
Submitted:
08 August 2023
Posted:
09 August 2023
You are already at the latest version
Abstract
Chronic kidney disease (CKD) is a progressive condition of kidney dysfunction due to diverse causes of injury. In healthy kidneys, protein-bound uremic toxins (PBUTs) are cleared from the systemic circulation by proximal tubule cells through the concerted action of plasma membrane transporters that facilitate their urinary excretion, but the endogenous metabolites are hardly removed with kidney dysfunction and may contribute to CKD progression. Accumulating evidence suggests that senescence of kidney tubule cells influences kidney fibrosis, the common endpoint for CKD with an excessive accumulation of extracellular matrix (ECM). Senescence is a special state of cells characterized by permanent cell cycle arrest and limitation of proliferation, which promotes fibrosis by releasing senescence-associated secretory phenotype (SASP) factors. The accumulation of PBUTs in CKD causes oxidative stress and increases the production of inflammatory (SASP) factors that could trigger fibrosis. Recent studies provide some clues that PBUTs may also promote senescence in kidney tubular cells. This review provides an overview on how senescence contributes to CKD and the involvement of PBUTs in this process, and how kidney senescence can be studied, Finally, some suggestions for future therapeutic options for CKD while targeting senescence are given.
Keywords:
chronic kidney disease
; uremic toxins
; renal tubular transport
; extracellular matrix remodeling
; apoptosis resistance
; inflammatory response
; senescence-associated secretory phenotype factors
1. Introduction
Kidney fibrosis leads to organ failure by an excessive accumulation of extracellular matrix (ECM), which is the common endpoint for a variety of progressive chronic kidney diseases (CKD) [1]. Senescence is a special form of permanent cell cycle arrest, which limits proliferation and is highly related to inflammation and fibrosis. Senescent cells exacerbate these processes by releasing senescence-associated secretory phenotype (SASP) factors, which are of pro-inflammatory and pro-fibrotic nature [2]. Uremic toxins are metabolites that accumulate during kidney disease. Protein-bound uremic toxins (PBUTs) are mostly less than 500 Da, but are poorly removed with kidney dysfunction as they are tightly bound to plasma proteins and can also hardly cross dialyzer membranes [3,4]. PBUTs, like indoxyl sulfate (IS) and p-cresol sulfate (PCS), accumulate in CKD, maintaining and reinforcing CKD and kidney fibrosis [5,6]. Recent studies reported that IS and PCS activate the renal RAAS/TGF-β pathway and induce epithelial mesenchymal transition (EMT) [6]. EMT is a common process during fibrosis and concerns the loss of a differentiated epithelial-like state of cells (e.g., cell-to-cell junctions) to acquire a more mesenchymal-like phenotype (e.g., enhanced ECM expression) [7]. Senescence and EMT are both characterized by cells dedifferentiation, loss of epithelial phenotype, cell cycle arrest, and negative effects on surrounding cells [8]. IS triggers senescence [9] and induces EMT with ECM (i.e., α-SMA) deposition [10], which suggests that PBUTs may induce kidney fibrosis by propagating senescence. However, the crosstalk between PBUTs-related fibrosis and senescence-related fibrosis remains unclear. Here, we provide some mechanistic insight in how PBUTs could promote kidney fibrosis by accelerating senescence.
2. The mechanisms of kidney fibrosis
Kidney fibrosis is induced by abnormal accumulation of ECM, that often initiates as the result of a wound healing response. The response is orchestrated by complex activities of different cells, including macrophages and T cells, epithelial cells, myofibroblasts and endothelial cells. Four major phases are involved in this process: 1) primary injury that initiates a fibrotic response, 2) the activation of effector cells triggering the fibrosis signalling (e.g. TGF-β signalling), 3) production of ECM, and 4) deposition of ECM that promotes tissue fibrosis and eventually leads to kidney failure [1].
2.1. Main signaling of fibrosis
Three main signaling pathways are involved in fibrosis: transforming growth factor (TGF)-β, wingless/Int (WNT), and yes-associated protein (YAP)/transcriptional coactivator with PDZ-binding motif (TAZ) signalling pathways [11]. TGF-β signals through both canonical (Smad-based) and non-canonical (non-Smad-based) pathways; Smad-based TGF-β signalling plays a central role in the development of renal fibrosis; non-Smad-based profibrotic actions of TGF-β signalling are regulated by interactions with other signalling pathways [12]. The WNT signaling pathway is activated by secreted lipid modified proteins of the WNT family. Activation of WNT signaling stabilizes β-catenin; the nuclear translocation of β-catenin initiates transcription of fibrotic genes such as collagen and fibronectin [13,14]. YAP and TAZ are major players of the Hippo pathway that is involved in organ development, epithelial homeostasis, tissue regeneration, wound healing and immune modulation; ECM stiffening promotes the nuclear activity of YAP/TAZ, which in turn, promotes development of a fibrotic cellular phenotype, including increasing the expression of connective tissue growth factor (CTGF) and plasminogen activator inhibitor 1 (PAI-1) [15,16,17]. These three signaling pathways show a cross-talk during fibrosis. Their mechanisms range from modulating the availability of growth factors and the availability of membrane bound receptors to nuclear entry and activation of transcription factors [11]. Recent studies revealed that TGF-β and WNT signaling are also related to senescence [18,19].
2.1. ECM in kidney fibrosis
The ECM is a non-cellular component of tissue that provides essential structural support for cellular constituents, and acts as an active component in cell signaling. It is composed of water, proteins, and polysaccharides, responsible for cell–cell communication, cell adhesion, and cell proliferation [20,21]. There are two main types of ECM: interstitial connective tissue matrix (e.g., collagen I and fibronectin) and the basement membrane (e.g., collagen IV and laminins) [22]. Interstitial connective tissue matrix is responsible for tissue structure, while the basement membrane underlies or surrounds most tissues, including epithelial and endothelial tissues, and interact with cells (Figure 1) [22,23]. Three histologically distinct compartments with a variety of ECMs are affected in kidney fibrosis: the glomeruli, tubulointerstitium, and vasculature (Table 1) [24]. As a result of ECM remodeling, the deposition of matrix proteins is observed in kidney fibrosis (Table 1).
2.2. ECM remodeling
ECM remodeling is referred to as a balance between degradation and production of ECM. When the balance is disrupted [24], a positive feedback loop resulting in increased ECM production drives the development of fibrosis [26]. The cleavage of ECM by different proteases is the main process during the remodelling, and includes matrix metalloproteinases (MMPs), adamalysins, meprins and metalloproteinase inhibitors (reviewed in REF. [22]). MMPs are the main enzymes involved in ECM degradation and remodelling. MMPs can cleave ECM components and activate other MMPs and proteins. Various cytokines (interleukin [IL], tumor necrosis factor [TNF]) and growth factors (epidermal growth factor [EGF], transforming growth factor [TGF]) may be involved in the gene expression of MMPs at the transcription level [27]. Adamalysins include a disintegrin and metalloproteinases (ADAMs) and ADAMs with a thrombospondin motif (ADAMTS); Adamalysins contain twenty-one ADAM, and nineteen ADAMTS proteins, shedding of various substrates including adhesion ligands, growth factors and their receptors, and cytokines [28]. Meprins are the only astacin proteinases that can be bound to membranes or secreted as soluble factors; meprin subunits cleave a variety of biologically active peptides, many cytokines and chemokines, leading to an alteration in biological function/activity of those factors/proteins [29]. Tissue inhibitor of metalloproteinases (TIMP) is an endogenous inhibitor of MMPs and adamalysins. Each TIMP specifically binds to their target MMPs or adamalysins, regulating the production/deposition of various ECM components, such as collagens, fibronectin and laminins [30].
3. Senescence
Senescence is a special form of permanent cell cycle arrest, which limits cellular proliferation. It was first reported as a loss of replicative capacity in cultured human fibroblasts in 1961 [31]. Senescent cells are currently regarded as a potentially important contributor to different types of diseases [32], including aging-related diseases [33], kidney [34] and pulmonary disease [35]. Some senescent cells can be cleared by immune cells, through chemo-attracting of immune cells, followed by tissue regeneration, which is called acute (short-term) senescence; while chronic (long-term) senescent cells accumulate and create a lesion, aggravating the pathology [36,37]. Major types of senescence are highlighted as replicative senescence (RS), oncogene-induced senescence (OIS), and stress-induced (premature) senescence (SIS) (Figure 2). RS is linked to telomere shortening that is associated with cell division. This type of senescence is a consequence of activating a DNA-damage response (DDR), which is induced by short telomeres through the induction of the cell cycle inhibitor p21, arresting proliferation [38,39,40,41].
OIS refers to cell cycle arrest by aberrant activation of oncogenic signaling, which promotes the initiation and development of cancer [42]. OIS can be caused by numerous oncogenes including constitutively active variants in the RAS/MAPK pathway (RAS-induced senescence, RIS) as well as in the PI3K/AKT pathway (AKT-induced senescence, AIS), the former undergoes a DDR, while the latter is independent of DDR [43]. SIS appears after exposing cells to chemical or physical stresses, including radiation waves, hydrogen peroxide and chemotherapeutic agents [44], leading to cellular stress, increased reactive oxygen species (ROS) generation and subsequent DNA damage, eventually contributing to senescence [37,44].
3.1. Mechanisms of senescence
As discussed, senescence is triggered by various stressors including DNA damage, mitochondrial dysfunction, metabolism and cell stress [2,45,46]. Most of them accompany the DDR outcomes, followed by activation of the cell cycle arrest and the release of SASP factors [47,48].
3.1.1. Cell cycle arrest
Cell cycle arrest in senescence is largely mediated via the p53/p21CIP1/WAF1 (p21) and p16Ink4a (p16)/pRb checkpoint pathways controlled by DDR [49,50], which are independent processes in senescence induction. p53/p21 is activated when DDR occurs, promoting a p21-dependent G0/G1 cell-cycle arrest [51,52]. p16 suppresses Retinoblastoma 1 (pRb), and prevents the action of the cyclin dependent kinases, which induces a G1 cell cycle arrest [53]. Acute DNA damage drives cell cycle arrest via the p53/p21 pathway, while chronic DNA damage followed by the induction of the p16/pRB pathway maintains cell cycle arrest and senescence [54]. As a key mediator of cell cycle arrest, some studies also demonstrated that p21 can be upregulated via a p53-independent mechanism [55,56]. Checkpoint signaling pathways are associated to p53-mediated apoptosis [57]. During DDR, the abnormal expression of p53 may further lead to apoptosis resistance.
3.1.2. Apoptosis resistance
Senescent cells are resistant to apoptosis [58], via intrinsic and extrinsic pathways. The intrinsic pathway refers to the mitochondrial pathway of apoptosis, related to mitochondrial outer membrane permeabilization (MOMP) [59]. In this pathway, MOMP and the release of cytochrome c are required to trigger apoptosis, and involve Bcl-2 and caspase family proteins [60,61]. The Bcl-2 family is divided into three main groups: anti-apoptotic (Bcl-2, Bcl-xl, and Mcl-1), pro-apoptotic (Bax and Bak), and pro-apoptotic BH3-only (Bim, Bid, Bad and Puma) proteins [62]. The balance between Bcl-2 family members of the pro-apoptotic and anti-apoptotic determines the threshold in MOMP for apoptosis. Caspase proteins are downstream players of MOMP in the intrinsic apoptosis pathway [63]. After the activation of Bax-Bak-dependent MOMP, cytochrome c is released from the mitochondria stimulating the activation of caspase-9 and its downstream executioners, caspases-3 and -7 to initiate apoptosis [61]. The extrinsic pathway is initiated via death receptors that bind death ligands secreted by other cells (e.g., macrophages and natural killer cells), activating caspase-8 and its downstream executioner caspases-3 to initiate apoptosis [59]. Natural ligands, including TNF, Fas-L and TRAIL, are known to bind to their receptors TNFR1, TNFR2, Fas and TRAIL-R to activate caspase-8 [64]. Caspase-8 activation can lead to cleavage of Bid to tBid and initiates the mitochondria-mediated intrinsic apoptosis pathway [59]. Accumulation of dysfunctional mitochondria in senescent cells has been reported [65]. Senescent cells are in a primed apoptotic state, triggered by the abnormal regulation of anti-apoptotic and pro-apoptotic Bcl-2 family proteins, keeping cells alive without undergoing proliferation or apoptosis [66]. SASP factors, such as TNF-α [67,68], released from senescent cells also play a role in the extrinsic apoptosis pathway. This kind of regulation finally inhibits the activation of executioner caspase-3, leading to apoptosis resistance and chronic senescence.
3.1.3. SASP factors
SASP factors are related to a DDR and are generally proinflammatory and/or profibrotic compounds, including numerous cytokines, chemokines, growth factors, and matrix-metalloproteinases (MMPs) [2,69]. Several reports described that SASP factors are not only responsible for the maintenance and reinforcement of senescence, but also key players during its transmission [70]. Cytokines, such as IL-6 and IL-8, are well proven to play such critical role in stress-induced senescence [71,72,73]. IL-6 maintains senescence through the p53/p21 pathway [74,75]. This role of IL-6 in senescence is shared by IL-8, which is expressed as a function of IL-6 [72]. Both cytokines are regulated by IL-1α [76]. Nucleotide-binding oligomerization domain (NOD)-like receptor 3 (NLRP3) inflammasome is upregulated in senescence, which leads to expression of IL-1α and IL-1β, resulting in the upregulation of SASP factors and reinforcement of senescence in a paracrine manner [77]. Chemokine signalling is also reported as being responsible for reinforcing growth arrest by the CXCR2 receptor and CXCR2-binding chemokines [71]. Chemokines including CCLs and CXCLs are involved in stress(radiation)-induced senescence, thus leading to fibrosis [78].Chemokine signalling also plays a role in OIS; senescent cells increase the survival of cancer cells via CXCL12/CXCR4 signalling, leading the collective invasion in thyroid cancer [79]. Growth factors like CTGF and TGF-β induce senescence and are accompanied with the upregulation of IL-6 and IL-8, thus reinforcing paracrine senescence [80,81]. TGF-β induces CTGF expression through activation of Smad3 and p53 [82,83], inducing cell cycle arrest and contributing to senescence [84]. Accumulation of MMPs is also observed in senescence [85]. MMPs shed ectodomains of cell surface receptors and activate other SASP factors, hence promoting senescence via paracrine signaling [86].
3.2. Senescence and fibrosis
Senescence contributes to fibrosis in multiple organs [87,88,89] and is considered to be a result of the release of SASP factors and the pathways triggered by them (Figure 3).
TGF-β signaling controls cell proliferation and survival, regulating apoptosis and senescence [84], and initiates fibrosis through the canonical Smad signaling and Smad-independent signaling pathways, with subsequent ECM deposition [90]. CTGF has been shown to contribute to TGF-β signaling through the extracellular signal-regulated kinase (ERK), ADAM17, ribosomal S6 kinases 1 (RSK1) and CCAAT/enhancer-binding protein β (C/EBPβ) signaling pathway in human lung epithelial cells [82,91]. CTGF is necessary for the TGF-β induced phosphorylation of Smad1 and Erk1/2, but it is not needed for the activation of the Smad3 pathway [92]. Proinflammatory mediators like IL-6 and IL-1β are also involved in fibrosis. IL-6 shifts acute inflammation into a chronic fibrosis state by regulating MMPs and the TGF-β pathway [93,94]. IL-1β augments TGF-β1-induced EMT through MAPK signaling pathways [95], which may be dependent on IL-17A [96]. MMPs release ectodomains of cell surface receptors and activate other SASP factors [86], thus regulating ECM production and promoting EMT and kidney fibrosis [22]. Other SASP factors like CCL2 and PAI-1 are also important players in fibrosis, exerting their effect through chemokine and TGF-β signaling, respectively [97,98].
4. Protein-bound uremic toxins: drivers of senescence and kidney fibrosis
Uremic toxins are endogenous metabolites that are being excreted into the urine through glomerular filtration and active transport by the proximal epithelial cells [99]. In kidney disease, uremic toxins management is compromised, which leads to systemic accumulation of the toxins and activation of inflammation and oxidative stress. Furthermore, uremic toxins can induce profibrotic effects, promoting progression of kidney damage [100]. Uremic toxins are divided in three distinct groups: 1) small-water soluble compounds (molecular weight <500 Da, e.g. creatinine, urea and uric acid); 2) middle molecules (peptides with molecular weight >500 Da, e.g. IL-6, IL-8 and TNF-α); and 3) protein bound uremic toxins (PBUTs; molecular weight mostly <500 Da, e.g. indoxyl sulfate, p-cresyl sulfate and p-cresol)[99,101]. Small-water soluble compounds are hydrophilic, which pass through the glomerular barrier and can be removed easily by dialysis [102,103]. Most of middle molecules are peptides and difficult to remove in the process of dialysis unless the dialyzer pore size is large enough [104]. PBUTs are removed by proximal tubule cells in healthy kidneys through active secretion involving transporter proteins, but poorly removed with kidney dysfunction [105]. Current dialysis therapy is limited, because the high binding to plasma proteins, with albumin being the primary carrier protein, and only a small free fraction is available for transfer across dialyzer membranes [3,4]. PBUTs accumulate systemically but also in kidney tissue where they can induce oxidative stress and stimulate production of inflammatory factors, which might be a trigger for fibrosis [106]. PBUTs induce ROS production and enhance oxidative stress and IL-1β (SASP) expression in kidney proximal tubule cells [107]. Furthermore, it has been reported that PBUTs induce TGF-β and WNT signalling that promote ECM remodelling [108,109]. IS and PCS induce EMT by activating the renal TGF-β signalling [6], and contribute to ECM remodelling by upregulating MMP2 and MMP9 in an EGF receptor-dependent manner [110]. As discussed, TGF-β and WNT signalling are also related to senescence [18,19], which might suggest that PBUTs can be drivers of senescence and kidney fibrosis.
5. Protein-bound uremic toxins promote fibrosis by accelerating senescence
Kidney failure is accompanied with PBUTs accumulation [103,111], which leads to ECM remodelling and fibrosis [26]. In turn, PBUTs accumulation promotes kidney disease and fibrosis by oxidative stress and inflammation [106]. The accumulation of PBUTs occurs in a time- and stage-dependent manner during CKD. As the loss of kidney function in CKD is progressive and irreversible, advanced CKD has more severe uremic toxin plasma levels [5] that potentially can induce senescence [9,112]. Chronic senescence promoted by external factors (e.g. ionizing radiation, exposure to toxicants or heat stress) may develop in a time-dependent manner [113,114,115]. This suggests that PBUTs may also promote senescence time-dependently during CKD, progressing the disease and reinforcing fibrosis (Figure 4). In CKD animal models, it was shown that the accumulation of PBUTs correlated to fibrosis outcome and/or senescence phenotype (Table 2). We, therefore, hypothesize that PBUTs may promote kidney fibrosis by accelerating senescence, possibly via mitochondrial dysfunction, cell cycle arrest, and the production of SASP factors.
5.1. PBUTs accelerate senescence via mitochondrial dysfunction?
Different types of senescence have been reported to have increased ROS and mitochondrial dysfunction [65], which influences intrinsic apoptosis pathway by abnormal expression of Bcl-2 family and caspase family markers, which in turn, maintain and reinforce senescence [128]. Overproduction of ROS during cell stress leads to mitochondrial dysfunction after kidney injury [129], which is promoted by PBUTs accumulation [130]. A cocktail of PBUTs, consisting of IS, PCS, indoxyl-β-glucuronide, p-cresyl glucuronide, indol-3-acetic acid, hippuric acid, kynurenic acid, and l-kynurenine, have shown to promote ROS production and to upregulate IL-6 in proximal tubule epithelial cells [107,131]. In addition ROS-induced senescence was shown to require mammalian target of rapamycin (mTOR) activation [132], and accumulated IS promoted renal fibrosis via mTOR under CKD conditions [121]. Furthermore, the class I phosphoinositide 3-kinase (PI3K) signalling regulates and activates mTOR [133]. PCS activates NADPH oxidase through a mechanism that involves the PI3K signalling, inducing ROS production and TGF-β1 secretion [134]. PBUTs, like IS, PCS and hippuric acid influenced apoptosis by causing an imbalance of caspase-3, caspase-9, Bcl-2, and Bax in hepatocytes, with marked ROS generation and mitochondrial damage [135]. Although there is a lack of evidence showing that PBUTs inhibit apoptosis, IS and PCS increased the expression of the anti-apoptotic genes Bcl-2, Bcl-xl and Bax in proximal tubule cells [136], which is also observed in senescent cells [58,115].
5.2. PBUTs accelerate senescence via cell cycle arrest?
Cell cycle arrest is necessary for the repair of DNA damage after injury [137], which generally occurs in senescence and is a critical factor for fibrosis development [138]. DDR is a cause of cell cycle arrest mediated by the p53/p21 and p16/pRb pathways [48]. ROS triggers DDR and DDR promotes ROS production by activating its downstream effectors, including p53 and p21 [139]. Recent research has suggested that PBUTs may accelerate senescence via cell cycle arrest and inhibition of cell proliferation [140,141]. Others have suggested that PCS and IS upregulate p21 and increase the number of cells positive for senescence-associated beta-galactosidase [9]. IS also promotes p53 expression, stimulating the expression of TGF-β1 and ECM deposition [142].
5.3. PBUTs accelerate senescence via SASP factors?
During CKD progression, the inflammatory (SASP) factors released activate different pathways and initiate various processes, including senescence and EMT in tubular epithelial cells [140,143]. As discussed, PBUTs accumulation-induced inflammation might be one reason for senescence development. SASP factors like IL-6, TGF-β1 and CXCL10 were reported to be increased in proximal tubule cells after the treatment with the PBUTs IS and PCS [136]. Furthermore, ROS overproduction can activate the NLRP3 inflammasome, which cleaves pro-caspase-1 and pro-interleukin-1β (IL-1β) into the proinflammatory factors caspase-1 and IL-1β, thus promoting fibrosis [144]. A cocktail of PBUTs (IS, PCS, indoxyl-β-glucuronide, p-cresyl glucuronide, indol-3-acetic acid, hippuric acid, kynurenic acid, and l-kynurenine) has been shown to promote the NLRP3 inflammasome-mediated IL-1β production via oxidative stress and NF-κB signalling [107]. Interestingly, the NLRP3 inflammasome/ IL-1β also promotes cellular senescence [145]. SASP factors reinforce senescence and induce senescence transmission or paracrine senescence, which is regulated by the inflammasome [77]. Therefore, PBUTs may play an important role during CKD to promote paracrine senescence and senescence transmission.
5. Conclusions and future therapeutic perspectives
PBUTs may promote senescence in CKD through the release of SASP factors (e.g., IL-6 and IL-1β) and common senescence markers (e.g., p21 and Laminb1), and trigger oxidative stress possibly causing mitochondrial dysfunction, promoting an inflammatory response and increased resistance to cell death. As SASP factors are typically profibrotic and proinflammatory mediators, a novel treatment strategy of CKD could be inhibiting the related signaling, thus suppressing SASP expression. Potential novel agents already exist for this, including anti-fibrotic agents (e.g., TGF-β inhibitor; pirfenidone) and anti- inflammatory agents (e.g., the anti-TNF-α monoclonal antibody, infliximab) [146]. Moreover, the NF-κB inhibitor resveratrol and mTOR inhibitor rapamycin are also recognized as SASP inhibitors (called senomorphics), reducing the development of cellular senescence [147,148], which could represent possibilities for CKD treatment as well [149]. In addition, strategies to inhibit senescence phenotypes by promoting cell cycle process and cell death signaling, such as an inhibitor of p21 and/or promoter of caspase proteins, could be treatment options. Considering that chronic senescent cells cannot be cleared by the immune cells, strengthening the immune system by increasing the binding affinity of the involved membrane receptors is another approach to more efficiently clear senescent cells [8]. Advanced cell therapy, such as chimeric antigen receptor (CAR) T cell, may be employed to specifically target senescent cells by recognizing appropriate antigens [36,150]. Finally, identifying and targeting most relevant and specific senescence-associated markers by means of gene therapy could be a valid approach to be investigated in the future for combatting kidney senescence [151].
Author Contributions
Conceptualization, Y.Y., M.M. and R.M.; methodology, Y.Y., M.M. and R.M.; writing original draft preparation, Y.Y.; writing review and editing, Y.Y., M.M. and R.M.; supervision, M.M. and R.M.; funding acquisition, Y.Y. and R.M. All authors have read and agreed to the published version of the manuscript.
Funding
This study is supported by the China Scholarship Council (No. 201806910081) and by the Dutch Kidney Foundation (CP1805).
Data Availability Statement
Data sharing not applicable.
Conflicts of Interest
The authors declare no conflict of interest and funders had no role in the design of the study, in the writing of the manuscript or in the decision to publish.
References
- Rockey, D.C., P.D. Bell, and J.A. Hill, Fibrosis--a common pathway to organ injury and failure. N Engl J Med, 2015. 372(12): p. 1138-49. [CrossRef]
- Hernandez-Segura, A., J. Nehme, and M. Demaria, Hallmarks of Cellular Senescence. Trends Cell Biol, 2018. 28(6): p. 436-453. [CrossRef]
- Masereeuw, R., The Dual Roles of Protein-Bound Solutes as Toxins and Signaling Molecules in Uremia. Toxins (Basel), 2022. 14(6). [CrossRef]
- Maheshwari, V., et al., Removal of Protein-Bound Uremic Toxins Using Binding Competitors in Hemodialysis: A Narrative Review. Toxins (Basel), 2021. 13(9). [CrossRef]
- Nigam, S.K. and K.T. Bush, Uraemic syndrome of chronic kidney disease: altered remote sensing and signalling. Nat Rev Nephrol, 2019. 15(5): p. 301-316. [CrossRef]
- Sun, C.Y., S.C. Chang, and M.S. Wu, Uremic toxins induce kidney fibrosis by activating intrarenal renin-angiotensin-aldosterone system associated epithelial-to-mesenchymal transition. PLoS One, 2012. 7(3): p. e34026. [CrossRef]
- Singh, M., et al., EMT: Mechanisms and therapeutic implications. Pharmacol Ther, 2018. 182: p. 80-94. [CrossRef]
- Faheem, M.M., et al., Convergence of therapy-induced senescence (TIS) and EMT in multistep carcinogenesis: current opinions and emerging perspectives. Cell Death Discov, 2020. 6: p. 51. [CrossRef]
- Kamprom, W., et al., P-cresol and Indoxyl Sulfate Impair Osteogenic Differentiation by Triggering Mesenchymal Stem Cell Senescence. Int J Med Sci, 2021. 18(3): p. 744-755. [CrossRef]
- Kim, S.H., et al., Indoxyl sulfate-induced epithelial-to-mesenchymal transition and apoptosis of renal tubular cells as novel mechanisms of progression of renal disease. Lab Invest, 2012. 92(4): p. 488-98. [CrossRef]
- Piersma, B., R.A. Bank, and M. Boersema, Signaling in Fibrosis: TGF-beta, WNT, and YAP/TAZ Converge. Front Med (Lausanne), 2015. 2: p. 59. [CrossRef]
- Meng, X.M., D.J. Nikolic-Paterson, and H.Y. Lan, TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol, 2016. 12(6): p. 325-38. [CrossRef]
- Rim, E.Y., H. Clevers, and R. Nusse, The Wnt Pathway: From Signaling Mechanisms to Synthetic Modulators. Annu Rev Biochem, 2022. 91: p. 571-598. [CrossRef]
- Tan, R.J., et al., Wnt/β-catenin signaling and kidney fibrosis. Kidney Int Suppl (2011), 2014. 4(1): p. 84-90. [CrossRef]
- Dey, A., X. Varelas, and K.L. Guan, Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat Rev Drug Discov, 2020. 19(7): p. 480-494. [CrossRef]
- Liu, F., et al., Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol, 2015. 308(4): p. L344-57. [CrossRef]
- Zhao, B., et al., TEAD mediates YAP-dependent gene induction and growth control. Genes Dev, 2008. 22(14): p. 1962-71. [CrossRef]
- Luo, C., et al., Wnt9a Promotes Renal Fibrosis by Accelerating Cellular Senescence in Tubular Epithelial Cells. J Am Soc Nephrol, 2018. 29(4): p. 1238-1256. [CrossRef]
- Tominaga, K. and H.I. Suzuki, TGF-β Signaling in Cellular Senescence and Aging-Related Pathology. Int J Mol Sci, 2019. 20(20). [CrossRef]
- Genovese, F., et al., The extracellular matrix in the kidney: a source of novel non-invasive biomarkers of kidney fibrosis? Fibrogenesis & tissue repair, 2014. 7(1): p. 4 %@ 1755-1536. [CrossRef]
- Frantz, C., K.M. Stewart, and V.M. Weaver, The extracellular matrix at a glance. J Cell Sci, 2010. 123(Pt 24): p. 4195-200. [CrossRef]
- Bonnans, C., J. Chou, and Z. Werb, Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol, 2014. 15(12): p. 786-801. [CrossRef]
- Jayadev, R. and D.R. Sherwood, Basement membranes. Curr Biol, 2017. 27(6): p. R207-R211.
- Bulow, R.D. and P. Boor, Extracellular Matrix in Kidney Fibrosis: More Than Just a Scaffold. J Histochem Cytochem, 2019. 67(9): p. 643-661. [CrossRef]
- Ariza de Schellenberger, A., et al., The Extracellular Matrix as a Target for Biophysical and Molecular Magnetic Resonance Imaging, in Quantification of Biophysical Parameters in Medical Imaging, I. Sack and T. Schaeffter, Editors. 2018, Springer International Publishing: Cham. p. 123-150.
- Herrera, J., C.A. Henke, and P.B. Bitterman, Extracellular matrix as a driver of progressive fibrosis. J Clin Invest, 2018. 128(1): p. 45-53. [CrossRef]
- Peng, W.J., et al., Matrix metalloproteinases: a review of their structure and role in systemic sclerosis. J Clin Immunol, 2012. 32(6): p. 1409-14. [CrossRef]
- Przemyslaw, L., et al., ADAM and ADAMTS family proteins and their role in the colorectal cancer etiopathogenesis. BMB Rep, 2013. 46(3): p. 139-50. [CrossRef]
- Aan, G.J., et al., Differences in protein changes between stress-induced premature senescence and replicative senescence states. Electrophoresis, 2013. 34(15): p. 2209-17. [CrossRef]
- Khokha, R., A. Murthy, and A. Weiss, Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat Rev Immunol, 2013. 13(9): p. 649-65. [CrossRef]
- Hayflick, L. and P.S. Moorhead, The serial cultivation of human diploid cell strains. Experimental cell research, 1961. 25(3): p. 585-621. [CrossRef]
- He, S. and N.E. Sharpless, Senescence in Health and Disease. Cell, 2017. 169(6): p. 1000-1011. [CrossRef]
- Childs, B.G., et al., Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med, 2015. 21(12): p. 1424-35. [CrossRef]
- Docherty, M.H., et al., Cellular Senescence in the Kidney. J Am Soc Nephrol, 2019. 30(5): p. 726-736. [CrossRef]
- Wang, Z.N., et al., Potential Role of Cellular Senescence in Asthma. Front Cell Dev Biol, 2020. 8: p. 59. [CrossRef]
- Kobbe, C.v., Targeting senescent cells: approaches, opportunities, challenges. Aging 2019. 11: p. 18. [CrossRef]
- Munoz-Espin, D. and M. Serrano, Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol, 2014. 15(7): p. 482-96. [CrossRef]
- Marcotte R, W.E., Replicative Senescence Revisited. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences, 2002. 57(7): p. B257-B269. [CrossRef]
- d'Adda di Fagagna, F., et al., A DNA damage checkpoint response in telomere-initiated senescence. Nature, 2003. 426(6963): p. 194-8. [CrossRef]
- Herbig, U., et al., Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol Cell, 2004. 14(4): p. 501-13. [CrossRef]
- Rossiello, F., et al., Telomere dysfunction in ageing and age-related diseases. Nat Cell Biol, 2022. 24(2): p. 135-147. [CrossRef]
- Liu, X.L., J. Ding, and L.H. Meng, Oncogene-induced senescence: a double edged sword in cancer. Acta Pharmacol Sin, 2018. 39(10): p. 1553-1558. [CrossRef]
- Zhu, H., et al., Oncogene-induced senescence: From biology to therapy. Mech Ageing Dev, 2020. 187: p. 111229. [CrossRef]
- Suzuki, M. and D.A. Boothman, Stress-induced premature senescence (SIPS)--influence of SIPS on radiotherapy. J Radiat Res, 2008. 49(2): p. 105-12. [CrossRef]
- Chaib, S., T. Tchkonia, and J.L. Kirkland, Cellular senescence and senolytics: the path to the clinic. Nat Med, 2022. [CrossRef]
- Krizhanovsky, R.S.a.V., Cell Senescence, DNA Damage, and Metabolism. Antioxidants & Redox Signaling, 2021. 34(4): p. 324-334. [CrossRef]
- Huang, W., et al., Cellular senescence: the good, the bad and the unknown. Nat Rev Nephrol, 2022. [CrossRef]
- Jackson, S.P. and J. Bartek, The DNA-damage response in human biology and disease. Nature, 2009. 461(7267): p. 1071-8. [CrossRef]
- Kumari, R. and P. Jat, Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front Cell Dev Biol, 2021. 9: p. 645593. [CrossRef]
- Mijit, M., et al., Role of p53 in the Regulation of Cellular Senescence. Biomolecules, 2020. 10(3). [CrossRef]
- Ceccaldi, R., et al., Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell, 2012. 11(1): p. 36-49.
- Ou, H.L. and B. Schumacher, DNA damage responses and p53 in the aging process. Blood, 2018. 131(5): p. 488-495. [CrossRef]
- Rayess, H., M.B. Wang, and E.S. Srivatsan, Cellular senescence and tumor suppressor gene p16. Int J Cancer, 2012. 130(8): p. 1715-25. [CrossRef]
- Sperka, T., J. Wang, and K.L. Rudolph, DNA damage checkpoints in stem cells, ageing and cancer. Nat Rev Mol Cell Biol, 2012. 13(9): p. 579-90. [CrossRef]
- Ruan, B., et al., NVP-BEZ235 inhibits thyroid cancer growth by p53- dependent/independent p21 upregulation. Int J Biol Sci, 2020. 16(4): p. 682-693. [CrossRef]
- Zhang, Y., et al., DNMT3a plays a role in switches between doxorubicin-induced senescence and apoptosis of colorectal cancer cells. Int J Cancer, 2011. 128(3): p. 551-61. [CrossRef]
- Pietenpol, J. and Z. Stewart, Cell cycle checkpoint signaling: Cell cycle arrest versus apoptosis. toxicology, 2002. 181: p. 475-481. [CrossRef]
- Childs, B.G., et al., Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep, 2014. 15(11): p. 1139-53. [CrossRef]
- D'Arcy, M.S., Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell Biol Int, 2019. 43(6): p. 582-592. [CrossRef]
- Ngoi, N.Y.L., et al., Targeting Mitochondrial Apoptosis to Overcome Treatment Resistance in Cancer. Cancers (Basel), 2020. 12(3). [CrossRef]
- Van Opdenbosch, N. and M. Lamkanfi, Caspases in Cell Death, Inflammation, and Disease. Immunity, 2019. 50(6): p. 1352-1364.
- Anantram, A. and M. Degani, Targeting cancer's Achilles' heel: role of BCL-2 inhibitors in cellular senescence and apoptosis. Future Med Chem, 2019. 11(17): p. 2287-2312. [CrossRef]
- Shalini, S., et al., Old, new and emerging functions of caspases. Cell Death Differ, 2015. 22(4): p. 526-39. [CrossRef]
- Carneiro, B.A. and W.S. El-Deiry, Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol, 2020. 17(7): p. 395-417. [CrossRef]
- Korolchuk, V.I., et al., Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? EBioMedicine, 2017. 21: p. 7-13. [CrossRef]
- Fan, Y., et al., Senescent Cell Depletion Through Targeting BCL-Family Proteins and Mitochondria. Front Physiol, 2020. 11: p. 593630. [CrossRef]
- Guo, Q., et al., Tumor Necrosis Factor-alpha (TNF-α) Enhances miR-155-Mediated Endothelial Senescence by Targeting Sirtuin1 (SIRT1). Med Sci Monit, 2019. 25: p. 8820-8835. [CrossRef]
- Li, P., et al., The inflammatory cytokine TNF-α promotes the premature senescence of rat nucleus pulposus cells via the PI3K/Akt signaling pathway. Scientific Reports, 2017. 7(1): p. 42938. [CrossRef]
- Birch, J. and J. Gil, Senescence and the SASP: many therapeutic avenues. Genes Dev, 2020. 34(23-24): p. 1565-1576. [CrossRef]
- Kuilman, T. and D.S. Peeper, Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer, 2009. 9(2): p. 81-94. [CrossRef]
- Acosta, J.C., et al., Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell, 2008. 133(6): p. 1006-18. [CrossRef]
- Kuilman, T., et al., Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell, 2008. 133(6): p. 1019-31. [CrossRef]
- You, K., et al., Moderate hyperoxia induces senescence in developing human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol, 2019. 317(5): p. L525-l536. [CrossRef]
- Li, Y., et al., Interleukin-6 Knockout Inhibits Senescence of Bone Mesenchymal Stem Cells in High-Fat Diet-Induced Bone Loss. Front Endocrinol (Lausanne), 2020. 11: p. 622950. [CrossRef]
- Effenberger, T., et al., Senescence-associated release of transmembrane proteins involves proteolytic processing by ADAM17 and microvesicle shedding. FASEB J, 2014. 28(11): p. 4847-56. [CrossRef]
- Orjalo, A.V., et al., Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci U S A, 2009. 106(40): p. 17031-6. [CrossRef]
- Acosta, J.C., et al., A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol, 2013. 15(8): p. 978-90. [CrossRef]
- Su, L., et al., Potential role of senescent macrophages in radiation-induced pulmonary fibrosis. Cell Death Dis, 2021. 12(6): p. 527. [CrossRef]
- Kim, Y.H., et al., Senescent tumor cells lead the collective invasion in thyroid cancer. Nat Commun, 2017. 8: p. 15208. [CrossRef]
- Jun, J.I. and L.F. Lau, CCN2 induces cellular senescence in fibroblasts. J Cell Commun Signal, 2017. 11(1): p. 15-23. [CrossRef]
- Fan, C., et al., TGF-β induces periodontal ligament stem cell senescence through increase of ROS production. Mol Med Rep, 2019. 20(4): p. 3123-3130. [CrossRef]
- Ou, S.C., et al., TGF-β Induced CTGF Expression in Human Lung Epithelial Cells through ERK, ADAM17, RSK1, and C/EBPβ Pathways. Int J Mol Sci, 2020. 21(23). [CrossRef]
- Li, X., et al., DsbA-L mediated renal tubulointerstitial fibrosis in UUO mice. Nat Commun, 2020. 11(1): p. 4467. [CrossRef]
- Zhang, Y., P.B. Alexander, and X.F. Wang, TGF-beta Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb Perspect Biol, 2017. 9(4). [CrossRef]
- Wang, X.-H., et al., GATA4 promotes the senescence of nucleus pulposus cells via NF-κB pathway. Archives of Gerontology and Geriatrics, 2022. 101: p. 104676. [CrossRef]
- Levi, N., et al., The ECM path of senescence in aging: components and modifiers. FEBS J, 2020. 287(13): p. 2636-2646. [CrossRef]
- Hernandez-Gonzalez, F., et al., Cellular Senescence in Lung Fibrosis. Int J Mol Sci, 2021. 22(13). [CrossRef]
- Zhang, M., et al., Hepatic stellate cell senescence in liver fibrosis: Characteristics, mechanisms and perspectives. Mech Ageing Dev, 2021. 199: p. 111572. [CrossRef]
- Chen, M.S., R.T. Lee, and J.C. Garbern, Senescence mechanisms and targets in the heart. Cardiovasc Res, 2022. 118(5): p. 1173-1187. [CrossRef]
- Kim, K.K., D. Sheppard, and H.A. Chapman, TGF-β1 Signaling and Tissue Fibrosis. Cold Spring Harb Perspect Biol, 2018. 10(4).
- Yanagihara, T., et al., Connective-Tissue Growth Factor Contributes to TGF-β1-induced Lung Fibrosis. Am J Respir Cell Mol Biol, 2022. 66(3): p. 260-270. [CrossRef]
- Nakerakanti, S.S., A.M. Bujor, and M. Trojanowska, CCN2 is required for the TGF-β induced activation of Smad1-Erk1/2 signaling network. PLoS One, 2011. 6(7): p. e21911.
- Fielding, C.A., et al., Interleukin-6 signaling drives fibrosis in unresolved inflammation. Immunity, 2014. 40(1): p. 40-50. [CrossRef]
- Epstein Shochet, G., et al., TGF-β pathway activation by idiopathic pulmonary fibrosis (IPF) fibroblast derived soluble factors is mediated by IL-6 trans-signaling. Respir Res, 2020. 21(1): p. 56. [CrossRef]
- Zhang, S., et al., IL-1β augments TGF-β inducing epithelial-mesenchymal transition of epithelial cells and associates with poor pulmonary function improvement in neutrophilic asthmatics. Respir Res, 2021. 22(1): p. 216. [CrossRef]
- Wilson, M.S., et al., Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J Exp Med, 2010. 207(3): p. 535-52. [CrossRef]
- Xu, L., D. Sharkey, and L.G. Cantley, Tubular GM-CSF Promotes Late MCP-1/CCR2-Mediated Fibrosis and Inflammation after Ischemia/Reperfusion Injury. J Am Soc Nephrol, 2019. 30(10): p. 1825-1840. [CrossRef]
- Gifford, C.C., et al., PAI-1 induction during kidney injury promotes fibrotic epithelial dysfunction via deregulation of klotho, p53, and TGF-β1-receptor signaling. Faseb j, 2021. 35(7): p. e21725. [CrossRef]
- Vanholder, R., et al., Review on uremic toxins: classification, concentration, and interindividual variability. Kidney Int, 2003. 63(5): p. 1934-43. [CrossRef]
- Rocchetti, M.T., et al., Protein-Bound Uremic Toxins and Immunity, in Cytotoxic T-Cells: Methods and Protocols, M. Gigante and E. Ranieri, Editors. 2021, Springer US: New York, NY. p. 215-227. [CrossRef]
- Fujii, H., S. Goto, and M. Fukagawa, Role of Uremic Toxins for Kidney, Cardiovascular, and Bone Dysfunction. Toxins (Basel), 2018. 10(5). [CrossRef]
- Vanholder, R., et al., Biochemical and Clinical Impact of Organic Uremic Retention Solutes: A Comprehensive Update. Toxins (Basel), 2018. 10(1). [CrossRef]
- Chen, J.H. and C.K. Chiang, Uremic Toxins and Protein-Bound Therapeutics in AKI and CKD: Up-to-Date Evidence. Toxins (Basel), 2021. 14(1). [CrossRef]
- Chmielewski, M., et al., The peptidic middle molecules: is molecular weight doing the trick? Semin Nephrol, 2014. 34(2): p. 118-34. [CrossRef]
- Smith, H.W., The kidney: structure and function in health and disease. 1951: Oxford University Press, USA.
- Chao, C.T. and C.K. Chiang, Uremic toxins, oxidative stress, and renal fibrosis: an interwined complex. J Ren Nutr, 2015. 25(2): p. 155-9. [CrossRef]
- Mihajlovic, M., et al., Protein-Bound Uremic Toxins Induce Reactive Oxygen Species-Dependent and Inflammasome-Mediated IL-1beta Production in Kidney Proximal Tubule Cells. Biomedicines, 2021. 9(10). [CrossRef]
- Sun, B., et al., Hippuric Acid Promotes Renal Fibrosis by Disrupting Redox Homeostasis via Facilitation of NRF2-KEAP1-CUL3 Interactions in Chronic Kidney Disease. Antioxidants (Basel), 2020. 9(9). [CrossRef]
- Zhang, H., et al., Indoxyl sulfate accelerates vascular smooth muscle cell calcification via microRNA-29b dependent regulation of Wnt/β-catenin signaling. Toxicol Lett, 2018. 284: p. 29-36. [CrossRef]
- Sun, C.Y., et al., Protein-bound uremic toxins induce tissue remodeling by targeting the EGF receptor. J Am Soc Nephrol, 2015. 26(2): p. 281-90. [CrossRef]
- Schafer, M.J., et al., Targeting Senescent Cells in Fibrosis: Pathology, Paradox, and Practical Considerations. Curr Rheumatol Rep, 2018. 20(1): p. 3. [CrossRef]
- Niwa, T. and H. Shimizu, Indoxyl sulfate induces nephrovascular senescence. J Ren Nutr, 2012. 22(1): p. 102-6. [CrossRef]
- Fletcher-Sananikone, E., et al., Elimination of Radiation-Induced Senescence in the Brain Tumor Microenvironment Attenuates Glioblastoma Recurrence. Cancer Res, 2021. 81(23): p. 5935-5947. [CrossRef]
- Hu, X. and H. Zhang, Doxorubicin-Induced Cancer Cell Senescence Shows a Time Delay Effect and Is Inhibited by Epithelial-Mesenchymal Transition (EMT). Med Sci Monit, 2019. 25: p. 3617-3623. [CrossRef]
- Yang, Y., et al., A Human Conditionally Immortalized Proximal Tubule Epithelial Cell Line as a Novel Model for Studying Senescence and Response to Senolytics. Front Pharmacol, 2022. 13: p. 791612. [CrossRef]
- Chang, J.F., et al., Therapeutic Targeting of Aristolochic Acid Induced Uremic Toxin Retention, SMAD 2/3 and JNK/ERK Pathways in Tubulointerstitial Fibrosis: Nephroprotective Role of Propolis in Chronic Kidney Disease. Toxins (Basel), 2020. 12(6). [CrossRef]
- Tolle, M., et al., Uremic mouse model to study vascular calcification and "inflamm-aging". J Mol Med (Berl), 2022. 100(9): p. 1321-1330. [CrossRef]
- Li, X., et al., Effects of Uremic Clearance Granules on p38 MAPK/NF-κB Signaling Pathway, Microbial and Metabolic Profiles in End-Stage Renal Disease Rats Receiving Peritoneal Dialysis. Drug Des Devel Ther, 2022. 16: p. 2529-2544. [CrossRef]
- Wang, X., et al., Aberrant gut microbiota alters host metabolome and impacts renal failure in humans and rodents. Gut, 2020. 69(12): p. 2131-2142. [CrossRef]
- Huang, Y., et al., Indoxyl sulfate induces intestinal barrier injury through IRF1-DRP1 axis-mediated mitophagy impairment. Theranostics, 2020. 10(16): p. 7384-7400. [CrossRef]
- Nakano, T., et al., Indoxyl Sulfate Contributes to mTORC1-Induced Renal Fibrosis via The OAT/NADPH Oxidase/ROS Pathway. Toxins (Basel), 2021. 13(12). [CrossRef]
- Chen, C., et al., Yishen-Qingli-Huoxue formula attenuates renal fibrosis by inhibiting indoxyl sulfate via AhR/snai1 signaling. Phytomedicine, 2023. 108: p. 154546. [CrossRef]
- Hsieh, Y.H., et al., Rosmarinic acid ameliorates renal interstitial fibrosis by inhibiting the phosphorylated-AKT mediated epithelial-mesenchymal transition in vitro and in vivo. Food Funct, 2022. 13(8): p. 4641-4652. [CrossRef]
- Cai, H., et al., Lindera aggregata intervents adenine-induced chronic kidney disease by mediating metabolism and TGF-β/Smad signaling pathway. Biomed Pharmacother, 2021. 134: p. 111098. [CrossRef]
- Kim, H., et al., Lactobacillus acidophilus KBL409 Reduces Kidney Fibrosis via Immune Modulatory Effects in Mice with Chronic Kidney Disease. Molecular Nutrition & Food Research, 2022. 66(22): p. 2101105. [CrossRef]
- Barba, C., et al., A low aromatic amino-acid diet improves renal function and prevent kidney fibrosis in mice with chronic kidney disease. Sci Rep, 2021. 11(1): p. 19184. [CrossRef]
- Sun, C.Y., et al., p-Cresol Sulfate Caused Behavior Disorders and Neurodegeneration in Mice with Unilateral Nephrectomy Involving Oxidative Stress and Neuroinflammation. Int J Mol Sci, 2020. 21(18). [CrossRef]
- Miwa, S., et al., Mitochondrial dysfunction in cell senescence and aging. J Clin Invest, 2022. 132(13). [CrossRef]
- Zhao, M., et al., Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics, 2021. 11(4): p. 1845-1863. [CrossRef]
- Rossi, M., et al., Protein-bound uremic toxins, inflammation and oxidative stress: a cross-sectional study in stage 3-4 chronic kidney disease. Arch Med Res, 2014. 45(4): p. 309-17. [CrossRef]
- Mihajlovic, M., et al., Role of Vitamin D in Maintaining Renal Epithelial Barrier Function in Uremic Conditions. Int J Mol Sci, 2017. 18(12). [CrossRef]
- Correia-Melo, C., et al., Mitochondria are required for pro-ageing features of the senescent phenotype. Embo j, 2016. 35(7): p. 724-42. [CrossRef]
- Dibble, C.C. and L.C. Cantley, Regulation of mTORC1 by PI3K signaling. Trends Cell Biol, 2015. 25(9): p. 545-55. [CrossRef]
- Watanabe, H., et al., p-Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Int, 2013. 83(4): p. 582-92. [CrossRef]
- Deng, M., et al., Short-Chain Fatty Acids Alleviate Hepatocyte Apoptosis Induced by Gut-Derived Protein-Bound Uremic Toxins. Front Nutr, 2021. 8: p. 756730. [CrossRef]
- Sun, C.Y., H.H. Hsu, and M.S. Wu, p-Cresol sulfate and indoxyl sulfate induce similar cellular inflammatory gene expressions in cultured proximal renal tubular cells. Nephrol Dial Transplant, 2013. 28(1): p. 70-8. [CrossRef]
- Branzei, D. and M. Foiani, Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol, 2008. 9(4): p. 297-308. [CrossRef]
- Yang, L., et al., Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med, 2010. 16(5): p. 535-43, 1p following 143. [CrossRef]
- Passos, J.F., et al., Feedback between p21 and reactive oxygen production is necessary for cell senescence. Molecular Systems Biology, 2010. 6(1): p. 347. [CrossRef]
- Yang, Y., et al., The Uremic Toxin Indoxyl Sulfate Accelerates Senescence in Kidney Proximal Tubule Cells. Toxins, 2023. 15(4): p. 242. [CrossRef]
- Li, L., et al., Protein-bound P-cresol inhibits human umbilical vein endothelial cell proliferation by inducing cell cycle arrest at G(0)/G(1). Am J Transl Res, 2017. 9(4): p. 2013-2023.
- Shimizu, H., et al., Indoxyl sulfate enhances p53-TGF-β1-Smad3 pathway in proximal tubular cells. Am J Nephrol, 2013. 37(2): p. 97-103. [CrossRef]
- Humphreys, B.D., Mechanisms of Renal Fibrosis. Annu Rev Physiol, 2018. 80: p. 309-326. [CrossRef]
- Liu, Y., et al., The NLRP3 inflammasome in fibrosis and aging: The known unknowns. Ageing Research Reviews, 2022. 79: p. 101638. [CrossRef]
- Romero, A., et al., Pharmacological Blockade of NLRP3 Inflammasome/IL-1β-Positive Loop Mitigates Endothelial Cell Senescence and Dysfunction. Aging Dis, 2022. 13(1): p. 284-297. [CrossRef]
- Yan, M.T., C.T. Chao, and S.H. Lin, Chronic Kidney Disease: Strategies to Retard Progression. Int J Mol Sci, 2021. 22(18). [CrossRef]
- Latorre, E., et al., Small molecule modulation of splicing factor expression is associated with rescue from cellular senescence. BMC Cell Biology, 2017. 18: p. 1-15. [CrossRef]
- Herranz, N., et al., mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol, 2015. 17(9): p. 1205-17. [CrossRef]
- Knoppert, S.N., et al., Cellular Senescence and the Kidney: Potential Therapeutic Targets and Tools. Front Pharmacol, 2019. 10: p. 770. [CrossRef]
- June, C.H., et al., CAR T cell immunotherapy for human cancer. Science, 2018. 359(6382): p. 1361-1365. [CrossRef]
- Dunbar, C.E., et al., Gene therapy comes of age. Science, 2018. 359(6372): p. eaan4672. [CrossRef]
Figure 1.
Composition of ECM (reproduced with permission from [25]). (A) The basic subdivision of the ECM into (B) basement membrane and (C) interstitial matrix is shown along with major structural components (collagen and elastin), as well as the background matrix made up of proteoglycans and hyaluronan [25].
Figure 1.
Composition of ECM (reproduced with permission from [25]). (A) The basic subdivision of the ECM into (B) basement membrane and (C) interstitial matrix is shown along with major structural components (collagen and elastin), as well as the background matrix made up of proteoglycans and hyaluronan [25].
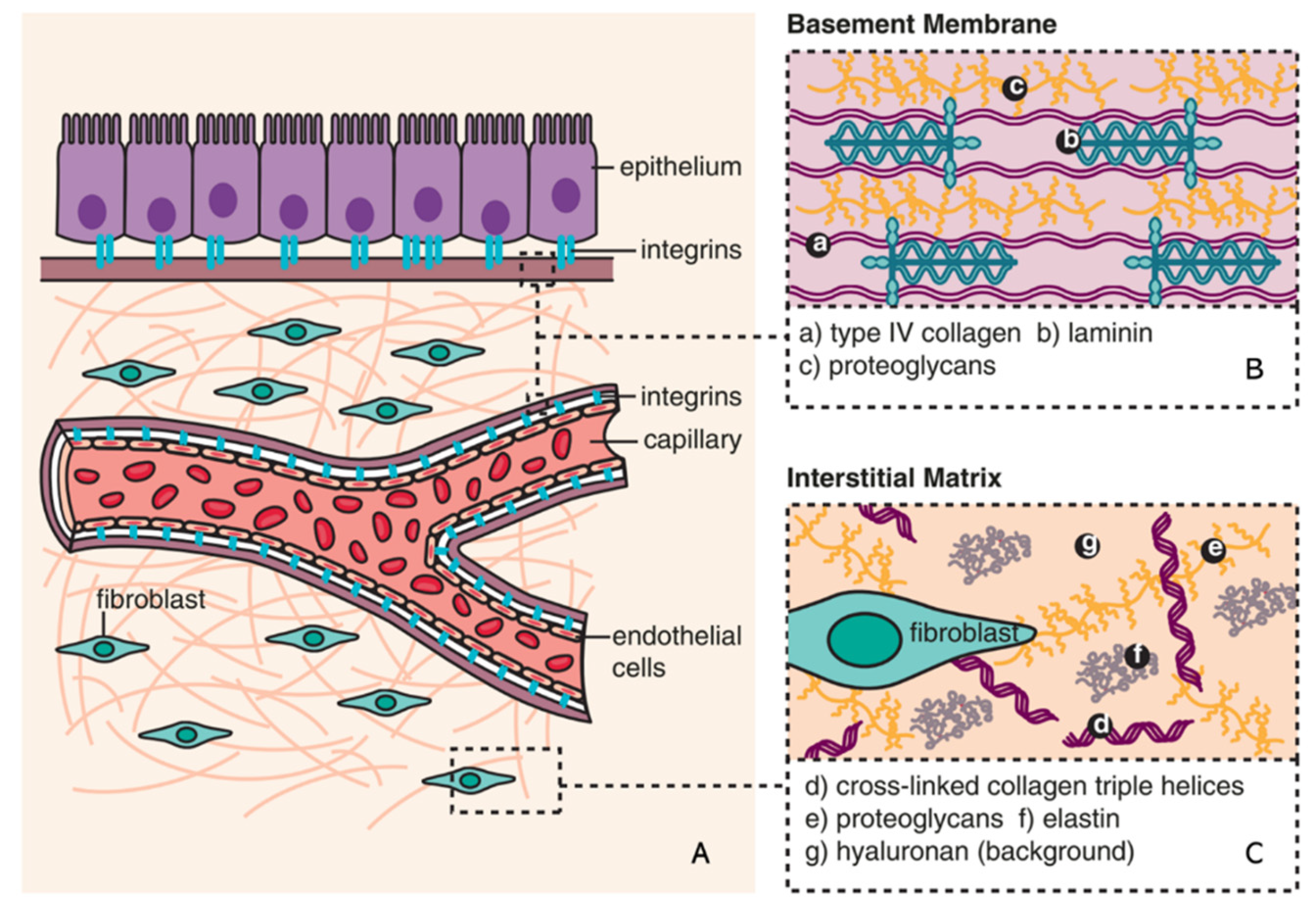
Figure 2.
Major types of senescence. Three main types of senescence are identified. Replicative senescence (RS) links to telomere shortening that is associated with cell division. Oncogene-induced senescence (OIS) refers to cell cycle arrest by aberrant activation of oncogenic signalling, which promotes the initiation and development of cancer. Stress-induced (premature) senescence (SIS) appears after exposing cells to chemical or physical stresses. Accumulation of long-term senescent cells leads to chronic senescence.
Figure 2.
Major types of senescence. Three main types of senescence are identified. Replicative senescence (RS) links to telomere shortening that is associated with cell division. Oncogene-induced senescence (OIS) refers to cell cycle arrest by aberrant activation of oncogenic signalling, which promotes the initiation and development of cancer. Stress-induced (premature) senescence (SIS) appears after exposing cells to chemical or physical stresses. Accumulation of long-term senescent cells leads to chronic senescence.
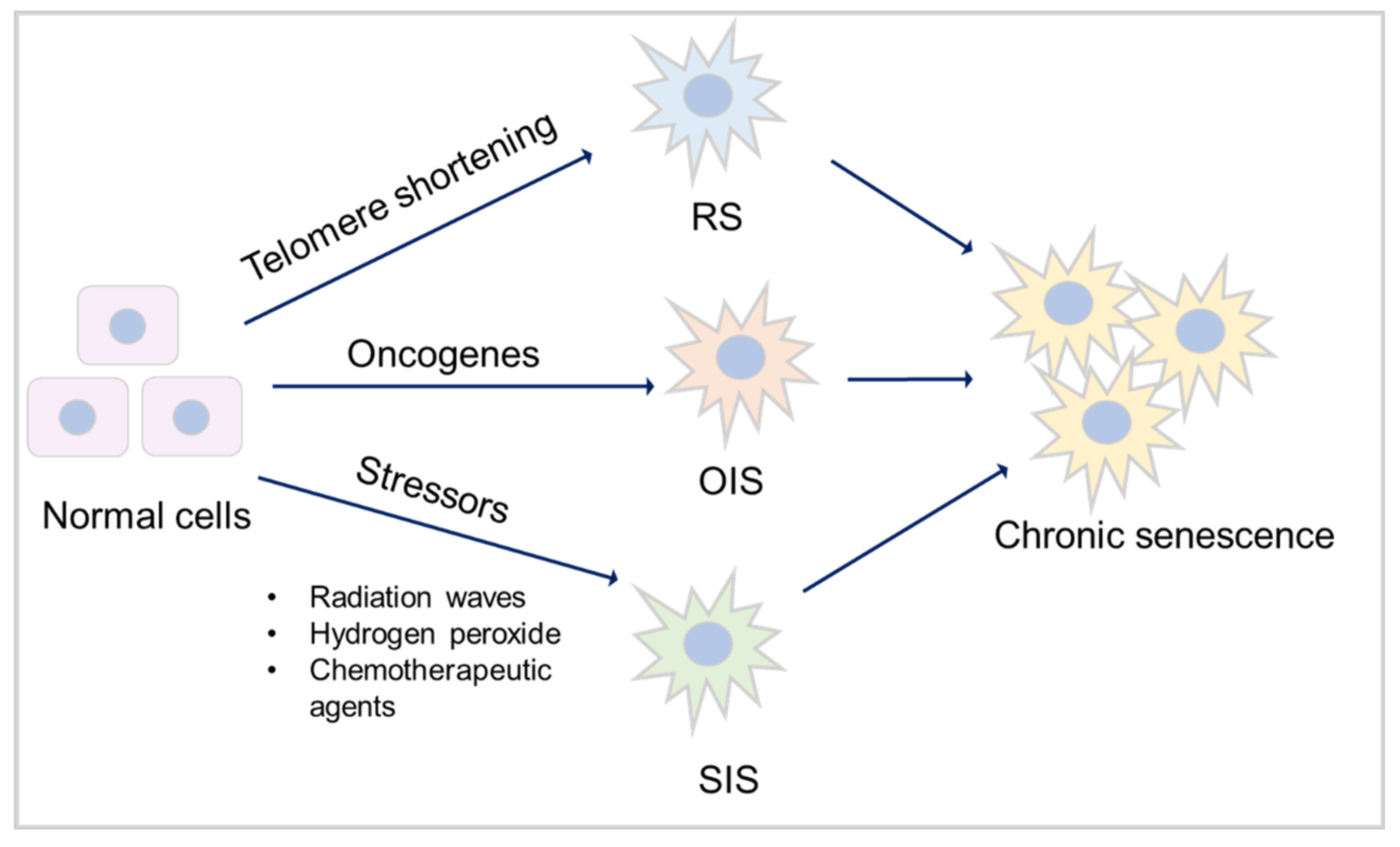
Figure 3.
The mechanism of senescence in kidney fibrosis. Senescence is initiated by various stimulations (e.g. ionizing radiation, exposure to toxicants, and heat stress), which triggers DDR. This, on the one hand, induces mitochondrial dysfunction, resulting in abnormal expression of Bcl-2 family proteins, eventually leading to apoptosis resistance and promotion of senescence. On the other hand, DDR mediates cell cycle arrest via p53/ p21 and p16/pRb checkpoint pathways, which also results in senescence. Senescent cells show a downregulation of LaminB1 and SA-β-gal. SASP factors, including profibrotic markers (TGF-β and CTGF), proinflammatory markers (IL-6 and IL-1β), and ECM remodelling proteases (MMPs) expressed by senescent cells promote ECM deposition (α-SMA, fibronectins and collagens), finally leading to kidney fibrosis.
Figure 3.
The mechanism of senescence in kidney fibrosis. Senescence is initiated by various stimulations (e.g. ionizing radiation, exposure to toxicants, and heat stress), which triggers DDR. This, on the one hand, induces mitochondrial dysfunction, resulting in abnormal expression of Bcl-2 family proteins, eventually leading to apoptosis resistance and promotion of senescence. On the other hand, DDR mediates cell cycle arrest via p53/ p21 and p16/pRb checkpoint pathways, which also results in senescence. Senescent cells show a downregulation of LaminB1 and SA-β-gal. SASP factors, including profibrotic markers (TGF-β and CTGF), proinflammatory markers (IL-6 and IL-1β), and ECM remodelling proteases (MMPs) expressed by senescent cells promote ECM deposition (α-SMA, fibronectins and collagens), finally leading to kidney fibrosis.
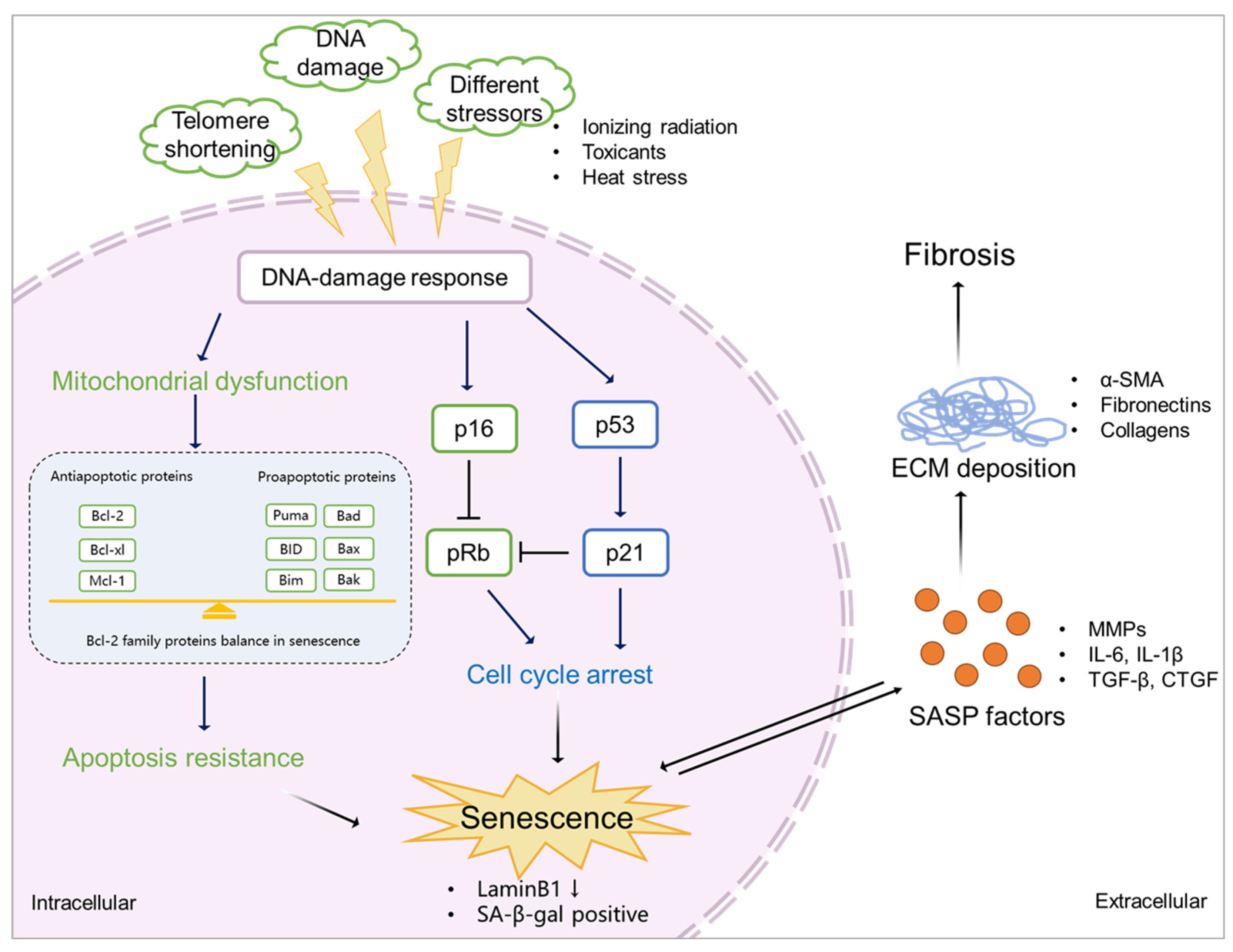
Figure 4.
Proposed scheme of PBUTs promoting kidney fibrosis by accelerating senescence. After reaching the cells, PBUTs promote ROS production, triggering DDR and mitochondrial dysfunction, inducing apoptosis inhibition, cell cycle arrest and the production of SASP factors, thus promoting senescence. Senescent cells show a downregulation of LaminB1 and SA-β-gal activity. SASP factors expressed by senescent cells promote ECM deposition, leading to kidney fibrosis.
Figure 4.
Proposed scheme of PBUTs promoting kidney fibrosis by accelerating senescence. After reaching the cells, PBUTs promote ROS production, triggering DDR and mitochondrial dysfunction, inducing apoptosis inhibition, cell cycle arrest and the production of SASP factors, thus promoting senescence. Senescent cells show a downregulation of LaminB1 and SA-β-gal activity. SASP factors expressed by senescent cells promote ECM deposition, leading to kidney fibrosis.
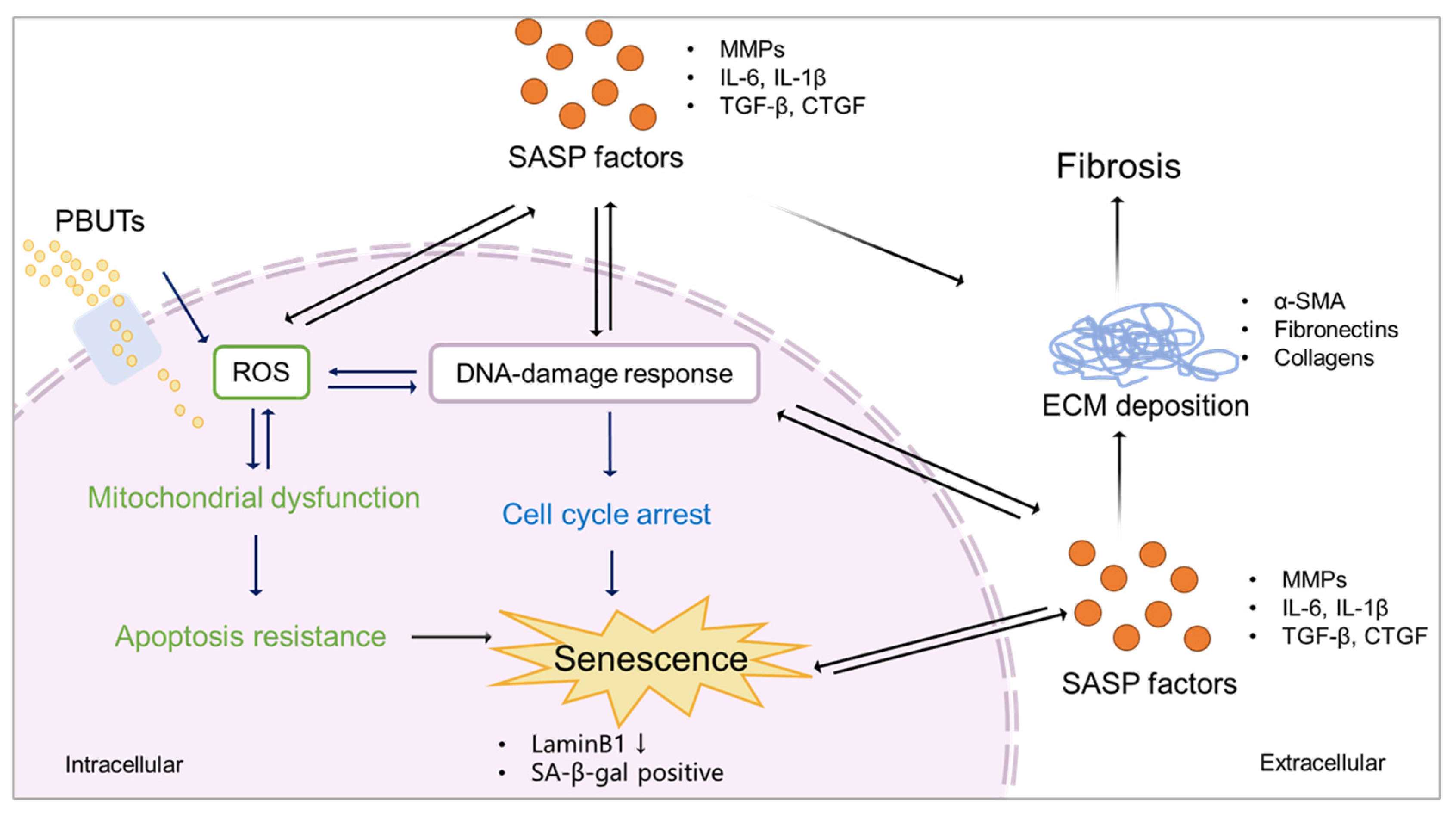

Table 1.
ECM in kidney fibrosis (adapted from [24]).
Table 1.
ECM in kidney fibrosis (adapted from [24]).
| Compartment | ECM in healthy kidney | Increased ECM in kidney fibrosis |
|---|---|---|
| Glomeruli | Mesangial Matrix: collagen IV, V, fibronectin, nidogen, laminin. | Nodular mesangial sclerosis: collagen I, III, IV, V, fibronectin, nidogen, laminin, decorin, biglycan. |
| Glomerular basement membrane: collagen I, III, VI, IV, VII, XV, XVII, agrin, perlecan, nidogen, laminin. | Focal segmental glomerulosclerosis: collagen III, IV, heparan sulfate proteoglycans. | |
| Bowman's capsule: collagen IV, laminins, nidogen, heparan sulfate proteoglycans. | Thickening of glomerular basement membrane: collagen I, III, VI, IV, VII, XV, XVII, perlecan, nidogen, laminin. | |
| Bowman's capsule: collagen IV and heparan sulfate proteoglycans. | ||
| Tubulointerstitium | Tubular basement membrane: collagen IV, agrin, perlecan, laminin. |
Thickening of tubular basement membrane: collagen IV, perlecan; |
| Interstitium: collagen I, II, III, V, VI, VII, XV, fibronectin, biglycan, decorin, versican. | Interstitial fibrosis: collagen I, II, III, V, VI, VII, XV, fibronectin, biglycan, decorin, ersican. | |
| Capillary basement membrane: N/A. | Thickening and multilayering of capillary basement membrane: N/A. |
|
| Vasculature | Intima with internal elastic lamina: elastin, perlecan, agrin, collagen XVIII, versican, biglycan, decorin. | Neointima: versican, collagen XVIII, agrin, perlecan. |
| Media with external elastic lamina: collagen I, III, XVII, elastin, agrin, perlecan, decorin, verslcan. | Intima with internal elastic lamina: elastin, perlecan, agrin, collagen XVIII, versican. | |
| Adventitia: collagen I, III, fibronectin, elastin. | Media with external elastic lamina: elastin, collagen XVII, agrin, perlecan,versican. | |
| Perivascular fibrosis (Thickening of adventitia): N/A. |
Table 2.
Overview of CKD animal models that reported PBUTs accumulation, fibrosis outcome, or senescence phenotype.
Table 2.
Overview of CKD animal models that reported PBUTs accumulation, fibrosis outcome, or senescence phenotype.
| CKD model | Species | PBUTs | Fibrosis/EMT markers | Senescence markers/ SASP factors | Invloved pathways/mechanism | Reference |
|---|---|---|---|---|---|---|
| Aristolochic acids-induced | Mouse | PCS, IS | α-SMA, collagen I, α-1 and IV | NR | TGF-β signaling | [116] |
| Adenine-induced | Mouse | NR | Collagen (masson staining) | p21, Il-6, and Il-1β | chronic inflammation | [117] |
| 5/6 nephrectomy | Rat | NR | Collagen (masson staining) | TNF-α, IL1β, and IL-6 | p38 MAPK/NF-κB signaling pathway | [118] |
| Ischemia-reperfusion injury | Mouse | NR | Collagen (masson staining), fibronectin and α-SMA | SA–β-gal, p16, p19, p53, p21, MMP-7, PAI-1 and TGF-β1 | WNT and TGF-β signaling | [18] |
| Adenine-induced | Mouse | PCS, IS, and hippuric acid | Collagen (masson staining) and α-SMA | NR | gut microbiota | [119] |
| IS-injected mouse and unilateral nephrectomy | Mouse | IS | ZO-1, occludin,claudin-1, and claudin-2 | TNF-α, IL-1β and IL-6 | mitochondrial dysfunction and mitophagy impairment | [120] |
| Adenine-induced | Mouse | IS | α-SMA, E-cadherin, and collagen I | TNF-α and IL-6 | mTOR activation | [121] |
| Adenine-induced | Rat | IS | fibronectin, collagen I, α-SMA, vimentin, and E-cadherin | NR | EMT | [122] |
| Unilateral ureteral obstruction (UUO) | Mouse | IS | Collagen (masson staining), α-SMA, collagen I, fibronectin, vimentin, and E-cadherin | TGF-β1 | EMT | [123] |
| Adenine-induced | Rat | PCS, IS, hippuric acid,p-cresyl glucuronide, and indol-3-acetic acid | NR | TGF-β1 | TGF-β signaling | [124] |
| Adenine-induced | Mouse | PCS | NR | TNF-α and IL-6 | NLRP3 inflammasome pathway | [125] |
| Adenine-induced | Mouse | PCS, IS and p-cresyl glucuronide | collagen α-1 type 1 | TGF-β1, TNF-α, MCP-1 and IL-6 | production of uremic toxins, and inflammation | [126] |
| Unilateral nephrectomy | Mouse | PCS | NR | p38 and IL-1β | Oxidative stress and inflammation | [127] |
| 5/6 nephrectomy | Rat | Hippuric acid | α-SMA, vimentin, and collagen I | MMP9 and TIMP1 | Oxidative stress and TGF-β signaling | [108] |
| NR – Not Reported. | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



