
Submitted:
11 August 2023
Posted:
14 August 2023
You are already at the latest version
Abstract
As environmental regulations become stricter, weight- and cost-effective fiber-reinforced polymer com-posites are being considered as alternative materials in the automobile industry. Rapid impregnation of the resin into the reinforcing fibers is critical during liquid composite molding, and the optimization of resin impregnation is related to the cycle time and quality of the products. In this review, various resins capable of rapid impregnation, including thermoset and thermoplastic resins, are discussed for manu-facturing fiber-reinforced composites used in the automobile industry, along with their advantages and disadvantages. Finally, vital factors and perspectives for developing rapidly impregnated resin-based fiber-reinforced composites for automobile applications are discussed
Keywords:
Rapid impregnating
; Thermoset resins
; Thermoplastic resins
; Fiber reinforced composites
1. Introduction
Recently, lightweight automobiles have been increasingly used to save energy and reduce pollution in light of strict regulations. It has been reported that reducing the weight by 10 wt% could decrease fuel consumption by 6–8% and reduce CO2 emissions [1]. The key to solving this problem is to replace metal components with high-performance fiber-reinforced polymer-based composites. Polymer composites, including thermosets and thermoplastic composites, have been used in panels, modules, structures, and other parts of automobiles after being reinforced with continuous or discontinuous fibers using liquid composite molding (LCM) processes, such as resin transfer molding (RTM), vacuum infusion, and reaction injection molding [2]. Takahashi et al. [3] reported that the weight of carbon fiber reinforced polymer (CFRP) composites is 1/3 that of steel panels, while the flexural strength of CFRPs is approximately three times higher.
Fiber-reinforced thermoset composites (FRTSCs) generally exhibit good mechanical properties, thermal stability, and dimensional stability. Moreover, thermoset resins have a relatively low viscosity compared to thermoplastic resins, which is an important factor in the RTM process. However, the cycle time of the thermoset resin molding process is longer than that of thermoplastic resins because of the curing time. Traditionally, depending on the part size and geometry, the cycle time of standard RTM is 30–60 min with a 10–20 bar injection pressure, whereas that of high-pressure resin transfer molding is less than 10 min with an injection pressure of 20–120 bar and a pressure of up to 150 bar in the mixing head. In the automobile industry, cost and cycle time reduction are key issues; thus, research on fast-curing thermoset resins, such as fast-curing epoxy and endo-dicyclopentadiene, has been conducted.
Fiber-reinforced thermoplastic composites (FRTPCs) are widely used because of their high processability and recyclability. However, the high viscosity of thermoplastic resins requires high temperatures and pressures for the materials to be impregnated with fiber reinforcements. For example, thermosetting resins, such as epoxy, can be impregnated by fiber reinforcements and cured below 200 ℃; whereas thermoplastic resins must be heated above the melting temperature, that is typically well above 200 ℃, and impregnated by high pressures (10–50 bar). Moreover, the high pressure and viscosity of the resins may cause a misalignment of the fiber reinforcements. Therefore, research has been conducted to develop rapid-impregnation resins at low manufacturing temperatures and pressures.
This review provides a comprehensive overview of rapid-impregnation low-viscosity resins commonly used in the automobile industry, including thermosets and thermoplastic resins. Various reactive processes and parameters affecting the preparation and properties of the FRTSCs and FRTPCs are discussed. The status of related research and future perspectives are also addressed.
2. Rapid Impregnating Thermoset Resins and Their Fiber-Reinforced Composites
Thermoset resins, such as epoxy, polyester, and vinyl ester, are used in a wide range of automobile parts, such as headlamp housings, battery covers, and frames for windows or sunroofs. Thermoset composites have excellent dimensional and chemical stabilities and high impact strengths, which are necessary for the interior and exterior parts of automobiles. In this section, thermoset resins and their fiber-reinforced composites are discussed.
2.1. Epoxy
Epoxy resins have been used in the automobile industry since the 1980s owing to their superior mechanical properties, low shrinkage and creep, and outstanding chemical resistance. A representative commercial epoxy resin is the epoxy-dicyandiamide system, which has the drawbacks of relatively high viscosity and long curing time. Therefore, many strategies have been implemented to develop low-viscosity, fast-curing epoxy resins, which can be obtained by adding glycol diglycidyl ether (GDE) series. For example, a low-viscosity acrylate-based epoxy resin (AE)/ GDE system was developed by Yang et al., and its rheological behavior is shown in Figure 1A [4]. Seraji et al [5,6,7] developed a rapid-curing epoxy amine resin with low viscosity, which consists of diglycidyl ether of bisphenol F, epoxy phenolic novolac resin, diethyl toluene diamine, and 2-ethyl-4-methylimidazole. The resin system exhibited good thermal and mechanical properties, and superior flame retardancy. Based on these trends, low-viscosity fast-curing epoxy resins were obtained using the synthesized epoxies. Two resins, diglycidyl ether of ethoxylated bisphenol-A (BPA) with two and six oxyethylene units (DGEBAEO-2 and DGEBAEO-6), respectively, were synthesized and characterized; the curing exothermic enthalpy decreased with increasing oxyethylene units (Figure 1B) [8]. The viscosities of the blends decreased with increasing the DGEBAEO-6 content. In addition, difunctional aromatic epoxy-divinylbenzene dioxide, which has been synthesized by epoxidizing divinylbenzene using the metal acetylacetone compound grafted Fe3O4 particles as the catalyst, had low molecular weight and viscosity as well as excellent thermal (Tg was approximately 201 °C) and mechanical properties (tensile strength was 131.99 MPa). Wu, Xiankun, et al. [9] and Chen et al. [10] developed a series of epoxy systems with soft butyl glycidyl ether and rigid nano silica with viscosity lower than 600 mPa·s, providing excellent processing performance for the large-scale production of composites for automobile manufacturing. In addition, this system demonstrated improvements in tensile strength and modulus, as well as in elongation at break. Wang et al. [11] reported an epoxy resin-1-(cyanoethyl)-2-ethyl-4-methylimidazol system. The epoxy cures in a few minutes at 120 ℃ with acceptable pot life and low water absorption.
The reaction time decreased with the addition of the various particles. Chikhi et al. [12] developed a modified epoxy resin using liquid rubber (ATBN). All reactivity characteristics (gel time, temperature, curing time, and exothermic peaks) decreased. The addition of ATBN led to a decrease in either the glass transition temperature or the stress at break, accompanied by an increase in the elongation at break and the appearance of yielding. Zhang et al. [13] designed a tetrafunctional eugenol-based epoxy resin with a cyclosiloxane structure. Allyl glycidyl ether was selected as the reference compound to generate a silylation epoxy resin. The viscosity of the silicone-containing tetrafunctional epoxy monomers (< 0.315 Pa·s) was significantly lower than that of conventional oil-based epoxy resin (14.320 Pa·s) (Figure 1C).
2.2. Polyester
Low-viscosity polyester resins can be obtained by particle synthesis and can be applied in the fields of environmentally friendly coatings, toughening, and reinforcing unsaturated polymers.
Chen et al. [14] prepared a series of silica particles with different sizes and surface groups through the sol–gel process of tetraethyl orthosilicate and directly introduced them into polyester polyol resins via in situ polymerization. The resulting nanocomposites exhibited lower viscosities than the resins obtained using the blending method. Viscosity increased as the particle concentration increased (Figure 2A). Zhang et al. [17] examined a low-viscosity unsaturated hyperbranched polyester resin (< 10 000 cP) using a synthetic method involving the reaction between maleic anhydride monoisooctyl alcohol ester and hydroxyl-ended hyperbranched polyester resin prepared from phthalic anhydride and trimethylolpropane. Zhou et al. [18] synthesized a series of unsaturated polyester resins with low viscosities (< 300 mPa·s) for vacuum infusion molding process by simply controlling the amount of alcohol used in the reactants. Yuan et al. [19] developed a series of low-viscosity transparent UV-curable polyester methacrylate resins derived from renewable biologically fermented lactic acid (LA), reducing the viscosity from 34,620 mPa⋅s to 160–756 mPa⋅s by random copolymerization of LA and-caprolactone.
The curing time can be reduced by using various solvents and applying microwaves. Nasr and Abdel-Azim [20] investigated unsaturated polyester resin, where styrene, methyl ethyl keton peroxide (MEKP), and cobalt octoate were selected as solvent (monomer), catalyst, and accelerator, respectively. A significant decrease in the curing time occurred when the cobalt octoate concentration was increased to 0.02 wt%. Furthermore, the curing time decreased when the catalyst concentration from zero to 2 wt%. Mo et al. [15] applied microwave irradiation to the curing of an unsaturated polyester resin with the addition of CaCO3 particles and showed that microwave irradiation heated the unsaturated polyester resin evenly and rapidly, causing a chain growth reaction, thus greatly shortening the curing time (Figure 2B). Chirayil et al. [16] prepared nanocellulose reinforced unsaturated polyester composites by mechanical mixing. The curing time required for gelation in the nanocellulose-filled unsaturated polyester was shorter than that in the neat resin, indicating the catalytic action of nanocellulose in the curing reaction (Figure 2C).
2.3. Vinyl Ester
The low viscosity, rapid curing at room temperature, and relatively low cost of vinyl ester resins have led to their extensive use as matrix materials in reinforced composites. Highly viscous vinyl ester resins can be obtained by using a dispersant and various acids to reduce their surface activities.
Yong and Hahn [21] conducted a rheological analysis of SiC nanoparticle-filled vinyl ester resin systems using the Bingham, power law, Herschel–Bulkley, and Casson models. The incompatibility between hydrophilic SiC and a hydrophobic vinyl ester resin can act as the driving force for the formation of SiC aggregates, even at low particle loading (< 0.04 volume fraction), resulting in high viscosity of the resin. The optimum fractional weight percentage of dispersants (wt% dispersant/wt% SiC) for dispersion stabilization is 1–3% for particles in the 0.1–3-μm range, and it can be proposed: the addition of a dispersant at the optimum dosage lowers the viscosity of SiC/vinyl ester suspensions by 50% (Figure 3A).Gaur et al. [22] obtained the zero-shear viscosity of vinyl ester resins containing styrene (40 wt%) as the reactive diluent. The curing of vinyl ester resins can be controlled by reacting the epoxy novolac resin with methacrylic acid. They found that the curing and decomposition behavior of vinyl ester resins worsened with an increase in methacrylic acid content (11, 22, 32, 38, and 48 mg KOHg-1 solid). The cured product with the lowest acid value was the most thermally stable product. Cook et al. [23] analyzed the gel time and reaction rate of a vinyl ester resin and found that the cobalt species played a dual role in initializing the formation of radicals from MEKP and destroying primary and polymeric radicals. Based on these results, the reaction rate (determined by differential scanning calorimetry, (DSC)) increased and the gel time decreased with increasing concentrations of MEKP. However, the cobalt octoate cocatalyst retards the reaction rate, except at very low concentrations. The gel time decreased for all increases in MEKP and cobalt octoate concentrations. Curing of a vinyl ester resin with modified silicone-based additives was developed by Mazali et al. [24]. Silicone-based additives were used to modify the properties of the vinyl ester resin. For the resin cured in the absence of N,N-dimethylaniline, the silicone-based additives acted as retardants of the curing reaction, which is a typical diluent effect, whereas in the presence of this promoter, the reaction enthalpy and rate improved.
The viscosity of the vinyl ester resin could be reduced by increasing the reactive diluent content. Rosu et al. [25] found that a linear correlation between the reactive diluent content and the logarithm of viscosity, showing that the presence of reactive diluents accelerated the curing reaction and diminished the gel time. Dang et al. [26] proposed reinforcements for a comonomer vinyl ester (cVE) resin at different weight fractions up to 2% via a direct polymerization process with a eutectic gallium–indium (EGaIn) alloy and graphene nanoplatelets, showing that sub-micron sized EGaIn (≤1 wt%) could promote the curing reaction of cVE without changing the curing mechanism (Figure 3B).
2.4. Polydicyclopentadiene (p-DCPD)
Dicyclopentadiene (DCPD) is a commercially available monomer that is derived from low-viscosity petrochemicals. Polydicyclopentadiene (PDCPD) is a highly crosslinked polymer formed by ring-opening metathesis polymerization (ROMP) of its monomer precursor. Exothermic characteristics were observed during the polymerization process because of the relief of the ring strain energy initiated by the transition-metal/alkylidene complexes. Several studies investigated the effects of these catalysts.
Li et al. [27] conducted the ROMP of DCPD using the catalyst systems WCl6–Et2AlCl, (WCl6–PhCOMe)–Et2AlCl, and their polystyrene-supported counterparts. The acetophenone-modified catalyst system exhibited better catalytic properties than the unmodified system. Moreover, as the polymer yield of ROMP increased, the mechanical properties of notched impact strength (NIS) and tensile strength (TS) of the synthesized PDCPD increased. Kessler et al. [28] conducted the curing kinetics of PDCPD prepared via ROMP with three different concentrations of Grubbs’ catalyst using differential scanning calorimetry (Figure 4A). The catalyst concentration had a large effect on the curing kinetics, showing the activation energy increased significantly for 30 ℃. Yang and Lee [29] investigated the curing kinetics of endo-dicyclopentadiene (DCPD) with two types of Grubbs’ catalysts, the 1st and 2nd generation, using dynamic DSC at different heating rates.
Experimental DSC data obtained at different heating rates were used to evaluate the kinetic parameters of the model-free isoconversional and model-fitting methods. In the single DSC exotherm of the 1st generation system (Figure 4B (i)), the appearance of a shoulder above the single exotherm of the 2nd generation system (Figure 4B (ii)) suggests that reaction mechanisms other than ROMP of the norbornene and cyclopentene units may be involved in this catalyst system. The 2nd generation catalyst system showed a slower initiation rate but a faster polymerization rate than the 1st generation.
Yang and Lee [30] also studied two Grubbs’ catalysts that exhibited apparent differences in the isothermal curing of endo-dicyclopentadiene (endo-DCPD) by ROMP using the 1st and 2nd generation Grubbs’ catalysts as polymerization initiators. The 2nd generation catalyst was more efficient than the 1st generation catalyst in terms of catalytic activity, as evidenced by the reaction rates and fractional conversions (Figure 5A).
Yoo et al. [31] obtained the curing kinetics of endo-DCPD using isothermal differential scanning calorimetry by experimentally acquiring the kinetic parameters according to model-fitting approaches. With the rapid curing of DCPD, a decelerator was included in the manufacturing process. Therefore, the effect of the decelerator was investigated using the curing kinetics of endo-DCPD with different amounts of decelerator solutions, which showed that the decelerator delayed the reaction and slowed the curing process (Figure 5B and C).
3. Rapid Impregnating Thermoplastic Resins and Their Fiber-Reinforced Composites
Recently, thermoplastic composites (TPCs) have partially replaced thermosetting composites and light-metal materials, and are widely used in various fields. The market value of TPCs worldwide increases every year, from 28 billion U.S. dollars in 2019 to an estimated value of 36 billion U.S. dollars by 2024, owing to their high toughness, faster manufacturing process, high processability and recyclability, and welding, etc. [38]. However, the high melting viscosities of thermoplastic polymers require high processing temperature and pressure to fully impregnate fibers and reduce the defects of products [39]. Subsequently, in situ polymerization methods for fiber-reinforced TPCs have been developed using low-viscosity monomers or oligomeric precursors, such as caprolactam [40-43], laurolactam [44,45], methylmethacrylate (MMA) [46], and cyclic butylene terephthalate (CBT) [47,48], to fabricate fiber-reinforced polyamide 6 (PA6), polyamide 12 (PA12), polymethylmethacrylate (PMMA), and polybutylene terephthalate (PBT) composites, respectively. These monomers or oligomeric precursors are polymerized via the addition of catalysts and activators. Table 1 lists several processing parameters and applications of commonly used monomers or oligomeric precursors with low viscosities that are suitable for LCM. In this section, we introduce PA6, PA12, PMMA, and PBT thermoplastic composites and provide an overview of thermoplastic composites fabricated via in situ polymerization during LCM. Moreover, the effects of reactive processing parameters on the mechanical properties are discussed.
3.1. Polyamide 6 (PA6)
PA6 was synthesized by the anionic ring-opening polymerization of ε-caprolactam, which is a crystalline cyclic amide with a melting temperature of 70 ℃, and polymerized at 130–170 ℃ in the presence of catalyst and activator [32] (Figure 6). PA6 based fiber reinforced composites can be fabricated within 3–60 min, depending on the type and amount of the catalyst and activator used. Ahmadi et al. [49] suggested that the correct ratio of monomer, catalyst, and activator is a key component of anionic-caprolactam polymerization, leading to the lowest monomer residue and the best properties of the PA6 samples. In addition, polymerization time directly affects the production cycle and cost. Therefore, our previous research [42] focused on the effect of polymerization temperature on the degree of polymerization and polymerization time to produce perfect products with the shortest molding cycle time. The results showed that the polymerization and crystallization of PA6 occurred simultaneously during heating. As the heating rate increased, the crystallinity decreased but the degree of polymerization increased. Furthermore, the viscosity of ε-caprolactam varied almost linearly with time in the early stage, whereas it increased exponentially from 20 s after the start of polymerization, indicating the injection molding cycle time. Ben et al. fabricated glass and carbon fiber hybrid PA6 composites (Figure 7) with A (caprolactam and activator) and B (caprolactam and catalyst) mixtures via vacuum-assisted resin transfer molding (VaRTM) to evaluate their mechanical properties when applied to automobile structures [41]. The results showed that the bending, tensile, and compressive strengths of the hybrid-fiber-reinforced PA6 were 594, 315, and 297 MPa, respectively, which were comparable to those of the hybrid-fiber-reinforced fast-curing epoxy (597, 327, and 318 MPa, respectively). However, the flammability of polyamides, which is a key issue in the automobile industry, limits their widespread application. The main challenges are the inhibition of in situ polymerization in the presence of flame retardants and the insolubility of flame retardants owing to the filtration of reinforced fibers [50]. In addition, various types of flame retardants and technologies suitable for flame-retardant PA6 composite systems are summarized.
Figure 6.
Schematic of anionic ring open polymerization of PA6.
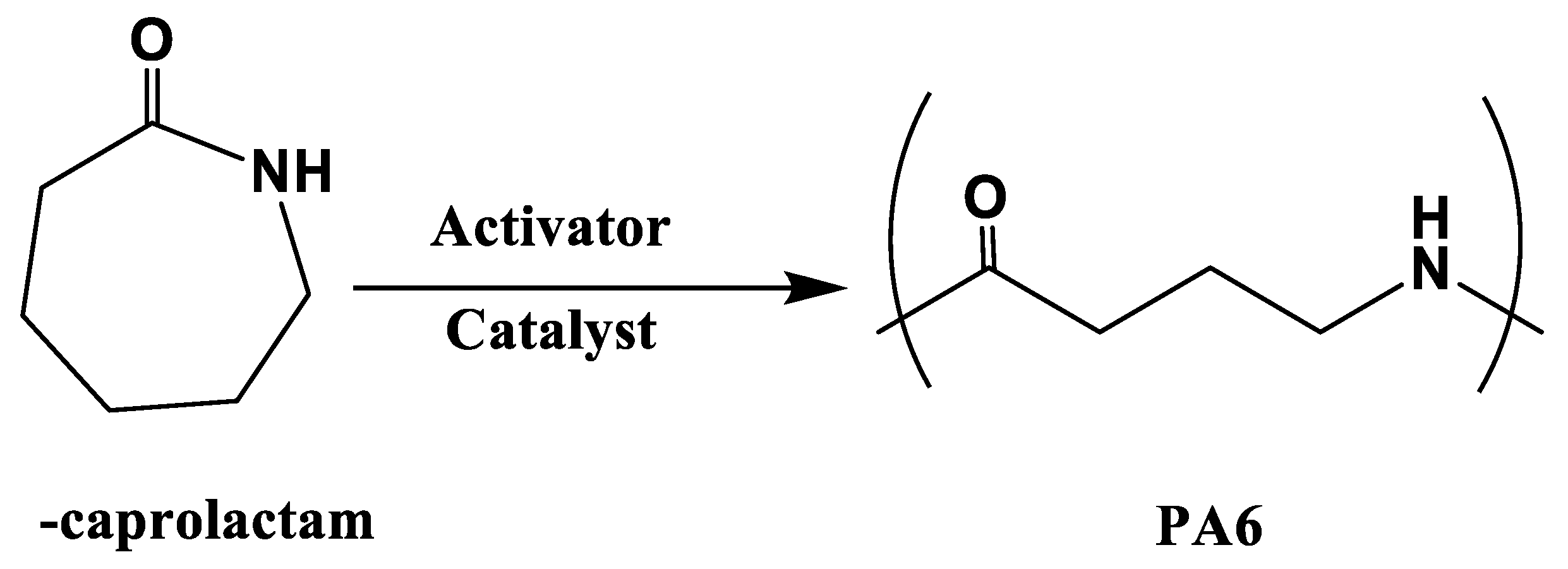
Figure 7.
Cross sections of both plates (a) hybrid fiber reinforced thermoplastic (HFRTP), (b) hybrid fibers reinforced plastic (HFRP). Reproduced with permission [41]. Copyright 2015, Elsevier.
Figure 7.
Cross sections of both plates (a) hybrid fiber reinforced thermoplastic (HFRTP), (b) hybrid fibers reinforced plastic (HFRP). Reproduced with permission [41]. Copyright 2015, Elsevier.

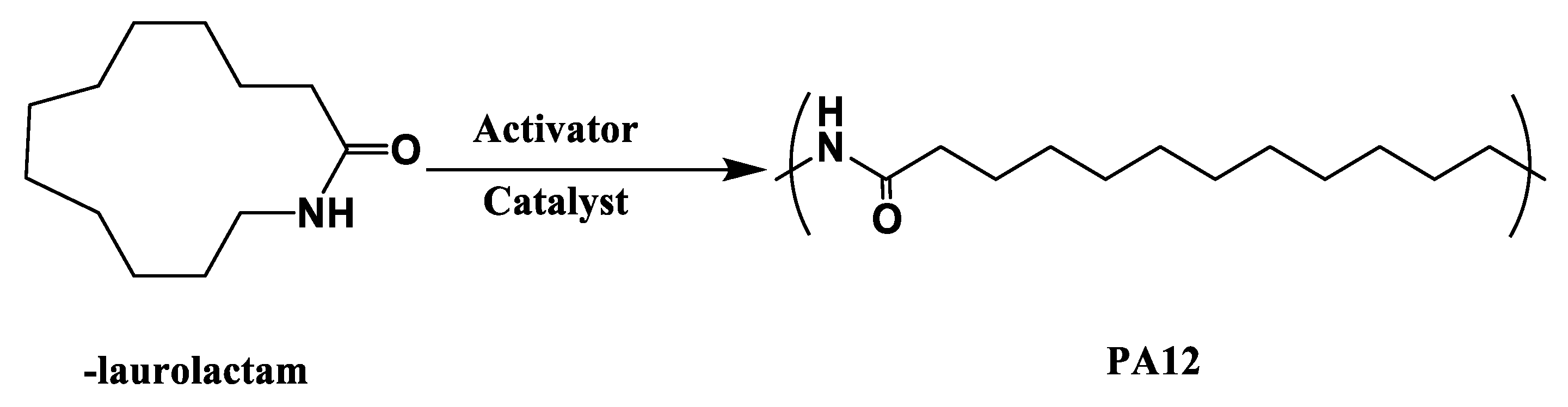
3.2. Polyamide 12 (PA12)
PA12 is called nylon 12 and is synthesized by anionic ring-opening polymerization of-laurolactam, as shown in Figure 8. ω-laurolactam has low initial viscosity above its melting point at 153 ℃, facilitating an easy and complete impregnation of fibers in the mold. Similar to PA6, PA12-based fiber-reinforced composites can be fabricated using LCM processes, such as thermoplastic resin transfer molding (T-RTM). The desired injection temperature is 170–205 ℃, and the polymerization starts at 180–250 ℃ after introducing the catalyst and initiator. Mairtin et al. [45] developed carbon fiber-reinforced PA12 composites with a 60% carbon fiber volume fraction, which exhibited high tensile strength (788.3 MPa) and high compression strength (365.7 MPa). It is also reported that the polymerization time is related to polymerization temperature, that is, it takes 8.5 min and 20 min at the mold temperatures of 240 ℃ and 200 ℃, respectively.
Figure 8.
Schematic of anionic ring open polymerization of PA12.
As listed in Table 1, PA12 is commonly used in fuel filter housing and fuel pipe connectors, which are close to the engine and are exposed to fuel and high service temperatures. Therefore, the fuel uptake and aging behavior are important factors. Wei et al. [51] found that pure PA12 showed fast and remarkably high fuel uptake when exposed to a mixture of ethanol and gasoline at 120 ℃; however, lower uptake was observed for glass fiber-reinforced PA12 composites. As shown in Figure 9, the PA12 and glass-fiber-reinforced PA12 composites gradually changed color from white to yellow as the exposure time increased, owing to the oxidation of PA12, and cracks in PA12 were larger than those in glass-fiber-reinforced PA12, indicating a suppression effect of glass fiber on fuel uptake.
Figure 9.
Images of (a) PA12 and (c) glass fiber-reinforced PA12 composites (from left to right, samples aged for 0, 150, 300, 500, and 700 h), and SEM images of the surface of (b) 150 h-aged PA12 sample and (d) 150 h-aged glass fiber reinforced PA. Reproduced with permission [52]. Copyright 2022, Springer Nature.
Figure 9.
Images of (a) PA12 and (c) glass fiber-reinforced PA12 composites (from left to right, samples aged for 0, 150, 300, 500, and 700 h), and SEM images of the surface of (b) 150 h-aged PA12 sample and (d) 150 h-aged glass fiber reinforced PA. Reproduced with permission [52]. Copyright 2022, Springer Nature.

In addition, PA12 can be recycled and used in automobiles. It has been reported that an automobile fuel-line clip produced with recycled PA12 through a selective laser sintering method provides an 8% reduction in life-cycle global warming potential and life-cycle primary energy demand compared to conventional PA66 [52] thus improving sustainability.
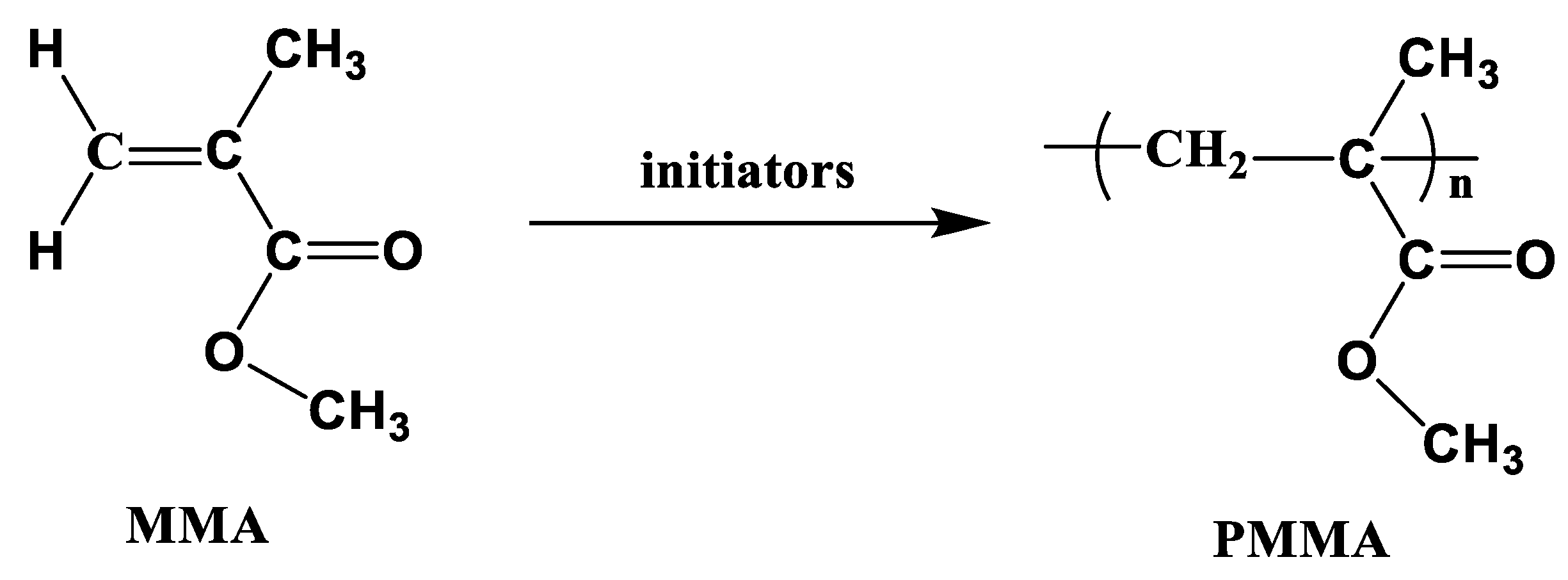
3.3. Polymethyl Methacrylate (PMMA) (Elium®)
PMMA is extensively used in the automobile industry to produce various parts and components, such as external, rear, and indicator light covers; decorative trims; ambient lighting; door entry strips; and automobile glazing, owing to its light weight, high scratch resistance, and low stress birefringence. As shown in Figure 10, PMMA was synthesized via free radical vinyl polymerization of methylmethacrylate (MMA) in the presence of peroxide initiators. The melting temperature of MMA is -48 ℃ and it is polymerized at relatively low temperature (120–160 ℃); however, the boiling temperature of MMA is 100 ℃, which makes it boil easily and cause voids in the final products. Moreover, a long cycle time (> 900 min) was required to fully polymerize MMA below its boiling temperature.
Figure 10.
Schematic of vinyl polymerization of PMMA.
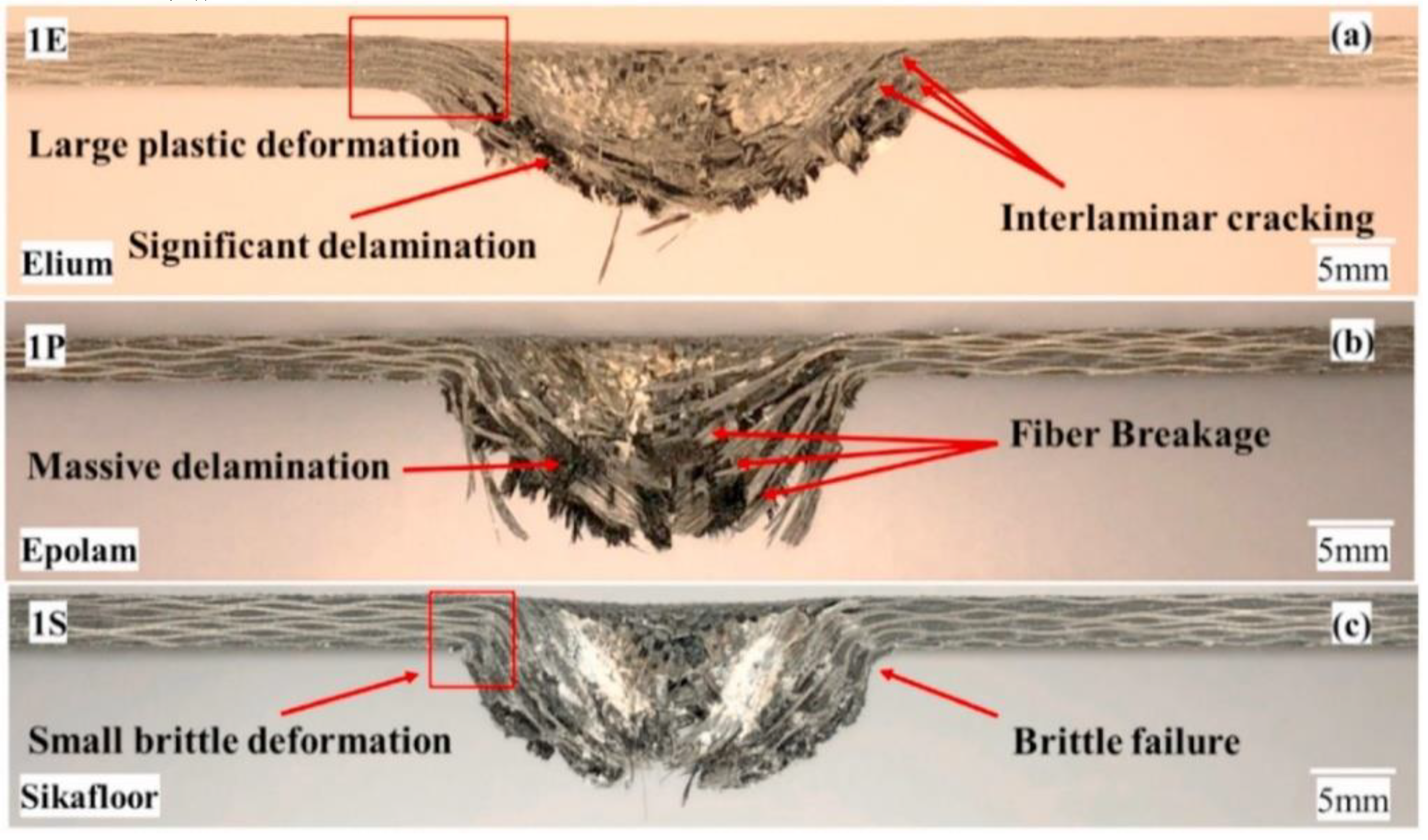
Recently, a novel liquid-reactive MMA has been developed by Arkema, named Elium®, which has low viscosity and low process temperature at room temperature with the addition of a dibenzoyl peroxide initiator [35]. Elium® can also be used to impregnate fibers through the LCM process, same as the traditional MMA. Several studies have been conducted to evaluate the mechanical properties. Kazemi et al. [53] studied the dynamic response of carbon fiber-reinforced Elium® and carbon fiber-reinforced epoxy (Epolam, Sikafloor) under low-velocity impact tests, demonstrating the higher plasticity of elium-based composites compared to epoxy-based composites, resulting in lower structural loss and lower absorbed energy. In addition, many studies have reported good mechanical properties of Elium®-based fiber-reinforced composites, such as good toughness, flexural and tensile strength, and welding performance, etc. [54,55,56].
Figure 11.
The comparison of cross-sectional observations of (a) the carbon-fiber reinforced Elium® and (b-c) thermosetting epoxy (Epolam, Sikafloor) laminates at 20 J. Reproduced with permission [54]. Copyright 2021, Elsevier.
Figure 11.
The comparison of cross-sectional observations of (a) the carbon-fiber reinforced Elium® and (b-c) thermosetting epoxy (Epolam, Sikafloor) laminates at 20 J. Reproduced with permission [54]. Copyright 2021, Elsevier.
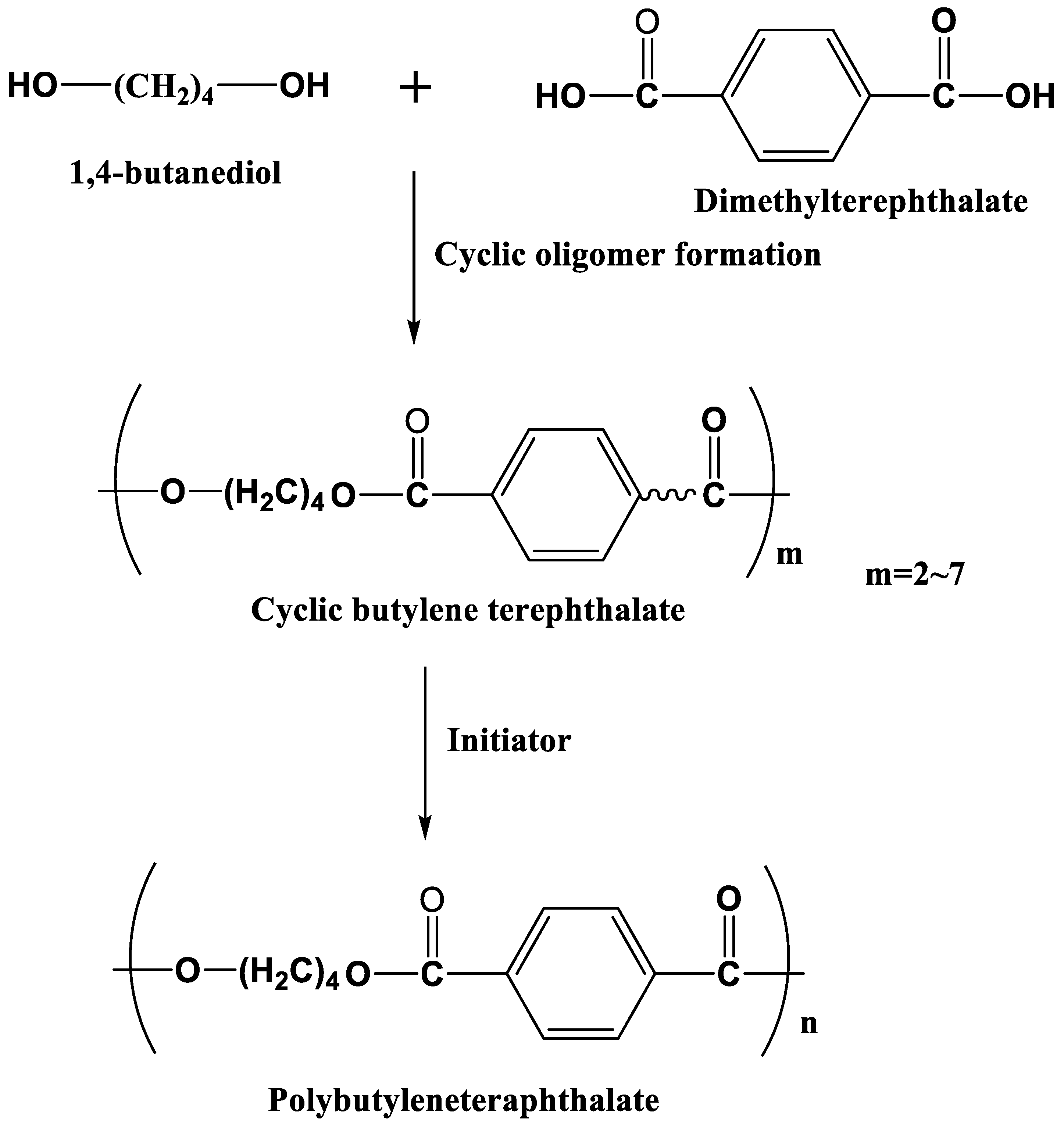
3.4. Polybutyleneteraphthalate
PBT is widely used in the automobile industry owing to its high stiffness and strength. 1,4 butanediol and dimethyltetrephthalate are used as monomers to produce macrocyclic oligomers of CBT with two to seven repeat units, [57], followed by the polymerization of semicrystalline PBT in the presence of an initiator (Figure 12). The initial viscosity of CBT is 20 mPa⸱s at 190 ℃, which is suitable for LCM processing such as RTM [58]. It is reported that PBT polymerized from CBT via RTM is more brittle than that from conventional PBT due to the high crystallinity of the polymerized PBT [59], and its toughness could be improved by the addition of nanoparticles [60,61] and fibers [62], etc. Baets et al. found that the addition of 0.05–0.1 wt% of multi-walled carbon nanotubes (MWCNTs) could increase the toughness, stiffness, and strength of PBT composites [63]. They also prepared polycaprolactone blended CBT/glass fiber composites to improve the toughness of composites [63]. Yang et al. [64] found that woven carbon fabric and glass fabric hybrid PBT composites, which are fabricated via a vacuum assisted prepreg process, have higher impact resistance than that of PBT/carbon fiber (CF) composites, although the presence of fibers may reduce the conversion of CBT. Furthermore, non-isothermal production processes, solvent blending, the addition of plasticizers, and chemical modification can enhance the toughness of CBT composites [47,65,66].
In addition, PBT can be recycled by depolymerization into CBT or monomers (1,4 butanediol and dimethyltetrephthalate), which exhibit properties comparable to those of baseline materials. Cao et al. [67] prepared super tough PBT/MWCNT/epoxidized elastomer composites with excellent mechanical properties for a wide range of PBT applications in the automobile industry.
Figure 12.
Schematic of anionic ring open polymerization of PBT.
4. Conclusion and Outlook
Rapid-impregnating resins for fiber-reinforced composites are discussed as alternatives to high-performance metal components. An overview of suitable rapid impregnating resins with low viscosity is introduced, and the differences between thermoset and thermoplastic composites are identified.
Thermoset resins, such as epoxy, polyester, vinyl ester, and DCPD, have excellent dimensional and chemical stabilities and high impact strength. The epoxy-dicyandiamide system, as a representative commercial epoxy resin, has the drawbacks of relatively high viscosity and long curing time. Therefore, many strategies have been implemented to develop low-viscosity, fast-curing epoxy resins, such as the addition of a GDE series and synthesized epoxies. The reaction time decreased with the addition of the various particles. The reinforcement of low-viscosity unsaturated polyester resins has also been introduced. Low-viscosity polyester resins can be obtained by particle synthesis. The curing time can be reduced by using various solvents and applying microwaves. Low viscosity, coupled with a rapid curing rate at room temperature and the relatively low cost of vinyl ester resins, can be obtained using dispersants and various acids to reduce the surface-active properties.
Regarding the thermosetting resins, PA-6, PA-12, PMMA (Elium®), and PBT were introduced, which have high melting viscosities and require a high processing temperature and pressure to fully impregnate the fibers and reduce defects in the products. Therefore, in situ polymerization methodologies for fiber-reinforced thermoplastic composites with low viscosities have been developed that are suitable for liquid molding processes.
Overall, extensive studies have been conducted on the characterization, analysis, and simulation of rapid-impregnating resin-based fiber-reinforced composites. However, large-scale production of such composites has been rare. Therefore, future research should focus on the large-scale production of composites for the automobile industry, reduction in their manufacturing time, and improvement in their performance.
Author Contributions
Conceptualization, M.-X.L.; data curation, H.-L.M. and Y.R.; writing—original draft preparation, M.-X.L., H.-L.M., S.K.L.; writing—review and editing, M.-X.L., H.-L.M., S.K.L., W. Z. and S.W.C.; visualization, H.-L.M. and Y.R.; supervision, S.W.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant funded by the Korean government (MOTIE) (20213000000020, Development of core equipment and evaluation technology for construction of subsea power grid for offshore wind farm).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- C. Annandarajah, A. Langhorst, A. Kiziltas, D. Grewell, D. Mielewski and R. Montazami, Materials, 2019, 12.
- J. Takahashi and T. Ishikawa, International Conference on Textile Composites, Leuven, 2013.
- J. Takahashi, K. Uzawa and T. Matsuo, American Society for Composites-25th Annual Technical Conferenc, 2010.
- Y. Yang, Y.-F. Zhao, J.-Y. Wang, C. Zhao, L. Tong, X.-Y. Liu, J.-H. Zhang and M.-S. Zhan, J. Appl. Polym. Sci., 2016, 133.
- S. M. Seraji, H. Gan, S. R. Swan and R. J. Varley, React. Funct. Polym., 2021, 164.
- S. M. Seraji, H. Gan, S. Issazadeh and R. J. Varley, Macromol. Chem. Phys., 2021, 222.
- S. M. Seraji, H. Gan, N. D. Le, J. Zhang and R. J. Varley, Polym. Int., 2022, 71, 1320–1329.
- X. Yang, W. Huang and Y. Yu, J. Appl. Polym. Sci., 2012, 123, 1913–1921.
- X. Wu, C. A. Xu, M. Lu, X. Zheng, Y. Zhan, B. Chen, K. Wang and H. Meng, High Perform. Polym., 2023, 35, 153–165.
- H. Chen, Q. Lian, W. Xu, X. Hou, Y. Li, Z. Wang and Y. Liu, J. Appl. Polym. Sci., 2021, 138.
- H. Wang, H. Wang and G. Zhou, Polym. Int., 2011, 60, 557–563.
- Chikhi, S. Fellahi and M. Bakar, Eur. Polym. J., 2002, 38, 251–264.
- Z. Zhang, R. Pinnaratip, K. G. Ong and B. P. Lee, J. Appl. Polym. Sci., 2020, 137’.
- Y. C. Chen, S. X. Zhou, G. D. Chen and L. M. Wu, Prog. Org. Coat., 2005, 54, 120–126.
- Q. Mo, Y. Huang, L. Ma, W. Lai, Y. Zheng, Y. Li, M. Xu and Z. Huang, Polymers, 2022, 14.
- C. J. Chirayil, L. Mathew, P. A. Hassan, M. Mozetic and S. Thomas, Int. J. Biol. Macromol., 2014, 69, 274–281.
- D. Zhang, J. Wang, T. Li, A. Zhang and D. Jia, Chem. Eng. Technol., 2011, 34, 119–126.
- L. Zhou, R. Zhou, J. Zuo, S. Tu, Y. Yin and L. Ye, Mater. Res. Express, 2019, 6.
- Z. Yuan, Q. Liu, X. Pan, J. Wang, M. Jin and J. Li, Prog. Org. Coat., 2021, 157.
- E. S. Nasr and A.-A. A. Abdel-Azim, Polym. Adv. Technol., 1992, 3, 407–411.
- V. Yong and H. T. Hahn, J. Appl. Polym. Sci., 2006, 102, 4365–4371.
- B. Gaur and J. S. P. Rai, Polym. Plast. Technol. Eng., 2006, 45, 197–203.
- W. D. Cook, G. P. Simon, P. J. Burchill, M. Lau and T. J. Fitch, J. Appl. Polym. Sci., 1997, 64, 769–781.
- C. A. I. Ittner Mazali and M. I. Felisberti, Eur. Polym. J., 2009, 45, 2222–2233.
- L. Rosu, C. N. Cascaval and D. Rosu, J. Optoelectron. Adv. Mater., 2006, 8, 690–693.
- T. K. M. Dang, M. Nikzad, T. V. Khanh, S. Masood, C. K. Nguyen and I. Sbarski, Polymers, 2022, 14.
- H. Li, Z. Wang and B. He, J. Mol. Cat. A Chem., 1999, 147, 83–88.
- M. R. Kessler and S. R. White, J. Polym. Sci. A Polym. Chem., 2002, 40, 2373–2383.
- G. Yang and J. K. Lee, Thermochim. Acta, 2013, 566, 105–111.
- G. Yang and J. K. Lee, Ind. Eng. Chem. Res., 2014, 53, 3001–3011.
- H. M. Yoo, J. H. Jeon, M.-X. Li, W. I. Lee and S. W. Choi, Compos. B Eng., 2019, 161, 439–454.
- K. van Rijswijk and H. E. N. Bersee, Compos. A, 2007, 38, 666–681.
- P. Rosso, K. Friedrich, A. Wollny and R. Mülhaupt, J. Thermoplast. Compos. Mater., 2005, 18, 77–90.
- Y. Wei, M. Cui, Z. Ye and Q. Guo, J. Clean. Prod., 2021, 291, 125246’.
- M. Bodaghi, C. H. Park and P. Krawczak, Front. Mater., 2022, 9.
- N. T. Tran and N. T. H. Pham, Int. J. Polym. Sci., 2021, 2021, 1–7.
- M. M. Davoodi, S. M. Sapuan, D. Ahmad, A. Aidy, A. Khalina and M. Jonoobi, Mater. Lett., 2012, 67, 5–7.
- J. Observer, JEC Group, Paris, 2020.
- E. Akca and A. Gursel, Periodicals of engineering and natural, Sciences (PEN), 2015, 3.
- G. Ben, A. Hirabayashi, K. Sakata, K. Nakamura and N. Hirayama, Sci. Eng. Compos. Mater., 2015, 22, 633–641.
- G. Ben and K. Sakata, Compos. Struct., 2015, 133, 1160–1167.
- M. X. Li, D. Lee, G. H. Lee, S. M. Kim, G. Ben, W. I. Lee and S. W. Choi, Polymers, 2020, 12.
- M. X. Li, H. L. Mo, Y. Ren and S. W. Choi, Coatings, 2022, 12.
- L. Zingraff, V. Michaud, P.-E. Bourban and J.-A. E. Månson, Compos. A, 2005, 36, 1675–1686.
- P. Ó Máirtín, P. McDonnell, M. T. Connor, R. Eder and C. M. Ó Brádaigh, Compos. A, 2001, 32, 915–923.
- M. Banerjee, S. Sain, A. Mukhopadhyay, S. SenGupta, T. Kar and D. Ray, J. Appl. Polym. Sci., 2014, 131, n/a–n/a.
- T. Abt and M. Sánchez-Soto, Crit. Rev. Solid State Mater. Sci., 2017, 42, 173–217.
- C. Yan, L. Liu, Y. D. Zhu, H. B. Xu and D. Liu, J. Thermoplast. Compos. Mater., 2018, 31, 181–201.
- S. Ahmadi, J. Morshedian, S. A. Hashemi, P. J. Carreau and W. Leelapornpisit, Iran. Polym. J., 2010, 19, 229–240.
- Z. Kovács, Á. Pomázi and A. Toldy, Polym. Degrad. Stab., 2022, 195.
- X. F. Wei, K. J. Kallio, R. T. Olsson and M. S. Hedenqvist, npj Mater. Degrad., 2022, 6.
- D. He, H. C. Kim, R. De Kleine, V. K. Soo, A. Kiziltas, P. Compston and M. Doolan, J. Ind. Ecol., 2022, 26, 1378–1388.
- M. E. Kazemi, L. Shanmugam, A. Dadashi, M. Shakouri, D. Lu, Z. Du, Y. Hu, J. Wang, W. Zhang, L. Yang and J. Yang, Compos. B Eng., 2021, 207.
- S. K. Bhudolia, G. Gohel, L. K. Kah Fai and R. J. Barsotti, Mater. Lett., 2020, 264.
- S. K. Bhudolia, G. Gohel, K. F. Leong and R. J. Barsotti, Materials, 2020, 13.
- W. Obande, C. M. O. Ó Brádaigh and D. Ray, Compos. B Eng., 2021, 215.
- D. J. Brunelle, J. E. Bradt, J. Serth-Guzzo, T. Takekoshi, T. L. Evans, E. J. Pearce and P. R. Wilson, Macromolecules, 1998, 31, 4782–4790.
- Z. A. M. Ishak, K. G. Gatos and J. Karger-Kocsis, Polym. Eng. Sci., 2006, 46, 743–750.
- H. Parton, J. Baets, P. Lipnik, B. Goderis, J. Devaux and I. Verpoest, Polymer, 2005, 46, 9871–9880.
- J. Baets, A. Godara, J. Devaux and I. Verpoest, Compos. A, 2008, 39, 1756–1761.
- G. Broza, M. Kwiatkowska, Z. Rosłaniec and K. Schulte, Polymer, 2005, 46, 5860–5867.
- Y. Swolfs, L. Gorbatikh and I. Verpoest, Compos. A, 2014, 67, 181–200.
- J. Baets, A. Godara, J. Devaux and I. Verpoest, Polym. Degrad. Stab., 2010, 95, 346–352.
- B. Yang, Z. Q. Wang, L. M. Zhou, J. F. Zhang and W. Y. Liang, Compos. Struct., 2015, 132, 464–476.
- T. Abt, M. Sánchez-Soto, S. Illescas, J. Aurrekoetxea and M. Sarrionandia, Polym. Int., 2011, 60, 549–556.
- J. Baets, J. Devaux and I. Verpoest, Adv. Polym. Technol., 2010, 29, 70–79.
- Cao, P. W. Xu, B. G. Wu, M. Hoch, P. J. Lemstra, W. J. Yang, W. F. Dong, M. L. Du, T. X. Liu and P. M. Ma, Compos. Sci. Technol., 2020, 190.
Figure 1.
(A) Effect of glycol diglycidyl ether (GDE) content on the rheological behavior of acrylate-based epoxy resin (AE)-40/LPA system (i) 40 ℃, (ii) 50 ℃, (iii) 80 ℃, (iv) 20 phr GDE content. Reproduced with permission [4]. Copyright 2015, John Wiley and Sons. (B) DSC curves for (i) three neat epoxy resins and (ii) DGEBA/DGEBAEO-6 cured by DDM. Reproduced with permission [5]. Copyright 2011, John Wiley and Sons. (C) Rheological behaviors of epoxies (without curing agent) (i) Change in viscosity with shear rate from 0.01 to 60 s-1 at 25 ℃, (ii) Change in viscosity with temperature from 25 to 100 ℃ at a shear rate of 60 s-1. Reproduced with permission [6]. Copyright 2022, John Wiley and Sons.
Figure 1.
(A) Effect of glycol diglycidyl ether (GDE) content on the rheological behavior of acrylate-based epoxy resin (AE)-40/LPA system (i) 40 ℃, (ii) 50 ℃, (iii) 80 ℃, (iv) 20 phr GDE content. Reproduced with permission [4]. Copyright 2015, John Wiley and Sons. (B) DSC curves for (i) three neat epoxy resins and (ii) DGEBA/DGEBAEO-6 cured by DDM. Reproduced with permission [5]. Copyright 2011, John Wiley and Sons. (C) Rheological behaviors of epoxies (without curing agent) (i) Change in viscosity with shear rate from 0.01 to 60 s-1 at 25 ℃, (ii) Change in viscosity with temperature from 25 to 100 ℃ at a shear rate of 60 s-1. Reproduced with permission [6]. Copyright 2022, John Wiley and Sons.
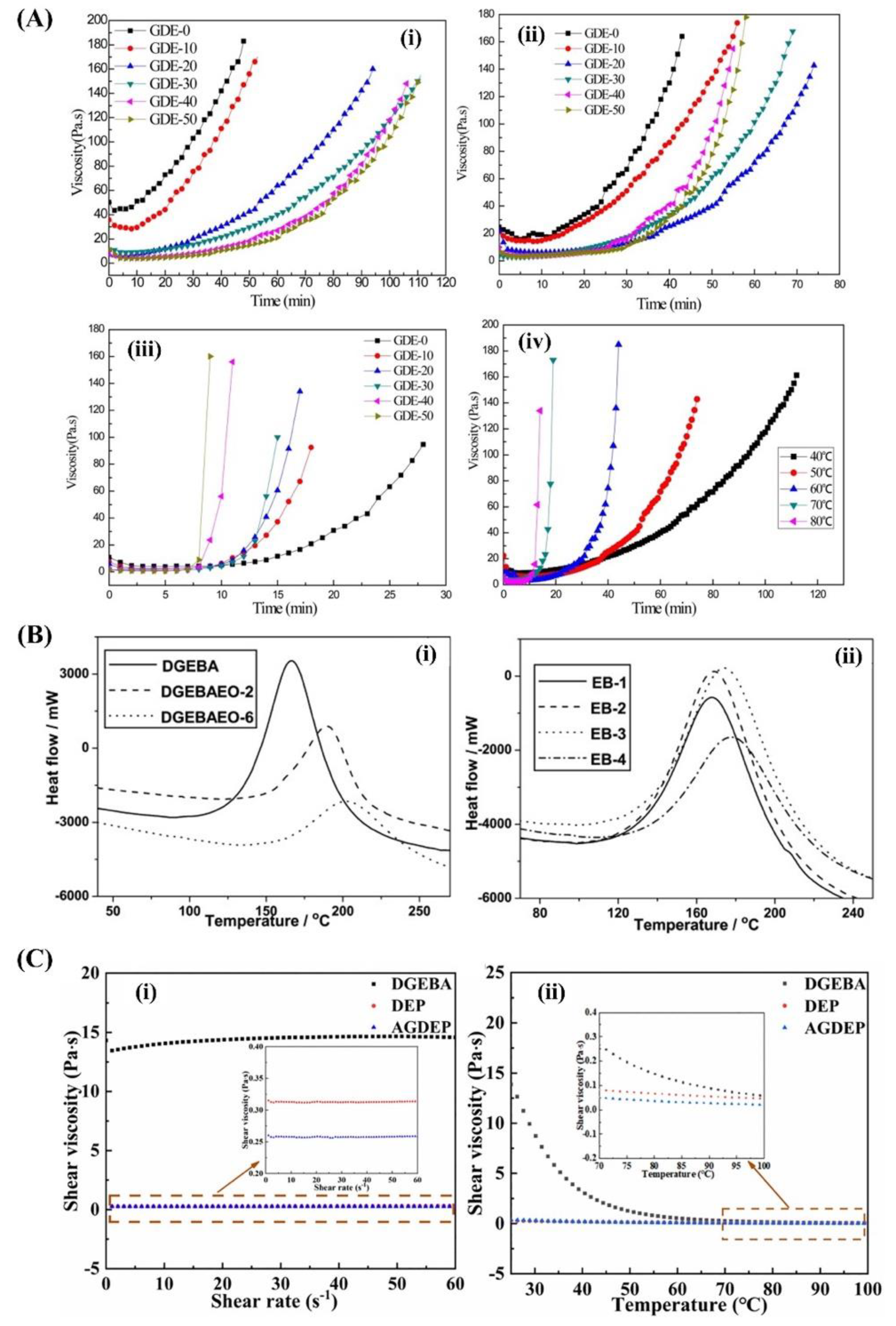
Figure 1.
(A) Effect of silica content on the viscosity of nanocomposite resins embedded by silica sol S2 or S9. Reproduced with permission [7]. Copyright 2005, Elsevier. (B) DSC curves of liquid UPR and cured samples with microwave curing and thermal curing, respectively. Reproduced with permission [15]. Copyright 2022, MDPI. (C) Variation of viscosity with time for NC filled composites. Reproduced with permission [16]. Copyright 2014, Elsevier.
Figure 1.
(A) Effect of silica content on the viscosity of nanocomposite resins embedded by silica sol S2 or S9. Reproduced with permission [7]. Copyright 2005, Elsevier. (B) DSC curves of liquid UPR and cured samples with microwave curing and thermal curing, respectively. Reproduced with permission [15]. Copyright 2022, MDPI. (C) Variation of viscosity with time for NC filled composites. Reproduced with permission [16]. Copyright 2014, Elsevier.
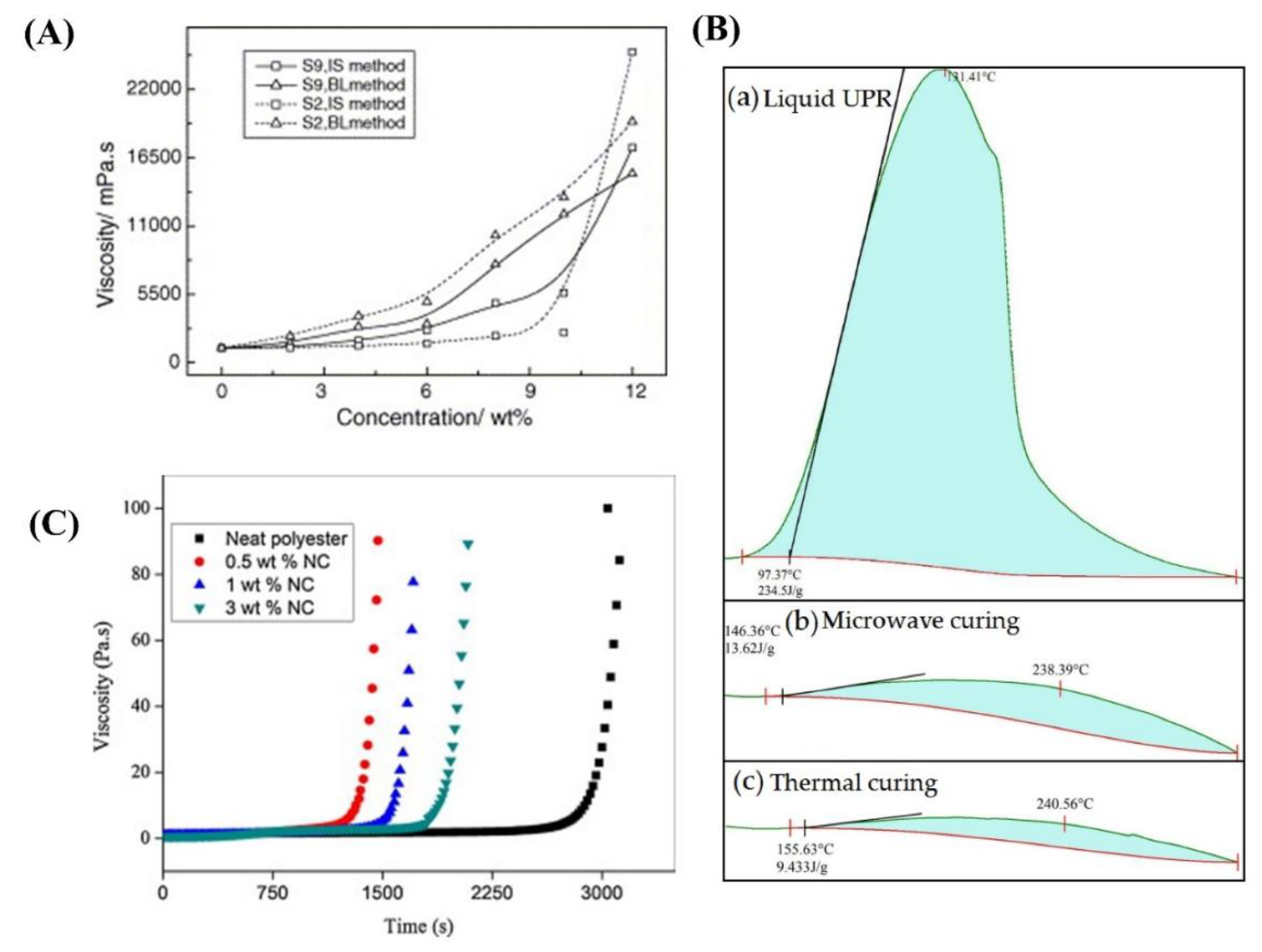
Figure 3.
(A) (i) Viscosity curves of SiC/vinyl ester resin systems with and without MPS/W966. (ii) Viscosity curves of SiC/vinyl ester resin systems with and without 1-octanol. Reproduced with permission [21]. Copyright 2006, John Wiley and Sons. (B) The DSC graphs (i), degree of conversion at 60 ℃, (ii), graphs of TGA (iii), and DTG (iv) at the heating rate of 5 ℃/min of the LM filled and unfilled comonomer vinyl ester composites. Reproduced with permission [26]. Copyright 2022, MDPI.
Figure 3.
(A) (i) Viscosity curves of SiC/vinyl ester resin systems with and without MPS/W966. (ii) Viscosity curves of SiC/vinyl ester resin systems with and without 1-octanol. Reproduced with permission [21]. Copyright 2006, John Wiley and Sons. (B) The DSC graphs (i), degree of conversion at 60 ℃, (ii), graphs of TGA (iii), and DTG (iv) at the heating rate of 5 ℃/min of the LM filled and unfilled comonomer vinyl ester composites. Reproduced with permission [26]. Copyright 2022, MDPI.
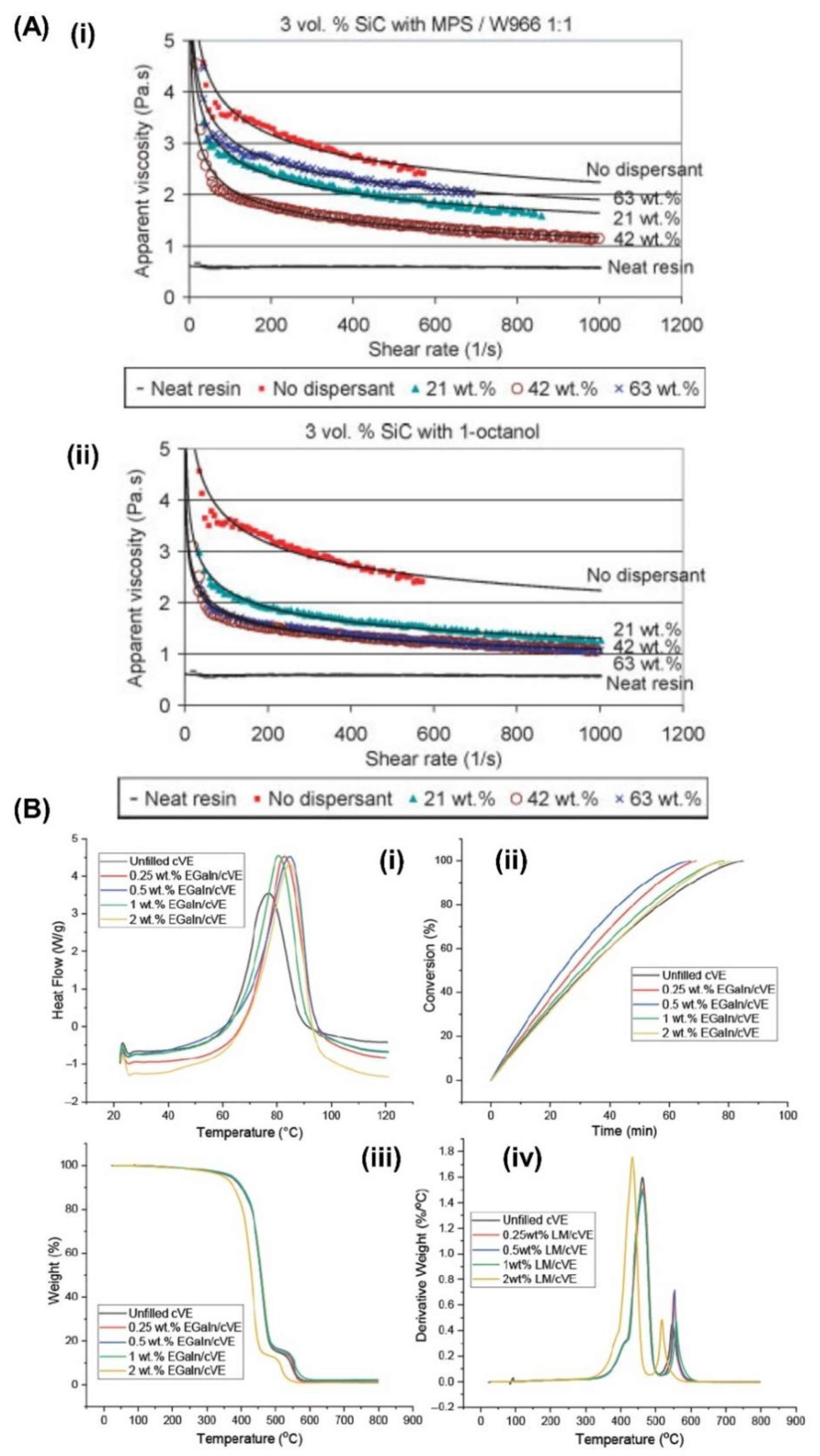
Figure 4.
(A) The DSC curves for (i) low concentration (ii) medium-concentration, and (iii) high-concentration DCPD and Grubbs’ catalyst samples, (iv) predictions for isothermal curing at 30°C based on the model-free isoconversional method for low, medium, and high catalyst concentrations. Reproduced with permission [28]. Copyright 2002, John Wiley and Sons. (B) DSC scans at different heating rates for endo-DCPD with (i) the 1st generation and (ii) the 2nd generation Grubbs’ catalysts (inset shows the shoulder region). Reproduced with permission [29]. Copyright 2013, Elsevier.
Figure 4.
(A) The DSC curves for (i) low concentration (ii) medium-concentration, and (iii) high-concentration DCPD and Grubbs’ catalyst samples, (iv) predictions for isothermal curing at 30°C based on the model-free isoconversional method for low, medium, and high catalyst concentrations. Reproduced with permission [28]. Copyright 2002, John Wiley and Sons. (B) DSC scans at different heating rates for endo-DCPD with (i) the 1st generation and (ii) the 2nd generation Grubbs’ catalysts (inset shows the shoulder region). Reproduced with permission [29]. Copyright 2013, Elsevier.
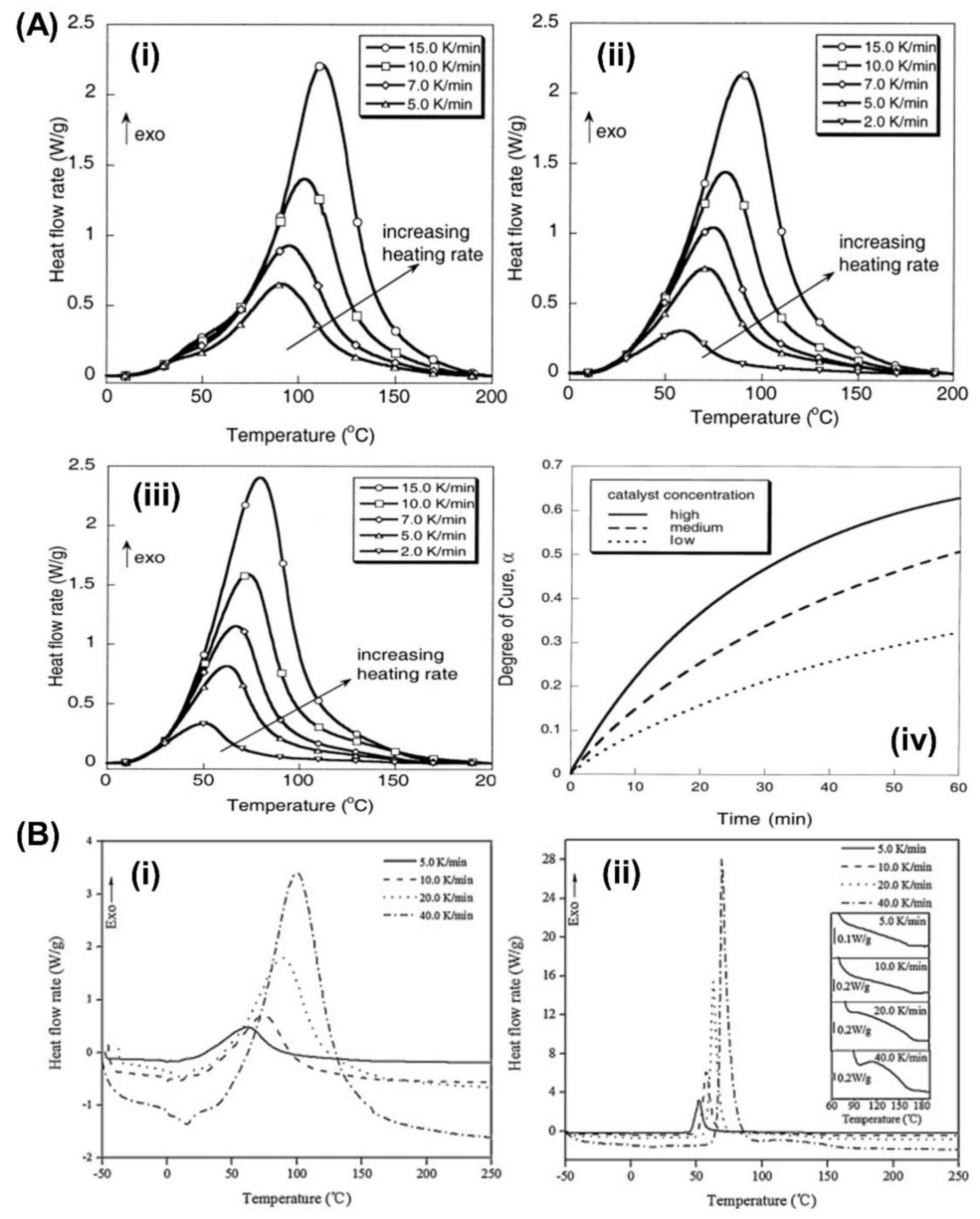
Figure 5.
(A) Dynamic DSC scans following the isothermal cure of endo-DCPD with (i) the 1st generation and (ii) the 2nd generation Grubbs' catalysts. (iii) Activation energy as a function of the fractional conversion for endo-DCPD with the 1st and the 2nd generation Grubbs' catalysts. Reproduced with permission [30]. Copyright 2014, American Chemical Society. (B) Dynamic DSC curve with endo-DCPD with different amounts of decelerators. Reproduced with permission [31]. Copyright 2019, Elsevier. (C) Isothermal DSC profiles of endo-DCPD with different amounts of decelerators: (i) 0.5 wt mass %, (ii) 1.0 wt mass %, (iii) 1.5 wt mass %, (iv) 2.0 wt mass %. Reproduced with permission [32]. Copyright 2019, Elsevier.
Figure 5.
(A) Dynamic DSC scans following the isothermal cure of endo-DCPD with (i) the 1st generation and (ii) the 2nd generation Grubbs' catalysts. (iii) Activation energy as a function of the fractional conversion for endo-DCPD with the 1st and the 2nd generation Grubbs' catalysts. Reproduced with permission [30]. Copyright 2014, American Chemical Society. (B) Dynamic DSC curve with endo-DCPD with different amounts of decelerators. Reproduced with permission [31]. Copyright 2019, Elsevier. (C) Isothermal DSC profiles of endo-DCPD with different amounts of decelerators: (i) 0.5 wt mass %, (ii) 1.0 wt mass %, (iii) 1.5 wt mass %, (iv) 2.0 wt mass %. Reproduced with permission [32]. Copyright 2019, Elsevier.


Table 1.
Processing temperatures and processing times of various monomers.
| Monomer | Processing temp. (℃) |
Processing Time (min) |
Application | Ref. | |
| Thermoset | Epoxy | 120 | 180 | Aerospace, marine, automobile, construction |
6, 8,9 |
| Polyester | 40-80 | 120 | Construction, marine, chemical |
18 | |
|
Vinyl ester |
50-200 | < 240 | Coatings, printed circuit boards, building materials, automotive parts, and fiber-reinforced composites |
22,23 | |
| p-DCPD | 50-90 | 2 | Sporting goods, automotive industries as well as military and aerospace applications | 31 | |
| Thermoplastic | PA-6 | 130-170 | 3-60 | Wipers, gears, bearings, and weather proof coatings |
32 |
| PA-12 | 180-240 | with cooling process | Fuel pipes, fuel filters, high pressure oil pipes, gears, brake hoses |
33,34 | |
|
PMMA (Elium®) |
80-160 (room temp.~) |
@low emp.: > 900 @high temp.: Boiling of monomer |
Decorative trims; interior lighting; door entry strips | 32,35 | |
| PBT | 180-250 | < 30 | Door handles, bumpers | 36,37 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



