
Submitted:
29 September 2023
Posted:
03 October 2023
You are already at the latest version
Abstract
Immunotherapy with stereotactic body radiotherapy (SBRT), low-dose antiangiogenics, immune adjuvants, nanomedicine, or other combinations will likely play a lead role in managing malignancies soon. Presently, its benefit extends to about 12% to 20% of patients, in the background of possible recurrence eventually and the toxicities, including in non-responders. Stereotactic body radiotherapy (SBRT) not only eliminates the indexed lesions, but recently, the evidence is accumulating about its systemic abscopal effects. It needs to be evolved to a dependable in situ therapeutic vaccine production and to activate short- and long-term memory immune lymphocytes. The in-situ immune cascade happens with the ability of SBRT to generate Tumor-specific neoantigens and neoepitopes, which enhance the activation of antigen-presenting cells, invoke and restore the competence of Tumor-infiltrating lymphocytes, resulting in cancer cell lysis. This cell lysis, in turn, can generate further neoantigens and epitopes, imparting a virtuous cycle that is the hallmark of effective vaccines. Additionally, SBRT can synergistically enhance the other in situ vaccination strategies, concurrent in vitro vaccines or nanomedicines, making it a primary tool for sensitizing even the “cold” tumors for immunotherapy. The present proposed hypotheses, primarily based on a preclinical literature review, focus on the critical aspects of the in-situ vaccination generation capability of SBRT when used in pulsed, cyclical, or intermittent endothelial-sparing single dose schedules along with immunotherapy and other supportive measures. This schedule is categorized as in situ vaccine dose (ISVD) radiotherapy (RT) (distinguishable from the standard therapeutic SBRT schedule) in metastatic cancer settings after standard /first-line therapies.
Keywords:
in-situ-vaccination
; Immunotherapy
; cyclical-intermittent-SBRT
; vascular-normalization
Introduction
Over the decades, enough effective therapies have been available, the latest being immunotherapy (IMT). However, there are two root causes: one, intrinsic heterogeneity and repopulation of resistant clones (1), and the second, “rewiring” of inexhaustible evolutionary web-like pathways of immune escape mutations. The primary resistance is consequent to genomic complexity, and the process of secondary adaptive resistance following therapy is the result of poorly understood rapid dynamic rewiring of the transcriptional network of complex crosstalk feedback leading to survival by adapting to the presence of a drug, circumventing the initial challenge (2). In both primary and secondary resistance processes, in many patients, either the short term or long term, the tumor mass transforms to resistant clonal types in treatment-failed patients. Given the dynamicity of cancer cells, therapy-resistant population evolution with evolving mutations, a technique that kills these molecularly-evolving cancer cells and induces adaptive/dynamic immunogenicity could be the answer for making the cancer unlethal.
Cancer is a byproduct of persistent, progressive, pathological proliferation of normal cells that have gone out of control of the regular immune system. Therefore, the two essential components of the treatment approach should be to remove/kill the proliferating cells and restore the immune control mechanism. The first component was addressed over the decades by classical therapies of surgery, radiotherapy (RT), and chemotherapy (CT). The immune mechanism was too complex to be managed entirely by these three methods, and although there was encouraging improvement in the cure over time, a significant population still developed recurrence, and a sense of cancer as a fatal disease on diagnosis persists. The recent rise of immunotherapy has changed immune modulation strategies considerably, and optimizing this modality to make cancer a chronic illness is within the realm of possibilities.
Therefore, in the long term, a strategy to have a durable and consistent method is empowering the innate immune network to constantly engage inside the aberrant cell through surface receptors and eliminate them at the first sign of repopulation. Thus, the potential way to eliminate cancer as a fatal disease is by taking the leaf out of control of infectious disease and strategizing a way to develop a therapeutic vaccination methodology. In vitro vaccination technology for cancer has come a long way. Yet, since the cancer cells are evolving continuously, it is necessary to have in place the in-vivo technique alone or supplement the in-vitro process to keep an “internal watch” on the dynamic flow of developing mutation. In addition to the ability to eliminate tumor mass, RT simultaneously generates tumor-specific antigens and invokes adaptive immunity, making it an “in-situ vaccine” initiator (3). Several studies have demonstrated the effectiveness of in-situ vaccination procedures by intratumoral administration of drugs (4). However, stereotactic body radiotherapy (SBRT) is the other method that directly causes significant cell kill, invokes in-vivo immunity via immunological cancer cell death, and releases the tumor-specific antigens (neoantigens) and neoepitopes (5). The neoantigen is a cancer-induced unique mutational short peptide sequence-specific protein, and neoepitope is a self-defined nine to ten amino acid sequence frame that can fit into cell surface Major Histocompatibility Complex (MHC) molecule as well as Human Leukocyte Antigen (HLA). Neoantigens help to distinguish cancer cells from normal cells. Neoepitopes, linked to the tumor mutational burden, correlate with response to immunotherapy (6). SBRT’s precision, technological adaptability to changing Tumor morphological-phenotypic changes of cancer, and non-invasiveness make it convenient to give it repetitively and ease clinical development (7), as long as the total equivalent dose is within the tolerance limits. Thus, the second action of SBRT, in addition to an established therapeutic modality in indicated conditions, could also act as a potential immunogenic tool that is yet to be explored to its full potential to come into clinical practice routinely. The following review is a compilation of literature about its in situ vaccine extractor capability with proposed strategies to optimize it. The SBRT cancer cell kill and its professional phagocytosis lead to secondary effects of reduced interstitial pressure, improved lymphatic drainage for the process of presenting neoantigens, improved oxygenation with accompanying vascular normalization, shift towards immune favorable tumor microenvironment (TME) milieu, synergic with other anticancer therapies are dealt in detail by the present author elsewhere. The present article combines all these complex concepts discussed in the earlier publications to give an algorithm for workable validation (8, 9, 10). The current review and hypothesis paper deals specifically with the ability of SBRT as an effective, dependable in-vivo anticancer vaccination tool when used intermittently/cyclically in a pulsed manner alone or along with other in-vitro and intra-tumoral procedures.
Hypothesis and Theory
1. The Hypothesis—Pulsed/Cyclical/Intermittent In Situ Vaccine Dose RT (ISVD RT)
The role of SBRT as a potential method in enhancing local immunostimulant milieu and possible abscopal effect in the distant organs with its ability to generate tumor-specific neoantigens leading to the bystander effect, improving the TME to further this effect and synergism of this in combination with immunotherapy is gaining momentum (11). Until now, all the clinical trials are being tried in using SBRT in its conventional fractions as a course in different fractionation schedules, and although the result has been encouraging, it has yet to make a dramatic change in outcomes of advanced cancer. Most studies highlighted that it is vital to use the endothelial-sparing vascular non-disruptive SBRT dose of 5–10 Gy (12). Classical radiotherapy has been tried, especially in its formative years, in various fractionation schedules of ascending, descending, concomitant boost, sequential boost, and hypofractionation schedules, primarily to handle hypoxic/anoxic resistant populations of cancer cells and thus improve the results.
The present author’s Hypothesis, which has two components, states that the standard course of SBRT, as routinely practiced today, when split into single doses given as a pulsed approach in a cyclical fashion before each cycle of immunotherapy (+/-CT, targeted therapy), generates repeated pulses of tumor-specific neoantigens/neoepitopes dynamically priming the antigen-presenting cells (APCs) (fulfilling one of the fundamentals of vaccination and boost doses) and is categorized as ISVD RT schedule. This intermittent In-Situ Vaccination Dose (ISVD) radiotherapy (RT) schedule, along with supporting strategies, fundamentally improves oxygenation-immune-phenotypic conditions within the tumor with every dose and is anticipated to precipitate the cascading effect on eliminating local immunosuppressive TME, enhances abscopal impact, and establish effective functioning of anticancer immune memory cells for a lifetime.
The premise is based on generating in vivo stimulation of antigen-presenting cells by SBRT as an ISVD RT to target continually mutating cancer cell surface antigens dynamically over time, arming/rearming the antigen APCs multiple times adoptively. This premise contrasts the presently clinically established SBRT therapeutic schedule of giving one to eight fractions as a single course, exhausting the potential at one-course-single-time enhancement of arming the antigen-presenting cells. Naturally, this ISVD RT approach is proposed as a clinical trial for patients with metastatic cancer progression after being failed on standard therapy/IMT. Subsequently, continued immunotherapy/targeted therapy is expected to have enhanced action given the presence of induced memory cells and is likely to lead to a significant increase in the control rate and application of immunotherapy in a broader population. This approach has the added advantage of “titrating” the number of fractions of SBRT to limit the toxicities of combined SBRT and IMT, where some long-term effects are still unknown.
1.1. The Basis for the Hypothesis
Limited clinical data: There is almost non-existent clinical data in the literature regarding giving radiotherapy intermittently cyclically like in the immunotherapy chemotherapy schedule. Prolonging the period of radiotherapy delivery is technically considered an overall time beyond a certain period, which results in inferior results in radical treatment approaches (13). In certain situations, a weekly schedule is practiced in the selected population group, keeping in mind limiting the number of hospital visits is considered similar in survival, local control, and acceptable side effects compared to standard RT in carcinoma breast (14). Hence, the proposed dose schedule of cyclical RT, matching the dose schedule of immunotherapy with or without chemo-targeted therapies, needs to be validated in animal studies. This dose schedule of SBRT can be considered an ISVD RT schedule to differentiate from the established therapeutic SBRT dose schedules. The objective of this approach is palliative intent to prolong the treatment response/quality of life in patients who failed standard IMT/CT therapies with or without RT/SBRT. To distinguish the two, throughout the article, when the term SBRT is used, it refers to the standard SBRT dose schedule, and ISVD RT is employed when explaining the SBRT dose in the context of the proposed Hypothesis.
Preclinical study: Sezen et al. (15) hypothesized that stereotactic radiation when given in pulses, would increase the repertoire of thymus lymphocytes (T) cell priming with incremental building up of immune memory against cancer cells. To prove the Hypothesis, the authors used a combination of immunotherapy and pulsed RT in 12 Gy x 2 fractions schedules two weeks apart in the lung adenocarcinoma murine model. The results showed enhanced antitumor efficacy in primary tumor sites, delayed tumor growth, and improved survival in the combined treatment group. Additionally, the pulsed treatment group showed increased cluster differentiation 4 (CD4+) effector memory cells compared to the single-cycle RT group, validating targeting multiple points of tumor immune evasion. The authors also suggest that pulsed RT has advantages for multiple metastatic situations regarding efficacy and safety (15). Moore et al. also found that radiation dosing with PULSAR type in a mouse model is more complimentary in combination with single-agent IMT in the preclinical model than traditional daily fractions (16). He K et al. (17) hypothesized that a single cycle of RT delivered to a limited number of metastases is rarely sufficient for systemic control, and several rounds of RT akin to IMT cycles have a greater probability of amplifying the adaptive immune response all in terms of T cell repertoire, high-affinity antibodies and in invoking memory cells. Further, they proposed “pulsed-RT” with three RT cycles given a month apart to 2-4 lesions (17).
The present author proposes that effective in situ vaccine creation is a complex process, and it requires not only pulsed delivery of ISVD RT but the adoption of various supporting concepts, as detailed below, to have a consistent, definitive, and effective in situ effect.
2. Immune Features to Be Exploited for Successful In Situ Vaccination: Neoantigens, Neoepitopes, Tumor Mutational Web, and Immune Escape Prevention
Types of tumor antigens are Tumor-specific antigens (TSAs), which are either oncovirus antigens or neoantigens of genomic mutations produced by cancer cells, and Tumor-associated antigens (TAAs) produced both by cancer cells and healthy cells. TSAs related to a total number of mutations per analyzed tumor genomic region defining total mutational burden (TMB), if low, is associated with a weak response to immunotherapy (IMT). TSAs generate tumor response, and TAAs can induce “on-target” toxicity reactions within the same structure or “off-target” in similar organs/structures elsewhere. Most tumor antigens are non-mutant and considered self-proteins by the immune system “drown” the impact of TSAs. However, despite being overwhelmed by nonspecific antigens, the significant advantage of in situ neoantigens is the relative ease of their generation (7). Fitting into the fundamental principles of vaccination, antigens, when presented repeatedly, unleashes adaptive immune responses (18).
In addition to neoantigens, related immune-enhancing molecules like epitopes are involved in the immune response. Radiation therapy also enhances the generation of neoepitopes and neoantigens, producing an entire repertoire of cluster differentiation 8 (CD8+) T cells (19).
Understanding the complexity of antitumor immune response (20) is essential to designing any strategy, which includes the creation/extraction of neoantigen of intracellular origin by effective cancer cell kill; migration of neoantigens to lymph nodes which also depends on interstitial pressure (ISP) and opening up of collapsed lymphatics; neoantigen incorporation to APCs; presentation of neoantigen by APCs to T lymphocytes (depends on various immune promoting cells and pathways); activation of T effector cells, thus expanding T cytotoxic cells immunosurveillance; infiltration of these activated T cytotoxic cells - Tumor-infiltrating cells (TILs) to TME (influenced by vascular integrity, ISP, elimination of immunosuppressive cell lineage in the TME); and interaction between T cytotoxic CD8+ cells and cancer cells mediated via T-cell receptor (TCR) and major histocompatibility (MHC)-type I molecules or tumor epitopes (21). A deficiency in any of these steps leads to ineffective therapy and recurrence/progression of the disease. SBRT can play a role in these steps in combination with other local or systemic therapies and immunotherapy. The process can be furthered by increasing the harvest of tumor-specific antigens by local combination therapies with SBRT, given its ability to act synergistically, bearing in mind the possibility of overlapping short and long-term toxicities. Over and above, it can invoke self-sustaining memory-capable virtual cycles. The process includes enhancing, more specifically, type-1 T helper (Th1) and type 2 T helper (Th2) cells that activate immune stimulatory macrophages and B-cells, respectively. Negating the TME immunosuppressive cycles and stimulation of TME immunity pathways (20) requires well-designed combination strategies.
Neoantigens are the product of cancer-induced mutations that ordinarily elicit an immune response but, over time, might provide “fuel to the fire” of cancer evolution en route to the immune escape. However, novel peptides neoantigens can also be presented on the cell surface, recognizing the cancer cells as “non-self,” leading to lysis, referred to as immune editing. The probability of immune editing to and fro can lead to the fluctuation of contraction or expansion of the cancer cell population, leading to subclones. Over time, a large load of neoantigens leads to evolution and immune escape (22) not recognized by innate immunity. After immunotherapy with a high rate of cell death, the net result could be a high tumor mutational burden with severe neoantigen depletion, making cells resistant to checkpoint inhibitors despite the initial high tumor mutational burden. The clonal escape mutations continue to be “antigen warm” (high neoantigen burden), with several sub-clonal neoantigens leading to immune escape. With rapid shrinkage of the active clonal population, passive clones continued growing progressively, pruning the neoantigens and turning them into immune-cold tumors. In the post-immunotherapy doses, the immune landscape differs from the original tumor, with new clonal emergence requiring the following/additional lines of therapy (22). Since the total mutational burden is proportional to the neoantigen production (23), proliferated subclones can be expected to have reestablished modified neoantigens.
Individual tumor trunk and heterogeneous branch mutations are present in all regions or at least two areas of the tumor, respectively, whereas private branch mutations are unique to one part (24). In a study, branch and private branch mutations representing the intratumor heterogeneity of resistant subclones comprise about 40% of hepatocellular carcinoma (25). Radiotherapy enhances the response to IMT by targeting trunk mutations (26).
Mutations outside the anchor position interact with peptide/MHC complex with TCR, leading to recognition of naïve T cells, while mutations in the anchor position potentially create high-affinity recognition of mutated specific neoepitopes. The authors propose the bioinformatic evaluations of entire neoepitope spectra to identify targets for the tailored therapy. However, vaccination or personalized adoptive cytotoxic T lymphocytes (CTL) transfer treatments favor the evolution of escape variants by loss of global MHC expression. Systemic Natural Killer (NK) cell activators and Chimeric antigen receptor (CAR) T immune cells may preferentially recognize MHC-negative tumor cells for elimination. The other combination option is the surgical excision or cytoreduction for the resistant residual lesion (27).
CAR-T adoptive cell therapy has surfaced as a breakthrough immunotherapy in cancer, especially in hematological malignancies. However, in solid tumors, antigen escape, toxic reactions, abnormal vascularization furthering tumor hypoxia, and insufficient infiltration of CAR-T cells leading to immunosuppression are the limiting factors (28). These inadequacies can be obviated by the proposed strategy described in the present article by titrating the RT dose per fraction to toxicity spread over time. Since the mechanism action is due to interferon-gamma (INF-γ), tumor necrosis factor-alpha (TNF-α), perforin, and granzyme-induced cell kill, independent of antigen-derived peptides and class I molecules of the MHC complex interactions, acting by recognizing and binding to proteins or gangliosides on the cancer cell surface. RT facilitates CAR-T cell homing by pro-inflammatory actions, migration of CAR-T cells along with reoxygenation by increasing integrins intercellular adhesion molecules–1 (ICAM-1), vascular cell adhesion molecule–1 (VCAM-1) expressions in endothelial cells, activating complementary endogenous several antigen-specific responses and converting TME immunosuppressive to immune promoting cells. Correspondingly, CAR-T cells sensitize and retarget the development of radioresistant cells. In vivo RT dose of as low as two Gy per fraction has shown expansion of CAR-T cells. However, another preclinical study in pancreatic cancer has found this schedule insufficient. Although existing data for combined RT and IMT favors 8 Gy x 3 fractions, in a preclinical glioblastoma study, 4 Gy x 1 fraction did extend the survival, and a certain number showed complete regression (28).
“Cold” or even an immune “desert” can be reprogrammed to hot tumors by targeting hypoxia. Hypoxia is the foundation on which immune resistance rests due to countless alterations in the metabolites, TME progressive acidosis, over-expression of immune checkpoints, cancer-favorable TME immune depletion, as well as hypoxia-induced tumor-promoting autophagy (18) resulting in plexiform of resistant pathways.
3. Optimizing ISVD RT-Generated Neoantigen, Neoepitope, and In Situ Vaccination
Two primary effects of Immunogenic cell death with inflammatory cascade and phenotypic changes give the capacity SBRT to extract needed neoantigens and possible impact in eliminating immunosuppressive cells in the TME for in situ therapeutic vaccine effect. This neoantigens release leads to dendritic cell (DC) cell maturation, antigen cross-presentation, and antitumor T cell response diversification. Single dose fraction eventually acts as an immune activator with initial lymphocyte depletion. Also, type I interferon phenotypic effects peak at 8 to 12 Gy single fraction through the cyclic synthase interferon genes (cGAS/STING) pathway. In addition to inducing a cluster of differentiation (CD8+) T cell response by radiation-generated neoantigens, RT also stimulates immune response via ferroptosis, necroptosis, and pyroptosis (18).
A higher dose per fraction of SBRT (>12 Gy per fraction) increases the expression of three prime repair exonucleases gene (TREX1), which reduces the accumulation of cytoplasmic dsDNA, impacting IFN-ϒ production negatively (29).
Radiation no doubt modulates the tumor microenvironment at low doses (1–4 Gy) (30). Yet, the present author has not gone into detail given the main focus on antigenicity and adjuvanticity of SBRT, which tends to be significant in the dose per fraction range of 6 to 10 Gy as per endothelial tolerance. However, a higher dose per fraction of 12 Gy or more needs to be relooked in the future because of the higher antigenicity of that schedule in case of clinical applicability of improving endothelial cell tolerance by dual recombination (31) or any other technology.
Two important specific points were demonstrated by Lussier et al. in their preclinical poorly antigenic IMT not responding cell lines study. One, non-curative doses of radiation can induce MHC-I neoepitopes, leading to tumor lysis populations of CD8+ T cells that sensitize low mutation burden cold tumors to IMT. Two, the delivery of neoantigen generation immunogenic dose of RT (4 to 9 Gy) throughout the tumor population, initially having a sub-clonal immune response, may drive spreading epitope resulting in rejection of mutationally heterogeneous tumor (32).
The other critical reversal process is the impact on phenotypic changes. Antigen-specific CD8+ T cell phenotypic analysis showed RT-enhanced antigen-experienced T cells and effector memory T cells. RT activates protein kinase C (PKC), mitogen-activated protein kinases (MAPK), and master immune response regulator nuclear factor Kappa B, which are signal transduction pathways as well as transcription factors, making them susceptible to immunotherapy response via phenotypic changes. RT up-regulates MHC and increases expression of specific tumor model antigenic epitopes presentation to MHC on the cell surface, resulting in enhanced memory phenotypic CD8+ T cells (33).
Radiation can also immunologically facilitate T-cell-mediated killing by altering the biology of surviving cells (34). Also, it can be hypothesized that SBRT spares irradiation of draining lymph node regions where antigen presentation occurs (33).
Along with RT, the additional strategies are interleukin-15 (IL-15) agonism (known activator of NK and CD8+ cells) augmenting the expansion antitumor memory CD8+ T cells; intratumoral depletion of regulatory T (Treg) cells by targeted glucocorticoid-induced tumor necrosis factor-receptor–related (GITR) agonism; downregulation of myeloid-derived suppressor (MDSC) cells by molecular restructuring to increase antigen recognition combining with anti-programmed cell death ligand-1 (PDL 1) as well as ipilimumab, leading to positive feedback with further destruction of Myeloid-Derived Suppressor cells (MDSCs). Immunogenic cell death by RT increase in damage-associated molecular patterns (DAMPs) made up of group box-1 protein (HMGB1) impacting toll-like receptor-4 (TLR-4) (thus dendritic cells), which facilitates more extensive antigen presentation due to inhibition of intracellular antigen degradation and adenosine triphosphate (ATP) stimulating aggregation of dendritic cells for antigen presentation in the tumor. Natural Killer (NK) cells capable of spontaneously targeting cancer cells are not limited by the need for antigen presentation or MHC pathway to activate adaptive cell-mediated immunity pronounced in conventional doses and mixed results with SBRT due to competing effects (35).
Timing with IMT is the most critical of all the factors discussed here. RT can Damage immune cells locally immediately. It can increase immune suppressive cells, signal pathways, and immune suppressive factors (36). Hence, synchronizing SBRT and IMT sequences is critical.
According to the concept of “reoxygenation utilization rate,” in hypo-fractionated radiotherapy, a single fraction cannot utilize the phenomenon of reoxygenation, depriving the applicability of one of the five radiobiological principles for an increase in efficacy of fractionated radiation, unlike in a situation where oxygen utilization goes up to 87% with 6 to 8 fractions with an ensuing enhanced response. In the hypofractionated schedule, reoxygenation for that dose will likely be complete in 48-72 hours (37). There is a general understanding that SBRT after IMT is less effective, and the optimum time may be anywhere from 48 hours to less than seven days (12, 38). IMT comparing administration on days 6-10 was more effective after RT in a mouse model than days 1-5 or 11-15, according to study results by Morris et al. They surmised that T cell response reflects a delayed reaction to RT (39). Considering the critical aspect of reoxygenation and initial TME lymphocyte depletion recovery, the least time interval after SBRT could be 48 hours.
4. Factors Mitigating the Local Response and Abscopal Effect and Countermeasures
The next critical factor is the mitigation of “immunosuppressive recoil.” The high dose schedule of SBRT does invoke reactive effects from the untreated tumor load, either from imageable cancer mass or subclinical disease. This immunosuppressive “backfire” could be one of the reasons for the abscopal effect’s rarity that raised much hope initially based on anecdotal reports, especially with the introduction of immunotherapy. Determination of optimal dose/fractionation schemes, optimal time points for integrating RT plus IMT, and predictability of efficacy is yet to reach a consensus (40). Therefore, several supportive issues must be resolved along with the SBRT-IMT combination to induce significant abscopal effects.
4.1. Hypoxia/Anoxia
Hypoxia has been the bugbear of radiotherapy resistance with increased mutations from its inception. These higher mutation rates and burdens can source more neoantigens for immune checkpoint inhibition. Additionally, hypoxia is the fertile ground for immunosuppressive cell types such as Tregs and MDSCs maturing to M2-polarized Tumor-associated macrophages (TAMs), TH2 polarized Dendritic cells (DCs), downregulation of MHC class-I molecules plus NK cell-activating ligands and upregulation PD-L1 on tumor cells that can be countered by combinatorial immunotherapy and SBRT. Thus, hypoxia is responsible for immune tolerance via multiple mechanisms (41). In normoxic tumors, immunogenic cell death with upregulation of MHC class-I, the release of immune stimulatory DAMPS, activation of TLRs, new TAAs, maturation of DCs along with upregulation of MHC-class II leading to T cell priming in the draining lymph node facilitate tumor lysis by cytotoxic T cells NK cells on traveling back to the tumor. From intensely immunosuppressive intrinsically radioresistant hypoxic tumors, reoxygenation with fractionated radiotherapy in combination with immunotherapy enhances local control and abscopal effects (42).
Additionally, cell kill of susceptible cells, decreased ISP, increased immune cell infiltration, and increased IMT penetration for subsequent doses complete the positive effects of cyclical SBRT. Vascular normalization and use of drugs facilitating macrophage-mediated professional phagocytosis, e.g., oligonucleotides C phosphate G (CpG) to reduce the interstitial pressure (43) concurrent to ISVD RT, is essential. A single dose of ~8–10 Gy is the lower threshold for significant endothelial apoptosis (44) and, hence, a step away from the aggravation of hypoxia, converting durable response to short-term control (31).
4.2. Immunosuppressive Recoil – Tregs, BMDCs
Zachary S. Morris et al., in their mouse tumor model, proved that contrary to their initial Hypothesis, untreated second Tumor-induced Tregs mediate immune suppression of indexed primary lesion treated by RT combined with an intralesional agent for vaccination effect was countered by Treg-depleting anti-CTLA-4 restoring the vaccination effect. The findings are very revealing that “tumor-specific,” Treg-dependent (not ruling out Treg-independent other mechanisms like MDSCs), suppressive effect is exerted by the untreated lesions, referred to by the authors as “concomitant immune tolerance.” Although the feedback immunosuppressive cascade was lesser in the case of small untreated lesions (45), one can expect this recoil suppressive mechanism to be systemic, impacting even the invisible large numbers of lesions (total tumor cell burden) until proven otherwise. Therefore, “concomitant immune tolerance” could be one of the reasons for the abscopal effect not living up to its expectations all along. Morris et al. also demonstrated that immunosuppressive effects were seen in 12x1 Gy fraction and 8 Gy x 3 fractions, even though the latter is considered more immunogenic (45), indicating the complex nature of the immune playout.
Immunosuppressive BMDCs, the first influx happening in 3 to 5 days and the second wave after two weeks, is the other mitigator of SBRT systemic and local effects, which is more than double with 15 Gy, compared to 8 Gy per fraction. This tumor-promoting influx is effectively blocked by stromal cell-derived factor 1 (SDF-1)/chemokine receptor 4 (CXCR4) when given immediately after SBRT (12).
4.3. Activation of Trex-1 Pathway
Countering by avoiding/blocking these immune-mitigating pathways with the available strategies is required for optimum local and abscopal effects. SBRT doses above 12 -18 Gy showed a Trex-1 exonuclease-induced immunosuppressive effect, unlike at lower doses per fraction for long-term control. The latter propagated DC maturation and immune cell priming, as demonstrated by Demaria et al. and others, although some studies showed better tumor control with a 15 Gy single dose (29, 46), which needs to be validated in the long-term result.
4.4. Other Immunosuppressive Recoil Pathways
Radiation-induced downregulation of cluster differentiation 47 (CD47) has a DC phagocytosis suppression effect (29). One arm of the binary complex role of autophagy is the degradation of MHC I in T cells and DCs, MHC II in MDSCs, and impaired PD-L1 degradation, all causing deficient neo antigen presentation and impaired T cell action (20), which needs further understanding.
A significant transforming growth factor beta (TGF-β) linked immune-hostile TME disruption with 12 Gy per dose has a minor impact with 6 Gy dose of SBRT (12).
Antihistamine and antifibrotic drug tranilast is a TGF-β inhibitor that can assist in the suppleness of extracellular matrix (ECM) for cancer cell inhibitory TME. Combining the monoclonal DC101 (VEGFR antibody) and an anti-TGF-β1 antibody helps normalize and reduce collagen density. There is unlikely clinical benefit from targeting fibroblastic activation protein (FAP) alone in the TME (47), the other factor responsible for reduced local immunity.
Immunogenic cell death (ICD), a hallmark of the SBRT dose schedule, is a type of cell death where it releases damage-associated molecular patterns (DAMPs), the translocation of the ER chaperone calreticulin and ATP, which in turn responsible for recruiting and activation DCs and phagocytic macrophages happens. However, rapid accumulation of catabolic product of ATP adenosine suppresses DCs and immune cells promote Tregs and favors TAMs transition to the M2 phenotype through TGF-β. These changes can be countered by a bifunctional fusion protein, which can block both the PD-L1 and TGF-β pathways (47).
5. Improving In Situ Vaccination Long-Term Memory- Enduring Immunogenicity
Immunologically cold tumors had a ten-fold increase in CD8+ infiltrates with combined intralesional plus RT therapy, which jumped to 18-fold when anti-cytotoxic T-lymphocyte associated protein 4 (anti-CTLA-4) was added (45).
RT, chemotherapy, and IMT can generate persistent, long-lived, self-renewable memory T cells. The same thing holds suitable for CAR-T cell therapy (48). Claire C. Baniel et al. (49) hypothesized that a combination of RT, intratumoral immune cytokine, and immune checkpoint blocker as an in-situ vaccine primes memory B cells responsible for a persistent humoral response without further stimulation. However, the exact role needs to be clarified. Surgery does not display immunogenic memory against the rechallenge in the animal models (49), indicating rethinking in the future may be required about the sequencing of the surgery after invoking memory cells by SBRT with or without intralesional therapy bringing in situ vaccination preceding the surgery. The most significant advantage of surgery would be eliminating the final group of immune intransigent cold residue for cure or long-term control. Evidence is needed whether intermittent repeated SBRT with or without intratumoral techniques can also keep pace with the molecular mutations, improving the B cell memories exponentially.
Baniel et al. observed an increase in memory-derived antitumor IgG antibodies even before engraftment on rechallenge, indicating antitumor activity even before the macroscopic manifestation of cancer (49). Thus, to overcome Treg propagated immunosuppressive effect by the larger nonindexed lesions (45), SBRT plus or minus intratumoral agents along with checkpoint inhibitors can be invoked with non-curative dose as “vaccination generators” when the clinical extent of disease (non-oligometastatic) does not permit the inclusion of all lesions. The other option would be combining systemic targeted radionuclide therapy alone (to suppress “concomitant immune tolerance”), alternating or in combination with SBRT for in-situ vaccination approach (30).
The Imt, on average, has benefits in 12% of patients, indicating a significant group of primary and secondary resistance, along with a burden of a large proportion with toxicity risk. Resistance can dominate at the TME level even when antigen-specific T cells are high in circulation. The primary problem is poor neoantigen availability with low TMB tumors, leading to suboptimal immune priming of APCs. Both types of resistance need to be overcome by the use of immune stimulatory agonists promoting stimulatory interferon genes (STING) and Toll-like receptor (TLR) signaling (29).
Radiotherapy causes gene upregulation, augmented protein degradation, and mToR-regulated translation, increasing the peptide pool combined with increased MHC class I expression, resulting in more antigenic peptide presentation for recognition ending in enhanced T cell receptor engineering (TCR) repertoire. In addition to this induction of tumor antigenicity, especially of immunologically cold tumors, SBRT can cause “adjuvanticity” by invoking regulated immunological cell death, triggering inflammatory signals by stressed or dying cells resulting in the damage-associated molecular patterns (DAMPs) release encompassing ATP, HMGB1, and calreticulin; activation of interferon genes STING triggering the transcription of Type I IFNs to recruit DCs for maturation which helps in naïve T cell cross-priming after migrating into the lymph nodes. The adjuvanticity is critical for bridging adaptive and innate cell responses (29).
The other dimension is in combination with other immunogenic local therapies. Intratumoral saponin-Based Adjuvants; TLRS agonists – CpG, an analog of synthetic double-strand ribonucleic acid (dsRNA) polyinosinic-polycytidylic acid (Poly-IC), feline Mcdonough sarcoma -like tyrosine kinase-3 ligands (FMS - FLT3L), TLR7/8 agonist Imiquimod, nanoparticles (28); Oncolytic virus, oncolytic peptides; thermal therapy; stimulator of interferon genes (STING) agonists; melphalan; rose bengal disodium (PV-10), are other potential combinations tried with immunotherapy to improve antigenicity and adjuvanticity. However, the limitations of these therapies are accessibility for administration, especially repeated injections, unequal distribution within the tumor (29), and disruption of the vasculature. Some of these disadvantages can be amenably overcome by nanomedicine technology.
All the above in the background of normalization / oxic conditions of the vasculature a. using timed low-dose antiangiogenics, b. by limiting inflammation and keeping the interstitial compartment supple.
6. Expanding SBRT Immunological Cascade by Adjunctives and Immune Adjuvants
The effectiveness of any vaccine against infectious disease is based not only on the antigens but also on immune adjuvants. In-situ vaccination in cancer tumor nidus itself supplies the antigen (50), the mutation prevalent de novo, which can be released by primary and booster effects of ISVD RT, and the antitumor effects can be expanded disproportionately by systemic or local immune adjuvants. This approach obviates the need to identify and harvest all neoantigens as and when they develop, enhances the synergy of SBRT and immunotherapy when combined, minimizes immune escape, and reduces toxicities, including financial. Immune adjuvants act as slow-release systems in the nidus, ensuring a continuum of stimulation to the immune system (51) when homed in or tagged. One technology suggested is leading and activating DCs combined with antigens generated from thermal tumor ablation, which later acts as a nidus for slow-release sustainable immune stimulation for the drainage lymph nodes. However, there is a possibility of rebound recovery of cancer cells at the edge in the sublethal temperature zone (51). Since tumor control with vascular disruption with ablative doses of stereotactic radiosurgery (SRS) and other methods is more likely to be a short-term benefit (31), timed repeated noninvasive delivery of ISVD RT with non-vascular disruptive dose is likely to maintain long-term immune-stimulation without the risk of rebound aberrant vascularization and repopulation at the edge of ablation that is seen in vascular disruptive methods/drugs. Another advantage of ISVD RT is dynamic matching with the evolved mutations de novo. Dendritic cell growth factor, feline Mcdonough sarcoma (FMS), like tyrosine kinase 3 (Flt3) ligand, demonstrated an abscopal effect (3). The Autophagy process has binary roles as immune-promoter or suppressor, depending on the TME. In the immunoprotection role, autophagy, influencing APCs, T-cells, macrophages, MHC I cross-presentation of autophagy-dependent neoantigens, and MHC-I, MHC-II in DCs and creates T-cell memory (20). Autophagy can help in the extraction of antigens when lysosomal enzymes isolate and degrade intracellular antigens under the action of autophagolysosome to several peptides, which get transferred to APC cell surface to get integrated with MHC II in the presence of protein molecules such as calreticulin. Induction of the mitophagy of defective mitochondria pathway in tumor cells that lack signal transducer and transcription-3 (STAT3) activator leads to enhanced antigen presentation (20). Also, autophagy-originated newly developed epitopes improve antigen availability (20).
7. Minimizing Toxicities with ISVD RT
Since SBRT has the propensity to invoke an inflammatory cascade via the arm of DAMPS and pattern recognition receptors (PRR), it potentially can cause severe immediate and long-term side effects. With progress in immunotherapy, we expect dramatic improvement in the long-term survivors, giving importance to avoiding fibrosis and maintaining the extracellular matrix (ECM) suppleness is as important as the cure.
RT can potentially cause long-term severe complications due to progressive senescent changes on endothelial stem cells and fibrosis of ECM, preventing immunogenic crosstalk (52). RT and IMT must be considered a “double-edged sword” that needs careful planning to avoid synergistic adverse effects because of the higher risk in combination RT + IMT therapy arm. The adverse events can be less when the interval between RT and immunotherapy is higher with certain types of IMTs where both tumor cells and normal tissues develop immune responses rather than in cancer cells alone (53).
The literature review discussed above can be classified into four intertwining categories integrated into the present Hypothesis. Theme one is the measures and pathways that enhance the impact of ISVD RT; second is mitigating/ reversing the immune suppressive recoil factors with ISVD RT; third is overcoming the immune escape by invoking long-term memory cells, and the fourth critical approach is minimizing the toxicities of combination therapies by ISVD RT titration by basically keeping the ECM supple without excessive inflammatory changes.
8. Hypothesis-Validation-Algorithm for Pulsed/Cyclical/Intermittent ISVD RT with Supporting Stratagems (Figure 1, Figure 2, Figure 3 and Figure 4)
Radiotherapy has been tried in various fractionated schedules but probably not like chemotherapy in weekly/3 weekly cycles for a prolonged time. Based on the literature reviewed above, the (present) author proposes using the standard SBRT schedule in divided fractions as ISVD RT, each of the fractions delivered before a cycle of immunotherapy with or without other combination therapies, with a minimum of 3-week intervals. The ISVD cycles can be continued till the disappearance of gross disease or the appearance of unacceptable acute toxicities. After completion of the ISVD RT dose fractions, based on the total dose limits, the IMT/targeted therapy or its combinations can be continued as per the existing protocol. In this proposal, radiotherapy is suggested to be used in a non-curative single dose (17) [Lussier DM] as a prime modality for in-situ vaccine “extraction” repeatedly, adapting to the evolution of subclone repopulations as a priming dose before each cycle of immunotherapy with or without chemo/targeted therapies. Other local combination therapies, e.g., nanomedicines and intralesional drugs known to induce in situ vaccination response that can widen the intensity of the scope of antigen generation, along with ISVD RT, are open to further investigative trials. The total dose and the number of intermittent cyclical ISVD RT should be within the tolerance limits of combination therapies by the “titration” approach. New criteria for time-dose-fractionation for normal tissue tolerance may be evolved following conventional knowledge and accumulated experience.
Figure 1.
Theoretical Model - Multistep staged SBRT as an in-situ vaccination modality. Intermittent /Repetitive SBRT to harvest the changing spectrum of mutating neoantigens for improved immunotherapy and innate immunity. Subsequent doses of SBRT may act as a “boost” to reinforce the vaccine effect, which is fundamental in any vaccination program. SBRT = stereotactic body radiotherapy; W=week; APCs = Antigen-presenting cells; NA = Neoantigen; NE = Neoepitope.
Figure 1.
Theoretical Model - Multistep staged SBRT as an in-situ vaccination modality. Intermittent /Repetitive SBRT to harvest the changing spectrum of mutating neoantigens for improved immunotherapy and innate immunity. Subsequent doses of SBRT may act as a “boost” to reinforce the vaccine effect, which is fundamental in any vaccination program. SBRT = stereotactic body radiotherapy; W=week; APCs = Antigen-presenting cells; NA = Neoantigen; NE = Neoepitope.

Figure 2.
Use of SBRT in cyclical approach for “unmasking” and “rewiring” the mutational. pathways, sustaining the immunotherapy response, and invoking/incorporating innate immune mechanisms. Adding neoadjuvant(s) and mitigating concomitant immune resistance to amplify the virtuous cycle would be an additional stratagem. SBRT = stereotactic body radiotherapy; CTLs = Cytotoxic T lymphocytes; TME = Tumor Micro Environment;.
Figure 2.
Use of SBRT in cyclical approach for “unmasking” and “rewiring” the mutational. pathways, sustaining the immunotherapy response, and invoking/incorporating innate immune mechanisms. Adding neoadjuvant(s) and mitigating concomitant immune resistance to amplify the virtuous cycle would be an additional stratagem. SBRT = stereotactic body radiotherapy; CTLs = Cytotoxic T lymphocytes; TME = Tumor Micro Environment;.
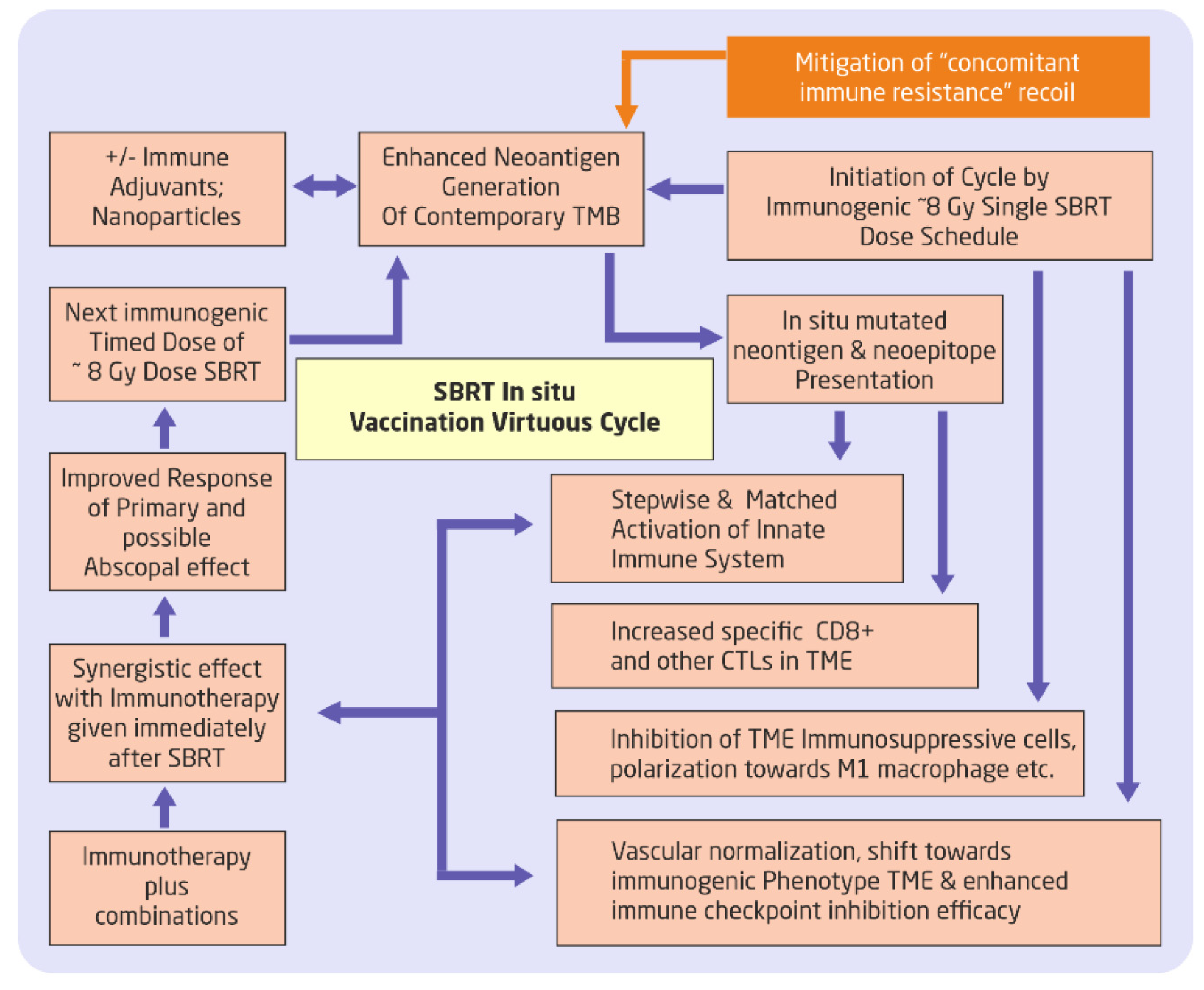
Figure 3.
Optimization of three fundamental immune modulation factors by the immunogenic dose of 8 Gy SBRT in the background of vascular normalization will boost the in situ self-sustaining vaccination effects as the mutational changes progress. With the proposed approach, the toxicity of combined modality is expected to decrease steeply along with the ability to titrate the total dose of SBRT cycles depending on the acute toxicities and anticipated to reduce the tumor volume to be treated for subsequent cycles of SBRTs. SBRT = Stereotactic Body Radiotherapy.
Figure 3.
Optimization of three fundamental immune modulation factors by the immunogenic dose of 8 Gy SBRT in the background of vascular normalization will boost the in situ self-sustaining vaccination effects as the mutational changes progress. With the proposed approach, the toxicity of combined modality is expected to decrease steeply along with the ability to titrate the total dose of SBRT cycles depending on the acute toxicities and anticipated to reduce the tumor volume to be treated for subsequent cycles of SBRTs. SBRT = Stereotactic Body Radiotherapy.
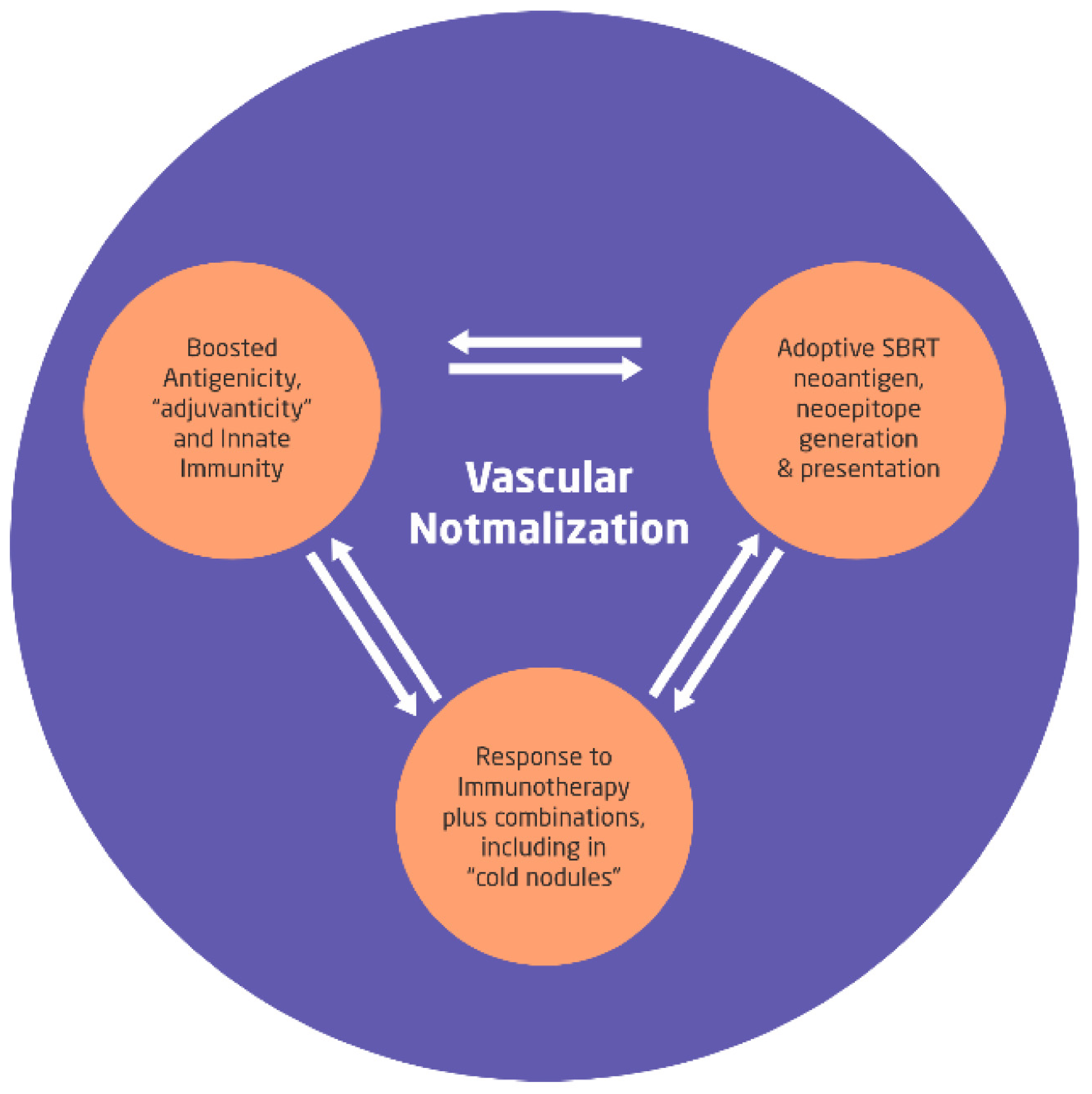
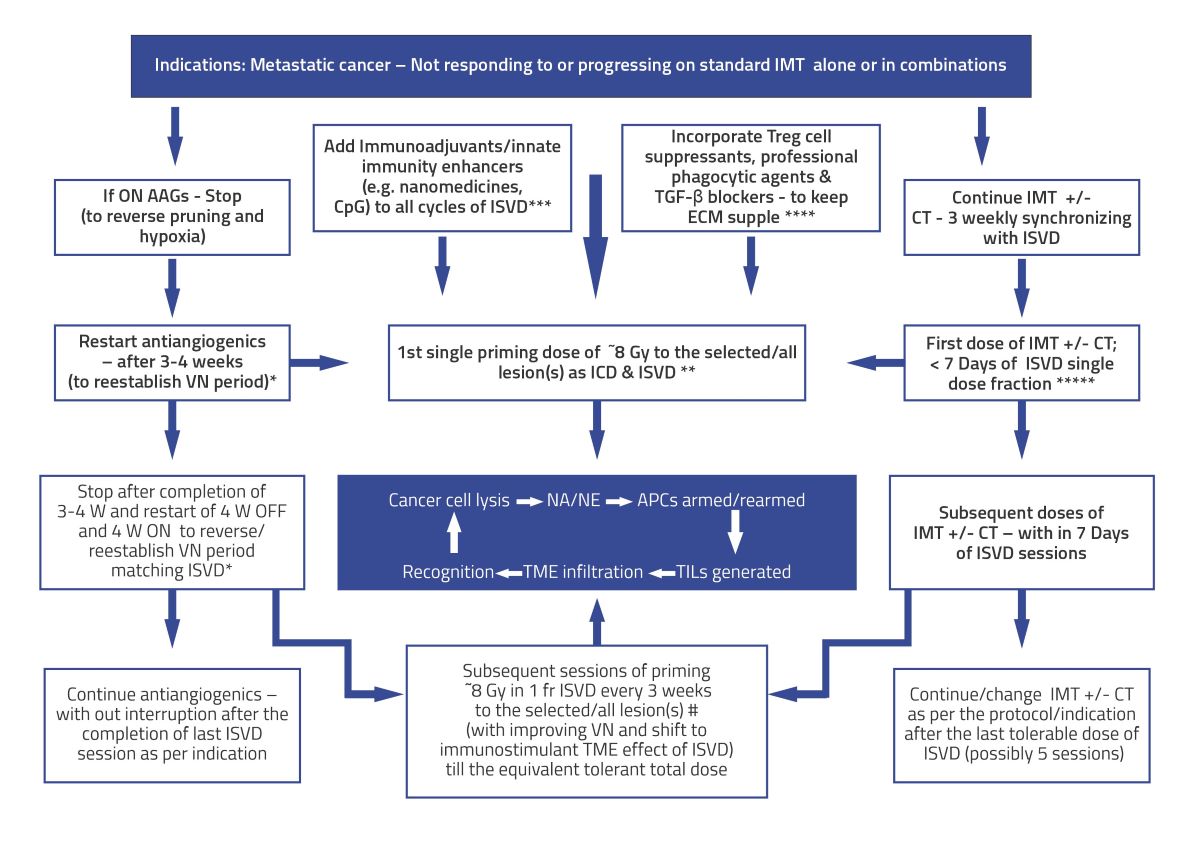
Figure 4.
Algorithm for validating Hypothesis using Pulsed/Intermittent /Repetitive situ vaccination dose (ISVD) RT in metastatic cancer after first-line radical therapies. The objective is to harvest the changing spectrum of mutating neoantigens/neoepitopes for improved immunotherapy and innate immunity. Also, ISVD “unmasks” and “rewires” the mutational pathways de novo, sustaining the immunotherapy response and invoking/incorporating/enhancing natural/innate immune mechanisms. Subsequent doses of ISVD synchronized with immunotherapy or its combinations may act as a “boost” to reinforce the vaccine effect, which is fundamental in any vaccination program. ISVD = In Situ Vaccination Dose RT; W = Week; AAGs = Antiangiogenics; ICD = immunogenic cell death; VN = vascular normalization; TME = Tumor Micro Environment; APCs = Antigen-presenting cells; ICD = Immunogenic cell death.; NA = Neoantigens; NE = Neoepitopes; TILs = Tumor-infiltrating lymphocytes; IMT = immunotherapy; CT = chemotherapy; CpG = cytosine-phosphate-guanine oligo deoxy nucleotide, commonly used adjuvant for innate immune stimulation; * = add-on - cyclical AAGs; ** = Immunogenic ISVD single dose; *** & **** = combination immune enhancing and crosstalk options; ***** = Synergistic IMT and ISVD strategy; # = “hottest higher SUV value” volume/lesions ISVD RT likely to have better cell kill and more significant neoantigen/neoepitope generation; Central blue box = Representation of the virtuous cycle with ISVD RT pulsed dose.
Figure 4.
Algorithm for validating Hypothesis using Pulsed/Intermittent /Repetitive situ vaccination dose (ISVD) RT in metastatic cancer after first-line radical therapies. The objective is to harvest the changing spectrum of mutating neoantigens/neoepitopes for improved immunotherapy and innate immunity. Also, ISVD “unmasks” and “rewires” the mutational pathways de novo, sustaining the immunotherapy response and invoking/incorporating/enhancing natural/innate immune mechanisms. Subsequent doses of ISVD synchronized with immunotherapy or its combinations may act as a “boost” to reinforce the vaccine effect, which is fundamental in any vaccination program. ISVD = In Situ Vaccination Dose RT; W = Week; AAGs = Antiangiogenics; ICD = immunogenic cell death; VN = vascular normalization; TME = Tumor Micro Environment; APCs = Antigen-presenting cells; ICD = Immunogenic cell death.; NA = Neoantigens; NE = Neoepitopes; TILs = Tumor-infiltrating lymphocytes; IMT = immunotherapy; CT = chemotherapy; CpG = cytosine-phosphate-guanine oligo deoxy nucleotide, commonly used adjuvant for innate immune stimulation; * = add-on - cyclical AAGs; ** = Immunogenic ISVD single dose; *** & **** = combination immune enhancing and crosstalk options; ***** = Synergistic IMT and ISVD strategy; # = “hottest higher SUV value” volume/lesions ISVD RT likely to have better cell kill and more significant neoantigen/neoepitope generation; Central blue box = Representation of the virtuous cycle with ISVD RT pulsed dose.
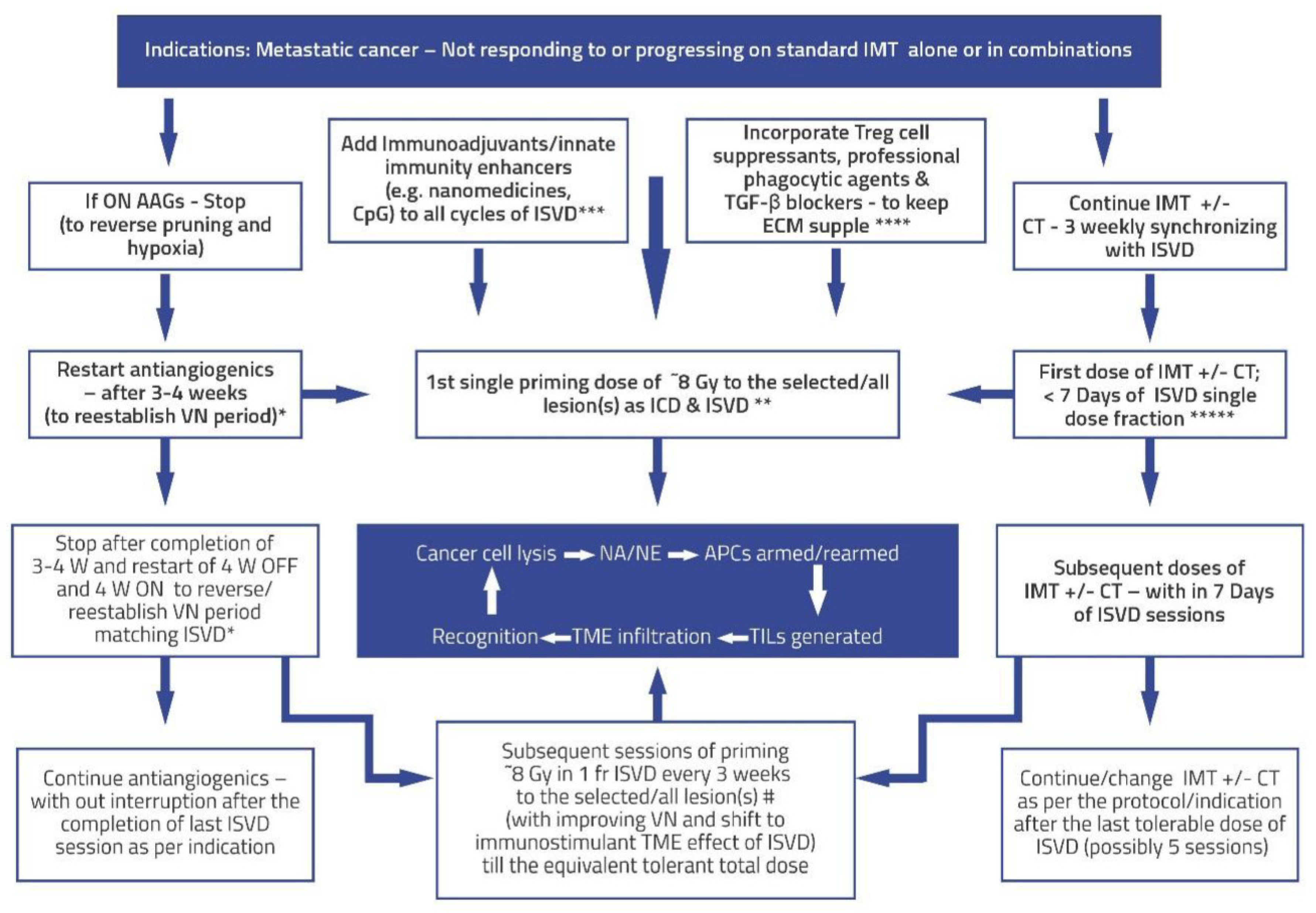
The following are some points projected in Figure 4. The algorithm for validating these points (an essential component for any hypothesis) surrounding the ISVD RT hypothesis is enumerated in Figure 4.
- The indications for this approach would be cancers with disseminated metastatic disease not responding or progressing on IMT+/- CT/targeted therapy, +/- SBRT. The ISVD RT schedule proposed here will be used to cause local response (both direct and immunological) and induce a consistent, predictable abscopal effect. Expansion of further indications depends on the preclinical and clinical trials with the proposed approach.
- The first step in improving the effects of ISVD RT is the normalization of vasculature to reduce hypoxia and sensitize the cancer cells to radiation. Presently, one of the best-known vascular normalization groups of drugs is antiangiogenics, the effects of which usually last 28 days, called the “normalization window,” and reversible by giving drug holidays of about one month in between each cycle. After this window period, on the continuation of antiangiogenics, there will be excessive-extensive pruning of blood vessels, thickened pericyte, increased deposition in the ECM, and development of alternative angiogenic pathways such as vessel co-option with progressive worsening drug-resistant hypoxia. Reintroduction of the same or similar antiangiogenics reestablishes vascular normalization. Also, a lower dose range of antiangiogenics therapy can be more effective than the higher dose due to reduced excessive thickness of pericyte coverage and better oxygen diffusion (54, 55). Theoretically, this cycle can be repeated (Figure 1, Figure 2 and Figure 3). Anti-VEGF drugs also cause restoration of the vessel patency with improved blood flow and repair pericytes with a baseline phenotype within seven days. Regression on stopping the antiangiogenic drug is restored upon restarting the drug (56).
- In the present day, the understanding of radiotherapy is undergoing a sea change, with irradiated Tumors becoming an immunogenic hub for cancer elimination, radiotherapy acting as an efficient in situ vaccine generator, and a systemic disease modifier to reject the metastases. Successful radiotherapy provides lifetime immunological memory (cryptic vaccine); in others, dormancy and immune editing lead to immune escape (3). The pro-immunogenic effects of radiotherapy can be harnessed effectively by correcting the simultaneous mitigation of immunosuppressive “recoil.” In immunotherapy-resistant patients, radiotherapy can restore the response and are amenable to more complex immunotherapy manipulations (3).
- SBRT (unlike conventional RT), although it produces enhanced cell kill per fraction dose along with neoantigen generation, also induces immune resistance “recoil” by at least four major mechanisms: a. Vascular disruption when the SBRT dose is >10 Gy per fraction (12); b. Treg cell proliferation and activation (35); c. Two peaks of BMDCs mobilization, one immediately, the other at two weeks (12); and 4. Activation of Trex-1 pathway (29). Overall, dose per fraction of < = 10 Gy per fraction mitigates almost all these immune resistance recoil pathways on each dose of ISVD RT and forms one of the foundational basis of the proposed strategy.
- IMT can sensitize tumors for subsequent radiotherapy, reducing the required radiation dose (57), supporting the concept of ISVD RT presented here. Thus, with the proposed schedule synchronizing the proliferating cancer cells to susceptible cell cycle phases, RT, chemotherapy (CT), and IMT sensitize each other during systemic therapies. Additionally, with the ISVD RT approach, one controls dose modification and radiation volume depending on the interim response.
- Enhancing the vascular normalization, professional phagocytosis, decreasing the ISP, improving neoantigen lymphatic drainage, mitigating nonspecific antigen flood, and the importance of supple extracellular matrix for effective in-situ immune response are discussed elsewhere (10).
- Avoiding RT dose wastage is possible by not delivering during the hypoxic phase but timing it with each improved degree of oxygenation expected with ongoing tumor response during the ISVD RT. Also, the approach can enhance innate immune mechanisms by decreasing tumor burden and increasing the lymphocytes, possibly arming the memory cells against future mutations, recurrence, and second malignancies.
- Regarding CAR-T cell therapy (28), even though the appropriate dose per fraction needs to be decided for combined ISVD RT and CAR-T cell therapy, the proposed principles in this article of single dose fractions delivered before each cycle of CAR-T cell therapy over a while, keeping watch on the toxicities can be followed in future trials.
- A study indicates that even a single immunogenic radiation dose may be sufficient for a response (32) supporting the ISVD RT approach with a multiplier effect.
- In patients with disseminated metastases, since the technique is explicitly aimed at “in situ vaccine generation,” one could select safer locations of metastases away from critical areas for ISVD RT delivery, partial volumes (58), and low dose areas near the vital structures. In situ vaccination of SBRT effects may be diversified, intensified, amplified, and sustained indefinitely (with activation of memory T cells) by cyclical ISVD RT, keeping the total equivalent dose within acceptable toxicity levels by adopting the titration principle.
- The points to be considered to reduce the possible toxicities of combined therapy are: a). Limit the dose per fraction to less than 10 Gy and as low as 5 Gy per fraction to avoid vascular endothelial disruption and senescence, which causes fibrosis in the long term. b). Titration of dose per fraction and total dose of SBRT: Reduce the subsequent dose per fraction in case of the appearance of higher-grade acute toxicities or skip the further fractions altogether. It can be reintroduced with later IMT cycles in case of the persistence of residual lesions if not operable. c) Prevention: i). Selecting the non-critical areas of metastases for antigen generation strategy when selected lesions are treated in disseminated metastases. ii). Limitation of the radiation volume in large lesions or the one near the critical structures by adopting sub-volume treatment techniques described by Tubin et al. (58). iii). Incorporate trials to maintain the suppleness of ECM by TGF-β blockers (52) or adopt embryonic stem cell reversal techniques to normalize the treated area (59).
- Conventional dose fractionation schedule has taught us that cells become resistant if radiation overall time is too prolonged. SBRT can create both antigenicity and adjuvanticity (29). Hence, ISVD RT here is primarily used for its ability to facilitate immunotherapy tumor response under these circumstances. However, considering the time gap in the pulsed approach, TDF model redesigning is necessary to arrive at a maximum total dose that can be used for ISVD RT.
- According to the present author, analyzing and forming comprehensive combinations for the constantly shifting trunk and branch mutational pattern can be obviated by ISVD RT delivered in divided doses targeting the prevailing trunk mutations just before each cycle of IMT/targeted therapies without excluding other potential in situ local treatments or in vitro approaches. Nanomedicines can potentially bridge the gap in all steps of in situ vaccination strategies (60).
- Radiotherapy is usually synergistic and rarely antagonistic with other therapies as long as there are no overlapping significant toxicities. Therefore, as long as vascular integrity is maintained, ISVD RT and different local therapy combinations and immunotherapies are potentially synergistic.
- Using initiation of immune memory cells by SBRT and immunotherapy and not with surgery in an animal model (49), surgery can be sequenced after in situ immunization schedules (akin to total neoadjuvant protocol in carcinoma rectum).
- IMT, with or without other combinations or targeted therapies, can be continued after the ISVD RT course, per the protocol. The initiation and induced clone of memory cells by ISVD RT is expected to facilitate the continuation of standard therapies. In the KEYNOTE-01 trial in non-small cell lung cancer, even at a median of 9.5 months after RT immunotherapy had longer OS and PFS, indicating persistent synergistic benefit within the limitations of a retrospective review of the study (38), indicating the role of memory cells.
Summary & Limitations of the article
Therapeutic SBRT is given as a course in divided fractions, mostly in consecutive days, never tried in divided doses like chemotherapy/ immunotherapy course in clinical practice, and hence needs to be validated in preclinical trials. However, few animal trial results are available. There is a theoretical possibility that during the intervals of ISVD RT doses, repopulation of radioresistant cells can take place. Since ISVD RT is done under the cover of immunotherapy and its combinations with consequent synergistic cancer cell kill, repopulation is expected to be taken care of. Although gaps in the ISVD RT may theoretically encourage an increase in metastases, mouse models have shown pulsed RT with immunotherapy showing lower lung metastases and an increase in survival (15).
In cancer, a high neoantigen burden results in immune escape. Immune cells are prevented from dialogue or “looking” inside the cancer cells with increasing mutational burden, which becomes the characteristic of cold nodules to immunotherapy. Two steps in the immune escape are – 1. Accumulation of neoantigens and absence of immune editing of mutations. 2. Shutting off the receptor prevents immune cell dialogue inside the cancer cells. The signaling network inside the cell gets altered, adopting multiple pathways.
Additionally, resistance develops through several mechanisms. A. Mutations in the existing pathways. B. Rewiring bypassing the blocked pathways. C. Invoking evolutionary new pathways. Therefore, to overcome these changes, the proposed strategy of cyclical ISVD RT facilitates the new approach of treating with the existing drugs/ methods, keeping abreast of the mutational changes of cancer in situ. ISVD RT avoids repeated testing to identify evolving clones with manifestations of resistance and limits the invitro/intratumoral approaches to the bare essentials.
In conclusion, four conditions for cyclical/intermittent single-dose ISVD RT success are: a). Generating significant neoantigens/neoepitopes for dynamic/adoptive in situ vaccination effect; b). To counter the “immunosuppressive recoil” of “concomitant immune tolerance” demonstrated by the non-irradiated lesions (39); c). Expanding the four-dimensional effect of neoantigens quantity, widening the spectrum of antigens in different mutational types, intensifying antigenicity, and adapting to the trunk, branch, and private mutational evolution over time; d). Expanding the repertoire and repeated stimulations – booster vaccine effects and eliciting plus enhancing innate immune memory cells pathways. Both quantity and density of activated CD8+ cells (36) mattes in immune therapy-induced response. e). Vascular normalization is critical for actualizing the SBRT immunological effect (55). Figure 4 is the algorithm to validate all these factors. Since the algorithm is complex and multifactorial, using the multiarm-multistage (MAMS) clinical trials method may be necessary, where multiple experimental treatments testing against a control arm within the same trial have several advantages over time and resource consumption when done as separate trials (61).
The Preprint draft version of the present article is documented (62). Steps that optimize SBRT in the overall context of cancer reversal and the need for vascular normalization as a central theme for CONVERGENT research were published by Swamy K earlier (10).
Funding
no funding.
Conflict of Interest
No conflict of interest.
References
- Mansoori, B. Mohammadi A, Davudian S, Shirjang S, Baradaran B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv Pharm Bull. 2017 Sep; 7(3): 339–348. [CrossRef]
- Cremers C G, Nguyen L K. Network rewiring, adaptive resistance and combating strategies in breast cancer. Cancer Drug Resist 2019;2:1106-26. [CrossRef]
- Formenti SC, Demaria S. Radiotherapy to convert the tumor into an in situ vaccine. J Radiat Res. 2012;53(4):545-556. [CrossRef]
- Hammerich L, Binder A, Brody J D. In situ vaccination: Cancer immunotherapy both personalized and off-the-shelf. MOLECULAR ONCOLOGY 9 (2015) 1966 e1981. [CrossRef]
- Eric, C. Ko E C, Benjamin K T, Formenti S C. Generating antitumor immunity by targeted radiation therapy: Role of dose and fractionation. Advances in Radiation Oncology (2018) 3, 486-493. [CrossRef]
- Richard G, Princiotta M F , Bridona D, Martin W D, Steinberg G D, and De Groot A S. Neoantigen-based personalized cancer vaccines: the emergence of precision cancer immunotherapy EXPERT REVIEW OF VACCINES 2022, VOL. 21, NO. 2, 173–184. [CrossRef]
- Schumacher TN, Schreiber RD. Neo-antigens in cancer immunotherapy. Science (2015) 348(6230):69–74. [CrossRef]
- Swamy, K. Vascular-immuno-phenotypic (VIP) model for locally advanced and oligo-metastatic cancer: A hypothesis. Med Hypotheses. 2021;152:110618. [CrossRef]
- Swamy, K. Stereotactic Body Radiotherapy Immunological Planning—A Review With a Proposed Theoretical Model. Front Oncol. 2022;12:729250. [CrossRef]
- Swamy, K. Vascular normalization and immunotherapy: Spawning a virtuous cycle. Front Oncol. 2022;12:1002957. [CrossRef]
- Goedegebuure RSA, de Klerk LK, Bass AJ, Derks S, Thijssen VLJL. Combining radiotherapy with antiangiogenic therapy and immunotherapy; a therapeutic triad for cancer? Front Immunol (2019) 9:3107. [CrossRef]
- Martinez-Zubiaurre I, Chalmers AJ, Hellevik T. Radiation Induced transformation of immunoregulatory networks in the tumor stroma. Front Immunol (2018) 9:167. [CrossRef]
- Soyfer, V. , Geva, R., Michelson, M. et al. The impact of overall radiotherapy treatment time and delay in initiation of radiotherapy on local control and distant metastases in gastric cancer. Radiat Oncol 9, 81 (2014). [CrossRef]
- Sanz J, Zhao M, Rodríguez N, Granado R, Foro P, Reig A, Membrive I, Algara M. Once-Weekly Hypofractionated Radiotherapy for Breast Cancer in Elderly Patients: Efficacy and Tolerance in 486 Patients. Biomed Res Int. 2018 Mar 15;2018:8321871. [CrossRef]
- Sezen D, Barsoumian HB, He K, Hu Y, Wang Q, Abana CO, Puebla-Osorio N, Hsu EY, Wasley M, Masrorpour F, Wang J, Cortez MA and Welsh JW (2022) Pulsed radıotherapy to mıtıgate hıgh tumor burden and generate ımmune memory. Front. Immunol. 13:984318. [CrossRef]
- Moore C, Hsu CC, Chen WM, Chen BPC, Han C, Story M, et al. Personalized ultrafractionated stereotactic adaptive radiotherapy (PULSAR) in preclinical models enhances single-agent immune checkpoint blockade. Int J Radiat Oncol Biol Phys (2021) 110(5):1306–16. [CrossRef]
- He K, Barsoumian HB, Sezen D, Puebla-Osorio N, Hsu EY, Verma V, et al. Pulsed radiation therapy to improve systemic control of metastatic cancer. Front Oncol (2021) 11:737425. [CrossRef]
- Benoit A, Vogin G, Duhem C, Berchem G, and Janji B. Lighting Up the Fire in the Microenvironment of Cold Tumors: A Major Challenge to Improve Cancer Immunotherapy. Cells. 2023;12(13):1787. [CrossRef]
- Ko EC, Formenti SC. Radiation therapy to enhance tumor immunotherapy: a novel application for an established modality. Int J Radiat Oncol Biol Phys. 2019;104(1):3-4. [CrossRef]
- Koustas E, Trifylli EM, Sarantis P, Papadopoulos N, Papanikolopoulos K, Aloizos G, et al. Exploiting Autophagy-Dependent Neoantigen Presentation in Tumor Microenvironment. Genes (Basel). 2023;14(2):474. [CrossRef]
- Wegiel B, Vuerich M, Daneshmandi S, Seth P. Metabolic switch in the tumor microenvironment determines immune responses to anticancer therapy. Front Oncol (2018) 8:284. 10.3389/fonc.2018.00284] [Schaaf MB, Garg AD and Agostinis P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Disease. (2018) 9:115. [CrossRef]
- Lakatos E, Williams MJ, Schenck RO, Cross WCH, Househam J, Zapata L, et al. Evolutionary dynamics of neoantigens in growing tumors. Nat Genet. 2019;51(5):734-742. [CrossRef]
- Iliadi C, Verset L, BouchartC, Martinive P, Gestel DV, Mohammad Krayem M. The current understanding of the immune landscape relative to radiotherapy across tumor types. Front Immunol. 2023;14:1148692. [CrossRef]
- Wuensch KE, Tan AC. Trunk or branch? Identifying and targeting intratumoral heterogeneity in hepatocellular carcinoma using genomics and patient-derived primary cancer cells. Transl Cancer Res. 2017;6(Suppl 5):S903-S915. [CrossRef]
- Gao Q, Wang ZC, Duan M, Lin YH, Zhou XY, Worthley DL. Cell culture system for analysis of genetic heterogeneity within hepatocellular carcinoma and response to pharmacological agents. Gastroenterology. 2017 Jan;152(1):232-242. [CrossRef]
- Kievit H, Muntinghe-Wagenaar MB, Hijmering-Kappelle LBM, B I Hiddinga BI, Ubbels JF, Wijsman R et al. Safety and tolerability of stereotactic radiotherapy combined with durvalumab with or without tremelimumab in advanced non-small cell lung cancer, the phase I SICI trial. [CrossRef]
- Wirth TC, Kühnel F. Neoantigen Targeting—Dawn of a New Era in CancerImmunotherapy? Front Immunol. 2017 Dec 19;8:1848. [CrossRef]
- Zhong L, Li Y, Muluh T, Wang Y. Combination of CAR-T cell therapy and radiotherapy: Opportunities and challenges in solid tumors (Review). Oncol Lett. 2023;25(4):13867. [CrossRef]
- Appleton E, Hassan J, Chan Wah Hak C, Sivamanoharan N, Wilkins A, Samson A, et al. Kickstarting Immunity in Cold Tumours: Localised Tumour Therapy Combinations With Immune Checkpoint Blockade. Front Immunol. 2021;12:754436. [CrossRef]
- Jagodinsky JC, Morris ZS. Priming and propagating antitumor immunity: Focal hypofractionated radiation for in situ vaccination and systemic targeted radionuclide theranostics for immunomodulation of tumor microenvironments. Semin Radiat Oncol. 2020;30(1):46-54. [CrossRef]
- Moding EJ, Castle KD, Perez BA, Oh P, Min HD, Norris H, et al. Tumor cells, but not endothelial cells, mediate eradication of primary sarcomas by stereotactic body radiation therapy. Sci Transl Med. 2015;7(278):278ra34. [CrossRef]
- Lussier DM, Alspach E, Ward JP, Miceli AP, Runci D, White JM, et al. Radiation-induced neoantigens broaden the immunotherapeutic window of cancers with low mutational loads. Proc Natl Acad Sci U S A. 2022;119(23):e2102611118. [CrossRef]
- Sharabi AB, Nirschl CJ, Kochel CM, Nirschl TR, Francica BJ, Velarde E, et al. Stereotactic radiation therapy augments antigen-specific PD-1-mediated antitumor immune responses via cross-presentation of tumor antigen. Cancer Immunol Res. 2015;3(4):345-355. [CrossRef]
- Ahmed MM, Hodge JW, Guha C, Bernhard EJ, Vikram B, Coleman CN Cancer Immunol Res. 2013 Nov;1(5):280-4. [CrossRef]
- Nelson BE, Adashek JJ, Sheth AA, Subbiah V. Predicting the Abscopal Effect: Associated Tumor Histologic Subtypes and Biomarkers. Mol Cancer Ther. 2023;22(1):16-24. [CrossRef]
- Jiaqiang Wang J, Ge H, Tian Z. Immunotherapy Plus Radiotherapy for the Treatment of Sarcomas: Is There a Potential for Synergism? Onco Targets Ther. 2023;16:385-397. [CrossRef]
- Shibamoto Y, Miyakawa A, Otsuka S, Iwata H. Radiobiology of hypofractionated stereotactic radiotherapy: what are the optimal fractionation schedules?Journal of Radiation Research, Vol. 57, No. S1, 2016, pp. [CrossRef]
- Breen WG, Leventakos K, Dong H, Merrell KW. Radiation and Immunotherapy: Emerging Mechanisms of Synergy. J Thorac Dis. 2020;12(Suppl 1):S41-S51. [CrossRef]
- Morris ZS, Guy EI, Francis DM, Gressett MM, Werner LR, Carmichael LL, et al. In situ tumor vaccination by combining local radiation and tumor-specific antibody or immunocytokine treatments. Cancer Res. 2016;76(13):3929-3941. [CrossRef]
- Liu, Y. , Dong, Y., Kong, L. et al. Abscopal effect of radiotherapy combined with immune checkpoint inhibitors. J Hematol Oncol 11, 104 (2018). [CrossRef]
- Fu Z, Mowday AM, Smaill JB, Hermans IF, Patterson AV. Tumour Hypoxia-Mediated Immunosuppression: Mechanisms and Therapeutic Approaches to Improve Cancer Immunotherapy. Cells. 2021 Apr 24;10(5):1006. [CrossRef]
- Eckert F, Zwirner K, Boeke S, Thorwarth D, Zips D, Huber SM. Rationale for combining radiotherapy and immune checkpoint inhibition for patients with hypoxic tumors. Front Immunol. 2019;10:407. [CrossRef]
- Li SY, Guo YL, Tian JW, Zhang HJ, Li RF, Gong P, Yu ZL. Antitumor Strategies by Harnessing the Phagocytosis of Macrophages. Cancers (Basel). 2023 May 11;15(10):2717. [CrossRef]
- Milano M T, Constine L S, Okunieff P. Normal tissue toxicity after small field hypofractionated stereotactic body radiation, Radiation Oncology3(1)(2008) 36–36. [CrossRef]
- Morris ZS, Guy EI, Werner LR, Carlson PM, Heinze CM, Kler JS, et al.Tumor-specific inhibition of in situ vaccination by distant untreated tumor sites. Cancer Immunol Res. 2017;5(7):517-523. [CrossRef]
- Demaria S, Karsten A, Pilones KA, Vanpouille-Box C, Golden EB, and Formenti SC. The Optimal Partnership of Radiation and Immunotherapy: from Preclinical Studies to Clinical Translation. RADIATION RESEARCH 182, 170–181 (2014). [CrossRef]
- Darragh LB, Oweida AJ, Karam SD. Overcoming resistance to combination radiation immunotherapy: A focus on contributing pathways within the tumor microenvironment. Front Immunol (2019) 9:3154. [CrossRef]
- Guan H, Wu Y, Li L, Yang Y, Qiu S, Zhao Z, et al. Tumorneoantigens: Novel strategies for application of cancer immunotherapy. Oncol Rep. 2021;46(3):9. [CrossRef]
- Baniel CC, Heinze CM, Hoefges A, SumiecEG, Hank JA, Peter M Carlson PM et al. In situ vaccine plus checkpoint blockade induces memory humoral response. Front Immunol. 2020;11:1610. [CrossRef]
- Sheen MR, Fiering S. In situ vaccination: Harvesting low hanging fruit on the cancer immunotherapy tree. WIREsNanomedNanobiotechnol. 2018;10(4):e1524. [CrossRef]
- Van Den Bijgaart RJE, Schuurmans F, Fütterer JJ, Verheij M, Cornelissen LAM, Adema GJ. Immune modulation plus tumor ablation: Adjuvants and antibodies to prime and boost antitumor immunity in situ. Front Immunol. 2021;12:617365. [CrossRef]
- Zhao Y, Yu X, Li J. Manipulation of immune-vascular crosstalk: New strategies towards cancer treatment. Acta Pharm Sin B. 2020;10(11):2018-2036. [CrossRef]
- Wirsdörfer F, De Leve S, Jendrossek V. Combining radiotherapy and immunotherapy in lung cancer: Can we expect limitations due to altered normal tissue toxicity? Int J Mol Sci. 2019;20(1):24. [CrossRef]
- Fukumura FD, Kloepper J, Amoozgar Z, Duda DG, Rakesh K. Jain. enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol (2018) 15(5):325–40. [CrossRef]
- Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, et al. Normalization of the Vasculature for Treatment of Cancer and Other Diseases. Physiol Rev. 2011;91(3):1071-1121. [CrossRef]
- Mancuso MR, Davis R, Norberg SM, O’Brien S, Sennino B, Nakahara T, et al. Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J Clin Invest (2006) 116(10):2610–21. [CrossRef]
- Gao Z, Zhao Q, Xu Y, Wang L. Improving the efficacy of combined radiotherapy and immunotherapy: Focusing on the effects of radiosensitivity. Radiat Oncol. 2023;17(1):1-13. [CrossRef]
- Tubin S, Popper HH, Brcic L. Novel Stereotactic Body Radiation Therapy (SBRT)-Based Partial Tumor Irradiation Targeting Hypoxic Segment of Bulky Tumors (SBRT-PATHY): Improvement of the Radiotherapy Outcome by Exploiting the Bystander and Abscopal Effects. Radiat Oncol. 2019.
- Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9(4):239-252. [CrossRef]
- Martin JD, Cabral H, Stylianopoulos T, Jain RK. Improving cancer immunotherapy using nanomedicines: progress, opportunities and challenges. Nat Rev Clin Oncol (2020) 17(4):251–66. [CrossRef]
- Magirr D., Jaki T., Whitehead J. A generalized Dunnett test for multi-arm multi-stage clinical studies with treatment selection. Biometrika. 2012;99(2):494–50.
- Swamy, K. Adoptive In Situ Vaccination with Cyclical Intermittent Stereotactic Body Radiotherapy (SBRT) and Immunotherapy – a Review and Proposed Strategy. Preprints 2023, 2023080648. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



