
Submitted:
10 August 2023
Posted:
11 August 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Covid-19 is one of humanity’s biggest threat in the 21st century with WHO figures reporting 636 million cases and up to 6.6 million deaths globally. SARS-CoV-2, the virus that causes the Covid-19 disease, is characterized by high mutation which contributes to its rapid spread. While several vaccines have been produced to minimize the severity of the coronavirus and diverse treatment regimens have been approved by the US FDA under Emergency Use Authorization (EUA), SARS-CoV-2 viral mutations continue to derail the efforts of scientists as the emerging variants evade the recommended therapies. Nonetheless, diverse computational models exist that offer an opportunity to overcome the barriers involved in developing new drugs. In this review, the focus is on the use of various virtual screening techniques like molecular docking, molecular dynamics simulations, QSAR, pharmacophore modeling, and homology modeling in repurposing SARS-CoV-2 therapeutics. The results have been promising from the computer-aided drug design (CADD) studies in suggesting potential compounds for treatment of Covid-19 and helping in bringing the pandemic under control.
Keywords:
Drug Discovery
; Drug Repurposing
; SARS-CoV-2
; COVID
; Molecular Docking
; QSAR
; Molecular Dynamic
; Virtual Screening
; Drug Design
1. Introduction
The Covid-19 pandemic still remains the world’s most serious public health threat since it first broke out in late 2019. Presently, the World Health Organization (WHO) coronavirus disease dashboard reports that the disease has resulted in more than 636 million infections and 6.6 million deaths [1]. The highly mutated nature of the coronavirus, which is an RNA class virus, means that SARS-CoV-2 changes at a rapid speed, thereby contributing to the significant threat the disease poses to mankind. The mutations in the virus have represented a monumental challenge to scientists as this means perpetually altering the treatment guidelines to display the most efficacious therapies [2]. This highly contagious viral illness continues to derail the substantial progress in clinical research towards a better understanding of SARS-CoV-2 as many countries still witness increased outbreaks attributed to the increasingly emerging mutant variants of the virus. Consequently, extensive global studies are still underway as researchers rush against time to find appropriate therapeutic agents to treat Covid-19 as the world still fights to overcome the pandemic.
To date, Covid-19 primary therapies are antivirals, anti-inflammatories, and respiratory treatments, with additional antibody therapies being an active and essential part of treating SARS-CoV-2 infection [3]. The first medication to be approved by the United States (US) Food and Drug Administration (FDA) for the treatment of Covid-19 was Remdesivir. It was approved on April 25, 2022, for treating both adults and children [4]. This medication was identified as a potential candidate that could be a therapeutic agent for Covid-19 owing to its inhibitory ability against SARS-CoV-2 in vitro [5]. Studies conducted on SARS-CoV-2 infected monkeys revealed that early administration of remdesivir significantly reduced the viral load and pulmonary damage; while studies from the National Institute for Allergy and Infectious Diseases (NIAID) and SIMPLE studies reenforced the FDA decision to issue an emergency use authorization (EUA) on May 1st, 2022 for severe hospitalized Covid-19 patients [6,7,8,9,10]. Other drugs approved by the FDA are the immune modulators baricitinib (Olumiant) and tocilizumab (Actemra) for certain adults hospitalized with Covid-19, and the oral antiviral pill Paxlovid (nirmatrelvir and ritonavir) (Figure 1) [11,12]. The use of computational software in the discovery of more efficacious therapeutic methods may prove to be useful in discovering novel drugs and drug combinations [13].
Figure 1.
Structure of Remdesivir, Baricitnib, Nirmatrelvir, and Ritonavir.
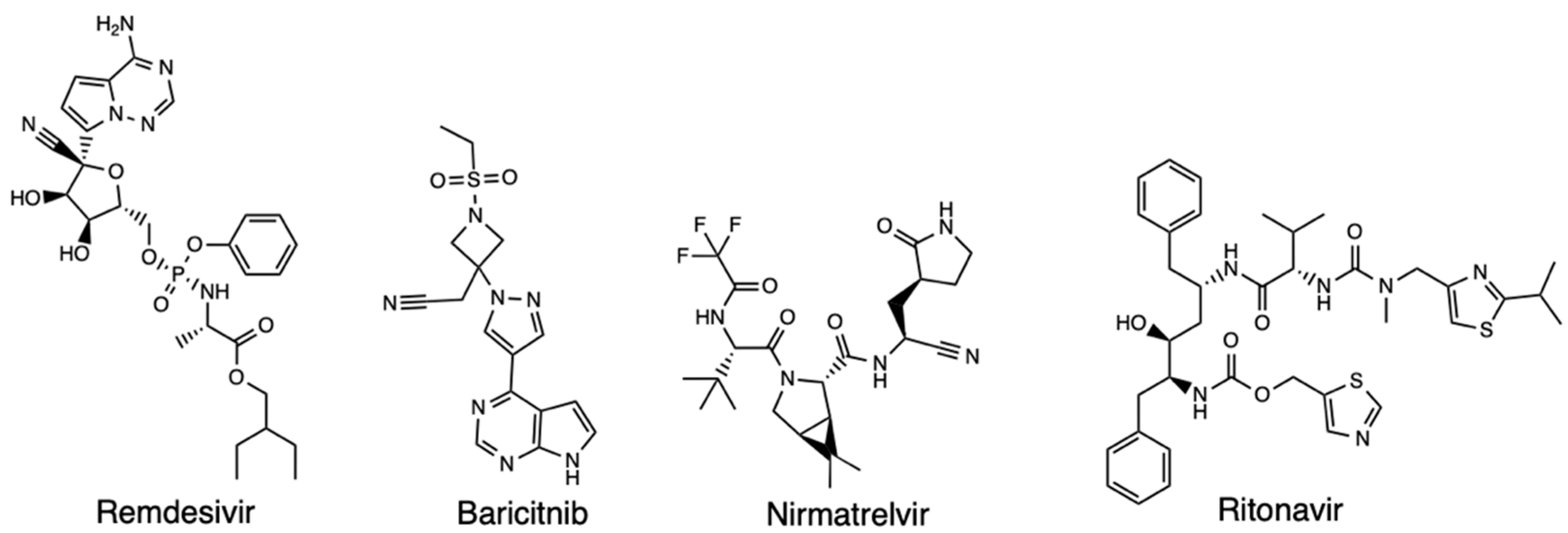
Since developing new drugs is time consuming, expensive, and limited by other factors such as lack of predictive ability of current animal models, the ability for drug repurposing and rediscovery provides a vital system in a health emergency such as coronavirus. Computational tools and the use of in silico methods through computer-aided drug design (CADD) has proven useful in predicting a drug’s quantum chemical properties efficiently. Through prioritizing potentially effective small molecules using techniques like molecular docking simulations and network-based repurposing [14,15]. Indeed, docking has been applied in proposing the efficacy of statins, with molecular docking aiding in identifying remdesivir and proposing it as a potentially effective drug due to its ability to target the RNA-dependent RNA polymerase (RdRp) [16,17]. Hence, exploring the use of computational software in finding Covid-19 therapeutics is a worthy cause as these tools provide insights about related molecular mechanisms as well as classifying drugs and predicting target specificity.
2. Computational Discovery of Covid-19 Therapeutics
Although several vaccines have been developed and approved to manage the spread of SARS-CoV-2, the highly mutated nature of the coronavirus as well as evidence of breakthrough infections by the different variants of the virus have necessitated the use of the advanced computational methods that are becoming increasingly powerful in biomedical research and pharmaceutical drug discovery [18]. Researchers have carried out intensive and extensive research on the coronavirus proteins and three-dimensional (3D) structures of the major SARS-CoV-2 proteins resolved and deposited in the Protein Data Bank (PDB). These structures provide a useful foundation for discovering and designing drugs based on structure-based computations (structure-based drug design [SBDD]) [19]. There are also ligand-based drug design (LBDD) methods that target the main protease (Mpro), such as pharmacophore modeling, quantitative structure-activity relationships (QSARs), and homology modeling, all of which have been useful in screening potential candidates for treating Covid-19 [20,21]. In this review, the focus is on how various CADD methods have been used and the drugs that have been developed and repurposed through virtual screening (VS) for use as SARS-CoV-2 therapeutics.
2.1. Molecular Docking
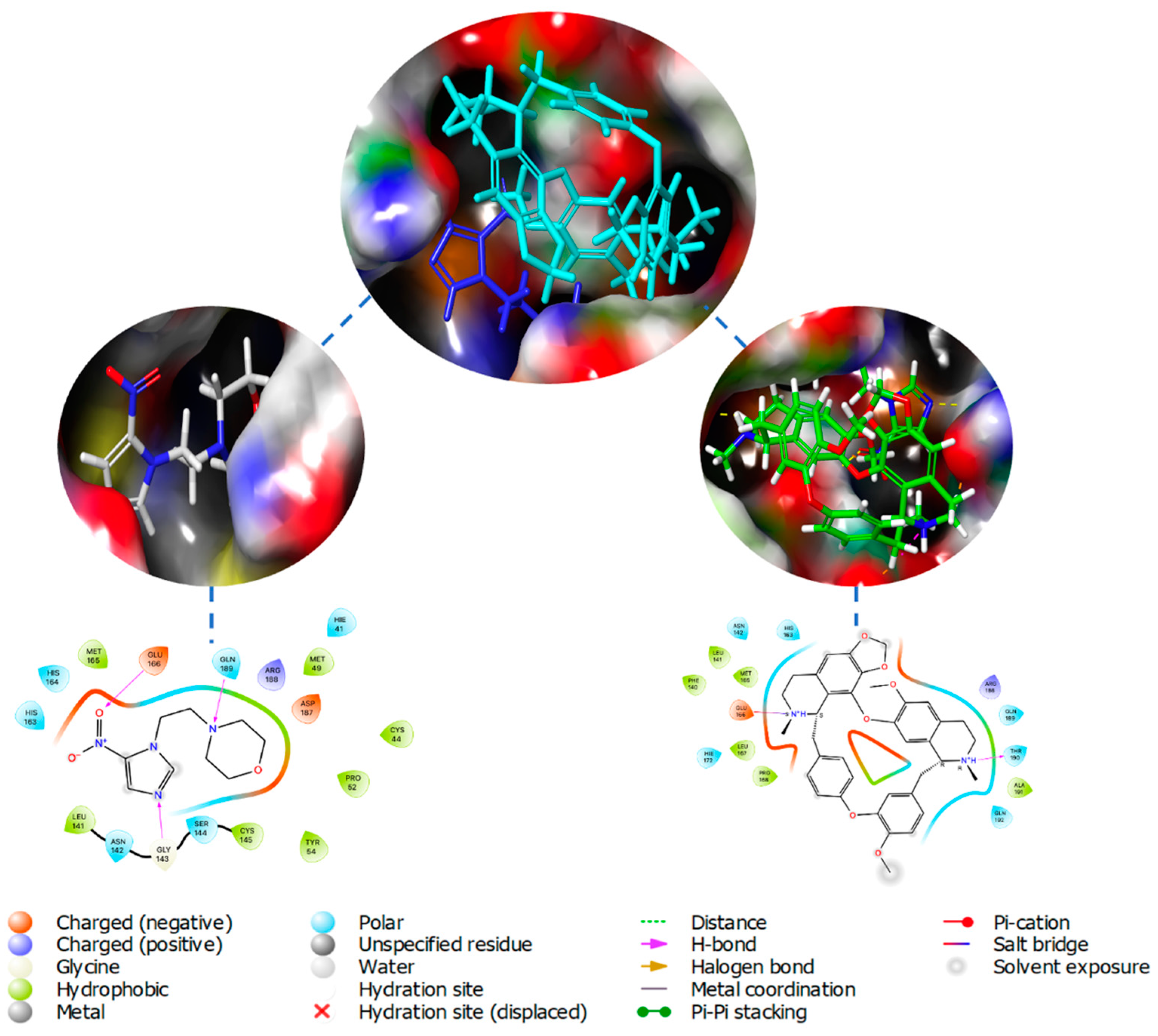
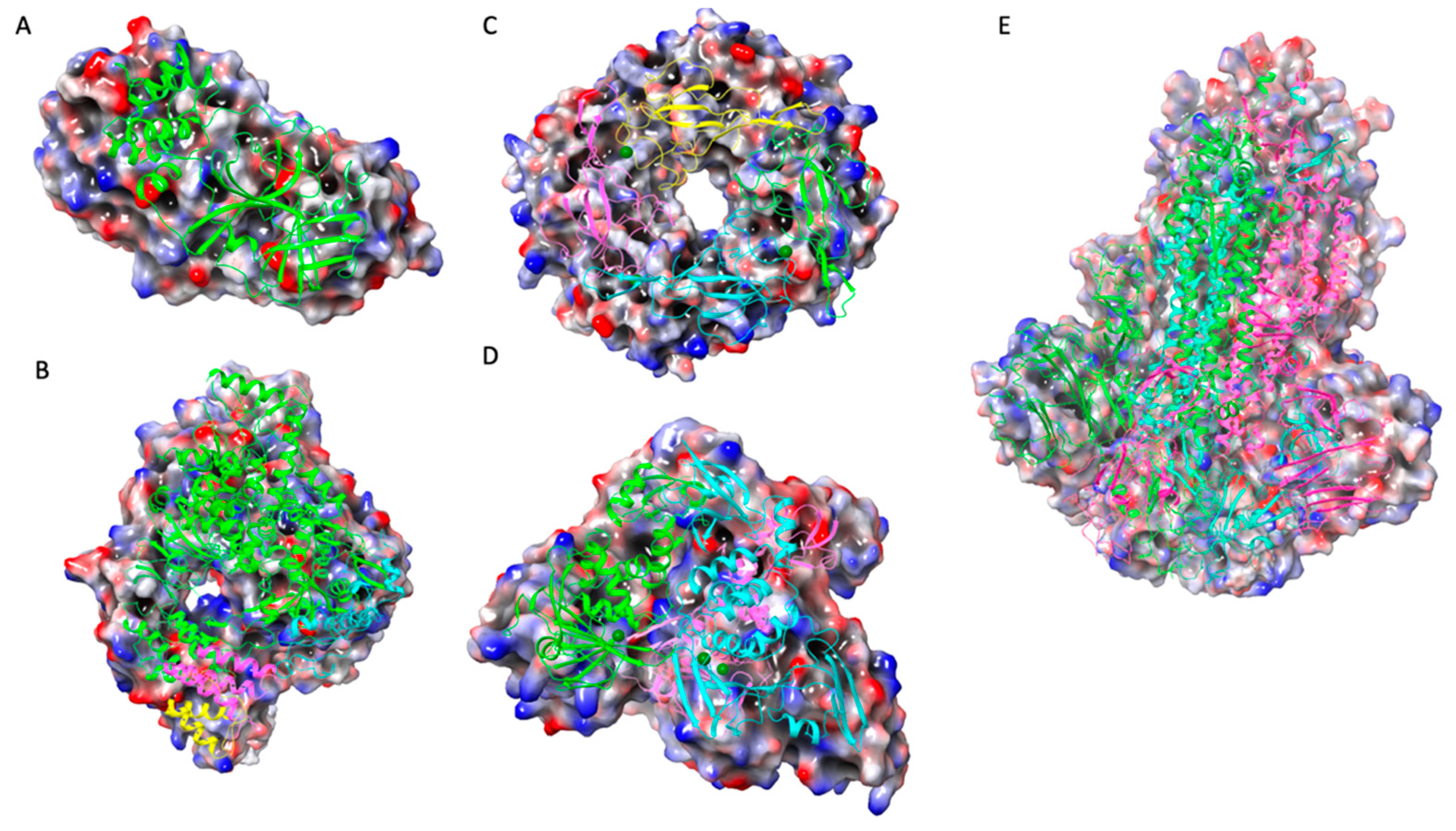
Following the sequencing of the SARS-CoV-2 genome, it became possible to identify and characterize the proteins of the virus, with Mpro identified as a potential therapeutic target due to its essential nature for the processing of other viral proteins. The description of the crystal structures of Mpro and inhibitors opened further opportunities to describe additional compounds inhibiting these proteins, with a molecular docking-based virtual screening (VS) performed against a library of experimental and approved drugs [22]. In this screening, the top 10 hits listed included pictilisib, Ergoloid mesylates, Cefuroxime, Nimorazole, Lumacaftor, Nilotinib, and Cepharanhine (Figure 2). The procedure entailed in the docking analysis for the Mpro structures PDB 6Y2E and 6LU7, after which molecular docking was performed and the 20 best conformations for each of the ligands inside the Mpro active cavity were analyzed with the ligand-protein interactions. The findings determined a higher binding affinity than the identified compounds for Mpro and could significantly control SARS-CoV-2 replication. Cephraranthine and Nimorazole (Figure 3) were supported by literature for further in vitro and in vivo evaluations as potential drugs, with pictilisib predicted to have the highest affinity for Mpro, followed by Nimorazole and Ergoloid mesylates.
Figure 2.
Structure of pictilisib, Ergoloid mesylates, Cefuroxime, Nimorazole, Lumacaftor, Nilotinib, and Cepharanhine.
Figure 2.
Structure of pictilisib, Ergoloid mesylates, Cefuroxime, Nimorazole, Lumacaftor, Nilotinib, and Cepharanhine.
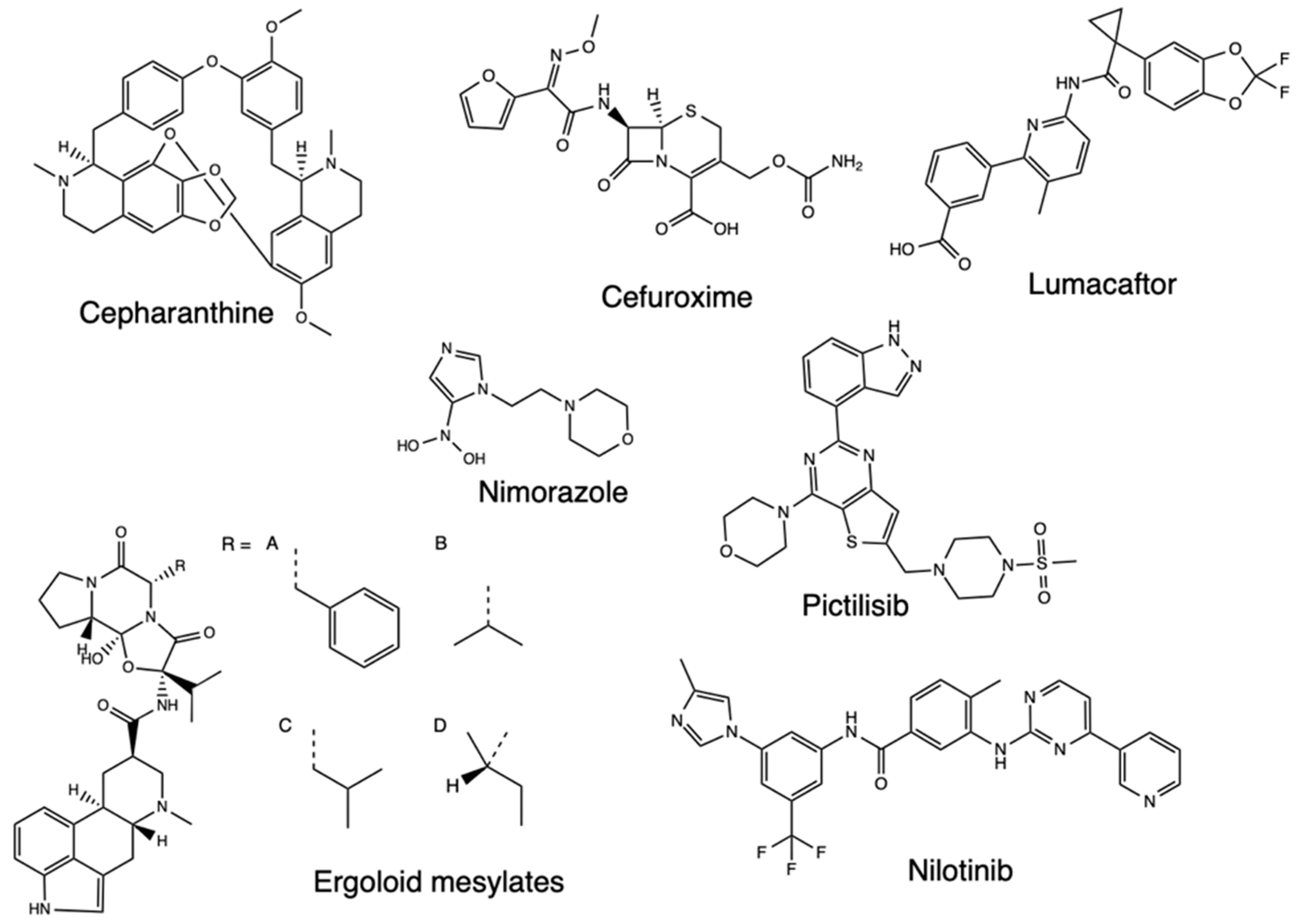
Figure 3.
Center Top: Overlay of docked Nimorazole (Blue) and Cephraranthine (Cyan) inside binding pocket of Mpro (PDB 6LU7); Left: Nimorazole inside binding pocket and ligand-protein interaction diagram; Right: Cephraranthine inside binding pocket and ligand-protein interaction diagram.
Figure 3.
Center Top: Overlay of docked Nimorazole (Blue) and Cephraranthine (Cyan) inside binding pocket of Mpro (PDB 6LU7); Left: Nimorazole inside binding pocket and ligand-protein interaction diagram; Right: Cephraranthine inside binding pocket and ligand-protein interaction diagram.
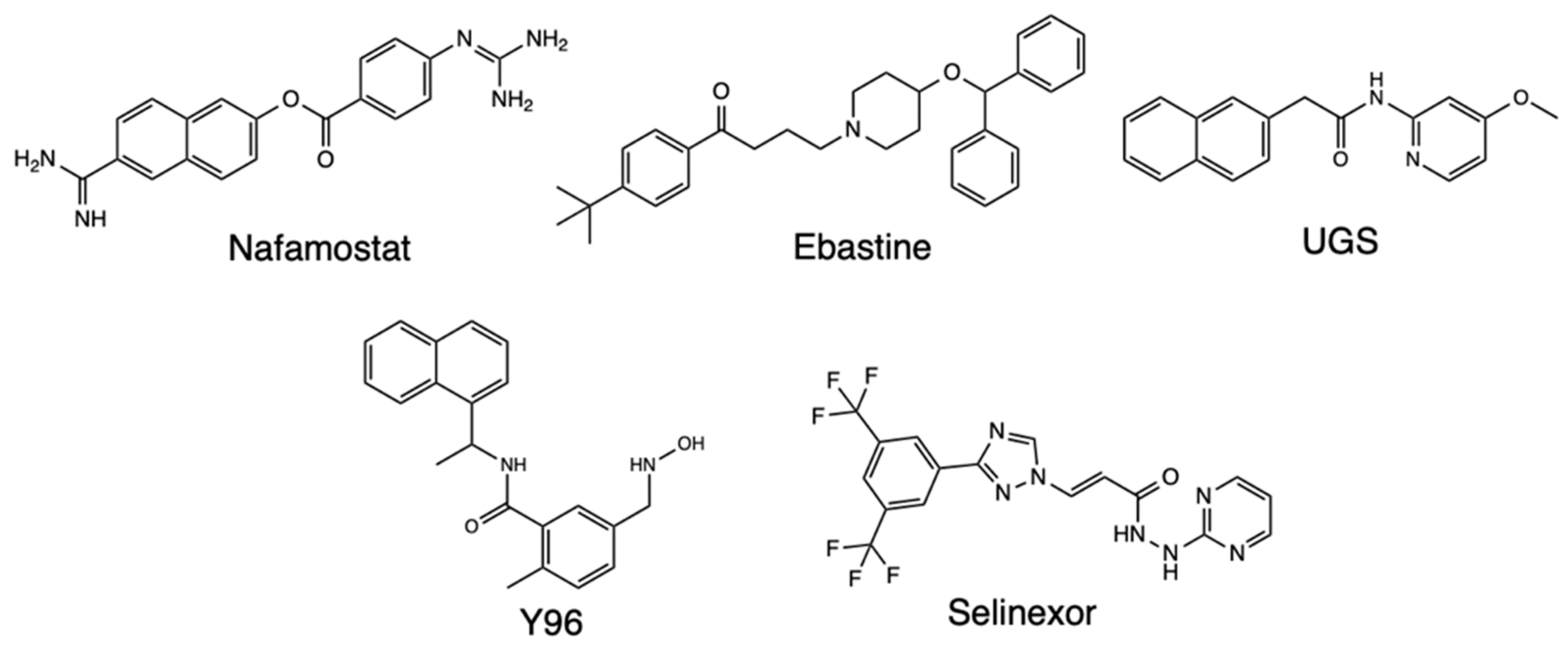
Molecular docking that targets the transmembrane receptor Neuropilin-1 (NRP-1) due to its role as a host cell entry factor for SARS-CoV-2 that causes the Covid-19 disease has also been performed. One such study recognized that molecular compounds capable of interfering with SARS-CoV-2 binding to NRP-1 are potential antiviral drug candidates, based on a library of 1167 compounds which had been analyzed previously in Covid-related studies [23] (Figure 4). The virtual screening analysis procedure was carried out on AutoDock Vina targeting a library of compounds to find potent NRP-1 receptor inhibitors. Consequently, the Nafamostat drug was found to have the highest binding affinity to the NRP-1 protein as its binding energy was -8.6 kcal/mol. It also showed that the drug is stabilized by four hydrogen bonds, and is the second ranking compound with a binding energy of -8.4 kcal/mol were Y96 and Selixenor. The other ligands found to have favorable binding energies were Ebastine and UGS, with all the top five chemicals depicting better binding affinities than the reference compounds EG00229 and EG01377 (Figure 5). Hence, the screened compounds showed potential as good candidates for inhibiting the interaction between the spike (S) protein and NRP-1 receptor.
Figure 4.
PDB 2QQI of NRP-1 receptor visualized (A); Natamostat (B), Y96 (C), and Selinexor (D) visualized inside binding pocket; Natamostat (E), Y96 (F), and Selinexor (G) ligand protein interaction.
Figure 4.
PDB 2QQI of NRP-1 receptor visualized (A); Natamostat (B), Y96 (C), and Selinexor (D) visualized inside binding pocket; Natamostat (E), Y96 (F), and Selinexor (G) ligand protein interaction.
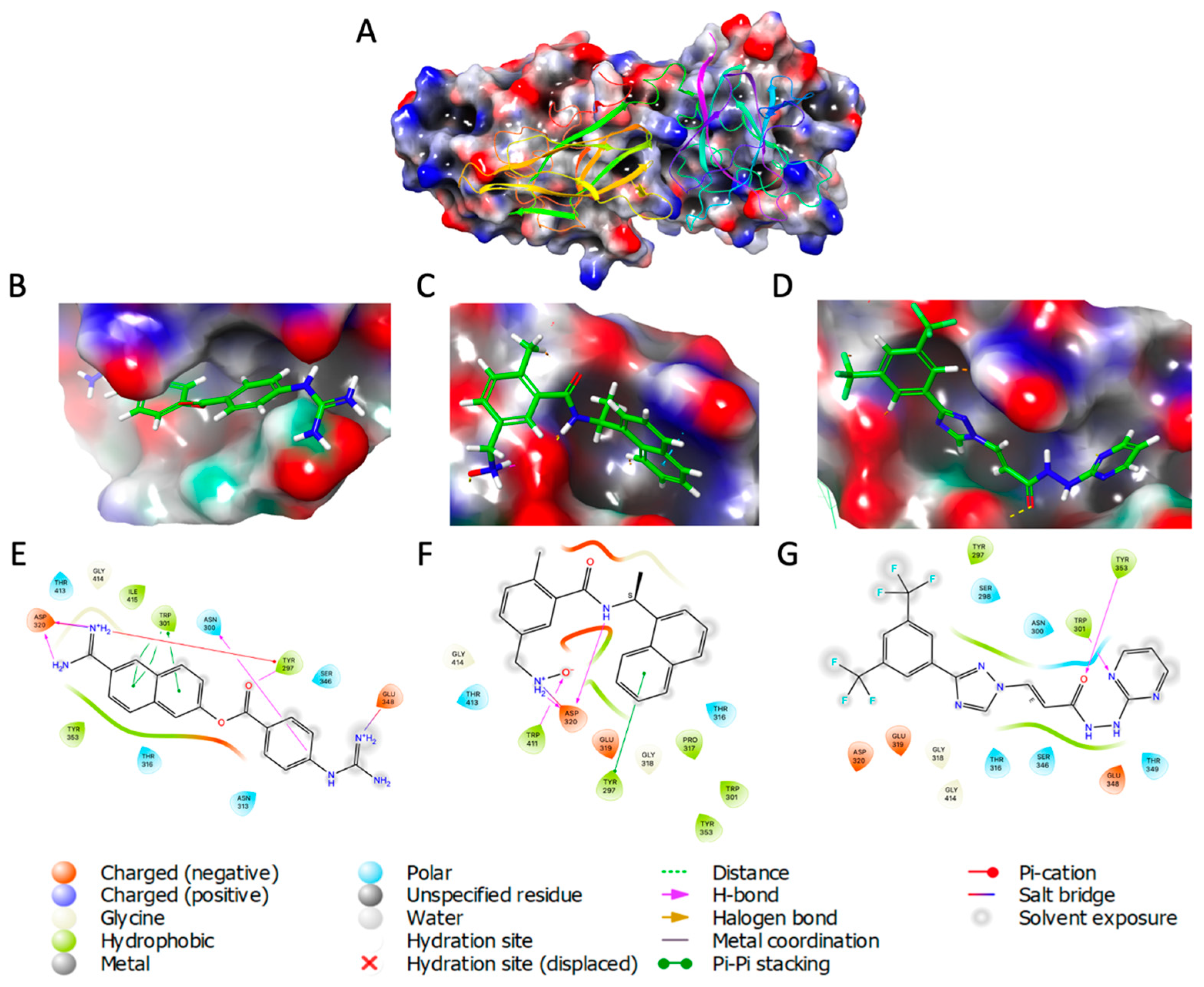
Figure 5.
Structure of Nafamostat, Ebastine, UGS, Y26, and Selinexor.
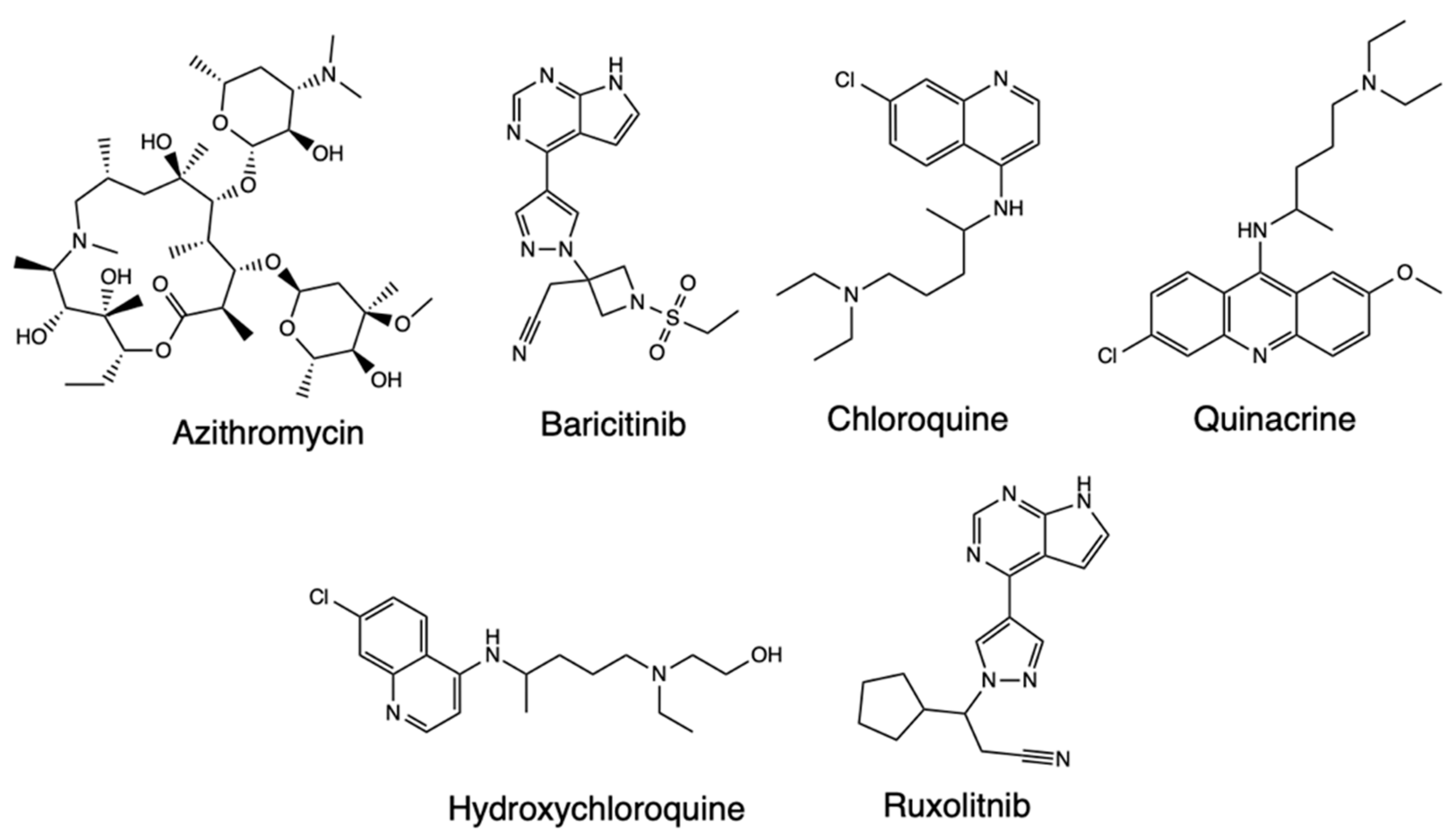
In silico molecular docking studies have also been performed to evaluate the molecular interactions of drugs with therapeutic indications for Covid-19 treatment, such as Azithromycin, Hydroxychloroquine, and Baricitinib, and similarly structured drug compounds like chloroquine, Quinacrine, and Ruxolitinib based on docking from the Mpro protein [24] (Figure 6). The inhibitors and the SARS-CoV-2 main protease were docked and the most favorable ones were represented by the lowest free-bond energy and the molecule’s preference for the same bond site. From the analysis of the molecular simulations, it was evident all inhibitors are linked to the same enzyme site, more isolated in Mpro’s domain III, and distant in the binding site of the N3 protease inhibitor. Azithromycin showed six interactions with Covid-19, one being the conventional hydrogen bond type with Leu272 and one of the alkyl type with Leu287 amino acids. Meanwhile, the receptor-ligand complex formed with baricitinib exhibited ten interactions with the amino acid residues of the enzymes, three conventional hydrogen bond types, and one each with Lys137, Asp197, and Leu287. The chloroquine inhibitor also interacted with Mpro, while hydroxychloroquine and chloroquine did not show adequate binding energy with the enzyme. The docking routines further indicated that quinacrine formed three interactions with the target protein, and ruxolitinib also interacted with the enzyme. Overall, the molecular docking simulations identified the formation of nine hydrogen bonds with the analyzed inhibitors, which were classified as hydrogen bonds covalent with chloroquine, baricitinib, and quinacrine while being moderate and mostly electrostatic with azithromycin, hydroxychloroquine, and ruxolitinib. Thus, baricitinib, quinacrine, and azithromycin were proposed as capable of being used alone or as combined therapeutics for Covid-19.
Figure 6.
Structure of Azithromycin, Baricitinib, Chloroquine, Quinacrine, Hydroxychloroquine, and Ruxolitnib.
Figure 6.
Structure of Azithromycin, Baricitinib, Chloroquine, Quinacrine, Hydroxychloroquine, and Ruxolitnib.
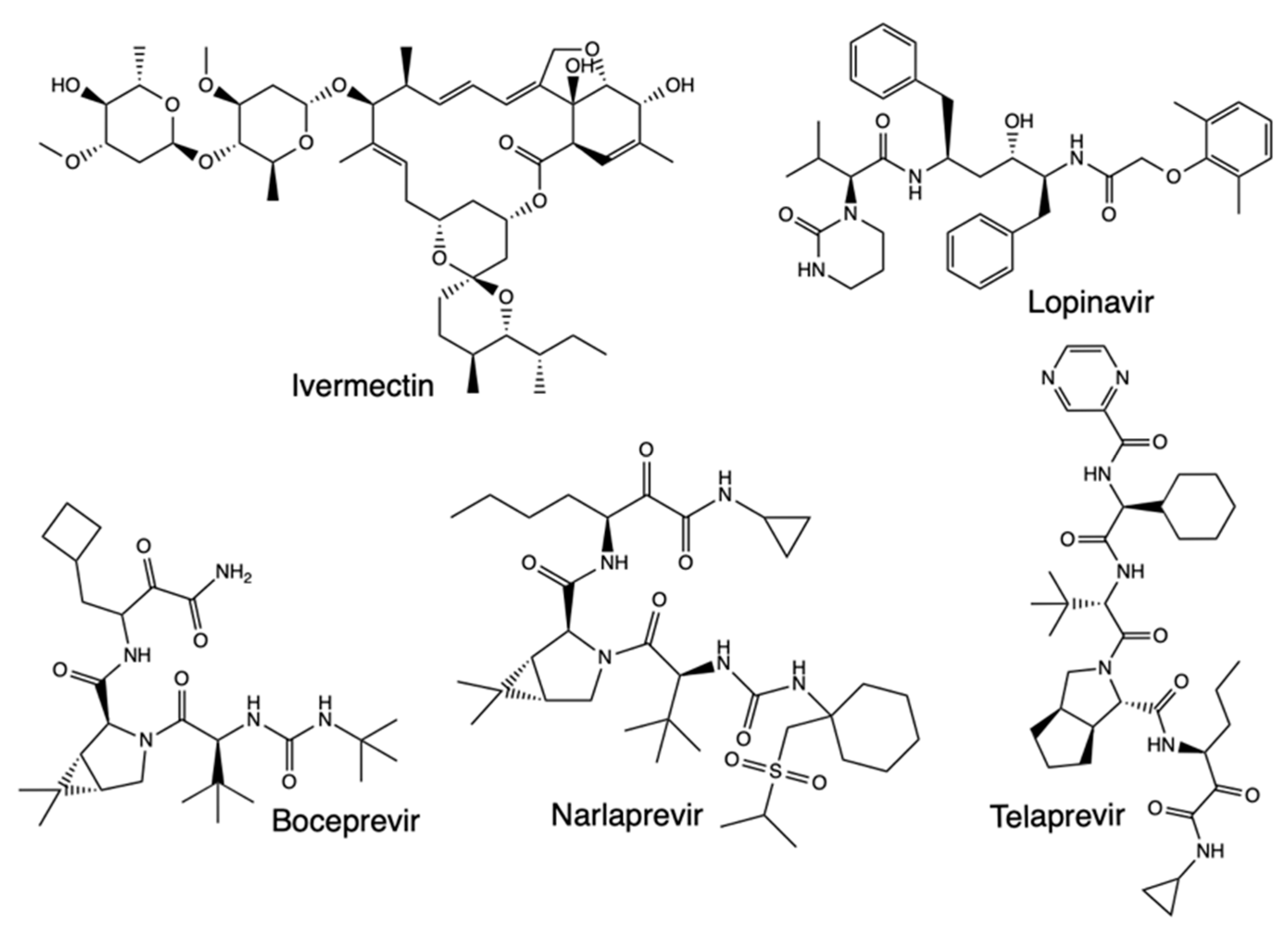
An earlier molecular docking study was done for repurposed therapeutics such as chloroquine, ivermectin, remdesivir, hydroxychloroquine, and favipiravir. They were screened with different SARS-CoV-2 target proteins including RdRp, spike (S protein) proteins, viral proteases, nucleoproteins, membrane protein (M) protein), nucleocapsid protein (N protein), and nsp14 (exoribonuclease) (Figure 7) [25]. After retrieving the proteins, molecular docking was performed on the MVD 6.0 platform and the potential binding sites (cavities) identified with the detection algorithm. The results indicated ivermectin had the highest binding affinity to the predicted active site of the S glycoprotein and the protein-ligand interactions, while favipiravir had the lowest binding affinity and protein-ligand interactions, while remdesivir showed considerable outcomes. As for the binding interactions with RdRp, remdesivir had the highest binding affinity and protein-ligand interactions, and ivermectin showed considerable results.
Figure 7.
(A) PDB 6Y2E (B) PDB 6M71 (C) PDB 6VY0 (D) PDB 6W9C (E) PDB 6VXX.
Furthermore, the docking scores with nsp14 indicated that ivermectin had the highest binding affinity and protein-ligand interactions. Remdesivir had relatively high results, while favipiravir showed the lowest values. Meanwhile, binding interactions with Mpro using lopinavir as a reference drug showed it had the highest binding scores, with remdesivir ranking with the uppermost values, followed by ivermectin, just as hydroxychloroquine and chloroquine while favipiravir did not depict considerable Mpro-binding. In sum, both ivermectin and remdesivir had high binding affinities to different viral proteins, making them the potent drug candidates against SARS-CoV-2. In another molecular docking study, screening of rutin, a natural compound, ritonavir, emetine, hesperidin, lopinavir, and indinavir showed all the molecules could bind near the crucial catalytic residues of Mpro, making them worthy of further analysis as SARS-CoV-2 therapeutic candidates [53]. Molecular docking studies have further revealed drugs like boceprevir, telaprevir, and narlaprevir, which are clinically approved for hepatitis C virus, also bind on the active site of SARS-CoV-2 Mpro (Figure 8) [60].
Figure 8.
Structure of Ivermectin, Lopinavir, Boceprevir, Narlaprevir, and Telaprevir.
2.2. Quantitative Structure-Activity Relationships (QSAR)
Studies have been done using QSAR methods to develop phosphorus-based drugs which have good inhibitory activity against Mpro, which are based on synthesizing and characterizing remdesivir derivatives as part of introducing a group of inhibitors to the coronavirus [26]. QSAR techniques were used to investigate the biological activities of the selected compounds, involving N-morphplinemethylenephosphonicacid, N-piperidinemethylenephosphonicacid, N-piperidinecarboxylicacidmethylenephosphonicacid, 4-(2-methylenephosphonicacidpiperazine) ethanol, and 4-ethylpiperazinemethylenephosphonicacid, among other ligands. After docking with Auto Dock Tools (ADT), the QSAR calculations were carried out to study the activity of phosphonates in inhibiting Covid-19, with a series of mono, bis, and tetra phosphonates selected to investigate their Mpro active site (S4) binding affinity. To build QSAR models, numerical descriptors of a set of inhibitors were built, with the descriptors representing quantitative properties dependent on the molecule’s structure. From the results, it was shown that the behavior of bisphosphonates in the QSAR model indicated that structural descriptors like non-H bonds (nB0), Narumi simple topological index (SNar), and number of hydrogen bond acceptors (nHAcc) are more momentous than electronic descriptors. Accordingly, the nB0 and SNar descriptors were two of the effective factors of the model, meaning that the non-hydrogen bonds and molecule typology are important in the inhibition mechanism against SARS-CoV-2.
A QSAR study based on the simplified molecular-input line-entry system (SMILES) strings of 32 bicycloproline derivatives has also been applied in the discovery of Covid-19 therapeutics, with the strings applied in calculating 0D, 1D, and 2D molecular descriptors [27]. Similar SMILES notation has been used to reveal new compounds with a potential 3C-like protease (3CLpro) inhibition activity in rediscovering and repurposing SARS-CoV-2 drugs [34,35,36]. In the reported study, new molecular scaffolds were searched using only the common 2D chemical structure of the congener series chosen for the study, with the authors using the process to identify new chemical scaffolds with the potential to help develop new Mpro inhibitors. The dataset preparation relied on 32 bicycloproline derivatives synthesized and tested for Mpro inhibition using a transgenic mouse model, deriving from the telaprevir and boceprevir molecules that can inhibit Mpro [28]. Boceprevir and telaprevir are protease inhibitors approved for the treatment of hepatitis C virus, and following in vivo studies, showed that six compounds had protective effects on cells against viral infection with high potency, while two showed strong antiviral activity in a mouse model [30,31]. With QSAR combined with other computational models like molecular docking, MD simulations, and free binding energy MM/PBSA, the MI-09 and MI-30 compounds facilitated a computational protocol built to design new derivatives with higher inhibitory activities on Mpro in SARS-CoV-2 [32]. Through virtual screening, it was revealed that a bicyclic moiety with a three-membered ring was important in the therapeutic potential at one binding pocket.
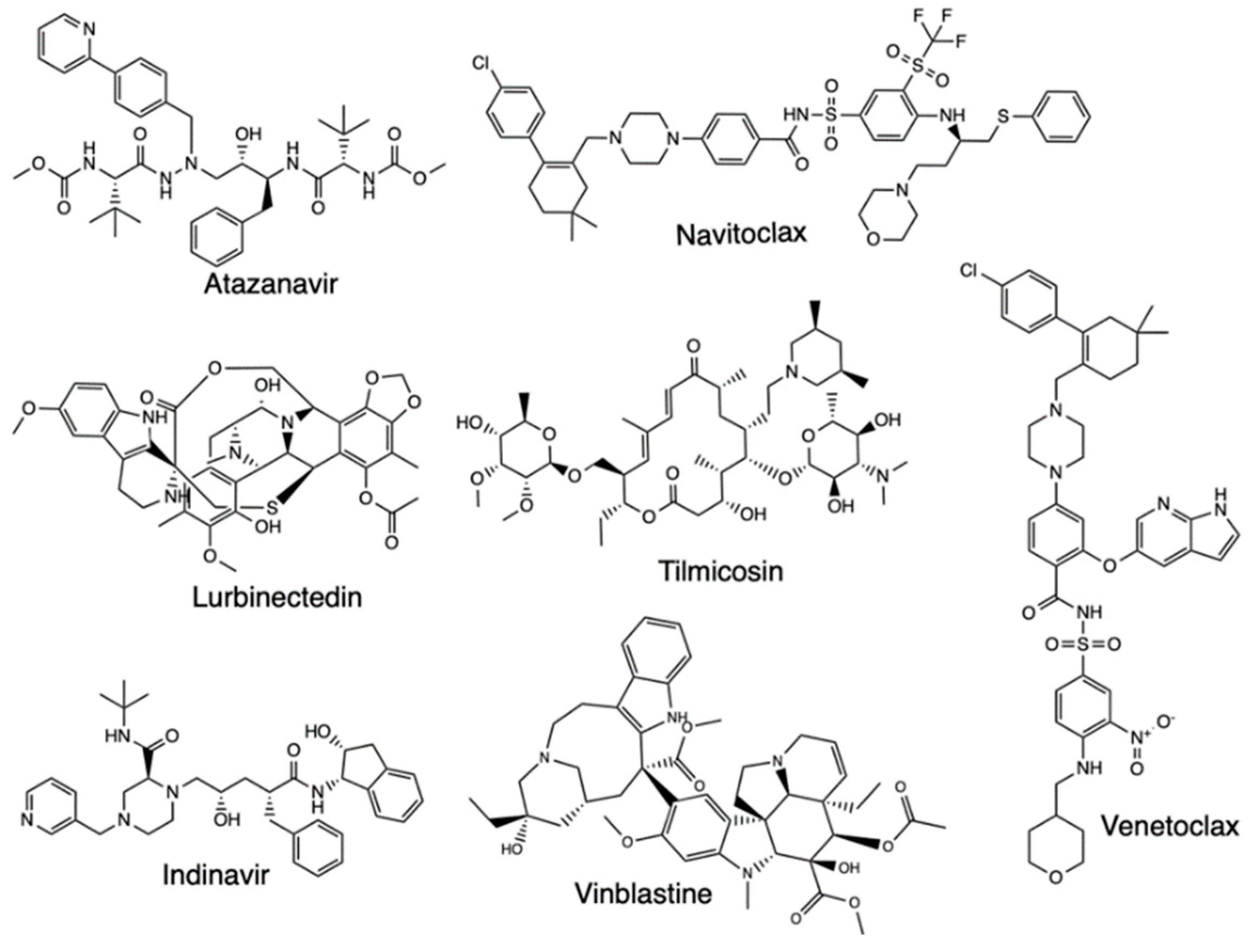
With Mpro proposed as a major drug target for Covid-19, studies have further developed QSAR models of the Mpro inhibitors whose inhibitory activity has been tested against the protease, with virtual screening then carried out for all drugs in the DrugBank database. One such study focused on 42 compounds as consensus computational hits, with three tested, which are cenicriviroc, proglumetacin, and sufugolix, employed in a docking validation run (Figure 9) [33]. Based on the outcome of the study, the three compounds were found to be active, with sufugolix and cenicriviroc having AC50 of 12.6 μM and 8.9 μM respectively, and proglumetacin tested twice independently had two AC50 of 12.6 μM and 8.9 μM. The other tested molecules, atazanavir, lurbinectedin, barasertib, indinavir, tilmicosin, vinblastine, navitoclax, and venetoclax were found to be inactive, with the overall results proving QSAR importance in finding Covid-19 treatment based on the ligands’ activity against Mpro (Figure 10).
Figure 9.
Structure of Proglumetacil, Cenicriviroc, and Sufugolix.
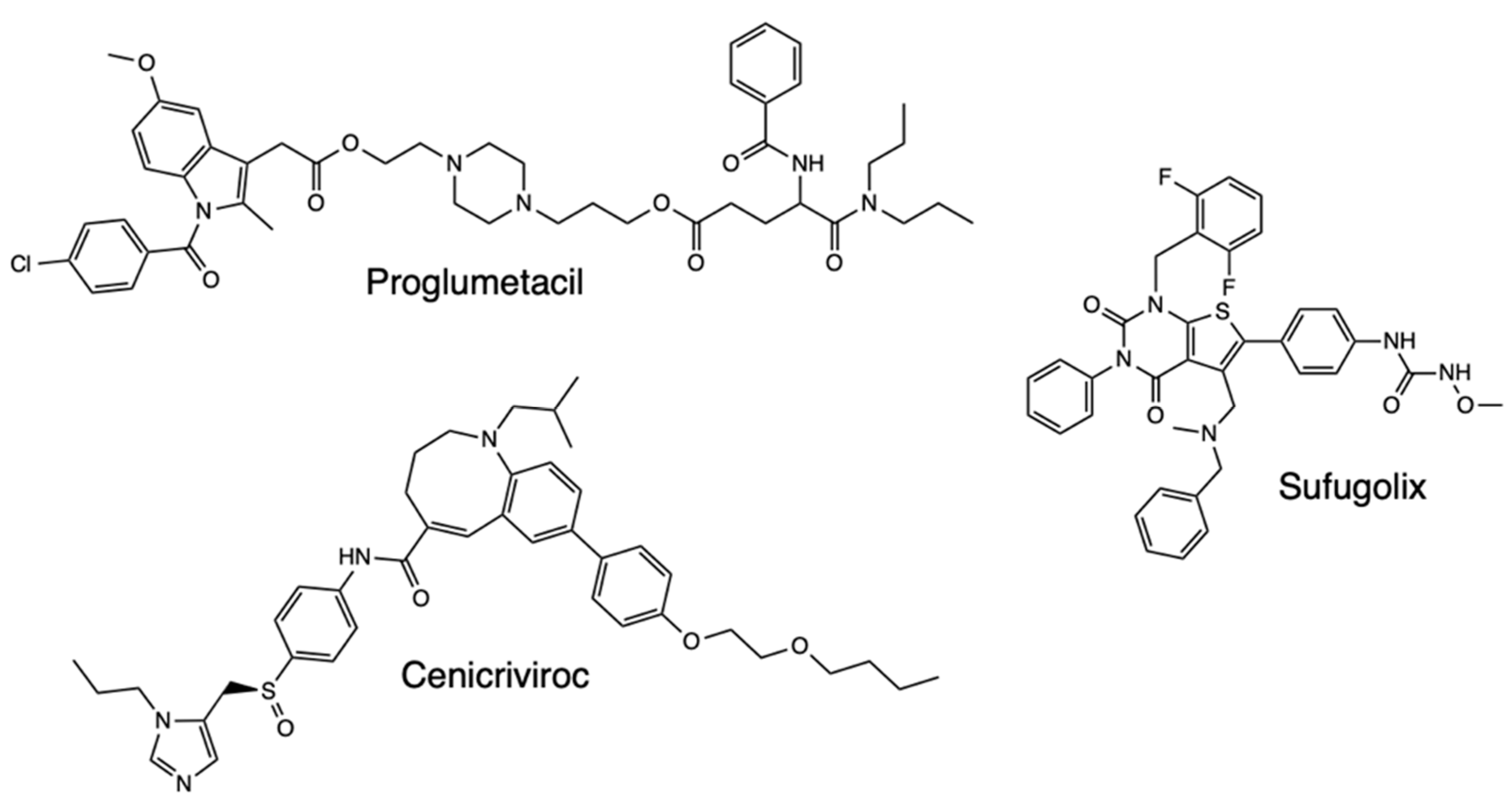
Figure 10.
Structure of Atazanavir, Navitoclax, Lurbinectedin, Tilmicosin, Indinavir, Vinblastine, and Venetoclax.
Figure 10.
Structure of Atazanavir, Navitoclax, Lurbinectedin, Tilmicosin, Indinavir, Vinblastine, and Venetoclax.
2.3. Molecular Dynamics (MD) Simulations
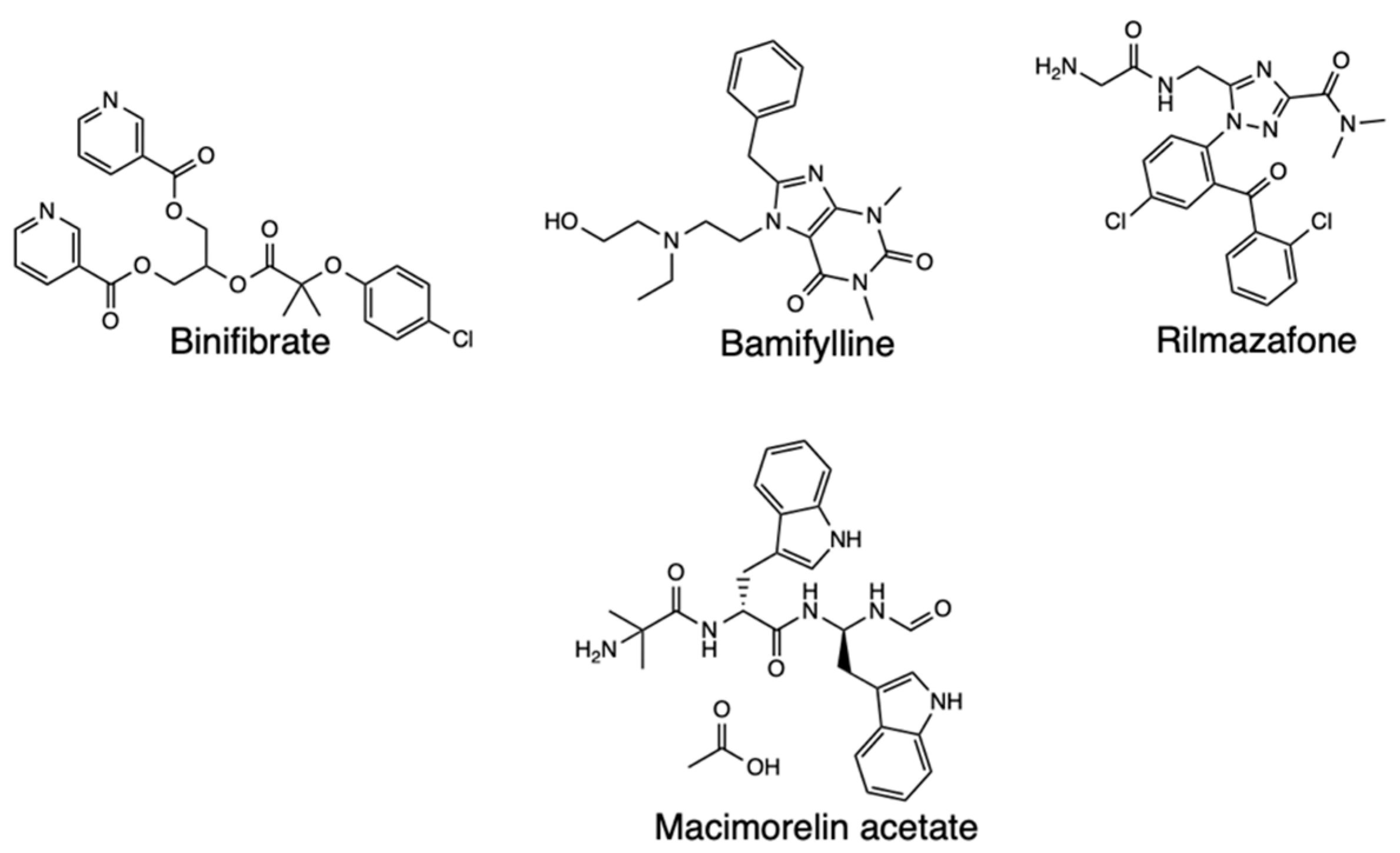
Molecular dynamics (MD) simulations have been applied in combination with other computational methods to investigate drug repurposing against SARS-CoV-2 targeting Mpro, with one such study applying MD simulations to explore the stability of the interactions between the selected drugs and the target [37]. The Desmond tool of Schrodinger helped determine the stability binding of the selected treatments with Mpro in an explicit solvent system, with the docked poses of protein ligand complexes serving as input structures. Subsequently, the binding energy of individual complexes was determined using MM-GBSA method, and MD simulations performed on the top 4 hits based on the results of the binding free energies, which were Binifibrate, Bamifylline, Macimorelin Acetate, and Rilmazafon in complex with Mpro (Figure 11). The Root Mean Square Deviation (RMSD) was then used to express the conformational changes of the protein and ligand from the initial structure. The results indicated that the four complexes subjected to MD simulations recorded protein RMSD deviations below 3 Å as compared with the initial frame. In studying globular protein conformations, RMSD of the C alpha atomic coordinates are used to measure the similarity in the 3-D structure, and a large value indicates dissimilarity while zero means identical conformation [38]. A value of 3 Å is acceptable for globular proteins, while large deviations mean large conformational changes of the protein during the simulation, hence, an unstable protein-ligand complex. As for the four complexes in the experiment, the protein RMSD deviations were below 3 Å compared to the initial frame. Binifibrate and Bamifylline specifically did not exhibit large deviations from the initial position, meaning their binding is stable, but Macimorelin acetate and Rilmazafone had larger RMSDs and had less stable binding. The major hydrogen bonds ensuring the stability of Binifibrate and Bamifylline persisted throughout the MD simulations and this meant the two drugs had better binding affinity towards the Mpro active site.
Figure 11.
Structure of Binifibrate, Bamifylline, Rilmazafone, and Macimorelin acetate.
MD simulations have further been used to screen Licorice for its action against SARS-CoV-2, with the study obtaining the small molecule-protein complexes by molecular docking for the initial structures for all-atom MD simulations and MMGBSA used to calculate the binding free energy [39]. The MD simulations results showed RMSD fluctuations of Glyasperin F and Glycyrol were within 4 Å, implying the system was less kinetic. The MMGBSA calculations also indicated Glyasperin F had the highest binding energy value, and the binding energies of the complexes mostly contributed by van der Waals energy and electrostatic energy. Further analysis revealed the drug small molecules and protein complexes could maintain a very stable binding state and exert pharmacological effects in treating SARS-CoV-2. Another study using MD simulations also screened Hydroxyethylamine (HEA) analogs to assess their action against 3CLpro protein targets of SARS-CoV-2, and the MD simulations was performed for the complex structure of 3CLpro receptor with the selected approved drug, indinavir, and designed novel HEA compound using Desmond software [40]. The aim was to evaluate the stability of binding for the ligand-protein complex. The RMSD values of the protein-ligand complex were found to be below 2 Å, and the presence of water molecules within the protein’s binding site suggested stability of the complex, which meant suitability of the compound as a potent candidate for further in vitro and in vivo studies for SARS-CoV-2 therapeutics.
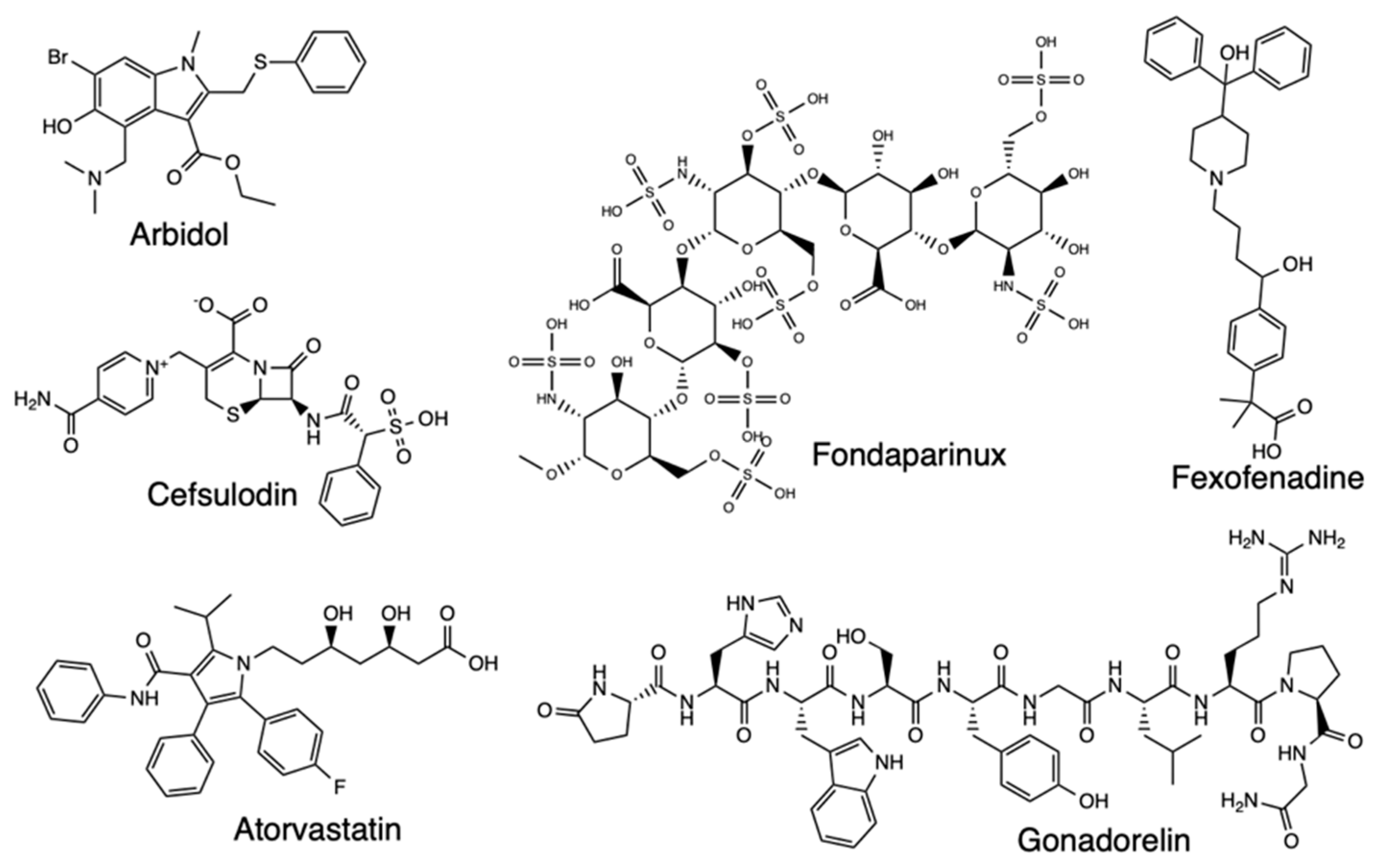
There are several other MD simulation studies for potential SARS-CoV-2 drugs, with one such exploring bioactive molecules from Triphala, an Ayurvedic herbal formulation that could inhibit Mpro [41]. The focus of this study was on the top four compounds with the best binding energies, terflavin A, corilagin, chebulagic acid, and chebulinic acid, which were further assessed for flexibility and stability at Mpro’s binding site and MM-PBSA calculations done to determine the binding free energies of the protein-ligand complexes (Figure 12). The Mpro-Corilagin and Mpro-Chebulagic acid complexes showed RMSD stability from the beginning until 100nm, meaning they are stable, Mpro-Terflavin A fluctuated at the beginning and at 100 ns, and Mpro-Chebulinic acid fluctuated from the beginning but stabilized after 70nm until 100nm. Therefore, the overall RMSD results confirmed corilagin, chebulagic acid, and chebulinic acid had stable binding of the ligands in the Mpro active site, meaning Triphala formulation is a promising Mpro inhibitor in SARS-CoV-2. Another research screened Lopinavir and Ritonavir using all-atom MD simulations to assess interaction with the residues at the active site of SARS-CoV2 3CLpro, with four residues found for lopinavir and nine residues for ritonavir that were important for binding to 3CLpro [42]. Atomistic MD simulations have also screened Arbidol binding and inhibition of SARS-CoV-2, with a focus on the receptor-binding domain (RBD)/angiotensin-converting enzyme 2 (ACE2) interface [43]. The RBD/ACE2-arbidol complex stability during simulations was evaluated and the results showed stabilization of Arbidol at RBD-ACE2 complex, which also formed favorable interactions with both RBD and ACE2. Arbidol also exhibited a higher binding affinity for RBD than ACE2, and the findings further suggested Arbidol interferes with viral binding to host cells, making the compound derivatives potential candidates as treatment for SARS-CoV-2. Studies on Arbidol have also been carried out for its binding to S protein, and the results indicated Arbidol action by effectively blocking or impeding trimerization of SARS-CoV-2 spike glycoprotein [44]. The S2 protein binding screening and assessment of the ACE2-RBD complex stability have further been carried out for Cefsulodin, Nilotinib, gonadorelin, fondaparinux, atorvastatin, and fexofenadine, with promising results for Covid-19 therapeutics [45,46,47,48] (Figure 13). Lisinopril and Alacepril have also been evaluated with MD simulations and shown to interact with human angiotensin-converting enzyme 2 (hACE2) due to the best binding affinity and favorable RMSD values [49]. Likewise, Nsp16/nsp10 inhibition has been explored through a pharmacoinformatics study seeking to identify naphthyridine and quinoline derivatives as potential inhibitors of these proteins, with MD simulations revealing stable protein-ligand complexes [52].
Figure 12.
Structure of Glyasperin F, Glycyrol, Chebulagic acid, Terflavin A, Corilagin, and Chebulinic acid.
Figure 12.
Structure of Glyasperin F, Glycyrol, Chebulagic acid, Terflavin A, Corilagin, and Chebulinic acid.
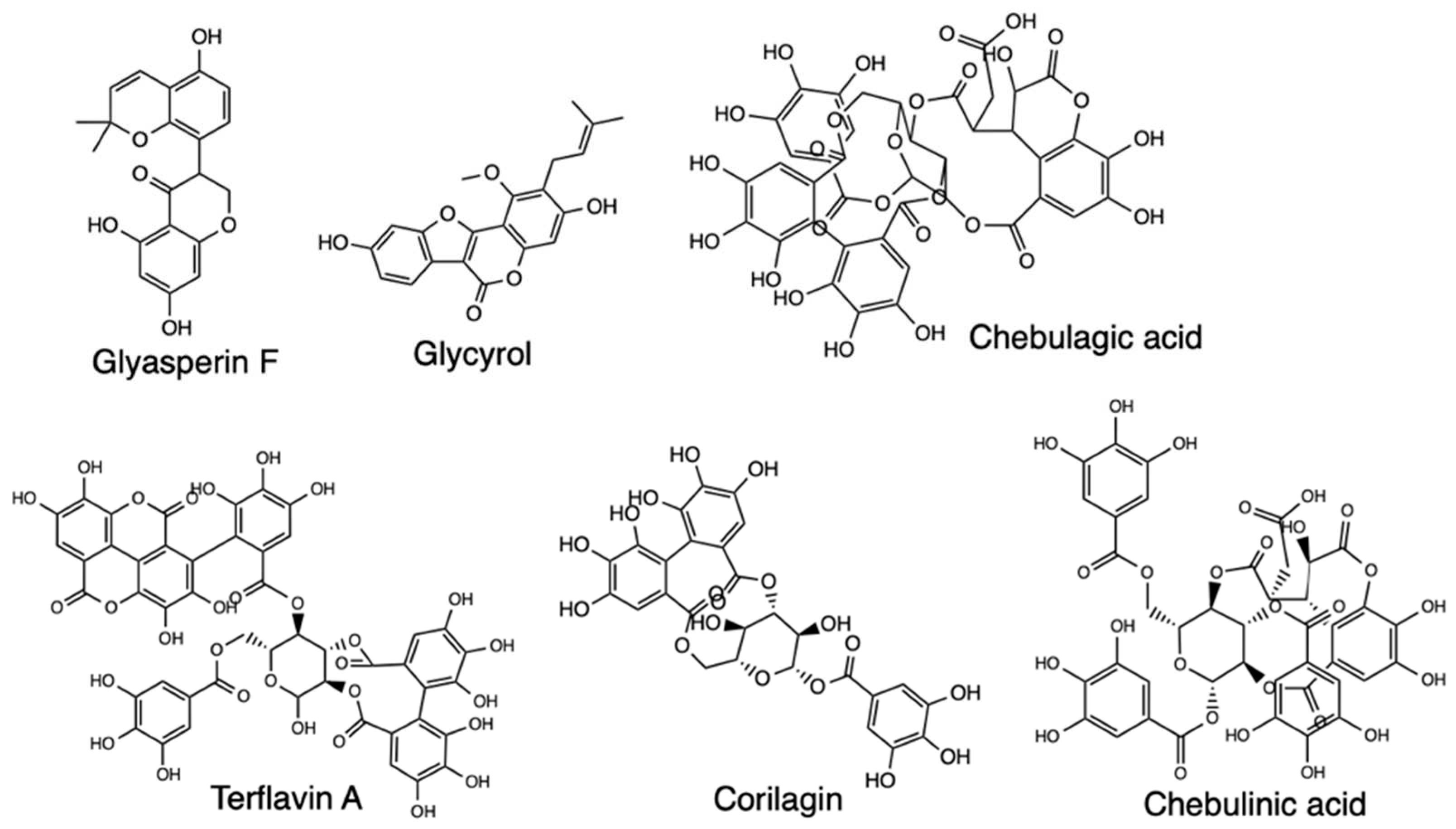
Figure 13.
Structure of Arbidol, Cefsulodin, Atorvastatin, Fondaparinux, Gonadorelin, and Fexofenadine.
Figure 13.
Structure of Arbidol, Cefsulodin, Atorvastatin, Fondaparinux, Gonadorelin, and Fexofenadine.
2.4. Pharmacophore Modeling
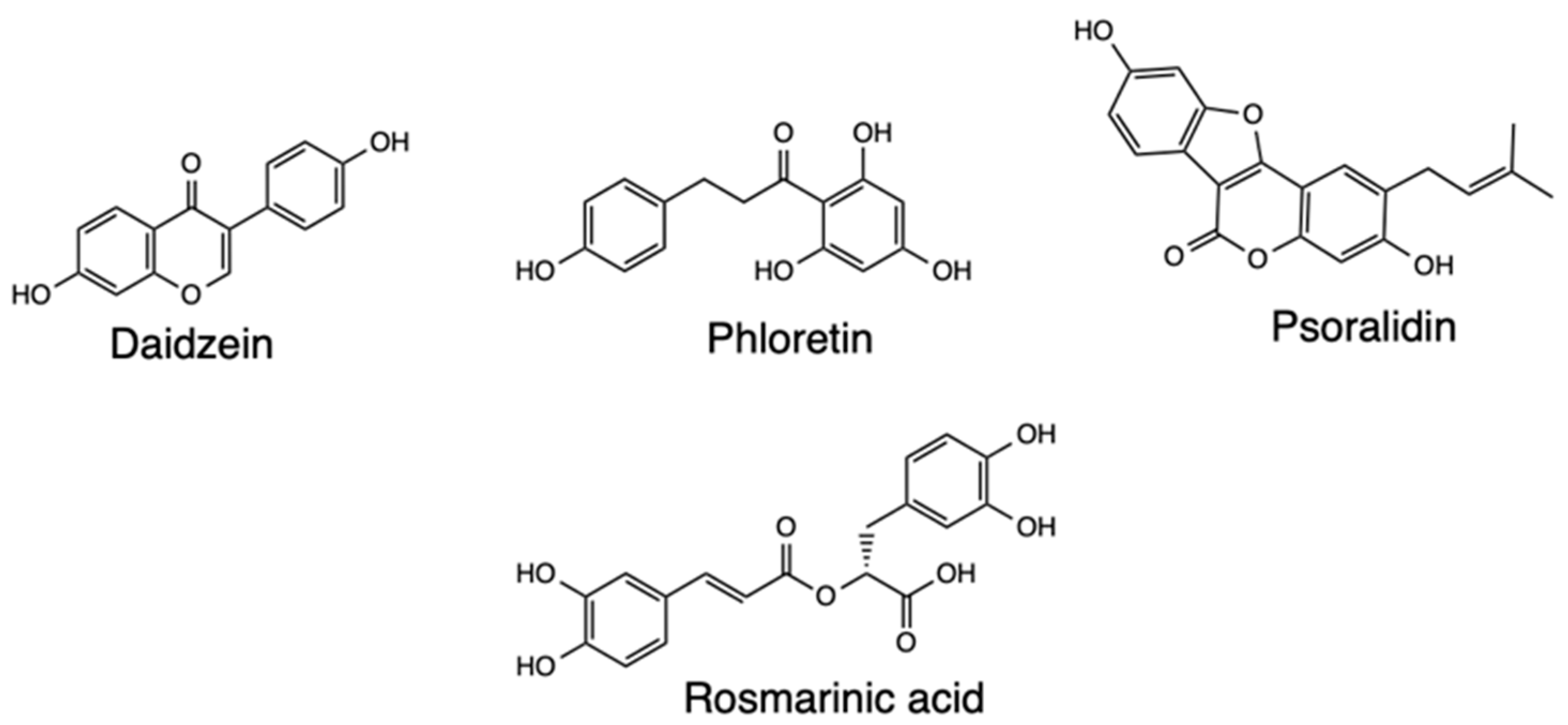
Pharmacophore modeling has been useful in researching Covid-19 therapeutics, such as has been applied with Mpro to identify FDA-approved drugs and hits from natural products as potential treatment of SARS-CoV-2. One such study utilized Mpro to develop a pharmacophore model consisting of a hydrogen bond acceptor, donor, and hydrophobic features, which was combined with virtual screening undertaken using the ZINC database [50]. Ligand pharmacophore mapping was used to extract and filter 208,000 hits by applying the lead-like properties, resulting in minimization of hits to the top 200, and simultaneous docking carried out for 200 hits and 28 hits from the experiments selected owing to the promising predicted pharmacodynamic and pharmacokinetic characteristics. Based on the findings, Daidzin depicted hydrogen bond interactions with amino acids in the binding site, also with hydrophobic interactions. The other drugs screened were phloretin, rosmarinic acid, and psoralidin, all of which showed hydrophobic contacts with the amino acids, with all the drugs anticipated to be effective natural inhibitors of Mpro (Figure 14).
Figure 14.
Structure of Daidzein, Phloretin, Psoralidin, and Rosmarinic acid.
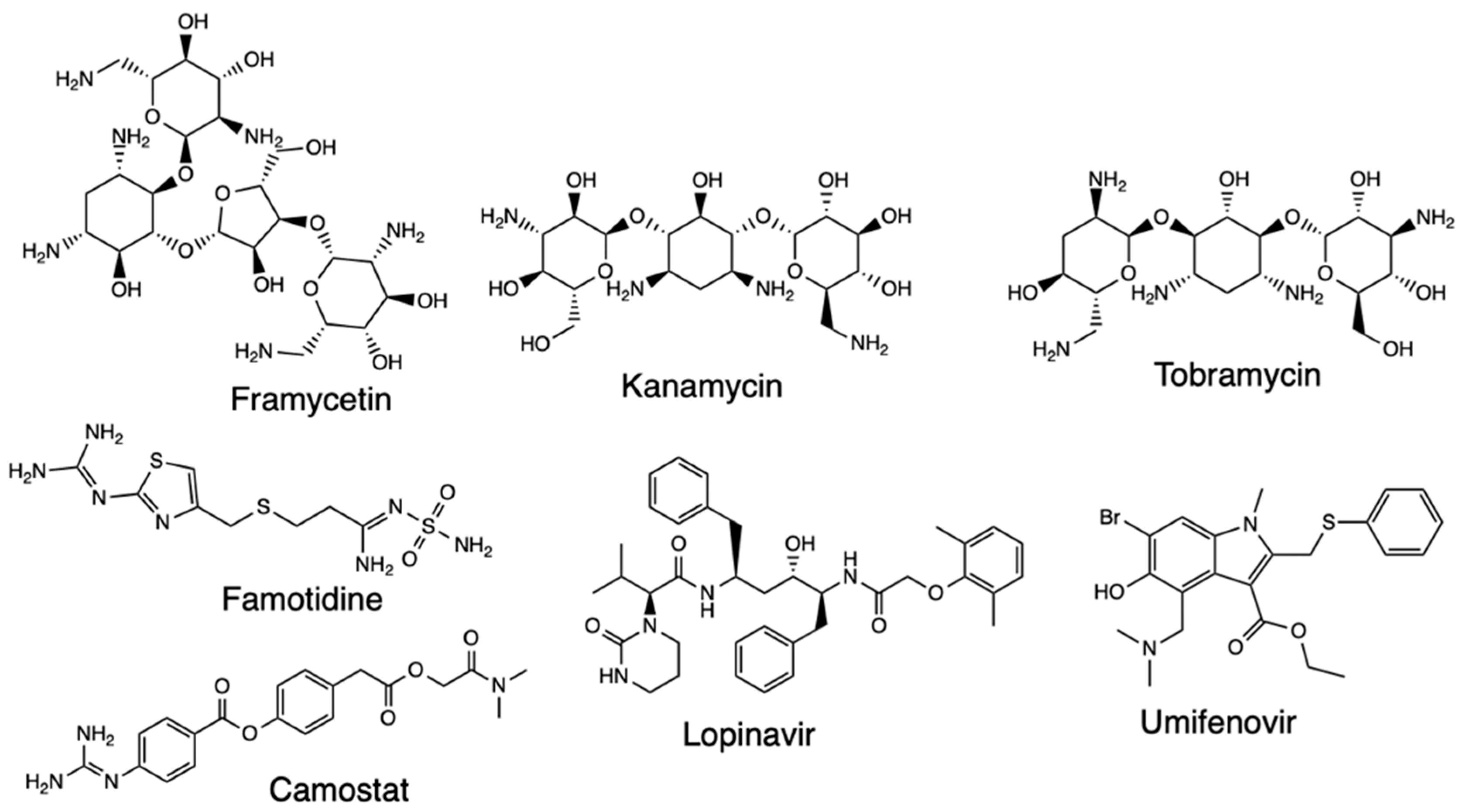
Another pharmacophore modeling study was applied on the DrugBank compounds targeting the nonstructural proteins 16/10 (nsp16/nsp10) complex, following a structure-based pharmacophore model generated and used to screen the database that resulted in three compounds [51]. The co-crystallized compound S-adenosylmethionine (SAM) was used as the reference compound, and hydroxychloroquine and remdesivir employed for comparative docking. Framycetin, kanamycin, and tobramycin were the three compounds retrieved as potential candidate drugs due to their higher dock score than the reference compound, and the molecules also formed hydrogen bonds with residues on the binding sites. The three compounds demonstrated the pharmacophore features of SAM and depicted key residue interactions and stable MDS results, which imply they are potential candidates as SARS-CoV-2 therapeutics.
Research has been conducted focusing on the CD146 receptor (human basigin) due to its presence on the host cell leading to SARS-CoV-2 infection and how drugs altering the formation of CD147 and S protein complex could inhibit the coronavirus replication [54]. This was based on an e-pharmacophore model developed using the receptor-ligand cavity of CD147 protein and further mapped against FDA-approved drugs. Eleven drugs were used for screening, and the results of the e-pharmacophore model revealed 19 non-bonded interactions found between the ligand and the receptor and 10 pharmacophores generated. The drugs that fitted to the pharmacophore pockets were famotidine, lopinavir, hydroxychloroquine, ritonavir, fluvoxamine, camostat, and umifenovir (Figure 15). From the subsequent docking results, ritonavir had the higher CDOCKER energy than the other compounds. Telaprevir has also been explored with the co-crystal structure of Mpro where the receptor-ligand pharmacophore models were developed and validated with pharmit [55]. Screening was done on the ZINC database and decoy compounds used for comparison, and the results of the pharmacophore modeling study showed that telaprevir formed hydrogen bonds with residues of the catalytic dyad and could be a candidate for SARS-CoV-2 therapeutics.
Figure 15.
Structure of Framycetin, Kanamycin, Tobramycin, Camostat, Lopinavir, and Umifenovir.
2.5. Homology Modeling
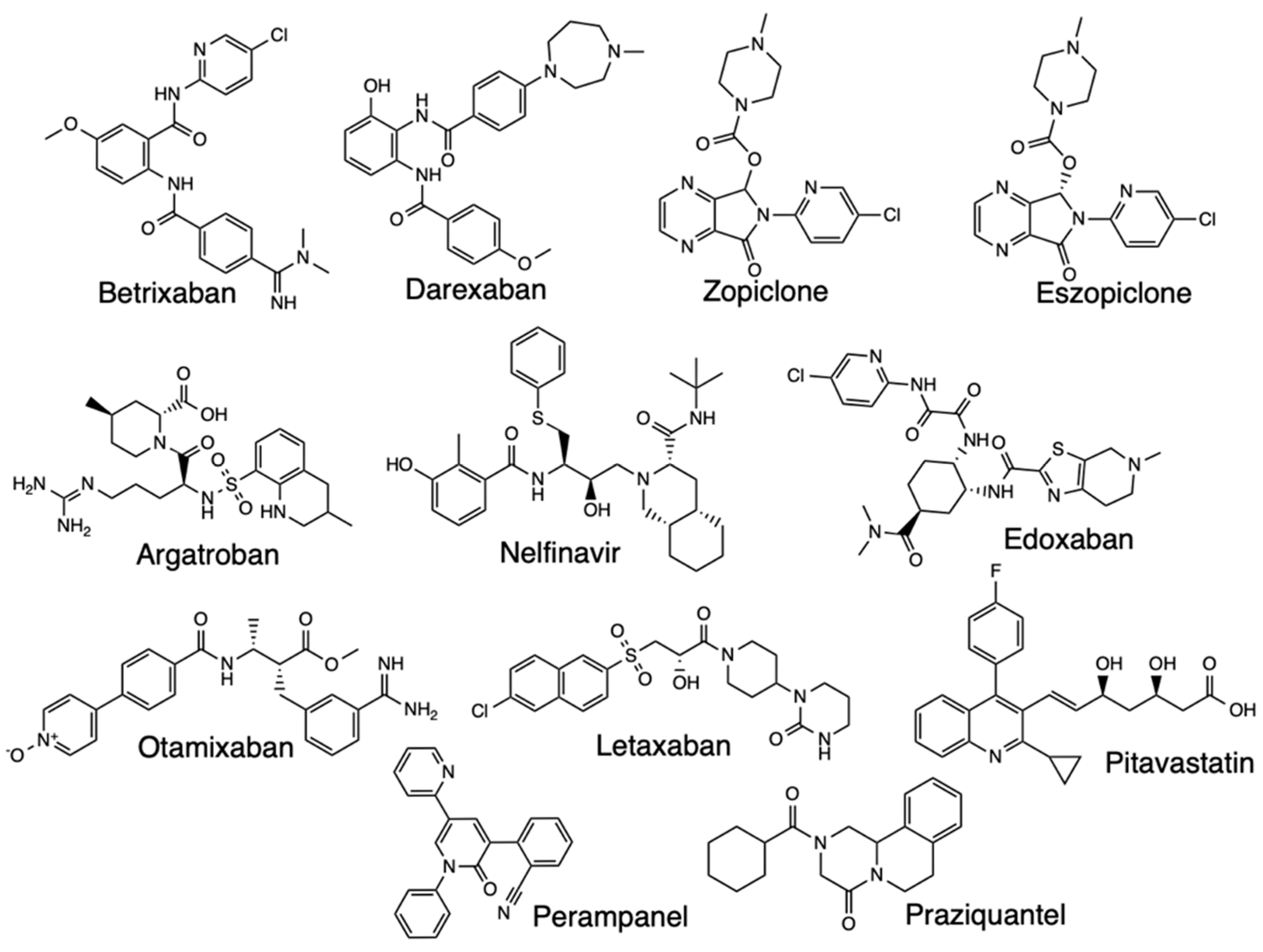
Homology modeling studies have been important in discovering treatment options for SARS-CoV-2, such as the research reported in a preprint focused on identifying repurposing candidates for the transmembrane serine protease family member II (TMPRSS2) [56]. The seven homology models used were created representing an ensemble of structures with an average RMSD of 1.27 Å and a maximum RMSD of 1.675 Å. Based on the findings, Camostat showed a median binding energy, Nafamostat ranked third, and the accompanying docking studies indicated key electrostatic interactions between the docked chemical structure of Otamixaban, the best scoring ligand, and residues in the binding pocket of a TMPRSS2 homology model. Argatroban, Otamixaban, Letaxaban, Edoxaban, Betrixaban, Darexaban, and Nafamostat all ranked highly across a majority of model structures and clustered with known active ligands. The known inhibitors confirmed results of providing a positive control, and the uniformly low docking scores meant the protocol generated homology models and docking results consistent with other completed studies.
Homology modeling, combined with other computational methods, have screened Neflinavir as a potential Mpro inhibitor in SARS-CoV-2, a study based on SARS Mpro structures aiding to build the homology models then docking 1903 small molecule drugs to the models [57]. Nelfinavir, perampanel, zopiclone, pitavastatin, eszeopiclone, and praziquantel were shown to exhibit good docking scores and binding modes, but MM/GBSA and SIE calculations proposed nelfinavir as a candidate that might be active against SARS-CoV-2 and the rest had moderate activities [58] (Figure 16). Tests of nelfinavir against SARS-CoV-2 infected cells demonstrated high antiviral activity and is a capable Mpro inhibitor [59].
Figure 16.
Structures of Betrixaban, Darexaban, Zopiclone, Eszopiclone, Argatroban, Nelfinavir, Edoxaban, Otamixaban, Letaxaban, Pitavastatin, Perampanel, and Praziquantel.
Figure 16.
Structures of Betrixaban, Darexaban, Zopiclone, Eszopiclone, Argatroban, Nelfinavir, Edoxaban, Otamixaban, Letaxaban, Pitavastatin, Perampanel, and Praziquantel.
3. Conclusions
Covid-19 remains humanity’s greatest threat in the 21st century, and though various vaccines have been developed to manage the spread of infections, the challenge still persists to get the pandemic under control. The existing computational drug repurposing and redesign models offer an opportunity for developing new drug candidates for treating SARS-CoV-2. Compounds that act on the Mpro active sites as well as inhibition of NRP-1 receptors and S protein have been screened using molecular dynamics simulations, molecular docking, QSAR, pharmacophore, and homology modeling studies and there is evidence that there are potent candidates for further analysis in vitro and in vivo.
Author Contributions
C.R.W. and G.C.L prepared and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no funding.
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Data Availability Statement
Not applicable
Acknowledgments
Figures 3, 4, and 7 were generated using Schrödinger Suites [61–64]
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| EUA Emergency Use Authorization |
| CADD Computer-aided Drug Design |
| WHO World Health Organization |
| FDA Food and Drug Administration |
| NIAID National Institute for Allergy and Infectious Diseases |
| RdRp RNA-dependent RNA polymerase |
| 3D Three-dimensional |
| PDB Protein Data Bank |
| SBDD Structure-based drug design |
| LBDD Ligand-based drug design |
| Mpro Main protease |
| QSAR Quantitative structure-activity relationships |
| VS Virtual screening |
| NRP-1 Neuropilin-1 |
| ADT Auto Dock Tools |
| nB0 Non-H bond |
| SNar Narumi simple topological index |
| nHAcc Number of hydrogen bond acceptors |
| SMILES Simplified molecular-input line-entry system |
| 3CLpro 3C-like protease |
| RMSD Root Mean Square Deviation |
| MD Molecular Dynamic |
| ACE2 Angiotensin-converting enzyme 2 |
| hACE2 Human angiotensin-converting enzyme 2 |
References
- Kalita, P., Tripathi, T. and Padhi, A.K., 2023. Computational Protein Design for COVID-19 Research and Emerging Therapeutics. ACS Central Science, 9(4), pp.602-613. [CrossRef]
- Cascella, M., Rajnik, M., Aleem, A., Dulebohn, S. and Di Napoli, R., 2023. Features, evaluation, and treatment of coronavirus (COVID-19). StatPearls.
- Niknam, Z., Jafari, A., Golchin, A., Danesh Pouya, F., Nemati, M., Rezaei-Tavirani, M. and Rasmi, Y., 2022. Potential therapeutic options for COVID-19: an update on current evidence. European Journal of Medical Research, 27, pp.1-15. [CrossRef]
- Chera, A. and Tanca, A., 2022. Remdesivir: the first FDA-approved anti-COVID-19 Treatment for Young Children. Discoveries, 10(2). [CrossRef]
- Beigel, J.H., Tomashek, K.M., Dodd, L.E., Mehta, A.K., Zingman, B.S., Kalil, A.C., Hohmann, E., Chu, H.Y., Luetkemeyer, A., Kline, S. and Lopez de Castilla, D., 2020. Remdesivir for the treatment of Covid-19—Final Report. New England Journal of Medicine, 383(19), pp.1813-1826.
- Rezagholizadeh, A., Khiali, S., Sarbakhsh, P. and Entezari-Maleki, T., 2021. Remdesivir for treatment of COVID-19; An updated systematic review and meta-analysis. European Journal of Pharmacology, 897, p.173926. [CrossRef]
- Abd-Elsalam, S., Salama, M., Soliman, S., Naguib, A.M., Ibrahim, I.S., Torky, M., Abd El Ghafar, M.S., Abdul-Baki, E.A.R.M. and Elhendawy, M., 2022. Remdesivir efficacy in COVID-19 treatment: A randomized controlled trial. The American Journal of Tropical Medicine and Hygiene, 106(3), p.886. [CrossRef]
- Gupte, V., Hegde, R., Sawant, S., Kalathingal, K., Jadhav, S., Malabade, R. and Gogtay, J., 2022. Safety and clinical outcomes of remdesivir in hospitalised COVID-19 patients: a retrospective analysis of active surveillance database. BMC Infectious Diseases, 22(1), p.1. [CrossRef]
- WHO Solidarity Trial Consortium, 2022. Remdesivir and three other drugs for hospitalised patients with COVID-19: Final results of the WHO Solidarity randomised trial and updated meta-analyses. The Lancet, 399(10339), pp.1941-1953. [CrossRef]
- Balakrishnan, V. and Lakshminarayanan, K., 2021. Screening of FDA approved drugs against SARS-CoV-2 main protease: coronavirus disease. International journal of peptide research and therapeutics, 27(1), pp.651-658.
- US Food and Drug Administration (FDA), 2023, June 28. Know your treatment options for Covid-19. https://www.fda.gov/consumers/consumer-updates/know-your-treatment-options-covid-19.
- Toussi, S.S., Hammond, J.L., Gerstenberger, B.S. and Anderson, A.S., 2023. Therapeutics for COVID-19. Nature Microbiology, 8(5), pp.771-786.
- Muratov, E.N., Amaro, R., Andrade, C.H., Brown, N., Ekins, S., Fourches, D., Isayev, O., Kozakov, D., Medina-Franco, J.L., Merz, K.M. and Oprea, T.I., 2021. A critical overview of computational approaches employed for COVID-19 drug discovery. Chemical Society Reviews, 50(16), pp.9121-9151.
- Saloni, Kumari, D., Ranjan, P. and Chakraborty, T., 2022. A computational study of potential therapeutics for COVID-19 invoking conceptual density functional theory. Structural Chemistry, 33(6), pp.2195-2204. [CrossRef]
- Napolitano, F., Xu, X. and Gao, X., 2022. Impact of computational approaches in the fight against COVID-19: An AI guided review of 17 000 studies. Briefings in bioinformatics, 23(1), p.bbab456. [CrossRef]
- Ghosh, D., Ghosh Dastidar, D., Roy, K., Ghosh, A., Mukhopadhyay, D., Sikdar, N., Biswas, N.K., Chakrabarti, G. and Das, A., 2022. Computational prediction of the molecular mechanism of statin group of drugs against SARS-CoV-2 pathogenesis. Scientific Reports, 12(1), p.6241.
- Singh, P.K., Pathania, S. and Rawal, R.K., 2020. Exploring RdRp–remdesivir interactions to screen RdRp inhibitors for the management of novel coronavirus 2019-nCoV. SAR and QSAR in Environmental Research, 31(11), pp.857-867. [CrossRef]
- Chang, C.C., Hsu, H.J., Wu, T.Y. and Liou, J.W., 2022. Computer-aided discovery, design, and investigation of COVID-19 therapeutics. Tzu-Chi Medical Journal, 34(3), p.276. [CrossRef]
- Wu, C., Liu, Y., Yang, Y., Zhang, P., Zhong, W., Wang, Y., Wang, Q., Xu, Y., Li, M., Li, X. and Zheng, M., 2020. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharmaceutica Sinica B, 10(5), pp.766-788.
- Dong, J., Varbanov, M., Philippot, S., Vreken, F., Zeng, W.B. and Blay, V., 2023. Ligand-based discovery of coronavirus main protease inhibitors using MACAW molecular embeddings. Journal of Enzyme Inhibition and Medicinal Chemistry, 38(1), pp.24-35. [CrossRef]
- Sharma, V., Wakode, S. and Kumar, H., 2021. Structure-and ligand-based drug design: Concepts, approaches, and challenges. Chemoinformatics and bioinformatics in the pharmaceutical sciences, pp.27-53.
- Alméciga-Díaz, C.J., Pimentel-Vera, L.N., Caro, A., Mosquera, A., Castellanos Moreno, C.A., Manosalva Rojas, J.P. and Díaz-Tribaldos, D.C., 2020. Virtual screening of potential inhibitors for SARS-CoV-2 main protease. Preprints.org, . [CrossRef]
- Charoute, H., Elkarhat, Z., Elkhattabi, L., El Fahime, E., Oukkache, N., Rouba, H. and Barakat, A., 2022. Computational screening of potential drugs against COVID-19 disease: The Neuropilin-1 receptor as molecular target. VirusDisease, 33(1), pp.23-31. [CrossRef]
- Marinho, E.M., de Andrade Neto, J.B., Silva, J., da Silva, C.R., Cavalcanti, B.C., Marinho, E.S. and Júnior, H.V.N., 2020. Virtual screening based on molecular docking of possible inhibitors of Covid-19 main protease. Microbial Pathogenesis, 148, p.104365.
- Eweas, A.F., Alhossary, A.A. and Abdel-Moneim, A.S., 2021. Molecular docking reveals ivermectin and remdesivir as potential repurposed drugs against SARS-CoV-2. Frontiers in Microbiology, 11, p.3602.
- Gholivand, K., Barzegari, A., Mohammadpanah, F., Yaghoubi, R., Roohzadeh, R. and Valmoozi, A.A.E., 2022. Synthesis, characterized, QSAR studies and molecular docking of some phosphonates as COVID-19 inhibitors. Polyhedron, 221, p.115824. [CrossRef]
- Costa, A.S., Martins, J.P.A. and de Melo, E.B., 2022. SMILES-based 2D-QSAR and similarity search for identification of potential new scaffolds for development of SARS-CoV-2 MPRO inhibitors. Structural Chemistry, 33(5), pp.1691-1706.
- Qiao, J., Li, Y.S., Zeng, R., Liu, F.L., Luo, R.H., Huang, C., Wang, Y.F., Zhang, J., Quan, B., Shen, C. and Mao, X., 2021. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science, 371(6536), pp.1374-1378.
- Butt, A.A. and Kanwal, F., 2012. Boceprevir and telaprevir in the management of hepatitis C virus–infected patients. Clinical infectious diseases, 54(1), pp.96-104. [CrossRef]
- Li, L. and Huang, S., 2021. Newly synthesized Mpro inhibitors as potential oral anti-SARS-CoV-2 agents. Signal Transduction and Targeted Therapy, 6(1), p.138.
- Fischer, C. and Feys, J.R., 2023. SARS-CoV-2 Mpro inhibitors: Achieved diversity, developing resistance and future strategies. Future Pharmacology, 3(1), pp.80-107.
- La Monica, G., Bono, A., Lauria, A. and Martorana, A., 2022. Targeting SARS-CoV-2 main protease for treatment of COVID-19: Covalent inhibitors structure–activity relationship insights and evolution perspectives. Journal of medicinal chemistry, 65(19), pp.12500-12534.
- Alves, V.M., Bobrowski, T., Melo-Filho, C.C., Korn, D., Auerbach, S., Schmitt, C., Muratov, E.N. and Tropsha, A., 2021. QSAR modeling of SARS-CoV Mpro inhibitors identifies sufugolix, cenicriviroc, proglumetacin, and other drugs as candidates for repurposing against SARS-CoV-2. Molecular informatics, 40(1), p.2000113.
- Ničkčović, V.P., Nikolić, G.R., Nedeljković, B.M., Mitić, N., Danić, S.F., Mitić, J., Marčetić, Z., Sokolović, D. and Veselinović, A.M., 2022. In silico approach for the development of novel antiviral compounds based on SARS-COV-2 protease inhibition. Chemical Papers, 76(7), pp.4393-4404. [CrossRef]
- Sharma S., Khan Y., Saini K., Bhatia V., 2022). Role of QSAR in filling in the gaps of CoVID-19 Therapeutics. Chem Informatics, 8 (5).
- Oubahmane, M., Hdoufane, I., Delaite, C., Sayede, A., Cherqaoui, D. and El Allali, A., 2023. Design of Potent Inhibitors Targeting the Main Protease of SARS-CoV-2 Using QSAR Modeling, Molecular Docking, and Molecular Dynamics Simulations. Pharmaceuticals, 16(4), p.608.
- Arun, K.G., Sharanya, C.S., Abhithaj, J., Francis, D. and Sadasivan, C., 2021. Drug repurposing against SARS-CoV-2 using E-pharmacophore based virtual screening, molecular docking and molecular dynamics with main protease as the target. Journal of Biomolecular Structure and Dynamics, 39(13), pp.4647-4658.
- Maiorov, V.N. and Crippen, G.M., 1994. Significance of root-mean-square deviation in comparing three-dimensional structures of globular proteins. Journal of molecular biology, 235(2), pp.625-634. [CrossRef]
- Cao, J.F., Gong, Y., Wu, M., Yang, X., Xiong, L., Chen, S., Xiao, Z., Li, Y., Zhang, L., Zan, W. and Zhang, X., 2022. Exploring the mechanism of action of licorice in the treatment of COVID-19 through bioinformatics analysis and molecular dynamics simulation. Frontiers in pharmacology, 13, p.1003310. [CrossRef]
- Kumar, S., Sharma, P.P., Shankar, U., Kumar, D., Joshi, S.K., Pena, L., Durvasula, R., Kumar, A., Kempaiah, P., Poonam and Rathi, B., 2020. Discovery of new hydroxyethylamine analogs against 3CLpro protein target of SARS-CoV-2: Molecular docking, molecular dynamics simulation, and structure–activity relationship studies. Journal of Chemical Information and Modeling, 60(12), pp.5754-5770.
- Rudrapal, M., Celik, I., Khan, J., Ansari, M.A., Alomary, M.N., Alatawi, F.A., Yadav, R., Sharma, T., Tallei, T.E., Pasala, P.K. and Sahoo, R.K., 2022. Identification of bioactive molecules from Triphala (Ayurvedic herbal formulation) as potential inhibitors of SARS-CoV-2 main protease (Mpro) through computational investigations. Journal of King Saud University-Science, 34(3), p.101826.
- Nutho, B., Mahalapbutr, P., Hengphasatporn, K., Pattaranggoon, N.C., Simanon, N., Shigeta, Y., Hannongbua, S. and Rungrotmongkol, T., 2020. Why are lopinavir and ritonavir effective against the newly emerged coronavirus 2019? Atomistic insights into the inhibitory mechanisms. Biochemistry, 59(18), pp.1769-1779. [CrossRef]
- Padhi, A.K., Seal, A., Khan, J.M., Ahamed, M. and Tripathi, T., 2021. Unraveling the mechanism of arbidol binding and inhibition of SARS-CoV-2: Insights from atomistic simulations. European journal of pharmacology, 894, p.173836.
- Vankadari, N., 2020. Arbidol: A potential antiviral drug for the treatment of SARS-CoV-2 by blocking trimerization of the spike glycoprotein. International Journal of Antimicrobial Agents, 56(2), p.105998.
- Deganutti, G., Prischi, F. and Reynolds, C.A., 2021. Supervised molecular dynamics for exploring the druggability of the SARS-CoV-2 spike protein. Journal of computer-aided molecular design, 35, pp.195-207.
- Razizadeh, M., Nikfar, M. and Liu, Y., 2021. Small molecule therapeutics to destabilize the ACE2-RBD complex: A molecular dynamics study. Biophysical Journal, 120(14), pp.2793-2804. [CrossRef]
- Kumar, V., Liu, H. and Wu, C., 2021. Drug repurposing against SARS-CoV-2 receptor binding domain using ensemble-based virtual screening and molecular dynamics simulations. Computers in Biology and Medicine, 135, p.104634.
- Pirolli, D., Righino, B., Camponeschi, C., Ria, F., Di Sante, G. and De Rosa, M.C., 2023. Virtual screening and molecular dynamics simulations provide insight into repurposing drugs against SARS-CoV-2 variants Spike protein/ACE2 interface. Scientific Reports, 13(1), p.1494.
- Al-Karmalawy, A.A., Dahab, M.A., Metwaly, A.M., Elhady, S.S., Elkaeed, E.B., Eissa, I.H. and Darwish, K.M., 2021. Molecular docking and dynamics simulation revealed the potential inhibitory activity of ACEIs against SARS-CoV-2 targeting the h ACE2 receptor. Frontiers in Chemistry, 9, p.661230.
- Saeed, M., Saeed, A., Alam, M.J. and Alreshidi, M., 2021. Receptor-based pharmacophore modeling in the search for natural products for COVID-19 Mpro. Molecules, 26(6), p.1549. [CrossRef]
- Rampogu, S. and Lee, K.W., 2021. Pharmacophore modelling-based drug repurposing approaches for SARS-CoV-2 therapeutics. Frontiers in Chemistry, 9, p.636362.π.
- Aldahham, B.J., Al-Khafaji, K., Saleh, M.Y., Abdelhakem, A.M., Alanazi, A.M. and Islam, M.A., 2022. Identification of naphthyridine and quinoline derivatives as potential Nsp16-Nsp10 inhibitors: a pharmacoinformatics study. Journal of Biomolecular Structure and Dynamics, 40(9), pp.3899-3906. [CrossRef]
- Das, S., Sarmah, S., Lyndem, S. and Singha Roy, A., 2021. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. Journal of Biomolecular Structure and Dynamics, 39(9), pp.3347-3357.
- Pandit, N.K., Mann, S.S., Mohanty, A. and Meena, S.S., 2023. e-Pharmacophore modeling and in silico study of CD147 receptor against SARS-CoV-2 drugs. Genomics & Informatics, 21(2).
- Halimi, M. and Bararpour, P., 2022. Natural inhibitors of SARS-CoV-2 main protease: structure based pharmacophore modeling, molecular docking and molecular dynamic simulation studies. Journal of Molecular Modeling, 28(9), p.279.
- Rensi, S., Keys, A., Lo, Y.C., Derry, A., McInnes, G., Liu, T. and Altman, R., 2020. Homology modeling of TMPRSS2 yields candidate drugs that may inhibit entry of SARS-CoV-2 into human cells. ChemRxiv.
- Xu, Z., Peng, C., Shi, Y., Zhu, Z., Mu, K., Wang, X. and Zhu, W., 2020. Nelfinavir was predicted to be a potential inhibitor of 2019-nCov main protease by an integrative approach combining homology modelling, molecular docking and binding free energy calculation. BioRxiv, pp.2020-01.
- Yang, H. and Yang, J., 2021. A review of the latest research on M pro targeting SARS-COV inhibitors. RSC Medicinal Chemistry, 12(7), pp.1026-1036.
- Sargolzaei, M., 2021. Effect of nelfinavir stereoisomers on coronavirus main protease: Molecular docking, molecular dynamics simulation and MM/GBSA study. Journal of Molecular Graphics and Modelling, 103, p.107803. [CrossRef]
- Mengist, H.M., Dilnessa, T. and Jin, T., 2021. Structural basis of potential inhibitors targeting SARS-CoV-2 main protease. Frontiers in Chemistry, 9, p.622898.
- Schrödinger Release 2023-3: Maestro, Schrödinger, LLC, New York, NY, 2023.
- Schrödinger Release 2023-3: LigPrep, Schrödinger, LLC, New York, NY, 2023.
- Friesner, R. A.; Murphy, R. B.; Repasky, M. P.; Frye, L. L.; Greenwood, J. R.; Halgren,T. A.; Sanschagrin, P. C.; Mainz, D. T., "Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes," J. Med. Chem., 2006, 49, 6177–6196 . [CrossRef]
- Schrödinger Release 2023-3: Glide, Schrödinger, LLC, New York, NY, 2023.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



