Submitted:
07 August 2023
Posted:
08 August 2023
You are already at the latest version
Abstract
This paper presents ConF, a novel deep learning model designed for accurate and efficient prediction of non-coding RNA families. NcRNAs are essential functional RNA molecules involved in various cellular processes, including replication, transcription, and gene expression. Identifying ncRNA families is crucial for comprehensive RNA research, as ncRNAs within the same family often exhibit similar functionalities. Traditional experimental methods for identifying ncRNA fam-ilies are time-consuming and labor-intensive. Computational approaches relying on annotated secondary structure data face limitations in handling complex structures like pseudoknots and have restricted applicability, resulting in suboptimal prediction performance. To overcome these chal-lenges, ConF integrates mainstream techniques such as residual networks with dilated convolutions and cross multi-head attention mechanisms. By employing a combination of dual-layer convolu-tional networks and BiLSTM, ConF effectively captures intricate features embedded within RNA sequences. This feature extraction process leads to significantly improved prediction accuracy compared to existing methods. Experimental evaluations conducted on a ten-fold publicly available dataset demonstrate the superiority of ConF in terms of accuracy, sensitivity, and other perfor-mance metrics. Overall, ConF represents a promising solution for accurate and efficient ncRNA family prediction, addressing the limitations of traditional experimental and computational methods.
Keywords:
non-coding RNA
; deep learning
; Gene expression
1. Introduction
RNA is a biopolymer composed of four nucleotides: adenine (A), uracil (U), gua-nine (G), and cytosine (C) [1]. Functionally, RNA can be categorized into coding RNA and non-coding RNA (ncRNA). While ncRNAs are derived from ncRNA genes, they do not encode proteins [2]. Nevertheless, they play significant roles in various cellular processes [3] and diseases [4] through mechanisms such as replication, transcription, and gene expression [5,6]. Extensive transcriptomics and bioinformatics studies have identified thousands of ncRNAs in humans, classified based on their functionality and length. Examples of ncRNA categories include microRNA, ribosomal RNA (rRNA), ri-bozymes, small nuclear RNA (snRNA) including small nucleolar RNA (snoRNA), transfer RNA (tRNA), Intron_RNA, internal ribosome entry site (IRES), Leader, and riboswitch. These ncRNAs exert crucial functions in organisms. For instance, snRNA processes heteronuclear RNA within the cell nucleus, regulates transcription factors, and maintains telomeres [7]. Ribozymes, serving as RNA enzymes in organs, facilitate the connection of amino acids during protein synthesis. tRNA acts as a physical bridge between messenger RNA (mRNA) and amino acid sequences [8]. Intron_RNA, tran-scribed from intron genes, engages in extensive internal interactions post-RNA tran-scription and aids in the proper ordering of exons [9,10]. IRES facilitates the binding of the ribosome to mRNA, initiating protein translation and synthesis [11]. The Leader represents the upstream portion of the start codon in mRNA and assumes an im-portant role in regulating mRNA transcription [12]. Riboswitches are regulatory seg-ments within mRNA that can adopt specific conformations to modulate mRNA tran-scription processes [13]. Consequently, ncRNAs hold a critical position in organisms and represent indispensable constituents in intricate biological activities.
Most notably, the majority of RNA in higher organisms is non-coding RNA (ncRNA) that lacks protein-coding capacity [14]. While ncRNA was once considered to be a byproduct of RNA polymerase transcription without any biological function [15], an increasing body of research has demonstrated that ncRNA participates in a wide range of intracellular biological processes and plays a critical regulatory role in organ-ismal growth, development, and apoptosis [16,17]. Furthermore, ncRNA has been found to be closely associated with a variety of complex human diseases [18,19]. As such, research into the complex and important functions of ncRNA has become a cru-cial component in unraveling the mysteries of life [20]. Regrettably, the instability and diversity of ncRNA present significant challenges to the study of its function. However, studies have indicated that ncRNAs from the same family exhibit similar functions [21], suggesting that identifying their families can provide preliminary insights into the function of ncRNAs and guide further experimental validation of their functions.
Currently, there are two main categories of methods for identifying ncRNAs: ex-perimental-based methods and computational-based methods. Each method has its principles, advantages, and disadvantages, which are discussed below.
1.1. Tranditional Experiment-based approach
The first experimental-based method involves using chemical or enzyme reagents for ncRNA sequencing, where classification and identification are based on the size of ncRNAs [22]. This method is relatively simple and independent of ncRNA structure, as it does not require reverse transcription of cDNA. However, it relies on gel electrophoresis, which requires a sufficient abundance of the target ncRNA for visible bands to form on the gel. Hence, it is less effective for ncRNAs with low abundance. The second method involves generating cDNA libraries through reverse transcription to identify ncRNAs. This method allows for the creation of specific cDNA libraries tailored to identify particular functional categories of ncRNAs. However, the efficiency of reverse transcription can be affected by the structure and modifications of ncRNAs, leading to incomplete reverse transcription and the inability to identify all ncRNAs from specific families in the library. Base loss during reverse transcription can also impact identification performance. Microarray analysis is the third method used to identify ncRNAs by probing their binding. This approach enables the rapid and simultaneous identification of multiple types of ncRNAs, even at lower concentrations. It has become a widely used method in transcription detection in research. However, the preparation of sample ncRNAs and microarrays with probes can be challenging. The fourth method involves using the SELEX technique [23], where ncRNAs are identified by forming ribonucleoprotein particles with specific proteins. This technique can generate ncRNAs from all genes in an organism, regardless of their abundance in the cell. However, it involves complex and time-consuming procedures.
These experimental-based methods share common disadvantages, including complexity, high costs, and limitations in meeting the demands of high-throughput ncRNA identification.
1.2. Machine learning-based approach
Owing to the industry's pressing need for efficient and expeditious ncRNA recognition, computational methods have come to the fore. These computational approaches primarily encompass two principal categories. The first method is based on sequence alignment. Infernal is a typical method based on sequence alignment [24]. It first uses secondary structure data to annotate the consistency of ncRNA sequences within the same family. Then, it builds covariance models (CM) based on Stochastic Context-Free Grammars (SCFGs) using the annotated sequence data. Finally, these covariance models are utilized to accurately identify ncRNA families. The second method is based on structural features, primarily leveraging the conservation principle of secondary structures in the same ncRNA family for identification. This type of method starts by using RNA secondary structure prediction tools such as mfold [25] and Ipknot [26] to predict the secondary structure. Then, algorithms are designed to learn the structural features for ncRNA identification. GraPPLE [27], RNAcon [28], nRC [29], and ncRFP [30] are representative methods in this field. Among them, GraPPLE utilizes global graph features of ncRNA secondary structures and designs an SVM method. RNAcon extracts 20 types of secondary structure graph features and employs a random forest method. nRC uses the Moss method [31] and one-hot encoding of ncRNA structural features, followed by a deep learning model based on convolutional neural networks. ncRFP simplifies the process by automatically extracting features from ncRNA sequences for predicting ncRNA families. Although these models can predict ncRNAs, there is still room for improvement in terms of accuracy and other metrics.
In addition, the Transformer model has gained widespread recognition as a highly influential deep learning algorithm [32]. It has attracted significant attention in the field of natural language processing in recent years. The introduction of the Transformer model has addressed the limitations of conventional Seq2Seq models and has demonstrated remarkable performance in various tasks such as machine translation, text summarization, and dialogue generation. By introducing multi-head self-attention mechanisms, the Transformer model allows for parallelized training, enabling efficient processing of input sequences and capturing the sequential relationships among words, thus improving overall accuracy. This has resulted in rapid expansion of Transformer-based algorithms across diverse domains including computer vision and bioinformatics. For instance, in computer vision, the Visual Transformer (VIT) algorithm has successfully applied the Transformer model to achieve state-of-the-art performance in image classification tasks [33], thereby showcasing the exceptional robustness of the Transformer model. In the field of bioinformatics, the AlphaFold [34] model, a deep learning-based protein structure prediction model, has leveraged various neural network structures, including the Transformer, to deliver outstanding results. Furthermore, Transformer-based algorithms have also demonstrated promising outcomes in tasks such as RNA secondary structure prediction and drug molecule screening and design, showcasing their efficacy in these domains.
This study specifically investigates the potential of utilizing the Transformer model for extracting RNA sequence features within the domains of bioinformatics and drug molecule design. Augmenting the performance of our model in this study entails capitalizing on the inherent capabilities of the attention mechanism and feature compression within the framework of the Transformer model. This study presents a novel deep learning-based approach for classifying non-coding RNA families. The proposed method utilizes a k-mer technique to represent features, thereby enhancing the accuracy of RNA sequence recognition. The RNA sequences are then fed into CNN (convolutional neural network) and BiLSTM (bidirectional long short-term memory) models, enabling the extraction of structural and sequential feature relationships within the sequences. To focus on important information and adjust the weights of key details, an MLP module with an integrated attention mechanism is employed to map the features onto a new feature space. The core component of the model consists of a residual network model that incorporates multi-scale CNN modules and attention mechanism-based feature alignment. The multi-scale CNN modules are capable of capturing structural features from diverse scales, thereby providing the model with a more comprehensive understanding of RNA structural characteristics. Additionally, by utilizing the attention mechanism as a residual module, the model can retain shallow features while capturing module variances. The performance of the proposed model is evaluated using ten publicly available datasets. Experimental results demonstrate its significant advantages over alternative algorithms, underscoring its potential in the accurate prediction of non-coding RNA families.
2. Materials and Methods
In order to investigate the practical performance of the model proposed in this study, publicly available datasets comprising 13 distinct ncRNAs were employed as experimental materials. The experimental results were comprehensively compared with those of benchmark algorithms, revealing Drawing upon the unique characteristics of RNA sequences, this study proposes a novel multi-scale residual network model for the prediction of non-coding RNA families. The model incorporates Bidirectional Long Short-Term Memory (BiLSTM), attention mechanisms, and dilated convolutions to capture the inherent complexities of RNA data.To address potential errors in the RNA dataset, a 2-mer approach is employed. Additionally, a feature representation method utilizing word embeddings with an embedding dimension of 16 and a length of 224 is adopted. The BiLSTM and convolutional neural network (CNN) modules are then applied to extract initial features from both the RNA sequence and structure, effectively augmenting the input dimensionality of the model. These extracted features are concatenated to form a sequence of dimensions . To facilitate the learning of intricate and abstract representations, non-linear feature mapping and transformation are achieved through fully connected layers.
Attention mechanisms are employed to compute the disparities between preceding features and those generated by the multi-layer perceptron (MLP), enabling the model to capture abstract information while preserving the original features. Block1, a CNN module encompassing multiple scales, is designed to encompass a convolutional module with a scale of 16, as well as two dilated convolution modules featuring convolution window sizes of 10 and 18, respectively. The integration of attention mechanisms allows for the computation of differences between shallow and deep networks, with the outcomes being added to the shallow network to mitigate overfitting risks associated with excessive network depth.Downsampling is achieved through positional data reshaping, enhancing the thickness of feature representations while maintaining the integrity of the original features. Consequently, the length of the sequence is halved, with the embedding dimension doubled. Block2 inherits the same parameters as Block1 but possesses twice the number of filters. It further extracts global information from the RNA and leverages attention mechanisms to calculate disparities. Ultimately, prediction is performed through fully connected layers and the softmax activation function.
2.1. dataset
The data used in this study was obtained from the Rfam database [35]. Rfam is a comprehensive collection of RNA families, providing a valuable resource for the analysis of non-coding RNA (ncRNA) sequences. The database contains 13 distinct types of ncRNAs, encompassing microRNAs, 5S_rRNA, 5.8S_rRNA, ribozymes, CD-box, HACA-box, scaRNA, tRNA, Intron_gpI, Intron_gpII, IRES, leader, and riboswitch.
The dataset employed in this study consists of 6320 non-redundant ncRNA sequences. Among these, the IRES family comprises 320 sequences, while the remaining families each contain 500 sequences. To train the model effectively, a ten-fold cross-validation methodology was employed during the model training phase. This approach involves splitting the data for each ncRNA family into two subsets: a training set and a test set.For each fold of the cross-validation, 5688 RNA data points were allocated as the training set, while the remaining data points were designated as the test set. By repeating this process ten times, ten sets of training and test data were generated, allowing for robust evaluation and validation of the model's performance.
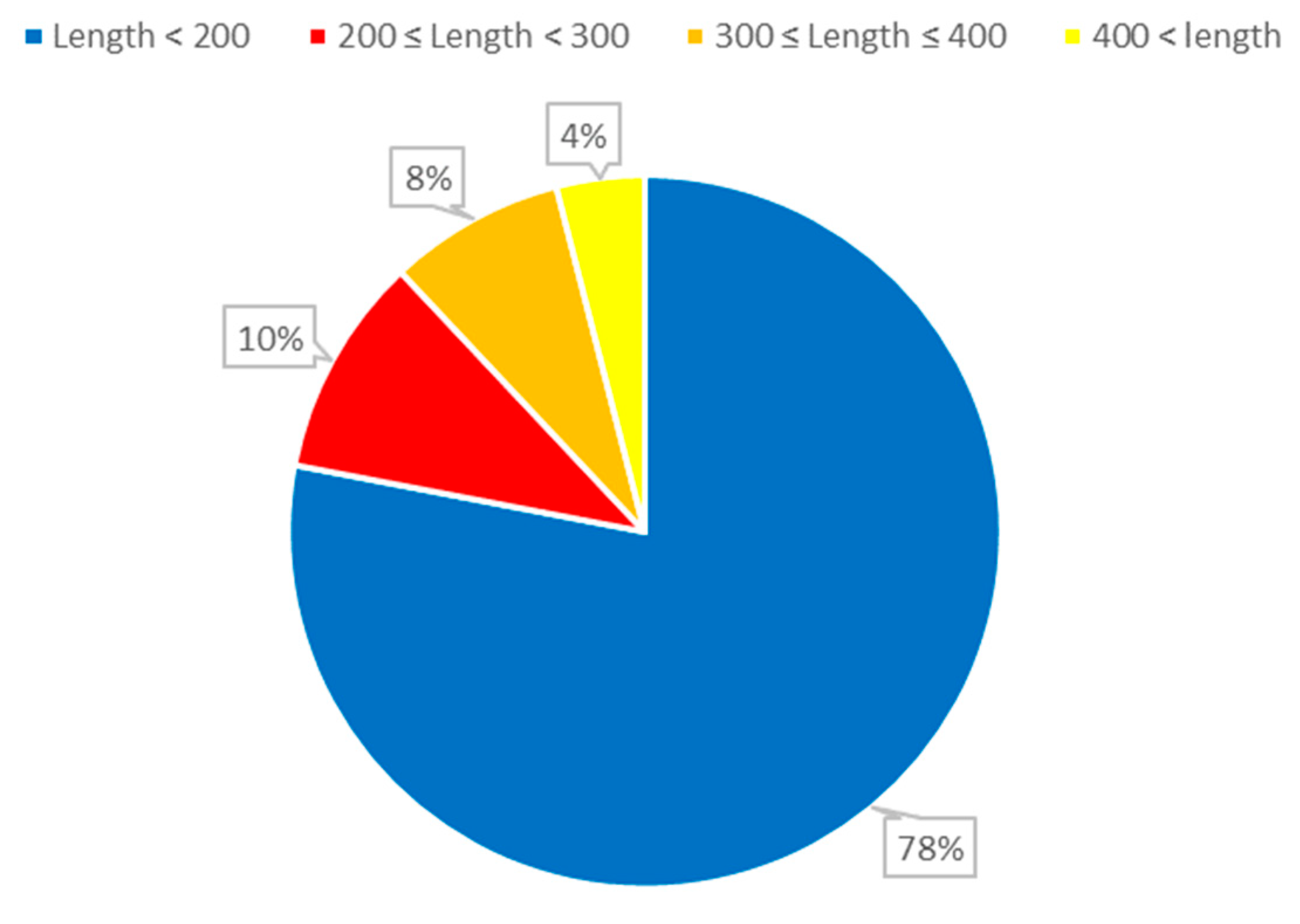
Figure 1.
The distribution of ncRNA sequence lengths. The length of RNA refers to the number of nucleotides (AUCG) present in the RNA sequence.
Figure 1.
The distribution of ncRNA sequence lengths. The length of RNA refers to the number of nucleotides (AUCG) present in the RNA sequence.
2.2. RNA Representation Method
Better feature representation contributes to more accurate differentiation of RNA families, thereby improving the predictive performance of the model. In this study, we utilized a feature representation method based on k-mer and embedding. Firstly, we selected the k-mer method to process RNA sequences, using 2-mers for RNA feature representation. Subsequently, we applied word embedding techniques to convert the RNA sequences into vector representations based on their frequency, facilitating the processing and training of the network model. The advantages of using k-mer method in RNA and DNA research include:
- Dimensionality reduction: RNA sequences are often very long, and analyzing the raw sequence data may result in a high-dimensional feature space, leading to the curse of dimensionality. Representing RNA sequences as k-mer sequences significantly reduces the dimensionality of the feature space, thus reducing computational complexity and improving processing efficiency.
- Capturing contextual information: Word embedding maps discrete symbol sequences (such as k-mer) into a continuous vector space, where symbols with similar contexts have similar embedding representations. By converting k-mer sequences into word embedding vectors, we can capture contextual information in RNA sequences, including the associations between nucleotides. This is important for many machine learning and deep learning algorithms, as they can utilize these embedding vectors to infer the functional and structural information of RNA sequences.
2.3. Neural Network Architecture
2.3.1. Convolutional neural network In ConF
Convolutional neural networks (CNNs) apply convolutional operations to RNA data using convolutional kernels and employ activation functions to introduce non-linear computations, thus increasing their expressive capacity. The resulting feature maps are then produced as inputs for the subsequent layers. CNNs commonly comprise multiple layers of convolutional layers, where the lower layers mainly extract low-level features from the input data, while the higher layers combine these low-level features to extract higher-level abstract features. The operation of a convolutional kernel in the -th layer can be represented by the following equation:
The notation used is as follows: represents the convolutional kernel at position in layer l, represents the feature map of the th layer, represents the bias, and f denotes the activation function. The Convolutional kernel, typically smaller than the input data, performs convolutional calculations on a subset of nodes within the input data known as the "receptive field." This strategy enables the effective extraction of local features from the input data, leading to improved accuracy. Moreover, the convolutional kernel can slide across all positions of the input data, with shared weights during each convolutional operation. This weight sharing mechanism reduces the number of parameters in the network, enhancing the scalability of the network model.
2.3.2. Cross multi-head self-attention in ConF
The primary function of the Cross Multi-Head Attention mechanism is to facilitate cross-interaction between two distinct feature sets, enabling each feature to consider information from the other feature set. This mechanism aims to enhance the capture of interactions and correlations between the features. In this paper, the Cross Multi-Head Attention mechanism is employed to handle comparisons between different blocks, with each input possessing its unique feature representation.
Specifically, the Cross Attention mechanism enables interaction between features at various levels. At each level, the attention mechanism calculates the similarity between the two feature sets and subsequently computes a weighted sum of elements in each set based on the similarity weights. This process allows each element within each feature set to incorporate information from all elements in the other feature set, resulting in a more effective capture of their associations and interactions.
The Multi-Head Attention mechanism (MHA) receives three vectors as inputs: the query vector, the key vector, and the value vector. Given a query vector, MHA calculates weighted sums of the key vectors, with the weights determined by the similarity between the query and key vectors. The resulting weighted sum is then multiplied by the value vector to generate the output. Common similarity calculation methods include dot product or bilinear calculations. The multi-head mechanism of MHA significantly enhances the expressive capacity of the model and enables it to learn more diverse and complex features. The formula for Multi-Head Attention is as follows:
Where Q, K, V represent the query matrix, key matrix and value matrix respectively, represent the weight matrices of the query matrix, key matrix and value matrix respectively, represents the output weight matrix, h represents the number of heads, represents the output of the i-th head, and Concat represents the concatenation operation.
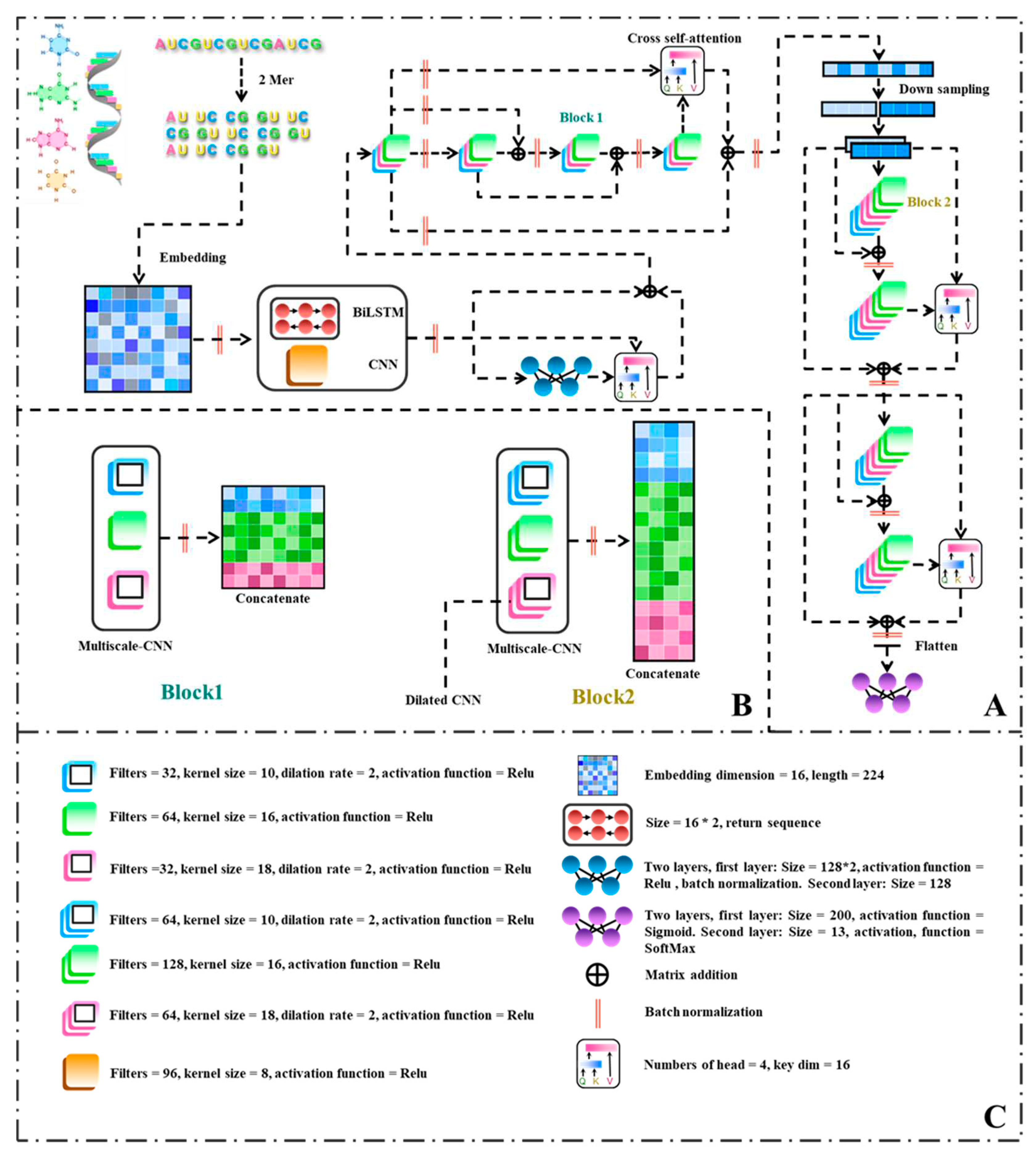
Figure 2.
Flowchart of the ConF algorithm: (A) Overall architecture of the algorithm; (B) Internal structures of Block 1 and Block 2; (C) Parameters used in each module of the algorithm along with their explanations.
Figure 2.
Flowchart of the ConF algorithm: (A) Overall architecture of the algorithm; (B) Internal structures of Block 1 and Block 2; (C) Parameters used in each module of the algorithm along with their explanations.
2.3.3. BiLSTM in ConF
Long Short-Term Memory (LSTM) has proven to be an effective model for handling long-range dependencies in sequential data. RNA sequences, being context-sensitive data, exhibit a strong correlation between the profile information of each target base and its surrounding context. In this study, LSTM is selected as the fundamental network for extracting target bases and their contextual features and subsequently encoding them. The operation of LSTM begins from one end of the sequence data and progresses to the other end. However, a unidirectional LSTM can only capture information from a single side of the target base. To overcome this limitation and capture contextual information from both sides, this study adopts Bidirectional LSTM (Bi-LSTM) to extract and learn the features of target bases and their corresponding sequence patterns.
Bi-LSTM is designed to extract and learn features from the input data, facilitating the creation of a model that encodes each base along with its contextual information in a consistent format. Bi-LSTM is composed of two LSTM networks: a forward LSTM network with 16 hidden nodes that records the contextual features of the target base's left side, progressing from left to right, and a backward LSTM network with 16 hidden nodes that records the contextual features of the target base's right side, progressing from right to left. Following the processing stage, the outputs of the two LSTMs are concatenated. The final output of the BiLSTM model can only be obtained when all time steps have been computed. At each base position, Bi-LSTM generates two hidden states. By combining these two hidden states at the target base, the encoded data (1x32) representing the target base and its contextual features is derived and subsequently outputted.
In the LSTM formula, represents the output of the forget gate, which determines which information should be forgotten from the cell state. represents the output of the input gate, which determines which new information should be stored in the cell state. ot represents the output of the output gate, which determines which information in the cell state should be output. The formula for LSTM calculation is as follows:
Here, is the forget gate, is the input gate, is the new candidate value, is the cell state, is the output gate, and is the hidden state. σ is the sigmoid function and tanh is the hyperbolic tangent function. , , , , , , and are all learnable parameters.
2.3.4. Residual Structure in ConF
In this study, a residual structure was introduced into the proposed model to extract features from the output data learned in the first part and classify them. The incorporation of residual connections allows information to selectively bypass certain layers in the neural network, facilitating the flow of information. The residual structure makes this choice more direct and easier for the network to learn. ResNet, a specialized type of convolutional neural network, employs residual blocks as fundamental units. By utilizing shortcut connections between the input and output layers of the residual block, it combines the input data with the mapped data to generate the output data, ensuring that each residual block in the network incorporates the original input information. This not only improves the model's trainability but also effectively mitigates the degradation issue that can arise with deeper network architectures. Typically, ResNet comprises a specific number of residual blocks, where the input data is denoted as , the mapping of the residual block is represented as , and the output is obtained by the sum of the mapping and the input, i.e., . In ResNet, when adding a new residual block as the network becomes saturated, the mapping function can be set to zero, which research has demonstrated to facilitate the implementation of an identity mapping compared to regular convolutional networks.
Capitalizing on the favorable performance and ease of training afforded by residual architectures, this study introduces an innovative paradigm of residual blocks. The blocks of the model incorporate multiple convolutional windows of varying sizes, allowing for the extraction of a broader range of structural features compared to conventional residual network modules. Additionally, the residual component employs a cross multi-head attention mechanism, which, as opposed to the traditional element-wise addition, enables the model to capture feature disparities across different modules more effectively, thereby enhancing the extraction of intrinsic features in RNA.
3. Results and discussion
In order to investigate the practical performance of the model proposed in this study, publicly available datasets comprising 13 distinct ncRNAs were employed as experimental materials. The experimental results were comprehensively compared with those of benchmark algorithms, revealing substantial advantages. This chapter provides a visual demonstration of the comprehensive outstanding performance of the proposed algorithm, emphasizing its commendable predictive capabilities across multiple evaluation metrics.
3.1. Evaluation metrics
In order to assess the overall performance of each method across various aspects, this study utilizes accuracy, sensitivity, precision, and F1-score as the evaluation metrics for comparing algorithm performance. The specific calculation methods for accuracy, sensitivity, precision, and F1-score are described below. In this context, TP, TN, FP, and FN represent the counts of true positive, true negative, false positive, and false negative, respectively, for the different methods evaluated using the 10-fold cross-validation test set.
3.2. Comprehensive performance evaluation
The datasets utilized in this study encompassed 13 distinct ncRNA families and served as the basis for comparing the performance of the conF model with three benchmark models: RNAcon, nRC, and ncRFP. Comparative analysis of the experimental results reveals that the proposed conF model outperforms the three benchmark algorithms in terms of accuracy, sensitivity, precision, and F1-score.Taking a vertical perspective, accuracy and F1-score offer a comprehensive assessment of the model's performance. Regarding accuracy, the conF model demonstrates superiority of 0.5831, 0.2608, and 0.1596 over the other three models, respectively. In terms of F1-score, the conF model exhibits a superiority of 0.6051, 0.2678, and 0.1673 over the other three models, respectively. These findings indicate that the conF model significantly surpasses the benchmark algorithms in accuracy and F1-score, underscoring its overall superior performance. Notably, the conF model also exhibits noticeable advantages in sensitivity and precision compared to other algorithms, suggesting its capability to detect a greater number of ncRNA families and accurately filter out irrelevant RNA sequences, thereby enhancing prediction precision.
Taking a horizontal perspective, the conF model attains the highest performance in terms of accuracy and precision, reaching an exceptionally high value of 0.9568. Additionally, it achieves the best performance in terms of F1-score and sensitivity, surpassing the benchmark algorithms with values of 0.9556 and 0.9553, respectively.

Table 1.
Performance comparison of each method.
| Model | Accuracy | Sensitivity | Precision | F1-score |
|---|---|---|---|---|
| ConF | 0.9568 | 0.9553 | 0.9568 | 0.9556 |
| RNAcon | 0.3737 | 0.3732 | 0.4497 | 0.3505 |
| nRC | 0.6960 | 0.6889 | 0.6878 | 0.6878 |
| ncRFP | 0.7972 | 0.7878 | 0.7904 | 0.7883 |
The best value in each column is bolded.
3.3. Performance comparison of diferent families
In order to assess the overall performance of each method across various aspects, this study utilizes accuracy, sensitivity, precision, and F1-score as the evaluation metrics for comparing algorithm performance. The specific calculation methods for accuracy, sensitivity, precision, and F1-score are described below. In this context, TP, TN, FP, and FN represent the counts of true positive, true negative, false positive, and false negative, respectively, for the different methods evaluated using the 10-fold cross-validation test set.
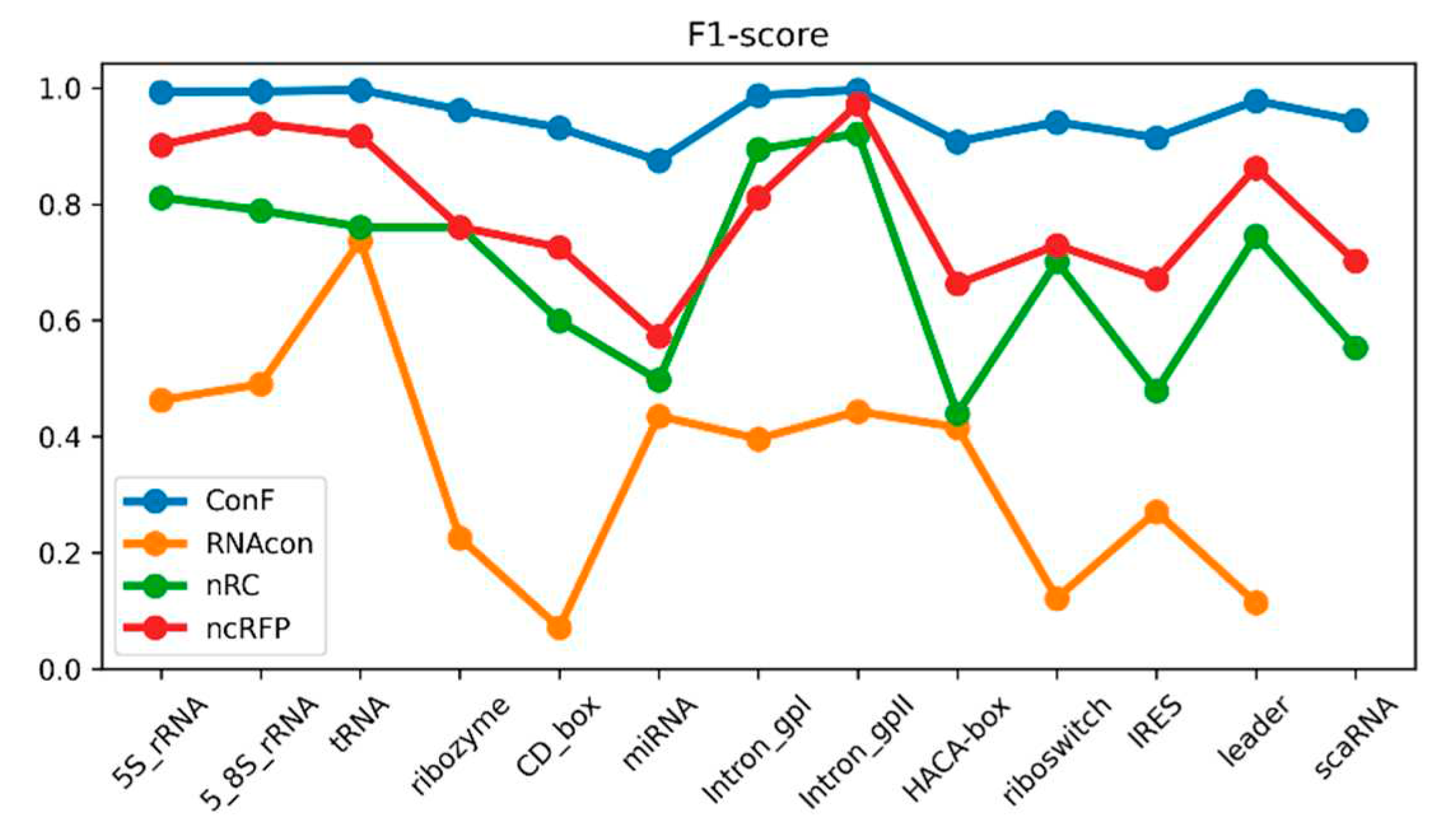
Figure 3.
This is a performance comparison among different families. The blue curve, pale green curve, green curve, and yellow curve represent the performance of RNAcon, nRC, ncRFP, and ConF, respectively.
Figure 3.
This is a performance comparison among different families. The blue curve, pale green curve, green curve, and yellow curve represent the performance of RNAcon, nRC, ncRFP, and ConF, respectively.
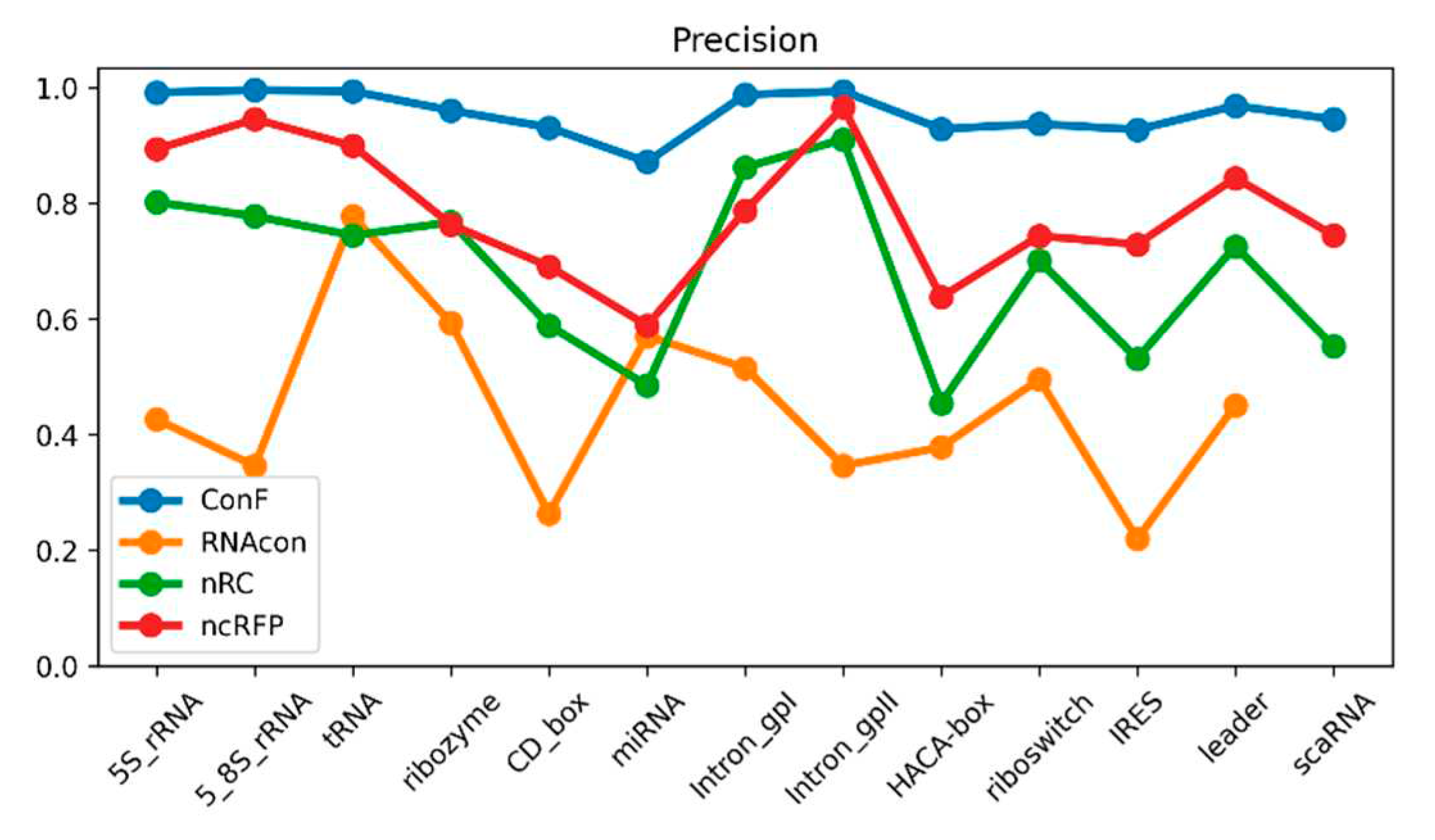
Figure 4.
This is a performance comparison among different families. The blue curve, pale green curve, green curve, and yellow curve represent the performance of RNAcon, nRC, ncRFP, and ConF, respectively.
Figure 4.
This is a performance comparison among different families. The blue curve, pale green curve, green curve, and yellow curve represent the performance of RNAcon, nRC, ncRFP, and ConF, respectively.
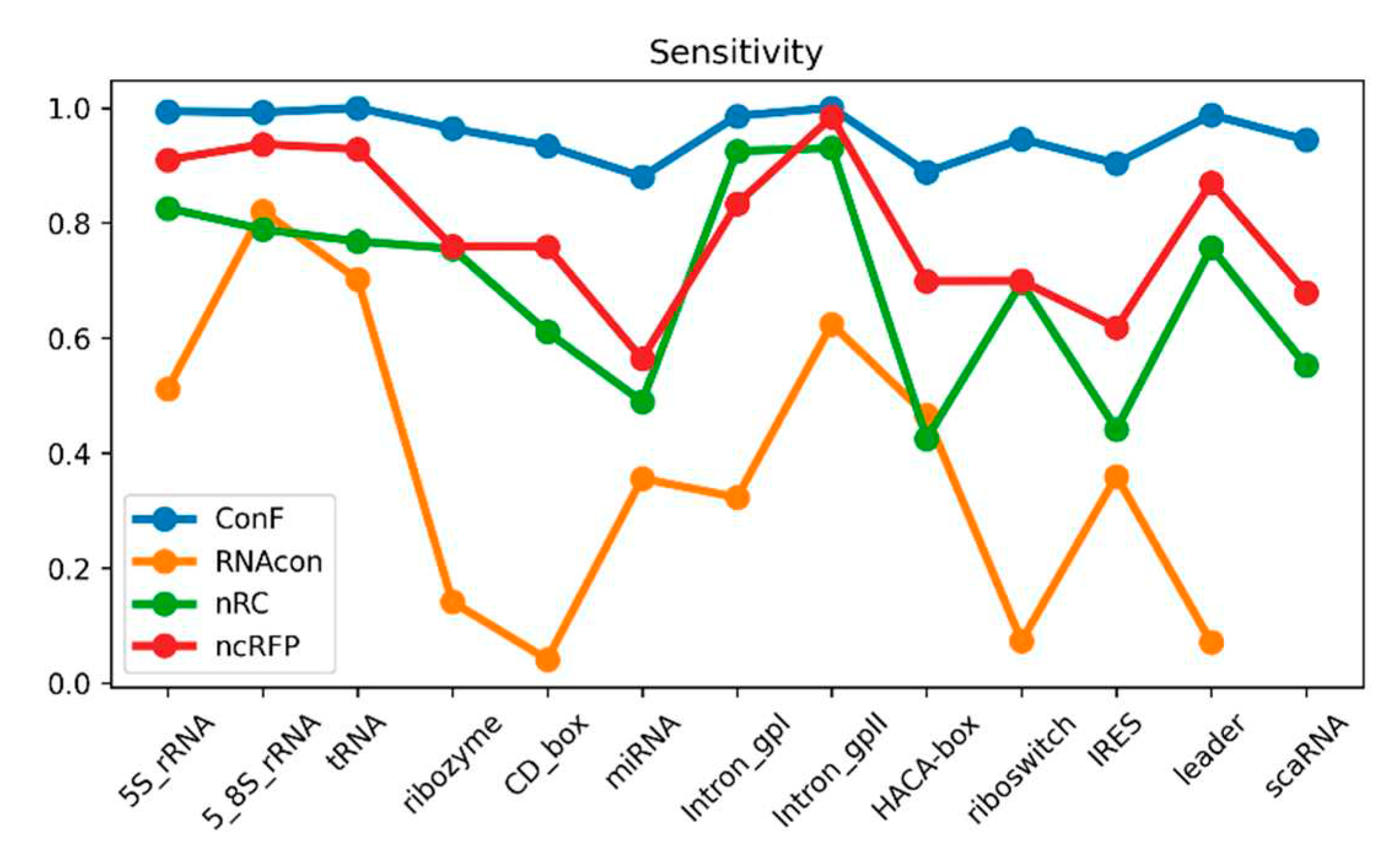
Figure 5.
This is a performance comparison among different families. The blue curve, pale green curve, green curve, and yellow curve represent the performance of RNAcon, nRC, ncRFP, and ConF, respectively.
Figure 5.
This is a performance comparison among different families. The blue curve, pale green curve, green curve, and yellow curve represent the performance of RNAcon, nRC, ncRFP, and ConF, respectively.
3.4. Performance Testing Based on Different Embedding Methods
The selection of RNA representation plays a crucial role in preserving its inherent features, consequently impacting the performance of RNA category prediction models. Experimental findings reveal that varying the lengths of k-mer methods results in diverse outcomes. In our study, we employed the single, 2-mer, and 3-mer sequence segmentation methods for testing purposes. Overall, the model exhibited the highest mean accuracy when k=2, surpassing the accuracy by 0.265% for k=1 and 0.314% for k=3.
Figure 3.
Performance Comparison of different Encoding Methods. The three boxplots in the figure represent the accuracy distributions of three feature representation methods in a 10-fold cross-validation. The blue box, orange box, and gray box respectively represent the accuracies obtained using the independent segmentation, 2-mer, and 3-mer methods for RNA sequence representation.
Figure 3.
Performance Comparison of different Encoding Methods. The three boxplots in the figure represent the accuracy distributions of three feature representation methods in a 10-fold cross-validation. The blue box, orange box, and gray box respectively represent the accuracies obtained using the independent segmentation, 2-mer, and 3-mer methods for RNA sequence representation.

3.5. Correlation Analysis
The correlation matrix in Figure 3 illustrates the relationships between F1-scores of 13 ncRNA types predicted by the conF algorithm. Each cell in the matrix represents the correlation coefficient between the predicted F1-score of an ncRNA and its corresponding ncRNA category. Higher values closer to 1 indicate a stronger positive correlation, while values closer to -1 suggest a stronger negative correlation.
For example, the correlation coefficient of 0.54 between 5S_rRNA and tRNA indicates a positive correlation, implying that these two RNA categories exhibit similar features that allow the model to make positively correlated predictions. Conversely, the correlation coefficient of -0.34 between 5S_rRNA and ribozyme suggests a weak correlation, indicating that the model struggles to extract relevant features distinguishing these two ncRNA categories.
Moreover, there are variations in the correlation of F1-score prediction values across different ncRNA categories. Comparing 5S_rRNA with Intron_gpI shows a relatively low correlation, whereas the correlation between 5S_rRNA and tRNA is high. This discrepancy suggests that the correlation between the same ncRNA type and different ncRNA types likely varies, possibly due to the model's bias in feature extraction and the inherent differences in ncRNA characteristics. These correlation coefficients could potentially be used to study feature similarity and functional similarity between RNA categories.
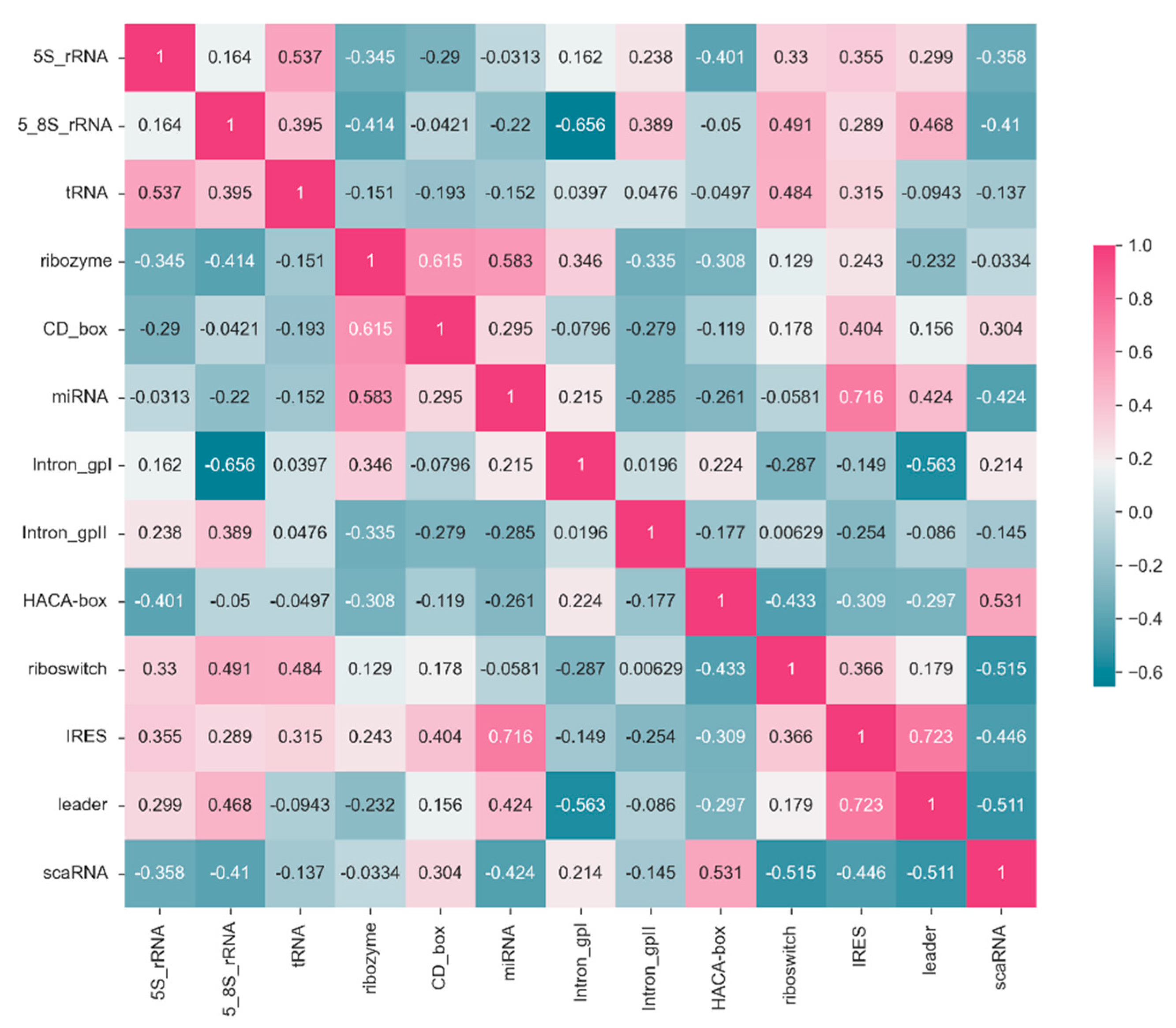
Figure 4.
Based on the ConF algorithm, a correlation matrix of precision for each RNA family in a 10-fold test. Each cell in the matrix represents the correlation of classification precision between two RNA families. The correlation ranges from 1 to -0.6, where cells closer to magenta indicate stronger correlation, while cells closer to navy blue indicate weaker correlation.
Figure 4.
Based on the ConF algorithm, a correlation matrix of precision for each RNA family in a 10-fold test. Each cell in the matrix represents the correlation of classification precision between two RNA families. The correlation ranges from 1 to -0.6, where cells closer to magenta indicate stronger correlation, while cells closer to navy blue indicate weaker correlation.
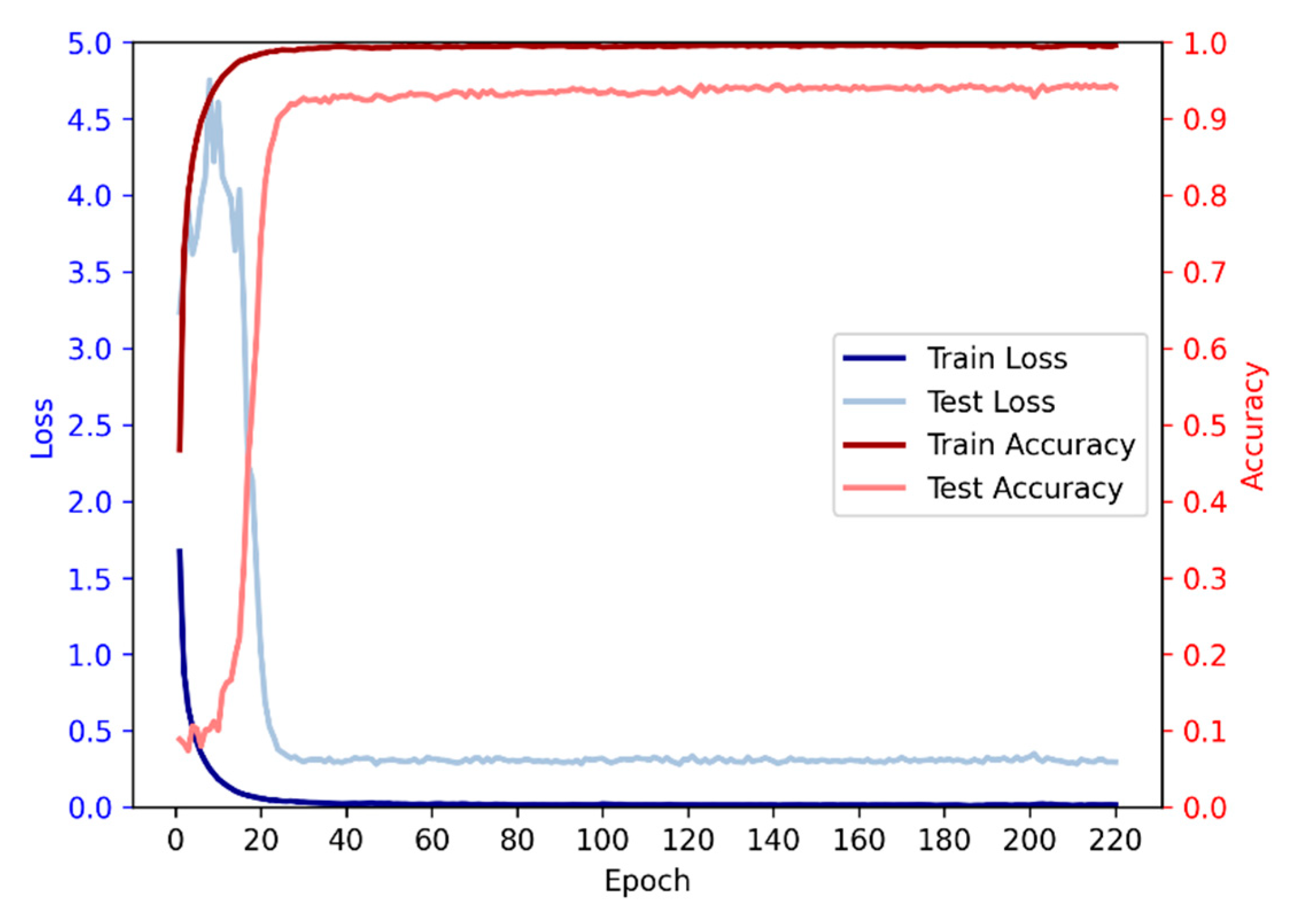
3.6. Relationship between Iterations and Performance
Based on the given data, we conducted an analysis on the relationship between the number of iterations and accuracy. It is evident that as the number of iterations increases, the accuracy initially exhibits a rising trend followed by a subsequent decline. During the initial iterations, there is a rapid growth in accuracy, surpassing 90% by the 28th iteration. Subsequently, the rate of accuracy improvement slows down, accompanied by a deceleration in loss reduction, while still maintaining an overall upward trend. However, beyond a certain number of iterations, a slight decrease in accuracy is observed, although the overall trend remains positive, indicating a continued increase in accuracy.
Figure 5.
The average accuracy and loss of each epoch in 10-fold.
4. Conclusions
ncRNA family identification has emerged as a prominent and challenging problem in RNA research in recent years. It plays a crucial role in unraveling the intricate functions of RNA and contributing to advancements in the life sciences. Although traditional biological experiments yield relatively accurate results, they are constrained by high costs, lengthy experimental cycles, and an inability to handle large-scale data predictions. These inherent limitations necessitate the development of computationally efficient methods to address this issue. In this study, we propose a deep learning-based approach, named the ConF model, for predicting the classification of non-coding RNA families. The ConF model aims to overcome the performance and applicability limitations observed in existing algorithms. By employing attention mechanisms, convolutional methods, and other techniques, the ConF model effectively extracts informative features from ncRNA sequences, thereby enhancing prediction accuracy. Furthermore, the ConF model solely relies on sequence data, enabling its broad applicability in scenarios with minimal data requirements. Experimental results demonstrate substantial performance improvements compared to several state-of-the-art approaches. Consequently, the ConF algorithm presents a promising solution for predicting RBP binding sites, offering potential support for functional studies and medical research related to non-coding RNA.
Supplementary Materials
The dataset used in this study is publicly available at https://github.com/FROZEN160/RNA-Family."
References
- Cerón-Carrasco, J.P.; Requena, A.; Perpète, E.A.; Michaux, C.; Jacquemin, D. Double proton transfer mechanism in the adenine–uracil base pair and spontaneous mutation in RNA duplex. Chem Phys Lett. 2009, 484, 64–68. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, H.; Zhang, D.; Qiu, J.; Yang, J.; Wang, K.; Zhu, L.; Fan, J.; Yang, J. A review on recent computational methods for predicting noncoding RNAS. BioMed Res Int. 2017, 2017, 1–14. [Google Scholar] [CrossRef]
- Meyers, B.C.; Matzke, M.; Sundaresan, V. The RNA world is alive and well. Trends Plant Sci. 2008, 13, 311–313. [Google Scholar] [CrossRef]
- Wang, W.-T.; Han, C.; Sun, Y.-M.; Chen, T.-Q.; Chen, Y.-Q. Noncoding RNAS in cancer therapy resistance and targeted drug development. J Hematol Oncol. 2019, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of novel genes coding for small expressed RNAS. Science. 2001, 294, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Mason, M.; Schuller, A.; Skordalakes, E. Telomerase structure function. Curr Opin Struct Biol. 2011, 21, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum Mol Genet. 2006, 15 (suppl_1), 17–29. [Google Scholar] [CrossRef]
- Scott, W.G. Ribozymes. Curr Opin Struct Biol. 2007, 17, 280–286. [Google Scholar] [CrossRef]
- Sharp, S.J.; Schaack, J.; Cooley, L.; Burke, D.J.; Soil, D. Structure and transcription of eukaryotic TRNA gene. Crit Rev Bio-chem. 1985, 19, 107–144. [Google Scholar] [CrossRef]
- Michel, F.; Ferat, J.-L. Structure and activities of group II introns. Annu Rev Biochem. 1995, 64, 435–461. [Google Scholar] [CrossRef] [PubMed]
- Baird, S.D.; Turcotte, M.; Korneluk, R.G.; Holcik, M. Searching for IRES. RNA. 2006, 12, 1755–1785. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Hou, Y.; Miranda, L.; Campbell, D.A.; Sturm, N.R.; Gaasterland, T.; Lin, S. Spliced leader RNA trans-splicing in dino-flagellates. Proc Natl Acad Sci. 2007, 104, 4618–4623. [Google Scholar] [CrossRef] [PubMed]
- Nudler, E.; Mironov, A.S. The riboswitch control of bacterial metabolism. Trends Biochem Sci. 2004, 29, 11–17. [Google Scholar] [CrossRef]
- Mattick, J.S. Non-coding RNAs: the architects of eukaryotic complexity. EMBO Reports, 2001, 2, 986–991. [Google Scholar] [CrossRef]
- Zhou, H. Long-chain non-coding RNA AC007392.4 Empirative study on the biological function of regulating tongue squamous cell carcinoma [D]. Southern Medical University, 2016.
- Gabory, A.; Jammes, H.; Dandolo, L. The H19 locus: role of an imprinted non-coding RNA in growth and development. Bioessays, 2010, 32, 473–480. [Google Scholar] [CrossRef]
- Chand Jha, U.; Nayyar, H.; Mantri, N.; et al. Non-Coding RNAs in Legumes: Their Emerging Roles in Regulating Biotic/Abiotic Stress Responses and Plant Growth and Development. Cells, 2021, 10, 1674. [Google Scholar] [CrossRef]
- Chen, W.; Liu, D.; Li, Q.Z.; et al. The function of ncRNAs in rheumatic diseases. Epigenomics, 2019, 11, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Taft, R.J.; Pang, K.C.; Mercer, T.R.; et al. Non-coding RNAs: regulators of disease. The Journal of Pathology: A Journal of the Pathological Society of Great Britain and Ireland, 2010, 220, 126–139. [Google Scholar] [CrossRef]
- Wang, J.; Samuels, D.C.; Zhao, S.; et al. Current research on non-coding ribonucleic acid (RNA). Genes, 2017, 8, 366. [Google Scholar] [CrossRef]
- Will, S.; Reiche, K.; Hofacker, I.L.; et al. Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Computational Biology, 2007, 3, e65. [Google Scholar] [CrossRef]
- Hüttenhofer, A.; Vogel, J. Experimental approaches to identify non-coding RNAs. Nucleic Acids Research, 2006, 34, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.L.; Yan, P.K. Research progress of SELEX technology and its application. Chinese Modern Doctor, 2008, 46, 55–57. [Google Scholar]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Research, 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Sato, K.; Kato, Y.; Hamada, M.; et al. IPknot: fast and accurate prediction of RNA secondary structures with pseudoknots using integer programming. Bioinformatics, 2011, 27, i85–i93. [Google Scholar] [CrossRef] [PubMed]
- Childs, L.; Nikoloski, Z.; May, P.; et al. Identification and classification of ncRNA molecules using graph properties. Nucleic Acids Research, 2009, 37, e66–e66. [Google Scholar] [CrossRef]
- Panwar, B.; Arora, A.; Raghava, G.P. Prediction and classification of ncRNAs using structural information. BMC Genomics, 2014, 15, 1–13. [Google Scholar] [CrossRef]
- Fiannaca, A.; Rosa, M.L.; Paglia, L.L.; et al. nRC: non-coding RNA Classifier based on structural features. Biodata Mining, 2017, 10, 27. [Google Scholar] [CrossRef]
- Wang, L.; et al. ncRFP: A Novel end-to-end Method for Non-Coding RNAs Family Prediction Based on Deep Learning. IEEE/ACM Trans Comput Biol Bioinform 2021, 18, 784–789. [Google Scholar] [CrossRef]
- Borgelt, C.; Meinl, T.; Berthold, M. Moss: a program for molecular substructure mining. In Proceedings of the 1st International Workshop on Open-Source Data Mining: Frequent Pattern Mining Implementations; pp. 6–15.
- Ashish, V.; Noam, S.; Niki, P.; Jakob, U.; Llion, J.; Aidan, N.G.; Lukasz, K.; Illia, P. “Attention Is All You Need” CoRR abs/1706.03762 (2017).
- Alexey, D.; Lucas, B.; Alexander, K.; Dirk, W.; Xiaohua, Z.; Thomas, U.; Mostafa, D.; Matthias, M.; Georg, H.; Sylvain, G.; Jakob, U.; Neil, H. “An Image is Worth 16x16 Words: Transformers for Image Recognition at Scale,” in ICCV, pp.9992-pp10002, 2021.
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.; Bridgland, A.; Penedones, H.; Petersen, S.; Simonyan, K.; Crossan, S.; Kohli, P.; Jones, D.T.; Silver, D.; Kavukcuoglu, K.; Hassabis, D. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Burge, S.W.; Bateman, A.; Daub, J.; Eberhardt, R.Y.; Eddy, S.R.; Floden, E.W.; Gardner, P.P.; Jones, T.A.; Tate, J.; et al. Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 2015, 43, 130–137. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



