
Submitted:
11 August 2023
Posted:
15 August 2023
You are already at the latest version
Abstract
Hepatitis B Virus (HBV) is a challenge for global health services, affecting millions and leading hundreds to end-stage liver disease each year. This comprehensive review explores the interactions between HBV and the host, examining their impact on clinical outcomes. HBV infection encompasses a spectrum of severity, ranging from acute hepatitis B to chronic hepatitis B, which can potentially progress to cirrhosis and hepatocellular carcinoma (HCC). Occult hepatitis B infection (OBI), characterized by low HBV DNA levels in hepatitis B surface antigen-negative individuals, can reactivate and cause acute hepatitis B. The identification of diverse HBV genotypes reveals distinct geographical distributions and associations with clinical outcomes. Moreover, single nucleotide polymorphisms (SNPs) within the host genome have been linked to several clinical outcomes, including cirrhosis, HCC, OBI, hepatitis B reactivation, and spontaneous clearance. The immune response plays a key role in controlling HBV infection by eliminating infected cells and neutralizing HBV in the bloodstream. Furthermore, HBV can modulate host metabolic pathways involved in glucose and lipid metabolism and bile acid absorption, further influencing disease progression. HBV clinical outcomes correlate with three levels of viral adaptation. In conclusion, the clinical outcomes of HBV infection could result from complex immune and metabolic interactions between the host and HBV. These outcomes can vary among populations and are influenced by HBV genotypes, host genetics, environmental factors, and lifestyle. Understanding the degrees of HBV adaptation is essential for developing region-specific control and prevention measures.
Keywords:
Hepatitis B virus
; HBV genotype H
; immune response
; metabolic interaction
; clinical outcome
; viral adaptation
1. Introduction
Hepatitis B Virus (HBV) infection remains a significant challenge for healthcare services worldwide. In 2019, approximately 296 million people had evidence of chronic hepatitis B, and 820 000 died due to complications associated with hepatitis B [1]. HBV is highly infectious and can be transmitted from mother to child, by sexual contact or contaminated materials [2]. While most cases of HBV infection are asymptomatic, around 90% of those infected in childhood and 5% of those infected in adulthood may develop chronic hepatitis B [2]. Without proper management, HBV infection can lead to serious liver complications, such as liver cirrhosis and hepatocellular carcinoma (HCC) [3].
In contrast to chronic hepatitis B, acute hepatitis B occurs within six months of exposure to the virus and refers to the initial phase of HBV infection [4]. Symptoms of acute hepatitis B include loss of appetite, fatigue, abdominal pain, jaundice, nausea, vomiting, fever, and dark urine [4]. While most individuals with acute HBV infection recover fully within a few months due to an efficient immune response that clears the virus [4], others may experience symptoms ranging from moderate to severe.
The most severe form of acute hepatitis B is fulminant hepatitis B, which affects about 1%-2% of people with hepatitis B [5,6]. Fulminate hepatitis B is rare, but when it occurs, it can progress rapidly to life-threatening liver failure. Common complications associated with fulminant hepatitis B include encephalopathy, coagulopathy, and multiorgan failure [7]. These complications are attributed to the host’s exaggerated immune response, which rapidly impairs the basic functions of the liver, such as detoxification, protein synthesis, and metabolism [7,8]. Dysfunction of these essential body processes can affect other organs (kidney, lung, heart, and brain), leading to abnormal personality changes, confusion, abnormal bleeding, easy bruising, prolonged bleeding time, and in severe cases, coma [7,8]. Without specialized medical care, the survival rate of patients with fulminant hepatitis B is poor (~25%), and liver transplantation could be the best hope for survival [8].
On the other hand, occult hepatitis B infection (OBI) is characterized by the presence of low levels of HBV DNA in the liver or peripheral blood of people who test negative for hepatitis B surface antigen (HBsAg) [9]. In OBI, the virus can pass undetected within cells without causing significant liver damage [10]. This characteristic favors the long-term survival of both the host and HBV. However, under certain circumstances, such as immune suppression or chemotherapy, OBI can reactivate, leading to acute hepatitis B [11].
2. HBV genotypes and human populations
Due to the high genetic variability, HBV has been classified into ten genotypes (A to J) and more than 40 sub-genotypes [12]. HBV genotypes A, B, C, and D were discovered simultaneously in 1988, and four years later, the genotypes E and F were proposed [12,13]. Subsequently, genotypes G and H were discovered in 2000 and 2002, respectively [14,15]. In 2000, an aberrant genotype with evidence of recombination between genotypes A and C was identified [17]. These HBV sequences were classified as genotype I in 2008 [18]. Finally, in 2009, genotype J was identified in an 88-year-old patient diagnosed with HCC [19]. With the discovery of all HBV genotypes, it was possible to identify that each has a different geographical distribution and shows a close relationship with its host populations [20,21]. Genotype A is prevalent in Europe and Eastern-Southern Africa. Genotypes B and C are commonly found in Asia, including China, Japan, Korea, and Australia. Genotype D is predominant in the Mediterranean, Arab countries, India, and Russia [20,21]. Genotype E is mainly found in West-Central Africa, while genotype F predominates in Central and South America [23]. Genotype G is mainly detected in the risk group of men who have sex with men (MSM) from Mexico, the United States, France, and the Netherlands [23,24]. Genotype H is endemic to the Mesoamerican region, including Mexico and Central America [25]. Genotypes I and J have also been reported in Southeast Asia [18,19].
In each region, HBV genotypes have been linked to several clinical outcomes. In Spain, genotype A has a low antiviral response, while genotypes B and C have been associated with HCC in Asia [26,27]. In an Alaska native population, the sub-genotype F1 increased 12-fold the risk of HCC compared with genotypes B and D [28]. In Europe and Asia, genotype D has been linked with the worst rates of acute, fulminate, and chronic liver disease compared with genotype A [29]. In Mexico, patients infected with the genotype A2 were more susceptible to resistance mutations than genotypes non-A [30]. In a small population residing in the Brazilian Amazon, individuals with genotype D exhibited a 7.44-fold higher risk of developing advanced liver disease than genotypes A and F [31]. In contrast, most people are asymptomatic to the infection caused by genotype F1b in the Colombian Amazon region [32]. In Mexico, genotype H infection is often asymptomatic without significant clinical or laboratory manifestations of liver disease. Also, genotype H has been associated with low viral loads and OBI, particularly in the indigenous people of Mexico [10,23]. HCC is rare among Mexican patients with genotype H [33], but when it is found in another region it has been linked to HCC, for example, in Japan [34], suggesting that genotype H has sustained a closer adaptative relationship with the Mexican population.
3. SNPs and HBV clinical outcomes
In addition to HBV genotypes, some variations in the patient’s genome, known as single nucleotide polymorphisms (SNPs), have been linked to different clinical outcomes, such as the risk of cirrhosis, HCC, OBI, hepatitis B reactivation, and clearance (Table 1). Cirrhosis-associated SNPs are in genes that participate in extracellular matrix production, immune response, intracellular adhesion, and signaling pathways [35,36,37,38]. A study in China associated SNPs at codons R241-E469 of the Intercellular adhesion molecule-1 (ICAM-1) gene with decompensated cirrhosis [35]. Also, the haplotype A874 and A2109 in Gamma Interferon (IFN-γ) gene increases 1.5-fold the risk of cirrhosis in the Chinese population [36]. A multivariate analysis found that Korean patients with detectable HBV viral load and L/L genotype in the Transforming growth factor (TGF)-b1 gene are at risk of developing cirrhosis [37]. In another Chinese study, the presence of rs4796793C-rs2293152G-rs1053004T haplotype in Signal Transducer and Activator of Transcription 3 (STAT3) significantly increases the risk of cirrhosis [38].
Among genes that increase susceptibility to HCC in chronic HBV patients are STAT4, complement component (C2), Protein Phosphatase 1 Catalytic Subunit Beta (PPP1CB), tumor suppressor protein (p53), MDM2 proto-oncogene (MDM2), DEP domain containing 5 (DEPDC5), X-chromosome long arm band 22.1 (Xq22.1), and CD33 molecule (SIGLEC3 or CD33), all studies were conducted in Asian populations [39,40,41,42,43,44,45]. Based on the odds ratio (OR) value, the most important SNPs associated with HCC in HBV Korean patients are rs1042522 in p53 and rs2279744 in MDM2, which increases 3.59- and 4.27-fold the risk of HCC, respectively [45]. Combined, the homozygotes MDM2C/C (p53: rs1042522) and G/G (MDM2: rs2279744) had a 20.78-fold higher risk of HCC than the rest of the genotypes [45].
Several SNPs have been analyzed in OBI cases and controls. However, only variations in HLA have shown a strong association with OBI. A study found that the T allele of HLA-DPA1 (rs3077) increases 6.12-fold the risk of OBI in the Indonesian population [46]. In northwest China, HLA-C*07:01, B*44:03, DRB1*07:01, and DQB1*02:02 were related to OBI with OR values of 4.7, 2.1, 2.0, and 1.9, respectively [47]. After achieving a sustained viral response, some HBV patients may develop reactivation. This can be triggered by immunosuppressive therapy, autoimmune diseases, and organ transplants [48]. It has been reported that 51.9% of patients with chronic hepatitis B can present reactivation after antiviral treatment [49]. HBV reactivation can be silent, but in patients with cirrhosis, it indicates a poor prognosis [49]. Currently, the AA genotype of HLA-DPB2 (rs872956) has been associated with HBV reactivation in Japanese treated with immunosuppressive therapy (OR: 8.27, n=42) [50]. In lymphoma patients (n=14), the SNPs in IL-13 (rs1295686) were associated with the reappearance of HBsAg in Chinese patients [51].
On the other hand, among the genes associated with spontaneous clearance of HBV infection are Sodium taurocholate cotransporting polypeptide (NTCP), Toll-like receptor (TLR), interferon-induced helicase C domain-containing protein 1 (IFIH1), microRNA 219-1, IL, and HLA [52,53,54,55,56,57]. Of these, one of the most studied is the SNPs in NTCP; this protein is a transporter of bile acids in the liver and functions as an entry receptor for HBV. NTCP is encoded by the Solute Carrier Family 10 member 1 (SLC10A1) gene [52]. A study in Taiwan found that the A allele SLC10A1 rs2296651 is associated with HBsAg seronegativity, while the AG or AA alleles are protective factors against HCC [57]. Based on a meta-analysis of 9 case-control studies, the A allele of the SLC10A1 rs2296651 shows strong evidence of spontaneous clearance [58].
4. HBV life cycle and immune response
The control of HBV infection by immune response is a complex process that includes eliminating infected cells and neutralizing HBV in the bloodstream by antibodies (Figure 1). During the initial phase of infection, HBV enters hepatocytes (liver cells) through receptor-mediated endocytosis, utilizing the NTCP receptor [59]. Once inside the hepatocyte, the virus undergoes uncoating, releasing its DNA into the cytoplasm. The viral DNA is then transported to the nucleus and converted into covalently closed circular DNA (cccDNA), which can integrate into the host genome [60]. The HBV genome serves as a template for the transcription of several messenger RNAs (mRNAs), which are transported to the cytoplasm to be translated into proteins. Some of these proteins contribute to assembling new viral particles, while others are targeted for degradation within the proteasome [61,62]. The resulting peptides can be recognized by pattern recognition receptors (PRRs), initiating a signaling cascade that leads to the production of Interferons (IFNs) (Figure 2a) [63]. IFNs activate immune response cells such as phagocytes and natural killer cells, while the peptides recognized by histocompatibility complexes class I (MHC-I) activate CD8 T cells [64,65]. Immune response cells attack infected hepatocytes releasing cytokines (Granzymes, perforin, soluble Fas ligand (SFasL)), which induce the programmed cell death through cytoplasmic and nuclear pathways (Figure 2a) [66]. Simultaneously, circulating phagocytes capture and destroy the HBVs in the bloodstream. In this case, the resulting peptides are presented by MHC-II to CD4 T cells [67]. This interaction stimulates the production of interleukin-21 (IL-21) in CD-4 T cells, particularly in follicular Helper T cells (TfH) [68]. IL-21 induces the expression of B lymphocyte-induced maturation protein-1 (BLIMP-1), which is a transcription factor key for plasma cell differentiation. Also, IL-21 promotes the generation of long-lived plasma cells in the bone marrow and continues to secrete antibodies for extended periods [69]. Among the most important antibodies produced by plasmatic cells are Immunoglobulin (Ig) M and IgG, which neutralize the hepatitis B core antigen (HBcAg); IgG anti-Hepatitis B e Antigen (IgG anti-HBe), and IgG anti-Hepatitis B surface Antigen (IgG anti-HBs) (Figure 2b) [70]. Overall, the immune response plays a crucial role in the outcome of HBV infection, determining whether it is cleared or progresses to chronic infection. Furthermore, the balance between the immune response and the low levels of viral replication could promote the long-term coexistence of HBV and its host.
5. HBV and metabolism
The liver performs vital metabolic processes in the body, and hepatocytes are the primary target of HBV. Consequently, the HBV genome or viral proteins can inevitably interact at a molecular level with metabolic pathways. Evidence suggests that HBV can modulate directly or indirectly key enzymes involved in glucose metabolism, lipid metabolism, and bile acid uptake (Figure 3) [77].
A study in vitro showed that Hepatitis B x protein (HBx) can up-regulate the expression of a long non-coding RNA Urothelial Carcinoma Associated 1 (LncRNA UCA1), which induces the expression of hexokinase [72,73]. In primary rat hepatocytes, phosphofructokinase transcription increased 72 hours post-transfection with HBx [74]. A proteomic analysis found that the enzymes glyceraldehyde-3-phosphate dehydrogenase (GAPDH), pyruvate kinase (PK), phosphoglycerate kinase (PGK), and lactate dehydrogenase (LDH) were significantly up-regulated in HepG2 cells after transfection with different HBx genotypes [75]. These enzymes are crucial to carry out glycolysis in the liver. HBx can also affect gluconeogenesis through overexpression of the phosphoenolpyruvate carboxylase (PEPCK) and Glucose-6-phosphatase (G6PC), that catalyze the conversion of oxaloacetate to phosphoenolpyruvate and the hydrolysis of glucose-6-phosphate to glucose, respectively. Overexpression of these enzymes caused hyperglycemia in HBx transgenic mice [76]. In a yeast-based expression system, it has been observed that the expression of the preS2 mRNA (code HBsAg) may be reduced or suppressed when glucose concentrations reach 2% (Figure 2b) [77]. These findings suggest that the levels of HBsAg may be difficult to detect under high glucose conditions. HBx can also induce the transcription of the glucose-6-phosphate dehydrogenase gene (G6PD) [78], which is a key enzyme in the pentose phosphate pathway (PPP). The PPP is important for generating nicotinamide adenine dinucleotide phosphate (NADPH) and ribose-5-phosphate, contributing to redox balance, energy production, and nucleotide biosynthesis [71].
In addition to its impact on glucose metabolism, HBV infection is associated with lipid droplet accumulation in hepatocytes. In cell culture, this phenomenon has been studied through the interaction of HBx with lipogenic genes, such as liver X receptor (LXR), sterol regulatory element-binding protein-1c (SREBP-1c), fatty acid synthase (FAS), and peroxisome proliferator-activated receptor (PPARs). In 2008, it was discovered that HBx can directly bind to LXRα isoform [79]. Once activated, the LXR is transported to the nucleus, where it activates the transcription of genes with the sequence AGGTCA [80]. Among them is the SREBP-1c, which acts as a transcription factor for genes responsible for synthesizing fatty acids, such as FAS and PPARs [81]. The FAS cytoplasmic complex converts acetyl-CoA and malonyl-CoA to palmitic acid, utilizing NADPH as a cofactor [81]. Palmitic acid is one of the most important fatty acids that make up triglycerides. On the other hand, PPAR upregulation can induce the expression of the enzyme serine-palmitoyl-transferase (SPT) that synthesizes sphingolipids [82]. Sphingolipids serve as structural components of the cell membrane and play a crucial role in maintaining the integrity of various organelles, including the endoplasmic reticulum, Golgi apparatus, mitochondria, and lysosomes [82]. Other enzymes induced by PPAR-α include carnitine palmitoyl transferase A1 (CPT1A), acyl-CoA dehydrogenase (ACADM), and peroxisomal acyl-CoA oxidase (ACOX1). These enzymes play crucial roles in β-oxidation, the process involved in the breakdown of fatty acids into acetyl-CoA molecules. Acetyl-CoA can enter the citric acid cycle or serve as substrates for the FAS complex [83]. Finally, the PPAR-ß/ isoform can induce the expression of the CD36 receptor and the phospholipid transfer protein (PLTP). CD36 facilitates the uptake of fatty acids and lipoproteins, while PLTP is involved in the transfer of phospholipids between lipoprotein particles [83,84].
HBV can impact bile acid metabolism through its interaction with NTCP, a protein involved in the reabsorption of bile acids. NTCP serves as a receptor for HBV to enter hepatocytes [59], and this interaction can disrupt the normal uptake and recycling of bile acids within the liver. As a result, there can be an increase in the levels of bile acids in the serum of patients [85]. This symptom was analyzed in a cohort of patients with hepatitis B, finding that high levels of five bile acids (glycocholic acid, glycochenodeoxycholic acid, taurocholic acid, taurochenodeoxycholic acid, and glycoursodeoxycholic acid) were associated with the progression of cirrhosis [86]. These findings suggest that serum bile acid levels could be helpful as biochemical markers of fibrosis progression in patients with hepatitis B.
HBV can modulate hepatocyte metabolism to create a favorable environment for its replication. HBx is the most important protein involved in the regulation of metabolism. The modulation of glucose metabolism creates an environment rich in glucose, energy, and nucleotides. At the same time, the control of lipid metabolism provides the necessary lipids to form new viral envelopes but promotes the accumulation of lipid droplets in hepatocytes.
6. The theory of viral adaptation
HBV-related viruses have been detected in various animal species, such as fish, amphibians, reptiles, rodents, bats, and primates [87]. The origin of these viruses can be traced back approximately 400 million years [87]. A multidisciplinary archaeological study recently identified traces of HBV genotypes in ten millennia-old human remains [88]. During this time, humans and HBV genotypes have been subjected to selective pressures shaping their coevolutionary dynamic. Humans have experienced different environmental conditions, infectious diseases, diets, and social interactions [89]. These pressures can influence human genetic diversity, metabolism, immunity, and infection susceptibility [89]. Simultaneously, HBV adapts to the selective pressures imposed by the host, leading to a complex interaction (HBV/human) that could determine its current distribution, epidemiology, and clinical outcomes.
In 2014, HBV and humans’ different degrees of adaptation were proposed [90]. This hypothesis suggests that the severity of liver disease tends to be lower as the degree of adaptation increases (Figure 4) [90]. HBV-host adaptations can be grouped into incomplete, semi-complete, and complete [90]. The lowest degree of adaptation is known as “incomplete adaptation.” Probably caused by a very recent interaction between the virus and its host population [91]. Incomplete adaptation can manifest in two ways. Firstly, in cases of fulminant hepatitis, HBV infection triggers a hyperimmune response that severely impairs liver function. This state is considered ineffective for HBV survival because it relies on the host to exist. Fulminant hepatitis represents an extreme form of incomplete adaptation. Another form of incomplete adaptation can be the cases of acute hepatitis B when the immune response efficiently eliminates the HBV DNA genomes without allowing the virus to develop a favorable intracellular environment for virus/host coexistence [6,7]. Both acute and fulminant hepatitis B are not commonly observed in human populations, suggesting that most individuals may exhibit another type of adaptation [90]. The second degree of adaptation is “semi-complete adaptation,” observed in patients with chronic hepatitis B [90]. Although this type of infection does not immediately compromise the host’s life, prolonged exposure can lead to the development of fibrosis, cirrhosis, and hepatocellular carcinoma [2,92]. Different HBV genotypes, particularly genotypes B and C, have a higher risk of hepatocellular carcinoma [27,28], indicating a potential disruption in the adaptive process regarding metabolic or immunological interaction. Finally, the highest degree of adaptation is exemplified by OBI [90], where patients have low HBV viral load (<200 IU/mL) and absence of HBsAg [9,10]. This state is likely achieved by integrating the HBV genome into host cells, stability of cccDNA, and immune tolerance [93]. It allows for a homeostasis state between the human host and the specific HBV genotype, resulting in asymptomatic infection for many years [90]. This type of adaptation has been observed in the native population of Mexico infected by genotype H [94]. The molecular dating estimates that genotype H has been present in the Mexican population for at least 2070 years [95]. Another virus with a similar adaptation may be the sub-genotype F1b in the Amerindian population of the Amazon basin. These observations suggest that complete adaptation could develop between an endemic HBV genotype and its respective native population in each region [90].
The balance between viral replication and immune control is crucial for the HBV to persist within the host without causing excessive liver damage. This balance can be disrupted by several factors, including viral mutations, a patient’s immune status, antiviral medications, exposure to toxins, co-infections, and lifestyle [97]. An additional significant factor to consider is the migration of HBV genotypes. As mentioned above, genotype H is less aggressive in native Mexican populations. However, when it migrates to a new population, such as Japan, it can cause acute hepatitis B [98] or liver cancer [34].
In contrast to HBV, the infection caused by the hepatitis C virus (HCV) is considered more severe due to its high mortality rate (3.45 deaths per 100,000 people) compared to HBV (0.45 deaths per 100,000 people) [99]. Among the attributable fraction contributing to the incidence of liver cancer, 50% is related to hepatitis C, whereas 15% is related to hepatitis B [100]. Approximately 55%-85% of adults with hepatitis C develop the chronic form, whereas less than 5% of adults with hepatitis B develop chronic infection [101]. This difference may indicate that spontaneous clearance is more common in acute HBV than in acute HCV infection. Based on the adaptation theory to these findings, it can be inferred that HCV exhibits a lower degree of adaptation than HBV, likely due to its recent introduction to human populations. A molecular dating study suggests HCV originated in the Old World approximately 3,000 years ago [102]. However, HBV is estimated to have circulated in human populations for at least ten millennia [88]. The lower degree of adaptation of HCV to humans may explain why it is more likely to cause serious health problems, such as chronic infection, liver cancer, and death.
In conclusion, the clinical outcomes of HBV infection could result from complex immune and metabolic interactions between the host and HBV. These outcomes can vary among populations and are influenced by several factors, including the HBV genotype, the genetic makeup of the population, exposure to toxins, co-infections, comorbidities, antiviral medications, diets, and lifestyle. The different types of HBV infection suggest three distinct degrees of viral adaptation. Understanding the degrees of adaptation of HBV genotypes in different populations is essential to develop the most appropriate control and prevention measures.
Author Contributions
Conceptualization, A.J.-A., S.R. and A.P.; Methodology, A.J.-A. and S.L.-M.; Writing—Original Draft Preparation, A.J.-A.; Writing—Review and Editing, A.J.-A., S.R., S.L.-M. and A.P.; Supervision, A.P.; Project Administration, A.P.; All authors have read and agreed to the published version of the manuscript.
Funding
This research was partly funded by Consejo Nacional de Ciencia y Tecnología (CONACYT), Grant Number CONACYT PN-2017-01-5254 to AP and Programa de Fortalecimiento de Institutos, Centros y Laboratorios de Investigación 2021, Universidad de Guadalajara.
Conflicts of Interest
The authors declare that the research was conducted without any commercial or financial relationships that could be considered a potential conflict of interest.
References
- World Health Organization. Hepatitis B, 2022; p. 4. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-b (accessed on 13 June 2023).
- Jeng, W.J.; Papatheodoridis, G.V.; Lok, A.S.F. Hepatitis B. Lancet 2023, 401, 1039-1052. [CrossRef]
- Weisberg, I.S.; Brown, R.S., Jr.; Sigal, S.H. Hepatitis B and end-stage liver disease. Clin Liver Dis 2007, 11, pp. 893-916. [CrossRef]
- Schillie, S.; Vellozzi, C.; Reingold, A.; Harris, A.; Haber, P.; Ward, J.W.; Nelson, N.P. Prevention of Hepatitis B Virus Infection in the United States: Recommendations of the Advisory Committee on Immunization Practices. MMWR Recomm Rep 2018, 67, pp. 1-31. [CrossRef]
- Lee, W.M. Acute liver failure. N Engl J Med 1993, 329, pp. 1862-1872. [CrossRef]
- Petrosillo, N.; Ippolito, G.; Solforosi, L.; Varaldo, P.E.; Clementi, M.; Manzin, A. Molecular epidemiology of an outbreak of fulminant hepatitis B. J Clin Microbiol. 2000, 38, pp. 2975-2981. [CrossRef]
- Ichai, P.; Samuel, D. Management of Fulminant Hepatitis B. Curr Infect Dis Rep 2019, 21, 25. [CrossRef]
- Ostapowicz, G.; Fontana, R.J.; Schiodt, F.V.; Larson, A.; Davern, T.J.; Han, S.H.; McCashland, T.M.; Shakil, A.O.; Hay, J.E.; Hynan, L.; et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002, 137, pp. 947-954. [CrossRef]
- Pollicino, T.; Raimondo, G. Occult hepatitis B infection. J Hepatol 2014, 61, pp. 688-689. [CrossRef]
- Panduro, A.; Maldonado-Gonzalez, M.; Fierro, N.A.; Roman, S. Distribution of HBV genotypes F and H in Mexico and Central America. Antivir Ther 2013, 18, pp. 475-484. [CrossRef]
- Raimondo, G.; Filomia, R.; Maimone, S. Therapy of occult hepatitis B virus infection and prevention of reactivation. Intervirology 2014, 57, pp. 189-195. [CrossRef]
- Kafeero, H.M.; Ndagire, D.; Ocama, P.; Kato, C.D.; Wampande, E.; Walusansa, A.; Kajumbula, H.; Kateete, D.; Ssenku, J.E.; Sendagire, H. Mapping hepatitis B virus genotypes on the African continent from 1997 to 2021: a systematic review with meta-analysis. Sci Rep 2023, 13, 5723. [CrossRef]
- Okamoto, H.; Tsuda, F.; Sakugawa, H.; Sastrosoewignjo, R.I.; Imai, M.; Miyakawa, Y.; Mayumi, M. Typing hepatitis B virus by homology in nucleotide sequence: comparison of surface antigen subtypes. J Gen Virol 1988, 69 ( Pt 10), pp. 2575-2583. [CrossRef]
- Norder, H.; Hammas, B.; Lofdahl, S.; Courouce, A.M.; Magnius, L.O. Comparison of the amino acid sequences of nine different serotypes of hepatitis B surface antigen and genomic classification of the corresponding hepatitis B virus strains. J Gen Virol 1992, 73 ( Pt 5), pp. 1201-1208. [CrossRef]
- Stuyver, L.; De Gendt, S.; Van Geyt, C.; Zoulim, F.; Fried, M.; Schinazi, R.F.; Rossau, R. A new genotype of hepatitis B virus: complete genome and phylogenetic relatedness. J Gen Virol 2000, 81, pp. 67-74. [CrossRef]
- Arauz-Ruiz, P.; Norder, H.; Robertson, B.H.; Magnius, L.O. Genotype H: a new Amerindian genotype of hepatitis B virus revealed in Central America. J Gen Virol 2002, 83, pp. 2059-2073. [CrossRef]
- Hannoun, C.; Norder, H.; Lindh, M. An aberrant genotype revealed in recombinant hepatitis B virus strains from Vietnam. J Gen Virol 2000, 81, pp. 2267-2272. [CrossRef]
- Olinger, C.M.; Jutavijittum, P.; Hubschen, J.M.; Yousukh, A.; Samountry, B.; Thammavong, T.; Toriyama, K.; Muller, C.P. Possible new hepatitis B virus genotype, southeast Asia. Emerg Infect Dis 2008, 14, pp. 1777-1780. [CrossRef]
- Tatematsu, K.; Tanaka, Y.; Kurbanov, F.; Sugauchi, F.; Mano, S.; Maeshiro, T.; Nakayoshi, T.; Wakuta, M.; Miyakawa, Y.; Mizokami, M. A genetic variant of hepatitis B virus divergent from known human and ape genotypes isolated from a Japanese patient and provisionally assigned to new genotype J. J Virol 2009, 83, pp. 10538-10547. [CrossRef]
- Velkov, S.; Ott, J.J.; Protzer, U.; Michler, T. The Global Hepatitis B Virus Genotype Distribution Approximated from Available Genotyping Data. Genes 2018, 9. [CrossRef]
- Osiowy, C.; Coffin, C.; Andonov, A. Review of Laboratory Tests used in Monitoring Hepatitis B Response to Pegylated Interferon and Nucleos(t)ide Analog Therapy. Curr Treat Options Infect Dis 2016, 8, pp. 177-193. [CrossRef]
- Alvarado-Mora, M.V.; Pinho, J.R. Distribution of HBV genotypes in Latin America. Antivir Ther 2013, 18, pp. 459-465. [CrossRef]
- Roman, S.; Panduro, A. HBV endemicity in Mexico is associated with HBV genotypes H and G. World J Gastroenterol 2013, 19, pp. 5446-5453. [CrossRef]
- Shah, A.A.; Bodewes, R.; Reijnen, L.; Boelsums, T.; Weller, C.M.; Fanoy, E.B.; Veldhuijzen, I.K. Outbreaks of mumps genotype G viruses in the Netherlands between October 2019 and March 2020: clusters associated with multiple introductions. BMC Infect Dis 2021, 21, 1035. [CrossRef]
- Jose-Abrego, A.; Roman, S.; Laguna-Meraz, S.; Rebello-Pinho, J.R.; Justo Arevalo, S.; Panduro, A. Tracing the evolutionary history of hepatitis B virus genotype H endemic to Mexico. Front Microbiol 2023, 14, 1180931. [CrossRef]
- Bottecchia, M.; Madejon, A.; Sheldon, J.; Garcia-Samaniego, J.; Barreiro, P.; Soriano, V. Hepatitis B virus genotype A2 harbours an L217R polymorphism which may account for a lower response to adefovir. J Antimicrob Chemother 2008, 62, pp. 626-627. [CrossRef]
- Chan, H.L.; Hui, A.Y.; Wong, M.L.; Tse, A.M.; Hung, L.C.; Wong, V.W.; Sung, J.J. Genotype C hepatitis B virus infection is associated with an increased risk of hepatocellular carcinoma. Gut 2004, 53, pp. 1494-1498. [CrossRef]
- Gounder, P.P.; Bulkow, L.R.; Snowball, M.; Negus, S.; Spradling, P.R.; McMahon, B.J. Hepatocellular Carcinoma Risk in Alaska Native Children and Young Adults with Hepatitis B Virus: Retrospective Cohort Analysis. J Pediatr 2016, 178, pp. 206-213. [CrossRef]
- Shi, Y.H. Correlation between hepatitis B virus genotypes and clinical outcomes. Jpn J Infect Dis 2012, 65, pp. 476-482. [CrossRef]
- Jose-Abrego, A.; Roman, S.; Rebello-Pinho, J.R.; Gomes-Gouvea, M; Panduro, A. High Frequency of Antiviral Resistance Mutations in HBV Genotypes A2 and H: Multidrug Resistance Strains in Mexico. J Clin Transl Hepatol 2023, pp. 1-12. [CrossRef]
- Roca, T.P.; Villar, L.M.; Nogueira Lima, F.S.; Vasconcelos, M.P.A.; Borzacov, L.M.P.; Silva, E.C.E.; Lago, B.V.D.; Silva, M.; Botelho Souza, L.F.; Salcedo, J.M.V.; et al. Genomic Variability of Hepatitis B Virus Circulating in Brazilian Western Amazon. Viruses 2022, 14. [CrossRef]
- di Filippo Villa, D.; Cortes-Mancera, F.; Payares, E.; Montes, N.; de la Hoz, F.; Arbelaez, M.P.; Correa, G.; Navas, M.C. Hepatitis D virus and hepatitis B virus infection in Amerindian communities of the Amazonas state, Colombia. Virol J 2015, 12, 172. [CrossRef]
- Roman, S.; Panduro, A.; Aguilar-Gutierrez, Y.; Maldonado, M.; Vazquez-Vandyck, M.; Martinez-Lopez, E.; Ruiz-Madrigal, B.; Hernandez-Nazara, Z. A low steady HBsAg seroprevalence is associated with a low incidence of HBV-related liver cirrhosis and hepatocellular carcinoma in Mexico: a systematic review. Hepatol Int 2009, 3, pp. 343-355. [CrossRef]
- Oba, U.; Koga, Y.; Hoshina, T.; Suminoe, A.; Abe, K.; Hayashida, M.; Taguchi, T.; Hara, T. An adolescent female having hepatocellular carcinoma associated with hepatitis B virus genotype H with a deletion mutation in the pre-S2 region. J Infect Chemother 2015, 21, pp. 302-304. [CrossRef]
- Zhang, X.Q.; Hong, X.J.; Bai, X.J. Susceptibility to active decompensated cirrhosis is associated with polymorphisms of intercellular adhesion molecule-1 (ICAM-1) in chronic HBV carriers. J Viral Hepat 2008, 15, pp. 173-178. [CrossRef]
- Sun, Y.; Lu, Y.; Xie, L.; Deng, Y.; Li, S.; Qin, X. Interferon gamma polymorphisms and hepatitis B virus-related liver cirrhosis risk in a Chinese population. Cancer Cell Int 2015, 15, 35. [CrossRef]
- Yu, S.K.; Kwon, O.S.; Jung, H.S.; Bae, K.S.; Kwon, K.A.; Kim, Y.K.; Kim, Y.S.; Kim, J.H. Influence of transforming growth factor-beta1 gene polymorphism at codon 10 on the development of cirrhosis in chronic hepatitis B virus carriers. J Korean Med Sci 2010, 25, pp. 564-569. [CrossRef]
- Yan, X.H.; Wu, J.L.; Yu, R.; Ma, X.H.; Li, Q.F.; Xie, R.F. Associations between gene polymorphisms of signal transducer and activator of transcription 3 and the susceptibility to hepatitis B virus related liver cirrhosis. Zhonghua Yu Fang Yi Xue Za Zhi 2022, 56, pp. 185-191. [CrossRef]
- Jiang, D.K.; Ma, X.P.; Wu, X.; Peng, L.; Yin, J.; Dan, Y.; Huang, H.X.; Ding, D.L.; Zhang, L.Y.; Shi, Z.; et al. Genetic variations in STAT4, C2, HLA-DRB1 and HLA-DQ associated with risk of hepatitis B virus-related liver cirrhosis. Sci Rep 2015, 5, 16278. [CrossRef]
- Liu, W.; Ma, N.; Zhao, D.; Gao, X.; Zhang, X.; Yang, L.; Liu, D. Correlation between the DEPDC5 rs1012068 polymorphism and the risk of HBV-related hepatocellular carcinoma. Clin Res Hepatol Gastroenterol 2019, 43, pp. 446-450. [CrossRef]
- Mai, H.; Xie, H.; Hou, J.; Chen, H.; Zhou, B.; Hou, J.; Jiang, D. A Genetic Variant of PPP1CB Influences Risk of Hepatitis B Virus-Related Hepatocellular Carcinoma in Han Chinese: A Pathway Based Analysis. J Hepatocell Carcinoma 2021, 8, pp. 1055-1064. [CrossRef]
- Zeisel, M.B.; Guerrieri, F.; Levrero, M. Host Epigenetic Alterations and Hepatitis B Virus-Associated Hepatocellular Carcinoma. J Clin Med 2021, 10. [CrossRef]
- [43] Jiang, J.H.; Gao, Q.; Shen, X.Z.; Yu, Y.; Gu, F.M.; Yan, J.; Pan, J.F.; Jin, F.; Fan, J.; Zhou, J.; et al. An X-chromosomal association study identifies a susceptibility locus at Xq22.1 for hepatitis B virus-related hepatocellular carcinoma. Clin Res Hepatol Gastroenterol 2013, 37, 586-595. [CrossRef]
- Tsai, T.Y.; Huang, M.T.; Sung, P.S.; Peng, C.Y.; Tao, M.H.; Yang, H.I.; Chang, W.C.; Yang, A.S.; Yu, C.M.; Lin, Y.P.; et al. SIGLEC-3 (CD33) serves as an immune checkpoint receptor for HBV infection. J Clin Invest 2021, 131. [CrossRef]
- Yoon, Y.J.; Chang, H.Y.; Ahn, S.H.; Kim, J.K.; Park, Y.K.; Kang, D.R.; Park, J.Y.; Myoung, S.M.; Kim, D.Y.; Chon, C.Y.; et al. MDM2 and p53 polymorphisms are associated with the development of hepatocellular carcinoma in patients with chronic hepatitis B virus infection. Carcinogenesis 2008, 29, pp. 1192-1196. [CrossRef]
- Mardian, Y.; Yano, Y.; Wasityastuti, W.; Ratnasari, N.; Liang, Y.; Putri, W.A.; Triyono, T.; Hayashi, Y. Genetic polymorphisms of HLA-DP and isolated anti-HBc are important subsets of occult hepatitis B infection in Indonesian blood donors: a case-control study. Virol J 2017, 14, 201. [CrossRef]
- Wang, T.; Shen, C.; Chen, L.; Liu, S.; Ji, Y. Association of human leukocyte antigen polymorphisms with occult hepatitis B virus infection in a Shaanxi Han population. J Gene Med 2017, 19. [CrossRef]
- Wang, Y.; Han, S.B. Hepatitis B Reactivation: A Review of Clinical Guidelines. J Clin Gastroenterol 2021, 55, pp. 393-399. [CrossRef]
- Zhu, Y.; Li, H.; Wang, X.; Zheng, X.; Huang, Y.; Chen, J.; Meng, Z.; Gao, Y.; Qian, Z.; Liu, F.; et al. Hepatitis B Virus Reactivation Increased the Risk of Developing Hepatic Failure and Mortality in Cirrhosis With Acute Exacerbation. Front Microbiol 2022, 13, 910549. [CrossRef]
- Matsuda, H.; Hiramatsu, K.; Akazawa, Y.; Nosaka, T.; Saito, Y.; Ozaki, Y.; Hayama, R.; Takahashi, K.; Naito, T.; Ofuji, K.; et al. Genetic polymorphism and decreased expression of HLA class II DP genes are associated with HBV reactivation in patients treated with immunomodulatory agents. J Med Virol 2018, 90, pp. 712-720. [CrossRef]
- Hsiao, L.T.; Wang, H.Y.; Yang, C.F.; Chiou, T.J.; Gau, J.P.; Yu, Y.B.; Liu, H.L.; Chang, W.C.; Chen, P.M.; Tzeng, C.H.; et al. Human Cytokine Genetic Variants Associated With HBsAg Reverse Seroconversion in Rituximab-Treated Non-Hodgkin Lymphoma Patients. Medicine 2016, 95, e3064. [CrossRef]
- Lee, H.W.; Park, H.J.; Jin, B.; Dezhbord, M.; Kim, D.Y.; Han, K.H.; Ryu, W.S.; Kim, S.; Ahn, S.H. Effect of S267F variant of NTCP on the patients with chronic hepatitis B. Sci Rep 2017, 7, 17634. [CrossRef]
- Wu, J.F.; Chen, C.H.; Ni, Y.H.; Lin, Y.T.; Chen, H.L.; Hsu, H.Y.; Chang, M.H. Toll-like receptor and hepatitis B virus clearance in chronic infected patients: a long-term prospective cohort study in Taiwan. J Infect Dis 2012, 206, pp. 662-668. [CrossRef]
- Yao, Y.; Shen, Y.; Shao, H.; Liu, Y.; Ji, Y.; Du, G.; Ye, X.; Huang, P.; Chen, H. Polymorphisms of RIG-I-like receptor influence HBV clearance in Chinese Han population. J Med Virol 2021, 93, pp. 4957-4965. [CrossRef]
- Shen, Z.; Yang, H.; Yang, S.; Wang, W.; Cui, X.; Zhou, X.; Liu, W.; Pan, S.; Liu, Y.; Zhang, J.; et al. Hepatitis B virus persistence in mice reveals IL-21 and IL-33 as regulators of viral clearance. Nat Commun 2017, 8, 2119. [CrossRef]
- Wang, L.; Zou, Z.Q.; Wang, K. Clinical Relevance of HLA Gene Variants in HBV Infection. J Immunol Res 2016, 2016, 9069375. [CrossRef]
- Hu, H.H.; Liu, J.; Lin, Y.L.; Luo, W.S.; Chu, Y.J.; Chang, C.L.; Jen, C.L.; Lee, M.H.; Lu, S.N.; Wang, L.Y.; et al. The rs2296651 (S267F) variant on NTCP (SLC10A1) is inversely associated with chronic hepatitis B and progression to cirrhosis and hepatocellular carcinoma in patients with chronic hepatitis B. Gut 2016, 65, pp. 1514-1521. [CrossRef]
- Hu, P.; Liu, J.; Zhang D. Association of NTCP Gene Polymorphisms and Spontaneous Clearance of Hepatitis B Virus in Asia: A Meta-Analysis. Hepat Mon 2019; 19: pp. 2-8. [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012, 1, e00049. [CrossRef]
- Yang, W.; Summers, J. Integration of hepadnavirus DNA in infected liver: evidence for a linear precursor. J Virol 1999, 73, 9710-9717. [CrossRef]
- Xia, Y.; Guo, H. Hepatitis B virus cccDNA: Formation, regulation and therapeutic potential. Antiviral Res 2020, 180, 104824. [CrossRef]
- Kim, J.H.; Kang, S.; Kim, J.; Ahn, B.Y. Hepatitis B virus core protein stimulates the proteasome-mediated degradation of viral X protein. J Virol 2003, 77, pp. 7166-7173. [CrossRef]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 2009, 22, pp. 240-273, Table of Contents. [CrossRef]
- Li, Q.; Sun, B.; Zhuo, Y.; Jiang, Z.; Li, R.; Lin, C.; Jin, Y.; Gao, Y.; Wang, D. Interferon and interferon-stimulated genes in HBV treatment. Front Immunol 2022, 13, 1034968. [CrossRef]
- Liu, Q.; Zheng, Y.; Yu, Y.; Tan, Q.; Huang, X. Identification of HLA-A*0201-restricted CD8+ T-cell epitope C(6)(4)(-)(7)(2) from hepatitis B virus core protein. Int Immunopharmacol 2012, 13, pp. 141-147. [CrossRef]
- Lee, J.Y.; Chae, D.W.; Kim, S.M.; Nam, E.S.; Jang, M.K.; Lee, J.H.; Kim, H.Y.; Yoo, J.Y. Expression of FasL and perforin/granzyme B mRNA in chronic hepatitis B virus infection. J Viral Hepat 2004, 11, pp. 130-135. [CrossRef]
- Schreiber, S.; Honz, M.; Mamozai, W.; Kurktschiev, P.; Schiemann, M.; Witter, K.; Moore, E.; Zielinski, C.; Sette, A.; Protzer, U.; et al. Characterization of a library of 20 HBV-specific MHC class II-restricted T cell receptors. Mol Ther Methods Clin Dev 2021, 23, pp. 476-489. [CrossRef]
- [68] Asao, H. Interleukin-21 in Viral Infections. Int J Mol Sci 2021, 22. [CrossRef]
- [69] Wang, S.; Wang, J.; Kumar, V.; Karnell, J.L.; Naiman, B.; Gross, P.S.; Rahman, S.; Zerrouki, K.; Hanna, R.; Morehouse, C.; et al. IL-21 drives expansion and plasma cell differentiation of autoreactive CD11c(hi)T-bet(+) B cells in SLE. Nat Commun 2018, 9, 1758. [CrossRef]
- Schillie, S.; Murphy, T.V.; Sawyer, M.; Ly, K.; Hughes, E.; Jiles, R.; de Perio, M.A.; Reilly, M.; Byrd, K.; Ward, J.W.; et al. CDC guidance for evaluating health-care personnel for hepatitis B virus protection and for administering postexposure management. MMWR Recomm Rep 2013, 62, pp. 1-19.
- Shi, Y.X.; Huang, C.J.; Yang, Z.G. Impact of hepatitis B virus infection on hepatic metabolic signaling pathway. World J Gastroenterol 2016, 22, pp. 8161-8167. [CrossRef]
- Hu, J.J.; Song, W.; Zhang, S.D.; Shen, X.H.; Qiu, X.M.; Wu, H.Z.; Gong, P.H.; Lu, S.; Zhao, Z.J.; He, M.L.; et al. HBx-upregulated lncRNA UCA1 promotes cell growth and tumorigenesis by recruiting EZH2 and repressing p27Kip1/CDK2 signaling. Sci Rep 2016, 6, 23521. [CrossRef]
- Li, Z.; Li, X.; Wu, S.; Xue, M.; Chen, W. Long non-coding RNA UCA1 promotes glycolysis by upregulating hexokinase 2 through the mTOR-STAT3/microRNA143 pathway. Cancer Sci 2014, 105, 951-955. [CrossRef]
- Lamontagne, R.J.; Casciano, J.C.; Bouchard, M.J. A broad investigation of the HBV-mediated changes to primary hepatocyte physiology reveals HBV significantly alters metabolic pathways. Metabolism 2018, 83, pp. 50-59. [CrossRef]
- Sadrolodabaee, L.; Low, T. K.; Feng, H.; Chen, W. N. Role of HBV Replication in Host Cell Metabolism: A Proteomics Analysis. Curr Proteomics 2013, 10, pp. 29-37. [CrossRef]
- Shin, H.J.; Park, Y.H.; Kim, S.U.; Moon, H.B.; Park, D.S.; Han, Y.H.; Lee, C.H.; Lee, D.S.; Song, I.S.; Lee, D.H.; et al. Hepatitis B virus X protein regulates hepatic glucose homeostasis via activation of inducible nitric oxide synthase. The Journal of biological chemistry 2011, 286, pp. 29872-29881. [CrossRef]
- Borchani-Chabchoub, I.; Mokdad-Gargouri, R.; Gargouri, A. Glucose dependent [correction of dependant] negative translational control of the heterologous expression of the preS2 HBV antigen in yeast. Gene 2003, 311, pp. 165-170. [CrossRef]
- Liu, B.; Fang, M.; He, Z.; Cui, D.; Jia, S.; Lin, X.; Xu, X.; Zhou, T.; Liu, W. Hepatitis B virus stimulates G6PD expression through HBx-mediated Nrf2 activation. Cell Death Dis 2015, 6, e1980. [CrossRef]
- Kim, K.; Kim, K.H.; Kim, H.H.; Cheong, J. Hepatitis B virus X protein induces lipogenic transcription factor SREBP1 and fatty acid synthase through the activation of nuclear receptor LXRalpha. Biochem J 2008, 416, pp. 219-230. [CrossRef]
- Willy, P.J.; Umesono, K.; Ong, E.S.; Evans, R.M.; Heyman, R.A.; Mangelsdorf, D.J. LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev 1995, 9, pp. 1033-1045. [CrossRef]
- DeBose-Boyd, R.A.; Ye, J. SREBPs in Lipid Metabolism, Insulin Signaling, and Beyond. Trends Biochem Sci 2018, 43, pp. 358-368. [CrossRef]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int J Mol Sci 2020, 21. [CrossRef]
- Wakil, S.J.; Abu-Elheiga, L.A. Fatty acid metabolism: target for metabolic syndrome. J Lipid Res 2009, 50 Suppl, pp. 138-143. [CrossRef]
- Marechal, L.; Laviolette, M.; Rodrigue-Way, A.; Sow, B.; Brochu, M.; Caron, V.; Tremblay, A. The CD36-PPARgamma Pathway in Metabolic Disorders. Int J Mol Sci 2018, 19. [CrossRef]
- Park, J.H.; Iwamoto, M.; Yun, J.H.; Uchikubo-Kamo, T.; Son, D.; Jin, Z.; Yoshida, H.; Ohki, M.; Ishimoto, N.; Mizutani, K.; et al. Structural insights into the HBV receptor and bile acid transporter NTCP. Nature 2022, 606, pp. 1027-1031. [CrossRef]
- Wang, X.; Xie, G.; Zhao, A.; Zheng, X.; Huang, F.; Wang, Y.; Yao, C.; Jia, W.; Liu, P. Serum Bile Acids Are Associated with Pathological Progression of Hepatitis B-Induced Cirrhosis. J Proteome Res 2016, 15, 1126-1134. [CrossRef]
- Lauber, C.; Seitz, S.; Mattei, S.; Suh, A.; Beck, J.; Herstein, J.; Borold, J.; Salzburger, W.; Kaderali, L.; Briggs, J.A.G.; et al. Deciphering the Origin and Evolution of Hepatitis B Viruses by Means of a Family of Non-enveloped Fish Viruses. Cell Host Microbe 2017, 22, pp. 387-399 e386. [CrossRef]
- Kocher, A.; Papac, L.; Barquera, R.; Key, F.M.; Spyrou, M.A.; Hubler, R.; Rohrlach, A.B.; Aron, F.; Stahl, R.; Wissgott, A.; et al. Ten millennia of hepatitis B virus evolution. Science 2021, 374, pp. 182-188. [CrossRef]
- Larsen, C.S. The past 12,000 years of behavior, adaptation, population, and evolution shaped who we are today. Proc Natl Acad Sci U S A 2023, 120, e2209613120. [CrossRef]
- Roman, S.; Jose-Abrego, A.; Fierro, N.A.; Escobedo-Melendez, G.; Ojeda-Granados, C.; Martinez-Lopez, E.; Panduro, A. Hepatitis B virus infection in Latin America: a genomic medicine approach. World J Gastroenterol 2014, 20, pp. 7181-7196. [CrossRef]
- Peacock, T.P.; Penrice-Randal, R.; Hiscox, J.A.; Barclay, W.S. SARS-CoV-2 one year on: evidence for ongoing viral adaptation. J Gen Virol 2021, 102. [CrossRef]
- Liang, T.J. Hepatitis B: the virus and disease. Hepatology 2009, 49, pp. 13-21. [CrossRef]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA Integration: Molecular Mechanisms and Clinical Implications. Viruses 2017, 9. [CrossRef]
- Roman, S.; Tanaka, Y.; Khan, A.; Kurbanov, F.; Kato, H.; Mizokami, M.; Panduro, A. Occult hepatitis B in the genotype H-infected Nahuas and Huichol native Mexican population. J Med Virol 2010, 82, pp. 1527-1536. [CrossRef]
- Jose-Abrego, A.; Roman, S.; Laguna-Meraz, S.; Rebello-Pinho, J.R.; Justo Arevalo, S.; Panduro, A. Tracing the evolutionary history of hepatitis B virus genotype H endemic to Mexico. Front Microbiol 2023, 14, 1180931. [CrossRef]
- Jaramillo, C.M.; de La Hoz, F.; Porras, A.; di Filippo, D.; Choconta-Piraquive, L.A.; Payares, E.; Montes, N.; Navas, M.C. Characterization of hepatitis B virus in Amerindian children and mothers from Amazonas State, Colombia. PloS one 2017.
- Wang, L.; Wang, K.; Zou, Z.Q. Crosstalk between innate and adaptive immunity in hepatitis B virus infection. World J Hepatol 2015, 7, 2980-2991. [CrossRef]
- Yamada, N.; Shigefuku, R.; Sugiyama, R.; Kobayashi, M.; Ikeda, H.; Takahashi, H.; Okuse, C.; Suzuki, M.; Itoh, F.; Yotsuyanagi, H.; et al. Acute hepatitis B of genotype H resulting in persistent infection. World J Gastroenterol 2014, 20, pp. 3044-3049. [CrossRef]
- Centers for disease control and prevention, 2020. The age-adjusted death rate for hepatitis C and Hepatitis B during 2020. Available online: https://www.cdc.gov/hepatitis/policy/npr/2022/reduce-reported-hepatitis-b-deaths.htm#:~:text=The%20age%2Dadjusted%20hepatitis%20B,the%20target%20rate%20of%200.42 (accessed on 4 July 2023).
- Centers for disease control and prevention, 2012. Viral Hepatitis and Liver Cancer. Available online: https://www.cdc.gov/nchhstp/newsroom/docs/factsheets/viral-hep-liver-cancer.pdf (accessed on 4 July 2023).
- World Health Organization, 2017. Guidelines on hepatitis B and C testing. Available online: https://www.who.int/publications/i/item/9789241549981(accessed on 4 July 2023).
- Forni, D.; Cagliani, R.; Pontremoli, C.; Pozzoli, U.; Vertemara, J.; De Gioia, L.; Clerici, M.; Sironi, M. Evolutionary Analysis Provides Insight Into the Origin and Adaptation of HCV. Front Microbiol 2018, 9, 854. [CrossRef]
Figure 1.
Hepatitis B virus cycle. 1) The cycle begins with the specific binding of HBV to its receptor, sodium-taurocholate co-transporting polypeptide (NTCP). This binding triggers the process of receptor-mediated endocytosis. 2) Inside the hepatocyte, the lysosomes break down the viral envelope and nucleocapsid. 3) Viral DNA is released into the cytoplasm. 4) Subsequently, the viral DNA is transported to the nucleus, where it undergoes repair and conversion to a more stable form called covalently closed circular DNA (cccDNA). 5) The cccDNA serves as a template for the synthesis of viral RNA. 6) After, ARNs are transported to the cytoplasm and the rough endoplasmic reticulum (RER) to facilitate the production of viral proteins. 7-8) The core protein or nucleoprotein assembles around a circularized RNA within the cytoplasm. 9) After as-sembly, transcription is carried out by the viral polymerase. 10-11) Within the RER, the viral particles are enveloped by a lipid membrane, which carries the Hepatitis B surface Antigen (HBsAg). 12) Finally, the mature virions are released into the bloodstream, accompanied by empty particles that serve as decoys for the immune response.
Figure 1.
Hepatitis B virus cycle. 1) The cycle begins with the specific binding of HBV to its receptor, sodium-taurocholate co-transporting polypeptide (NTCP). This binding triggers the process of receptor-mediated endocytosis. 2) Inside the hepatocyte, the lysosomes break down the viral envelope and nucleocapsid. 3) Viral DNA is released into the cytoplasm. 4) Subsequently, the viral DNA is transported to the nucleus, where it undergoes repair and conversion to a more stable form called covalently closed circular DNA (cccDNA). 5) The cccDNA serves as a template for the synthesis of viral RNA. 6) After, ARNs are transported to the cytoplasm and the rough endoplasmic reticulum (RER) to facilitate the production of viral proteins. 7-8) The core protein or nucleoprotein assembles around a circularized RNA within the cytoplasm. 9) After as-sembly, transcription is carried out by the viral polymerase. 10-11) Within the RER, the viral particles are enveloped by a lipid membrane, which carries the Hepatitis B surface Antigen (HBsAg). 12) Finally, the mature virions are released into the bloodstream, accompanied by empty particles that serve as decoys for the immune response.
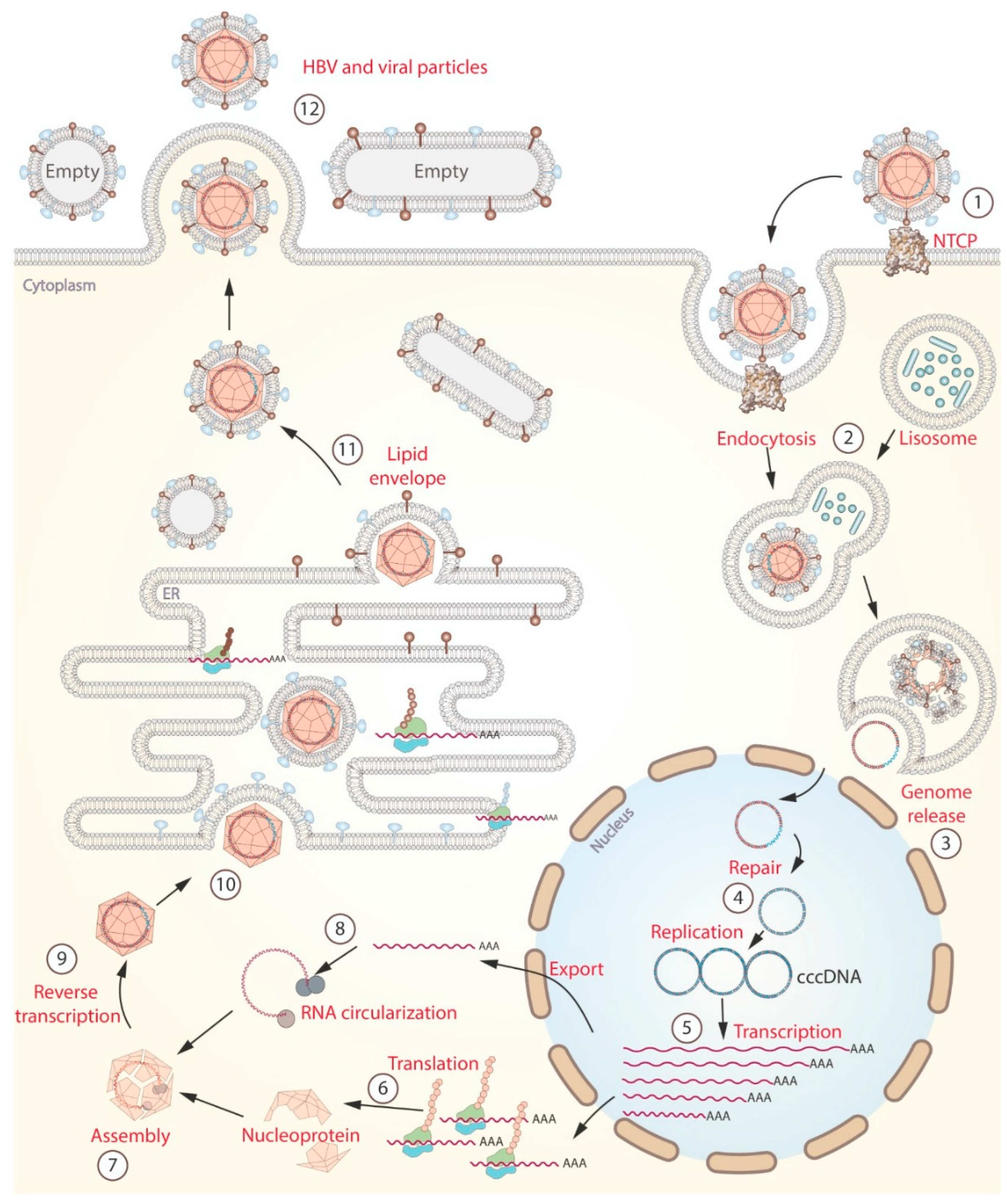
Figure 2.
Immune response during HBV infection. (A) shows the cytokine-mediated process of hepatocyte death. Numbers 1-10 indicate the cell activation process. Numbers 11-14 and 15-16 show the nuclear and cytoplasmic pathways (respectively) to activate apoptosis. (B) shows the neutralization of viral particles in the bloodstream. 1) capture and processing of HBV, 2) presentation of antigens, 3) release of cytokines (IL-21), 4) differentiating B cells into plasma cells. Finally, 5) production of primary antibodies against HBV infection.
Figure 2.
Immune response during HBV infection. (A) shows the cytokine-mediated process of hepatocyte death. Numbers 1-10 indicate the cell activation process. Numbers 11-14 and 15-16 show the nuclear and cytoplasmic pathways (respectively) to activate apoptosis. (B) shows the neutralization of viral particles in the bloodstream. 1) capture and processing of HBV, 2) presentation of antigens, 3) release of cytokines (IL-21), 4) differentiating B cells into plasma cells. Finally, 5) production of primary antibodies against HBV infection.
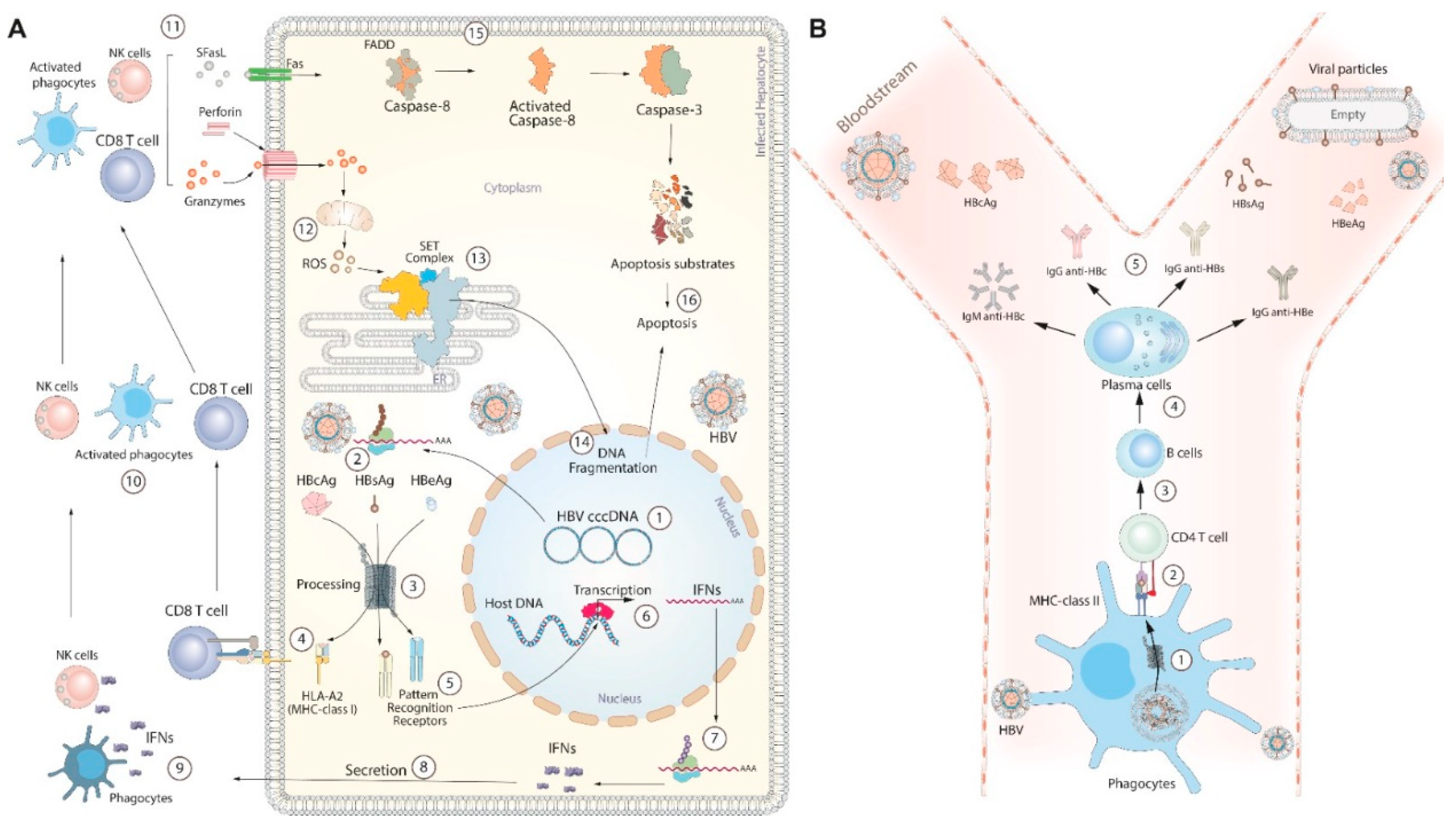
Figure 3.
HBV interaction with host metabolic pathways. 1-3) Metabolic control is mediated by the Hepatitis B protein X (HBx), which activates the expression of genes involved in 4-10) glucose metabolism (grey arrows), 11-18) lipid metabolism (blue arrows), and 19) bile acid uptake (red arrow). The enzymes or receptors highlighted in red represent the key proteins up-regulated by HBx. The orange arrows represent the main effects of metabolic control by HBV.
Figure 3.
HBV interaction with host metabolic pathways. 1-3) Metabolic control is mediated by the Hepatitis B protein X (HBx), which activates the expression of genes involved in 4-10) glucose metabolism (grey arrows), 11-18) lipid metabolism (blue arrows), and 19) bile acid uptake (red arrow). The enzymes or receptors highlighted in red represent the key proteins up-regulated by HBx. The orange arrows represent the main effects of metabolic control by HBV.
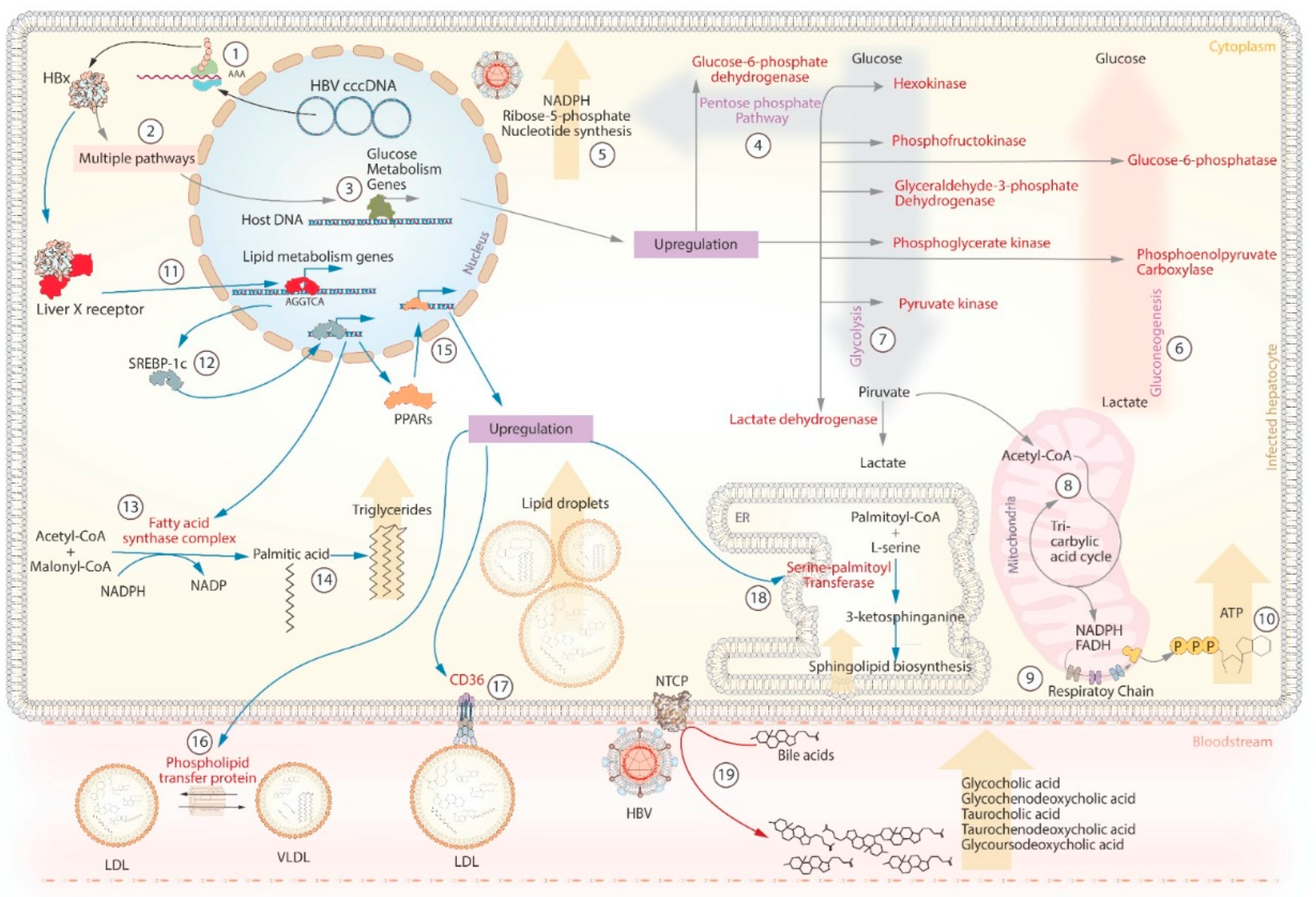
Figure 4.
The theory of viral adaptation. The three degrees of viral adaptation are shown on the left side. Each column indicates the representative clinical outcomes, viral load dynamics during infection, and immune and metabolic interactions. On the right, the potential factors that can break the levels of adaptation are represented.
Figure 4.
The theory of viral adaptation. The three degrees of viral adaptation are shown on the left side. Each column indicates the representative clinical outcomes, viral load dynamics during infection, and immune and metabolic interactions. On the right, the potential factors that can break the levels of adaptation are represented.
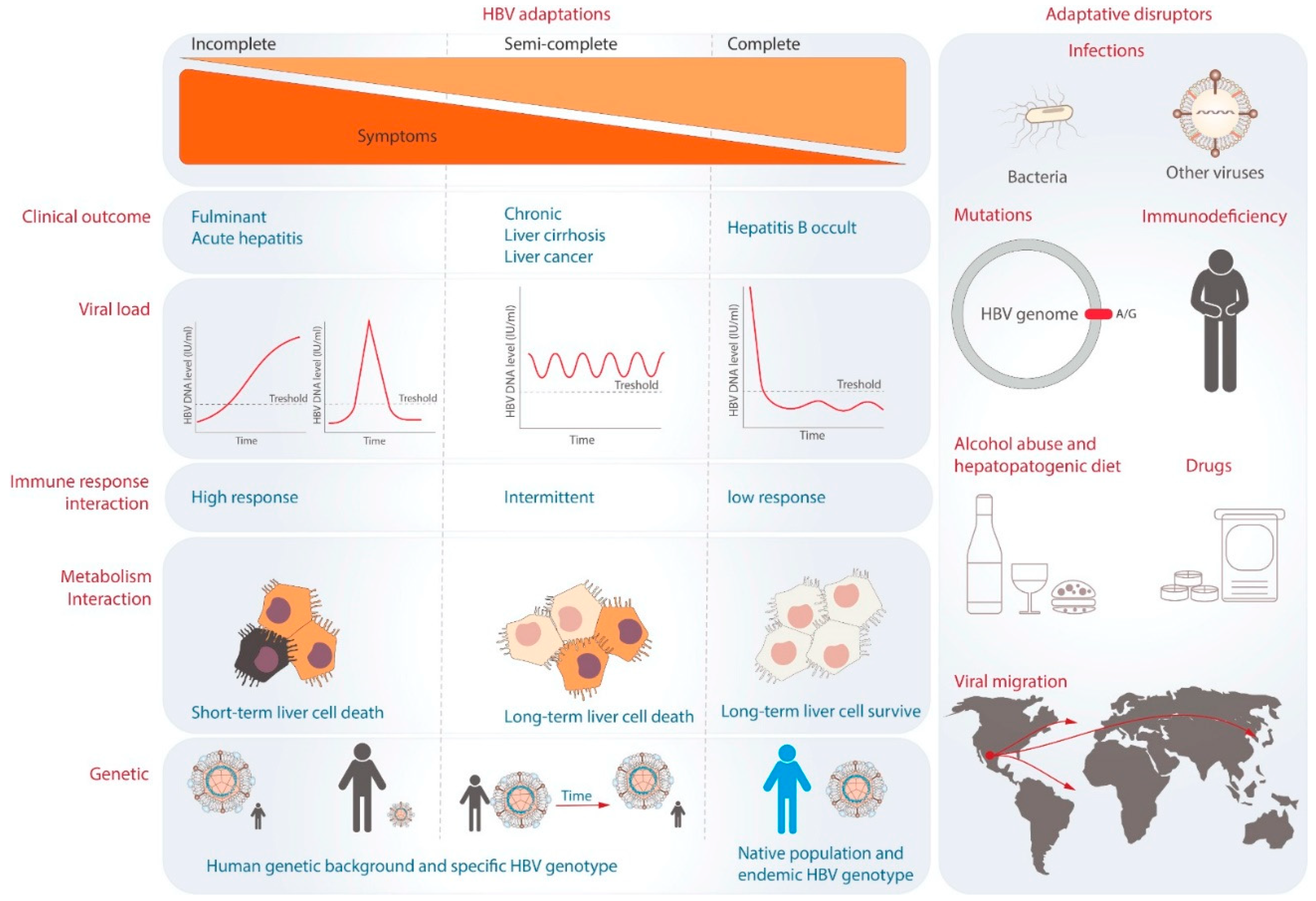

Table 1.
Single nucleotide polymorphism (SNP) associated with HBV clinical outcomes.
| Clinical outcome and associated genes | Population | SNP database | OR | 95% CI | Reference |
|---|---|---|---|---|---|
| Cirrhosis | |||||
| Intercellular adhesion molecule-1 (ICAM-1) | China | rs1799969 (A) | 4.197 | 2.550-7.074 | [35] |
| Interferon gamma (IFN-γ) | China | rs2430561 (A) + rs1861494 (A) | 1.485 | 1.065-2.070 | [36] |
| Transforming growth factor (TGF)-beta1 | Korea | rs1982073 (LL) | 3.408 | 1.279-9.085 | [37] |
| Signal transducer and activator of transcription 3 (STAT3) | China | rs4796793 (GG) | 2.17 | 1.11-4.23 | [38] |
| Signal transducer and activator of transcription 4 (STAT4) | China | rs7574865 (TG) | 1.17 | 1.03–1.34 | [39] |
| Complement component 2 (C2) | China | rs9267673 (TC) | 1.37 | 1.15–1.63 | [39] |
| Human leukocyte antigen (HLA)-DRB1 | China | rs2647073 (CA) | 1.63 | 1.29–2.06 | [39] |
| Human leukocyte antigen (HLA)-DRB1 | China | rs3997872 (AT) | 1.86 | 1.32–2.62 | [39] |
| Human leukocyte antigen (HLA)-DQ | China | rs9275319 (GA) | 1.32 | 1.06–1.64 | [39] |
| Hepatocellular carcinoma | |||||
| Protein Phosphatase 1 Catalytic Subunit Beta (PPP1CB) | China | rs13025377 (AA) | 1.54 | 1.22-1.95 | [41] |
| Mouse double minute 2 homolog (MDM2) | Korea | rs2279744 (G) | 4.27 | 2.23-8.20 | [45] |
| Tumor protein 53 (p53) | Korea | rs1042522 (Pro/Pro) | 3.59 | 1.77-7.31 | [45] |
| MDM2 + p53 | Korea | rs2279744 (G/G) + rs1042522 (Pro/Pro) | 20.78 | 5.25-82.36 | [45] |
| DEP Domain Containing 5 (DEPDC5) | China | rs1012068 (CC) | 2.397 | 1.251-4.595 | [40] |
| X-chromosome long arm band 22.1 (Xq22.1) | China | rs5945919 (AG) | 2.22 | 1.15-4.30 | [43] |
| CD33 molecule (SIGLEC3 or CD33) | Taiwan | rs12459419 (C) | 1.256 | 1.027-1.535 | [44] |
| Occult hepatitis B | |||||
| Human leukocyte antigen (HLA)-DPA1 | Indonesia | rs3077 (CC) | 6.12 | 1.30-28.85 | [46] |
| Human leukocyte antigen (HLA)-DPA1 | Indonesia | rs3077 (T) + rs3135021(G) + rs9277535 (A) | 4.9 | 1.12-21.52 | [46] |
| Human leukocyte antigen (HLA)-DQB1 | China | NA | 2.15 | 1.118-4.161 | [47] |
| Human leukocyte antigen (HLA)-HLA-C*07:01 | China | NA | 2.146 | 1.070-4.306 | [47] |
| Human leukocyte antigen (HLA)-B*44:03 | China | NA | 4.693 | 1.822-12.086 | [47] |
| Human leukocyte antigen (HLA)-DRB1*07:01 | China | NA | 1.919 | 1.188-3.101 | [47] |
| Human leukocyte antigen (HLA)-DQB1*02:02 | China | NA | 2.012 | 1.303-3.107 | [47] |
| HBV reactivation | |||||
| Human leukocyte antigen (HLA)-DPB2 | Japan | rs872956 (AA) | 8.277 | 1.540-51.550 | [50] |
| Interleukin 13 (IL-13) | Taiwan | rs1295686 (AA) | 4.683 | 1.030-19.156 | [51] |
| Clearance or protection | |||||
| Sodium taurocholate cotransporting polypeptide (NTCP) | Korea | rs2296651(CT) | 0.455 | 0.220-0.942 | [52] |
| Toll-like receptor (TLR5) | Taiwan | rs5744174 (T) | 1.32 | 1.03-1.69 | [53] |
| Interferon-induced helicase C domain-containing protein 1 (IFIH1) | China | rs2111485 (G) | 0.47 | 0.25-0.87 | [54] |
| DExD/H-box helicase 58 (DDX58) | China | rs3824456 (C) or rs2074160 (A) | 0.69 | 0.49-0.97 | [54] |
| Sodium taurocholate cotransporting polypeptide (NTCP) | Taiwan | rs2296651(AA) | 0.13 | 0.05-0.34 | [57] |
| NA: Not available; letters in parentheses indicate allele or genotype; OR: Odds ratio; 95% CI: 95% confidence interval | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



