
Submitted:
07 August 2023
Posted:
09 August 2023
You are already at the latest version
Abstract
To evaluate the relative importance of IGF-I expression in various cell types for endochon-dral ossification, we quantified the trabecular bone at the secondary spongiosa and epiphysis of the distal femur in 8-12-week-old male mice with a global knockout of the Igf-I gene as well as conditional deletion of the Igf-I gene in osteoblasts, chondrocytes, osteo-blasts/chondrocytes, and their corresponding control wild type littermates. The osteoblast-, chondrocyte- and osteoblast/chondrocyte-specific Igf-I conditional knockout mice were generated by crossing Igf-I floxed mice with Cre transgenic mice in which Cre expression is under the control of Col1α2 or Col2α1 promoter. We found that global disruption of Igf-I resulted in 80% and 70% reduction in bone size, which is defined as total volume, at the secondary spongiosa and epiphysis of the distal femur, respectively. Abrogation of Igf-I in Col1α2-producing osteoblasts, but not Col2α1-producing chondrocytes, decreased bone size by 25% at both the secondary spongiosa and epiphysis while deletion of the Igf-I globally or specifically in osteoblasts or chondrocytes reduced trabecular bone mass by 25%. By contrast, global Igf-I knockout but not conditional knockout of Igf-I in osteoblasts and/or chondrocytes reduced trabecular bone mass in the epiphysis. The reduced trabecu-lar bone mass at the secondary spongiosa in osteoblast- and/or chondrocyte-specific Igf-I conditional knockout mice is caused by reduced trabecular number and increased trabec-ular separation. Immunohistochemistry studies revealed that expression levels of chon-drocyte (COL10, MMP13) and osteoblast (BSP) markers were reduced in the secondary spongiosa and the epiphyses in the global Igf-I knockout mice. Our data indicate that local and endocrine IGF-I actions in bone are pleiotropic and dependent on cell type as well as the bone compartment where IGF-I acts.
Keywords:
IGF-I
; knockout
; bone
; chondrocyte
; osteoblast
; endochondral ossification
; epiphysis
; sec-ondary spongiosa
; bone mass
1. Introduction
Bone size and bone mineral density (BMD) are two key determinants of bone strength. Regarding the potential regulatory molecules that contribute to skeletal changes during postnatal growth, insulin-like growth factor 1 (IGF-I) has received considerable attention for several reasons. First, we have previously demonstrated that bone size and BMD are severely compromised in mice with targeted disruption of the Igf-I gene [1]. Total BMD, femoral cortical BMD, and femur bone length were reduced by 68%, 29%, and 42%, respectively in the global Igf-I KO mice at 8 weeks of age [1]. Periosteal circumference of the femur was reduced by 46% as compared to the control WT mice. Deletion of Igf-I completely blunted the periosteal expansion during puberty. Second, targeted overexpression of Igf-I in osteoblasts increases peak BMD caused by increased activity of resident osteoblasts [2]. Femoral trabecular and cortical BMD were increased by 10% and 4%, respectively, in osteoblast specific Igf-I transgenic mice at 6 weeks of age. Femoral bone volume to total volume was increased by 28% [2]. Treating adult OVX rats with IGF-1 increased trabecular bone mass in the distal femoral metaphysis, epiphysis, and lumbar vertebral body [3]. Regarding the relevance of these findings to explain peak BMD variation in humans, we have shown that the serum level of IGF-I is increased during puberty and correlates with bone size and BMD [4]. Furthermore, the findings that both the variation in peak BMD and circulating levels of IGF-I are largely determined genetically provide evidence that the differences in IGF-I expression caused by gene polymorphism could, in part, contribute to peak BMD differences and, therefore, the risk of osteoporosis [5]. In terms of mechanisms for IGF-1 regulation of bone size and peak BMD, both endocrine and local autocrine/paracrine actions of IGF-I have been proposed [6,7]. Much of the circulating IGF-I is known to be produced primarily by liver hepatocytes which enter the blood circulation and acts as an endocrine hormone [6]. In mice with a liver-specific abrogation of Igf-I, the circulating IGF-I protein level was reduced by more than 75% of normal [8,9]. Despite the great reduction in the systemic level of IGF-I, hepatic Igf-I conditional KO mice grew normally. [8]. The appendicular skeletal growth of the liver-specific IGF-I conditional KO mice, as determined by body weight, body length, and femoral length, did not differ from wild-type littermates [8]. However, the adult axial skeletal growth and the cortical bone width were reduced in the liver-specific conditional KO mice [10]. By contrast, global deletion of the IGF-I gene in every cell caused a 20-40% reduction in femur length, size, and BMD [1]. Our studies and those of others strongly suggest that IGF-I produced locally by the cells that reside in bone acts in an autocrine/paracrine manner and is sufficient to support skeletal development and growth during puberty [11,12]. However, the relative contribution of the IGF-I produced by specific skeletal cell types to skeletal development remains unclear.
Recent studies have established that the increase in thyroid hormone levels during the prepubertal growth period is essential for the endochondral ossification that occurs at the epiphyses and secondary spongiosa of long bones. Thyroid hormone effects on endochondral bone formation are predicted to be mediated via activation of several growth factor signaling pathways. One such mediator of thyroid hormone effects is IGF-I which elicits both endocrine and local actions in different bone cell types [13]. To establish the relative importance of different sources of IGF-I in mediating skeletal growth, we performed microCT scanning to evaluate the trabecular bone phenotypes at the distal femoral epiphysis and secondary spongiosa of mice with global KO of the Igf-I gene as well as conditional KO of the Igf-1 in osteoblasts and/or chondrocytes generated by crossing cell-type specific Cre transgenic mice with Igf-I floxed mice.
2. Materials and Methods
2.1. Generation of Igf-I KO mice
Generation of Igf-I global and conditional KO mice is illustrated in Figure 1. The global IGF-I KO mice in which the exon 4 of Igf-I gene is disrupted in every cell in the body was generated as reported previously [1]. The osteoblast-specific Igf-I conditional KO mice in which the exon 4 of Igf-I gene is deleted only in type I collagen producing osteoblasts were generated by crossing Igf-I floxed mice with Col1α2-Cre mice as described [14]. The chondrocyte-specific Igf-I conditional KO mice in which the exon 4 of the Igf-I gene is disrupted only in type II collagen-expressing chondrocytes were produced by breeding Igf-I floxed mice with Col2α1-Cre mice [15]. The osteoblast- and chondrocyte-specific Igf-I double conditional KO mice in which the exon 4 of Igf-I is abrogated in both cell types were made by crossing Igf-I floxed mice with Cre double transgenic mice in which Cre expression is driven by regulatory elements of the Col1α2 and Col2α1 genes [14,15]. Cre negative, homozygous floxed mice were used as control WT mice. Cre positive, homozygous floxed mice were considered as conditional KO mice. In our previous studies, we have demonstrated that the osteoblast-specific Igf-1 cKO mice generated using A26 line of Col1α1-Cre also generated conditional mutants with normal skeletal structures at the expected ratio[14]. Col1α2-Cre expression was observed in the bone-forming region of the tibia (primarily osteoblasts) in newborn conditional mutants but not control mice. There was no measurable Cre expression in the livers of conditional mutant and control mice. Expression of IGF-I in the long bones of conditional mutants was reduced by 70% compared to control mice [14]. The chondrocyte-specific Igf-1 cKO mice generated using Col2α1-Cre were born normal at the expected ratio of 50% conditional mutant and 50% control. There were no significant differences in any of the growth or skeletal parameters at 2 weeks of age. Cre was expressed in primary chondrocytes but not in primary osteoblasts. IGF-I expression was reduced by 40% in the long bones but not in the kidney or liver of chondrocyte-specific Igf-I cKO mice [15]. Mice were housed at the Loma Linda Veterans Administration Healthcare System (LLVAHCS) with controlled temperature (22°C), illumination (14-h light, 10-h dark), and unrestricted food and water. All procedures were performed by a protocol (MOH0029/204) approved by the Institutional Animal Care and Use Committee of the LLVAHCS. Mice were anesthetized with isoflurane prior to ear punch and tail clipping for genotyping. Experimental mice were euthanized by exposure to carbon dioxide followed by cervical dislocation.
2.2. MicroCT evaluation
Mouse axial skeleton length were measured after euthanization. Femur length was measured after dissection of the femur and removal of the soft tissues prior to microCT scanning. The epiphysis and secondary spongiosa of the femurs of 12-week-old global Igf-I KO and 8-week-old osteoblast- and/or chondrocyte-specific Igf-I conditional KO male mice and their wild-type gender-matched littermates were scanned by X-ray at 55 kVp with a voxel size of 10.5 µm using vivaCT 40 microCT system from Scanco (Scanco Medical, Bruttisellen, Switzerland). The trabecular bone of the epiphysis was scanned from the top to the bottom of the femoral epiphysis. The femoral trabecular bone of the secondary spongiosa region started at 0.36 mm from the distal growth plate in the direction of the metaphysis and extended for 180 slices (1.89 mm) for WT control mice. Because the bone length in Igf-I KO mice was significantly changed, the location of slices selected for analyses was adjusted for bone length so that the analyzed regions of the bone samples were anatomically comparable. The starting point for analysis is calculated by the formula: bone length of mutant mouse/mean bone length of wild type mice X 0.36 mm. The number of slices analyzed is calculated by the formula: bone length of mutant mouse/mean bone length of wild type mice X 180 slices. Based on the calculation, the range of the number of slices analyzed was 110-180 for global KO and conditional KO mice. The average number of slices was, 110, 166, 180, and 155 for global, osteoblast-specific, chondrocyte-specific, and osteoblast/chondrocyte-specific conditional KO mice, respectively. Total volume (TV, mm3), bone volume (BV, mm3), Bone volume fraction (BV/TV, %), trabecular number (Tb. N, mm-1), trabecular thickness (Tb. Th, mm) and trabecular separation (Tb. Sp, mm) were evaluated as reported [16,17,18]. The cross-sectional area (CSA) of the femoral metaphyseal secondary spongiosa was calculated by dividing TV by the scan length (10.5 µm x slice number).
2.3. Immunohistochemistry
Frozen bone sections were prepared via a cryostat and pretreated with a blocking solution containing normal goat serum for 20 minutes, and then incubated with primary antibodies at dilution of 1:100 for COL10 (ab58632, abcam), MMP13 (nbp1-45723, Novus) and BSPII (a kind gift from Dr. Renny Franceschi, University of Michigan), respectively, for 30 minutes at room temperature. After 3 times washing with PBS, the sections were incubated with the secondary anti-mouse or anti-rabbit Dylight 488-fluorochrome labeled antibody at 1x pre-dilution (Vector Laboratories, Burlingame, CA) for another 30 minutes at room temperature. The sections were washed with PBS again and mounted with Vectashield mounting medium with DAPI (Vector Laboratories, Inc, Burlingame, CA).
2.4. Statistical Analysis
Data was analyzed with Student’s t-test. Values are presented as mean ± SEM (n = 6-9 male mice per genotype).
3. Results
3.1. Bone size at the epiphysis is reduced in mice with global and osteoblastic specific disruption of the Igf-I gene but not in chondrocyte-specific conditional KO mice
Body weight was reduced by 77%, 34%, 18%, and 35% in the global Igf-I KO, osteoblast-specific, chondrocyte-specific, and osteoblast/chondrocyte-specific Igf-I conditional KO mice, respectively, as compared to the control WT mice (Figure 2A, B, C, D). Body length was also diminished by 40%, 12%, and 17% in the global Igf-I KO, osteoblast-specific, and osteoblast/chondrocyte-specific Igf-I KO mice, respectively. Body length was not significantly different between chondrocyte specific conditional Igf-I conditional KO and control mice (Figure 2C). In concurrent with the reduced body length and femur length, the CSA of the femoral metaphyseal secondary spongiosa were also significantly decreased in the global Igf-I KO, osteoblast-specific, and osteoblast/chondrocyte-specific Igf-I KO mice (Figure 2A, B, D). The CSA of the femoral metaphyseal region was reduced by 68%, 29% and 33% in the global, osteoblast-specific, and osteoblast/chondrocyte-specific Igf-I KO mice, respectively.
MicroCT analyses unveiled that disruption of the Igf-I gene in every cell type in the global Igf-I KO mice resulted in a 70% reduction in bone size, which is defined as total volume, at the epiphysis of the distal femur. Trabecular bone volume adjusted to total volume (BV/TV) and volumetric BMD (vBMD) were reduced by 25% and 40%, respectively, at the epiphysis of the distal femur compared to wild type (WT) control mice. The reduced bone volume and vBMD were due to a significant reduction in trabecular thickness (Figure 3A, B). Deletion of Igf-I gene in type I collagen-producing osteoblasts but not type II collagen-producing chondrocytes diminished bone size by 25% at the epiphysis. Compared with littermate control siblings, mice with the conditional abrogation of the Igf-I gene in both osteoblasts and chondrocytes exhibited a 25% reduction in bone size at the epiphysis of the femur (Figure 3B).
3.2. Trabecular bone volume and vBMD are reduced at the secondary spongiosa of the distal femur in mice with disruption of Igf-I gene
Disruption of the Igf-I gene in every cell type exhibited an 80% reduction in bone width, as reflected by TV, while mice with deletion of the Igf-I gene in osteoblasts or in both osteoblasts and chondrocytes displayed a 25% reduction in bone size at the secondary spongiosa of the distal femur (Figure 4A, B). By contrast, specific KO of the Igf-I gene in chondrocytes did not change the bone size at the secondary spongiosa of the distal femur. Trabecular BV/TV and vBMD were also reduced by 25% and 40%, respectively, in the global Igf-I KO mice compared to the littermate control mice. Disruption of the Igf-I gene specifically in osteoblasts, chondrocytes, or both osteoblasts and chondrocytes, reduced trabecular BV/TV and vBMD by 25% (Fig 3B). The reduced trabecular bone mass in the global and osteoblast- and/or chondrocyte-specific Igf-I conditional KO mice is primarily caused by reduced trabecular number and increased trabecular separation. Trabecular thickness was reduced in the secondary spongiosa of the distal femur in the global Igf-I KO mice but not in the osteoblast-, chondrocyte- or osteoblast/chondrocyte-specific conditional KO mice. Immunofluorescent staining found that collagen 10 (COL10), matrix metallopeptidase 13 (MMP13), two markers of differentiating chondrocytes, but not bone sialoprotein II (BSPII), a marker of differentiating osteoblast, were expressed in growth plate chondrocytes as expected (Figure 5A, B, C, D). The expression of BSPII was restricted in the trabecular bone of the secondary spongiosa of the distal femur (Figure 5E, F). In accordance with the micro-CT findings, both COL10- and MMP13- expressing chondrocytes and BSPII-expressing osteoblasts were markedly reduced in the secondary spongiosa of the Igf-I KO males compared to control mice (Figure 5A, B, C, D, E, F).
4. Discussion
Although IGF-I is known to be produced by many cell types in the body, hepatocytes are the primary contributor to circulating IGF-I [8,9]. The IGF-I produced in the liver acts in an endocrine manner, circulating in the blood primarily as a ternary complex with acid labile subunit (ALS) and IGF binding protein-3 [19]. Growth hormone and thyroid hormones are major regulators of IGF-I expression in hepatocytes [20,21]. Growth hormone deficiency in childhood had decreased BMD, and growth hormone replacement in these children increased bone growth and bone strength [22]. There is also a positive correlation between serum IGF-I and BMD in humans [23,24]. Lower levels of serum IGF-I in women are associated with increased osteoporotic fractures [25]. Local IGF-I expression is induced by systemic hormones such as growth hormone and thyroid hormone, as well as local growth factors, including BMP-7 and TGFβ1, to act in an autocrine/paracrine manner [13,20,26]. Both endocrine and local IGF-I actions have been implicated in promoting skeletal growth [1,15]. In terms of the relative role of circulating versus locally produced IGF-I in regulating bone growth and trabecular bone mass, previous studies have shown that the liver-specific KO of Igf-I in mice caused more than an 80% reduction in circulating IGF-I level and an increase in growth hormone. Still, these mice developed normally [8]. The circulating IGF-I level was further reduced in the mice lacking both liver-derived IGF-I and ALS, causing a reduction in bone size [27,28]. But the reduction in bone size in Igf-I and Als double KO mice was much smaller than the global Igf-I KO mice, suggesting locally produced IGF-I plays a pivotal role in promoting normal bone growth during development. However, the importance of IGF-I produced by different cell types and tissue compartments in bone in mediating skeletal growth has not been fully elucidated.
In this study, we hypothesized that circulating IGF-I and locally produced IGF-I in various cell types contribute differently to endochondral ossification between the metaphysis (primary ossification center) and the epiphysis (secondary ossification center) of the long bones. While both primary and secondary ossification centers are formed via endochondral ossification, important differences exist between them including the time at which they occur [29]. In previous studies, we found that thyroid hormone is essential for initiation and progression of secondary ossification center at the epiphysis. Since thyroid hormone has been shown to stimulate IGF-I expression [13], we evaluated if trabecular bone formation at the epiphysis also IGF-I dependent, as in the case of metaphysis. To test the different role of IGF-I in the endochondral bone formation, we performed microCT scans to evaluate the trabecular bone phenotypes at the distal femoral epiphysis and the secondary spongiosa of male mice with a global KO of the Igf-I gene as well cKO of the Igf-I gene in osteoblasts and/or chondrocytes, and their corresponding control WT littermates. We only analyzed males because female mice have estrous cycles that cause a significant variation on bone growth and remodeling. Consistent with a previous study [27], global Igf-I KO mice exhibited a more significant reduction in bone size, as evidenced by reduced body weight, body length, femur length, and femur cross sectional area, compared to osteoblast and chondrocyte double conditional KO mice. We observed that disruption of the Igf-I gene in every cell type in the global IGF-I KO mice resulted in an 80% and 70% reduction in bone size at the secondary spongiosa and the epiphysis of the distal femur, respectively. Deleting the Igf-I gene in type I collagen-producing osteoblasts but not type II collagen-producing chondrocytes in mice decreased bone size by 25% at both secondary spongiosa and the epiphysis of the femur. Abrogation of the Igf-I gene globally or specifically in osteoblasts or chondrocytes reduced trabecular bone mass by 25%. The reduced trabecular bone mass in the global and osteoblast- and/or chondrocyte-specific Igf-I conditional KO mice is primarily caused by reduced trabecular number and increased separation. Trabecular thickness was reduced in both secondary spongiosa and epiphysis of global Igf-I KO mice but not in the Igf-I conditional KO mice. In congruence with the findings by micro-CT scanning, the differentiation of both osteoblasts and chondrocytes was severely compromised as evidenced by reduced expression of COL10 and MMP13 in chondrocytes of the secondary spongiosa and diminished BSPII staining in the distal metaphysis of the femur in global Igf-I KO mice.
Collagen 2a1 is expressed in the cartilaginous primary spongiosa, but not the secondary spongiosa where the trabecular bone volume and BMD were significantly reduced in the chondrocyte specific conditional KO mice. The question is how disruption of chondrocyte produced IGF-I affects trabecular bone formation in the secondary spongiosa region of the femur. In our previous study, we found that growth plate hypertrophic chondrocytes can transdifferentiate to produce osteoblasts at the primary spongiosa region [29]. Therefore, the amount of trabecular bone changes in the secondary spongiosa may reflect direct effects of Igf-I gene disruption in chondrocytes influencing chondrocyte differentiation into osteoblasts, and thereby bone formation at the primary spongiosa. Our data suggest a larger role for local IGF-I than endocrine IGF-I in promoting bone development and growth. Locally, osteoblast-derived IGF-I, but not chondrocyte-derived IGF-I, regulates bone size. By contrast, osteoblast-derived IGF-I and chondrocyte-derived IGF-I are equally important in regulating trabecular bone mass at the secondary spongiosa but not at epiphysis. Interestingly, KO of Igf-I globally but not locally in mice reduces trabecular bone mass at the epiphysis. Our study supports our previous studies that circulating IGF-I is a major contributor of the epiphysis ossification. Thus, global and conditional disruption of the Igf-1 gene in osteoblasts and/or chondrocytes in mice leads to a discovery of cell type- and tissue compartment-specific effects of IGF-I in bone as summarized in Table 1.
Other studies have evaluated the role of IGF-I expressed in various bone cell types in regulating skeletal growth, repair and remodeling [30]. IGF-I expressed in osteoblasts or osteocytes, but not liver hepatocytes has been shown to be indispensable for mechanical loading-induced bone formation [31–33]. While osteocyte-derived IGF-I plays an important role skeletal development [34], surprisingly disruption of Igf1 gene in osteocytes did not impede but promoted fracture callus remodeling as well as bone repletion response in mice [35,36]. In our previous study, we found that disruption of Igf1 gene in chondrocytes led a significant reduction in cortical bone size measured by peripheral quantitative computed tomography at the mid-diaphysis of femur [15]. By contrast, there was no change in CSA as measured by microCT at the epiphysis of chondrocyte specific Igf1 conditional KO mice in the present study. These data are consistent with complex roles for IGF-I produced by different bone cell types in regulating bone metabolism.
The mechanism by which IGF-I regulates cell- and compartment-specific actions in bone is unknown. While we did not determine the expression level of the IGF-I receptor in osteoblasts, chondrocytes, and other tissues in this study, the IGF-I receptor is expressed widely in many tissues in the body. Whether disruption of Igf-1 expression in osteoblasts and/or chondrocytes produces a compensatory increase in IGF-I receptor expression remains to be determined. Besides systemic hormones and local growth factors, mechanical strain is an important regulator of Igf-1 expression in bone cells [30]. Besides, IGF-I’s actions are controlled by IGF-binding proteins and their proteases [31]. Thus, the local actions of IGF-I in bone are likely subject to regulation by a variety of signals to meet the demands of the growing skeleton.
The limitations of this study include: 1) Failure to include female gender to confirm the cell-type and compartment-specific effects of IGF-I in female mice; and 2) Lack of cortical bone analyses to determine the role of IGF-I expressed in chondrocytes and osteoblasts in cortical bone volume regulation.
Author Contributions
Conceptualization, S.M.; methodology, C.K.; data curation, S.P., C.K., and W.X.; writing-original draft preparation, W.X. and S.M.; writing, review and editing, W.X. and S.M.; supervision, S.M.; funding acquisition, S.M. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by National Institutes of Health grant R01 AR048139 (to S.M.). The Department of Veterans Affairs in Loma Linda, California provided facilities to carry out this work. S.M. is a recipient of Senior Research Career Scientist Award (BX005381) from the Department of Veterans Affairs.
Institutional Review Board Statement
Institutional Review Board Statement: Animal procedures were performed by a protocol (MOH0029/214) approved by the Institutional Animal Care and Use Committee of the Jerry L. Pettis Memorial Veterans Affairs Medical Center, in accordance with the National Institutes of Health guidelines.
Informed Consent Statement
N/A.
Data Availability Statement
The raw datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
We thank Catrina Alarcon and Nancy Lowen for technical assistance and Gustavo A. Gomez for proofreading the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mohan S, Richman C, Guo R, Amaar Y, Donahue LR, Wergedal J, Baylink DJ: Insulin-like growth factor regulates peak bone mineral density in mice by both growth hormone-dependent and -independent mechanisms. Endocrinology 2003, 144(3):929-936. [CrossRef]
- Zhao G, Monier-Faugere MC, Langub MC, Geng Z, Nakayama T, Pike JW, Chernausek SD, Rosen CJ, Donahue LR, Malluche HH et al: Targeted overexpression of insulin-like growth factor I to osteoblasts of transgenic mice: increased trabecular bone volume without increased osteoblast proliferation. Endocrinology 2000, 141(7):2674-2682. [CrossRef]
- Bagi CM, Brommage R, Deleon L, Adams S, Rosen D, Sommer A: Benefit of systemically administered rhIGF-I and rhIGF-I/IGFBP-3 on cancellous bone in ovariectomized rats. J Bone Miner Res 1994, 9(8):1301-1312. [CrossRef]
- Libanati C, Baylink DJ, Lois-Wenzel E, Srinvasan N, Mohan S: Studies on the potential mediators of skeletal changes occurring during puberty in girls. J Clin Endocrinol Metab 1999, 84(8):2807-2814. [CrossRef]
- 5Gao ST, Lv ZT, Zhou CK, Mao C, Sheng WB: Association between IGF-1 polymorphisms and risk of osteoporosis in Chinese population: a meta-analysis. BMC Musculoskelet Disord 2018, 19(1):141. [CrossRef]
- Kineman RD, Del Rio-Moreno M, Sarmento-Cabral A: 40 YEARS of IGF1: Understanding the tissue-specific roles of IGF1/IGF1R in regulating metabolism using the Cre/loxP system. J Mol Endocrinol 2018, 61(1):T187-T198. [CrossRef]
- Wang Y, Bikle DD, Chang W: Autocrine and Paracrine Actions of IGF-I Signaling in Skeletal Development. Bone research 2013, 1(3):249-259. [CrossRef]
- Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B, LeRoith D: Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci U S A 1999, 96(13):7324-7329. [CrossRef]
- Sjogren K, Liu JL, Blad K, Skrtic S, Vidal O, Wallenius V, LeRoith D, Tornell J, Isaksson OG, Jansson JO et al: Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice. Proc Natl Acad Sci U S A 1999, 96(12):7088-7092. [CrossRef]
- Sjogren K, Jansson JO, Isaksson OG, Ohlsson C: A model for tissue-specific inducible insulin-like growth factor-I (IGF-I) inactivation to determine the physiological role of liver-derived IGF-I. Endocrine 2002, 19(3):249-256. [CrossRef]
- Lazowski DA, Fraher LJ, Hodsman A, Steer B, Modrowski D, Han VK: Regional variation of insulin-like growth factor-I gene expression in mature rat bone and cartilage. Bone 1994, 15(5):563-576. [CrossRef]
- Wang E, Wang J, Chin E, Zhou J, Bondy CA: Cellular patterns of insulin-like growth factor system gene expression in murine chondrogenesis and osteogenesis. Endocrinology 1995, 136(6):2741-2751. [CrossRef]
- Xing W, Govoni K, Donahue LR, Kesavan C, Wergedal J, Long C, Bassett JH, Gogakos A, Wojcicka A, Williams GR et al: Genetic evidence that thyroid hormone is indispensable for prepubertal IGF-I expression and bone acquisition in mice. J Bone Miner Res 2012, 27:1067-1079. [CrossRef]
- Govoni KE, Wergedal JE, Florin L, Angel P, Baylink DJ, Mohan S: Conditional deletion of IGF-I in collagen type 1{alpha}2 (Col1{alpha}2) expressing cells results in postnatal lethality and a dramatic reduction in bone accretion. Endocrinology 2007, 148(12):5706-5715. [CrossRef]
- Govoni KE, Lee SK, Chung YS, Behringer RR, Wergedal JE, Baylink DJ, Mohan S: Disruption of insulin-like growth factor-I expression in type IIalphaI collagen-expressing cells reduces bone length and width in mice. Physiol Genomics 2007, 30(3):354-362. [CrossRef]
- Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Muller R: Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res 2010, 25(7):1468-1486. [CrossRef]
- Xing W, Pourteymoor S, Mohan S: Ascorbic acid regulates osterix expression in osteoblasts by activation of prolyl hydroxylase and ubiquitination-mediated proteosomal degradation pathway. Physiol Genomics 2011, 43(12):749-757. [CrossRef]
- Xing W, Kim J, Wergedal J, Chen ST, Mohan S: Ephrin B1 regulates bone marrow stromal cell differentiation and bone formation by influencing TAZ transactivation via complex formation with NHERF1. Mol Cell Biol 2010, 30(3):711-721. [CrossRef]
- Hjortebjerg R, Flyvbjerg A, Frystyk J: Insulin growth factor binding proteins as therapeutic targets in type 2 diabetes. Expert Opin Ther Targets 2014, 18(2):209-224. [CrossRef]
- Rotwein P: Mapping the growth hormone--Stat5b--IGF-I transcriptional circuit. Trends Endocrinol Metab 2012, 23(4):186-193. [CrossRef]
- Rodriguez-Arnao J, Miell JP, Ross RJ: Influence of thyroid hormones on the GH-IGF-I axis. Trends Endocrinol Metab 1993, 4(5):169-173. [CrossRef]
- Boot AM, Engels MA, Boerma GJ, Krenning EP, De Muinck Keizer-Schrama SM: Changes in bone mineral density, body composition, and lipid metabolism during growth hormone (GH) treatment in children with GH deficiency. J Clin Endocrinol Metab 1997, 82(8):2423-2428. [CrossRef]
- Barrett-Connor E, Goodman-Gruen D: Gender differences in insulin-like growth factor and bone mineral density association in old age: the Rancho Bernardo Study. J Bone Miner Res 1998, 13(8):1343-1349. [CrossRef]
- Janssen JA, Burger H, Stolk RP, Grobbee DE, de Jong FH, Lamberts SW, Pols HA: Gender-specific relationship between serum free and total IGF-I and bone mineral density in elderly men and women. Eur J Endocrinol 1998, 138(6):627-632. [CrossRef]
- Sugimoto T, Nishiyama K, Kuribayashi F, Chihara K: Serum levels of insulin-like growth factor (IGF) I, IGF-binding protein (IGFBP)-2, and IGFBP-3 in osteoporotic patients with and without spinal fractures. J Bone Miner Res 1997, 12(8):1272-1279. [CrossRef]
- Bozic D, Grgurevic L, Erjavec I, Razdorov G, Brkljacic J, Orlic I, Plancak D: Effect of bone morphogenetic protein-7 on gene expression of bone morphogenetic protein-4, dentin matrix protein-1, insulin-like growth factor-I and -II in cementoblasts in vitro. Coll Antropol 2012, 36(4):1265-1271. [CrossRef]
- Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A: Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 1993, 75(1):59-72. [CrossRef]
- Powell-Braxton L, Hollingshead P, Warburton C, Dowd M, Pitts-Meek S, Dalton D, Gillett N, Stewart TA: IGF-I is required for normal embryonic growth in mice. Genes Dev 1993, 7(12B):2609-2617. [CrossRef]
- Aghajanian P, Mohan S: The art of building bone: emerging role of chondrocyte-to-osteoblast transdifferentiation in endochondral ossification. Bone research 2018, 6:19. [CrossRef]
- Chaqour B, Han JS, Tamura I, Macarak E: Mechanical regulation of IGF-I and IGF-binding protein gene transcription in bladder smooth muscle cells. J Cell Biochem 2002, 84(2):264-277. [CrossRef]
- Bulbring E, Kuriyama H: The action of catecholamines on guinea-pig taenia coli. Philos Trans R Soc Lond B Biol Sci 1973, 265(867):115-121. [CrossRef]
Figure 1.
Generation of global knockout (KO) and conditional KO (cKO) of Igf-I gene in osteoblasts, chondrocyte, and osteoblasts/chondrocytes. OB, osteoblast; CC, chondrocyte; OB/CC, osteoblast/chondrocyte.
Figure 1.
Generation of global knockout (KO) and conditional KO (cKO) of Igf-I gene in osteoblasts, chondrocyte, and osteoblasts/chondrocytes. OB, osteoblast; CC, chondrocyte; OB/CC, osteoblast/chondrocyte.
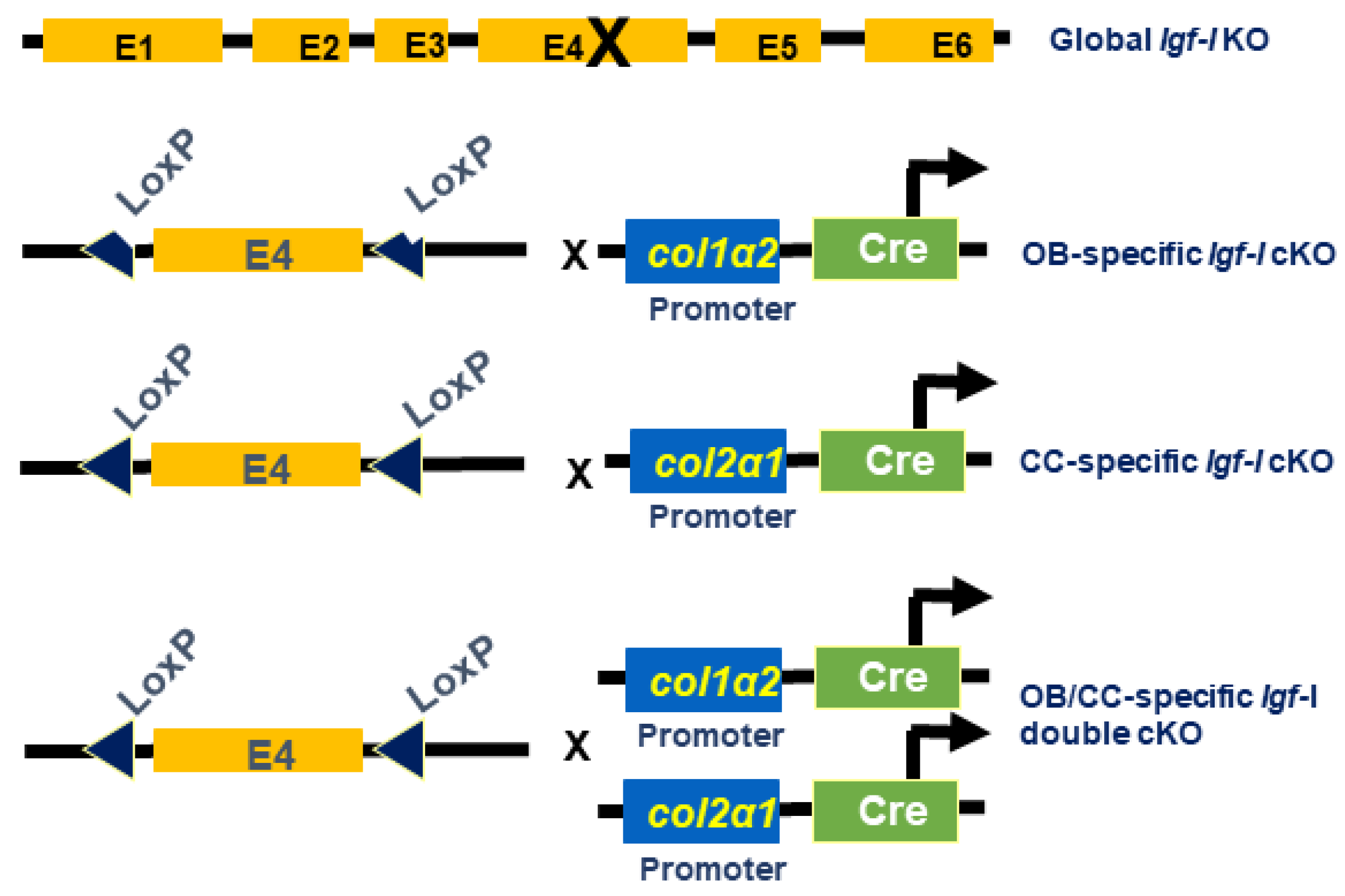
Figure 2.
Body weight and bone size were reduced in the global Igf-I and osteoblast-specific or osteoblast/chondrocyte-specific conditional KO mice. WT, wild type; KO, knockout (12-week-old) ; cKO, conditional KO (8-week-old); OB, osteoblast; CC, chondrocyte; CSA, cross sectional area. Values are Mean ± SEM (N = 6-9, males). A star (*) indicates p < 0.05. Stars (**) indicate p < 0.01.
Figure 2.
Body weight and bone size were reduced in the global Igf-I and osteoblast-specific or osteoblast/chondrocyte-specific conditional KO mice. WT, wild type; KO, knockout (12-week-old) ; cKO, conditional KO (8-week-old); OB, osteoblast; CC, chondrocyte; CSA, cross sectional area. Values are Mean ± SEM (N = 6-9, males). A star (*) indicates p < 0.05. Stars (**) indicate p < 0.01.
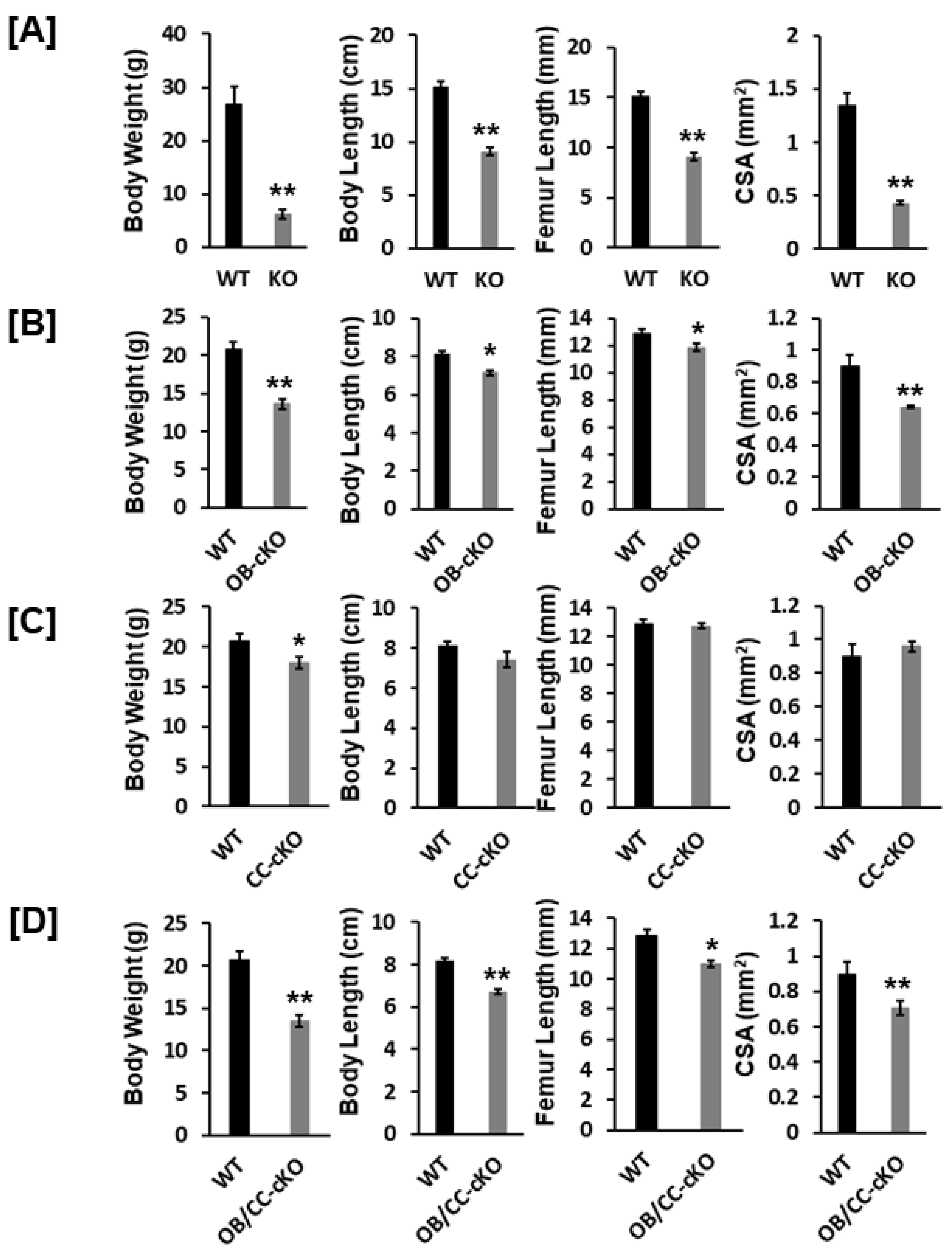
Figure 3.
Bone size at the epiphysis is reduced in mice with global and osteoblastic-specific conditional KO of Igf-I gene but not in chondrocyte-specific conditional KO mice. [A] MicroCT images of the trabecular bone of the epiphyses of the KO (12-week-old) and cKO mice (8-week-old). [B] Quantitative microCT data of the trabecular bone of the epiphysis in Figure 2A. TV, total volume; BV, bone volume; Tb. N, trabecular number; Tb.Th, trabecular thickness; Tb.Sp, trabecular spacing; vBMD, volumetric bone mineral density. Values are Mean ± SEM (N = 6-9, males). A star (*) indicates p < 0.05. Two stars (**) indicate p < 0.01.
Figure 3.
Bone size at the epiphysis is reduced in mice with global and osteoblastic-specific conditional KO of Igf-I gene but not in chondrocyte-specific conditional KO mice. [A] MicroCT images of the trabecular bone of the epiphyses of the KO (12-week-old) and cKO mice (8-week-old). [B] Quantitative microCT data of the trabecular bone of the epiphysis in Figure 2A. TV, total volume; BV, bone volume; Tb. N, trabecular number; Tb.Th, trabecular thickness; Tb.Sp, trabecular spacing; vBMD, volumetric bone mineral density. Values are Mean ± SEM (N = 6-9, males). A star (*) indicates p < 0.05. Two stars (**) indicate p < 0.01.
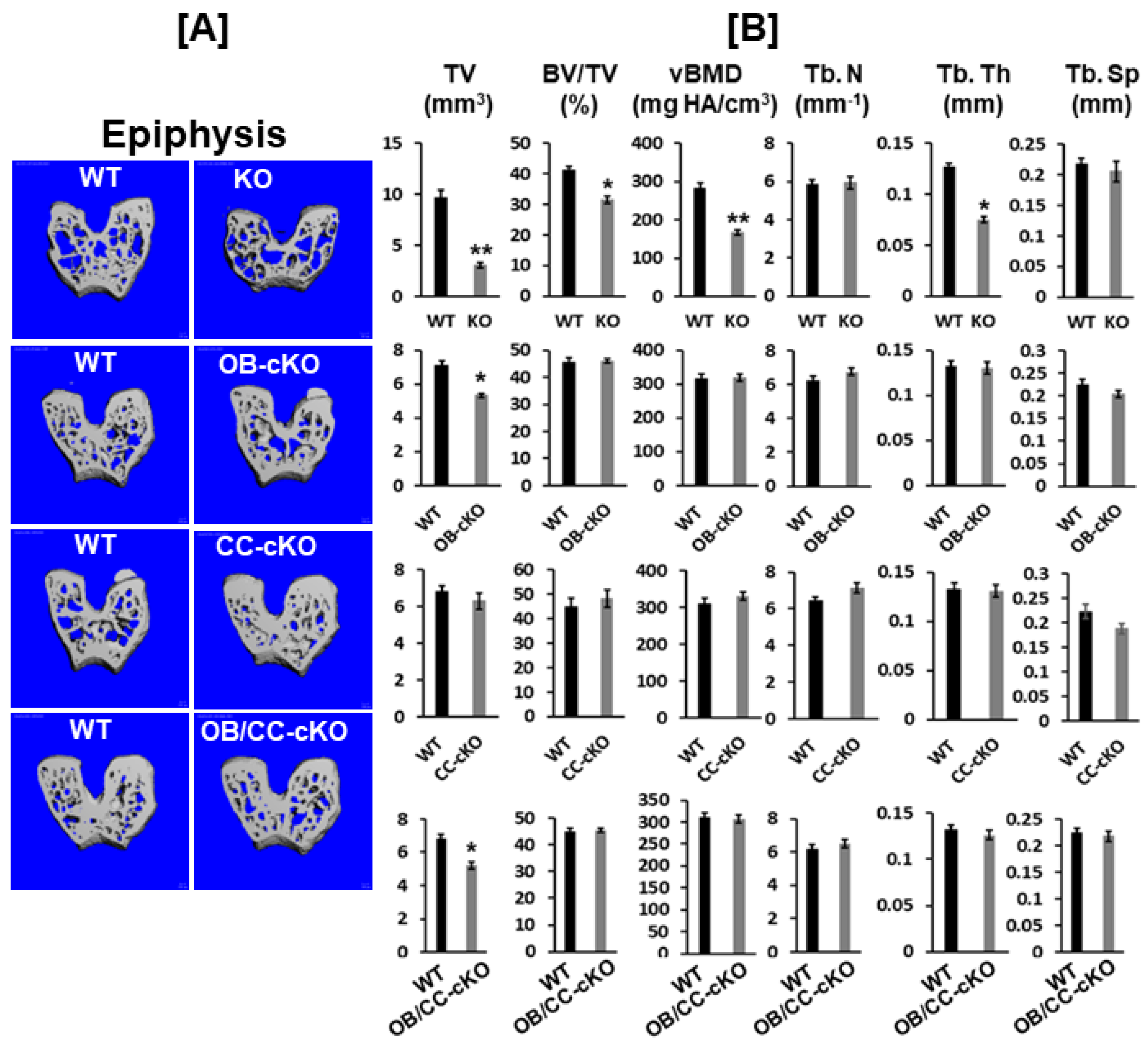
Figure 4.
Trabecular bone volume and vBMD are reduced at the secondary spongiosa of the distal femur in mice with disruption of the Igf-I gene. [A] MicroCT images of the trabecular bone of the secondary spongiosa of the KO (12-week-old) and cKO (8-week-old) mice. [B] Quantitative microCT data of the trabecular bone of the distal femur in Figure 2A. Values are Mean ± SEM (N = 6-9, males). A star (*) indicates p < 0.05. Two stars (**) indicate p < 0.01.
Figure 4.
Trabecular bone volume and vBMD are reduced at the secondary spongiosa of the distal femur in mice with disruption of the Igf-I gene. [A] MicroCT images of the trabecular bone of the secondary spongiosa of the KO (12-week-old) and cKO (8-week-old) mice. [B] Quantitative microCT data of the trabecular bone of the distal femur in Figure 2A. Values are Mean ± SEM (N = 6-9, males). A star (*) indicates p < 0.05. Two stars (**) indicate p < 0.01.
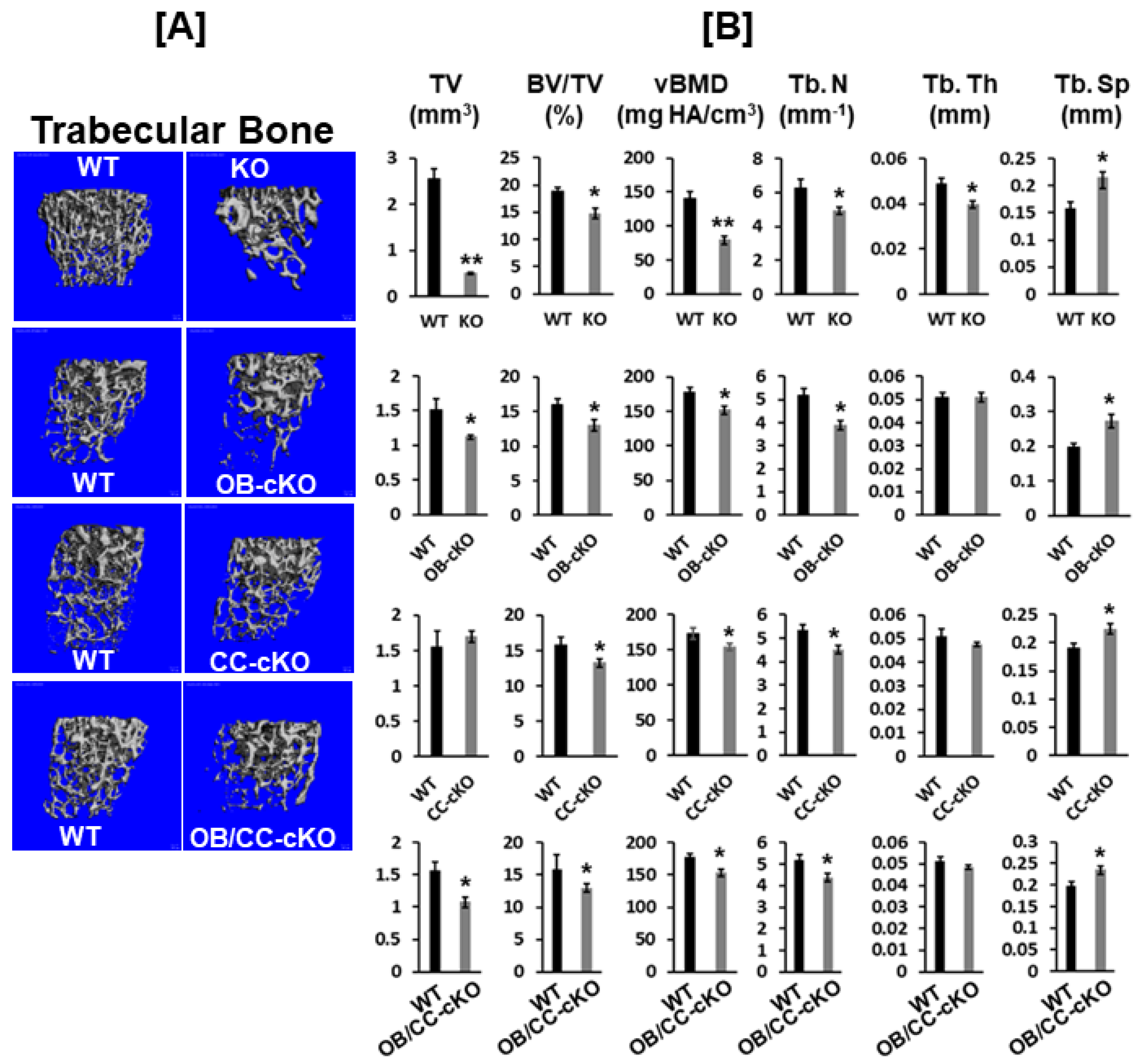
Figure 5.
Expression of COL10, MMP13, and BSPII expression was reduced in the distal femur of global Igf-I KO. Expression levels of collagen type 10 (COL10, matrix metallopeptidase 13 (MMP13), and bone sialoprotein II (BSPII) were analyzed by immunofluorescent staining. Nuclei were stained in blue with DAPI. Signals are shown in green. A, C, and E represent longitudinal sections of distal femur from wild type mice while B, D, and F represent longitudinal sections of distal femur from global Igf-I KO mice. GP, growth plate; SS: the secondary spongiosa. Arrows indicate differentiating chondrocytes expressing COL10 and MMP13 or osteoblasts expressing BSPII (Green).
Figure 5.
Expression of COL10, MMP13, and BSPII expression was reduced in the distal femur of global Igf-I KO. Expression levels of collagen type 10 (COL10, matrix metallopeptidase 13 (MMP13), and bone sialoprotein II (BSPII) were analyzed by immunofluorescent staining. Nuclei were stained in blue with DAPI. Signals are shown in green. A, C, and E represent longitudinal sections of distal femur from wild type mice while B, D, and F represent longitudinal sections of distal femur from global Igf-I KO mice. GP, growth plate; SS: the secondary spongiosa. Arrows indicate differentiating chondrocytes expressing COL10 and MMP13 or osteoblasts expressing BSPII (Green).
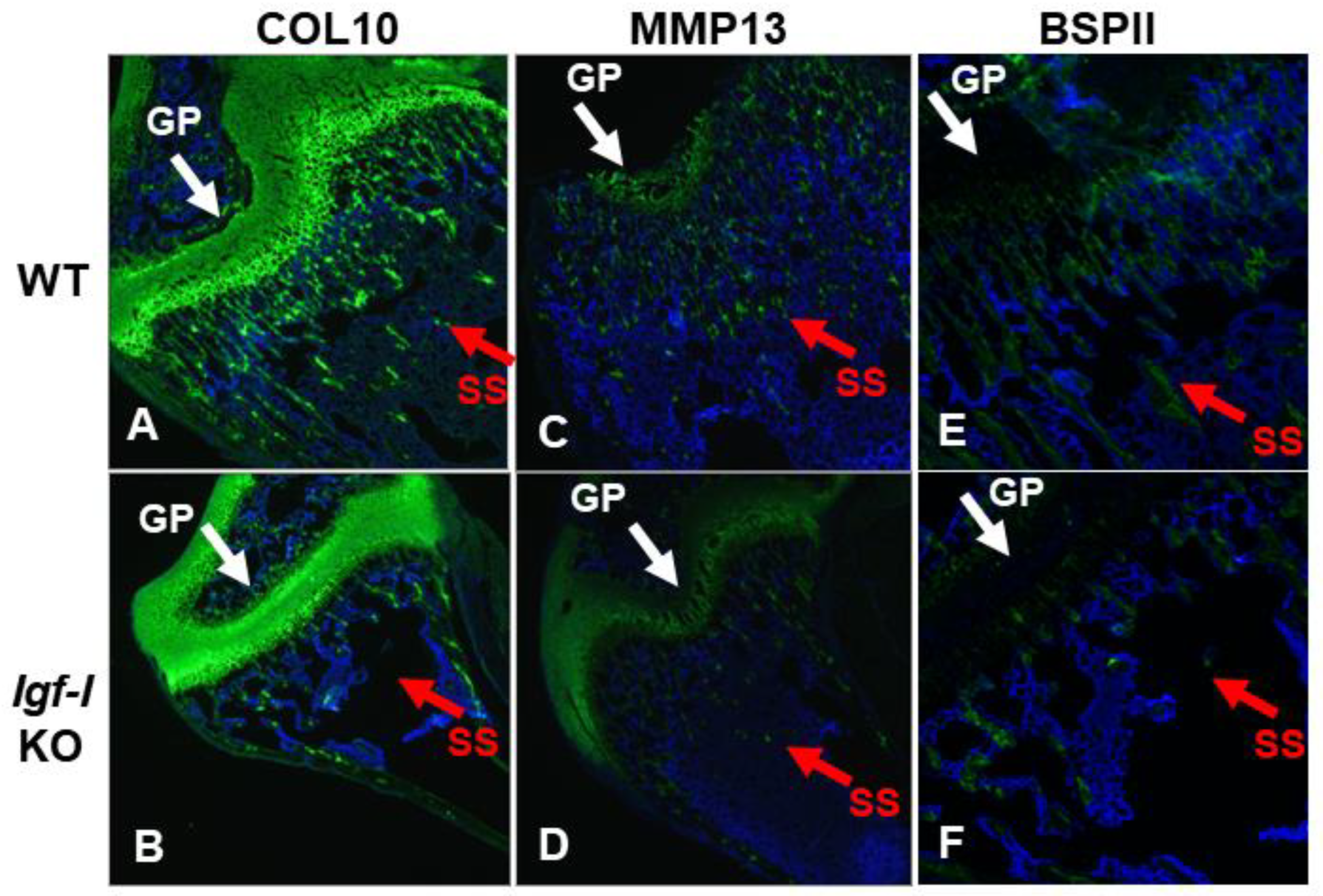

Table 1.
Summary of bone phenotypes of mice with deletion of Igf-I gene.
| KO Cell Type | Mechanism | Bone Size | Trabecular Bone |
|---|---|---|---|
| Every cell type (global) | Endocrine and local action | Reduced | Reduced |
| Osteoblasts | Local action | Reduced | Reduced |
| Chondrocytes | Local action | No change | Reduced |
| Osteoblasts/Chondrocytes | Local action | Reduced | Reduced |
| Hepatocytes | Local action | Reduced | No change |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



