
Submitted:
07 August 2023
Posted:
09 August 2023
You are already at the latest version
Abstract
Mitochondria are involved in the regulation of cellular energy metabolism, calcium homeostasis, and apoptosis. For mitochondrial quality control, dynamic processes, such as mitochondrial fission and fusion, are necessary to maintain shape and function. Disturbances of mitochondrial dynamics lead to dysfunctional mitochondria which contribute to the development and progression of numerous diseases, including Type 2 Diabetes (T2D). Compelling evidence put forward that mitochondrial dynamics play a significant role in the metabolism-secretion coupling of pancreatic β cells. The disruption of mitochondrial dynamics is linked with defects in energy production and increased apoptosis, ultimately impairing insulin secretion and β cell death. This review provides an overview of altered mitochondrial dynamics in pancreatic β cells using ge-netic/pharmacologic approaches, and how these affect insulin secretion.
Keywords:
diabetes
; mitochondrial dynamics
; glucose-stimulated insulin secretion
; pancreatic beta cell
; fusion
; fission
1. Introduction: Diabetes – A Global Epidemic
Global diabetes mellitus incidences are increasing tremendously in all age groups affecting the well-being and life quality of individuals [1]. Reports from the International Diabetes Federation stated that nearly 537 million people are suffering from diabetes and anticipatedto rise to 643 million by 2030 and 783 million by 2045 [2]. Diabetes is a multifactorial disease usually associated with persistently high glucose levels (hyperglycaemia), either due to impaired insulin secretion or action [3]. It is categorized into two distinct types: Type 1 (T1D) and Type 2 Diabetes (T2D), wherein T2D accounts for at least 90-95% of total diabetic cases. In people with T1D also known as early-childhood or juvenile diabetes, the own immune system obliterates insulin-producing pancreatic beta (β) cells. In contrast, T2D usually develops later and the patients are characterized by a blend of two metabolic dysfunctions: insulin resistance and inadequate insulin secretion [3,4]. Under conditions of insulin resistance, where insulin-dependent glucose uptake in peripheral organs is reduced, the β cells produce more insulin to compensate, leading to hyperinsulinemia [5]. Moreover, in some individuals, genetically compromised β cells fail to secrete an adequate amount of insulin, resulting in hyperglycaemia, the hallmark of T2D. Ultimately, β cells mass and functions decrease, further aggravating the pathology [6]. Whether the exhaustion of β cells is caused by elevated insulin production [7] or dedifferentiation due to other metabolic complications [8], remains to be determined. Epidemiological studies reveal that age, lifestyle, ethnicity, smoking, and obesity also contribute to T2D [4]. Diabetes co-morbidities such as cardiovascular diseases, peripheral vascular diseases, neuropathy, retinopathy, stroke, and nephropathy are the main reasons for mortality in T2D individuals [9,10]. Despite extensive research, the pathophysiology related to the progression and complications of T2D is not fully understood. Definitely, to comprehend the underlying mechanisms associated with the disease, it is required to understand the concept of glucose homeostasis (glycaemia).
1.1. Glucose Homeostasis
All mammalian cells require glucose as a metabolic substrate for energy production. Thus, it is necessary to sustain adequate levels of blood sugar in the range of 4-7 mmol/L. To maintain normal blood glucose levels, various hormones are released from the brain, liver, adipose tissues, pancreas, intestine, and muscles [3]. Among these, the endocrine pancreas plays a very critical role in regulating fuel storage by secreting several hormones. Mature endocrine cells, constituting only 2-3% of the total pancreatic volume, aggregate to form a discrete group of cells, which are termed the islets of Langerhans. These pancreatic islets consist of five types of endocrine cells: alpha-cells (15-20%) producing glucagon, beta-cells (65-80%) producing insulin and C-peptide, delta-cells (3-10%) producing somatostatin, gamma cells (3-5%) producing pancreatic polypeptide (PP) and epsilon-cells (<1%) producing ghrelin [11].Human islets display a unique architecture wherein all the endocrine cells are bordered by the blood vessels, β cells are mostly located in the center and α cells occupy the mantle position of the islet. As a result, the ratio of βcells to αcells isrelatively higherin the core as compared to the mantle part of the islets[12].The highly specialized blood supplypattern through the islet of Langerhansallowsthe ready exchange of molecules. The mass of βcells is maintained by the differentiation and replication of existing βcells, which is governedby the differentcellcycle machinery[13]. The islet cell organization of the diabetic patient and non-diabetic patient does not differ significantly.The quantitative analysis, however, demonstrated a substantial reduction inβcell mass relative to αcell mass in type 2 diabetic subjects[14].Two antagonistic hormones insulin and glucagon are crucial in sustainingthe blood sugarlevels in the body [15]. In response to low blood glucose levels (hypoglycaemia), glucagon promotes hepatic glucose release by glycogen breakdown and gluconeogenesis and enhances lipolysis in adipose tissue. In contrast, insulin has a counter-regulatory effect by reducing high blood glucose levels (hyperglycaemia). Insulin stimulates glycogen production (glycogenesis) and glucose uptake in the skeletal muscle,liver, and adipose tissues respectively. Alongside, hepatic glucogenesis and glycogenolysis are decreased and lipolysis is potently inhibited [15,16]. To sense the blood glucose level for regulation of glucose homeostasis, glucose is transported to and metabolized in β cells proportionally to the extracellular level [15,16,17].
1.2. Glucose-Stimulated Insulin Secretion
Glucose-Stimulated Insulin Secretion (GSIS) is a complex event modulated by the integration and interactions of multiple signal transductions in β cells [18]. Although glucose is the chief stimulator of insulin secretion, several hormones, nutrients, neural inputs, chemical messengers, and drugs modulate insulin release [18,19]. Following a meal, elevated blood glucose levels lead to glucose uptake via facilitated glucose transport into pancreatic β cells. In the cytoplasm, glucose is metabolized through glycolysis, thereby generating adenosine triphosphate (ATP), pyruvate, nicotinamide adenine dinucleotide (NADH), and water molecules [20,21]. The formed pyruvate enters the mitochondrial matrix, getsdecarboxylated, and converted into acetyl-Coenzyme A (CoA), which enters the tricarboxylic acid (TCA) cycle for the generation of reducing equivalents nicotinamide adenine dinucleotide (NADH), flavin adenine dinucleotide (FADH2), guanosine triphosphate (GTP) and carbon dioxide (CO2). The reduced electron carriers NADH and FADH transfer their high-energy electrons to the electron transport chain (ETC), which harvests the energy by step-wise transport to drive protons into intermembrane space, forming a proton gradient. The proton gradient is used to make ATP by driving the ATP synthase; the process known as oxidative phosphorylation (OXPHOS) [21,22,23]. Specifically, in β cells, the generated ATP elevates the cytoplasmic ATP/ADP ratio, which in turn closes the ATP-sensitive potassium channel, resulting in plasma membrane depolarization. Thisinitiates the opening of L-type voltage-gated calcium channels, facilitating calcium (Ca2+) ions influx into the cells, thereby triggering exocytosis of insulin granules [24,25]. GSIS is biphasic and has been considered as a combined effect of both triggering and amplification pathways. The triggering pathway initiates the first phase of insulin secretion lasting for 5-10 mins by the KATP-dependent mechanism. A sustained second phase is enabled by the amplification pathway, which augments GSIS by KATP-independent signals such as cyclic adenosine monophosphate (cAMP), cyclic guanosine triphosphate (cGTP), phospholipase C, glutamate, malonyl-CoA, and plasma membrane phosphoinositides [26,27]. From the above-described sequence of events, it is evident that the mitochondria of the pancreatic β cells control GSIS [28], in particular by transferring the stored energy of glucose to ATP, the main trigger of insulin secretion as represented in Figure 1.
2. Mitochondrial Dynamics in Diabetes
Mitochondria are organelles in eukaryotic cells comprising an inner and an outer membrane, separated by the intermembrane space. Beyond their primary role in fueling energy metabolism, they are also involved in several processes including, cell signaling, calcium homeostasis, and apoptosis [29]. A crucial role of mitochondria in metabolic disorders such as diabetes is evident from the fact that in β cells,around 80% of glucose oxidation takes place in mitochondria. Therefore, hampering the mitochondrial energy metabolism by blocking the ETC, for example, impairs GSIS [30]. The significance of mitochondria in metabolism-secretion coupling has further been illustrated in Rhocells devoid of mitochondrial DNA (mtDNA) that no longer respond to glucose. These cells exhibit higher NAD(P)H levels, which in turn inhibits the enzyme glyceraldehyde phosphate dehydrogenase but enhances lactate production by lactate dehydrogenase enzyme[31].The above findingshighlight the significance of hydrogen shuttle and mitochondrial respiration in re-oxidizing NAD formed during glycolysis in β cells. The effect of mitochondrial dysfunction is also demonstrated by mtDNA mutations, e.g the A3243G mutation in a transfer RNA (tRNA) causing a significant decline in both first and second-phase insulin secretion [32]. Further, the knockout of mitochondrial transcription factor A (Tfam) in β cells impairs OXPHOS and GSIS, resulting in diabetic mice [33]. The mutation m.8561C>G in the subunit ofmitochondrial ATP synthase (MT-ATP6/8) resulted in impaired complex assembly and decreased ATP production,causing peripheral neuropathy and diabetes mellitus[34].The above findings clearly demonstrate the critical role of mitochondria and their function in insulin secretion. Importantly, mitochondria exist as a dynamic reticular network that frequently undergoes repetitive cycles of fission and fusion in a regulated manner, referred to as “mitochondrial dynamics”. The mito-dynamics are regulatedby a group of highly conserved dynamin-related GTPases [29] as shown in Figure 2. In humans, mice, and rats, β cell mitochondria exist as densely interconnected tubules throughout the cytoplasm [35]. In the β cell, mitochondrial dynamics are compensating damaged and dysfunctional mitochondria by fusing them with functional ones. On the other hand, fission can drive the removal of damaged or non-functional mitochondria through mitophagy [36]. It is conceivable to state that a counterbalance between the two dynamic processes is required for normal mitochondrial functionality. Accumulatedpieces of evidence support the notion that disturbances in the tuning of the fusion/fission process in the pancreatic β cell led to the development and progression of diabetes in animal and human models. Islets isolated from Zucker diabetic fatty rats were found to be unresponsive to glucose with a reduced number of β cells and altered mitochondrial network. However, treatment with Troglitazone prevented mitochondrial alteration, loss of β cells, and henceforth impairment in GSIS [37]. Similarly, β cell mitochondria obtained from diabetic GotoKakizaki rats were found to be disconnected, swollen, and shorter [38], indicatingpossible disruption of mitochondrial structure. Islets obtainedfrom type 2 diabetic patients showed disrupted mitochondrial structure, reduced ATP levels, and decreased amount of insulin granules with consequent reduction of insulin secretion [39,40]. Given the evidence for the structure-function relationship of mitochondria, mitochondrial dynamics regulating proteins are certainly involved in the pathophysiology of diabetes. In the present review, we discuss the functional role of mitochondrial dynamics as a significant contributing factor to the perpetuation of normal pancreatic β cell function. The underlying mechanisms have been addressed by alteringthe expression of mitochondrial dynamics proteins using pharmacological and genetic tools, the latter either by overexpression or knockdown.
2.1. Mitochondrial Fusion and its Machinery
Mitochondrial fusion is considered the merging of the outer and inner mitochondrial membranes of two different mitochondria to form a larger unit. Fusion enables the rapid exchange of metabolites between neighboring mitochondria, thereby complementing impaired mitochondria and promoting their functionality [36]. Mitochondrial fusion is also required for the maintenance and distribution of mtDNA. Before entering the S phase of the cell cycle, hyperfusion of mitochondria takes place thereby increasing ATP production [36,41]. The three large dynamin-family GTPases responsible for mitochondrial fusion in mammals are Mitofusins 1 and 2 (Mfn1 and 2) and Optic atrophy 1 (Opa1) respectively [42].
2.1.1. Outer Membrane Fusion Proteins: Mitofusins (Mfn1/2)
Mitofusins 1 and 2 are embedded in the outer mitochondrial membrane (OMM) and tether mitochondria together by forming complexes in trans conformation [41,42]. Both proteins share structural homology but are functionally different. Mfn1 shows greater GTPase activity and more efficient fusion relative to Mfn2 [43]. However, Mfn1 and Mfn2 can functionally substitute each other and the lack of both mitofusins eliminates mitochondrial fusion. For instance, in fibroblasts, deletion of both Mfn1 and Mfn2 resulted in complete mitigation of mitochondrial fusion, and also the fibroblasts exhibited poor growth, decreased mitochondrial membrane potential, and reduced respiration [43,44].Genetic deletion of either mitofusin causes mitochondrial dysfunction and embryonic lethality [44]. For example, muscle cells lacking Mfn2 displayed a decrease in glucose oxidation, mitochondrial membrane potential, and mitochondrial respiration[45]. Overexpression of Mfn1T109A, a point mutation in the GTPase domain of Mfn1,acts as a dominant-negative (DN) and results in extreme fission[46].Missense mutations in Mfn2 are responsible for Charcot-Marie-tooth disorder type 2A2 (CMT2A2), a neurodegenerative disease [47]. Overexpression of wild-type Mfn1 (WT-Mfn1) shifts mitochondria towards fusion resulting in perinuclear mitochondrial aggregation in the pancreatic INS-1e cell culture model and primary β cells.This leads to increased lactate production in INS1e cells, shunting pyruvate away from complete oxidation in mitochondria, and decreased cellular ATP levels at both basal and stimulatory glucose concentration, causing impaired GSIS [48]. In a similar study, overexpression of WT-Mfn1 induced hypomotility and impaired functionality of mitochondria, attributed to Mfn1-induced mitochondrial aggregation, restricting the entrance into the subplasma membrane area and reducing mitophagy [49]. Adding further complexity to our understanding of mitodynamics in β cells, overexpression of the DN-Mfn1 results in small discrete mitochondria extensively distributed in the periphery due to disruption of mitochondria in the microtubule system. However, no statistically significant changes occurred in apoptosis, mitochondrial hyperpolarisation, and metabolism–secretion coupling [48,49]. Hence, mitochondrial fragmentation as such does not seem to affect insulin secretion, at least not in-vitro. In a recent study, Mfn1 or Mfn2 β cell-specific knockout mice exhibited normal glucose homeostasis with no changes in insulin secretion. In contrast, double knockout mice with β cell-specific Mfn1 and 2 deletions displayed glucose intolerance and impaired insulin secretion due to loss of mtDNA content. Interestingly, Tfam overexpression in fusion deficient β cells ameliorated the reduced mtDNA copy number but impaired GSIS. Furthermore, the pharmacologic agonism of mitofusin was capable of rescuing mtDNA content and GSIS in islets from db/db mice, a model of T2D [50]. Consistently, the pancreatic β cell-specific knockout of Mfn1/2 in mice (βMfn1/2 KO) led to several metabolic abnormalities, such as glucose intolerance, impaired glucose clearance, and a decrease in plasma insulin levels. βMfn1/2 KO mice displayed a higher degree of fragmented mitochondria and disrupted cristae structure. Under hyperglycaemic conditions, the KO mice showed a reduction of mitochondrial calcium accumulation and hyperpolarisation, associated with impaired GSIS [51,52] which could be rescued by treatment with glucagon-like peptide 1 (GLP1) or glucose-dependent insulinotropic peptide receptor (GIP) [52,53]. Indirect evidence was observed in Mfn1 KO mice, which displayed defective mitochondrial structure and flexibility in POMC neurons, and defective insulin secretion by pancreatic β cells [54].
2.1.2. Inner Membrane Fusion Protein: Optic Atrophy 1 (Opa1)
Opa1 is positioned in the inner mitochondrial membrane (IMM) through the intermembrane space of mitochondria [41,55]. It also promotes Mfn1-mediated mitochondrial fusion of the outer membrane [42,56]. The defect of Opa1 causes autosomal dominant optic atrophy, an inherited optic neuropathy condition [57]. As suggested by several in vitro cell culture studies, Opa1 plays a vitalrole in the intrinsic apoptotic pathway and the maintenance of mtDNA[58,59,60,61,62]. Possibly due to the loss of mtDNA, the reduction in mitochondrial fusion results in decreased oxidative phosphorylation [41,62]. In RIP2-Opa 1 KO islets, decreasedlevel and activity of electron transport chain complex IV resultedin impaired oxygen consumption rate, calcium signaling, and insulin secretion. The mice were found to be hyperglycaemic. Notably, however, the total amount of mtDNA remained unchanged in this case. In vivo, studies further confirmed that β cell-specific Opa1 knockout mice are hyperglycaemic and exhibited dismantled mitochondria with abnormal cristae structures. Additionally, these mice displayed reduced β cell proliferation, decreased ATP production, and GSIS [63]. Mild overexpression of Opa1 in INS1e cells led to more elongated and tubular mitochondria, whereas higher levels of Opa1 overexpression resulted in an increased number of fragmented mitochondria [64,65].
2.2. Mitochondrial Fission and its Machinery
Mitochondrial fission is defined as the division of mitochondria into two new organelles. Fission is necessary for growing cells to provide them with a sufficient number of mitochondria [41,42]. During cellular stress conditions, fission enables the removal of damaged mitochondria and promotes apoptosis, thus helping quality control [55,66,67]. Outer mitochondrial fission is mediated by conserved dynamin family GTPases, mitochondrial fission 1 protein (Fis1), and dynamin-related protein 1 (Drp1) [42].
2.2.1. Mitochondrial Fission 1 Protein (Fis1)
Fis1 induces fission by different mechanisms in eukaryotes. In yeast, Fis1 executes mitochondrial fragmentation by recruiting Drp1 homolog Dnm1p to the mitochondrial outer membrane (OMM) [42,55]. However, in humans, hFis1 promotes mitochondrial fragmentation by two different mechanisms: firstly, it is thought to have a similar role as the homolog Dnm1p, i.ethe mitochondrial recruitment of Drp1 and the initiation of fragmentation; secondly, it binds to pro-fusion proteins Mfn1, Mfn2, and Opa1, and blocks mitochondrial fusion by inhibiting the GTPase activity [41,68,69]. In addition, Fis1 has also been involved in apoptotic and autophagic pathways [70]. In primary pancreatic β cells and INS-1e cells, the alteration of fission by silencing Fis1 resulted in compromised mitophagy, accounting for the accumulation of dysfunctional mitochondria, reduced respiratory function, and GSIS. However, the Fis1 knockdown did not significantly alter mitochondrial morphology [65,71]. Intriguingly, in the reverse experiment of Fis1 overexpression, mitochondrial fragmentation was induced remarkably and resulted in a similar phenotype of bioenergetic dysfunction, i.e., increased lactate production, impaired glucose-induced hyperpolarisation and reduced ATP levels, accompanied by decreased cytosolic and mitochondrial calcium release, all accounting for impaired insulin secretion [48,71]. Schultz et al reported the reduced Fis1 expression in glucose unresponsive cells INS832/2 vs responsive cells INS832/13, alongside a greater number of elongated mitochondria in INS832/2 cells. Interestingly, lentiviral overexpression of Fis1 in INS832/2 cells induced a more homogenous mitochondrial network and enhanced insulin secretion. On the contrary, silencing of Fis1 in INS832/13 cells and primary mouse β cells increased mitochondrial elongation, decreasing GSIS. However, the expression levels of electron transport chain complexes and the ATP synthase were unaffected. Further overexpression of Fis1 in primary mouse β cells reduced insulin secretion and fragmented mitochondria. In contrast, Fis1 overexpression improved insulin secretion in INS832/13 cells. Furthermore, a stepwise increase of Fis1 levels in less glucose-responsive insulin-producing cell line RINm5F of the rat resulted in improved GSIS, whereas high overexpression was adverse, resulting in mitochondrial clusters and diminished GSIS [72]. The above finding confirms thesignificant role of Fis1 in regulating metabolism-secretion coupling in pancreatic β cells, although the effect depends on the expression levels of Fis1. However, Fis1 RNAi maintained mitochondrial dynamics by favoring fusion and preventing apoptosis [65].
2.2.2. Dynamin Related Protein 1 (Drp1)
In mammals, mitochondrial fragmentation is mainly mediatedby highly conserved GTPase protein, namely dynamin-related protein 1 (Drp1). Drp1 is distinctly expressed in various tissues; high levels are expressed in the brain, muscle, and endocrine tissues, moderate levels are found in the kidney, lung, pancreas, and liver, and low levels are detected in the ovaries [73,74]. There are four domains in Drp1: 1) the N-terminus GTPase domain,which forms a dimer, stabilizes the active sites, and stimulates GTPase activity, 2) the variable domain, which contains most of the post-translational modification sites, 3) helical middle assembly domain, promote Drp1 self-assembly into higher order structures, and a C-terminus GTPase effector domains, mediate both intra and intermolecular interactions. However, Drp1 lacks a C-terminus proline-rich domain and pleckstrin homology domain [74,75]. Drp1 consists of 21 exons and alternate splicing of exons 3, 16, and 17 gives rise to multiple isoforms with differential GTPase activity [76,77]. Different isoforms are expressed differently in various tissues. The longer Drp1 isoform is expressed predominantly in neurons and consists of distinctive polypeptide sequences within their GTPase and variable domain, called as A-insert (encode for exon 3) and the B-insert (encode for exon 16 and 17) respectively [76,77,78] . The widely expressed and shorter isoforms of Drp1 lack A insert and alternatively exclude either exon 16, 17, or both and differentially regulate the geometry and curvature of Drp1 on the fission sites [76,77]. Drp1 mostly resides in the cytoplasm and about 3% of the total protein dwells at the mitochondrial surface. Several cellular stimuli such as Ca2+ concentration and apoptosis, activate the recruitment of Drp1 to the OMM and self-associate with adapter proteins, mitochondrial fission factor (Mff), fission protein 1 (Fis1), mitochondrial dynamics protein of 49 and 51 KDa (Mid49, and Mid51) [42,69]. After association, Drp1 forms a higher-order assembly on prospective OMM fission sites, followed by GTP hydrolysis causing conformational changes and inducing mitochondrial fission [79].The incorporation of the dominant negative (DN) mutant of Drp1 K38A inhibits membrane constriction and block organelle fission [80]. In mammalian cells, inhibition of Mff or double knockdown of MiD49 and MiD51 decreases Drp1 translocation to mitochondria and promotes elongation [81,82]. It has also been reported that MiD49 and MiD51 proteins are capable of controlling mitochondrial fission independent of Fis1 and Mff [83]. Apart from regulating mitochondrial fission, Drp1 is also thought to be involved in mediating vesicle formation, endoplasmic reticulum morphology, and peroxisomal fission in mammals [84]. The function of the fission gene Drp1 is influenced by various posttranslational modifications such as phosphorylation, sumoylation, ubiquitination, and S-nitrosylation. These posttranslational modifications along with protein effectors are known to modulate the stability, localization, and GTPase activity of Drp1 in various physiological and pathological conditions [85]. Drp1 phosphorylation by the Cdk1/Cyclin B complex at serine 616 is necessary to trigger mitochondrial fission in mitotic cells to allow even mitochondrial distribution to progeny [86]. An increased level of phosphorylation at the Ser 616 site has been observed in Alzheimer’s patients [87]. The protein kinase (PKA) dependent Drp1 phosphorylation at Ser 616 enhances mitochondrial fission leading to hypertensive encephalopathy [88]. The cyclic adenosine monophosphate (cAMP)-mediated PKA-dependent Drp1 phosphorylation at Ser 637 attenuates Drp1 GTPase activity along with intermolecular interaction resulting in reduced mitochondrial fission [89]. Elongated mitochondria are spared during autophagy and maintain ATP production and cell viability, specifically during starvation [90]. On the contrary, calcineurin-dependent Drp1 dephosphorylation at Ser 637 boosts its recruitment to mitochondria and favors mitochondrial fission [91]. SUMO ligases like Sumo1, Ubc9, and MAPL mediates Drp1 sumoylation within the variable domain and exerts an effect on its interaction with the OMM or other proteins [92]. Overexpression of Sumo 1 stabilizes Drp1 on mitochondria and prevents its degradation thereby promoting mitochondrial fission [93]. SUMO protease SenP5 mediates desumoylation of Drp1 essential for the elimination of SUMO-2/3 conjugates of Drp1 [94]. MARCH5-dependent K63-linked ubiquitination stabilizes Drp1 on mitochondria, whereas parkin-mediated K48-linked ubiquitination triggers the proteasomal degradation of Drp1 [95,96]. Drp1 can also be modified by nitrosylation at Cys644. In Alzheimer’s patients, S-nitrosylation of Drp1 at Cys644 promotes mitochondrial fission, and neurotoxic events, however preventing S-nitrosylationby Cys644Ala mutation abrogated neurotoxicity [97]. In cardiomyocytes, O-linked-N-acetyl-glucosamine glycosylation (O-glcNAcylation) of Drp1 at residues T585 and T586 activates its recruitment to mitochondria and enhances fission [98]. The above findings certainly underscore the crucial role of Drp1 in regulating mitochondrial fission. Global Drp1-knockout mice were found to be embryonic lethal, due to the lack of mitochondrial fission [29]. Similarly, abnormal brain development was observed in newborn children with a heterozygous mutation in Drp1. Cells obtained from this patient displayed elongated and interconnected mitochondria [99]. All the above findings support the involvement of Drp1 in causing mitochondrial fission provoked by various cellular stimuli. Blocking of Drp1 function by RNAi or DN allele gives rise to elongated and interconnected mitochondria that result in degradation of mitochondrial mass. For instance, in HeLa cells, the knockdown of Drp1 induced a reduction in mtDNA and mitochondrial respiration [100]. Another study demonstrated that the downregulation of Drp1 prevented the decrease in mitochondrial membrane potential and the release of cytochrome c in COS-7 cells [65]. Whereas, in hippocampal neurons, it has been shown that loss of Drp1 function leads to misshaped synaptic vesicles [101].The indispensable role of Drp1 for mitochondrial fragmentation was also recognized in the research field of diabetes. Huang et.al reported alterations in mitochondrial morphology and a decrease in ATP production due to the abnormal increase of Drp1 expression in a mouse model of T2D [102]. Several knockdown and chemical inhibition studies in β cell lines and pancreatic islets have emphasized the role of Drp1-dependent mitochondrial fission in the regulation of insulin secretion. Glucose stimulation of INS1e cells induced reversible shortening of mitochondria and promoted insulin secretion. However, the suppression of the fission event by the dominant negative (DN) mutant DLP1-K38A eliminated glucose-induced morphological changes, increased proton leak, decreased ATP production, and consequently GSIS [103]. In a similar study by Twig et.al, inhibition of Drp1 by the DN mutant prevented mitochondrial autophagy, increased the accumulation of oxidized mitochondrial protein, and led to defective insulin secretion [65]. Further, the inhibition of fission by small hairpin Drp1 RNAs in INS1e cells and islets reduced the expression of mitochondrial fusion proteins, thereby shifting the mitochondrial morphology from moderate clusters to the elongated form. These morphological changes were responsible for reduced mitochondrial membrane potential, ATP production, and GSIS [104]. In NIT1 pancreatic β cell line, Drp1 knockdown caused impairment of GSIS, which was restored by SENP 2 overexpression [105]. In our previous work, we have demonstrated that the substrate supply upstream of the oxidative phosphorylation machinery is hampered by the pharmacologic silencing of Drp1 in MIN6 cells and mouse pancreatic islets [106]. Recently Bordtet.al suggested off-target effects of the Drp1 inhibitor Mdivi-1 (mitochondrial division inhibitor-1) on complex I of the ETC [107]. However, genetic silencing of Drp1 achieved similar results as pharmacology. The direct supply of exogenous pyruvate fully rescued the deficiency in oxidative phosphorylation, ATP levels, and GSIS [106], strongly suggesting that Drp1 silencing affects mainly substrate supply. While increasing Drp1 expression would have been a feasible route to improve insulin secretion, the transient Drp1 overexpression fails to rescue GSIS in Drp1-KD MIN6cells, which was due to drastically reducing insulin content [108]. The effect of inhibiting or blocking mitochondrial fission has also been interrogated in vivo by generating β cell-specific Drp1 knockout mice (β Drp1KO). The islets exhibited highly fused mitochondrial morphology and impaired second-phase insulin secretion with no alteration in oxygen consumption rates and calcium ion influx [109]. Comprehensively, in pancreatic β cell lines and islets, genetic or pharmacologic silencing of Drp1 caused impairment in insulin secretion due to decreased ATP-linked respiration and/or increased mitochondrial proton leak; whereas, in Drp1b KO islets oxygen consumption remained unchanged. These discrepancies in past data may be attributed to the difference in proliferative β cell lines and dormant mouse islets or the effect of chronic vs acute exposure. The effect of alteration of mitochondrial dynamic proteins on insulin secretion is summarized in the Figure 3. Genetic intervention of mitochondrial dynamic proteins and their effect on mitochondrial morphology and β cell function is summarized in Table 1.
3. Conclusions
T2D has emerged as one of the leading global health problems and is associated with insufficient insulin secretion from pancreatic β cells and peripheral insulin resistance. Mitochondria are highly dynamic organelles and play a vitalin maintainingenergy homeostasis. Mitochondrial morphological changes upon glucose stimulation are necessary for proper insulin secretion. Accumulating pieces of evidence supports the notion that mitochondrial fission and fusion cycles are essential for the metabolism-secretion coupling of pancreatic β cell both in vitro and in vivo. This review has provided an overview ofthe functional aspect of the alteration of mitochondrial dynamics protein on insulin secretion.
Author Contributions
All authors (U.D.K. and M.J.) contributed to the draft and editing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work is supported by Novo Nordisk Fonden (grant number 0059646).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Debono, M.; Cachia, E. The impact of diabetes on psychological well being and quality of life. The role of patient education. Psychology, Health and Medicine. 2007, 12, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.Y.; Hsu, D.Y.; Chou, C.H. Predicting the Onset of Diabetes with Machine Learning Methods. Journal of Personalized Medicine. 2023, 13, 406. [Google Scholar] [CrossRef] [PubMed]
- Baynes, H.W. Classification, pathophysiology, diagnosis and management of diabetes mellitus. J diabetes metab. 2015, 6, 1–9. [Google Scholar] [CrossRef]
- DeFronzo, R.A. Pathogenesis of type 2 diabetes mellitus. Medical clinics. 2004, 88, 787–835. [Google Scholar] [CrossRef]
- Weyer, C.; Tataranni, P.A.; Bogardus, C.; Pratley, R.E. Insulin resistance and insulin secretory dysfunction are independent predictors of worsening of glucose tolerance during each stage of type 2 diabetes development. Clinical Diabetology. 2001, 2, 167–172. [Google Scholar] [CrossRef]
- Kaiser, N.; Leibowitz, G. Failure of beta-cell adaptation in type 2 diabetes: lessons from animal models. Frontiers in Bioscience-Landmark. 2009, 14, 1099–1115. [Google Scholar] [CrossRef]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. β-cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003, 52, 102–110. [Google Scholar] [CrossRef]
- Migliorini, A.; Bade, E.; Lickert, H. Islet cell plasticity and regeneration. Molecular metabolism. 2014, 3, 268–274. [Google Scholar] [CrossRef]
- Donnelly, R.; Emslie-Smith, A.M.; Gardner, I.D.; Morris, A.D. Vascular complications of diabetes. Bmj. 2000, 320, 1062–1066. [Google Scholar] [CrossRef]
- Harding, J.L.; Pavkov, M.E.; Magliano, D.J.; Shaw, J.E.; Gregg, E.W. Global trends in diabetes complications: a review of current evidence. Diabetologia. 2019, 62, 3–16. [Google Scholar] [CrossRef]
- de Boer, P.; Giepmans, B.N. State-of-the-art microscopy to understand islets of Langerhans: what to expect next? Immunology and cell biology. 2021, 99, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Bosco, D.; Armanet, M.; Morel, P.; Niclauss, N.; Sgroi, A.; Muller, Y.D.; Giovannoni, L.; Parnaud, G.; Berney, T. Unique arrangement of α-and β-cells in human islets of Langerhans. Diabetes. 2010, 59, 1202–1210. [Google Scholar] [CrossRef]
- Cabrera, O.; Berman, D.M.; Kenyon, N.S.; Ricordi. C.; Berggren, P.O.;Caicedo, A. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proceedings of the National Academy of Sciences. 2006, 103, 2334–2339. [Google Scholar] [CrossRef]
- Cho, J.H.; Kim, J.W.; Shin, J.A.; Shin, J.; Yoon, K.H. β-cell mass in people with type 2 diabetes. Journal of diabetes investigation. 2011, 2, 6–17. [Google Scholar] [CrossRef]
- Tups, A.; Benzler, J.; Sergi, D.; Ladyman, S.R.; Williams, L.M. Central regulation of glucose homeostasis. Comprehensive physiology. 2011, 7, 741–764. [Google Scholar] [CrossRef]
- Marty, N.; Dallaporta, M.; Thorens, B. Brain glucose sensing, counterregulation, and energy homeostasis. Physiology. 2007, 22, 241–251. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Seeley, R.J.; Tschöp, M.H.; Woods, S.C.; Morton, G.J.; Myers, M.G.; D’Alessio, D. Cooperation between brain and islet in glucose homeostasis and diabetes. Nature. 2013, 503, 59–66. [Google Scholar] [CrossRef]
- Fu, Z.; Gilbert, E.R.; Liu, D. Regulation of insulin synthesis and secretion and pancreatic Beta-cell dysfunction in diabetes. Current diabetes reviews. 2013, 9, 25–53. [Google Scholar] [CrossRef] [PubMed]
- Seino, S. Cell signalling in insulin secretion: the molecular targets of ATP, cAMP and sulfonylurea. Diabetologia. 2012, 55, 2096–2108. [Google Scholar] [CrossRef]
- Herman, M.A.; Kahn, B.B. Glucose transport and sensing in the maintenance of glucose homeostasis and metabolic harmony. The Journal of clinical investigation. 2006, 116, 1767–1775. [Google Scholar] [CrossRef] [PubMed]
- Mertz, R.J.; Worley, J.F.; Spencer, B.; Johnson, J.H.; Dukes, I.D. Activation of Stimulus-Secretion Coupling in Pancreatic β-Cells by Specific Products of Glucose Metabolism: Evidence for privileged signaling by glycolysis. Journal of Biological Chemistry. 1996, 27, 4838–4845. [Google Scholar] [CrossRef]
- Fernie, A.R.; Carrari, F.; Sweetlove, L.J. Respiratory metabolism: glycolysis, the TCA cycle and mitochondrial electron transport. Current opinion in plant biology. 2004, 7, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Liemburg-Apers, D.C.; Imamura, H.; Forkink, M.; Nooteboom, M.; Swarts, H.G.; Brock, R.; Smeitink, J.A.; Willems, P.H.; Koopman, W.J. Quantitative glucose and ATP sensing in mammalian cells. Pharmaceutical research. 2011, 28, 2745–2757. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M.; Proks, P.; Smith, P.A.; Ämmälä, C.; Bokvist, K.; Rorsman, P. Stimulus–secretion coupling in pancreatic β cells. Journal of cellular biochemistry. 1994, 55, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Rutter, G.A.; Pullen, T.J.; Hodson, D.J.; Martinez-Sanchez, A. Pancreatic β-cell identity, glucose sensing and the control of insulin secretion. Biochemical Journal. 2015, 466, 203–218. [Google Scholar] [CrossRef]
- Henquin, J.C. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes. 2000, 49, 1751–1760. [Google Scholar] [CrossRef]
- Straub, S.G.; Sharp, G.W. Glucose-stimulated signaling pathways in biphasic insulin secretion. Diabetes/metabolism research and reviews. 2002, 18, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, B.A.; Li, C.; Soleimanpour, S.A. Mitochondrial regulation of β-cell function: maintaining the momentum for insulin release. Molecular aspects of medicine. 2015, 42, 91–104. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006, 125, 1241–1252. [Google Scholar] [CrossRef]
- Wiederkehr, A.; Wollheim, C.B. Impact of mitochondrial calcium on the coupling of metabolism to insulin secretion in the pancreatic β-cell. Cell calcium. 2008, 44, 64–76. [Google Scholar] [CrossRef]
- Kennedy, E.D.; Maechler, P.; Wollheim, C.B. Effects of depletion of mitochondrial DNA in metabolism secretion coupling in INS-1 cells. Diabetes. 1998, 47, 374–380. [Google Scholar] [CrossRef]
- De Andrade, P.B.; Rubi, B.; Frigerio, F.; Van den Ouweland, J.M.; Maassen, J.A.; Maechler, P. Diabetes-associated mitochondrial DNA mutation A3243G impairs cellular metabolic pathways necessary for beta cell function. Diabetologia. 2006, 49, 1816–1826. [Google Scholar] [CrossRef]
- Silva, J.P.; Köhler, M.; Graff, C.; Oldfors, A.; Magnuson, M.A.; Berggren, P.O.; Larsson, N.G. Impaired insulin secretion and β-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nature genetics. 2000, 26, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Kytövuori, L.; Lipponen, J.; Rusanen, H.; Komulainen, T.; Martikainen, M.H.; Majamaa, K. A novel mutation m. 8561C> G in MT-ATP6/8 causing a mitochondrial syndrome with ataxia, peripheral neuropathy, diabetes mellitus, and hypergonadotropic hypogonadism. Journal of neurology. 2016, 263, 2188–2195. [Google Scholar] [CrossRef]
- Dlasková, A.; Špaček, T.; Šantorová, J.; Plecitá-Hlavatá, L.; Berková, Z.; Saudek, F.; Lessard, M.; Bewersdorf, J.; Ježek, P. 4Pi microscopy reveals an impaired three-dimensional mitochondrial network of pancreatic islet β-cells, an experimental model of type-2 diabetes. Biochimica et Biophysica Acta -Bioenergetics. 2010, 1797, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Fusion and fission: interlinked processes critical for mitochondrial health. Annual review of genetics. 2012, 46, 265–287. [Google Scholar] [CrossRef]
- Higa, M.; Zhou, Y.T.; Ravazzola, M.; Baetens, D.; Orci, L.; Unger, R.H. Troglitazone prevents mitochondrial alterations, β cell destruction, and diabetes in obese prediabetic rats. Proceedings of the National Academy of Sciences. 1999, 96, 11513–11518. [Google Scholar] [CrossRef]
- Mizukami, H.; Wada, R.; Koyama, M.; Takeo, T.; Suga, S.; Wakui, M.; Yagihashi, S. Augmented β cell loss and mitochondrial abnormalities in sucrose-fed GK rats. VirchowsArchiv. 2008, 452, 383–392. [Google Scholar] [CrossRef]
- Anello, M.; Lupi, R.; Spampinato, D.; Piro, S.; Masini, M.; Boggi, U.; Del Prato, S.; Rabuazzo, A.M.; Purrello, F.; Marchetti, P. Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia. 2005, 48, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Vatamaniuk, M.; Huang, X.; Doliba, N.; Lian, M.M.; Frank, A.; Velidedeoglu, E.; Desai, N.M.; Koeberlein, B.; Wolf, B.; Barker, C.F. Structural and functional abnormalities in the islets isolated from type 2 diabetic subjects. Diabetes. 2004, 53, 624–632. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 2006, 22, 79–99. [Google Scholar] [CrossRef]
- Hoppins, S.; Lackner, L.; Nunnari, J. The machines that divide and fuse mitochondria. Annu. Rev. Biochem. 2007, 76, 751–780. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Eura, Y.; Mihara, K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. Journal of cell science. 2004, 117, 6535–6546. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. The Journal of cell biology. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Chen, H.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010, 141, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Santel, A.; Frank, S.; Gaume, B.; Herrler, M.; Youle, R.J.; Fuller, M.T. Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. Journal of cell science. 2003, 116, 2763–2774. [Google Scholar] [CrossRef]
- Züchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; Parman, Y. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nature genetics. 2004, 36, 449–451. [Google Scholar] [CrossRef]
- Park, K.S.; Wiederkehr, A.; Kirkpatrick, C.; Mattenberger, Y.; Martinou, J.C.; Marchetti, P.; Demaurex, N.; Wollheim, C.B. Selective Actions of Mitochondrial Fission/Fusion Genes on Metabolism-Secretion Coupling in Insulin-releasing Cells. Journal of Biological Chemistry. 2008, 283, 33347–33356. [Google Scholar] [CrossRef]
- Park, K.S.; Wiederkehr, A.; Wollheim, C.B. Defective mitochondrial function and motility due to mitofusin 1 overexpression in insulin secreting cells. The Korean Journal of Physiology & Pharmacology. [CrossRef]
- Sidarala, V.; Zhu, J.; Levi-D’Ancona, E.; Pearson, G.L.; Reck, E.C.; Walker, E.M.; Kaufman, B.A.; Soleimanpour, S.A. Mitofusin 1 and 2 regulation of mitochondrial DNA content is a critical determinant of glucose homeostasis. Nature Communications. 2022, 13, 2340. [Google Scholar] [CrossRef]
- Georgiadou, E.; Muralidharan, C.; Martinez, M.; Chabosseau, P.; Tomas, A.; Wern, F.Y.; Stylianides, T.; Rothery, S.M.; Di Gregorio, A.; Leclerc, I.; Ali, Y. Pancreatic beta cell selective deletion of mitofusins 1 and 2 (Mfn1 and Mfn2) disrupts mitochondrial architecture and abrogates glucose-stimulated insulin secretion in vivo. bioRxiv. 2020. [Google Scholar] [CrossRef]
- Dai, W.; Jiang, L. Dysregulated mitochondrial dynamics and metabolism in obesity, diabetes, and cancer. Frontiers in endocrinology. 2019, 10, 570. [Google Scholar] [CrossRef]
- Georgiadou, E.; Muralidharan, C.; Martinez, M.; Chabosseau, P.; Akalestou, E.; Tomas, A.; Wern, F.Y.; Stylianides, T.; Wretlind, A.; Legido-Quigley, C.; Jones, B. Mitofusins Mfn1 and Mfn2 Are Required to Preserve Glucose-but Not Incretin-Stimulated β-Cell Connectivity and Insulin Secretion. Diabetes. 2022, 71, 1472–1489. [Google Scholar] [CrossRef]
- Ramırez, S.; Gomez-Valades, A.G.; Schneeberger, M.; Varela, L.; Haddad-Tovolli, R.; Altirriba, J.; Noguera, E.; Drougard, A.; Flores-Martınez, A.; Imbernon, M.; Chivite, I. Mitochondrial dynamics mediated by mitofusin 1 is required for POMC neuron glucose-sensing and insulin release control. Cell Metab. 2017, 25, 1390–1399. [Google Scholar] [CrossRef]
- Benard, G.; Karbowski, M. Mitochondria fusion and fission. Cell Death. 2010, 22, 97. [Google Scholar]
- Cipolat, S.; de Brito, O.M.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proceedings of the National Academy of Sciences. 2004, 101, 5927–5932. [Google Scholar] [CrossRef]
- Alexander, C.; Votruba, M.; Pesch, U.E.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; Bhattacharya, S.S. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nature genetics. 2000, 26, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. Journal of Biological Chemistry. 2003, 278, 7743–7746. [Google Scholar] [CrossRef] [PubMed]
- Olichon, A.; Landes, T.; Arnauné-Pelloquin, L.; Emorine, L.J.; Mils, V.; Guichet, A.; Delettre, C.; Hamel, C.; Amati-Bonneau, P.; Bonneau, D.; Reynier, P. Effects of OPA1 mutations on mitochondrial morphology and apoptosis: relevance to ADOA pathogenesis. Journal of cellular physiology. 2007, 211, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Cipolat, S.; De Brito, O.M.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; Scorrano, L. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006, 126, 177–189. [Google Scholar] [CrossRef]
- Hudson, G.; Amati-Bonneau, P.; Blakely, E.L. , Stewart, J.D.; He, L.; Schaefer, A.M.; Griffiths, P.G.;Ahlqvist, K.;Suomalainen, A.;Reynier, P.; McFarland, R. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain. 2008, 131, 329–337. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Physiological functions of mitochondrial fusion. Annals of the New York Academy of Sciences. 2010, 1201, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wakabayashi, N.; Wakabayashi, J.; Tamura, Y.; Song, W.J.; Sereda, S. Clerc, P.Polster, B.M.; Aja, S.M.;Pletnikov, M.V.;Kensler, T.W. The dynamin-related GTPase Opa1 is required for glucose-stimulated ATP production in pancreatic beta cells. Molecular biology of the cell. 2011, 22, 2235–2245. [Google Scholar] [CrossRef]
- Molina, A.J.; Wikstrom, J.D.; Stiles, L.; Las, G.; Mohamed, H.; Elorza, A.; Walzer, G.; Twig, G.; Katz, S.; Corkey, B.E.; Shirihai, O.S. Mitochondrial networking protects β-cells from nutrient-induced apoptosis. Diabetes. 2009, 58, 2303–2315. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L. , Haigh, S.E.; Katz, S.; Las, G.;Alroy, J. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. The EMBO journal. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Shurland, D.L.; Ryazantsev, S.N.; van der Bliek, A.M. A human dynamin-related protein controls the distribution of mitochondria. The Journal of cell biology. 1998, 143, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Van Der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science. 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Yu, R.; Jin, S.B.; Lendahl, U.; Nistér, M.; Zhao, J. Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. The EMBO journal. 2019, 38, e99748. [Google Scholar] [CrossRef]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Molecular biology of the cell. 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Ihenacho, U.K.; Meacham, K.A.; Harwig, M.C.; Widlansky, M.E.; Hill, R.B. ; Mitochondrial fission protein 1: emerging roles in organellar form and function in health and disease. Frontiers in Endocrinology. 2021, 12, 660095. [Google Scholar] [CrossRef]
- Maechler, P. Mitochondrial function and insulin secretion. Molecular and cellular endocrinology. 2013, 379, 2–8. [Google Scholar] [CrossRef]
- Schultz, J.; Waterstradt, R.; Kantowski, T.; Rickmann, A.; Reinhardt, F.; Sharoyko, V.; Mulder, H.; Tiedge, M.; Baltrusch, S. Precise expression of Fis1 is important for glucose responsiveness of beta cells. J Endocrinol. 2016, 230, 81–91. [Google Scholar] [CrossRef]
- Smirnova, E.; Shurland, D.L.; Ryazantsev, S.N.; van der Bliek, A.M. A human dynamin-related protein controls the distribution of mitochondria. The Journal of cell biology. 1998, 143, 51–58. [Google Scholar] [CrossRef]
- Hoppins, S.; Lackne,r L. ; Nunnari, J. The machines that divide and fuse mitochondria. Annu. Rev. Biochem. 2007, 76, 751–780. [Google Scholar] [CrossRef]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; Van Der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Molecular biology of the cell. 2001, 12, 2245–2256. [Google Scholar] [CrossRef]
- Macdonald, P.J.; Francy, C.A.; Stepanyants, N.; Lehman, L.; Baglio, A.; Mears, J.A.; Qi, X.; Ramachandran, R. Distinct splice variants of dynamin-related protein 1 differentially utilize mitochondrial fission factor as an effector of cooperative GTPase activity. Journal of Biological Chemistry. 2016, 291, 493–507. [Google Scholar] [CrossRef]
- Strack, S.; Wilson, T.J.; Cribbs, J.T. Cyclin-dependent kinases regulate splice-specific targeting of dynamin-related protein 1 to microtubules. Journal of Cell Biology. 2013, 201, 1037–1051. [Google Scholar] [CrossRef]
- Uo, T.; Dworzak, J.; Kinoshita, C.; Inman, D.M.; Kinoshita, Y.; Horner, P.J.; Morrison, R.S. Drp1 levels constitutively regulate mitochondrial dynamics and cell survival in cortical neurons. Experimental neurology. 2009, 218, 274–285. [Google Scholar] [CrossRef]
- Mears, J.A.; Lackner, L.L.; Fang, S.; Ingerman, E.; Nunnari, J.; Hinshaw, J.E. Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nature structural & molecular biology. 2011, 18, 20–26. [Google Scholar] [CrossRef]
- Yapa, N.M.; Lisnyak, V.; Reljic, B.; Ryan, M.T. Mitochondrial dynamics in health and disease. FEBS letters. 2021, 595, 1184–1204. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. Journal of Cell Biology. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [PubMed]
- Richter, V.; Palmer, C.S.; Osellame, L.D.; Singh, A.P.; Elgass, K.; Stroud, D.A.; Sesaki, H.; Kvansakul, M.; Ryan, M.T. Structural and functional analysis of MiD51, a dynamin receptor required for mitochondrial fission. Journal of Cell Biology. 2014, 204, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S.; Elgass, K.D.; Parton, R.G.; Osellame, L.D.; Stojanovski, D.; Ryan, M.T. Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. Journal of Biological Chemistry. 2013, 288, 27584–27593. [Google Scholar] [CrossRef]
- Kamerkar, S.C.; Kraus, F.; Sharpe, A.J.; Pucadyil, T.J.; Ryan, M.T. Dynamin-related protein 1 has membrane constricting and severing abilities sufficient for mitochondrial and peroxisomal fission. Nature communications. 2018, 9, 5239. [Google Scholar] [CrossRef] [PubMed]
- Santel, A. Frank, S. Shaping mitochondria: The complex posttranslational regulation of the mitochondrial fission protein DRP1. IUBMB life. 2008, 60, 448–455. [Google Scholar] [CrossRef]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. Journal of Biological Chemistry. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.O.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. Journal of neuroscience. 2009, 29, 9090–9103. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Disatnik, M.H.; Shen, N.; Sobel, R.A.; Mochly-Rosen, D. Aberrant mitochondrial fission in neurons induced by protein kinase Cδ under oxidative stress conditions in vivo. Molecular biology of the cell. 2011, 22, 256–265. [Google Scholar] [CrossRef]
- Chang, C.R.; Blackstone, C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. Journal of Biological Chemistry. 2007, 282, 21583–21587. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.C.; Benedetto, G.D.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nature cell biology. 2011, 13, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Cereghetti, G.M.; Stangherlin, A.; De Brito, O.M.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proceedings of the National Academy of Sciences. 2008, 105, 15803–15808. [Google Scholar] [CrossRef]
- Figueroa-Romero, C.; Iñiguez-Lluhí, J.A.; Stadler, J.; Chang, C.R.; Arnoult, D.; Keller, P.J.; Hong, Y.; Blackstone, C.; Feldman, E.L. SUMOylation of the mitochondrial fission protein Drp1 occurs at multiple nonconsensus sites within the B domain and is linked to its activity cycle. The FASEB Journal. 2009, 23, 3917. [Google Scholar] [CrossRef] [PubMed]
- Harder, Z.; Zunino, R.; McBride, H. Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Current Biology. 2004, 14, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Zunino, R.; Schauss, A.; Rippstein, P.; Andrade-Navarro, M.; McBride, H.M. The SUMO protease SENP5 is required to maintain mitochondrial morphology and function. Journal of cell science. 2007, 120, 1178–1188. [Google Scholar] [CrossRef]
- Karbowski, M.; Neutzner, A.; Youle, R.J. The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. The Journal of cell biology. 2007, 178, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Song, P.; Du, L.; Tian, W.; Yue, W.; Liu, M.; Li, D.; Wang, B.; Zhu, Y.; Cao, C.; Zhou, J. Parkin ubiquitinates Drp1 for proteasome-dependent degradation: implication of dysregulated mitochondrial dynamics in Parkinson disease. Journal of Biological Chemistry. 2011, 286, 11649–11658. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science. 2009, 324, 102–105. [Google Scholar] [CrossRef]
- Gawlowski, T.; Suarez, J.; Scott, B.; Torres-Gonzalez, M.; Wang, H.; Schwappacher, R.; Han, X.; Yates, J.R.; Hoshijima, M.; Dillmann, W. Modulation of dynamin-related protein 1 (DRP1) function by increased O-linked-β-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes. Journal of Biological Chemistry. 2012, 287, 30024–30034. [Google Scholar] [CrossRef]
- Chang, C.R.; Manlandro, C.M.; Arnoult, D.; Stadler, J.; Posey, A.E.; Hill, R.B.; Blackstone, C. A lethal de novo mutation in the middle domain of the dynamin-related GTPase Drp1 impairs higher order assembly and mitochondrial division. Journal of Biological Chemistry. 2010, 285, 32494–32503. [Google Scholar] [CrossRef]
- Parone, P.A.; Da Cruz, S.; Tondera, D.; Mattenberger, Y.; James, D.I.; Maechler, P.; Barja, F.; Martinou, J.C. Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PloS one. 2008, 3, e3257. [Google Scholar] [CrossRef]
- Singh, M.; Denny, H.; Smith, C.; Granados, J.; Renden, R. Presynaptic loss of dynamin-related protein 1 impairs synaptic vesicle release and recycling at the mouse calyx of Held. The Journal of Physiology. 2018, 596, 6263–6287. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Wang, Y.; Gan, X.; Fang, D.; Zhong, C.; Wu, L.; Hu, G.; Sosunov, A.A.; McKhann, G.M.; Yu, H.; Yan, S.S. Drp1-mediated mitochondrial abnormalities link to synaptic injury in diabetes model. Diabetes. 2015, 64, 1728–1742. [Google Scholar] [CrossRef]
- Jhun, B.S.; Lee, H.; Jin, Z.G.; Yoon, Y. Glucose stimulation induces dynamic change of mitochondrial morphology to promote insulin secretion in the insulinoma cell line INS-1E. PloS one. 2013, 8, e60810. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, F.; Schultz, J.; Waterstradt, R.; Baltrusch, S. Drp1 guarding of the mitochondrial network is important for glucose-stimulated insulin secretion in pancreatic beta cells. Biochemical and biophysical research communications. 2016, 474, 646–651. [Google Scholar] [CrossRef]
- Nan, J.; Lee, J.S.; Moon, J.H.; Lee, S.A.; Park, Y.J.; Lee, D.S.; Chung, S.S.; Park, K.S. SENP2 regulates mitochondrial function and insulin secretion in pancreatic β cells. Experimental & Molecular Medicine. 2022, 54, 72–80. [Google Scholar] [CrossRef]
- Kabra, U.D.; Pfuhlmann, K.; Migliorini, A.; Keipert, S.; Lamp, D.; Korsgren, O.; Gegg, M.; Woods, S.C.; Pfluger, P.T.; Lickert, H.; Affourtit, C. Direct Substrate Delivery Into Mitochondrial Fission–Deficient Pancreatic Islets Rescues Insulin Secretion. Diabetes. 2017, 66, 1247–1257. [Google Scholar] [CrossRef]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Shealinna, X.G.; Francis, T.C. The putative Drp1 inhibitor mdivi-1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Developmental cell. 2017, 40, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Kabra, U.D.; Moruzzi, N.; Berggren, P.O.; Jastroch, M. Drp1 Overexpression Decreases Insulin Content in Pancreatic MIN6 Cells. International Journal of Molecular Sciences. 2022, 23, 12338. [Google Scholar] [CrossRef] [PubMed]
- Hennings, T.G.; Chopra, D.G.; DeLeon, E.R.; VanDeusen, H.R.; Sesaki, H.; Merrins, M.J.; Ku, G.M. In vivo deletion of β-cell Drp1 impairs insulin secretion without affecting islet oxygen consumption. Endocrinology. 2018, 159, 3245–3256. [Google Scholar] [CrossRef] [PubMed]
- Men, X.; Wang, H.; Li, M.; Cai, H.; Xu, S.; Zhang, W.; Xu, Y.; Ye, L.; Yang, W.; Wollheim, C.B.; Lou, J. Dynamin-related protein 1 mediates high glucose induced pancreatic beta cell apoptosis. The international journal of biochemistry & cell biology. 2009, 41, 879–890. [Google Scholar] [CrossRef]
- Peng, L.; Men, X.; Zhang, W.; Wang, H.; Xu, S.; Xu, M.; Xu, Y.; Yang, W.; Lou, J. Dynamin-related protein 1 is implicated in endoplasmic reticulum stress-induced pancreatic β-cell apoptosis. International journal of molecular medicine. 2011, 28, 161–169. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A schematic model showing the steps involved in glucose-stimulated insulin secretion by the pancreatic β cell.
Figure 1.
A schematic model showing the steps involved in glucose-stimulated insulin secretion by the pancreatic β cell.
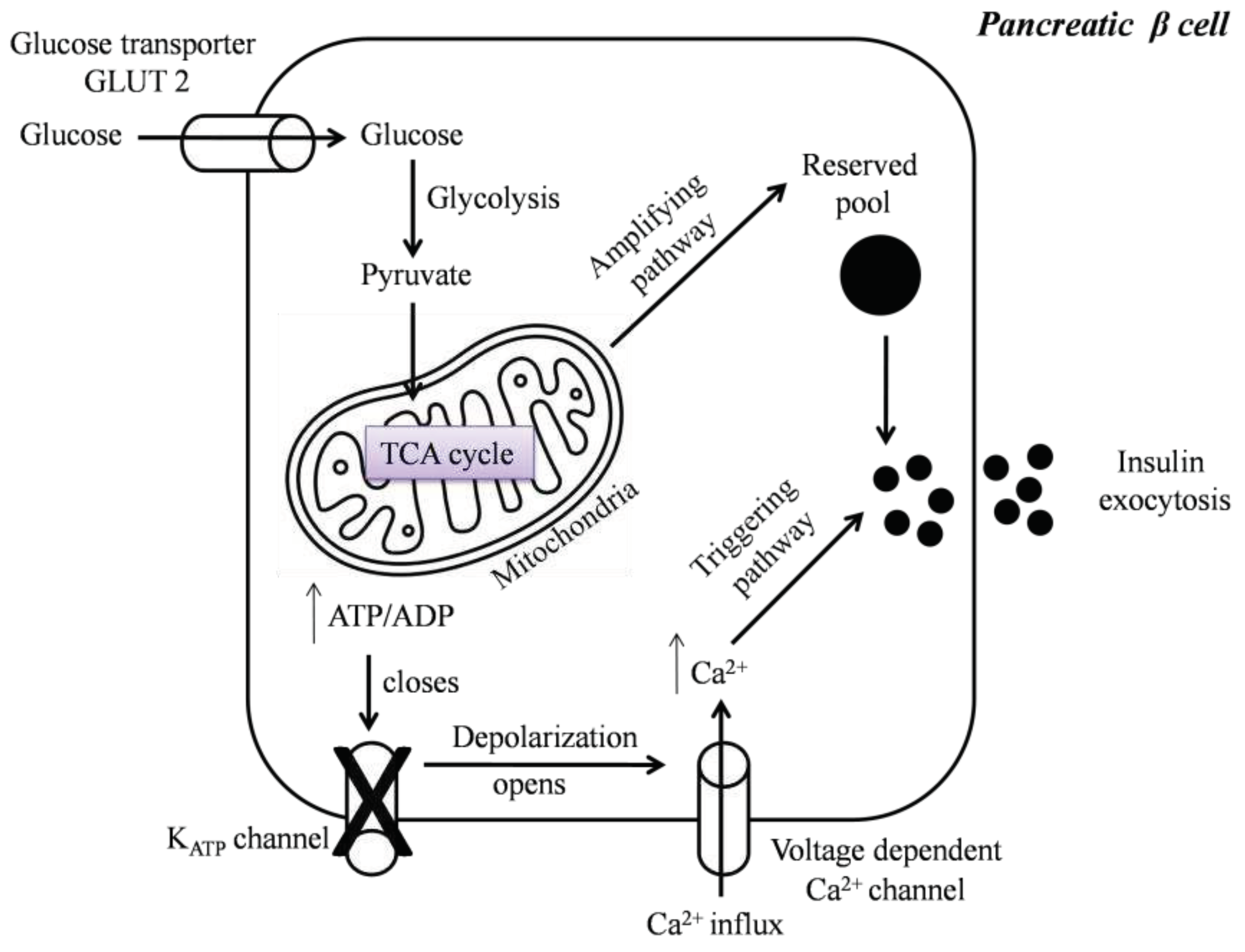
Figure 2.
Regulation of mitochondrial dynamics by fission and fusion genes.
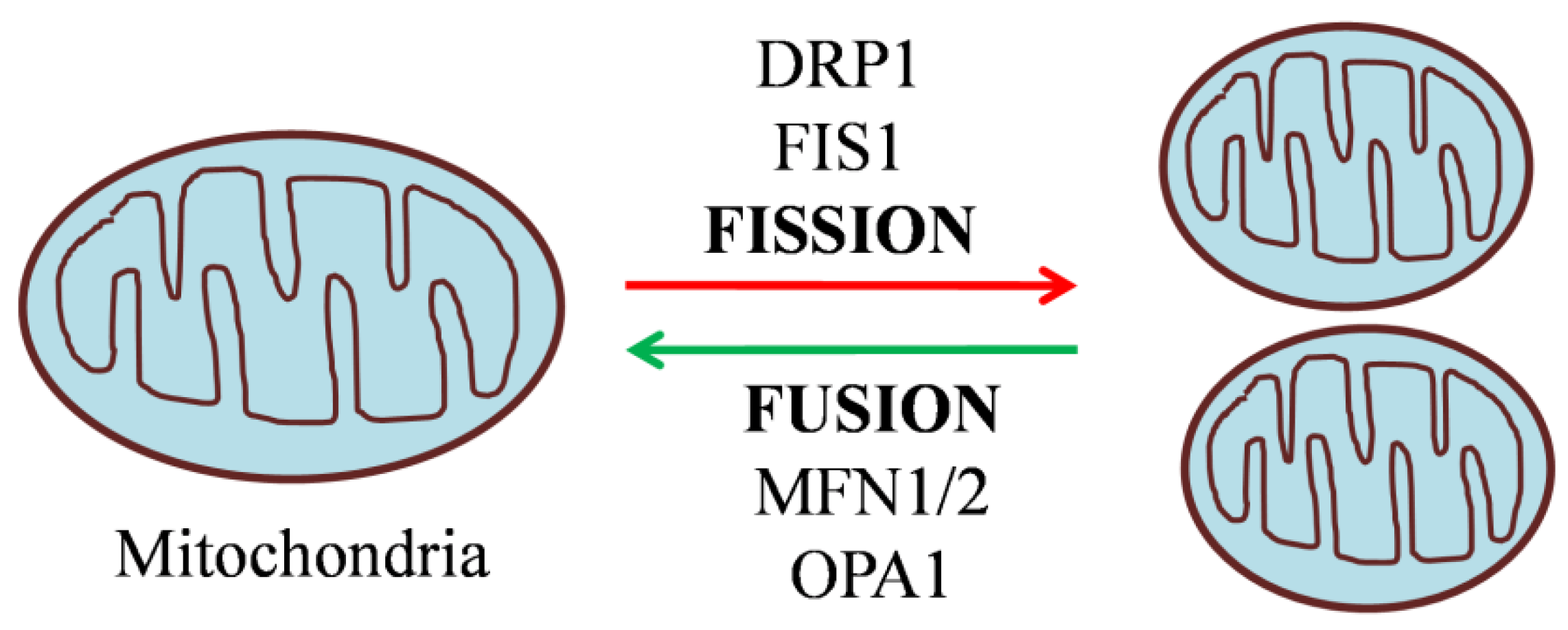
Figure 3.
Effect of manipulation of mitochondrial dynamic regulating players on insulin secretion. A: Mitofusins (Mfn1/2), B: Optic Atrophy 1 (Opa1), C: Mitochondrial Fission 1 Protein (Fis1), and D:Dynamin Related Protein 1 (Drp1).
Figure 3.
Effect of manipulation of mitochondrial dynamic regulating players on insulin secretion. A: Mitofusins (Mfn1/2), B: Optic Atrophy 1 (Opa1), C: Mitochondrial Fission 1 Protein (Fis1), and D:Dynamin Related Protein 1 (Drp1).
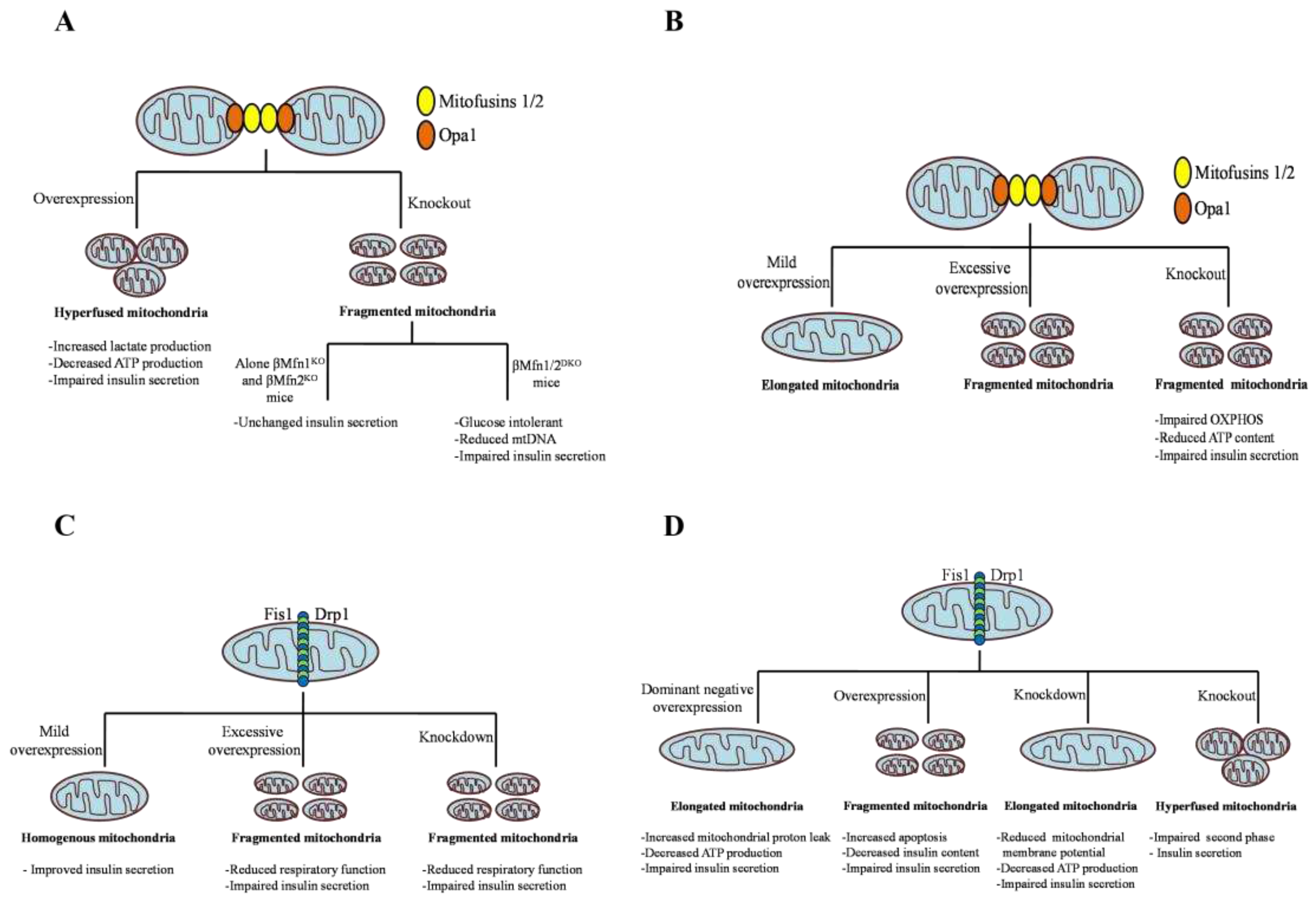

Table 1.
Manipulation of mitochondrial dynamics proteins in pancreatic β cells and islets and their effect on mitochondrial morphology and β cell function.
Table 1.
Manipulation of mitochondrial dynamics proteins in pancreatic β cells and islets and their effect on mitochondrial morphology and β cell function.
| Dynamin GTPase | Genetic intervention | Effect on mitochondrial morphology | Effect on β cell function | References |
|---|---|---|---|---|
| Mfn1/2 | Mfn1 overexpression in Ins1e cells and primary β cells | Hyperfused and aggregated | Increased lactate production, decreased cellular ATP levels, and impaired GSIS | [48] |
| Mfn1 overexpression in Ins1e cells | Hyperfused and aggregated | Loss of mitophagy, hypomotility, and impaired mitochondrial function and insulin secretion | [49] | |
| DN-Mfn1 overexpression in INS1e cells | Discrete |
No significant changes in apoptosis, mitochondrial hyperpolarisation, and metabolism–secretion coupling | [55,56] | |
| βMfn1/2KO mice |
Fragmented mitochondria, disrupted cristae shape and structures | Reduced Ca++ accumulation, mitochondrial membrane potential, β cell connectivity, and GSIS | [51,52,53] | |
| Alone βMfn1KO mice and βMfn2KO mice | Fragmented | Normal glucose homeostasis and no change in insulin secretion | [50] | |
| βMfn1/2DKO mice |
Fragmented | Glucose intolerant, reduced mtDNA content, and reduced insulin secretion | [50] | |
| Opa1 | RIP2-Opa1 KO β cells |
Fragmented mitochondria and abnormal cristae structures | Decreased complex IV level, impaired OXPHOS, decreased ATP content, reduced insulin secretion and cell proliferation | [63] |
| Mild overexpression in INS1e cell | Elongated | NA | [64,65] | |
| High overexpression in INS1e cell | Fragmented | NA | [64,65] | |
| Overexpression in primary β cell | Fragmented | NA | [64] | |
| Fis1 | Downregulation by RNAi in INS1e cells | Elongated |
Reduced mitophagy, respiratory functions, and GSIS |
[65] |
| Knockdown by shRNA INS832/13 and primary mouse β cells | Elongated |
Impaired GSIS but no changes in expression of OXPHOS complexes and ATP synthase activity | [72] |
|
| Overexpression in INS 1e cells | Fragmented, reduced volume, and swollen mitochondria | Increased lactate production, reduced mitochondrial energy metabolism, and impaired insulin secretion | [48] |
|
| Overexpression in primary mouse β cells | Fragmented | Reduced insulin secretion |
[72] |
|
| Overexpression in INS832/12 cells | Homogenous mitochondrial network | Enhanced insulin secretion |
[72] |
|
| Moderate overexpression in RINm5F cells | Homogenous mitochondrial network | Improved insulin secretion |
[72] |
|
| High overexpression in RINm5F cells | Clusters |
Reduced insulin secretion |
[72] |
|
| Drp1 | Drp1 DN (DLP1-K38A) overexpression in INS1e cells | Hyperfused | No significant change in GSIS; prevented apoptosis | [110] |
| Drp1 DN (DLP1-K38A) overexpression in INS1e cells | Swollen and elongated | Decreased mitochondrial autophagy, respiratory functions, and GSIS |
[64,65] | |
| Drp1 DN (DLP1-K38A) overexpression in INS1e cells | Elongated | Increased mitochondrial proton leak, decreased ATP production and GSIS |
[103] |
|
| Downregulation by shRNA in INS1e cells |
Elongated | Decreased mitochondrial membrane potential, reduced ATP production and GSIS |
[104] | |
| Knockdown in NIT-1cells |
Elongated | Impaired GSIS |
[105] | |
| Knockdown genetically or pharmacological inactivation by Mdivi1 in MIN6 cells and islets | Elongated |
Reduced mitochondrial ATP synthesis, and impaired GSIS due to compromised substrate delivery upstream of mitochondria | [106] | |
| β Drp1b-KO mice |
Hyperfused |
Normal oxygen consumption rate and calcium concentration but significantly impaired second-phase insulin secretion | [109] |
|
| Overexpression in INS1e cells |
Round and short | Increased cytochrome C release, and ROS production, no significant change in GSIS but tends towards decreased GSIS | [110,111] | |
| Overexpression in MIN6 cells | Fragmented | No effect on mitochondrial metabolism, impaired GSIS due to reduced insulin content | [108] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



