
Submitted:
08 August 2023
Posted:
09 August 2023
You are already at the latest version
Abstract
Over the last several decades, China has continuously introduced Duroc boars and used them as breeding boars. Although this crossbreeding method has increased pork production, it has affected pork quality. Nowadays, one of the primary goals of industrial breeding and production systems is to enhance the quality of meat. This research adds to our understanding of the molecular mechanisms that control the quality of pork and may be used as a guide for future efforts to enhance meat quality. We investigated the genetic mechanisms of cross-breeding for meat quality improvement by combining transcriptome and metabolome analysis, using Chinese native Jiaxing black (JXB) pigs and crossbred Duroc × Duroc × Berkshire × JXB (DDBJ) pigs. In the longissimus Dorsi muscle, the content of inosinic acid, polyunsaturated fatty acid, and amino acids was considerably higher in JXB pigs in contrast with that of DDBJ pigs, whereas DDBJ pigs have remarkably greater levels of polyunsaturated fatty acids than JXB pigs. Differentially expressed genes (DEGs) and differential metabolites were identified using transcriptomic and metabolomic KEGG enrichment analyses. Differential metabolites mainly include amino acids, fatty acids, phospholipids. In addition, we found several DEGs that may explain differences in meat quality between the two pig types, including genes associated with lipid metabolism (e.g., DGKA, LIPG, and LPINI), fatty acid metabolism (e.g., ELOVL5, ELOVL4, and ACAT2), and amino acid metabolism (e.g., SLC7A2, SLC7A4). Combined with the DEGS-enriched signaling pathways, the regulatory mechanisms related to amino acids, fatty acids, and phospholipids were mapped. The abundant metabolic pathways and DEGs may provide insight into the specific molecular mechanism that regulates meat quality. Optimizing the composition of fatty acids, phospholipids, amino acids, and other compounds in pork is conducive to improving meat quality. Overall, these findings will give useful information and further groundwork for enhancing the meat quality that may be achieved via hybrid breeding.
Keywords:
Transcriptomics
; Metabolomics
; Pork
; Meat quality
; Longissimus dorsi muscle
1. Introduction
Over half of the world’s pork is produced in China, making it the country with the biggest pig industry [1]. The focus of pig breeding efforts over the last several decades has been to increase pig productivity in key areas such as feed conversion, growth rate, lean meat content, and lowered backfat thickness and carcass amount of fat in the carcass. However, improvements in growth and carcass qualities have also influenced several characteristics of the meat’s quality, particularly intramuscular fat content, meat color, water holding capacity , and tenderness [2]. The market share in China and worldwide is dominated by Duroc × Landrace × Yorkshire (DLY) pigs due to their superior feed conversion, rapid growth, and high leanness [2]. However, inferior DLY pork meat can elicit consumer dissatisfaction [3,4]. In contrast, Chinese local pork has a perfect color, aroma, and taste and are preferred by consumers, and these pigs are characterized by good reproductive performance and adaptability. However, native pig breeds are also associated with low leanness, low feed conversion rates, and high feeding costs, thus, pig farmers frequently prefer DLY pigs. In the past decades, mass production was pursued at the expense of quality. As the consumers’ quality demands have increased, the consumption of meat has increasingly evolved from “quantity” toward “quality”, and local pigs have gradually received more attention [4]. In recent years, crossbreeding of imported pig breeds as sires and native sows was found to produce progeny combining quality and quantity traits.
Notably, poor quality pork not only affects taste but also increases the risk of cardiovascular disease in humans [5]. Therefore, the molecular mechanisms involved in meat quality have attracted research attention. Currently, the quantitative trait loci (QTL) database of pigs (https://www.animalgenome.org/) comprises a large number of QTL that are related to the quality of the meat, e.g., 890 QTL associated with intramuscular fat content, 831 QTL associated with the saturated fatty acid content, and 750 QTL associated with monounsaturated fatty acid content. Most meat quality traits are complex quantitative traits, and many studies have focused on additive genetic effects, neglecting non additive genetic effects. Non-additive effects will lead to nonlinear effects of phenotype. Understanding the mechanism of meat quality regulation will help researchers better understand the epistasis in non-additive effects. However, accurately determining the biomolecular mechanisms of meat quality is challenging, and new methods are warranted to assess meat quality traits in an accurate manner, thus improving productivity and meeting industry needs.
Multi-omics association analysis has been widely used in the fields of agricultural animal breeding [6,7,8]. The transcriptome can provide a comprehensive basis for elucidating processes of gene modulation transcriptionally; moreover, metabolomic analysis can inform on auxiliary metabolic changes caused by post-transcriptional modulation, which often represents a sequence of activities and alterations in the final stage of the gene transcription and translation [9]. In a previous study, transcriptome and targeted metabolome analyses were used to elucidate effects of bile acids on fat deposition and meat quality of lambs [10]. In a different study, the transcriptome was used to mine key candidate genes regulating pork quality [11], and transcriptome analyses were applied to reveal the effects of regulatory mechanisms of myoglobin content on Iberian pork color [12]. Furthermore, transcriptomic data from porcine subcutaneous and intramuscular fat suggested new directions to study the regulatory mechanisms of two important adipose tissues in pigs [9]. These studies contribute to a better understanding of meat quality regulation mechanisms through multi-omics.
Owing to its excellent and unique flavor, Jiaxing black (JXB) pork is very popular with consumers. Previous studies have shown that among the three breeds (JXB, Berkshire × JXB (BJ), and Duroc × Berkshire × JXB (DBJ)), DBJ pigs had the best carcass traits, while various aspects of JXB pigs’ meat quality were superior to those of DBJ and BJ pigs[13]. Therefore, from the perspective of economic efficiency and consumer demand, DBJ pigs can be used as a high-quality commercial breed, however, after several generations of breeding, DBJ pigs show segregation of hair color, which implies that the heritability of various traits of DBJ pigs is unstable. After we crossed Duroc sires with DBJ sows, trait heritability was stable in DDBJ pigs. Therefore, the meat quality of DDBJ and JXB pigs was analyzed comparatively, and comprehensive transcriptome and metabolome analyses were performed. The primary goal was to further optimize the meat quality of crossbred pigs with balanced genetic improvement and elucidate the potential genetic impacts of crossbreeding on improved pork quality2. Materials and methods
2.1. Ethics Statement
Zhejiang University’s Animal Protection Institution and Use Committee (Zhejiang, China) granted its approval to carry out experiments involving animals. The ethics committee number of this research institution is ZJU20170466.
2.2. Study animals and sample collection
Pigs used in this study were provided by Zhejiang Qinglian Food Co (Jiaxing, China),that supplied the pigs were utilized in this investigation. All pigs were kept in a pig house under similar environmental conditions, were fed the same basal diet (see Supporting Table S1), and had access to water ad libitum. ALL pigs were executed using 100V, 12S, 70HZ electrical stimulation. The farms are equipped with slaughterhouses and temperature control equipment to avoid transport stress and heat stress before slaughter. All pigs are slaughtered on the same day and in the same batch. 20 JXB pigs and 23 DDBJ (Figure 1a) pigs of similar age (280 ± 5 days) were selected for slaughter. A variety of organs and tissues, including the kidney, lung, small intestine, spleen, sebum, liver, heart, and longissimus dorsi (LD) muscle, were harvested, immediately transferred into lyophilization tubes, and preserved in liquid nitrogen.
2.3. Carcass characteristics
The weight (kg) of each pig was recorded before slaughter. The left side of the carcass was weighed in kilograms (kg) after the viscera, hooves, head, and tail had been removed (kidneys and slab oil were kept). Carcass length, carcass slant length, and loin eye muscle area (length × width × 0.7) were measured, followed by the separation of bone, meat, skin, and fat from the left carcass by professional dissectors for weighing and calculating the respective percentage (%) of carcass weight.
2.4. Content of inosine monophosphate d, intramuscular fat, and cholesterol
To determine inosine monophosphate (IMP), 0.4 g LD muscle was cut up and placed in liquid nitrogen for grinding. Subsequently, 20 mL of cold 5% perchloric acid was added, and the mixture solution was introduced into a volumetric flask with 5% perchloric acid to fix the volume. Following a 10-minute incubation period, 20 ml of supernatant was removed, the pH value was set to 6.5 by adding 1.25 mol/L NaOH, and the mixture solution was then introduced into a volumetric flask and fixed with distilled water. Then, 1.5 mL of the mixture was placed in a centrifuge tube and centrifugated at 13,000 rpm and 4°C for half an hour. Subsequently, the HPLC (high performance liquid chromatography) analysis was conducted by placing 1 mL of the supernatant in a chromatography bottle (Waters 515 HPLC, Waters, USA). The following chromatographic conditions were applied: Waters AtlantisTM dC18 column (5 μm, Φ 4.6 × 250 mm); mobile phase: 50 mmol/L pH 6.5 ammonium formate buffer solution (containing 5% methanol); flow rate: 1 mL/min; column temperature: 25 °C; injection volume: 10 μL; UV detection wavelength: 254 nm; run time: 8 min for the standard working solution and 12 min for the sample extract. The IMP content the sample (mg/g) was computed as per the corresponding values of peak areas of the two solutions using the equation Ci=Cs*Ai*V/As/m
Where the IMP content within the sample is symbolized by Ci (mg/g), the IMP working solution concentration is denoted by “Cs” (mg/mL), the sample’s IMP peak area response is indicated by “Ai”, the IMP standard working solution peak area response value is denoted by the “As”, the total volume of the sample is denoted by V (mL), and the mass of the sample is indicated by m (g).
The content of intramuscular fat (IMF), crude protein (CP), water, and cholesterol was determined by the Zhejiang Feed and Animal Nutrition Laboratory, in compliance with the National Standard Technical Specification for Determination of Pig Muscle Quality (GB/T 9695.7-2008). Cholesterol content was determined using corresponding kits (Nanjing Jiancheng Institute of Biological Engineering, TG, A110-1-1; TC, A111-1-1).
2.5. Amino acid and fatty acid content
The LD muscle samples, weighing 100 mg, were digested in 5 mL of 6 mol/L HCl solution. Subsequently, the digested solution was then transferred to a volumetric flask and diluted to 50 mL. The filtered sample was passed through a 0.22 μm aqueous phase filter into a centrifuge tube. A 2 mL aliquot of the filtrate was placed in an evaporating dish and evaporated at 60°C in a water bath. Then, 4 mL of 0.02 mol/L HCl solution was added, and the sample was dissolved. The samples were stored at 4°C for analysis using an ion-exchange amino acid analyzer (L8900, Hitachi, Tokyo, Japan).
For the fatty acid analysis, 150 mg of LD muscle samples were weighed and mixed with 4 mL of chloroacetic acid/anhydrous methanol (1:10, v/v), 1 mL of n-hexane, and 1 mL of internal standard solution. The mixture was heated in a water bath at 80°C for 2.5 hours. After cooling to room temperature, the solution was mixed with 5 mL of 7% K2CO3 solution and centrifuged at 1200g for 5 min. The supernatant was transferred to vials for analysis using a gas chromatograph (Agilent 1200 Series, Agilent, USA). Prior to GC analysis, each sample was filtered through a 0.22 nm filter membrane (model 7890 A, Agilent Technologies, Palo Alto, CA, USA). Finally, the fatty acid concentrations were analyzed using GC ChemStation software (Agilent Technologies, Palo Alto, CA, USA).
2.6. RNA sequencing
SAMN 19237330 is the accession number for our data uploaded to the NCBI. Accurate quantitative transcriptome sequencing analysis was performed by Beijing Allwegene Technology Co. (China). The RNA sequencing data output included sample extraction, sample detection, library construction, library inspection, and up-sequencing. RNA purity was assessed using a Nanodrop device (Thermo Fisher Scientific, USA). Subsequently, an Agilent 2100 instrument (Agilent, USA) was used to measure the length of RNA fragments. Next, enrichment was done with the aid of magnetic oligo(dT) beads, and mRNA was fragmented with the addition of a fragmentation buffer. Afterward, the first strand of complementary DNA (cDNA) was produced by the process of reverse transcription with the aid of base random primers (random hexamers), whereas the second strand of cDNA was produced with the addition of buffer, DNA polymerase I, and dNTPs. Subsequently, AMPure XP beads were used in the process of purifying the double-stranded cDNA. The double-stranded cDNA that had been purified was subjected to end repair, the addition of A, as well as the addition of splice. Thereafter, AMPure XP beads (Beckman Coulter, USA) were employed to determine the fragment size of the double-stranded cDNA. The construction of cDNA libraries was accomplished by means of PCR amplification. The constructed libraries were quality-screened before being 2 × 150-bp paired-end sequenced using an Illumina platform (Illumina, USA). Comparison and transcript splicing analyses were completed using Star and Cufflinks software programs, respectively, followed by quantitative analysis of all genes.
2.7. Metabolome analysis
In order to avoid the interference of the weight of the pigs on the experimental results, we selected 10 pigs (5 JXB pigs and 5 DDBJ pigs) with the closest body weight. The LD muscle samples were extracted from the -80°C freezer and slowly thawed at 4°C. An appropriate amount of sample was taken and added to a pre-cooled MeOH: ACN: H2O solution (v: v: v=2:2:1) containing internal standards and two steel beads. The tissue homogenization was carried out using a tissue grinder at 60 Hz for 120 seconds, followed by sonication for 10 minutes. The mixture was allowed to stand at 20°C for 1 hour and then centrifuged at 13,000 rpm for 15 minutes at 4°C. The upper clear liquid was collected and freeze-dried. For mass spectrometry analysis, an appropriate amount of ACN: H2O solution (v:v=1:1) was added to the dried samples, and the mixture was vortexed for 30 seconds and sonicated for 10 minutes. The solution was then centrifuged at 13,000 rpm for 15 minutes at 4°C. The upper clear liquid was transferred to an injection vial for subsequent LC-MS/MS analysis.
Extensive untargeted metabolome sequencing analysis was performed by Beijing allwegene Technology Co.Ten samples were selected and were assigned to two groups for metabolic analyses based on extensively untargeted metabolomics performed using ultra-performance liquid chromatography (UPLC)-tandem mass spectrometry (MS/MS)-based detection platform, a self-built database, and multivariate statistics. The mobile phase for LC-MS/MS analysis consisted of eluent A (0.1% formic acid in water) and eluent B (0.1% formic acid in methanol). A Waters ACQUITY UPLC HSS T3 C18 column (1.8 μm, 2.1 mm * 100 mm) was used for reversed-phase separation. The injection volume was 2 μL, the flow rate was set at 0.3 ml/min, and the column temperature was maintained at 50°C. The metabolites from the column were subjected to high-resolution mass spectrometry using Triple TOF 5600+ in both positive and negative ion modes. The detailed parameters were as follows: Ion Source Gas1: 50 psi, Ion Source Gas2: 50 psi, Curtain Gas: 35 psi, Source Temperature: 500°C, IonSapary Voltage Floating: 5500 V &- 4500V (positive & negative); Declustering Potential (DP): ±80V (positive & negative); TOF MS scan m/z range: 60-1200 Da, Product ion scan m/z range: 25-1200 Da, TOF MS scan accumulation time 0.25s/spectra, Product ion scan accumulation time 0.03 s/spectra; Two stage mass spectrometry was acquired using Information Dependent Acquisition (IDA) in High Sensitivity mode, with CE: 30V±15. Data acquisition was performed using UPLC (ExionLC AD, https://sciex.com.cn/) and MS/MS (QTRAP®, https://sciex.com.cn/). Using the in-house created database of targeted standards, qualitative analysis was carried out on the substances that were discovered by using retention time (RT), daughter ion pair data, and secondary spectral data. The program known as Analyst 1.6.3 was used in the processing of the mass spectrometry data.
2.8. Evaluation of variation in gene expression and metabolic profiles
The differential gene criterion was a q-value < 0.05. The criteria for differential metabolites were VIP ≥ 1 and fold change ≤ 0.5 and ≥ 2. Differential gene ontology (GO) enrichment analysis was conducted using GO seq, which uses the Wallenius non-central hypergeometric distribution. The KEGG database’s pathways were used in a hypergeometric test-based differential gene KEGG enrichment study.
The KEGG Compound database (http://www.kegg.jp/kegg/compound/) was used for the annotation of the metabolites that were found before mapping them to the KEGG Pathway database (http://www.kegg.jp/kegg/pathway.html). Hypergeometric tests were conducted to examine the significance of the pathways to which the substantially regulated metabolites were mapped before processing them via a metabolite set enrichment analysis.
2.9. Data analyses
We used GraphPad Prism 9.0 (GraphPad Software, San Diego, USA) to analyze the variations in the contents of amino acids and fatty acids in the two pig breeds LD muscle tissue. Student’s t-test was applied for variance analysis when the variances were equal, and otherwise, Welch’s test was used. All data from the study were show as means ± SEM(23 DDBJ pigs, 20 JXB pigs).
Using multiple hypothesis tests to correct BH (FDR correction with Benjamin/Hochberg), the FDR values were obtained for screening DEGs. Based on the OPLS-DA results, the obtained multivariate analysis of the variable importance in projection (VIP) of the OPLS-DA model can preliminarily screen metabolites with differences between different varieties. P-value that can be combined with univariate analysis (comparison between two groups: Student’s t test) (n=5). We performed Pearson’s and Spearman correlation analyses of variance for differential genes enriched in the relevant pathways with the corresponding differential metabolites. Correlations were determined with the cor function in R, whilst correlation heat maps were produced using the Hmisc package in R(https://cran.r-project.org/web/packages/Hmisc/index.html). Subsequently, network plots were produced using Cytoscape software [14].
3. Results
3.1. Quality and carcass characteristics of JXB and DDBJ pigs
Pigs of the JXB breed had LD muscles with elevated levels of inosine monophosphate , free amino acids, intramuscular fat, and crude protein in contrast with those of the DDBJ breed (p < 0.01, Figure 1b). In addition, when comparing DDBJ with JXB pigs, it was found that the former had remarkably greater cholesterol and water levels (p < 0.01, Figure 1 b). Additionally, JXB pigs exhibited considerably greater levels of monounsaturated fatty acids compared to DDBJ pigs, particularly in terms of C16:1 (p < 0.05), C18:1 (p < 0.01), and C22:1 (p < 0.01) (Figure 2 a). By contrast, polyunsaturated fatty acid levels were substantially reduced in JXB relative to DDBJ pigs (p < 0.01), where, C18:2, C18:3n3, C18:3n6, and C20:3n6 levels were remarkably elevated in DDBJ in contrast with JXB pigs (p < 0.01) and C20:2, C22:6n3, and C20:5 levels were remarkably elevated in JXB in contrast with DDBJ pigs (p < 0.01). Notably, unsaturated fatty acid levels were elevated in DDBJ as opposed to JXB pigs (Figure 2a).
The levels of essential amino acids (EAA) and non-EAA (NEAA) were considerably elevated in JXB in contrast with those of DDBJ pigs (p < 0.01). JXB pigs also had a remarkable higher level of fresh-flavor amino acids compared to DDBJ pigs (p < 0.01). Figure 2b depicts the variations in the contents of 17 specific amino acids. We also compared several carcass traits of the two pig lines (Supporting Figure S1).
3.2. Differentially expressed genes (DEGs) in LD muscle tissue
Our analysis revealed 2,959 DEGs, including 1,494 and 1,465 up-and-down-regulated genes, respectively (Supporting Figure S2a). Furthermore, KEGG enrichment classification showed predominant enrichment of DEGs in metabolism, genetic information processing, organic systems, environmental information processing, and cellular processes, among which KEGG pathways of the metabolism branch were the most enriched.The differential genes were enriched as follows; 33 in the nucleotide metabolism-associated pathways, 68 in the metabolism of cofactors and vitamin-related pathways, 75 in lipid metabolism-associated pathways, 112 in glycan biosynthesis and metabolism-associated pathways, 10 in energy metabolism-associated pathways, 53 in carbohydrate metabolism-associated pathways, and 67 in amino acid metabolism-associated pathways (Supporting Figure S2b). The functional entries annotated by GO enrichment were divided into biological processes, cellular components, and molecular functions, and the GO functional terms under these three categories are displayed in Supporting Figure S2c-d. In addition, we used the KEGG database to determine which pathways were enriched for the up-and-down-regulated DEGs in the JXB and DDBJ pigs (Figure 2c-d). The downregulated genes showed primary enrichment in metabolic pathways, vitamin B6 metabolism, purine metabolism, protein digestion and absorption, mannose and fructose metabolism, nucleotide sugar and amino sugar metabolism, and other pathways. The upregulated genes were primarily enriched in the parathyroid hormone synthesis, secretion, and action, FoxO signaling pathway, MAPK signaling pathway, and the adherens junction pathway.
3.3. LD muscle metabolome analysis
The grouped samples were subjected to principal component analysis (PCA) to test variability between groups and among samples within each group (Figure 3a). We classified differential metabolites (Figure 3b, Supporting Figure S3b-d). The top twenty pathways ranked by p-value from our KEGG pathway enrichment study of the differential metabolites were chosen (Supporting Figure S4a). Similarly, the top 20 metabolic pathways according to p-value were chosen for examination of total KEGG metabolic pathway alterations, and the differential enrichment score was reflection of the total alterations that occurred in all of a pathway’s metabolites (Figure 3c). Metabolic enrichment analysis indicated significant enrichment of the top 50 metabolic sets ranked by p-value, including D-arginine and D-ornithine metabolism, tryptophan metabolism, steroid biosynthesis, glycolysis/gluconeogenesis, inositol phosphate metabolism, mannose and fructose metabolism, thiamine metabolism, and pentose phosphate pathways (p < 0.05; Figure 3d). In addition, we performed HMDB functional annotation and enrichment of differential metabolites and found that they were significantly enriched in pathways such as thiamine metabolism, bile acid biosynthesis, and oxidation of branched-chain fatty acids (Supporting Figure S4c).
3.4. Combined transcriptomic and metabolomic analysis
We divided the list of meat quality-related KEGG pathways enriched through downregulated genes into three categories: synthesis and metabolism of amino acids pathways, fatty acid synthesis, and metabolic pathways, and sugar metabolism and synthesis pathways (Figure 4a). Among the meat quality-related KEGG pathways enriched through upregulated genes, four categories were identified: vitamin metabolism and synthesis pathways, fatty acid synthesis and metabolic pathways, synthesis and metabolism of amino acids pathways, and sugar metabolism and synthesis pathways (Figure 4b). The up- and downregulated differential genes in KEGG pathways related to fat metabolism and synthesis were subjected to Spearman correlation analysis with fatty acyl and glycerophospholipid metabolites, respectively (Figure 4c-d). The results showed that the downregulated genes SLC27A1, PLA2G6, AGPAT5, LTA4H, DGKA, ACOT4, ACOX2, PLA2G4E, LPIN3, ELOVL4, ENSSSCG00000026701 and others were significantly and positively correlated with unsaturated glycerophospholipid metabolites such as: lysophosphatidylcholine (LPC (22:5/0:0)), lysophosphatidylethanolamine (LPE (0:0/22:6)), LPE (22:6/0:0)), and lysophosphatidic acid (LPA (18:1/0:0)). These were significantly negatively correlated with saturated glycerophospholipid metabolites (LPE (14:0/0:0)). The upregulated genes AKR1B1, DGKD, PLPP1, LIPG, SORS1, CHKA, GPAT4, LCLAT1, LPGAT1, DGKH, ACO1, MMP3, and UBC were significantly and positively correlated with carnitine C5:1, carnitine C5:0, carnitine-2-methyl-C, LPE (14:0/0:00), carnitine C4:0, and carnitine isoC4:0; the DGKD, PLPP1, PTGS1, LIPG, SORBS1, ENSSSCG00000012257, MMP3, UBC, and GPX3 genes were significantly negatively correlated with carnitine C14:2, LPA(18:1/0:0), (+-)15-HETE, LPC(22:5/0:0), LPE(0:0/22:6), and LPE(22:6/0:0).
We compared the up- and downregulated differential genes in KEGG pathways associated with amino acid metabolism and synthesis with those of amino acids, amino acid derivatives, and small peptide metabolites by Spearman correlation analysis. Most downregulated genes had a substantial negative correlation with amino acids and small peptide metabolites, and most upregulated genes were shown to have a substantial positive association with amino acids and small peptide metabolites (Figure 5a-b). We also tested correlations of differential genes in pathways related to glucose metabolism and vitamin coenzyme metabolism with metabolites (Figure 4e). Genes including TPK1, PDXP, COASY, AOX1, FCSK, ENOSF1, AK5, ADH5, TKFC, and PMM1 were significantly correlated with d-fructose-6-phosphate and all-trans-13,14-dihydroretinol, among which TKFC, FPGT, FBP2, NT5E, and ADH5 were also significantly correlated with the thiamine content. The AOX1 gene in the retinol metabolism pathway was positively correlated with the metabolite all-trans-13,14-dihydroretinol; The TPK1 gene in the thiamine metabolism pathway was significantly positively correlated with the thiamine content.
3.5. Potential regulatory mechanisms and functions related to meat quality hub genes
After reviewing the literature and identifying relevant signaling pathways associated with the differential genes, we produced a differential gene metabolite regulation map based on phenotypic data and metabolome data for phospholipid class, fatty acid class, and amino acid class (Figs. 5c and 6a-b). Differentially expressed genes that played important roles in the biochemical steps of metabolism and synthesis explained some differences in phospholipid metabolites, amino acid metabolites, and fatty acyl metabolites between JXB pigs and DDBJ pigs (Supporting Figs. 5c and 6a-b). To identify new hub genes most strongly associated with pork quality regulation, differential genes were screened with the criteria foldchange > 2 and p < 0.05. In total, 444 DEGs were screened (217 upregulated differential genes and 227 downregulated genes; Figure 7a), and to further highlight the possible genes linked to meat quality, we collected data from the PubMed database with the keywords “pig” + “meat quality” + “gene name” (using the selected 444 genes of interest) and identified 56 differential genes. Subsequently, these 56 differential genes were subjected to Spearman correlation analysis with differential metabolites, and the results were visualized as a network plot (Figure 7b). The downregulated genes CHODL and DBX1 and upregulated genes FLNC and MAPK10 were selected according to the absolute value of correlation coefficient |r| > 0.8 and p < 0.05 (Figure 7c).
4. Discussion
The aim of the present study was to estimate the meat quality of Chines local pigs and its hybrid pigs. Furthermore, we performed a comprehensive analysis of the LD muscle quality of JXB and DDBJ pigs by combining transcriptome and metabolome analysis, several DEGs and metabolic regulatory pathways related to meat quality were identified, which explained the regulatory mechanism of meat quality differences between the two pig lines. The DEGs regulatory map was drawn according to the omics data, and key functional genes such as DBX1, CHODL, FLNC, and MAOK10 were mined.
Gene selection has made a significant contribution to the performance improvement of production animals. Notably, genetic improvement may be achieved through intravarietal selection (i.e., pure breeding) and cross breeding (i.e., using native or foreign genetic resources). Hybridization, in contrast with pure breeding, does not lead to genetic advancement (that is, the average value of the parental varieties is equivalent to the additive genetic benefit of hybrid animals) but may provide a variety of benefits, including the use of complementary features and heterosis effects [15]. Many studies have shown that cross-breeding can change pig meat quality traits, carcass traits, nutritional value, energy metabolism, and related gene expression [16]. Although Chinese purebred pigs typically exhibit good meat quality, BQ pigs produced by crossbreeding Bactrian terminal boars with native pigs (Qing Yu) show higher water-holding capacity [17]. In a previous study, pigs with intermediate features in most examined traits such as meat color, intramuscular fat, PH24, and back fat thickness were produced by crossing two local breeds with notable variances in several markers of carcass and meat quality traits [18]. Thus, crossbreeding remains an important option for improving the production of animal traits.
Phospholipids are the only intramuscular lipids that undergo pronounced changes during processing, and when the dry-cured ham is being processed, they serve as the primary substrate for fat decomposition, which may negatively impact the product’s quality [19]. Due to their high polyunsaturated fatty acid content, phospholipids are also vital substrates for lipid oxidation [19]. It is worth noting that to some extent, rancidity is caused by triglycerides and phospholipids. In particular, phospholipids are the first molecules to undergo oxidation, and their contribution to rancidity is by far the most significant. The degree of unsaturation in triglycerides determines the extent to which they contribute to the rancidity of meat. The oxidation of polyunsaturated fatty acids is inextricably linked to rancidity. DGKA can phosphorylate diacylglycerol (DAG) to form phosphatic acid, which is the precursor of glycerol phospholipid synthesis [20]. LIPG is a cell surface lipoprotein that can help with cell internalization of surface lipoprotein particles (HDL/LDL/VLDL) through endocytosis, leading to their decomposition. In addition, LIPG is capable of hydrolyzing high-density lipoprotein that is attached to the cell surface, resulting in the release of lipid precursors like LPE, LPC, and fatty acids [21]. LPIN1 and LPIN3 are involved in the generation of DAG from phosphatic acid [22] LPCAT4 promotes phosphatidylcholine (PC) to produce lysophosphatidylcholine [23]. Lysophosphatidylglycerol acyltransferase I (LPGAT1) is an sn-1 specific acyltransferase, which regulates the stearic acid/palmitic acid ratio of phosphatidylethanolamine (PE) and phosphatidylcholine and prefers stearyl coenzyme A to palmitoyl coenzyme A as the substrate. The lack of LPGAT1 leads to the substitution of stearate in phospholipids (including PE, dimethylphosphodiethanolamine, and PC) of the PE methylation pathway by palmitate. Two fatty acids are identified in phospholipids and are bound to the glycerol sn-1 and sn-2 carbons. Generally, saturated fatty acids, primarily palmitate (C16:0) or stearate (C18:0), are found at the sn-1 position. Furthermore, sn-2 mostly consists of unsaturated fatty acids, the composition of which is controlled by the process of fatty acid remodeling [24]. 3, Glycerol phosphate acyltransferase (GPAT) catalyzed the first step of de novo synthesis of triglycerides, and GPAT3 was identified as a triacylglycerol biosynthesis enzyme [25]. The different expressions of these up and down differentially expressed genes in DDBJ pigs and JXB pigs revealed the potential gene regulation mechanism of the difference in phospholipid content in their muscles.
Dietary saturated fatty acids may play a role in elevating human blood cholesterol content, which in turn raises the risk of developing cardiovascular disease. When determining the nutritional and health benefits of meat, the ratio of polyunsaturated fatty acids to free fatty acids in the meat is often regarded as the most important factor to consider [26]. Meat that has a sufficient ratio of n-3 to n-6 fatty acids is one way to increase one’s consumption of the healthful n-3 fatty acids via one’s diet, and this may lead to an overall improvement in health. As per previous research findings, n-3 polyunsaturated fatty acids, also known as PUFAs, may increase antioxidant capacity, insulin resistance, and the nutritional value of meat [27]. ALA (C18:3n3) is an essential fatty acid for humans. Other PUFAs can be synthesized through several desaturation extensions reactions, such as docosahexaenoic acid (C22:6n3) and EPA (C20:5n3) (M. Xu et al., 2022). C20:5n3 and C20:6n3 can reduce the risk of coronary heart disease. N-6 fatty acids have not been studied as extensively as n-3 fatty acids, and many of the conclusions that have been drawn about them are still up for debate. Fatty acid extentase 5 (ELOVL5) is a key enzyme for the endogenous synthesis of long-chain unsaturated fatty acids. It catalyzes the formation of n-3 and n-6 PUFA and is involved in the de novo synthesis of monounsaturated fatty acids [28]. Supplemental PUFA may prevent the fatty-tissue deterioration caused by ELOVL5 ablation (C20:4, n-6; DHA (C22:6, n-3)), as ELOVL5 is essential for the production of these PUFAs [29,30]. ELOVL4 (fatty acid elongation enzyme 4) is an endoplasmic reticulum-localized enzyme that facilitates the condensation process during the biosynthesis of C28-C38 ultra long-chain PUFAs (VLC-PUFA) and C28 and C30 saturated fatty acids (VLC-FA) [31,32]. However, ELOVL5 was upregulated in JXB pigs, and ELOVL4 was upregulated in DDBJ pigs, which explained why the SFA and PUFA (C<24) content of JXB pig tissue was higher than that in DDBJ pigs.
LDLR is a crucial regulator of cholesterol metabolism that affects the level of circulating low-density lipoprotein cholesterol (LDLc), and it is involved in the clearance of LDLc [33]. LDLR is significantly upregulated in JXB pigs, which explains why the cholesterol content in LD muscle tissue of DDBJ pigs was considerably elevated in contrast with that in JXB pigs. ACAT2 reduces LDLR levels and disturbs the cholesterol metabolism homeostasis, thereby inhibiting the differentiation of preadipocytes in pig muscle tissue [34]. ACAT2 gene expression was significantly upregulated in JXB pigs, thus we speculated whether inhibition of LDLR by ACAT2 could offset the high expression level of LDLR in JXB pigs. Whether ACAT2 can reduce LDLR levels needs further verification. In addition, cholesterol acyltransferase-1 (ACAT1) is localized in the mitochondria, which helps form cholesterol lipids from free cholesterol. Cholesterol lipids accumulate in the endoplasmic reticulum [35,36]. Compared with DDBJ pigs, ACAT1 was also highly expressed in the LD tissue of JXB pigs, which further explains the significant difference in cholesterol content in the LD muscle of the two pig breeds. The down-regulation of CPT2 will cause the β-oxidation process to be inhibited, and the accumulation of acylcarnitine was a surrogate marker for the down-regulation of CPT2 [37]. The CPT2 was considerably downregulated in the LD of JXB pigs, but more types of acylcarnitine metabolites were downregulated in the LD tissue of JXB pigs, which may require further absolute quantitative tests of acylcarnitine metabolites for confirmation.
Muscle protein quality is determined by the specific amino acid composition of muscle tissue. Generally, three primary processes are involved in the decomposition of proteins: fibrin breakdown from major muscle, polypeptide synthesis as substrates for peptidase, and free amino acid synthesis [38]. Numerous endogenous muscle proteolytic enzymes may be involved in the hydrolysis of meat protein, like calpain, dipeptidyl peptidase, cathepsin, and aminopeptidase [39]. Some free amino acids are considered precursors of flavor substances and can form flavor substances with soluble reducing sugars like fructose and glucose. Apart from alanine, the content of the other 16 amino acids in identified pig LD tissue was remarkably elevated in JXB as opposed to DDBJ pigs. Two distinct subfamilies are discernible within the solute carrier family 7 (SLC7): L-type amino acid transporters (LAT), comprising SLC7A15 and SLC7A5-13, and cationic amino acid transporters comprising SLC7A14 and SLC7A1-4. We found that the expression of the SLC7A1 gene was remarkably upregulated in the LD of JXB pigs, and the expression of the SLC7A2 and SLC7A4 genes was remarkably upregulated in the LD of DDBJ pigs. SLC7A1 can perform a major role in the transport of cationic amino acids in pig muscle tissue, and its high expression in JXB pigs may explain why the levels of its cationic amino acids (lysine, arginine, and histidine) were higher than those in DDBJ pigs. Glutamate transporters play an important role in terminating excitatory neurotransmission and providing glutamate for systemic cells for metabolic purpose [40]. The high level of glutamate found in the livers of JXB pigs may be attributed to the high expression level of the SLC1A1 gene, which encodes the high-affinity glutamate transporter EAAC1. The level of GLUL gene expression in the LD of JXB pigs was remarkably elevated in comparison to that of DDBJ pigs. The glutamine synthase encoded by the gene GLUL converts glutamate and ammonia into glutamine [41]. Therefore, the content of glutamine in the LD of JXB pigs was higher. In addition, we found that some genes involved in the synthesis and metabolism of amino acids, the tricarboxylic acid cycle, and other pathways were significantly up-regulated in the LD of JXB pigs, such as ME2, SDS, and ALDH18A1. These differential genes further explained the difference in amino acid content in LD muscle tissue between the two breed lines.
Muscle fiber characteristics and purine content also affect the taste and nutritional value of meat. For example, pig muscle with a higher proportion of large IIB fiber is tougher, lighter, and better marbling score than muscle with a higher proportion of small or normal IIB fiber [42]. Purine metabolism allows for the absorption and use of dietary purines, particularly bases, nucleosides, and nucleotides within the human body. Uricase can catalyze the oxidation of uric acid to allantoin. Humans with attenuated uricase activity have elevated uric acid levels in their blood [43]. Consequently, uric acid is the end product of the purine metabolic process, and once it has been produced, it cannot be decomposed further in the human body. Hyperuricemia and gout are two conditions that may be easily brought on by having a high uric acid concentration in the blood [44]. Therefore, low-purine pig breeds would be less detrimental to consumer health. In addition, we found that eritadenine was downregulated in JXB pigs, although the difference was not significant. Eritadenine had a hypolipidemic effect. Numerous studies examined hypolipidemic activity and the mechanism of eritadenine. However, considering the 160 million hyperlipidemic patients in China, comparatively few studies assessed the application of eritadenine. Therefore, further research and development of eritadenine have a broad market prospect.
5. Conclusions
We propose the abundant metabolic pathways and DEGs provide insight into the specific molecular mechanism that regulates meat quality. With further functional verification, these important DEGs may be used to make accurate predictions about the quality of the meat and to guide decision-making in slaughterhouses. Optimizing the composition of fatty acids, phospholipids, amino acids, and other compounds in pork is conducive to improving meat quality. Overall, these findings will give useful information and further groundwork for enhancing the meat quality that may be achieved via hybrid breeding.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, Q, Chen; Methodology, Q, Chen and L, Xiao; Software, Q, Chen, W, Zhang and Y, Wang; Validation, Q, Sun; Formal analysis, Q, Chen and W, Zhang; Investigation, G, Liu; Writing – original draft, Q, Chen; Supervision, Y, Pan, Q, Wang and J, Zhang; Project administration, F, Wu; Funding acquisition, J, Zhang.
Funding
This work was supported by funds from the Program of Breeding of Agricultural New Species (Livestock and Poultry) in Zhejiang [2021C02068-2].
Acknowledgments
We thank Fen Wu, Shuang Liu, Jiabao Sun, Caiyun Cao, Yuge Tan in our lab, and Xin Li, Qi Wang from Institute of Feed Science, Zhejiang University and all workers in the slaughterhouse of Zhejiang Qinglian Food Company for their participation in the sample collection, and phenotype measurements. We thank Bullet Edits Limited for providing language polishing.
Conflicts of Interest
The authors declare no competing financial interest.
Abbreviations
DLY: Duroc × Landrace × Yorkshire; JXB: Jiaxing black; DDBJ: Duroc × Duroc × Berkshire × JXB; DBJ: Duroc × Berkshire × JXB; BJ: Berkshire × JXB; LD: Longissimus Dorsi muscle; DEGs: Differentially expressed genes; IMP: Inosine monophosphate; QTL: Quantitative trait loci; UPLC: Ultra-performance liquid chromatography; GO: Gene ontology; EAA: Essential amino acids; non-EAA: NEAA; PCA: Principal component analysis; LPC: Lsophosphatidylcholine; LPE: Lysophosphatidylethanolamine; LPA: Lysophosphatidic acid; PE: Phosphatidylethanolamine; LPGAT1: Lysophosphatidylglycerol acyltransferase I; GPAT: Glycerol phosphate acyltransferase; PUFAs: Polyunsaturated fatty acids; LDLc: Low-density lipoprotein cholesterol; LAT: L-type amino acid transporters.
References
- Chen, Q.; Cai, J.; Hua, W.; Li, K.; Zhang, X.; Xiao, L.; Zhang, W.; Ni, Y.; Zhang, J. , Effect of a new tungsten trioxide-based bactericide on the environment of piggeries and piglet health. ENVIRONMENTAL TECHNOLOGY & INNOVATION 2022, 28. [Google Scholar]
- Hérault, F.; Damon, M.; Cherel, P.; Le Roy, P. , Combined GWAS and LDLA approaches to improve genome-wide quantitative trait loci detection affecting carcass and meat quality traits in pig. Meat science 2018, 135, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Faucitano, L.; Ielo, M. C.; Ster, C.; Lo Fiego, D. P.; Methot, S.; Saucier, L. , Shelf life of pork from five different quality classes. Meat science 2010, 84, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhou, L.; Zhang, J.; Liu, X.; Zhang, Y.; Cai, L.; Zha ng, W.; Cui, L.; Yang, J.; Ji, J.; Xiao, S.; Ai, H.; Chen, C.; Ma, J.; Yang, B.; Huang, L. , A large-scale comparison of meat quality and intramuscular fatty acid composition among three Chinese indigenous pig breeds. Meat science 2020, 168, 108182. [Google Scholar] [CrossRef]
- Kityo, A.; Lee, S. A.; Kang, D. , Total and cause-specific mortality associated with meat intake in a large cohort study in Korea. Frontiers in nutrition 2023, 10, 1138102. [Google Scholar] [CrossRef]
- Zhan, H.; Xiong, Y.; Wang, Z.; Dong, W.; Zhou, Q.; Xie, S.; Li, X.; Zhao, S.; Ma, Y. , Integrative analysis of transcriptomic and metabolomic profiles reveal the complex molecular regulatory network of meat quality in Enshi black pigs. Meat science 2022, 183, 108642. [Google Scholar] [CrossRef]
- Wang, X.; Xu, R.; Tong, X.; Zeng, J.; Chen, M.; Lin, Z.; Cai, S.; Chen, Y.; Mo, D. , Characterization of different meat flavor compounds in Guangdong small-ear spotted and Yorkshire pork using two-dimensional gas chromatography–time-of-flight mass spectrometry and multi-omics. LWT 2022, 169, 114010. [Google Scholar] [CrossRef]
- Tamura, Y.; Iwatoh, S.; Miyaura, K.; Asikin, Y.; Kusano, M. , Metabolomic profiling reveals the relationship between taste-related metabolites and roasted aroma in aged pork. LWT 2022, 155, 112928. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Y.; Wu, Z.; Xiong, X.; Zhang, J.; Ma, J.; Xiao, S.; Huang, L.; Yang, B. , Subcutaneous and intramuscular fat transcriptomes show large differences in network organization and associations with adipose traits in pigs. Science China. Life sciences 2021, 64, 1732–1746. [Google Scholar] [CrossRef]
- Zhang, B.; Sun, Z.; Yu, Z.; Li, H.; Luo, H.; Wang, B. , Transcriptome and targeted metabolome analysis provide insights into bile acids’ new roles and mechanisms on fat deposition and meat quality in lamb. Food Research International 2022, 162, 111941. [Google Scholar] [CrossRef]
- Zequan, X.; Yonggang, S.; Heng, X.; Yaodong, W.; Xin, M.; Dan, L.; Li, Z.; Tingting, D.; Zirong, W. , Transcriptome-based analysis of early post-mortem formation of pale, soft, and exudative (PSE) pork. Meat science 2022, 194, 108962. [Google Scholar] [CrossRef]
- Fernández-Barroso, M.; García-Casco, J. M.; Núñez, Y.; Ramírez-Hidalgo, L.; Matos, G.; Muñoz, M. , Understanding the role of myoglobin content in Iberian pigs fattened in an extensive system through analysis of the transcriptome profile. Animal genetics 2022, 53, 352–367. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Song, Q. Q.; Wu, F.; Zhang, J. Z.; Xu, M. S.; Li, H. H.; Han, Z. J.; Gao, H. X.; Xu, N. Y. , Evaluation of the four breeds in synthetic line of Jiaxing Black Pigs and Berkshire for meat quality traits, carcass characteristics, and flavor substances. Animal science journal = Nihon chikusan Gakkaiho 2019, 90, 574–582. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N. S.; Wang, J. T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. , Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Leroy, G.; Baumung, R.; Boettcher, P.; Scherf, B.; Hoffmann, I. , Review: Sustainability of crossbreeding in developing countries; definitely not like crossing a meadow…. Animal : an international journal of animal bioscience 2016, 10, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chai, J.; Luo, Z.; He, H.; Chen, L.; Liu, X.; Zhou, Q. , Meat and nutritional quality comparison of purebred and crossbred pigs. Animal Science Journal 2018, 89, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Yang, Y.; Liao, K.; Liu, B.; Chen, Y.; Shen, L.; Chen, L.; Jiang, A.; Liu, Y.; Li, Q.; Wang, J.; Li, X.; Zhang, S.; Zhu, L. , Genetic parameter estimation for reproductive traits in QingYu pigs and comparison of carcass and meat quality traits to Berkshire×QingYu crossbred pigs. Asian-Australasian journal of animal sciences 2020, 33, 1224–1232. [Google Scholar] [CrossRef]
- Martins, J. M.; Fialho, R.; Albuquerque, A.; Neves, J.; Freitas, A.; Nunes, J. T.; Charneca, R. , Growth, blood, carcass and meat quality traits from local pig breeds and their crosses. Animal : an international journal of animal bioscience 2020, 14, 636–647. [Google Scholar] [CrossRef]
- Gandemer, G. , Lipids in muscles and adipose tissues, changes during processing and sensory properties of meat products. Meat science 2002, 62, 309–321. [Google Scholar] [CrossRef]
- Purcell, A. B.; Voss, B. J.; Trent, M. S. , Diacylglycerol Kinase A Is Essential for Polymyxin Resistance Provided by EptA, MCR-1, and Other Lipid A Phosphoethanolamine Transferases. Journal of bacteriology 2022, 204, e0049821. [Google Scholar] [CrossRef]
- Yu, J. E.; Han, S. Y.; Wolfson, B.; Zhou, Q. , The role of endothelial lipase in lipid metabolism, inflammation, and cancer. Histology and histopathology 2018, 33, 1–10. [Google Scholar] [PubMed]
- Clifford, B. L.; Sedgeman, L. R.; Williams, K. J.; Morand, P.; Cheng, A.; Jarrett, K. E.; Chan, A. P.; Brearley-Sholto, M. C.; Wahlström, A.; Ashby, J. W.; Barshop, W.; Wohlschlegel, J.; Calkin, A. C.; Liu, Y.; Thorell, A.; Meikle, P. J.; Drew, B. G.; Mack, J. J.; Marschall, H. U.; Tarling, E. J.; Edwards, P. A.; de Aguiar Vallim, T. Q. , FXR activation protects against NAFLD via bile-acid-dependent reductions in lipid absorption. Cell metabolism 2021, 33, 1671–1684. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Tanaka, N.; Sato, M.; Kang, D. W.; Krausz, K. W.; Flanders, K. C.; Ikeda, K.; Luecke, H.; Wakefield, L. M.; Gonzalez, F. J. , TGF-β-SMAD3 signaling mediates hepatic bile acid and phospholipid metabolism following lithocholic acid-induced liver injury. Journal of lipid research 2012, 53, 2698–2707. [Google Scholar] [CrossRef]
- Xu, Y.; Miller, P. C.; Phoon, C. K. L.; Ren, M.; Nargis, T.; Rajan, S.; Hussain, M. M.; Schlame, M. , LPGAT1 controls the stearate/palmitate ratio of phosphatidylethanolamine and phosphatidylcholine in sn-1 specific remodeling. Journal of Biological Chemistry 2022, 298, 101685. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Li, J. L.; Li, D.; Tobin, J. F.; Gimeno, R. E. , Molecular identification of microsomal acyl-CoA:glycerol-3-phosphate acyltransferase, a key enzyme in de novo triacylglycerol synthesis. Proceedings of the National Academy of Sciences of the United States of America 2006, 103, 19695–19700. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, M. C.; Saadoun, A. , An overview of the nutritional value of beef and lamb meat from South America. Meat science 2014, 98, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Blanco Canalis, M. S.; Baroni, M. V.; León, A. E.; Ribotta, P. D. , Effect of peach puree incorportion on cookie quality and on simulated digestion of polyphenols and antioxidant properties. Food chemistry 2020, 333, 127464. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, J.; Cui, L.; Ma, J.; Chen, C.; Ai, H.; Xie, X.; Li, L.; Xiao, S.; Huang, L.; Ren, J.; Yang, B. , Genetic architecture of fatty acid composition in the longissimus dorsi muscle revealed by genome-wide association studies on diverse pig populations. Genetics, selection, evolution : GSE 2016, 48, 5. [Google Scholar] [CrossRef]
- Nitta, S.; Kandori, S.; Tanaka, K.; Sakka, S.; Siga, M.; Nagumo, Y.; Negoro, H.; Kojima, T.; Mathis, B. J.; Shimazui, T.; Miyamoto, T.; Matsuzaka, T.; Shimano, H.; Nishiyama, H. , ELOVL5-mediated fatty acid elongation promotes cellular proliferation and invasion in renal cell carcinoma. Cancer Science 2022, 113, 2738–2752. [Google Scholar] [CrossRef]
- Centenera, M. M.; Scott, J. S.; Machiels, J.; Nassar, Z. D.; Miller, D. C.; Zinonos, I.; Dehairs, J.; Burvenich, I. J. G.; Zadra, G.; Chetta, P. M.; Bango, C.; Evergren, E.; Ryan, N. K.; Gillis, J. L.; Mah, C. Y.; Tieu, T.; Hanson, A. R.; Carelli, R.; Bloch, K.; Panagopoulos, V.; Waelkens, E.; Derua, R.; Williams, E. D.; Evdokiou, A.; Cifuentes-Rius, A.; Voelcker, N. H.; Mills, I. G.; Tilley, W. D.; Scott, A. M.; Loda, M.; Selth, L. A.; Swinnen, J. V.; Butler, L. M. , ELOVL5 Is a Critical and Targetable Fatty Acid Elongase in Prostate Cancer. Cancer Research 2021, 81, 1704–1718. [Google Scholar] [CrossRef]
- Agbaga, M. P.; Brush, R. S.; Mandal, M. N.; Henry, K.; Elliott, M. H.; Anderson, R. E. , Role of Stargardt-3 macular dystrophy protein (ELOVL4) in the biosynthesis of very long chain fatty acids. Proceedings of the National Academy of Sciences of the United States of America 2008, 105, 12843–12848. [Google Scholar] [CrossRef] [PubMed]
- Logan, S.; Agbaga, M.-P.; Chan, M. D.; Kabir, N.; Mandal, N. A.; Brush, R. S.; Anderson, R. E. , Deciphering mutant ELOVL4 activity in autosomal-dominant Stargardt macular dystrophy. Proceedings of the National Academy of Sciences 2013, 110, 5446–5451. [Google Scholar] [CrossRef] [PubMed]
- Lebeau, P. F.; Byun, J. H.; Platko, K.; Saliba, P.; Sguazzin, M.; MacDonald, M. E.; Paré, G.; Steinberg, G. R.; Janssen, L. J.; Igdoura, S. A.; Tarnopolsky, M. A.; Wayne Chen, S. R.; Seidah, N. G.; Magolan, J.; Austin, R. C. , Caffeine blocks SREBP2-induced hepatic PCSK9 expression to enhance LDLR-mediated cholesterol clearance. Nature communications 2022, 13, 770. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Zhao, Y.; Yu, W.; Melak, S.; Niu, Y.; Wei, W.; Zhang, L.; Chen, J. , ACAT2 Is a Novel Negative Regulator of Pig Intramuscular Preadipocytes Differentiation. Biomolecules 2022, 12. [Google Scholar] [CrossRef]
- Chistiakov, D. A.; Melnichenko, A. A.; Myasoedova, V. A.; Grechko, A. V.; Orekhov, A. N. , Mechanisms of foam cell formation in atherosclerosis. Journal of molecular medicine (Berlin, Germany) 2017, 95, 1153–1165. [Google Scholar] [CrossRef]
- Rogers, M. A.; Chang, C. C. Y.; Maue, R. A.; Melton, E. M.; Peden, A. A.; Garver, W. S.; Lee, J.; Schroen, P.; Huang, M.; Chang, T. Y. , Acat1/Soat1 knockout extends the mutant Npc1 mouse lifespan and ameliorates functional deficiencies in multiple organelles of mutant cells. Proceedings of the National Academy of Sciences of the United States of America 2022, 119, e2201646119. [Google Scholar] [CrossRef]
- Fujiwara, N.; Nakagawa, H.; Enooku, K.; Kudo, Y.; Hayata, Y.; Nakatsuka, T.; Tanaka, Y.; Tateishi, R.; Hikiba, Y.; Misumi, K.; Tanaka, M.; Hayashi, A.; Shibahara, J.; Fukayama, M.; Arita, J.; Hasegawa, K.; Hirschfield, H.; Hoshida, Y.; Hirata, Y.; Otsuka, M.; Tateishi, K.; Koike, K. , CPT2 downregulation adapts HCC to lipid-rich environment and promotes carcinogenesis via acylcarnitine accumulation in obesity. Gut 2018, 67, 1493–1504. [Google Scholar] [CrossRef]
- Toldra, F. , The role of muscle enzymes in dry-cured meat products with different drying conditions. Trends in Food Science & Technology 2006, 17, 164–168. [Google Scholar]
- Zhang, W. A.; Xiao, S.; Samaraweera, H.; Lee, E. J.; Ahn, D. U. , Improving functional value of meat products. Meat science 2010, 86, 15–31. [Google Scholar] [CrossRef]
- Kanai, Y.; Clémençon, B.; Simonin, A.; Leuenberger, M.; Lochner, M.; Weisstanner, M.; Hediger, M. A. , The SLC1 high-affinity glutamate and neutral amino acid transporter family. Molecular Aspects of Medicine 2013, 34, 108–120. [Google Scholar] [CrossRef]
- Eelen, G.; Dubois, C.; Cantelmo, A. R.; Goveia, J.; Brüning, U.; DeRan, M.; Jarugumilli, G.; van Rijssel, J.; Saladino, G.; Comitani, F.; Zecchin, A.; Rocha, S.; Chen, R.; Huang, H.; Vandekeere, S.; Kalucka, J.; Lange, C.; Morales-Rodriguez, F.; Cruys, B.; Treps, L.; Ramer, L.; Vinckier, S.; Brepoels, K.; Wyns, S.; Souffreau, J.; Schoonjans, L.; Lamers, W. H.; Wu, Y.; Haustraete, J.; Hofkens, J.; Liekens, S.; Cubbon, R.; Ghesquière, B.; Dewerchin, M.; Gervasio, F. L.; Li, X.; van Buul, J. D.; Wu, X.; Carmeliet, P. , Role of glutamine synthetase in angiogenesis beyond glutamine synthesis. Nature 2018, 561, 63–69. [Google Scholar] [CrossRef]
- Lee, S.-H.; Kim, S.; Kim, J.-M. , Genetic correlation between biopsied and post-mortem muscle fibre characteristics and meat quality traits in swine. Meat science 2022, 186, 108735. [Google Scholar] [CrossRef] [PubMed]
- Oda, M.; Satta, Y.; Takenaka, O.; Takahata, N. , Loss of Urate Oxidase Activity in Hominoids and its Evolutionary Implications. Molecular Biology and Evolution 2002, 19, 640–653. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Huang, Y.; Ji, J.; Xiao, S.; Ma, J.; Huang, L. , Effects of breeds, tissues and genders on purine contents in pork and the relationships between purine content and other meat quality traits. Meat science 2018, 143, 81–86. [Google Scholar] [CrossRef] [PubMed]
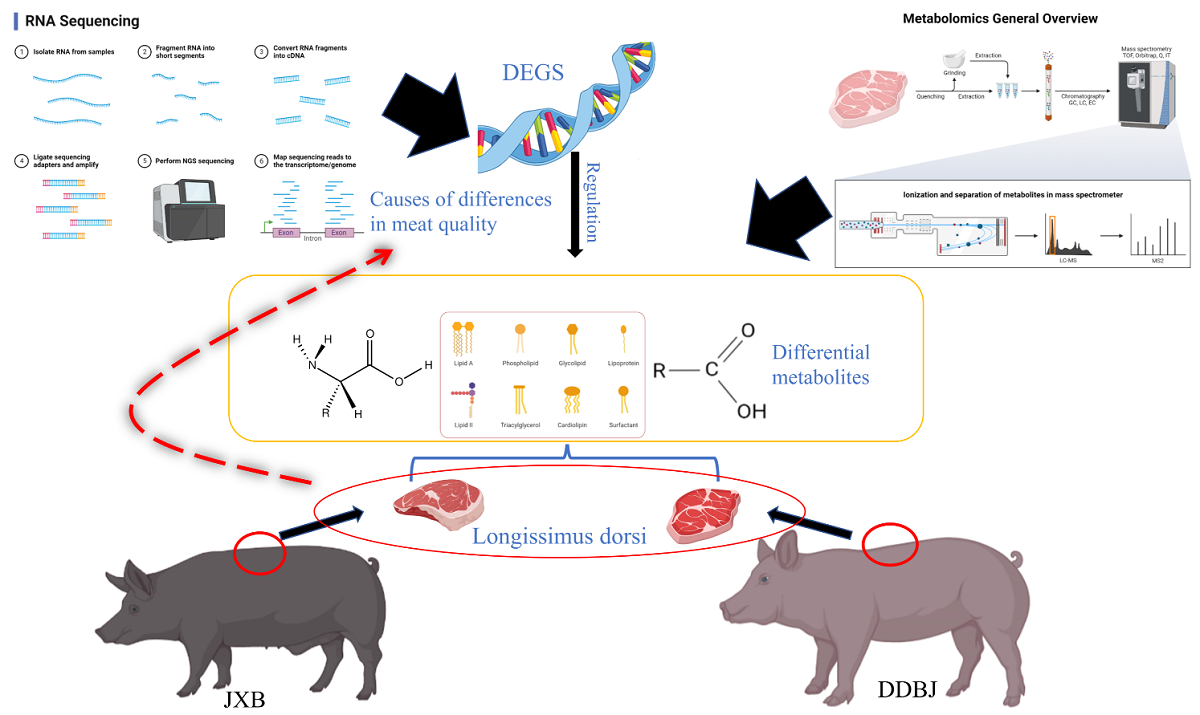
Figure 1.
Crossbreeding methods and meat quality indexes. (a). Diagram of the hybridization mode of Jiaxing black, Berkshire, and Duroc pigs to produce DDBJ pigs. The difference of inosinic monophosphate (IMP), free amino acid (FAA), cholesterol, intramuscular fat (IMF), crude protein (CP), and moisture in LD muscle between DDBJ pigs and JXB pigs; ** denotes significant difference at p < 0.01.
Figure 1.
Crossbreeding methods and meat quality indexes. (a). Diagram of the hybridization mode of Jiaxing black, Berkshire, and Duroc pigs to produce DDBJ pigs. The difference of inosinic monophosphate (IMP), free amino acid (FAA), cholesterol, intramuscular fat (IMF), crude protein (CP), and moisture in LD muscle between DDBJ pigs and JXB pigs; ** denotes significant difference at p < 0.01.
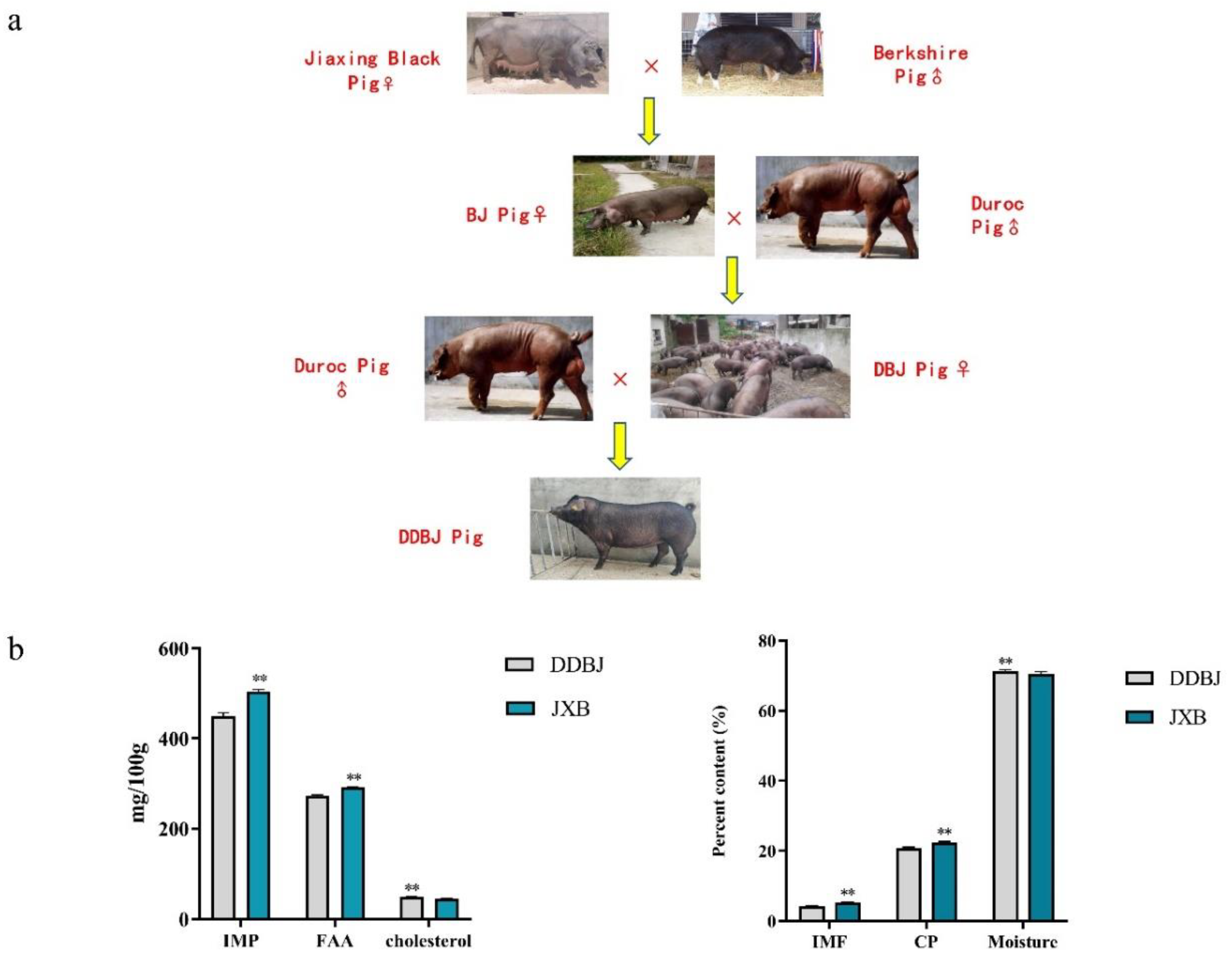
Figure 2.
Comparison of fatty acid and amino acid levels in LD muscle of DDBJ and JXB pigs, and KEGG enrichment analysis. (a). Comparison of 19 fatty acids in LD muscle of DDBJ and JXB pigs. SFA = saturated fatty acid; UFA = unsaturated fatty acid; MUFA = monounsaturated fatty acid; PUFA = polyunsaturated fatty acid; SFA = C14:0 + C16:0 + C17:0 + C18:0 + C20:0 + C22:0 + C24:0, UFA = C16:1 + C18:1 + C18:2 + C18:3n6 + C18:3n3 + C20:1 + C20:2 + C20:3n6 + C20:4n6 + C20:5 + C22:1 + C22:6 n3; MUFA = C16:1 + C18:1 + C20:1 + C22:1; PUFA = C18:2 + C18:3n6 + C18:3n3 + C20:3n6 + C20:4n6 + C20:5. (b). Comparison of 17 amino acid levels in LD muscle of DDBJ and JXB pigs. EAA = essential amino acids, EAAΣ = Thr + Val + Met + Ile + Leu; FFAA = fresh flavor amino acid, FFAAΣ = Ser + Glu + Pro + Gly + Ala, NEAA = non-essential amino acid, NEAA = Asp + Cys + Tyr, Σ= total amino acid. Asterisk denotes significant difference; * p < 0.05, ** p < 0.01. (c), (d). KEGG pathway enrichment, up- and downregulated differentially expressed genes (DEGs); shown are the first 20 pathways.
Figure 2.
Comparison of fatty acid and amino acid levels in LD muscle of DDBJ and JXB pigs, and KEGG enrichment analysis. (a). Comparison of 19 fatty acids in LD muscle of DDBJ and JXB pigs. SFA = saturated fatty acid; UFA = unsaturated fatty acid; MUFA = monounsaturated fatty acid; PUFA = polyunsaturated fatty acid; SFA = C14:0 + C16:0 + C17:0 + C18:0 + C20:0 + C22:0 + C24:0, UFA = C16:1 + C18:1 + C18:2 + C18:3n6 + C18:3n3 + C20:1 + C20:2 + C20:3n6 + C20:4n6 + C20:5 + C22:1 + C22:6 n3; MUFA = C16:1 + C18:1 + C20:1 + C22:1; PUFA = C18:2 + C18:3n6 + C18:3n3 + C20:3n6 + C20:4n6 + C20:5. (b). Comparison of 17 amino acid levels in LD muscle of DDBJ and JXB pigs. EAA = essential amino acids, EAAΣ = Thr + Val + Met + Ile + Leu; FFAA = fresh flavor amino acid, FFAAΣ = Ser + Glu + Pro + Gly + Ala, NEAA = non-essential amino acid, NEAA = Asp + Cys + Tyr, Σ= total amino acid. Asterisk denotes significant difference; * p < 0.05, ** p < 0.01. (c), (d). KEGG pathway enrichment, up- and downregulated differentially expressed genes (DEGs); shown are the first 20 pathways.
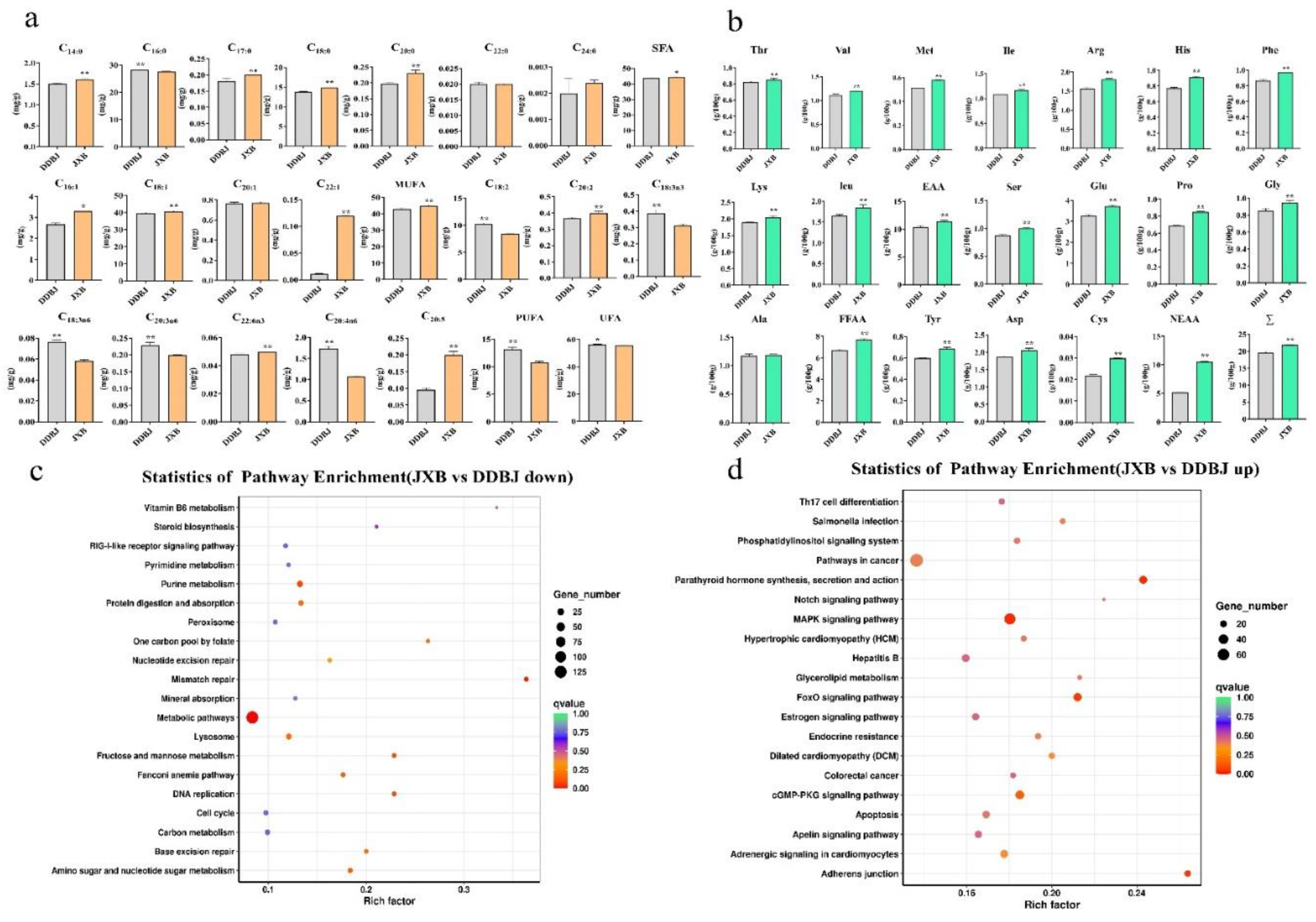
Figure 3.
Metabolic analysis results of LD muscle in DDBJ and JXB pigs (a). Grouping principal com ponent analysis; PC1 represents the first principal component, PC2 represents the second principal component, and the percentage represents the interpretation rate of the principal component to the dataset; each point in the figure represents a sample, samples of the same group are indicated by the same color, and “Group” is a group. (b). Differential metabolite scatter diagram, where each point represents a metabolite, and different colors represent distinct classifications. The abscissa denotes the logarithm (log2FC) of the relative content difference multiple of a substance in two sample groups. The size of the dot signifies the VIP value. (c). For the overall change analysis of the KEGG metabolic pathway, the ordinate represents the name of different pathways (sorted by p-values), and the abscissa indicates the DA score. The DA Score is the sum of all metabolite alterations throughout the metabolic pathway. An expression score of 1 shows an upregulation trend in the expression of all metabolites in this pathway, and a score of -1 signifies a downregulation trend in the expression of all of the detected metabolites within this pathway (d). Metabolite sets enrichment analysis; the ordinate highlights the name of the metabolic set (sorted by p-values), corresponding to the p-value of the metabolic set; the abscissa indicates fold enrichment.
Figure 3.
Metabolic analysis results of LD muscle in DDBJ and JXB pigs (a). Grouping principal com ponent analysis; PC1 represents the first principal component, PC2 represents the second principal component, and the percentage represents the interpretation rate of the principal component to the dataset; each point in the figure represents a sample, samples of the same group are indicated by the same color, and “Group” is a group. (b). Differential metabolite scatter diagram, where each point represents a metabolite, and different colors represent distinct classifications. The abscissa denotes the logarithm (log2FC) of the relative content difference multiple of a substance in two sample groups. The size of the dot signifies the VIP value. (c). For the overall change analysis of the KEGG metabolic pathway, the ordinate represents the name of different pathways (sorted by p-values), and the abscissa indicates the DA score. The DA Score is the sum of all metabolite alterations throughout the metabolic pathway. An expression score of 1 shows an upregulation trend in the expression of all metabolites in this pathway, and a score of -1 signifies a downregulation trend in the expression of all of the detected metabolites within this pathway (d). Metabolite sets enrichment analysis; the ordinate highlights the name of the metabolic set (sorted by p-values), corresponding to the p-value of the metabolic set; the abscissa indicates fold enrichment.
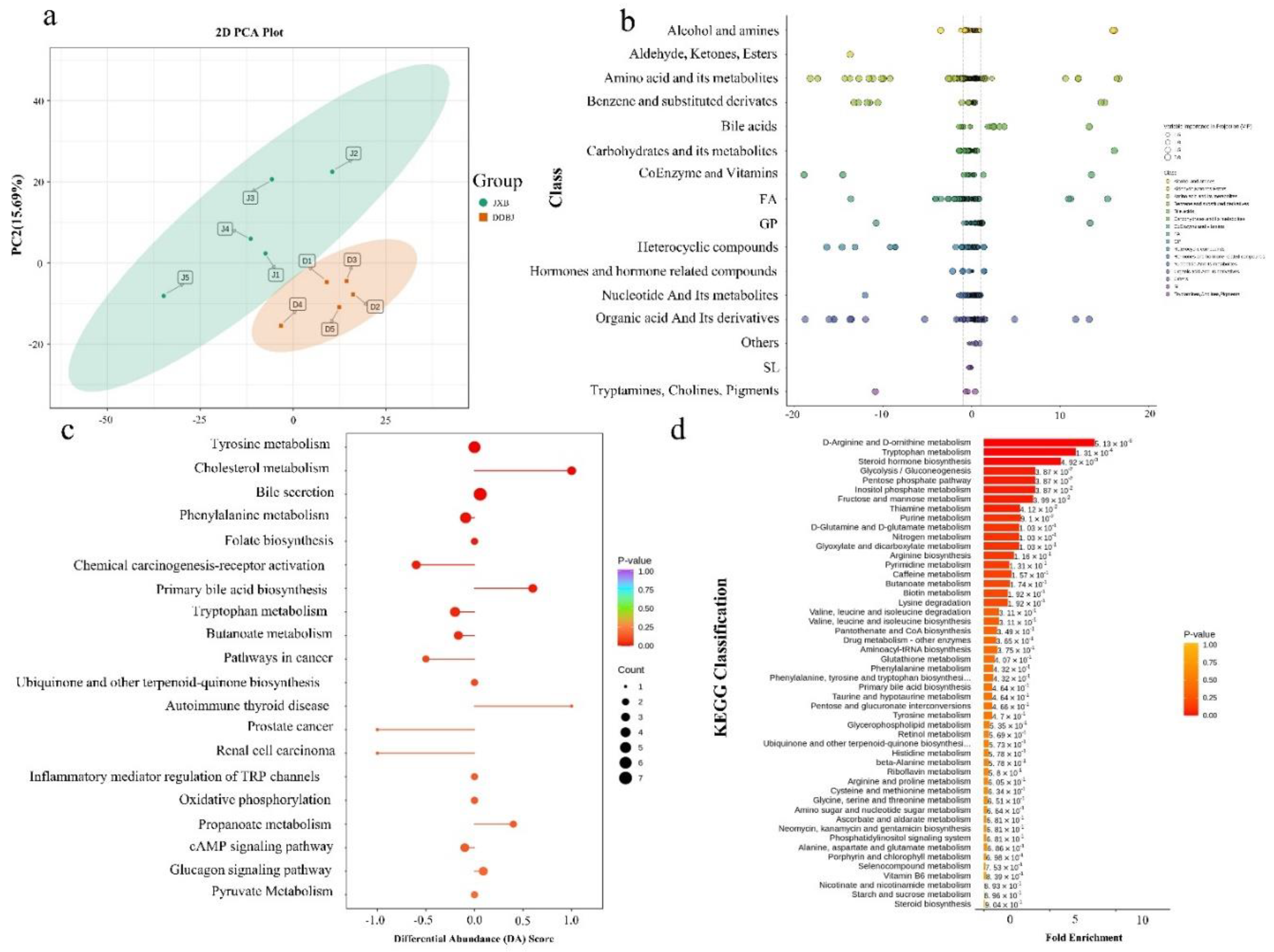
Figure 4.
The enrichment analysis of DEGs in meat quality regulation-related pathways and correlation analysis between DEGs and differential metabolites (Spearman correlation). (a), (b). Downregulation and upregulation of DEGs in KEGG enrichment pathway map; different colors represent different classified pathway sets, and bubble size indicates the number of DEGs. (c), (d) and (e). Correlation analysis of DEGs and differential metabolites. c, d: Spearman correlation analysis of DEGs with phospholipid metabolite and fatty acyl differential metabolite. e. Spearman correlation analysis of DEGs and differential metabolite such as vitamin coenzyme and carbohydrate related to sugar metabolism and vitamin metabolism, color depth represents correlation strength, positive and negative correlation are denoted by red and blue colors, correspondingly; asterisks denote the significant variation * p < 0.05, ** p < 0.01.
Figure 4.
The enrichment analysis of DEGs in meat quality regulation-related pathways and correlation analysis between DEGs and differential metabolites (Spearman correlation). (a), (b). Downregulation and upregulation of DEGs in KEGG enrichment pathway map; different colors represent different classified pathway sets, and bubble size indicates the number of DEGs. (c), (d) and (e). Correlation analysis of DEGs and differential metabolites. c, d: Spearman correlation analysis of DEGs with phospholipid metabolite and fatty acyl differential metabolite. e. Spearman correlation analysis of DEGs and differential metabolite such as vitamin coenzyme and carbohydrate related to sugar metabolism and vitamin metabolism, color depth represents correlation strength, positive and negative correlation are denoted by red and blue colors, correspondingly; asterisks denote the significant variation * p < 0.05, ** p < 0.01.
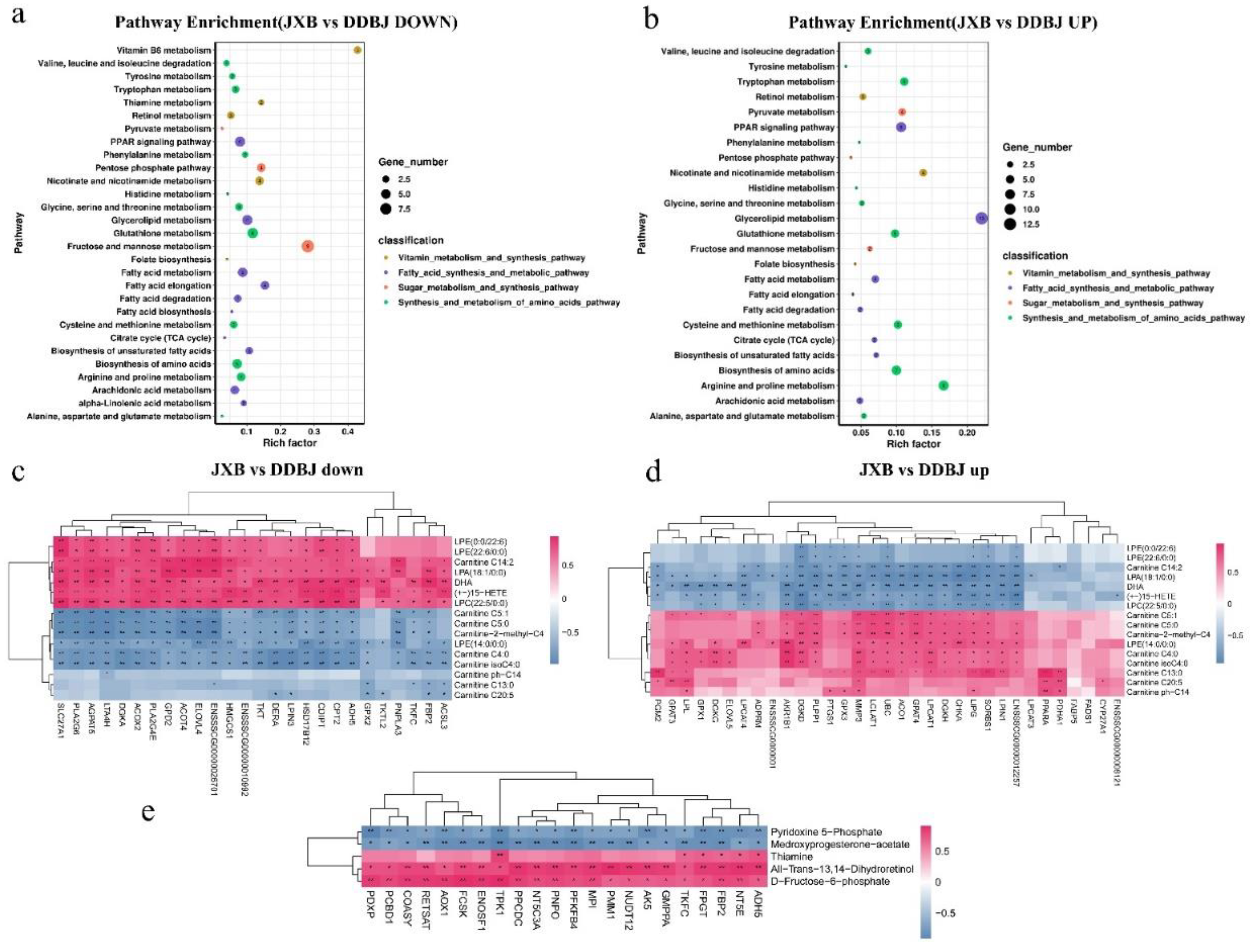
Figure 5.
Spearman correlation analysis of DEGs and small peptides in the pathway linked to amino acid synthesis and metabolism and the regulatory mechanism of compound metabolism. (a), (b). Spearman correlation analysis of up and downregulated DEGs and small peptides. (c). The regulation mechanism diagram of KEGG pathway enriched DEGs combined with phenotypic data and differential metabolites. Genes are indicated by rectangles (| log2 (fold change) | > log21, padj < 0.05), red and blue colors denote upregulated and downregulated genes, correspondingly; The yellow rectangle denotes the pathway, and the circle represents the differential metabolite (VIP > 1, | log2 (fold change) |>2, padj < 0.05); The yellow circle represents upregulation, green represents downregulation, the alternating yellow and green represents both up- and downregulation of such substances, the gray represents no detection, and the asterisk indicates significant differences; the dotted line represents an indirect reaction, while the solid line does not necessarily represent a direct reaction, but may also represent reaction conditions and directions. Abbreviations: SAM-e: s adenosylmethionine, BA: bile acid, TC: cholesterol, HDL: high-density lipoprotein, EMP: glycolysis, PA: phosphatic acid, FA: fatty acid, TAG: triglyceride, LPA: lysophosphatidic acid, LPG: lysophosphatidylcholine, LPE: lysophosphatidylethanolamine, TAG: triglyceride, DAGS: diacylglycerol, MAG: monoglyceride, GL: glycerol, G3P: 3, glycerol phosphate, P3G: triglyceride triphosphate, DHA: dihydroxyacetone phosphate, DF6P: 6, fructose phosphate, α- KGA: α- Ketoglutarate, E363: succinic acid, E297: fumaric acid, OAA: oxaloacetic acid, CA: citric acid EMP: glycolysis, PPP: pentose phosphate pathway, PC: phosphatidylcholine, PE: phosphatidylethanolamine.
Figure 5.
Spearman correlation analysis of DEGs and small peptides in the pathway linked to amino acid synthesis and metabolism and the regulatory mechanism of compound metabolism. (a), (b). Spearman correlation analysis of up and downregulated DEGs and small peptides. (c). The regulation mechanism diagram of KEGG pathway enriched DEGs combined with phenotypic data and differential metabolites. Genes are indicated by rectangles (| log2 (fold change) | > log21, padj < 0.05), red and blue colors denote upregulated and downregulated genes, correspondingly; The yellow rectangle denotes the pathway, and the circle represents the differential metabolite (VIP > 1, | log2 (fold change) |>2, padj < 0.05); The yellow circle represents upregulation, green represents downregulation, the alternating yellow and green represents both up- and downregulation of such substances, the gray represents no detection, and the asterisk indicates significant differences; the dotted line represents an indirect reaction, while the solid line does not necessarily represent a direct reaction, but may also represent reaction conditions and directions. Abbreviations: SAM-e: s adenosylmethionine, BA: bile acid, TC: cholesterol, HDL: high-density lipoprotein, EMP: glycolysis, PA: phosphatic acid, FA: fatty acid, TAG: triglyceride, LPA: lysophosphatidic acid, LPG: lysophosphatidylcholine, LPE: lysophosphatidylethanolamine, TAG: triglyceride, DAGS: diacylglycerol, MAG: monoglyceride, GL: glycerol, G3P: 3, glycerol phosphate, P3G: triglyceride triphosphate, DHA: dihydroxyacetone phosphate, DF6P: 6, fructose phosphate, α- KGA: α- Ketoglutarate, E363: succinic acid, E297: fumaric acid, OAA: oxaloacetic acid, CA: citric acid EMP: glycolysis, PPP: pentose phosphate pathway, PC: phosphatidylcholine, PE: phosphatidylethanolamine.
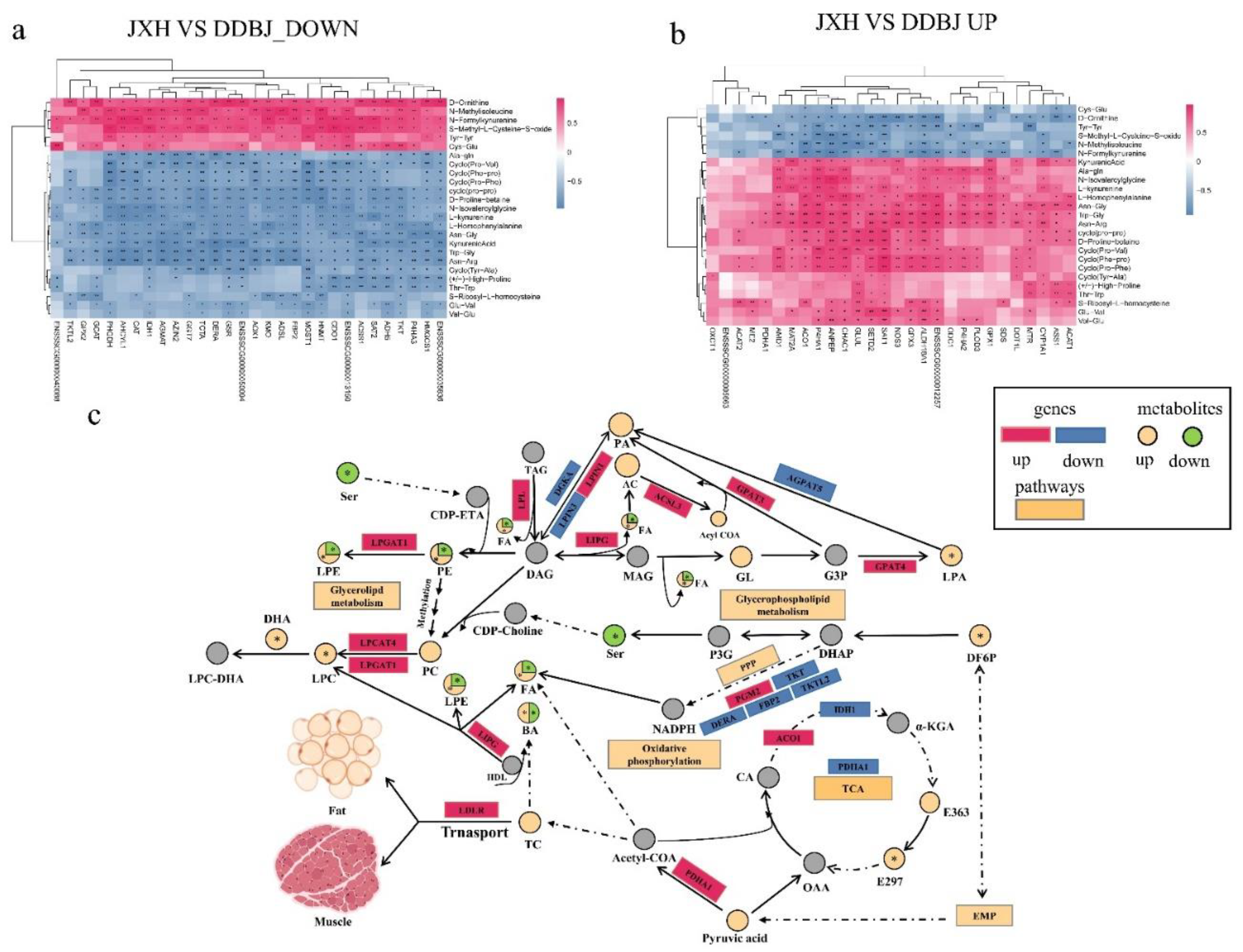
Figure 6.
Regulation mechanism diagram of DEGs and differential metabolites (fats and amino acids). (a). Amino acid anabolism regulation network diagram. (b). Fatty acid anabolism regulation diagram. AA: argininosuccinic acid. The yellow rectangle represents the regulatory pathway, the red and blue rectangles denote the upregulated and downregulated genes, correspondingly; the yellow and green circles denote the upregulated and downregulated differential metabolites, correspondingly, and the gray signifies the undetected metabolite.
Figure 6.
Regulation mechanism diagram of DEGs and differential metabolites (fats and amino acids). (a). Amino acid anabolism regulation network diagram. (b). Fatty acid anabolism regulation diagram. AA: argininosuccinic acid. The yellow rectangle represents the regulatory pathway, the red and blue rectangles denote the upregulated and downregulated genes, correspondingly; the yellow and green circles denote the upregulated and downregulated differential metabolites, correspondingly, and the gray signifies the undetected metabolite.
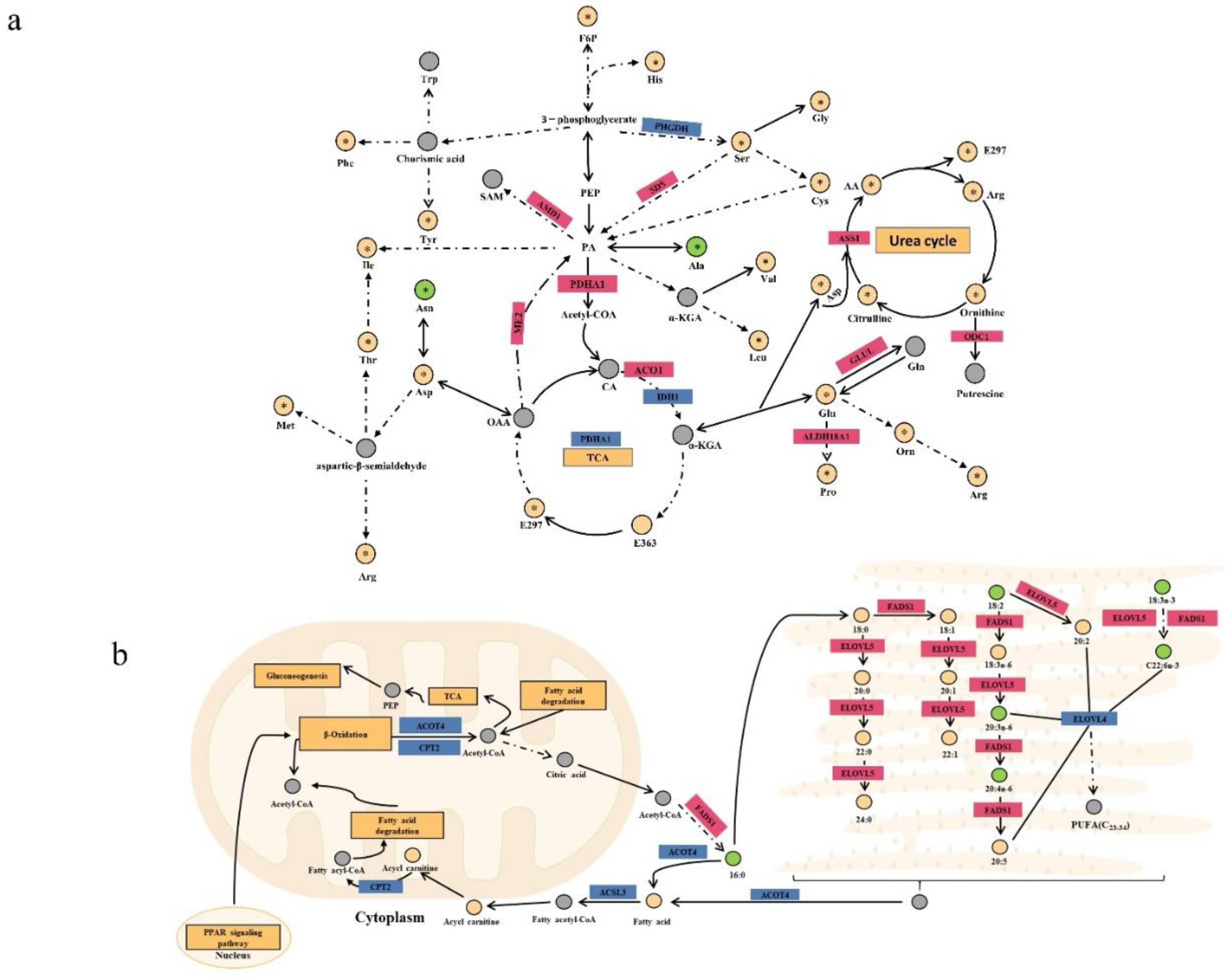
Figure 7.
Network diagram of DEGs and differential metabolites. (a). With p < 0.05, | log2 (fold change)|> 2 as the screening condition, the discovered differential genes included 217 upregulated and 227 downregulated genes. (b). The specific correlation coefficient and p-value of the differential gene metabolite correlation network are shown in the supply tables. (c). According to correlation analysis, four target genes and their related networks with metabolites were selected. Small circles represent differential genes, blue and red denote downregulated and upregulated DEGs, correspondingly, the hexagon represents differential metabolites, and different colors represent different types of differentials.
Figure 7.
Network diagram of DEGs and differential metabolites. (a). With p < 0.05, | log2 (fold change)|> 2 as the screening condition, the discovered differential genes included 217 upregulated and 227 downregulated genes. (b). The specific correlation coefficient and p-value of the differential gene metabolite correlation network are shown in the supply tables. (c). According to correlation analysis, four target genes and their related networks with metabolites were selected. Small circles represent differential genes, blue and red denote downregulated and upregulated DEGs, correspondingly, the hexagon represents differential metabolites, and different colors represent different types of differentials.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



