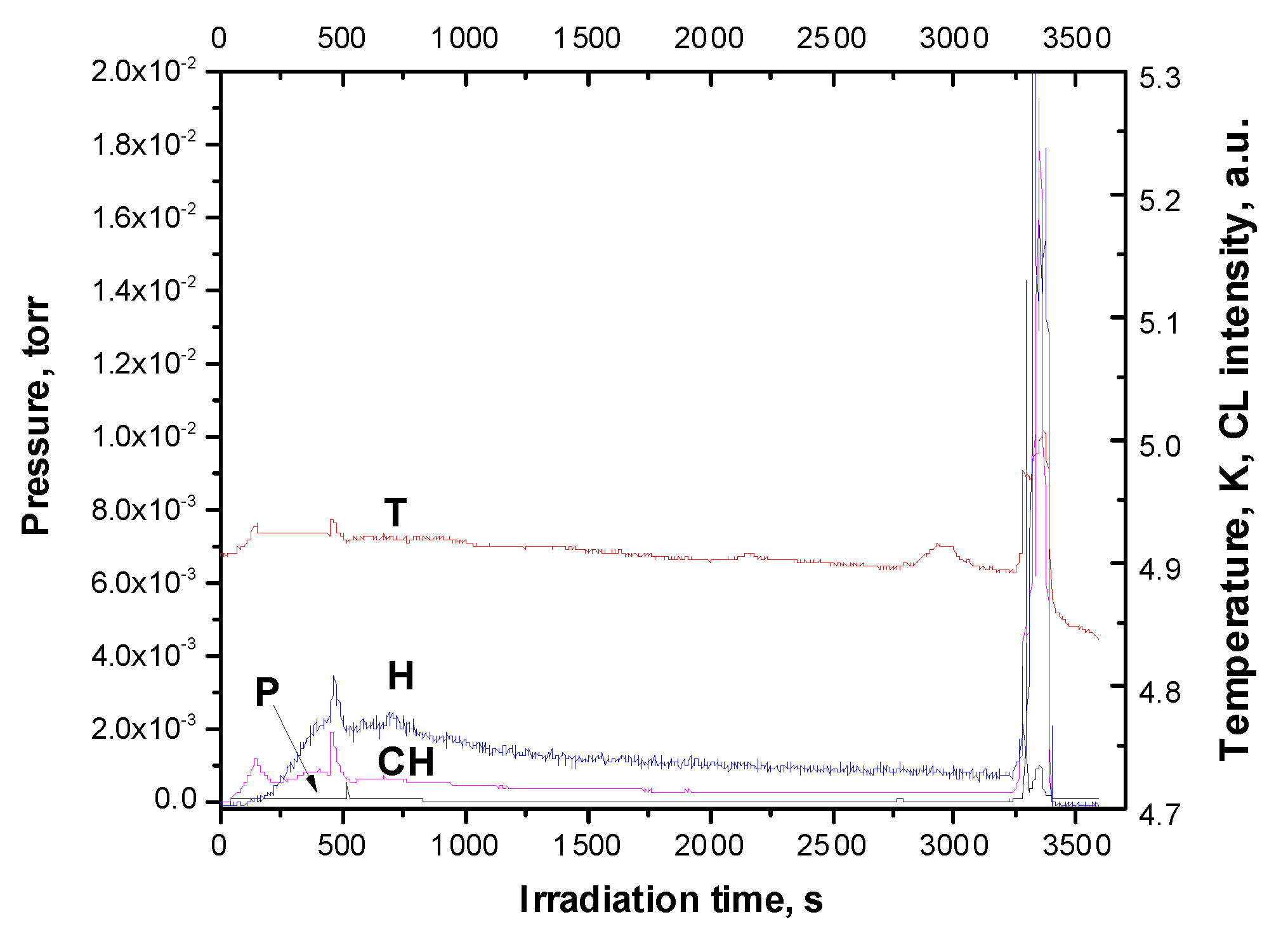
Figure 7.
Yields of optical emissions in the range of pressure burst: (a) Yield of the H band; (b) Yield of the CH band. Both yields are recorded simultaneously with pressure and temperature.
Figure 7.
Yields of optical emissions in the range of pressure burst: (a) Yield of the H band; (b) Yield of the CH band. Both yields are recorded simultaneously with pressure and temperature.
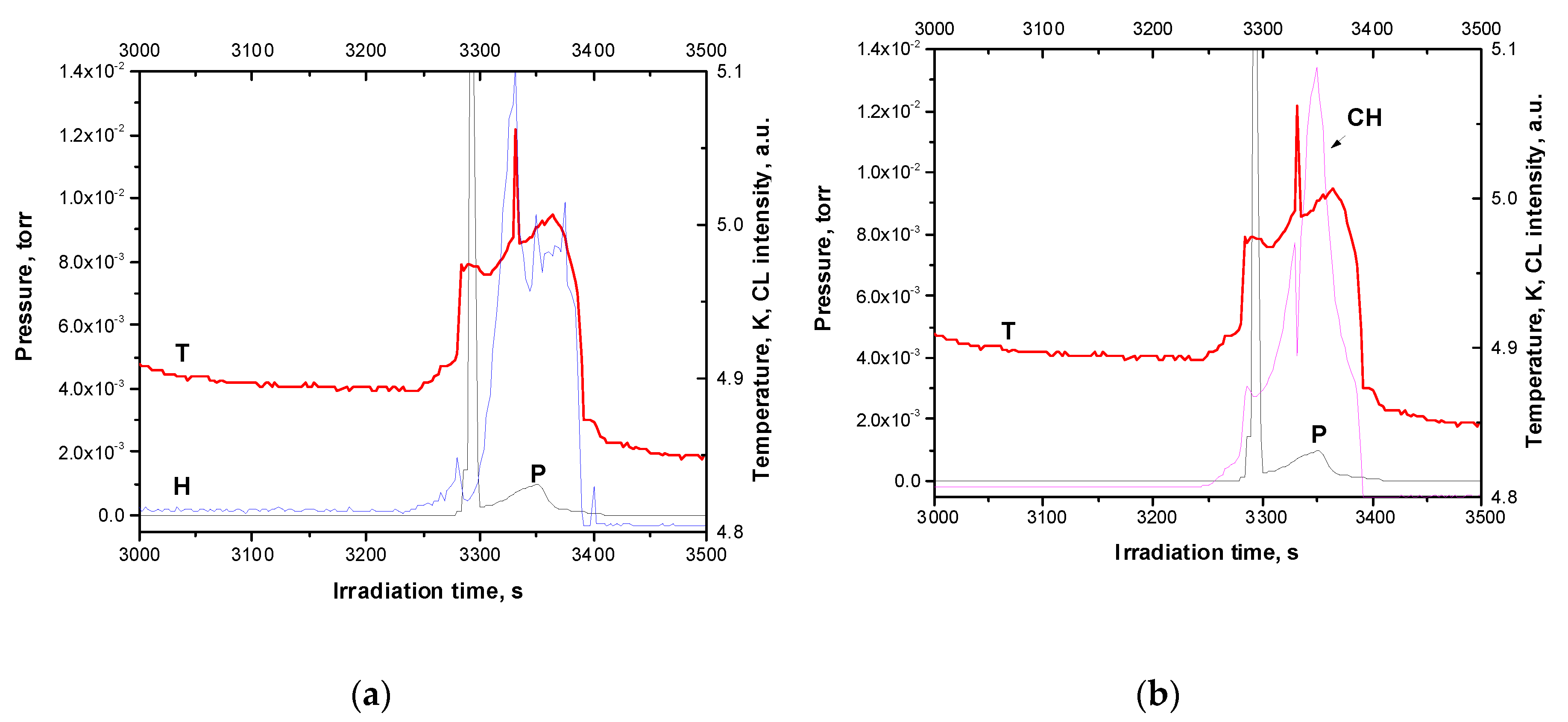
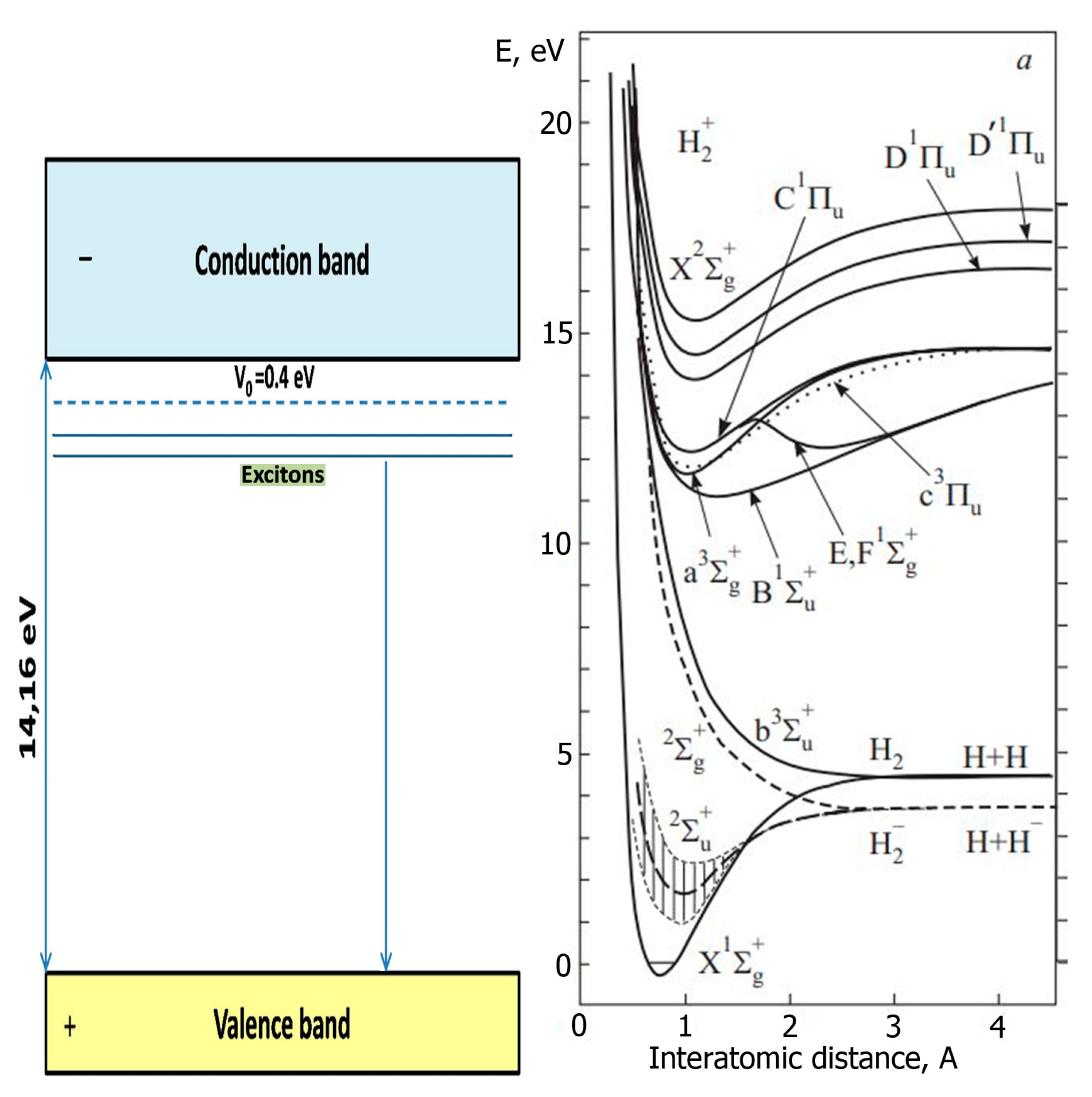
Figure 9.
Dose dependence of the H band detected in a correlated manner with the total yield of particle (P) from lightly doped Ar matrix (C=1% of CH4). Irradiation was performed with a 1.5 keV electron beam at current density of 3 mAcm-1.
Figure 9.
Dose dependence of the H band detected in a correlated manner with the total yield of particle (P) from lightly doped Ar matrix (C=1% of CH4). Irradiation was performed with a 1.5 keV electron beam at current density of 3 mAcm-1.
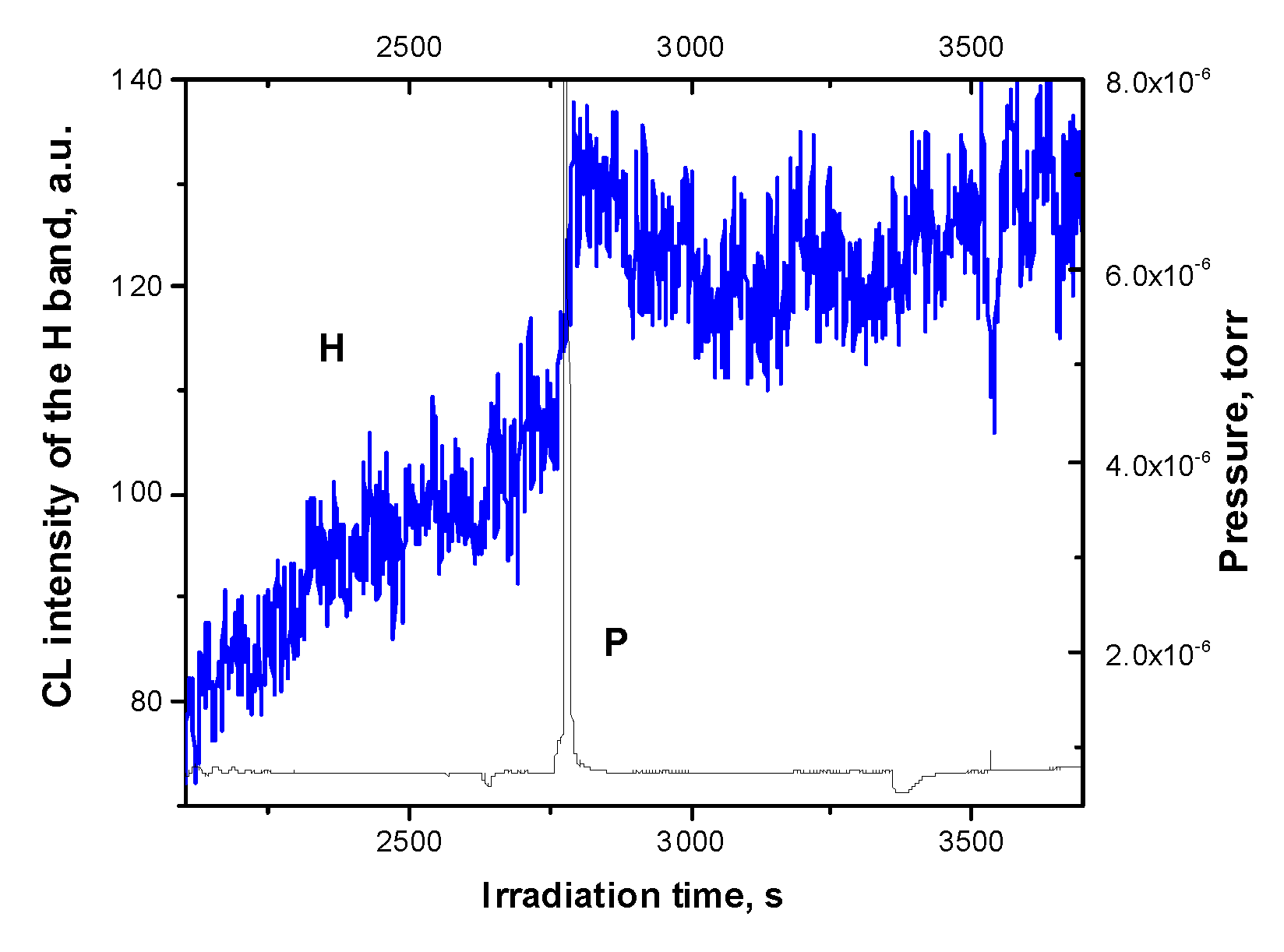

Table 1.
Ionization potentials and electron affinities of methane and products of its transformation.
Table 1.
Ionization potentials and electron affinities of methane and products of its transformation.
| Species | Ionization potential, eV | Electron affinity, eV |
|---|---|---|
| CH4 | 12.61 | unstable |
| CH3 | 9.84 | +0.09 |
| CH2 | 10.35 | +0.65 |
| CH | 10.64 | +1.24 |
| H | 13.59 | +0.75 |
| H2 | 15.43 | unstable |
| C | 11.26 | 1.26 |
1 The data are presented according to [64].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



