Submitted:
08 August 2023
Posted:
09 August 2023
You are already at the latest version
Abstract
Coptisine (Cop) exerts a neuroprotective effect in central nervous system disease, particularly ischemic stroke. However, its protective mechanism is still unclear. This study aimed to investigate the protective effect of Cop on cerebral ischemia-reperfusion (IR) rats with a middle cerebral artery occlusion model by integrating a gas chromatography-mass spectrometry (GC-MS) based metabolomics approach with biochemical assessment. Our results showed that Cop could improve neurobehavioral function and decrease the ischemia size in IR rats. In addition, Cop was found to decrease inflammatory mediators (e.g., prostaglandin D2 (PGD2) and tumor necrosis factor-α (TNF-α) and attenuate oxidative stress response (e.g., increase the superoxide dismutase (SOD) expression and decrease 8-iso-PGF2α level). Furthermore, GC-MS based cerebrospinal fluid (CSF) metabolomics analysis indicated that Cop influenced the level of glycine, 2,3,4-trihydroxybutyric acid, oleic acid, glycerol, and ribose during IR injury. Cop exhibited a good neuroprotective effect against cerebral IR injury and metabolic alterations which might be mediated through its antioxidant and anti-inflammatory property.
Keywords:
Cerebral ischemia-reperfusion
; Coptisine
; arachidonic acid metabolomics
; inflammation
; and oxidative stress
1. Introduction
Ischemic stroke is the second leading cause of death in people over 60 years[1]. Energy crisis induced by stroke triggers a series of events including neuroexcitotoxicity, mitochondrial dysfunction, elevated intracellular calcium levels, and activation of apoptotic genes. Currently, the first-choice treatment for ischemic stroke is thrombolysis, which restores blood flow by dissolving the clots and preserving the surrounding brain tissue[2]. However, thrombolysis fails to reverse the ischemic death of nerve cells and results in several side effects including a reduced level of consciousness and reduced language ability, and severe restrictions on patient fitness and therapeutic time window. Therefore, the need for more effective treatment and its corresponding mechanism has been highlighted.
Metabolomics is a thriving technology that is defined as the quantitative analysis of dynamic metabolic responses to pathophysiological stimuli or genetic modification, and it provides a new perspective on understanding of multifactorial mechanisms of disease and evaluated the effect of drug treatment or intervention. In recent, a liquid chromatography tandem mass spectrometry based-metabolomic approach was applied to investigate the protective mechanism of Bai-Mi-decoction on ischemic stroke rats, and found a total of 53 differential metabolites between ischemic stroke and control groups, of which 30 were significantly regulated by Bai-Mi-decoction intervention, e.g., phenylalanine, valine and LysoPI(18:0/0:0)[3]. Similarly, significantly differential metabolites were screened with untargeted metabolomics by cross-comparisons with pre- and posttreatment of edaravone under cerebral IR injury, involved in valine, leucine, and isoleucine biosynthesis, and phenylalanine, taurine, and hypotaurine metabolisms [4]. Lin et al applied metabolomic technique to find Gastrodia elata Blume prevent cerebral IR injury by targeting phenylalanine, pyrimidine, methionine, and sphingolipid metabolism [5]. Hence, metabolomics techniques are increasingly being used to study the protective mechanisms of pharmacological interventions on ischemic stroke
Cop is an effective component of the traditional Chinese medicine Coptidis Rhizoma, which exerts various pharmacological effects such as neuroprotection [6], anti-inflammatory [7, 8], antioxidant [9, 10], and anti-cancer [11, 12]. The effects of Cop on central nervous system disorders have recently attracted much interest. The neuro-defensive effects of Cop against excitotoxicity compound glutamate-induce PC12 cell damage and reduction in brain IR injury in rats were reported by Huang et al [13]. In SH-SY5 cells exposed to tert-butyl hydroperoxide, the neuroprotective effects of Cop were directly correlated with improved mitochondrial membrane potential and reduced apoptosis [14]. In addition, Cop operated through voltage and receptor calcium channels impeded extracellular calcium flooding, which alters a wide range of cellular functions and inevitably reduces neuronal survival [15]. Yu et al. demonstrated that Cop reduced neuronal loss and inhibited the activation of microglia and astrocytes, both of which are involved in the secretion of pro-inflammatory mediators [16]. These findings suggest that Cop may be a promising therapeutic agent for the treatment of central neurological diseases, e.g., stroke. However, further studies are needed to investigate the molecular mechanisms of Cop against cerebral ischemic stroke injury.
In this study, we used middle cerebral artery occlusion to establish IR injury and utilized GC-MS to detect untargeted metabolomics in the CSF between the control, cerebral IR injury and Cop-treated groups. The aim of our study was to reveal the molecular regulation mechanism of Cop in alleviating cerebral IR injury in rats.
2. Results
2.1. Protective Effect of Cop on Cerebral IR
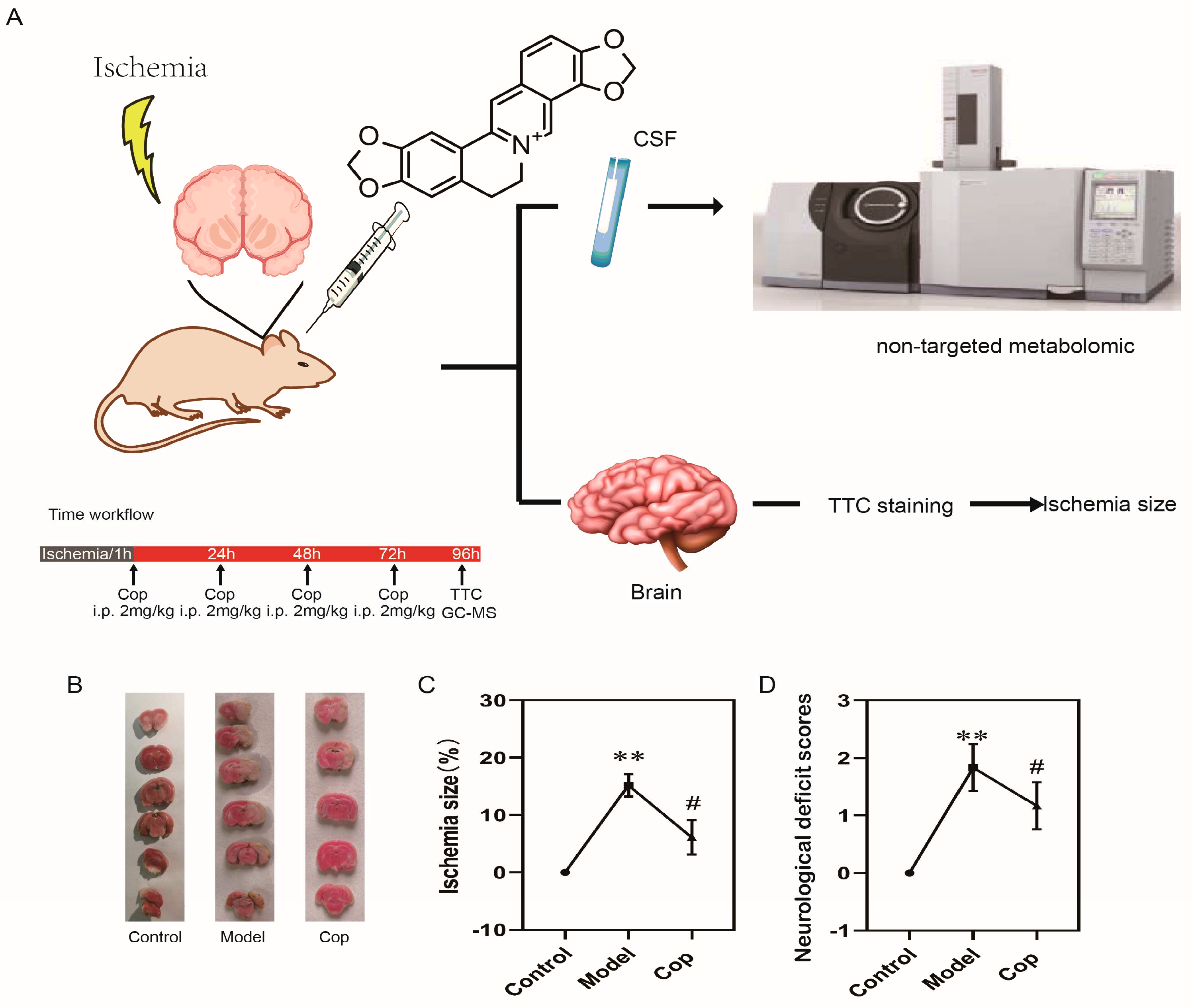
The experimental design of our study was depicted in Fig 1A. To confirm whether the cerebral IR rat model was successfully established, 2,3,5-triphenytetrazolium chloride staining and neurological deficit scores was performed with or without middle cerebral artery occlusion. From Fig 1B and 1C, we can clearly observe no cerebral ischemia lesion was showed in control group, whereas the ischemia size was apparently increased in cerebral IR group (19.40±2.47%, p<0.001). Meanwhile, the neurological deficit scores in model group (Fig 1D) were increased in comparison with control group (p<0.01). Our result suggested that the cerebral ischemic injury rat model was successfully established.
Subsequently, to investigate whether the protective effect of Cop against cerebral IR injury, we calculated cerebral ischemia size and neurological deficit scores with or without Cop-treated. A remarkably reduction in pale-colored region was observed in Cop-treated (2mg/kg) groups relative to model (in Fig 1B), and the cerebral ischemia size were significantly diminished after Cop treatment (7.65±2.50%, p<0.01 vs. model group). Neurological functional deficits were evaluated on 72h after surgery in rats, which could be prevented by Cop treatment (Fig 1D). These findings suggested that Cop possessed significant protective properties against cerebral IR injury in experimental animals.
Figure 1.
Coptisine effectively protected against cerebral IR injury in rat. (A) Flow chart of experimental protocol. (B) Representative TTC staining images at 96 h after reperfusion and quantitative analysis of infarct volume (n = 6). The ischemic portion appeared white and the non-ischemic portion appeared red.(C) Statistical results of ischemic size in rat. (D)Results of neurological deficit score in rat. Data were presented as mean±SD. ( # p < 0.05(model vs Cop), ** p < 0.01(control vs model)).
Figure 1.
Coptisine effectively protected against cerebral IR injury in rat. (A) Flow chart of experimental protocol. (B) Representative TTC staining images at 96 h after reperfusion and quantitative analysis of infarct volume (n = 6). The ischemic portion appeared white and the non-ischemic portion appeared red.(C) Statistical results of ischemic size in rat. (D)Results of neurological deficit score in rat. Data were presented as mean±SD. ( # p < 0.05(model vs Cop), ** p < 0.01(control vs model)).
2.2. Antioxidant and Anti-Inflammatory Effects of Cop in Response to Cerebral IR
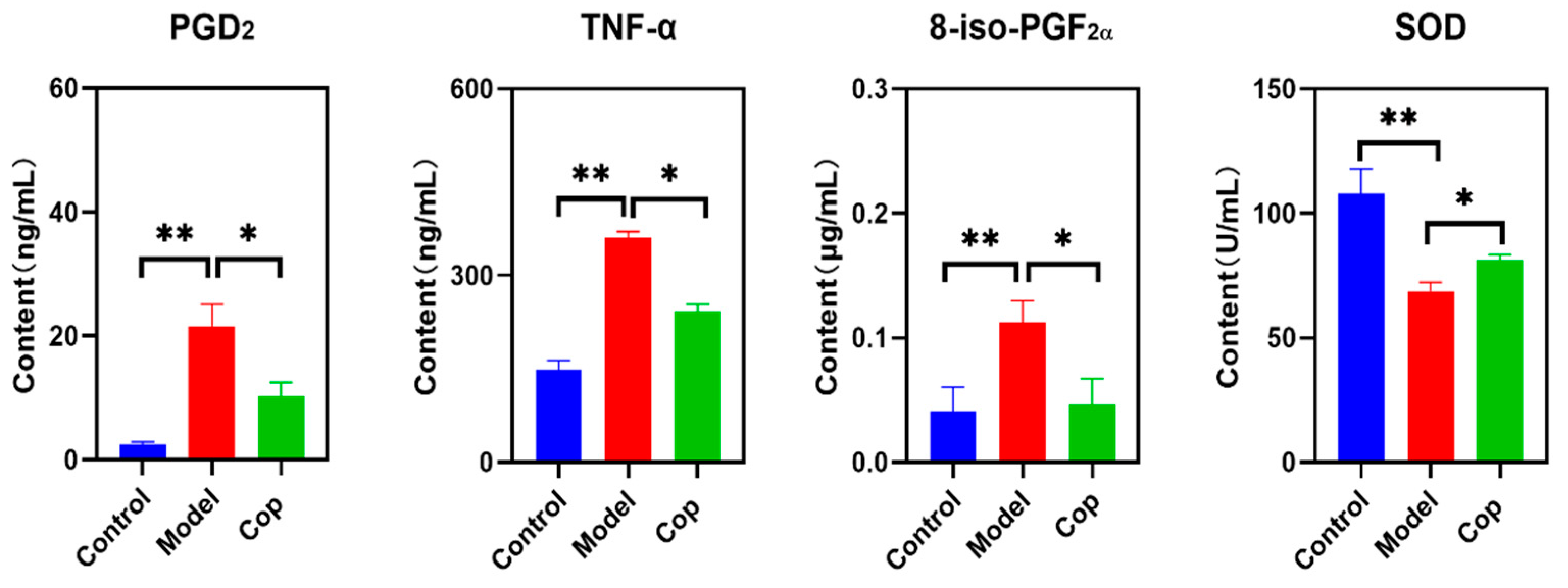
Enhancement in inflammation lead to cerebral tissue injury. To evaluate anti-inflammatory effect of Cop during cerebral IR injury, the brain levels of PGD2 and TNF-α were determined. In comparison with control group, high level of PGD2 and TNF-α displayed in cerebral IR group, but they were reversed when treated after Cop (Fig 2). In addition, upregulation in oxidative stress causes oxidative damage of proteins, lipids, and nucleic acids, and subsequently results in cell and tissue dysfunction, and cell death. Compared with control, the brain 8-iso-PGF2α in the cerebral IR group significantly increased to 0.113±0.017 μg/mL, while decrease in it was observed after Cop treatment (0.047±0.021 μg/mL). In contrast, reduced in SOD illustrated in cerebral IR group compared with control (107.80±10.05 U/mL vs 68.80±3.48 U/mL, p<0.01), but it was reversed when Cop exposure (81.24±2.19 U/mL, p value<0.05).
Figure 2.
Change in PGD2, TNF-α,8-iso-PGF2α and SOD among three groups. (*p value<0.05, ** p value<0.01).
Figure 2.
Change in PGD2, TNF-α,8-iso-PGF2α and SOD among three groups. (*p value<0.05, ** p value<0.01).
2.3. GC–MS Analysis of CSF Samples
In this study, we applied an untargeted GC-MS approach to investigate CSF metabolic profile among control, cerebral IR, and Cop-treated groups. From our result, more than 50 compound in CSF sample were separated and identified as shown in Tab S1. Quality control(n=6) were tested to verify the reliability of the method before the actual samples were measured. Six compounds (including amino acids, sugars, and fatty acids) in the quality control samples were selected as target of the method, who had different charge-to-mass ratio, chromatographic retention time, as well as satisfying conditions for better peak shape and higher peak intensities. As can be seen in Tab S2, the relative standard deviation values of the retention time and peak intensity of them were in the range of 0.013-0.037% and 2.584-5.873%, respectively. Our data indicated the method has good reproducibility and stability.
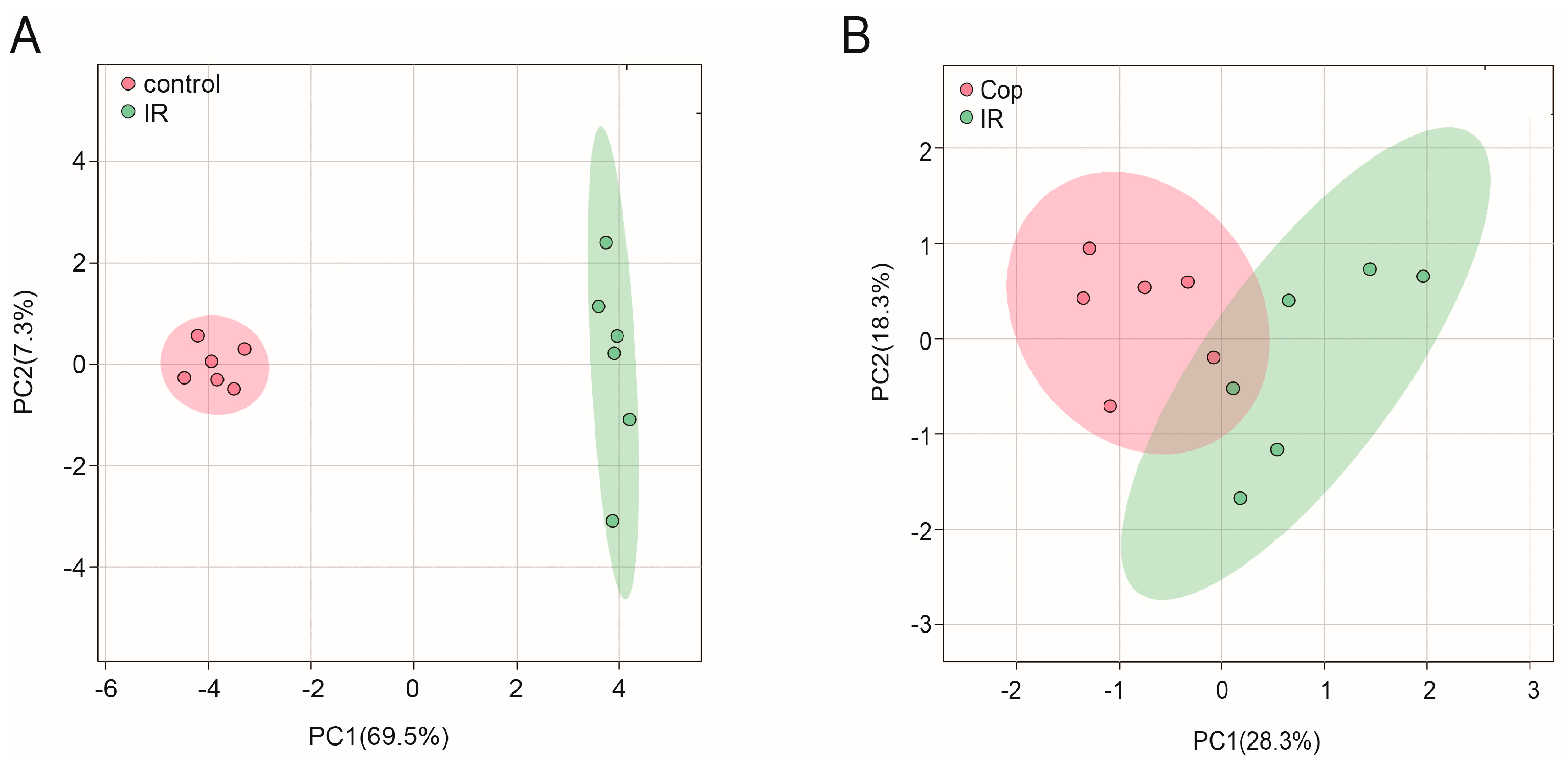
Figure 3.
Untargeted metabolomic analysis of CSF after cerebral IR and Cop-treated in rats. Plot of CSF PLS-DA between control and IR(A) group, IR and Cop-treated(B) group.
Figure 3.
Untargeted metabolomic analysis of CSF after cerebral IR and Cop-treated in rats. Plot of CSF PLS-DA between control and IR(A) group, IR and Cop-treated(B) group.
2.4. Screen in Metabolites from GC-MS Data as Potential Biomarkers between Control, Model, and Cop-Treated Groups
To initialize the systemic alteration of metabolites, a multivariate pattern discriminative analysis was conducted. A PLS-DA score plot of GC-MS data in CSF illustrated that control and cerebral IR injury can be well differentiated (Fig 3A), implying that the metabolic profile was significantly altered after cerebral IR. Meanwhile, Cop treated and untreated rats were always separated clearly (Fig 3B). These may indicate a broad role of Cop in treating cerebral IR injury.
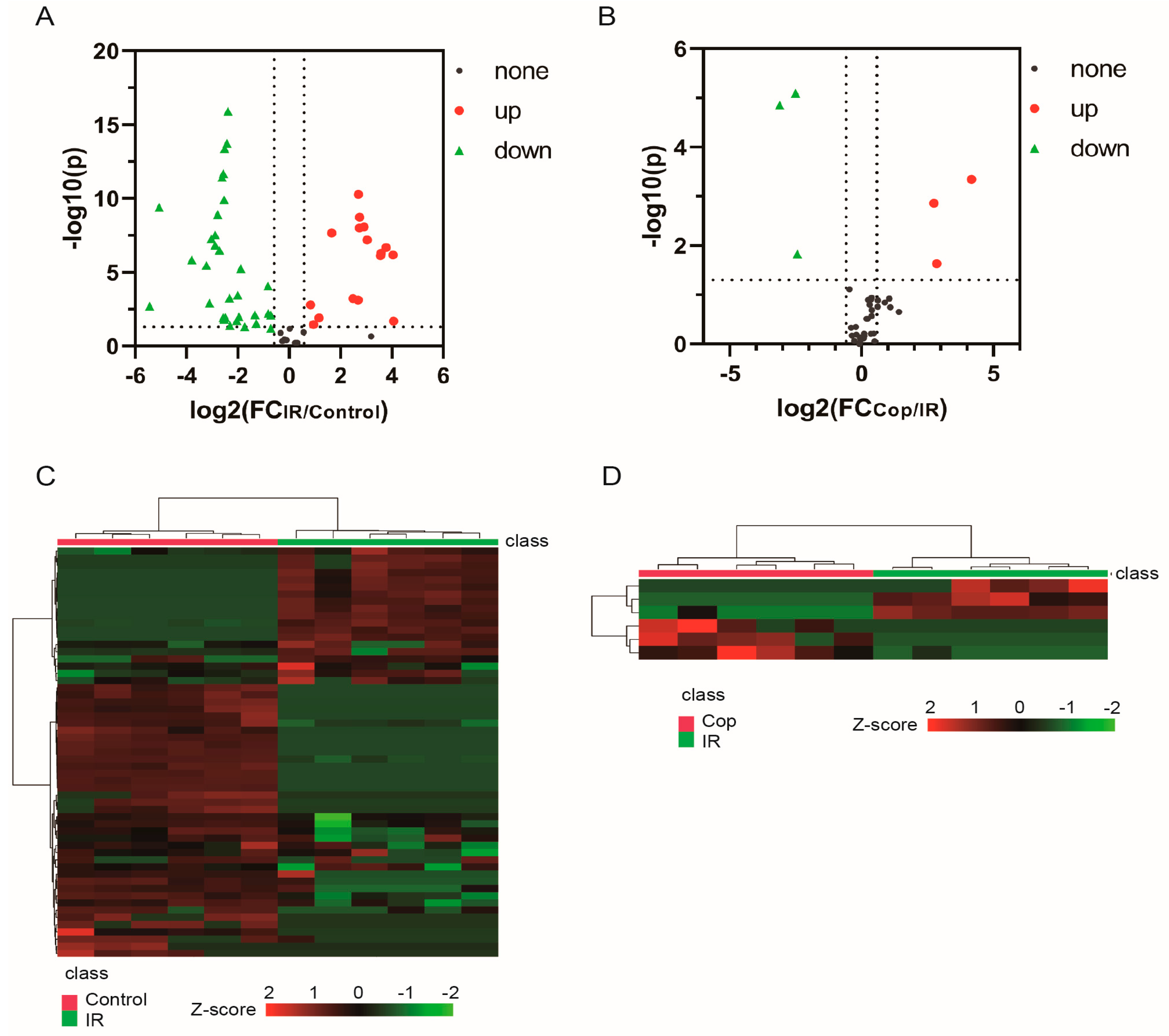
Subsequently, potential targets between cerebral IR and Cop-treated were screen by p-value < 0.05 in t-test and |FC|>1.5. Metabolite levels in response to cerebral IR and Cop-treated were visualized in heatmap that metabolites with warm color represented up-regulation and with cool color reflected down-regulation. There were 47 differential metabolites (16 increased and 31 decreased) were analyzed between IR and control groups in CSF, otherwise there were 6 differential metabolites in CSF (3 increased and 3 decreased) after Cop-treated. The volcano plot and heatmap also showed the expression level of differential metabolites among three groups in CSF (Fig 4). From the results above, significant differences of metabolites between control, cerebral IR and Cop treated were identified. Therefore, Cop may play a role in treatment of cerebral IR injury by altering the expression of certain metabolites.
Figure 4.
The volcano map and heatmap showed the differential compounds expression level between control and IR(A and C) , IR and Cop-treated(B and D). The red color indicates the upregulated compounds, while green color represents the downregulated compounds.
Figure 4.
The volcano map and heatmap showed the differential compounds expression level between control and IR(A and C) , IR and Cop-treated(B and D). The red color indicates the upregulated compounds, while green color represents the downregulated compounds.
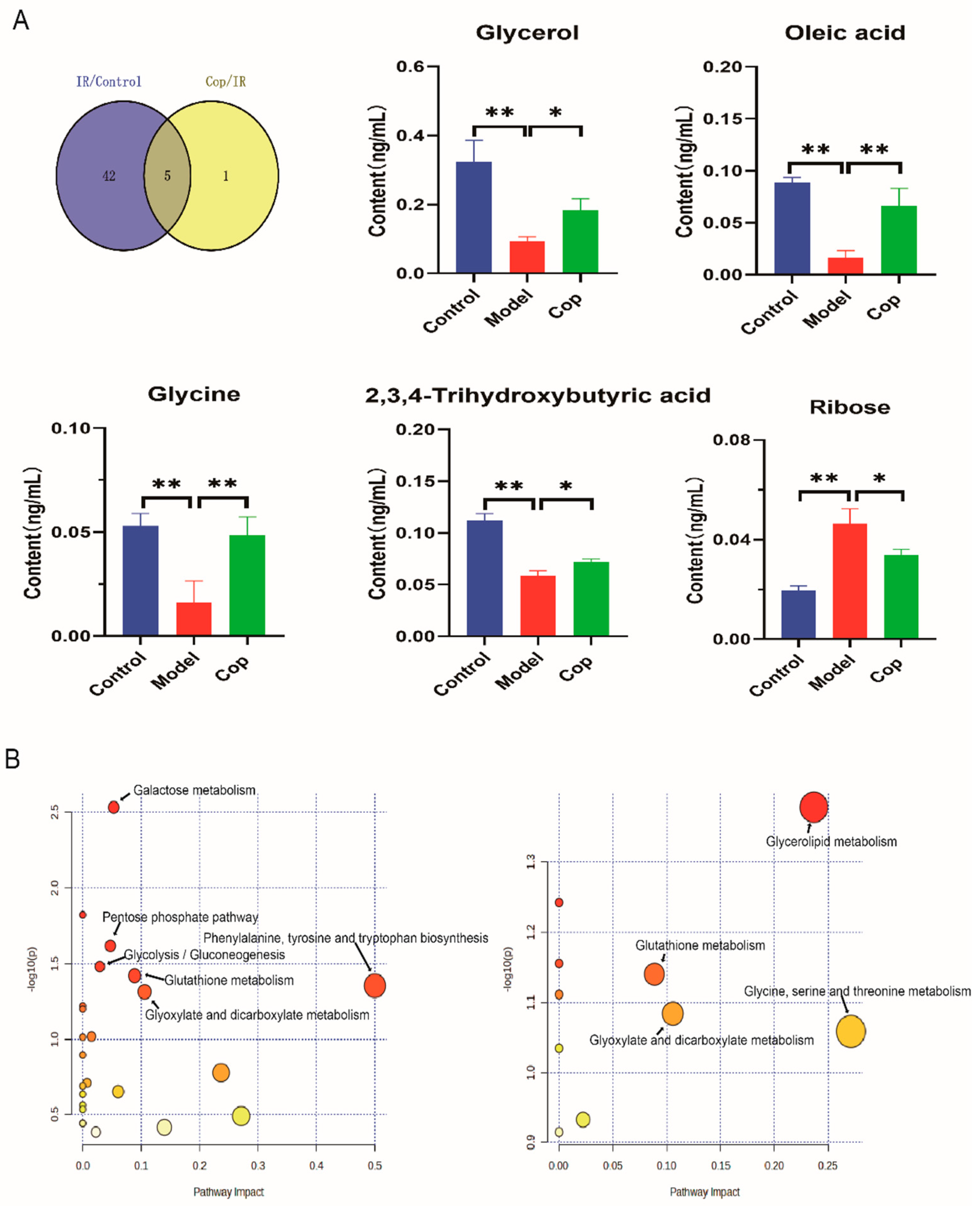
In order to evaluate the key metabolites which induced by cerebral IR and Cop-treated, we used a venn map to find the common metabolites between cerebral IR and Cop treated rat in CSF (Fig 5A). From our result, five common metabolites displayed between control, IR and Cop treated, including glycerol, oleic acid, glycine, ribose and 2,3,4-trihydroxybutyric acid (Fig 5).
2.5. Analysis of Metabolic Pathway Enrichment
To further explore the mechanisms responsible for cerebral IR and Cop treated, we carried out enrichment analysis and metabolic pathway analysis of marker using the MetaboAnalyst 5.0 platform. As can be seen in Fig 5B, CSF biomarkers of model and control enriched in the (1) Phenylalanine, tyrosine, and tryptophan biosynthesis;(2) Galactose metabolism (3) Pentose phosphate pathway; (4) Glycolysis / Gluconeogenesis; (5) Glutathione metabolism and (6) Glyoxylate and dicarboxylate metabolism. Additionally, the Glycerolipid metabolism was observed in the enrichment analysis between cerebral IR and Cop-treated..
Figure 5.
The unique differential compounds of different comparison groups. (A) The venn plot indicated the common different compounds as potential target among three groups and the bar chart revealed changes in their level among three groups. (* p value<0.05, * * p value<0.01) (B) Enrichment analysis of differential compounds between control and IR (left) , IR and Cop treated(right).
Figure 5.
The unique differential compounds of different comparison groups. (A) The venn plot indicated the common different compounds as potential target among three groups and the bar chart revealed changes in their level among three groups. (* p value<0.05, * * p value<0.01) (B) Enrichment analysis of differential compounds between control and IR (left) , IR and Cop treated(right).
3. Discussion
To our best knowledge, this is the first report that investigates the mechanism of Cop against cerebral IR by untargeted CSF metabolomes. In this study, we observed that Cop attenuated cerebral IR induced cerebral ischemia size by 2, 3, 5-triphenytetrazolium chloride staining, and decreased in neurological deficit function score, suggested that Cop exerted a neuroprotective role in cerebral IR. To investigate the protective mechanism of Cop against cerebral IR, biochemical indicators and an untargeted metabolomic experiment were carried out. In the process of cerebral IR, excessive in the release of pro-inflammatory factor PGD2, and TNF-α increases blood-brain barrier permeability, promoting the infiltration and aggregation of leukocytes thereby causing neuroinflammation [17, 18]. Our data revealed that Cop decreased the level of PGD2, and TNF-α in brain. Moreover, imbalance in oxidative stress induced cerebral IR injury were reported by previous studies. Changes in SOD and 8-iso-PGF2α present the oxidative stress state in vivo[19-21]. Our result showed that upregulation on 8-iso-PGF2α and downregulation on SOD expression displayed in cerebral IR compared with control, whereas their levels were reversed through Cop treatment. These results indicated that antioxidant and antiinflammation effect are the mechanism of Cop against cerebral IR.
A large body of literature reports about the metabolic changes after cerebral IR injury and attempts to find the corresponding potential therapeutic targets. However, there are relatively few studies on the response of the CSF metabolome to Cop during cerebral IR therapy. In our results, CSF five compounds (glycine, glycerol, oleic acid, and ribose) were discovered by untargeted metabolomic analysis. Interestingly, the role of these analytes in vivo has been reported to be closely associated with antiinflammation and antioxidant.
Glycine, a nerve inhibitory amino acid, exert neuroprotective properties. Goulart and their colleagues also reported that diminished in glycine was observed in ischemic stroke compared with control[22], which is similar with our studies. Supplement on glycine reduce the secretion of pro-inflammatory cytokine in PC12 cell with induction of oxygen-glucose deprivation[23]. Liu et al. demonstrated that glycine treated cerebral IR injury rat was able to suppress ischemia-mediated inflammation by improving M2 microglia polarization and inhibiting M1 microglia polarization by NF-κB p65/Hif-1ɑ signaling pathway[24]. Our result showed that high level of glycine was observed after Cop treatment.
Oleic acid is a major constituent of membrane phospholipids and is highly concentrated in myelin. Recent studies have demonstrated that oleic acid is necessary for the appropriate development and functioning of the brain. During brain development, oleic acid is used to synthesize myelin phospholipids and acts as a neurotrophic factor by promoting axonal and dendritic growth, enhancing neuronal migration and aggregation, and facilitating synapse formation[25]. Vascellari et al reported that decreased in oleic acid level was observed in Parkinson’s disease compared with health[26]. An in vitro study demonstrated that oleic acid attenuates the microglial inflammatory responses that promote neuronal death after cerebral ischemia[27]. Song et al reported that oleic acid significantly reduced the infarct volume after cerebral ischemia, prevent CA1 neuronal death and markedly attenuated the immunoreactivity of cyclooxygenase-2, inducible nitric oxide synthase and TNF-α[28]. Our data indicated that down-regulation in oleic acid level was illustrated in cerebral IR injury compared with control, while upregulation in it was observed when treated after Cop.
As an aldehyde containing pentose sugar, ribose participates in numerous biochemical processes and has an active role glycation of protein, producing advanced glycation end products that have severe cytotoxicity, which can lead to cell dysfunction and death. Zhang found ribose decreased the cell viability and increased lactate dehydrogenase release in mesangial cell, and lead to glutathione depletion that naturally occurring antioxidant present in cell and clear intracellular reactive oxygen species[29]. Yu et al. reported that decreased ribose markedly reduced advance glycation end products accumulation, Tau hyperphosphorylation, and neuronal death[30]. Moreover, ribose activated NLRP3 inflammasome and induced NF-κB pathway via advance glycation end products pathway were evidenced by Hong et al[31, 32]. NLRP3 inflammasome mediates the activation of caspase-1, which promotes the secretion of proinflammatory cytokines. Wu et al reported that Cop not only prevented NLRP3 inflammasome assembly by affecting the binding between pro-capase-1 and apoptosis-associated speck like protein containing a CARD, but also inhibited inflammasome priming by decreasing NLRP3 expresion through inactivation of NF-κB pathway[33]. Xiong et al reported that Cop decreased the level of TNF-α, IL-1β and IL-18, and significantly downregulated the protein expression levels of NLRP3, apoptosis-associated speck-like protein containing a CARD and caspase-1[34]. Our results showed that ribose levels were increased in the cerebral IR group and that Cop reversed it. We hypothesized Cop might reduce ribose levels to inhibit NF-κB inflammatory signaling caused by activation of the NLRP3 inflammasome, and specific mechanism need to be further studied.
Glycerol is a precursor for membrane phospholipid synthesis and the main lipid constituent of the myelin sheath. Stroke is associated with the disruption of the blood-brain-barrier, as evidenced by enhancing matrix metalloproteinase-9 activity and reducing zonula occludens-1 protein expression. Chang et al reported that glycerol reduced matrix metalloproteinase-9 activity and increased zonula occludens-1 protein expression in the stroke model[35]. Meanwhile, glycerol could also alleviate oxidative stress and neuroinflammation in stroke by NF-κB pathway. Previous studies have demonstrated that the increase in glycerol and the decrease in formate levels were detected in the ischemic brain treated with drug, and that drug-mediated increase in glycerol was associated with myelin formation in oligodendrocytes which serve to maintain and protect normal neuronal function[36]. Cop increased glycerol level in CSF, suggesting that it might be associated with anti-inflammation.
2,3,4-trihydroxybutyric acid (namely erythronic acid), which is commonly found in aqueous humor of the eye and in cerebrospinal fluid, derived from glycated proteins. Our result revealed that lower level of it displayed in cerebral IR group. Lower blood level of 2,3,4-trihydroxybutyric acid presented in patients with acute ischemic stroke compared with health[37], which is similar with our results. The role of 2,3,4-trihydroxybutyric acid in brain injury is still unclear, but it might be negatively associated with inflammation and oxidative stress. Huang et al reported that 2,3,4-trihydroxybutyric acid in hyperlipidemia acute pancreatitis decreased observably compare with the health control[38]. Moreover, 2,3,4-trihydroxybutyric acid is an oxidative product of erythritol, which has excellent hydroxyl radical scavenger properties. Jin et al reported that erythritol reversed lipid accumulation, and promote the production of heme oxygenase 1 and NQO1 antioxidant proteins and inhibit the expression of endoplasmic reticulum stress proteins GPR78, p-PERK and CHOP by nuclear factor E2-related factor pathway [39]. Both erythritol and 2,3,4-trihydroxybutyric acid have been identified as products when N-acetylglucosamine is oxidized NaOCl, that relates to reactive oxygen species degradation of connective tissue[40]. In our result, Cop treatment increased the level of 2,3,4-trihydroxybutyric acid. We hypothesis the regulation mechanism of Cop might be associated with antioxidant. Cop inhibited H2O2-induced cytotoxicity and DNA damage by blocking abnormal reactive oxygen species generation, and the activation of nuclear factor-erythroid-2-related factor 2 and heme oxygenase-1 was markedly promoted by Cop in the presence of H2O2[9]. Zhai and their colleagues also illustrated that the expression levels of nuclear factor-erythroid 2-related factor 2 and its targeted antioxidative genes heme oxygenase 1 and NADPH quinone oxidoreductase 1 after Cop treatment [41].
4. Materials and Methods
4.1. Chemical Reagents
Methanol was chromatographic grade and obtained from Merck (Germany). Cop(purity>98%) was purchase by Yirui Biotechnology Co.Ltd.(Chengdu, China), and Methoxyamine hydrochloride and N, O-bis (trimethylsilyl) trifuoroacetamide (1% trimethylchlorosilane) were supplied by Macklin (Shanghai, China). Heptadecanoic acid (purity >98%) as internal standard was obtained from Sigma-Aldrich (USA) and diluted with ethyl acetate at concentration of 100 μg/mL. A Milli-Q purification system (Billerica, MA, USA) was used to get ultrapure water.
4.2. Animal Care
The experiment protocol was authorized by Ethics Committee for Guangdong Medical laboratory animal center (Foshan, China) and conformed to internationally accepted standard. Eighteen male Sprague-Dawley rats (300-320g) was furnished by Guangdong Medical Laboratory Animal center and housed under controlled conditions with a 12h light-dark at 21±2℃, and with a relative humidity of 60%±5% for at least five days prior to experiment. They had free access to a standard rodent diet and tap water.
4.3. Transient Focal Cerebral IR and Animal Treatment
The intraluminal suture method was used to close the middle cerebral artery. First, rats were sedated with chloral hydrate (350mg/kg, i.p.). The origin of the middle cerebral artery was then blocked with a 3-0 monofilament after 2 cm skin dissection at the ventral muscle of the rat’s neck. After 60 min of ischemia, the monofilament was removed, and reperfusion was completed, followed by wound closure. During the operation and reperfusion, a heated blanket kept the rectal temperature at 37±0.5℃
After surgery, rats were randomly divided into two groups: IR (n=6) and IR after Cop-treatment (n=6). After 60 minutes of reperfusion, rats were given an intraperitoneal injection of Cop (2mg/kg/qd), while the control (Sham, n=6) and IR groups received saline instead.
4.4. Neurological Function Evaluation
The neurological function of the conscious rat was blindly assessed 72h after middle cerebral artery occlusion. Neurological deficits scores in rats were assess by an observed blinded to the group using a previously reported five-point scale[42] (score 0, no deficit being observed; score 1, failing to fully extend left forepaw; score 2, circling to the left side; score 3, falling to the left; score 4, with no walking and consciousness being depressed).
4.5. Sample Collection
After 96 h of cerebral IR injury, rats were given chloral hydrate(350mg/kg,i.p.) for anesthesia, then placed on a homemade device that bent their heads downward 45°, and a small midline incision between the ears was made to separate the muscles to expose the atlanto-occipital membrane located between the occipital bone and the upper cervical vertebrae. A butterfly needle (25 gauge×19mm) was used to aspirate clear CSF (nearly 50 μL) into the syringe until blood appeared and the clear CSF was transferred to an Eppendorf tube and stored at -80℃. Brain tissue (n=6/per group) was collected and cut into 6 coronal sections of 2 mm thickness, finally these sections was stained with 2% 2,3,5-triphenytetrazolium chloride to assess the size of cerebral IR injury.
4.6. Sample Preparation and GC-MS Analysis
Firstly, took 20 μL CSF and 15 μL heptadecanoic acid (100 μg/mL, internal standard solution) in 1 mL methanol, and the mixture was vortexed at 4℃ for 10 min, then centrifuged at 15,000 rpm for 5 min, and the supernatant was extracted into a glass bottle. Dried the supernatant with nitrogen. Dissolved the residue in the bottle with 50μL of 2% methoxyamine hydrochloride and react at 37℃ for 90 min, then add 50μL of N, O-bis (trimethylsilyl) trifuoroacetamide (1% trimethylchlorosilane) into the bottle and react at 70℃ for 60 min. Samples were stored at − 80℃ before analysis to prevent metabolite degradation.
The CSF metabolites were analyzed by GCMS-QP2010Ultra (Shimadzu, Japan), and compounds in samples were separated using a capillary column DB-5 (30 m × 0.25 mm × 0.25 μm, Agilent, USA). The mass spectrometer was operated in the electron impact mode at 70 eV. The injection volume was 1 μL per sample and the split mode was set to splitless. The temperature program was as follows: the inlet temperature was set at 230 °C. The initial temperature was held at 85 °C for 2 min, elevated to 150 °C at a rate of 5 °C/min, increased to 240 °C at a rate of 15 °C/min, raised to 280 °C by 40 °C/min, and then kept at 280 °C for 5 min. The temperature of transfer line and ion source was 250 °C and 200 °C respectively. Helium (99.999%) was used as the carrier gas, the flow control mode was constant pressure, and the set pressure was 100 kPa. The analyses were performed in the full scan mode, monitoring the mass range of the detector was set to m/z 50–650.
4.7. Qualitative Identification of Metabolites and Raw Data Preprocessing
Preliminary identification of compounds was performed firstly on NIST 11 library; the qualitative standard of the database was that the similarity was greater than 80%.
Divided the peak area of the compound by the peak area of heptadecanoic acid for standardization correction, performed logarithmic conversion and autoscaling to achieve normal distribution. After that, the corrected peak areas of all identified metabolites were used for further analysis.
4.8. Measurement of Brain PGD2, TNF-α, 8-iso-PGF2α and SOD,
The levels of PGD2 and 8-iso-PGF2α in brain were detected by liquid chromatography-tandem mass spectrometry as previous studies[43]. The level of TNF-α and SOD was calculated by commercial ELISA kit according to the instruction of manufacturer (Shanghai Enzyme-linked Biotechnology Co., Ltd., Shanghai, China)
4.9. Statistical Analysis
Data were presented as mean ± standard deviation. Metabolomic analysis were achieved using the online software MetaboAnalyst (http://www.metaboanalyst.ca/), including multivariate analysis (principal component analysis and partial least square discriminant analysis (PLS-DA) and cluster analysis. We used Student’s t-test (SPSS 19.0 software) to calculate the statistical significance. p value after the Benjamini–Hochberg false discovery rate adjustment was less than 0.05 that was considered significant difference..
5. Conclusions
In this study, Cop significantly reduced the ischemia size and improve neurological deficits scores after IR injury. Moreover, Cop inhibited the release of pro-inflammatory mediators (e.g., PGD2 and TNF-α) and ameliorate the oxidative stress (increase SOD expression and suppress 8-iso-PGF2α level). Furthermore, metabolomic analysis shown different CSF metabolic profile among control, IR and Cop treated groups. A total of five metabolites in CSF were identified as target and the related pathways analysis were further conducted. The combination of biochemical indicator and metabolomics methods revealed Cop exerted an antiinflammation and antioxidant effect against cerebral IR injury..
Author Contributions
Junjie Zhang, Chun Cai, and Hui Xu contribute to the study design. Junjie Zhang and Ao Qi conduct the research; Junjie Zhang and Lulu Liu analyzed data; Junjie Zhang, Chun Cai and Hui Xu wrote the paper; All authors read and approved the final manuscript.
Funding
This work was financially supported by the National Natural Science Foundation of China (C. Cai, Grant No. 21375209).
Institutional Review Board Statement
The animal experiment protocol was authorized by Ethics Committee for Guangdong Medical laboratory animal center (Foshan, China, approval No. B202101-11).
Data Availability Statement
The datasets used and/or analyzed in during the current study are available from the corresponding author on reasonable request.
Conflicts of Interest
The authors declare that they have no competing interests.
References
- Tsao CW, Aday AW, Almarzooq ZI, Anderson CAM, Arora P, Avery CL, Baker-Smith CM, Beaton AZ, Boehme AK, Buxton AE et al: Heart Disease and Stroke Statistics-2023 Update: A Report From the American Heart Association. Circulation 2023, 147, e93–e621.
- Marko M, Posekany A, Szabo S, Scharer S, Kiechl S, Knoflach M, Serles W, Ferrari J, Lang W, Sommer P et al: Trends of r-tPA (Recombinant Tissue-Type Plasminogen Activator) Treatment and Treatment-Influencing Factors in Acute Ischemic Stroke. Stroke 2020, 51, 1240–1247. [CrossRef] [PubMed]
- Yang L, Su X, Lu F, Zong R, Ding S, Liu J, Wilson G, Li L, Yang Y, Wang W et al: Serum and brain metabolomic study reveals the protective effects of Bai-Mi-Decoction on rats with ischemic stroke. Front Pharmacol 2022, 13, 1005301. [CrossRef] [PubMed]
- Ma HF, Zheng F, Su LJ, Zhang DW, Liu YN, Li F, Zhang YY, Gong SS, Kou JP: Metabolomic Profiling of Brain Protective Effect of Edaravone on Cerebral Ischemia-Reperfusion Injury in Mice. Front Pharmacol 2022, 13, 814942. [CrossRef]
- Lin B, Chen R, Wang Q, Li Z, Yang S, Feng Y: Transcriptomic and Metabolomic Profiling Reveals the Protective Effect of Acanthopanax senticosus (Rupr. & Maxim.) Harms Combined With Gastrodia elata Blume on Cerebral Ischemia-Reperfusion Injury. Front Pharmacol 2021, 12, 619076. [Google Scholar]
- Friedemann T, Ying Y, Wang W, Kramer ER, Schumacher U, Fei J, Schroder S: Neuroprotective Effect of Coptis chinensis in MPP[Formula: see text] and MPTP-Induced Parkinson's Disease Models. Am J Chin Med 2016, 44, 907–925. [CrossRef]
- Wang Y, Liu J, Huang Z, Li Y, Liang Y, Luo C, Ni C, Xie J, Su Z, Chen J et al: Coptisine ameliorates DSS-induced ulcerative colitis via improving intestinal barrier dysfunction and suppressing inflammatory response. Eur J Pharmacol 2021, 896, 173912. [CrossRef]
- Liu Y, Gong S, Li K, Wu G, Zheng X, Zheng J, Lu X, Zhang L, Li J, Su Z et al: Coptisine protects against hyperuricemic nephropathy through alleviating inflammation, oxidative stress and mitochondrial apoptosis via PI3K/Akt signaling pathway. Biomed Pharmacother 2022, 156, 113941. [CrossRef]
- Jo HG, Park C, Lee H, Kim GY, Keum YS, Hyun JW, Kwon TK, Choi YH, Hong SH: Inhibition of oxidative stress induced-cytotoxicity by coptisine in V79-4 Chinese hamster lung fibroblasts through the induction of Nrf-2 mediated HO-1 expression. Genes Genomics 2021, 43, 17–31. [CrossRef]
- Hu YR, Ma H, Zou ZY, He K, Xiao YB, Wang Y, Feng M, Ye XL, Li XG: Activation of Akt and JNK/Nrf2/NQO1 pathway contributes to the protective effect of coptisine against AAPH-induced oxidative stress. Biomed Pharmacother 2017, 85, 313–322. [CrossRef]
- Wen X, Zhang X, Qu S, Chen X, Liu C, Yang Y: Coptisine induces G2/M arrest in esophageal cancer cell via the inhibition of p38/ERK1/2/claudin-2 signaling pathway. Pharmazie 2021, 76, 202–207.
- Han B, Jiang P, Li Z, Yu Y, Huang T, Ye X, Li X: Coptisine-induced apoptosis in human colon cancer cells (HCT-116) is mediated by PI3K/Akt and mitochondrial-associated apoptotic pathway. Phytomedicine 2018, 48, 152–160. [CrossRef] [PubMed]
- Huang J, Sun C, Yao D, Wang CZ, Zhang L, Zhang Y, Chen L, Yuan CS: Novel surface imprinted magnetic mesoporous silica as artificial antibodies for efficient discovery and capture of candidate nNOS-PSD-95 uncouplers for stroke treatment. J Mater Chem B 2018, 6, 1531–1542. [CrossRef] [PubMed]
- Friedemann T, Schumacher U, Tao Y, Leung AK, Schroder S: Neuroprotective Activity of Coptisine from Coptis chinensis (Franch). Evid Based Complement Alternat Med 2015, 2015, 827308.
- Gong LL, Fang LH, Qin HL, Lv Y, Du GH: Analysis of the mechanisms underlying the vasorelaxant action of coptisine in rat aortic rings. Am J Chin Med 2012, 40, 309–320. [CrossRef]
- Yu D, Tao BB, Yang YY, Du LS, Yang SS, He XJ, Zhu YW, Yan JK, Yang Q: The IDO inhibitor coptisine ameliorates cognitive impairment in a mouse model of Alzheimer's disease. J Alzheimers Dis 2015, 43, 291–302.
- Iwasa K, Yamamoto S, Yamashina K, Yagishita-Kyo N, Maruyama K, Awaji T, Takei Y, Hirasawa A, Yoshikawa K: A peripheral lipid sensor GPR120 remotely contributes to suppression of PGD(2)-microglia-provoked neuroinflammation and neurodegeneration in the mouse hippocampus. J Neuroinflammation 2021, 18, 304.
- Zhang H, Wang L, Yang Y, Cai C, Wang X, Deng L, He B, Zhou W, Cui Y: DL-3-n-butylphthalide (NBP) alleviates poststroke cognitive impairment (PSCI) by suppressing neuroinflammation and oxidative stress. Front Pharmacol 2022, 13, 987293.
- Elesber AA, Best PJ, Lennon RJ, Mathew V, Rihal CS, Lerman LO, Lerman A: Plasma 8-iso-prostaglandin F2alpha, a marker of oxidative stress, is increased in patients with acute myocardial infarction. Free Radic Res 2006, 40, 385–391. [CrossRef]
- Wisniewski K, Popeda M, Price B, Bienkowski M, Fahlstrom A, Drummond K, Adamides AA: Glucose-6-phosphate dehydrogenase and 8-iso-prostaglandin F2alpha as potential predictors of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. J Neurosurg 2023, 1–10.
- Arteaga Cabeza O, Zhang Z, Smith Khoury E, Sheldon RA, Sharma A, Zhang F, Slusher BS, Kannan RM, Kannan S, Ferriero DM: Neuroprotective effects of a dendrimer-based glutamate carboxypeptidase inhibitor on superoxide dismutase transgenic mice after neonatal hypoxic-ischemic brain injury. Neurobiol Dis 2021, 148, 105201.
- Goulart VAM, Sena MM, Mendes TO, Menezes HC, Cardeal ZL, Paiva MJN, Sandrim VC, Pinto MCX, Resende RR: Amino Acid Biosignature in Plasma among Ischemic Stroke Subtypes. Biomed Res Int 2019, 2019, 8480468.
- Chen ZJ, Zhao XS, Fan TP, Qi HX, Li D: Glycine Improves Ischemic Stroke Through miR-19a-3p/AMPK/GSK-3beta/HO-1 Pathway. Drug Des Devel Ther 2020, 14, 2021–2031. [CrossRef] [PubMed]
- Liu R, Liao XY, Pan MX, Tang JC, Chen SF, Zhang Y, Lu PX, Lu LJ, Zou YY, Qin XP et al: Glycine Exhibits Neuroprotective Effects in Ischemic Stroke in Rats through the Inhibition of M1 Microglial Polarization via the NF-kappaB p65/Hif-1alpha Signaling Pathway. J Immunol 2019, 202, 1704–1714.
- Polo-Hernandez E, Tello V, Arroyo AA, Dominguez-Prieto M, de Castro F, Tabernero A, Medina JM: Oleic acid synthesized by stearoyl-CoA desaturase (SCD-1) in the lateral periventricular zone of the developing rat brain mediates neuronal growth, migration and the arrangement of prospective synapses. Brain Res 2014, 1570, 13–25.
- Vascellari S, Palmas V, Melis M, Pisanu S, Cusano R, Uva P, Perra D, Madau V, Sarchioto M, Oppo V et al: Gut Microbiota and Metabolome Alterations Associated with Parkinson's Disease. NA 2020, 5.
- Beaulieu J, Costa G, Renaud J, Moitie A, Glemet H, Sergi D, Martinoli MG: The Neuroinflammatory and Neurotoxic Potential of Palmitic Acid Is Mitigated by Oleic Acid in Microglial Cells and Microglial-Neuronal Co-cultures. Mol Neurobiol 2021, 58, 3000–3014. [CrossRef]
- Song J, Kim YS, Lee DH, Lee SH, Park HJ, Lee D, Kim H: Neuroprotective effects of oleic acid in rodent models of cerebral ischaemia. Sci Rep 2019, 9, 10732. [CrossRef]
- Zhang N, Zhao S, Hong J, Li W, Wang X: Protective Effects of Kaempferol on D-Ribose-Induced Mesangial Cell Injury. Oxid Med Cell Longev 2019, 2019, 7564207.
- Yu L, Chen Y, Xu Y, He T, Wei Y, He R: D-ribose is elevated in T1DM patients and can be involved in the onset of encephalopathy. Aging (Albany NY) 2019, 11, 4943–4969.
- Hong J, Bhat OM, Li G, Dempsey SK, Zhang Q, Ritter JK, Li W, Li PL: Lysosomal regulation of extracellular vesicle excretion during d-ribose-induced NLRP3 inflammasome activation in podocytes. Biochim Biophys Acta Mol Cell Res 2019, 1866, 849–860. [CrossRef] [PubMed]
- Hong J, Li G, Zhang Q, Ritter J, Li W, Li PL: D-Ribose Induces Podocyte NLRP3 Inflammasome Activation and Glomerular Injury via AGEs/RAGE Pathway. Front Cell Dev Biol 2019, 7, 259. [CrossRef] [PubMed]
- Wu J, Luo Y, Jiang Q, Li S, Huang W, Xiang L, Liu D, Hu Y, Wang P, Lu X et al: Coptisine from Coptis chinensis blocks NLRP3 inflammasome activation by inhibiting caspase-1. Pharmacol Res 2019, 147, 104348. [CrossRef] [PubMed]
- Xiong Y, Wei H, Chen C, Jiao L, Zhang J, Tan Y, Zeng L: Coptisine attenuates post-infectious IBS via Nrf2-dependent inhibition of the NLPR3 inflammasome. Mol Med Rep 2022, 26.
- Chang CY, Pan PH, Li JR, Ou YC, Liao SL, Chen WY, Kuan YH, Chen CJ: Glycerol Improves Intracerebral Hemorrhagic Brain Injury and Associated Kidney Dysfunction in Rats. Antioxidants (Basel) 2021, 10.
- Lee J, Shin JA, Lee EM, Nam M, Park EM: Noggin-mediated effects on metabolite profiles of microglia and oligodendrocytes after ischemic insult. J Pharm Biomed Anal 2023, 224, 115196. [CrossRef]
- Wang D, Kong J, Wu J, Wang X, Lai M: GC-MS-based metabolomics identifies an amino acid signature of acute ischemic stroke. Neurosci Lett 2017, 642, 7–13. [CrossRef]
- Huang JH, He D, Chen L, Dong CY, Zhang SH, Qin YH, Yu R, Ahmed R, Kuang JJ, Zhang XW: GC-MS based metabolomics strategy to distinguish three types of acute pancreatitis. Pancreatology 2019, 19, 630–637. [CrossRef]
- Jin M, Wei Y, Yu H, Ma X, Yan S, Zhao L, Ding L, Cheng J, Feng H: Erythritol Improves Nonalcoholic Fatty Liver Disease by Activating Nrf2 Antioxidant Capacity. J Agric Food Chem 2021, 69, 13080–13092. [CrossRef]
- den Hartog GJ, Boots AW, Adam-Perrot A, Brouns F, Verkooijen IW, Weseler AR, Haenen GR, Bast A: Erythritol is a sweet antioxidant. Nutrition 2010, 26, 449–458. [CrossRef]
- Zhai J, Li Z, Zhang H, Ma L, Ma Z, Zhang Y, Zou J, Li M, Ma L, Li X: Coptisine ameliorates renal injury in diabetic rats through the activation of Nrf2 signaling pathway. Naunyn-Schmiedeberg's Arch Pharmacol 2020, 393, 57–65. [CrossRef] [PubMed]
- Cai L, Yao ZY, Yang L, Xu XH, Luo M, Dong MM, Zhou GP: Mechanism of Electroacupuncture Against Cerebral Ischemia-Reperfusion Injury: Reducing Inflammatory Response and Cell Pyroptosis by Inhibiting NLRP3 and Caspase-1. Front Mol Neurosci 2022, 15, 822088. [CrossRef] [PubMed]
- Yamada M, Kita Y, Kohira T, Yoshida K, Hamano F, Tokuoka SM, Shimizu T: A comprehensive quantification method for eicosanoids and related compounds by using liquid chromatography/mass spectrometry with high speed continuous ionization polarity switching. J Chromatogr B Analyt Technol Biomed Life Sci 2015, 995–996, 74–84.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions, or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



