
Submitted:
28 August 2023
Posted:
30 August 2023
You are already at the latest version
Abstract
DNA damage repair lies at the core of all cells’ survival strategy, including cancerous. Therefore, targeting such repair mechanisms forms the major goal of cancer therapeutics. The mechanism of DNA repair has been tousled with the discovery of multiple kinases. Recent studies on Tousled like Kinases have brought significant clarity on the effectors of these kinases which stands to regulate DSB repair. In addition to their well-established role in the DDR and cell cycle checkpoint mediation after DNA damage or inhibitors of replication, their suspected involvement in the actual DSB repair process has more recently been strengthened by the important finding that TLK1 phosphorylates RAD54 and regulates some of its activities and localization in the cell. Earlier findings of its regulation of RAD9 during checkpoint deactivation as well as defined steps during NHEJ ends processing were earlier hints of its important involvement broadly in DSB repair. All this has opened up new avenues to target cancer cells in combination therapy with genotoxins and TLK inhibitors.
Keywords:
TLKs
; DNA repair
; HRR
; NHEJ
; replication
; DNA damage
; therapeutics
1. Introduction
Tousled like Kinases (TLKs) are Ser/Thr kinases which were discovered with potential functions in DNA replication and in DNA damage repair nexus in higher eukaryotes [1,2,3,4]. There are two homologs, Tousled like Kinases 1 and 2 (TLK1 and TLK2) which shares 94% amino acid sequence identity in the kinase domain and an overall 84% identity. There is significant overlap between the substrates of TLK1 and TLK2 so far obtained from in vitro mass-spectrometry in different studies [5]. However, distinct biological roles of the paralogs are forthcoming, as not all their substrates are common. Their function overall was addressed with viability studies with mouse embryos, which showed that TLK1 is not essential during the placental development stages whereas TLK2 is required for placental development. Rather, the biological role of TLK1 was implicated for sensitivity to DNA damage induction, more so in the conditional KO of both proteins. The most well-supported role of TLK1 is in DNA damage response and replication. TLK1 depletion has been shown to delay S-phase progression [6]. This is possible because TLK1 has been found to interact with human RAD9 during replication fork stalling. Phosphorylation of RAD9 (S328) by TLK1 has been shown to dissociate 9-1-1 complex thereby causing cytosolic localization of RAD9 and thus participate in the deactivation of the checkpoint after completion of DNA repair [7]. There has been increasing appreciation for structural domains in kinases that can act as scaffolding units to recruit substrates for their catalytic function. Interestingly TLK1 also possesses chaperone function that recruits RAD9 at the junction of dsDNA and ssDNA at double-strand break (DSB) site [8] Another downstream target of TLK1 is Asf1a/b which is phosphorylated at its C-terminus during S-phase and that leads to binding of H3-H4 tetramer that is assembled on newly replicated DNA. TLK1 can directly bind to chromatin and its interaction with chromatin has been also linked to replication stress. During replication stress TLK1 binding to chromatin decreases [8,9] TLK1B, a splice variant of TLK1 is expressed upon ionizing radiation (IR) exposure and provides radio-resistance to cells. TLK1B overexpression has been also shown to induce UV resistance [10]. TLK1 and TLK1B share the conserved kinase domain and therefore, TLK1 and TLK1B have considerable substrate overlap. One such substrate is Asf1b which is phosphorylated by TLK1B and has been shown to gain chromatin remodeling activity in cell extracts [10]. Similarly, TLK1 dependent phosphorylation of Asf1a leads to an increase in histone octamer recruitment at newly repaired DNA strands [11]. In another model organism T. brucei, TLK1 interacts with Asf1a and Asf1b to phosphorylate the histone chaperones which helps to maintain their activity [6].
2. Role of TLK1 in DSB repair
In vitro, Tousled like Kinase 1 can bind directly with many DNA damage- and DSB repair-related proteins like - NEK1, AKTIP, RAD54B, FANCM [12], and even APEX2 with its established role in synthetic lethality with BRCA2 deficiency [13]. A more specific role of TLK1 in regulating DSB repair via RAD54 has recently been discovered [4]. This is further addressed below, but RAD54 along with its partner RAD51 (the key recombinase of eukaryotes) performs much of the work in the HRR process, which has led to naming RAD54 the Swiss army knife of HRR [14] (Figure 1).
Upon DSB induction by IR, TLK1 is transiently inhibited via a Chk1 dependent mechanism, and hence depletion of Chk1 followed by irradiation did not decrease the TLK1 activity in HeLa cells. The fact that HeLa model cells lack functional p53 therefore suggests that, decrease in TLK1 activity post IR may be p53 independent [3]. Initially TLK1 was peptide mapped to be phosphorylated by Chk1 at Ser 695 that lies within the kinase domain [3] but later studies aligned Ser 743 as the site of phosphorylation that marks TLK1 inactivity [15,16]. The sub-cellular localization of full-length human TLK1 in HeLa cells is mostly nuclear with a diffused signal pattern [1]. Post DSB induction with Mitomycin C, we observed distinct TLK1 foci that partly colocalize with H2A.X foci 2hrs after DNA damage [4]. This suggests that TLK1 plays an important role in DSB repair as most probably it promotes convergence of a hub of factors that function downstream in repair pathways. RAD54 and RAD51 co-localizes to form foci with slightly delayed kinetics than those formed by TLK1 (approx. 4hrs post-induction)), and RAD54-51 foci persist for 10 h. An intriguing speculation is prompted by the pattern of activity of TLKs upon DNA damage, including IR, and the resulting effect on the phosphorylation of RAD54. As previously mentioned, TLK activity drops rapidly following IR [17] and recovers, actually over phosphorylates some of its substrates [4,8], after about one hour in most mammalian cells [8]. This is expected to result in a pattern of rapid de-phosphorylation followed by hyper-phosphorylation of its substrates, including RAD54. This was in fact observed for the specific pT700 site, in both HeLa and U2OS cells [4] which display an initial loss of phosphorylation signal, and then a progressive increase during repair. This is accompanied by the interesting pattern of relocalization of GFP-RAD54 and RAD51 (likely in a complex) from the cytoplasm to the nuclei. GFP-RAD54 shuttles back to the cytoplasm after about 10 hours, which coincides with the timing of completion of the repair of most DSBs [4].
It is a tempting speculation to imagine that RAD54 (and perhaps in association with an ‘inactive’ RAD51 in terms of DNA repair functions) is retained in a cytoplasmic complex by a ‘shuttling’ chaperone that relies in its interaction impinging on the phosphorylation state of RAD54. Following de-phosphorylation of RAD54 after DNA damage and temporary inactivation of TLK1, this results in the shuttling of the complex to the nuclei, where it is recruited to damage sites (namely DSBs) and performs its complex and rather lengthy process of HRR In time, the activity of TLK1 recovers, and re-phosphorylates RAD54, which may be a complex regulatory process accompanying various steps of the HRR (most importantly perhaps, branch migration), and finally its return to the cytoplasm after completion of repair. Suggestive evidence that this, in fact, occurs is provided by the fact that the hyper-phosphorylated form of pRAD54-T700 is all nuclear when analyzed by cellular fractionation without any detectable cytoplasmic signal despite the fact that some (pan) RAD54 is detectable in the cytoplasmic fraction [4]. The most likely involvement of TLK1 in these dynamic processes, apart from the highly specific role in the phosphorylation of T700, is the fact that if the cells are concomitantly treated with J54 (a highly specific TLK inhibitor [18]) during the recovery from IR damage, the GFP-RAD54 is then retained in the nuclei for much longer than the typical 10 hours of recovery, and does not relocate to the cytoplasm as seen for (J54) untreated cells [4]. The speculation is that high level of phosphorylation of pRAD54-T700 must persist during various aspect of the HRR process, perhaps starting from the D-loop formation and subsequently during branch migration with its nucleosomes displacement, but must be tuned down perhaps during RAD51 filament dissolution that must accompany the new synthesis of DNA by the repair polymerase complex using as template the intact homologous chromatid [19]. Perhaps the regulated phosphorylation and de-phosphorylation cycle of RAD54 by TLK1 act as molecular switches to ensure that the right extent of recombinational repair is achieved. Thus, preventing hyper-recombination that has its own significant drawbacks when viewed in the context of normal replication forks progression or regression/degradation recently found to depend on a new RAD54 function [20].
Another aspect worthy of some discussion and speculation is the possible nature of the two substrates of TLK1 that are known to be directly involved in aspects of DSB repair: RAD9, a sensor of DNA damage that travels along the chromatin to detect lesions (including DSBs), and RAD54 that is specifically recruited to DSBs to perform HRR. While formation of RAD54 IRIFs is detected after some time following IR [4], RAD9 on the other hand has been found to form foci rapidly post DNA damage independent of RAD52 group foci localization and this has been assigned to the checkpoint-related function of RAD9 [21] and potentially to a preferential involvement in NHEJ [22,23,24,25], where it is believed to work as the ‘clamp’ to ensure processivity of the repair polymerases to fill-in potentially incompatible ends to facilitate their ligation, a mechanism directly demonstrated in vitro in [26]. While the subject of repair type choice, which clearly involves the particular phase of the cell cycle, has already been much discussed and extensively reviewed [27], we would like to speculate on a possible role of TLK1 in coordinating the cell cycle checkpoint upon IR damage and the repair process itself, irrespective of whether through HRR or NHEJ.
Concurrent with recombinational repair, the DNA damage checkpoint is activated to slow down DNA replication and arrest cells before cell division (G2 phase) until the DNA lesion has been repaired. A functional checkpoint response, first identified in yeast, requires the ATR-related Mec1 kinase and its binding partner Ddc2 (homolog of human ATR-interacting protein, ATRIP), as well as the Rad9 checkpoint protein, the Rad53 (Checkpoint kinase 2 -CHK2) kinase, the Tel1 (ataxia telangiectasia mutated -ATM) kinase, the MRE11/Rad50/Xrs2 (MRX; MRN in humans) complex, the Rad24 (human RAD17)/Rfc2-5 clamp loader, and the Ddc1/Mec3/ Rad17 (human RAD9/HUS1/RAD1) DNA clamp ( [21,28]). These critical checkpoint functions that are largely mediated by ataxia telangiectasia and Rad53-related (ATR) (or its related ATM) and RAD9 to ensure the coordination of the cell cycle progression with sufficient time for repair and finally resumption of the cell cycle after deactivation of the checkpoint. TLK1 was found to be critically involved in this process albeit in higher eukaryotes. On one hand through its already discussed role on phosphorylation of RAD9-S328 during restoration of G2 progression after completion of repair [7] and on the other hand, by mediating the phosphorylation of NEK1-T141, an activating event, thus enabling this key kinase to perform its critical function as a co-activator of ATR upon various types of DNA damage [12,29]. The conservation of proteins related to the DDR and Repair between yeast and human made it so that a large number of studies that are (or were) genetically and biochemically much easier to do with yeast, were later mimicked with mammalian models. Of course, there are no TLKs in either S.cerevisiae or S. pombe, but for example Rad9 is highly conserved and its BRCT domain that interacts with TOPBP1 only when phosphorylated at S387 (human) [30] vs yeast [31], likely by Rad53, suggests very similar regulation of functions.
With different RAD proteins serving as TLK1 substrates, these observations suggest that TLK1 can interact and phosphorylate different RAD proteins and regulate DSB repair, via either HRR or NHEJ, in a concerted and perhaps sequential fashion.
3. Role of TLK1 in regulating HRR factors
Our recent study shows that TLK1 can phosphorylate RAD54 and regulate different stages of HRR. TLK1 phosphorylates RAD54 at both the N-terminal and C-terminal domains [4]. While the N-terminal domain has been shown to be a regulatory domain of RAD54, the C-terminal domain is a critical domain for contacting the double-strand DNA template and therefore serves as functional domain of the protein. Phosphorylation at both T41 and T59 of RAD54 can serve as an interacting platform for several known proteins of HRR for example, RAD51AP1, NUCKS1, CDK2 [32,33,34]. Interestingly, phosphorylation at the C-terminal domain (T700) can alter the intra-protein ionic environment within the Zn-finger like motif thereby causing a major change of interaction with its partner protein RAD51. Depleting TLK1, as shown by our previous study and others, exhibits a delayed S-phase progression phenotype [35,36]. HRR is a major participant of replication fork reversal. As recent studies have shown that RAD54 restrains replication fork progression when cells are stressed [20], the phosphorylation of RAD54 by TLK1 could play a significant role in modulating the dynamics of fork progression vs regression (or even reversal) as a major player for enacting accuracy and processivity at sites where DNA lesions are detected. An earlier explanation for the slow S-phase progression was attributed to a defect in chromatin assembly during the replication process due to the established role of TLKs as regulators of Asf1. However, it was also noted that the phenotypes from depletion of TLKs vs those observed with depletion of Asf1 are quite different with respect to the state of chromatin compaction and cell cycle progression [36], suggesting that TLKs rather perform more complex functions than simply regulating chromatin assembly. For instance, in our previous study, overexpression of a dominant-negative mutant of NEK1, which cannot be phosphorylated by TLK1, implements an intra S-phase checkpoint upon oxidative stress and may cause slow S-phase progression [12].
4. Role of TLK1 in eukaryotic recombination repair
TLK1 activity is important for Homologous recombination repair. Activation of TLK1 activity using Gallic acid has been shown to increase HRR activity in cells [37]. TLK1 depletion by siRNA or shRNA methods across different cell lines has been shown to decrease HRR efficiency in cells significantly [4,35]. In a complementary approach, inhibition of TLK1 with thioridazine, led to a much greater accumulation of γH2Ax foci due to unrepaired DSBs [38].
TLK1 interactome reveals that RIF1 is a possible in vivo target [39,40,41]. RIF1 acts at the decision-making junction of NHEJ vs HRR, downstream of 53BP1 [42,43]. It is known to turn on NHEJ while inhibiting 5’-end resections in HR. Asf1a/b (NTD, 1-154 a.a) has been found to be interacting with RIF1 (N-terminus, 967-1350 a.a) independent of its chaperone activity [44]. Although TLK1 interacts with both Asf1a/b and RIF1, it remains to be elucidated whether TLK1 can impinge on NHEJ vs HRR decision by regulating RIF1, and further if it is dependent on TLK1 kinase activity or its chaperone function. In mammalian cells, RIF1 binds to aberrant telomeres in an ATM-53BP1 dependent manner when telomeres are unprotected and recognized as sites of DNA damage [45]. TLK1 depletion has been shown to increase telomeric sister chromatid exchange, thus indicating a state of hyper-recombination [39]. This implies that TLK1 can regulate telomeric recombination as well, and possibly mediate chromosomes fusion during aberrant cancer hyper-recombination.
TLK1 has been found to interact with another human paralog of RAD54 i.e, RAD54B which in yeast (Rdh54) localizes to DNA damage foci in a RAD52 dependent manner [12,21]. In vitro TLK1 phosphorylates RAD54B at T73 (unpublished data from our lab). Interestingly, the localization of Rdh54 in kinetochores has been shown to be RAD52 independent. Since TLK1 has been shown to function in chromosome segregation in different organisms [35,46,47], it is speculated that TLK1 can regulate RAD54B functions in mitotic or meiotic events of chromosomes dynamics. TLK1 preferentially localizes to the nucleolus even without DNA damage induction in HeLa cells [4]. The nucleolus is the compartment that drives ribosomal biogenesis and therefore, it is speculated TLK1 may play a role in ribosome biogenesis or have a specific role in DSB repair at this site, known to be comprised of highly compacted chromatin and highly repetitive sequences that presents additional challenges [48].
5. TLKs and DNA damage and checkpoint functions
In addition to their functions in repair of DSBs, TLKs are clearly involved in the process of repair of other types of lesions such as UV-induced thymidine dimers (CPDs) [10]and cisplatin-induced ICLs [49]. While we still lack fundamental knowledge of their possible involvement in direct repair of these lesions, a possible explanation for their roles in the repair process, inferred through effects observed in knock-down and overexpression studies, have been tentatively attributed to their role in chromatin assembly and checkpoint functions. While several studies have described these activities, perhaps the most authoritative one was an unbiased siRNA-mediated screen study for kinases directly affecting potential regulators of recovery from DNA damage in U2OS cells, which positively identified TLK2 in modulating the DDR and G2 recovery. Specifically, these authors reported that TLK2 regulates the Asf1A histone chaperone in response to DNA damage and that its loss of function/depletion, resulting in subsequent Asf1A loss, produces a recovery defect. Both TLK2 and Asf1A were required to restore histone H3 incorporation into damaged chromatin. Failure to do so affected the expression of pro-mitotic genes and compromised the cellular competence to recover from damage-induced cell cycle arrests. Their results demonstrated that TLK2 promotes Asf1A functions during the DNA damage response in G2 to allow for proper restoration of chromatin structure at the break site and subsequent recovery from the arrest. Our early in vitro experiments support these findings, as we showed that TLK1B also affords protection against UV radiation, likely through its effects on chromatin remodeling during NER. We found that nuclear extracts isolated from TLK1B-containing mouse cells promote more efficient chromatin assembly than comparable extracts lacking TLK1B. TLK1B-containing extracts are also more efficient in repair of UV-damaged plasmid DNA assembled into nucleosomes. As already mentioned, one of the two earliest known substrates of TLK1 (or TLK1B) is the histone chaperone Asf1, and immuno-inactivation experiments suggest that TLK1B increases UV-repair through the action of Asf1 on chromatin assembly/disassembly [10].
Furthermore, in TLK1 overexpression studies in normal mouse mammary cells, we directly observed in vivo that whereas MM3MG-TLK1B overexpressing cells efficiently repaired almost all the CPDs by 12 hours, control MM3MG cells showed very poor levels of CPD removal, when assessed using the southwestern blotting technique [10]. Another method (with intact cells) was also used to compare the repair abilities of the two cell lines. For this purpose, we assessed the repair of episomal vectors in cells over a time course following exposure to UV. MM3MG cells (that were stably transfected with the empty BK-shuttle vector) and the MM3-TLK1B cells (carrying the TLK1B insert in the same vector) were exposed to 5 J/m2 of UV radiation, and allowed to recover for 0 to12 hours (Figure 3 in [10]). Episomes were extracted at various time points (0, 2, 4, 8 and 12 hours) and were either mock-treated or digested with T4 endonuclease V enzyme, which specifically cuts DNA at CPD-sites. Since the episomes are damaged on both strands at multiple sites, digestion with T4 endonuclease results in extensive cleavage of the recovered plasmid DNA, whereas in unirradiated (or fully repaired) cells T4 endonuclease leaves the plasmids intact. On comparison of the two cell lines (control and TLK1B overexpressing) it was seen that the majority of low molecular weight episomal DNA was recovered into repaired high molecular weight DNA in the MM3G-TLK1B cells between 8 and 12 hours, while the T4 endonuclease treated episomal DNA still appeared as a low molecular weight smear in the MM3MG cells. These two experiments strongly suggest that the MM3G-TLK1B cells repaired DNA faster than the control MM3MG cells. Perhaps, the explanation for these effects of TLK1B in UV-damage repair are much more complex than the simple attribution to its activity on chromatin remodeling, and rather implies that there are still direct targets/substrates of TLKs that are involved in the actual repair process, at least in the case for NER [10].
Beside the aforementioned established role of TLK1 in repair of Cyclo-Pyrimidine Dimers (CPDs), there is emerging evidence for a role in cisplatin-induced ICLs and base adducts, particularly inferred through the effects observed in knock-down studies of cancer cells. Cholangiocarcinoma is a particularly devastating carcinoma with limited treatment options. As Tousled-like kinase 1 (TLK1) is a serine/threonine protein kinase that regulates DNA replication and DNA repair pathways, these authors studied its possible role in sensitization to cisplatin, which is one of the few treatment options for this deadly disease, apart from gemcitabine [50]. First, these authors showed that TLK1 is abundantly expressed in cholangiocarcinoma as well as in cell lines derived from cholangiocarcinoma. Second, although siRNA knockdown of TLK1 did not affect the viability of cholangiocarcinoma cells, it sensitized these to cisplatin-induced apoptosis. Furthermore, immunofluorescence analysis of γH2AX foci showed that silencing of TLK1 enhanced DNA damage induced by cisplatin treatment. Their results clearly support the hypothesis that TLK1 plays a pivotal role in the repair of cisplatin-induced DNA damage.
6. Role of TLK1 in cancer
In different disease models like prostate cancer (PCa), glioblastoma (GBM), TLK1 depletion has been shown to elicit DNA damage response and therefore TLK1 forms a druggable target [51,52,53] (Figure 2). Human cancer data revealed frequent up-regulation of TLK genes and an association with poor patient outcome in multiple types of cancer, and depletion of TLK activity leads to increased replication stress and DNA damage in a panel of cancer cells [54]. GWAS analyses revealed that TLK1 mutations are rare in cancer but rather its overexpression is frequently linked to poor prognosis; this is particularly evident for patients with low Gleason scores (e.g, GS=6 - Ualcan.path.uab.edu/analysis page 2), which would otherwise be expected to fare better survival. Likewise, amplification of TLK2 has been reported as a frequent event in breast cancer [55], GBM [56], and neuroblastoma [57]. Our previous work shows that DNA damage activates the TLK1> NEK1> YAP1 axis, which either further elevates the apoptotic pathway in PCa [4] or can lead to compensatory adaptation to genotoxins [18]. Broadly, it can be said that TLKs can impact cancer ontology and/or progression by regulating genome and epigenome stability [5], as well as potentially suppressing aspects of innate immune signaling [39]. Specifically, on this important interplay of the critical relation between cancer progression and the immune response, it was reported that depletion of TLK activity, apart from causing replication stress, increased chromosomal instability and cell cycle arrest or death. These studies also showed that stalled forks in TLK depleted cells are processed by BLM, SAMHD1 and the MRE11 nuclease to generate ssDNA activating checkpoint signaling. Thus, TLK depletion ultimately impairs heterochromatin maintenance, inducing features of alternative lengthening of telomeres and increasing spurious expression of other repetitive elements, associated with impaired deposition of the histone variant H3.3 [5]. Moreover, TLK2 depletion culminated in a BLM- dependent, STING-mediated innate immune response, whereby in many human cancers, TLK1/2 expression correlated with signatures of chromosomal instability and anti-correlated with STING and innate and adaptive immune response signatures [39]. All this prompts an exciting but challenging new prospective view of how TLKs may operate on different, albeit still understudied, aspects of their involvement in cancer development and progression and as possible target in cancer immunotherapy and maybe in immune checkpoint regulation [58,59], recombination and cytokine signaling [5]. Further important genetic evidence comes from the identification of a novel heterozygous variant of TLK1 in a patient with a neuro-developmental and immunological syndrome. This mutation impaired kinase activity and caused alterations in genes involved in class switch recombination, suggesting potential involvement in central nervous system and immune system development [60].
7. Conclusion: Targeting TLK1 for cancer treatment
After the seminal discoveries that TLK1 has important modulatory role in the DDR and DNA repair, the quest turned rapidly into trying to identify specific TLK inhibitors to enhance the effectiveness of XRT or radiomimetic drugs. This led to the initial identification of certain phenothiazines (PTH) antipsychotics as surprisingly specific TLK inhibitors that in fact enhanced the killing of cancer cells, in vitro and xenografts, when combined to IR or doxorubicin [38]. A newer PTH scaffold, called J54, that substantially lacked anti-dopaminergic undesirable effect, showed very promising results in the regression of androgen-sensitive prostate cancer cells largely by passing the DDR and thereby enforcing entry unto catastrophic mitotic progression, even resulting in substantial tumor regression in SCID xenografts [18]. Other studies on structure/function of TLKs have revealed additional potential inhibitors [61]. Considering the availability of such drugs, of even greater importance now, is the rebound effort to target the activity of TLK1 on RAD9 and RAD54, with the obvious suggestion that both translesion (TSL) repair, single-strand gaps, and HRR can be simultaneously targeted. Various chemotherapeutic agents, therefore, fall under the umbrella of TLK inhibitors for their therapeutic potentiation. These include for example: topoisomerase poisons, bleomycin, MMC, PARPis, and cisplatin, all of which ultimately lead to the formation of SSBs and DSBs. For some of these (e.g., doxorubicin and cisplatin) direct evidence of synthetic lethality in combination with TLK inhibitors has already been verified [38,62]. Of particular note is the reported synthetic lethal interaction of combining knock-down of TLKs with PARP inhibitors [54], which could be expected to engender a double-hit on both NHEJ and HRR based on the already described functions of TLKs. Inhibiting TLK1 prior to radiation in diseases like GBM where base-excision repair is activated [53] or cholangiocarcinoma or prostate cancer where intra-strand crosslink repair is elevated [49] increases their chemo-sensitization, which suggests that TLK1 forms nexus of a vast spectrum of DNA repair mechanisms (Figure 2). In addition to these obvious actionable targets via inhibitors of TLK1 in DNA repair and checkpoint surveillance, more recent functions for TLK1 in disparate important oncogenic pathways, such as regulation of AKT (via AKTIP); the important pro-metastatic kinase MK5; and the ultimate effector of the Hippo pathway, YAP (via NEK1) are coming to light (rev. in [63]). In addition to these, a glimpse into a potentially important role of the TLK1>NEK1 interaction in an anti-apoptotic function related to VDAC1, a key regulator of mitochondrial permeability, was recently revealed in our studies [64], wherein we speculated that a limiting shuttling function of NEK1 between nuclei and mitochondria to regulate competing activities in either compartment, could be a critical decision point between implementing DNA repair vs. prompting apoptosis if the damage is too extensive. In all the important roles of TLKs in various aspects of oncogenic development and as possible targets for various cancer-directed therapies is slowly but surely becoming ‘untousled’.
Author Contributions
Conceptualization: IG and ADB; writing - original draft preparation and review and editing: IG and ADB; resources: ADB; project administration: ADB; funding acquisition: ADB. All authors have read and agreed to the published version of the manuscript.
Funding
Funding for preparation of this review was provided by a Louisiana CCRI grant LSU-2022-CCRI-6.
Institutional Review Board Statement
N/A.
Informed Consent Statement
N/A.
Data Availability Statement
No new unpublished data was included in this review.
Conflicts of Interest
The authors declare that no conflicts exist.
Abbreviations
DSB—double-strand break; DDR—DNA damage response; TLK1—tousled-like kinase 1; HRR—homologous recombination repair; NHEJ—non-homologous end joining.
References
- Silljé, H.H.W., Takahashi, K., Tanaka, K., Van Houwe, G. and Nigg, E.A. [1999] Mammalian homologues of the plant Tousled gene code for cell-cycle-regulated kinases with maximal activities linked to ongoing DNA replication. The EMBO Journal, 18, 5691-5702. [CrossRef]
- Li, Y., DeFatta, R., Anthony, C., Sunavala, G. and De Benedetti, A. [2001] A translationally regulated Tousled kinase phosphorylates histone H3 and confers radioresistance when overexpressed. Oncogene, 20, 726-738. [CrossRef]
- Groth, A., Lukas, J., Nigg, E.A., Silljé, H.H.W., Wernstedt, C., Bartek, J. and Hansen, K. [2003] Human Tousled like kinases are targeted by an ATM- and Chk1-dependent DNA damage checkpoint. The EMBO Journal, 22, 1676-1687. [CrossRef]
- Ghosh, I., Kwon, Y., Shabestari, A.B., Chikhale, R., Chen, J., Wiese, C., Sung, P. and De Benedetti, A. [2023] TLK1-mediated RAD54 phosphorylation spatio-temporally regulates Homologous Recombination Repair. Nucleic Acids Research. [CrossRef]
- Segura-Bayona, S. and Stracker, T.H. [2019] The Tousled-like kinases regulate genome and epigenome stability: implications in development and disease. Cell Mol Life Sci, 76, 3827-3841. [CrossRef]
- Li, Z., Gourguechon, S.p. and Wang, C.C. [2007] Tousled-like kinase in a microbial eukaryote regulates spindle assembly and S-phase progression by interacting with Aurora kinase and chromatin assembly factors. Journal of Cell Science, 120, 3883-3894. [CrossRef]
- Awate, S. and De Benedetti, A. [2016] TLK1B mediated phosphorylation of Rad9 regulates its nuclear/cytoplasmic localization and cell cycle checkpoint. BMC Mol Biol, 17, 3. [CrossRef]
- Sunavala-Dossabhoy, G. and De Benedetti, A. [2009] Tousled homolog, TLK1, binds and phosphorylates Rad9; tlk1 acts as a molecular chaperone in DNA repair. DNA Repair, 8, 87-102. [CrossRef]
- Klimovskaia, I.M., Young, C., Stromme, C.B., Menard, P., Jasencakova, Z., Mejlvang, J., Ask, K., Ploug, M., Nielsen, M.L., Jensen, O.N. et al. [2014] Tousled-like kinases phosphorylate Asf1 to promote histone supply during DNA replication. Nat Commun, 5, 3394. [CrossRef]
- Sen, S. and De Benedetti, A. [2006] TLK1B promotes repair of UV-damaged DNA through chromatin remodeling by Asf1. BMC Mol Biol., 7, 37. [CrossRef]
- Klimovskaia, I.M., Young, C., Strømme, C.B., Menard, P., Jasencakova, Z., Mejlvang, J., Ask, K., Ploug, M., Nielsen, M.L., Jensen, O.N. et al. [2014] Tousled-like kinases phosphorylate Asf1 to promote histone supply during DNA replication. Nature Communications, 5, 3394. [CrossRef]
- Singh, V., Connelly, Z.M., Shen, X. and De Benedetti, A. [2017] Identification of the proteome complement of humanTLK1 reveals it binds and phosphorylates NEK1 regulating its activity. Cell Cycle, 16, 915-926. [CrossRef]
- Mengwasser, K.E., Adeyemi, R.O., Leng, Y., Choi, M.Y., Clairmont, C., D'Andrea, A.D. and Elledge, S.J. [2019] Genetic Screens Reveal FEN1 and APEX2 as BRCA2 Synthetic Lethal Targets. Mol Cell, 73, 885-899.e886. [CrossRef]
- Kiianitsa, K., Solinger Jachen, A. and Heyer, W.-D. [2006] Terminal association of Rad54 protein with the Rad51–dsDNA filament. Proceedings of the National Academy of Sciences, 103, 9767-9772. [CrossRef]
- Elisabeth, K., Reinhard, K., Hak, C. and Debabrata, S. [2008] Sustained Metaphase Arrest in Response to Ionizing Radiation in a Non-small Cell Lung Cancer Cell Line. Radiation Research, 169, 46-58. [CrossRef]
- Kelly, R. and Davey, S.K. [2013] Tousled-Like Kinase-Dependent Phosphorylation of Rad9 Plays a Role in Cell Cycle Progression and G2/M Checkpoint Exit. PLOS ONE, 8, e85859. [CrossRef]
- Kodym, R., Mayerhofer, T. and Ortmann, E. [2004] Purification and identification of a protein kinase activity modulated by ionizing radiation. Biochem Biophys Res Commun., 313, 97-103. [CrossRef]
- Singh, V., Bhoir, S., Chikhale, R.V., Hussain, J., Dwyer, D., Bryce, R.A., Kirubakaran, S. and De Benedetti, A. [2020] Generation of phenothiazine with potent anti-TLK1 activity for prostate cancer therapy. Iscience, 23, 101474. [CrossRef]
- Adkins, N.L., Niu, H., Sung, P. and Peterson, C.L. [2013] Nucleosome dynamics regulates DNA processing. Nat Struct Mol Biol, 20, 836-842. [CrossRef]
- Uhrig, M.E., Sharma, N., Maxwell, P., Selemenakis, P. and Wiese, C. [2023] RAD54L regulates replication fork progression and nascent strand degradation in BRCA1/2-deficient cells. bioRxiv. [CrossRef]
- Lisby, M., Barlow, J.H., Burgess, R.C. and Rothstein, R. [2004] Choreography of the DNA Damage Response: Spatiotemporal Relationships among Checkpoint and Repair Proteins. Cell, 118, 699-713. [CrossRef]
- Delacroix, S., Wagner, J.M., Kobayashi, M., Yamamoto, K.-i. and Karnitz, L.M. [2007] The Rad9–Hus1–Rad1 (9–1–1) clamp activates checkpoint signaling via TopBP1. Genes & Development, 21, 1472-1477. [CrossRef]
- Zhu, A., Zhang, C.X. and Lieberman, H.B. [2008] Rad9 Has a Functional Role in Human Prostate Carcinogenesis. Cancer Research, 68, 1267-1274. [CrossRef]
- Post, S.M., Tomkinson, A.E. and Lee, E.Y.H.P. [2003] The human checkpoint Rad protein Rad17 is chromatin-associated throughout the cell cycle, localizes to DNA replication sites, and interacts with DNA polymerase ϵ. Nucleic Acids Research, 31, 5568-5575. [CrossRef]
- Lin, C.-Y., Chang, H.-H., Wu, K.-J., Tseng, S.-F., Lin, C.-C., Lin, C.-P. and Teng, S.-C. [2005] Extrachromosomal Telomeric Circles Contribute to Rad52-, Rad50-, and Polymerase δ-Mediated Telomere-Telomere Recombination in Saccharomyces cerevisiae. Eukaryotic Cell, 4, 327-336. [CrossRef]
- Canfield, C., Rains, J. and De Benedetti, A. [2009] TLK1B promotes repair of DSBs via its interaction with Rad9 and Asf1. BMC Mol Biol, 10, 110. [CrossRef]
- Brandsma, I. and Gent, D.C. [2012] Pathway choice in DNA double strand break repair: observations of a balancing act. Genome Integr, 3, 9. [CrossRef]
- Melo, J. and Toczyski, D. [2002] A unified view of the DNA-damage checkpoint. Curr Opin Cell Biol, 14, 237-245. [CrossRef]
- Liu, S., Ho, C.K., Ouyang, J. and Zou, L. [2013] Nek1 kinase associates with ATR-ATRIP and primes ATR for efficient DNA damage signaling. Proc Natl Acad Sci U S A, 110, 2175-2180. [CrossRef]
- Day, M., Rappas, M., Ptasinska, K., Boos, D., Oliver, A.W. and Pearl, L.H. [2018] BRCT domains of the DNA damage checkpoint proteins TOPBP1/Rad4 display distinct specificities for phosphopeptide ligands. eLife, 7, e39979. [CrossRef]
- Hammet, A., Magill, C., Heierhorst, J. and Jackson, S.P. [2007] Rad9 BRCT domain interaction with phosphorylated H2AX regulates the G1 checkpoint in budding yeast. EMBO Rep, 8, 851-857. [CrossRef]
- Goyal, N., Rossi, M.J., Mazina, O.M., Chi, Y., Moritz, R.L., Clurman, B.E. and Mazin, A.V. [2018] RAD54 N-terminal domain is a DNA sensor that couples ATP hydrolysis with branch migration of Holliday junctions. Nature Communications, 9, 34. [CrossRef]
- Maranon, D.G., Sharma, N., Huang, Y., Selemenakis, P., Wang, M., Altina, N., Zhao, W. and Wiese, C. [2020] NUCKS1 promotes RAD54 activity in homologous recombination DNA repair. Journal of Cell Biology, 219. [CrossRef]
- Selemenakis, P., Sharma, N., Uhrig, M.E., Katz, J., Kwon, Y., Sung, P. and Wiese, C. [2022] RAD51AP1 and RAD54L Can Underpin Two Distinct RAD51-Dependent Routes of DNA Damage Repair via Homologous Recombination. Frontiers in Cell and Developmental Biology, 10. [CrossRef]
- Sunavala-Dossabhoy, G., Li, Y., Williams, B. and De Benedetti, A. [2003] A dominant negative mutant of TLK1 causes chromosome missegregation and aneuploidy in normal breast epithelial cells. BMC Cell Biology, 4, 16. [CrossRef]
- Lee, S.-B., Segura-Bayona, S., Villamor-Payà, M., Saredi, G., Todd, M.A.M., Attolini, C.S.-O., Chang, T.-Y., Stracker, T.H. and Groth, A. Tousled-like kinases stabilize replication forks and show synthetic lethality with checkpoint and PARP inhibitors. Science Advances, 4, eaat4985. [CrossRef]
- Timiri Shanmugam, P.S., Nair, R.P., De Benedetti, A., Caldito, G., Abreo, F. and Sunavala-Dossabhoy, G. Tousled kinase activator, gallic acid, promotes homologous recombinational repair and suppresses radiation cytotoxicity in salivary gland cells. [CrossRef]
- Ronald, S., Awate, S., Rath, A., Carroll, J., Galiano, F., Dwyer, D., Kleiner-Hancock, H., Mathis, J.M., Vigod, S. and De Benedetti, A. [2013] Phenothiazine Inhibitors of TLKs Affect Double-Strand Break Repair and DNA Damage Response Recovery and Potentiate Tumor Killing with Radiomimetic Therapy. Genes Cancer, 4, 39-53. [CrossRef]
- Segura-Bayona, S., Villamor-Payà, M., Attolini, C.S., Koenig, L.M., Sanchiz-Calvo, M., Boulton, S.J. and Stracker, T.H. [2020] Tousled-Like Kinases Suppress Innate Immune Signaling Triggered by Alternative Lengthening of Telomeres. Cell Rep, 32, 107983. [CrossRef]
- Sukackaite, R., Cornacchia, D., Jensen, M.R., Mas, P.J., Blackledge, M., Enervald, E., Duan, G., Auchynnikava, T., Köhn, M., Hart, D.J. et al. [2017] Mouse Rif1 is a regulatory subunit of protein phosphatase 1 (PP1). Sci Rep, 7, 2119. [CrossRef]
- Yang, S., Liu, L., Cao, C., Song, N., Wang, Y., Ma, S., Zhang, Q., Yu, N., Ding, X., Yang, F. et al. [2018] USP52 acts as a deubiquitinase and promotes histone chaperone ASF1A stabilization. Nat Commun, 9, 1285. [CrossRef]
- Escribano-Díaz, C., Orthwein, A., Fradet-Turcotte, A., Xing, M., Young, Jordan T.F., Tkáč, J., Cook, Michael A., Rosebrock, Adam P., Munro, M., Canny, Marella D. et al. [2013] A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Molecular Cell, 49, 872-883. [CrossRef]
- Batenburg, N.L., Walker, J.R., Noordermeer, S.M., Moatti, N., Durocher, D. and Zhu, X.-D. [2017] ATM and CDK2 control chromatin remodeler CSB to inhibit RIF1 in DSB repair pathway choice. Nature Communications, 8, 1921. [CrossRef]
- Tang, M., Chen, Z., Wang, C., Feng, X., Lee, N., Huang, M., Zhang, H., Li, S., Xiong, Y. and Chen, J. [2022] Histone chaperone ASF1 acts with RIF1 to promote DNA end-joining in BRCA1-deficient cells. Journal of Biological Chemistry, 101979. [CrossRef]
- Xu, L. and Blackburn, E.H. [2004] Human Rif1 protein binds aberrant telomeres and aligns along anaphase midzone microtubules. Journal of Cell Biology, 167, 819-830. [CrossRef]
- Carrera, P., Moshkin, Y.M., Grönke, S., Silljé, H.H.W., Nigg, E.A., Jäckle, H. and Karch, F. [2003] Tousled-like kinase functions with the chromatin assembly pathway regulating nuclear divisions. Genes & Development, 17, 2578-2590. [CrossRef]
- Han, Z., Riefler, G.M., Saam, J.R., Mango, S.E. and Schumacher, J.M. [2005] The C. elegans Tousled-like Kinase Contributes to Chromosome Segregation as a Substrate and Regulator of the Aurora B Kinase. Current Biology, 15, 894-904. [CrossRef]
- Korsholm, L.M., Gál, Z., Nieto, B., Quevedo, O., Boukoura, S., Lund, C.C. and Larsen, D.H. [2020] Recent advances in the nucleolar responses to DNA double-strand breaks. Nucleic Acids Research, 48, 9449-9461. [CrossRef]
- Takayama, Y., Kokuryo, T., Yokoyama, Y., Ito, S., Nagino, M., Hamaguchi, M. and Senga, T. [2010] Silencing of Tousled-like kinase 1 sensitizes cholangiocarcinoma cells to cisplatin-induced apoptosis. Cancer Lett, 296, 27-34. [CrossRef]
- Lairmore, T.C., Abdulsattar, J., De Benedetti, A., Shi, R., Huang, S., Khalil, M.I. and Witt, S.N. [2023] Loss of tumor suppressor menin expression in high grade cholangiocarcinomas. BMC Research Notes, 16, 15. [CrossRef]
- Singh, V., Jaiswal, P.K., Ghosh, I., Koul, H.K., Yu, X. and De Benedetti, A. [2019] The TLK1-Nek1 axis promotes prostate cancer progression. Cancer Letters, 453, 131-141. [CrossRef]
- Singh, V., Jaiswal, P.K., Ghosh, I., Koul, H.K., Yu, X. and De Benedetti, A. [2019] Targeting the TLK1/NEK1 DDR axis with Thioridazine suppresses outgrowth of androgen independent prostate tumors. International Journal of Cancer, 145, 1055-1067.
- Ibrahim, K., Abdul Murad, N.A., Harun, R., Jamal, R., Ibrahim, K., Abdul Murad, N.A., Harun, R., Jamal, R., Ibrahim, K., Abdul Murad, N.A. et al. [2020] Knockdown of Tousled-like kinase 1 inhibits survival of glioblastoma multiforme cells. Int J Mol Med, 46, 685-699. [CrossRef]
- Lee, S.-B., Segura-Bayona, S., Villamor-Payà, M., Saredi, G., Todd, M.A.M., Attolini, C.S.-O., Chang, T.-Y., Stracker, T.H. and Groth, A. [2018] Tousled-like kinases stabilize replication forks and show synthetic lethality with checkpoint and PARP inhibitors. Science Advances, 4, eaat4985. [CrossRef]
- Kim, J.-A., Anurag, M., Veeraraghavan, J., Schiff, R., Li, K. and Wang, X.-S. [2016] Amplification of TLK2 Induces Genomic Instability via Impairing the G2–M Checkpoint. Molecular Cancer Research, 14, 920-927.
- Lin, M., Yao, Z., Zhao, N. and Zhang, C. [2019] TLK2 enhances aggressive phenotypes of glioblastoma cells through the activation of SRC signaling pathway. Cancer Biol Ther, 20, 101-108. [CrossRef]
- Van Roy, N., Vandesompele, J., Berx, G., Staes, K., Van Gele, M., De Smet, E., De Paepe, A., Laureys, G., van der Drift, P., Versteeg, R. et al. [2002] Localization of the 17q breakpoint of a constitutional 1;17 translocation in a patient with neuroblastoma within a 25-kb segment located between the ACCN1 and TLK2 genes and near the distal breakpoints of two microdeletions in neurofibromatosis type 1 patients. Genes Chromosomes Cancer, 35, 113-120. [CrossRef]
- Shaaban, M., Othman, H., Ibrahim, T., Ali, M., Abdelmoaty, M., Abdel-Kawi, A.R., Mostafa, A., El Nakeeb, A., Emam, H. and Refaat, A. [2020] Immune Checkpoint Regulators: A New Era Toward Promising Cancer Therapy. Curr Cancer Drug Targets, 20, 429-460. [CrossRef]
- Lentz, R.W., Colton, M.D., Mitra, S.S. and Messersmith, W.A. [2021] Innate Immune Checkpoint Inhibitors: The Next Breakthrough in Medical Oncology? Mol Cancer Ther, 20, 961-974. [CrossRef]
- Villamor-Paya, M., Sanchiz-Calvo, M., Smak, J., Pais, L., Sud, M., Shankavaram, U., Lovgren, A.K., Austin-Tse, C., Ganesh, V.S., Gay, M. et al. [2023] Identification of a de novo mutation in TLK1 associated with a neurodevelopmental disorder and immunodeficiency. medRxiv, 2023.2008.2022.23294267. [CrossRef]
- Mortuza, G.B., Hermida, D., Pedersen, A.-K., Segura-Bayona, S., López-Méndez, B., Redondo, P., Rüther, P., Pozdnyakova, I., Garrote, A.M., Muñoz, I.G. et al. [2018] Molecular basis of Tousled-Like Kinase 2 activation. Nature Communications, 9, 2535. [CrossRef]
- Bhoir, S. and De Benedetti, A. [2023] Targeting Prostate Cancer, the ‘Tousled Way&rsquo. International Journal of Molecular Sciences. [CrossRef]
- Khalil, M.I. and De Benedetti, A. [2022] Tousled-like kinase 1: a novel factor with multifaceted role in mCRPC progression and development of therapy resistance. Cancer Drug Resistance, 5, 93-101. [CrossRef]
- Singh, V., Khalil, M.I. and De Benedetti, A. [2020] The TLK1/Nek1 axis contributes to mitochondrial integrity and apoptosis prevention via phosphorylation of VDAC1. Cell Cycle, 19, 363-375. [CrossRef]
Figure 1.
Mechanistic role of TLK1 in DNA damage repair (DDR). TLK1 (light purple) can heterodimerize with TLK2 (cyan) in certain cell types and normal kinase activity of TLK1 is dependent on homodimerization/oligomerization and/or heterodimerization. The N-terminal region of TLK1/TLK2 is essential for higher order arrangement. In cells, when DSB is induced with IR, there is a transient decrease in TLK1 activity (red arrow) which is restored within an hour (in HeLa cells). The inhibition of TLK1 is dependent on ATM-Chk2 phosphorylation at S695 (or equivalent S457 of TLK1B, spliced isoform). Activation of TLK1 (dark purple) is dependent on its autophosphorylation (shown by circular arrow). When TLK1 is active following DSB damage (green arrow), TLK1 phosphorylates multiple DDR substrates. TLK1 can phosphorylate RAD54 particularly at T41, T59 and T700 which has been investigated recently [4]. During replication stress by hydroxyurea (HU) or oxidative damage by H2O2, TLK1 can phosphorylate Nek1 at T141 which leads to ATR-ATRIP complex (grey spheres) activation that function upstream of Chk1 (green sphere). Chk1 is phosphorylated within nucleus and inhibits replication, leads to fork stalling and arrests cell cycle at G1/S, intra-S phase and G2/M checkpoints. During HU dependent stress, RAD9 (light blue) is phosphorylated by TLK1 at S328 in 9-1-1 complex at replication fork. Phosphorylated RAD9 (dark blue), dissociates from 9-1-1 complex and shuttles to cytoplasm after recovery of DNA damage. Figures were designed in BioRender.com (26 July 2023).
Figure 1.
Mechanistic role of TLK1 in DNA damage repair (DDR). TLK1 (light purple) can heterodimerize with TLK2 (cyan) in certain cell types and normal kinase activity of TLK1 is dependent on homodimerization/oligomerization and/or heterodimerization. The N-terminal region of TLK1/TLK2 is essential for higher order arrangement. In cells, when DSB is induced with IR, there is a transient decrease in TLK1 activity (red arrow) which is restored within an hour (in HeLa cells). The inhibition of TLK1 is dependent on ATM-Chk2 phosphorylation at S695 (or equivalent S457 of TLK1B, spliced isoform). Activation of TLK1 (dark purple) is dependent on its autophosphorylation (shown by circular arrow). When TLK1 is active following DSB damage (green arrow), TLK1 phosphorylates multiple DDR substrates. TLK1 can phosphorylate RAD54 particularly at T41, T59 and T700 which has been investigated recently [4]. During replication stress by hydroxyurea (HU) or oxidative damage by H2O2, TLK1 can phosphorylate Nek1 at T141 which leads to ATR-ATRIP complex (grey spheres) activation that function upstream of Chk1 (green sphere). Chk1 is phosphorylated within nucleus and inhibits replication, leads to fork stalling and arrests cell cycle at G1/S, intra-S phase and G2/M checkpoints. During HU dependent stress, RAD9 (light blue) is phosphorylated by TLK1 at S328 in 9-1-1 complex at replication fork. Phosphorylated RAD9 (dark blue), dissociates from 9-1-1 complex and shuttles to cytoplasm after recovery of DNA damage. Figures were designed in BioRender.com (26 July 2023).
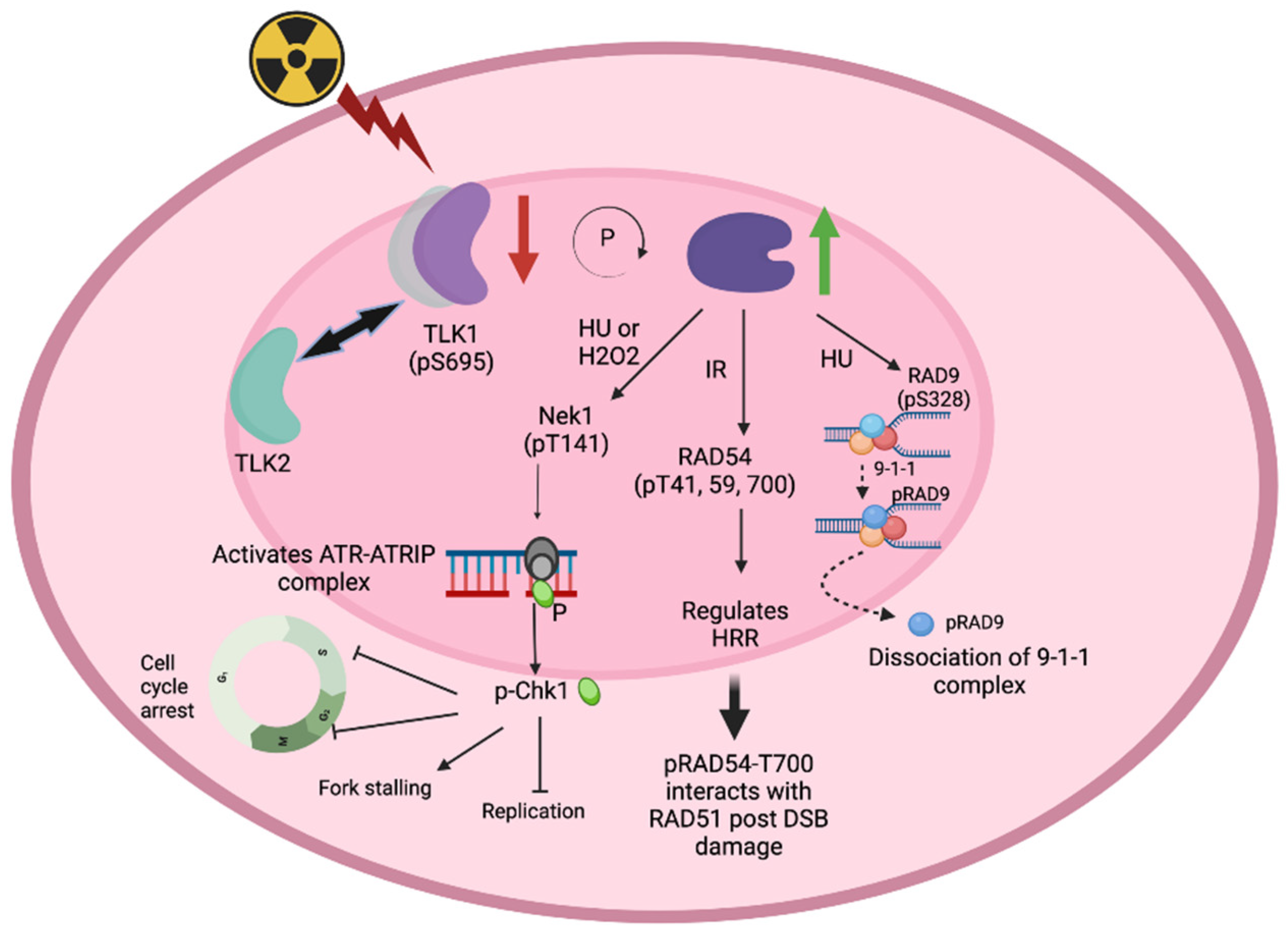
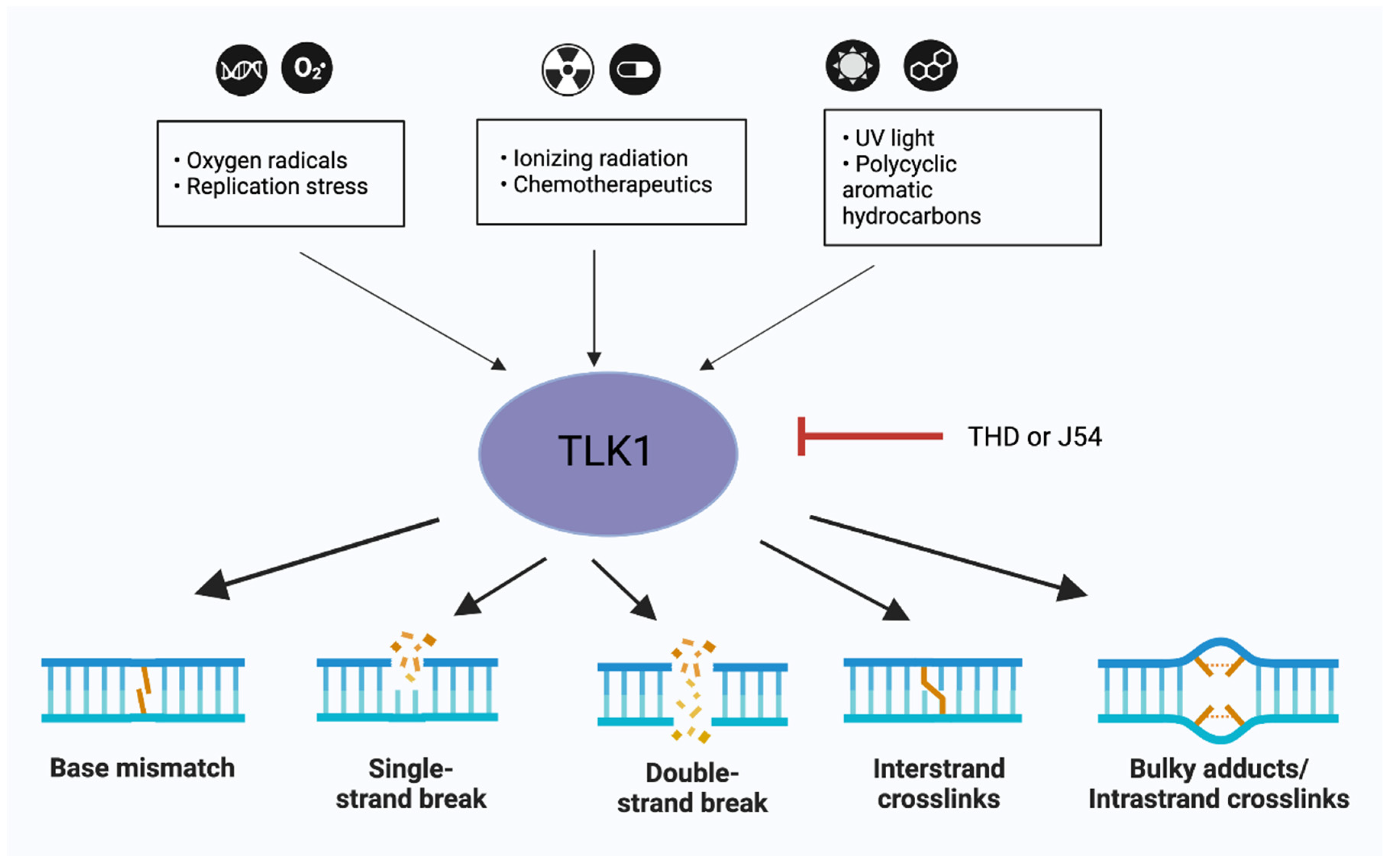
Figure 2.
TLK1 at the core of DNA repair mechanisms. TLK1 forms a therapeutic target in multiple tissue specific cancer models. Inhibiting TLK1 can affect the base-excision repair, single-strand break (SSB) repair, double-strand break (DSB) repair or Inter/Intra-strand crosslink (ICL) repair in cells. The small black arrows indicate DNA damaging agents which activate TLK1 while the bold black arrows indicate that inhibiting TLK1 affects the specific DNA repair pathways. Figures were designed in BioRender.com.
Figure 2.
TLK1 at the core of DNA repair mechanisms. TLK1 forms a therapeutic target in multiple tissue specific cancer models. Inhibiting TLK1 can affect the base-excision repair, single-strand break (SSB) repair, double-strand break (DSB) repair or Inter/Intra-strand crosslink (ICL) repair in cells. The small black arrows indicate DNA damaging agents which activate TLK1 while the bold black arrows indicate that inhibiting TLK1 affects the specific DNA repair pathways. Figures were designed in BioRender.com.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



