
Submitted:
10 August 2023
Posted:
14 August 2023
You are already at the latest version
Abstract
Littoraria melanostoma (Gray, 1839) is a most common species of gastropods in mangroves. They quickly respond during the early stage of mangrove restoration and usually form a dominant community within a certain period. We characterized the complete mitochondrial genome of the species. The whole mitogenome of L. melanostoma was 16,149 bp in length and its nucleotide composition showed high AT-content of 64.16%. It had 37 genes, including 13 protein-coding genes, two ribosomal RNA genes, and 22 transfer RNA genes, one control region between tRNA-Phe and COX3. The A/T compositions in the control region is 74.7%, respectively, and are much higher than the overall A/T composition of the mitochondrial genomes. The amino acid composition and codon usage of the mitochondrial genomes of from eight superfamilies of Littorinimorpha were analyzed, and the results showed that CUU (Leu), GCU (Ala), AUU (Ile), UCU (Ser), UUA (Leu), GUU (Gly) and UUU (Phe) are the commonly used codons in this order. The results of phylogenetic analysis were roughly consistent with morphological classification, which showed that L. melanostoma is closely related to L. sinensis, a rock-dwelling species that is widespread in the coastal intertidal zone of China. These results may provide a basis to understand the phylogeny and evolution of the Littorinimorpha.
Keywords:
mangrove
; mitochondrial genome
; Littoraria melanostoma
; phylogeny
; Littorinimorpha
1. Introduction
Mangroves are woody plant communities that become established on the intertidal flats of tropical and subtropical coasts. The harsh natural conditions of the coastal intertidal zone and the unique advantages of the transition zone between land and sea have produced the unique biome of the mangrove ecosystem. In addition, the resulting extremely rich biodiversity also plays a crucial role in the ecosystem. As one mollusk group in mangroves, gastropods play an important role in the detritus cycle and consume a large amount of plant tissue and humus [1].
Littorinidae, one of the major groups of arboreal gastropods in mangrove forests, adapt to the special environment of the coastal intertidal zone through numerous physiological and ecological manners, including their multiple reproductive means, which are known as a variable reproductive strategy [2]; complex food composition [3]; vertical climbing ability [4-6]; and variable shell color and shape [7]. Because gastropods have a limited ability to move, they generally only move on the same tree without external interference [8], and it is difficult for them to migrate from the intertidal zone to land. Consequently, they have a relatively fixed pattern of spatial distribution [9,10]. Simultaneous studies have shown that species of Littorinidae respond quickly to the restoration of mangrove vegetation, and they can form a dominant community in the early stage of vegetation restoration as exemplified by L. melanostoma [11]. Most of the Littorinidae in mangroves live on mangrove and salt marsh plants, driftwood, and stakes.
The family Littorinidae (Children, 1834) comprises more than 200 species that are common members of marine intertidal communities around the world, and most of them only live on mangrove plants[12]. Species of Littorinidae that have been reported in China include L. melanostoma, L. ardouiniana, L. intermedia, L. pallescens, L. articulata and L. brevicula [13-15]. L. melanostoma is often found in the branches and leaves of mangroves (Figure 1).
Morphological differences make the identification of gastropods confused, and the classification information is constantly adjusted. In recent years, molecular systematic studies were used to analyze the evolution and phylogeny of gastropods. Reid et al used nuclear 28S rRNA, mitochondrial 12S rRNA and COI to construct the phylogeny of family Littorininae [12]and genus Littoraria [16]. Li et al used complete mitochondrial genome sequences to analyze the phylogenic relationship between genus Littorina and Littoraria, but only four species were used in this study[17]. Phylogeny of Stromboidea, a superfamily in Littorinimorpha was studied based on 13 mitochondrial protein-coding genes[18]. By searching in Genbank, we found that the number of gastropods with available mitogenomes has increased, but there are few studies on the phylogenetic analyses that investigate relationships across the Littorinimorpha.
In this study, L. melanostoma, one of the most common gastropoda species in the mangrove wetlands of China, was studied for its molecular evolution and phylogeny. The mitochondrial genomes of L. melanostoma was sequenced, and the genome structure, base composition, codon usage, intergenic region, and codon preference of the mitochondrial genomes were analyzed. In addition, a phylogenetic tree of 30 species from 8 superfamilies of Littorinimorpha based on 13 PCGs was constructed using the maximum likelihood (ML) method. This study may increase our understanding of the phylogeny and evolution of Littorinimorpha.
2. Materials and methods
2.1. Sample collection and DNA extraction
Specimens of L. melanostoma was obtained from mangrove wetlands in Beihai, Guangxi, China (21.57°N, 109.16°E) and vouchered in the specimen room of the Guangxi Mangrove Research Center (Accession numbers LM#11-20, respectively). Since this species is unprotected invertebrates, no specific permission was required to collect samples from these locations. Total genomic DNA was obtained from the muscles of individuals using a QIAamp DNA Micro Kit (Qiagen, Hilden, Germany).
2.2. Sequencing, assembling and analysis
The gene cox1 was amplified using the universal primers LCO1490 and HCO2198 by standard PCR method [19]. The extracted DNA was sequenced using a NovaSeq 6000 platform (Illumina, San Diego, CA), and the mitogenome was assembled with NOVOPlasty v2.7.0 [20] and annotated with MitoZ v2.4 [21]. Protein-coding genes (PCGs) were determined by determining the open reading frames (ORFs) based on the invertebrate mitochondrial genetic code, and rRNAs and tRNAs were identified using the MITOS Web Server (http://mitos2.bioinf.uni-leipzig.de/index.py) [22]. The codon usage was calculated using MEGA 7.0 [23] . Strand bias was calculated using the following formulae: AT-skew = (A−T)/(A+T) and GC-skew = (G−C)/(G+C) [24]. The circular maps of the mitochondrial genomes were drawn using the online mitochondrial visualization tool Organellar Genome DRAW [25]. The nucleotide composition, codon usage, and comparative mitogenomic architecture tables for the two mitogenomes and data that were used to plot the relative synonymous codon usage (RSCU) figures were all calculated/created using PhyloSuite [26].
2.3. Phylogenetic analysis
The nucleotide sequences of the complete mt genomes from 30 species (Table 1), including five species from Littorinoidea, 24 species from other superfamilies of Littorinimorpha, and Ovatella vulcani as an outgroup, were downloaded from GenBank. A total of 30 amino acid sequences were aligned using MAFFT v.7.215 [27] and trimmed with trimAl v.1.4.1 [28] with the heuristic method ‘automated1.’ The phylogenetic tree was reconstructed using IQ-TREE v2 [29] based on ML with the partitioning method [30], and Branch support analysis was conducted using 10,000 ultrafast bootstrap replicates.
3. Results and discussion
3.1. Genome structure and organization
The complete L. melanostoma mitochondrial genome was 16,149 bp long, and it was uploaded to GenBank after annotation (ACCESSION ID: NC064398). The gene compositions of L. melanostoma was the same as most of the mitochondrial genomes of gastropods that have been published. Contained 37 genes, including 13 protein-coding, two rRNA and 22 tRNA genes [21,31] (Figure 2). According to the difference in G+T content, the two strands of mitochondrial DNA could be separated into a heavy strand (H strand) and a light strand (L strand). The 13 protein-coding genes in the mitochondrial genomes of L. melanostoma is located on the H strand, which is consistent with the findings of previous studies that showed that the mitochondrial genomes of Littorinidae, such as Littorina. fabalis, Littorina. obtusata and Littorina. saxatilis, harbor protein-encoding genes on the H strand [31]. Most genes are on the H strand except the eight tRNAs that are located on the L strand, and include trnM (CAU), trnY (GUA), trnC (GCA), trnW (UCA), trnQ (UUG), trnG (UCC), trnE (UUC), and trnT (UGU). The 13 PCGs include seven NADH dehydrogenase genes (complex I)—ND1, ND2, ND3, ND4, ND4L, ND5, and ND6; three cytochrome c oxidase genes (complex IV)—COX1, COX2, and COX3; two ATPase subunits (ATP6 and ATP8); and one cytochrome b gene.
Table 2 summarized the proportions of gene bases and protein-coding gene sequence bases in the complete mitochondrial genome sequences of L. melanostoma. The base composition of the L. melanostoma mitochondrial genome was 29.79% A, 34.37% T, 14.66% G and 21.18% C. The A+T content (64.16%) of its mitochondrial genes was higher than the G+C content (35.84%), and the A+T content of protein-coding genes was 62.39%. These results show L. melanostoma genomes display an obvious nucleotide composition that is biased to A+T, which is consistent with the other genomes of Littorinidae species that have been reported [31]. The base composition bias is usually reflected by AT skew and GC skew. The calculated AT skew and GC skew of the L. melanostoma mitochondrial genome were 0.071 and -0.182, respectively. These data indicate that the bases T and C appear more frequently than A and G in the mitochondrial genomes of L. melanostoma.
3.2. PCGs and codon usage
The nucleotide lengths of the 13 protein-coding genes of L. melanostoma is 11,034 bp, which encode 3,678 amino acid residues, respectively. Most protein-coding genes start with ATN and end with TAA or TAG codons (Table 3).
The codon usage of 13 protein-coding genes in the L. melanostoma mitochondrial genomes is shown in Table 3. In the L. melanostoma genome, only one gene (ND3) used ATA as the start codon, and two used ATT as the start codon, namely ND4 and ND5. The remaining 10 genes (COX1, COX2, ATP8, ATP6, ND1, ND6, CYTB, ND4L, COX3, and ND2) all used ATG as the start codon. ND4L, ND5 and ND2 used TAG, CTT and AAT as the stop codon, respectively, and these codons were each used by one gene only. There were 10 genes (COX1, COX2, ATP8, ATP6, ND1, ND6, CYTB, ND4, COX3, and ND3) that used TAA as the stop codon.
Figure 3 shows the amino acid composition and codon usage of the mitochondrial genomes of eight species from eight superfamilies. The results showed that CUU (Leu), GCU (Ala), AUU (Ile), UCU (Ser), UUA (Leu), GUU (Gly) and UUU (Phe) were the most commonly used codons. These observations suggest that there is a strong AT bias for protein-coding genes in the mitochondrial genomes of Littorinidae animals.
3.3. Ribosomal and transfer RNA genes
Two rRNA genes, l-rRNA and s-rRNA, are located between trnL (UAA) and trnV (UAA) and between trnV (UAA) and trE (UUC), respectively.
In the mitochondrial genome of L. melanostoma, l-rRNA was 1,419 bp, and s-rRNA was 901 bp (Table 3). A total of 22 tRNA genes was found in L. melanostoma, and its cloverleaf structures was 65 –72 bp.
3.4. Intergenic spaces and overlapping sequences
There were five overlapping gene regions in the mitochondrial genome of L. melanostoma, which ranged from 1 to 22 bp in length, and 30 intergenic regions, which ranged from 1 to 773 bp long. The longest intergenic region was located between trnF (GAA) and COX3 (Table 3).
3.5. Control regions
The control region (CR) of mitochondrial DNA is the primary non-coding region of the mitochondrial genome of animals, also known as the D-loop region, which is a key part for the replication and transcription of the mitochondrial genome and regulates the replication and transcription of the mitochondrial genome. During the process of evolution, since the selection pressure that acts on this region is relatively non-intrusive, the CR usually displays the largest sequence and variation in length, the highest rate of evolution, and is the most polymorphic in the mitochondrial genome [32]. However, since the UTR sequences of invertebrates are poorly conserved, there is no defined CR in their mitochondrial genomes[33]. For example, Marques studied the genomes of L. fabalis, L. obtusata, and L. saxatilis and found a region that contained some unique features, such as a non-coding region with a hairpin structure and a tandem repeat sequence, located between tRNA-Phe (trnF [GAA]) and COX3, and an AT content that was higher than the overall AT content in the mitochondrial genome. This region was then predicted as the CR. Similar to those results, we found a non-coding sequence that contained some unique features in the L. melanostoma genomes. It was between tRNA-Phe and COX3, and the AT content was 74.7%, respectively. This is much higher than the AT content of the mitochondrial genomes (64.16%). Thus, we consider that this region is a unique non-coding region of the Littoraria genus, which may play a regulatory role in the replication and transcription of the mtDNA of this genus.
3.6. Phylogenetic analyses
To further study the genetic background and taxonomic relationship of L. melanostoma, the complete mitochondrial genome sequences of L. melanostoma was compared with the complete mitochondrial genome sequences of 28 other species from 8 superfamily of the Littorinimorpha. Ovatella vulcani was utilized as an outgroup, and a phylogenetic tree was constructed based on 13 PCGs using IQ-TREE v2 with the ML method (Figure 4). It can be inferred from the phylogenetic tree that the rest of sequences were divided into four clades except for the outgroup. Among them, Littorinoidea and Naticoidea were grouped into one clade; nine species from Stromboidea, Tonnoidea and Cypraeoidea were grouped into one clade; the species from Rissooidea and Truncatelloidea were grouped into one clade, and the Vermetoidea species formed one separated clade. This phylogenetic relationship is consistent with the results using the traditional classification method. It is apparent from the phylogenetic tree that L. melanostoma is closely related to L. sinensis, which is a rock-dwelling species that is widespread in the coastal intertidal zone of China, just as the previous study[16].
4. Conclusions
In this study, the mitogenomes of L. melanostoma was sequenced, and 37 genes (13 PCGs, 22 tRNA genes and 2 rRNA genes) and one control region are located as typical of a Littorinoidea mitogenome. The ML phylogenetic relationships based on 13 PGs of the order Littorinimorpha were analyzed, indicating that the basis for the relationship based on a molecular analysis is consistent with that of the traditional morphological method.
Author Contributions
KC drafted the manuscript and performed data analysis. MLY collected and processed animal samples. XL designed and conceived the experiment and performed the data analysis. HSD and XL edited the manuscript. All authors read and approved the final manuscript.
Acknowledgments
We thank Mengling Liu from Marine Environment Monitoring Central Station of Guangxi for identifying animal samples. This study was supported by the National Natural Science Foundation of China [32060282], Special funding for Science & Technology bases and talents of Guangxi Province [AD20159032] and Open Research Fund Program of Guangxi Key Lab of Mangrove Conservation and Utilization [GKLMC-202102].
Conflicts of Interest
The authors declare there are no competing interests.
References
- Torres, P.; Alfiado, A.; Glassom, D.; Jiddawi, N.; Macia, A.; Reid, D.G.; Paula, J. Species composition, comparative size and abundance of the genus Littoraria (Gastropoda: Littorinidae) from different mangrove strata along the East African coast. Hydrobiologia 2008, 614, 339–351. [Google Scholar] [CrossRef]
- Berry, A.; Chew, E. Reproductive systems and cyclic release of eggs in Littorina melanostoma from Malayan mangrove swamps (Mollusca: Gastropoda). Journal of Zoology 1973, 171, 333–344. [Google Scholar] [CrossRef]
- Lee, O.H.; Williams, G.A.; Hyde, K.D. The diets of Littoraria ardouiniana and L. melanostoma in Hong Kong mangroves. Journal of the Marine Biological Association of the United Kingdom 2001, 81, 967–973. [Google Scholar] [CrossRef]
- Hamilton, P. Intertidal distribution and long-term movements of Littorina irrorata (Mollusca: Gastropoda). Marine Biology 1978, 46, 49–58. [Google Scholar] [CrossRef]
- Alfaro, A.C. Diet of Littoraria scabra, while vertically migrating on mangrove trees: Gut content, fatty acid, and stable isotope analyses. Estuarine, Coastal and Shelf Science 2008, 79, 718–726. [Google Scholar] [CrossRef]
- Little, C.; Stirling, P. Activation of a mangrove snail, Littorina scabra scabra (L.)(Gastropoda: Prosobranchia). Marine and Freshwater Research 1984, 35, 607–610. [Google Scholar] [CrossRef]
- Cook, L. Systematic effects on morph frequency in the polymorphic mangrove snail Littoraria pallescens. Heredity 1990, 65, 423–427. [Google Scholar] [CrossRef]
- Lee, O.H.; Williams, G.A. Spatial distribution patterns of Littoraria species in Hong Kong mangroves. Hydrobiologia 2002, 481, 137–145. [Google Scholar] [CrossRef]
- Blanco, J.F.; Cantera, J.R. The vertical distribution of mangrove gastropods and environmental factors relative to tide level at Buenaventura Bay, Pacific Coast of Colombia. Bulletin of marine science 1999, 65, 617–630. [Google Scholar]
- Reid, D.G. Habitat and zonation patterns of Littoraria species (Gastropoda: Littorinidae) in Indo-Pacific mangrove forests. Biological Journal of the Linnean Society 1985, 26, 39–68. [Google Scholar] [CrossRef]
- Chen, S.Y.; Chen, B.; Liao, J.J.; Chen, G.L.; Huang, Y.; Chen, G.C. Composition and distribution pattern of Littorinid snails in young rehabilitated mangroves. Chinese Journal of Ecology 2017, 36, 460. [Google Scholar]
- Reid, D.G.; Dyal, P.; Williams, S.T. A global molecular phylogeny of 147 periwinkle species (Gastropoda, Littorininae). Zoologica Scripta 2012, 41, 125–136. [Google Scholar] [CrossRef]
- Lai, T.H; He, B.Y. Studies on the macrobenthos species diversity for Guangxi mangrove areas. Guangxi Sciences 1998, 5, 166–172. [Google Scholar]
- Chen, G.; Ye, Y. Restoration of Aegiceras corniculatum mangroves in Jiulongjiang Estuary changed macro-benthic faunal community. Ecological Engineering 2011, 37, 224–228. [Google Scholar] [CrossRef]
- Bai, J.; Guo, Y.; Feng, J.; Ye, Y.; Li, J.; Yan, C.; Mao, S. The complete mitochondrial genome and phylogenetic analysis of Littorina brevicula (Gastropoda, Littorinidea). Mitochondrial DNA Part B 2020, 5, 2280–2281. [Google Scholar] [CrossRef] [PubMed]
- Reid, D.G; Dyal, P.; Williams, S. Global diversification of mangrove fauna: a molecular phylogeny of Littoraria (Gastropoda: Littorinidae). Molecular phylogenetics and evolution 2010, 55, 185–201. [Google Scholar] [CrossRef]
- Li, M. Y.; Li, Y. L.; Xing, T. F.; Liu, J. X. First mitochondrial genome of a periwinkle from the genus Littoraria: Littoraria sinensis. Mitochondrial DNA Part B 2019, 4, 4124–4125. [Google Scholar] [CrossRef]
- Irwin, A.R.; Strong, E.E.; Kano, Y.; Harper, E.M.; Williams, S.T. Eight new mitogenomes clarify the phylogenetic relationships of Stromboidea within the caenogastropod phylogenetic framework. Molecular Phylogenetics and Evolution 2021, 158, 107081. [Google Scholar] [CrossRef]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. 1994, 3, 294–299. [Google Scholar]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic acids research 2017, 45, e18–e18. [Google Scholar]
- Meng, G.; Li, Y.; Yang, C.; Liu, S. MitoZ: a toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic acids research 2019, 47, e63–e63. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: improved de novo metazoan mitochondrial genome annotation. Molecular phylogenetics and evolution 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular biology and evolution 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular biology and evolution 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic acids research 2013, 41, W575–W581. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Molecular ecology resources 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular biology and evolution 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Molecular biology and evolution 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nature methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Marques, J.P.; Sotelo, G.; Larsson, T.; Johannesson, K.; Panova, M.; Faria, R. Comparative mitogenomic analysis of three species of periwinkles: Littorina fabalis, L. obtusata and L. saxatilis. Marine genomics 2017, 32, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Song, W.T.; Gao, X.G.; Li, Y.F.; Liu, W.D.; Liu, Y.; He, C.B. Comparison of mitochondrial genomes of bivalves. Hereditas 2009, 31, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R.J.; Boore, J.; Brown, W. A novel mitochondrial genome organization for the blue mussel, Mytilus edulis. Genetics 1992, 131, 397–412. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
a. Specimen image of L. melanostoma; b. L. melanostoma inhabited on the leave of Avicennia marina (a common species of mangrove in China).
Figure 1.
a. Specimen image of L. melanostoma; b. L. melanostoma inhabited on the leave of Avicennia marina (a common species of mangrove in China).

Figure 2.
Circular maps of the mitogenomes of L. melanostoma.
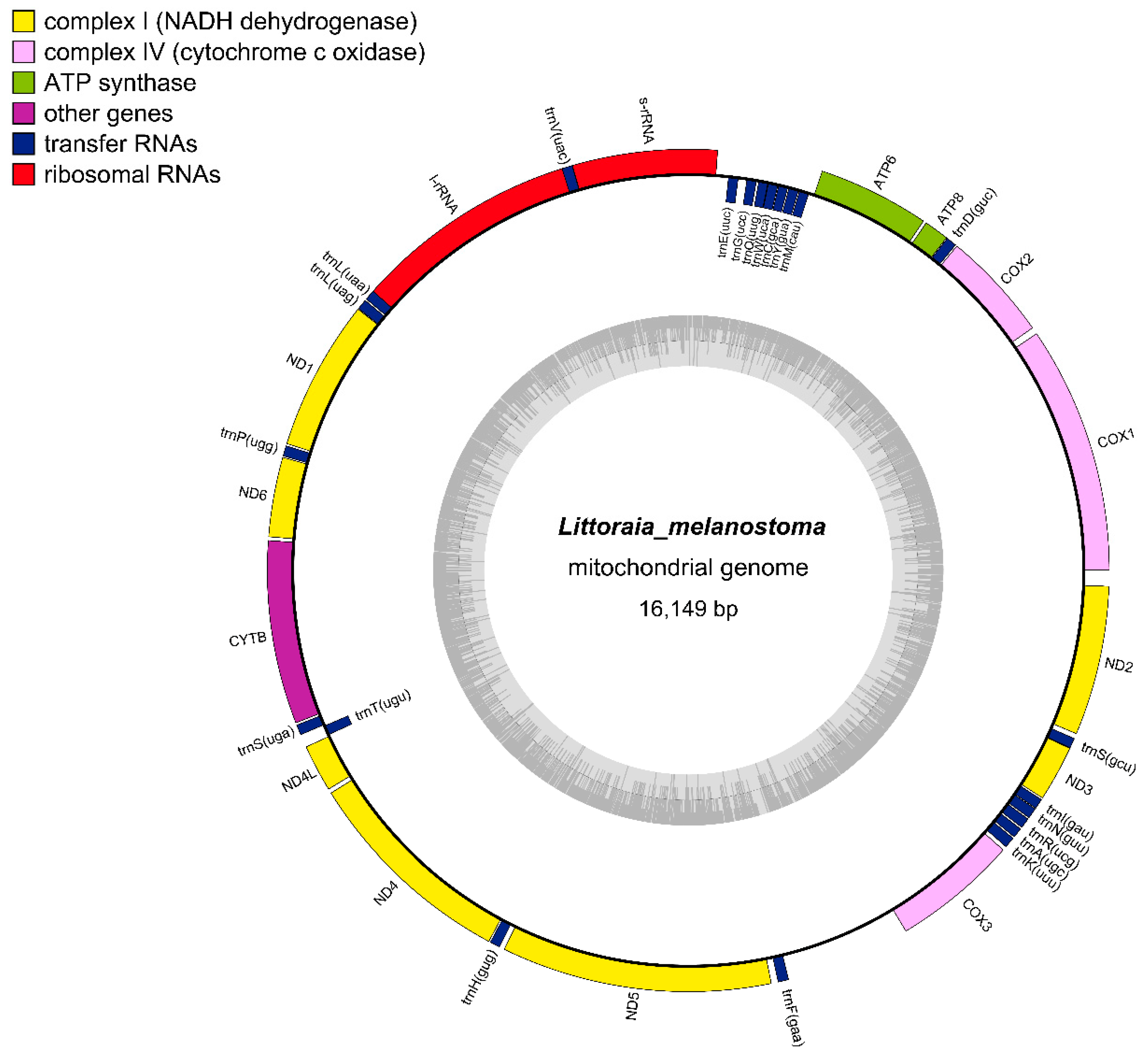
Figure 3.
Relative synonymous codon usage (RSCU) in the mitogenomes of eight species of Littorinimorpha.
Figure 3.
Relative synonymous codon usage (RSCU) in the mitogenomes of eight species of Littorinimorpha.
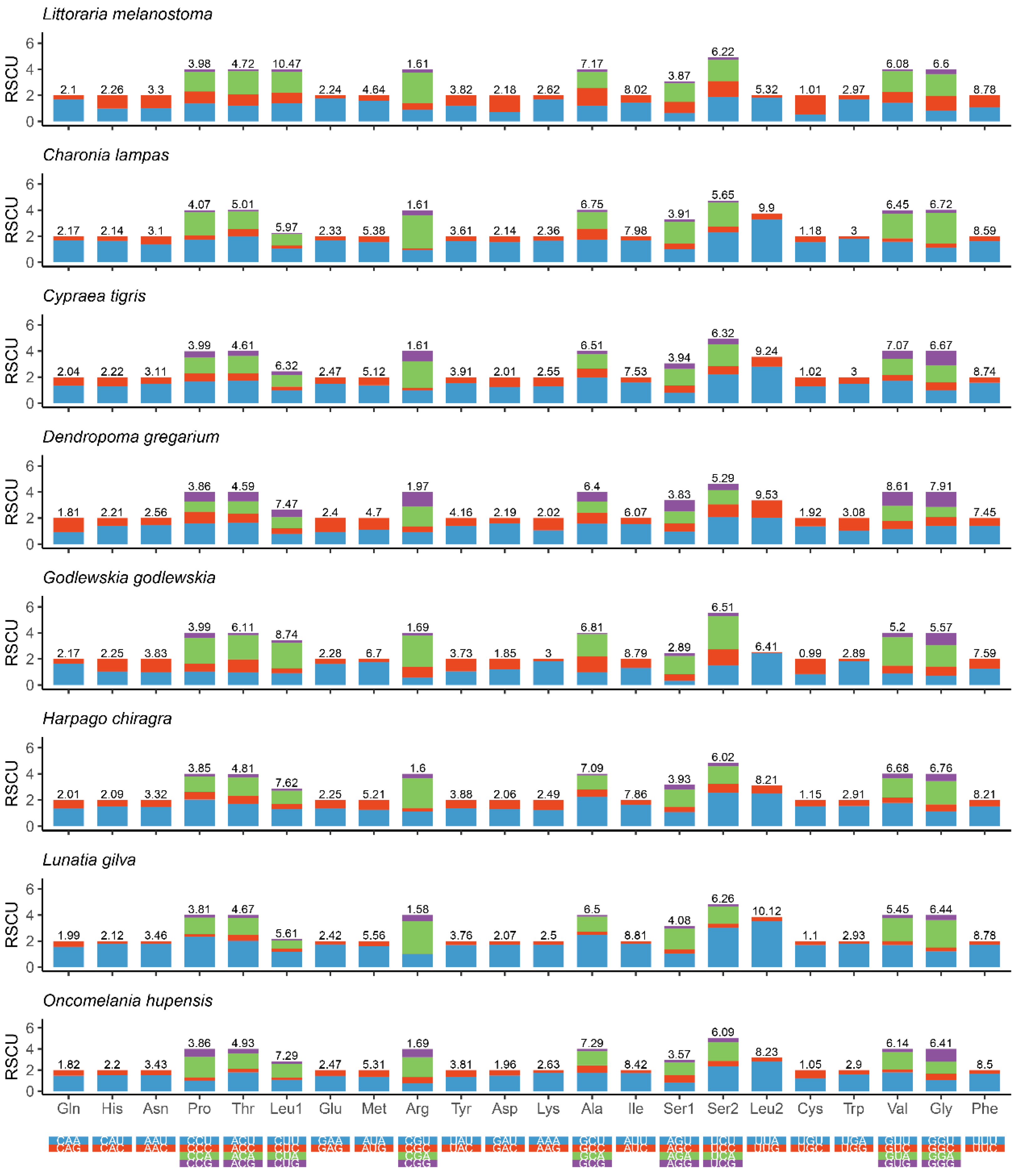
Figure 4.
Maximum likelihood phylogenetic tree inferred from 13 PCGs. SH-aLRT and UFBoot support values are given on nodes.
Figure 4.
Maximum likelihood phylogenetic tree inferred from 13 PCGs. SH-aLRT and UFBoot support values are given on nodes.
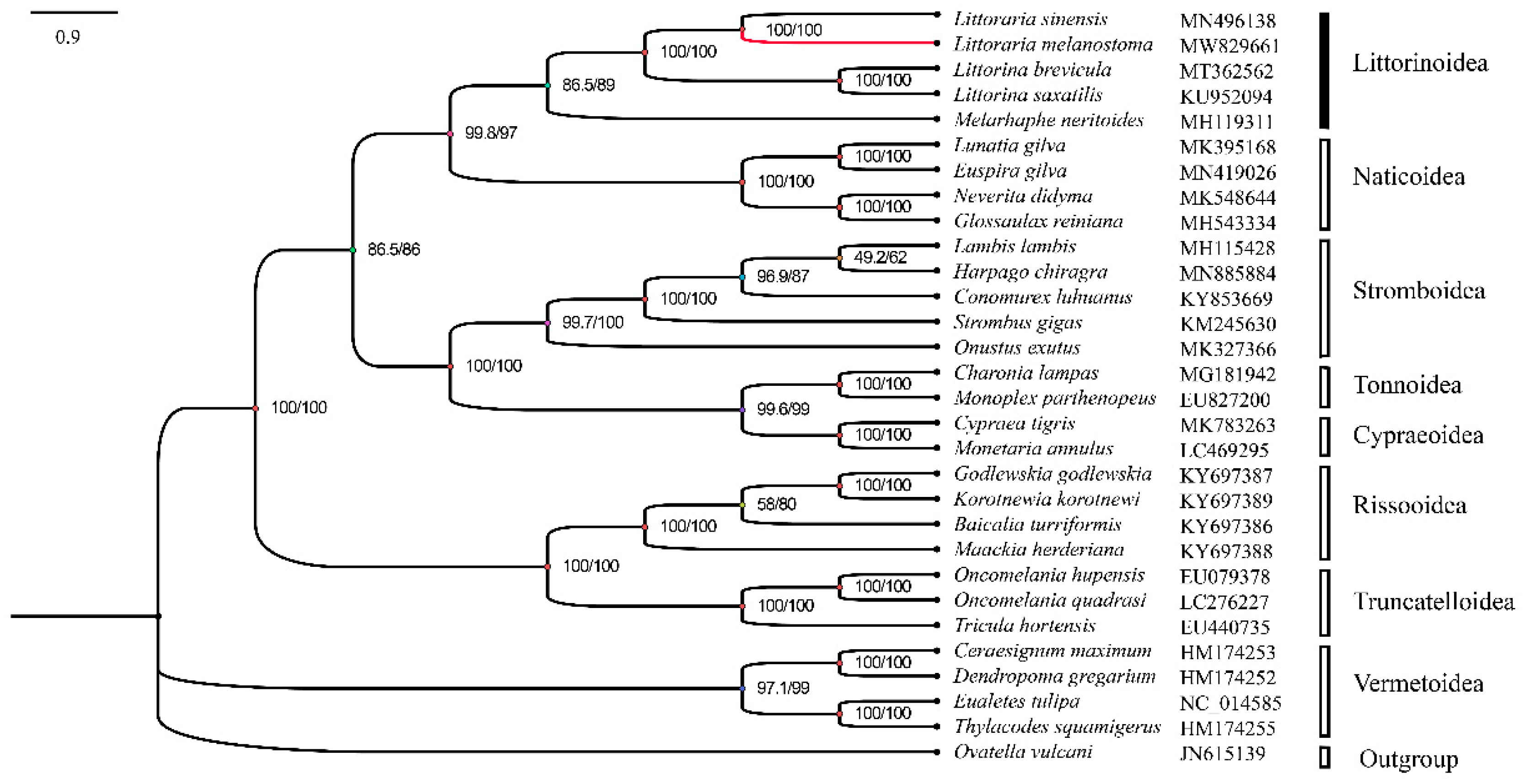

Table 1.
Classification and origins of the mitogenomic sequences used in this study.
| Taxonomy | Species | bp | Accession number |
| Littorinoidea | Littoraria melanostoma | 16,149 | NC064398 |
| Littorina brevicula | 16,356 | MT362562 | |
| Littorina saxatilis | 16,887 | KU952094 | |
| Littoraria sinensis | 16,420 | MN496138 | |
| Melarhaphe neritoides | 15,676 | MH119311 | |
| Stromboidea | Harpago chiragra | 16,404 | MN885884 |
| Lambis lambis | 15,481 | MH115428 | |
| Conomurex luhuanus | 15,799 | KY853669 | |
| Strombus gigas | 15,461 | KM245630 | |
| Cypraeoidea | Cypraea tigris | 16,177 | MK783263 |
| Monetaria annulus | 16,087 | LC469295 | |
| Naticoidea | Lunatia gilva | 16,139 | MK395168 |
| Euspira gilva | 16,119 | MN419026 | |
| Neverita didyma | 15,252 | MK548644 | |
| Glossaulax reiniana | 15,254 | MH543334 | |
| Xenophoroidea | Onustus exutus | 16,043 | MK327366 |
| Vermetoidea | Dendropoma gregarium | 15,641 | HM174252 |
| Ceraesignum maximum | 15,578 | HM174253 | |
| Thylacodes squamigerus | 15,544 | HM174255 | |
| Eualetes tulipa | 15,078 | NC_014585 | |
| Tonnoidea | Charonia lampas | 15,405 | MG181942 |
| Monoplex parthenopeus | 15,270 | EU827200 | |
| Truncatelloidea | Oncomelania hupensis | 15,191 | EU079378 |
| Oncomelania quadrasi | 15,184 | LC276227 | |
| Tricula hortensis | 15,179 | EU440735 | |
| Rissooidea | Godlewskia godlewskia | 15,224 | KY697387 |
| Baicalia turriformis | 15,127 | KY697386 | |
| Korotnewia korotnewi | 15,171 | KY697389 | |
| Maackia herderiana | 15,154 | KY697388 | |
| Outgroup | Ovatella vulcani | 14,274 | JN615139 |
Table 2.
Composition and base content of L. melanostoma protein coding genes.
| Littoraria melanostoma | ||||||||
| A % | T % | G % | C % | AT % | GC % | AT-Skew | GC-Skew | |
| Mitogenome | 29.79 | 34.37 | 14.66 | 21.18 | 64.16 | 35.84 | -0.071 | -0.182 |
| All PCGS | 27.77 | 34.62 | 15.15 | 22.46 | 62.39 | 37.61 | -0.110 | -0.194 |
| COX1 | 26.43 | 34.18 | 17.71 | 21.68 | 60.61 | 39.39 | -0.128 | -0.101 |
| COX2 | 28.97 | 30.57 | 17.76 | 22.71 | 59.53 | 40.47 | -0.027 | -0.122 |
| ATP8 | 32.70 | 36.48 | 10.69 | 20.13 | 69.18 | 30.82 | -0.055 | -0.306 |
| ATP6 | 26.44 | 36.06 | 13.22 | 24.28 | 62.50 | 37.50 | -0.154 | -0.295 |
| ND1 | 26.09 | 34.50 | 15.23 | 24.17 | 60.60 | 39.40 | -0.139 | -0.227 |
| ND6 | 27.31 | 34.94 | 13.05 | 24.70 | 62.25 | 37.75 | -0.123 | -0.309 |
| CYTB | 25.70 | 33.07 | 15.09 | 26.14 | 58.77 | 41.23 | -0.125 | -0.268 |
| ND4L | 25.70 | 33.07 | 15.09 | 26.14 | 68.01 | 31.99 | -0.125 | -0.268 |
| ND4 | 28.56 | 37.21 | 13.57 | 20.66 | 65.77 | 34.23 | -0.132 | -0.207 |
| ND5 | 30.15 | 33.13 | 13.35 | 23.36 | 63.29 | 36.71 | -0.047 | -0.273 |
| COX3 | 25.90 | 32.18 | 19.36 | 22.56 | 58.08 | 41.92 | -0.108 | -0.076 |
| ND3 | 27.35 | 39.32 | 15.10 | 18.23 | 66.67 | 33.33 | -0.180 | -0.094 |
| ND2 | 28.89 | 37.38 | 14.39 | 19.33 | 66.27 | 33.73 | -0.128 | -0.147 |
Table 3.
Mitogenomic organization of L. melanostoma.
| Position | Size(bp) | Intergenic nucleotides | Codon | Strand | ||||
| gene | From | To | Start | Stop | ||||
| Littoraia melanostoma | ||||||||
| 1 | COX1 | 1 | 1536 | 1536 | ATG | TAA | H | |
| 2 | COX2 | 1575 | 2261 | 687 | 38 | ATG | TAA | H |
| 3 | trnD(guc) | 2268 | 2336 | 69 | 6 | H | ||
| 4 | ATP8 | 2338 | 2496 | 159 | 1 | ATG | TAA | H |
| 5 | ATP6 | 2512 | 3207 | 696 | 15 | ATG | TAA | H |
| 6 | trnM(cau) | 3240 | 3306 | 67 | 32 | L | ||
| 7 | trnY(gua) | 3310 | 3377 | 68 | 3 | L | ||
| 8 | trnC(gca) | 3382 | 3446 | 65 | 4 | L | ||
| 9 | trnW(uca) | 3448 | 3514 | 67 | 1 | L | ||
| 10 | trnQ(uug) | 3514 | 3578 | 65 | -1 | L | ||
| 11 | trnG(ucc) | 3590 | 3656 | 67 | 11 | L | ||
| 12 | trnE(uuc) | 3710 | 3777 | 68 | 53 | L | ||
| 13 | s-rRNA | 3856 | 4756 | 901 | 78 | H | ||
| 14 | trnV(uac) | 4754 | 4822 | 69 | -3 | H | ||
| 15 | l-rRNA | 4801 | 6219 | 1419 | -22 | H | ||
| 16 | trnL(uaa) | 6210 | 6277 | 68 | -10 | H | ||
| 17 | trnL(uag) | 6284 | 6352 | 69 | 6 | H | ||
| 18 | ND1 | 6353 | 7291 | 939 | 0 | ATG | TAA | H |
| 19 | trnP(ugg) | 7301 | 7369 | 69 | 9 | H | ||
| 20 | ND6 | 7374 | 7871 | 498 | 4 | ATG | TAA | H |
| 21 | CYTB | 7890 | 9029 | 1140 | 18 | ATG | TAA | H |
| 22 | trnS(uga) | 9040 | 9107 | 68 | 10 | H | ||
| 23 | trnT(ugu) | 9111 | 9178 | 68 | 3 | L | ||
| 24 | ND4L | 9185 | 9481 | 297 | 6 | ATG | TAG | H |
| 25 | ND4 | 9505 | 10845 | 1341 | 23 | ATT | TAA | H |
| 26 | trnH(gug) | 10852 | 10918 | 67 | 6 | H | ||
| 27 | ND5 | 10947 | 12624 | 1678 | 28 | ATT | CTT | H |
| 28 | trnF(gaa) | 12663 | 12732 | 70 | 38 | H | ||
| CR | 12733 | 13505 | 773 | 0 | ||||
| 29 | COX3 | 13506 | 14285 | 780 | 773 | ATG | TAA | H |
| 30 | trnK(uuu) | 14307 | 14378 | 72 | 21 | H | ||
| 31 | trnA(ugc) | 14385 | 14451 | 67 | 6 | H | ||
| 32 | trnR(ucg) | 14459 | 14527 | 69 | 7 | H | ||
| 33 | trnN(guu) | 14533 | 14602 | 70 | 5 | H | ||
| 34 | trnI(gau) | 14604 | 14671 | 68 | 1 | H | ||
| 35 | ND3 | 14679 | 15029 | 351 | 7 | ATA | TAA | H |
| 36 | trnS(gcu) | 15029 | 15095 | 67 | -1 | H | ||
| 37 | ND2 | 15123 | 16053 | 931 | 27 | ATG | AAT | H |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



