
Submitted:
23 August 2023
Posted:
23 August 2023
You are already at the latest version
Abstract
Plasminogen (Plg) is the inactive form of plasmin (Plm) that exists in two major glycoforms, referred to as glycoforms I and II (GI and GII). In the circulation, Plg assumes an activation-resistant 'closed' conformation via interdomain interactions and is mediated by the lysine binding site (LBS) on the kringle (KR) domains. These inter-domain interactions can be readily disrupted when Plg binds to lysine/arginine residues on protein targets or free L-lysine and analogues. This causes Plg to convert into an 'open' form which is crucial for activation by host activators. In this study, we investigated how various ligands affect the kinetics of Plg conformational change using small-angle X-ray scattering (SAXS). We began by examining the open and closed conformations of Plg using size-exclusion chromatography (SEC) coupled with SAXS. Next, we developed a high throughput (HTP) 96-well SAXS assay setup. This method enables us to determine the Kopen value, which is used to compare ligands’ effect on Plg conformation directly. Based on our analysis using Plg GII, we have found that the Kopen for ε-aminocaproic acid (EACA) is approximately three times greater than that of Tranexamic acid (TXA), which is widely recognized as a highly effective ligand. We demonstrated further that Plg undergoes a conformational change when it binds to the C-terminal peptides of the inhibitor α2-antiplasmin and receptor Plg‑RKT. Our findings suggest that, besides the C-terminal lysine, internal lysine(s) are also necessary for the formation of open Plg. Finally, we compared the conformational changes of Plg GI and GII directly and found that the closed form of GI, which has an N-linked glycosylation, is less stable. To summarize, we have successfully determined the response of Plg to various ligand/receptor peptides by directly measuring the kinetics of its conformational changes.
Keywords:
SAXS
; conformational change
; plasminogen
; fibrinolysis
; structure-function
; lysine binding site
; lysine analogue
; kringle domain
Introduction
Fibrinolysis is essential for hemostasis and vascular patency. Plasmin (Plm) is the critical enzyme that breaks down fibrin clots. This enzyme also plays other vital roles in many physiological and pathological processes, including extracellular matrix degradation, tissue remodelling, wound healing, pathogen invasion and cancer migration (1,2).
Plasminogen (Plg) is the zymogen form of Plm. It is made up of 791 residues and seven domains, including an N-terminal Pan apple domain (PAp), five kringle domains (KR1-5) and a serine protease domain (SP). The Plg found in human plasma has two main glycoforms: GI and GII, of 92,000 and 89,000 Da, respectively. Both glycoforms are O-linked glycosylated at Thr346 and Ser248; GI has an additional N-linked glycan at Asn289 (6-8) in KR-3. The two glycoforms vary in half-life, substrate specificity, and target affinity and play different functional roles (9-11).
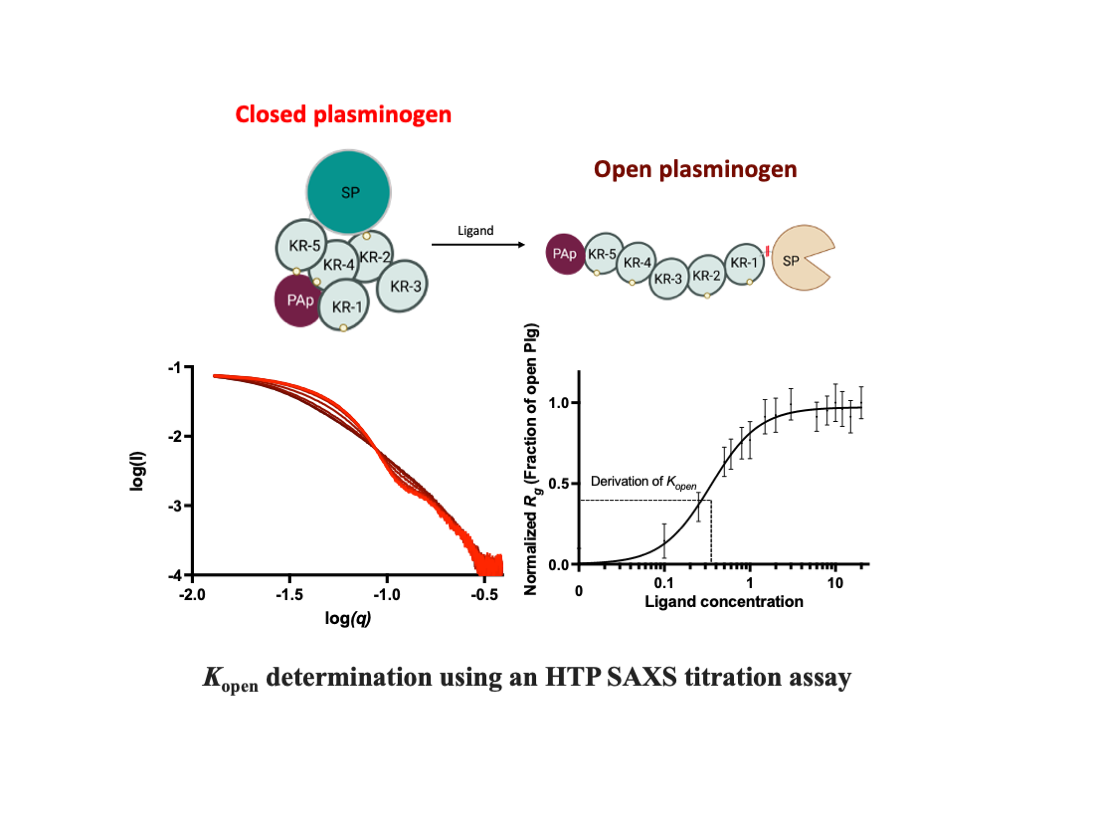
When circulating in the body, the full-length Plg forms a compact ‘closed’ conformation to prevent unintended activation and non-specific binding (Figure S1). We have shown by X-ray crystallography that the closed form is attained through multiple inter-domain interactions mediated by the lysine binding site (LBS) on the KR domains (3), where the PAp domain is indispensable (Figure S2). However, lysine or analogues in solution or on target receptors and substrates can interfere with these interactions and cause Plg to transition from a closed to an open conformation. When Plg assumes the relaxed and elongated “open” form, the activation loop, which is concealed (4) in the closed form, becomes exposed (Figure S1). Accordingly, Plg can be activated by the host tissue-type (tPA) and urokinase-type (uPA) Plg activators through a proteolytic cleavage between residues Arg561 and Val562 (5).
The transition from closed to open Plg involves a significant change in the shape of the molecule. This characteristic makes it an ideal candidate for biological small-angle x-ray scattering (SAXS) studies, allowing detailed analysis of the transition process and the flexibility profiles in the solution (15-19).
This paper presents a comprehensive analysis of the closed and open structures of Plg GII, a prominent glycoform found in human plasma.
In addition, we will detail the setup and validation of a high-throughput (HTP) SAXS assay that utilizes a 96-well plate and static mode for data collection. This method provides a reliable way to measure the kinetics of conformational transition in Plg directly. We used this method to compare the efficacy of different ligand-induced transformations in Plg GII conformation in solution, including L-lysine, lysine analogues EACA (ε-aminocaproic acid) and TXA (Tranexamic acid).
Due to their significant scattering-interfering signals, SAXS is not a suitable technique to study the conformational transitions of Plg when it binds to the macromolecular biological, such as fibrin, protease inhibitors, or cell surface receptors. In this study, we overcome this issue using peptides derived from two Plg binders: plasmin inhibitor α2-antiplasmin (α2AP) and Plg receptor Plg-RKT.
Several laboratories have utilized comparable techniques to study the transition of Plg from its closed to the open state. However, previous research was performed on preparations consisting of both GI and GII. Therefore, it remains to be investigated if there is any difference between conformational transition kinetics between GI and GII (4,12-14). Accordingly, we performed a thorough comparative analysis of Plg GI and GII and found that GII is more stable than GI, as expected.
Results
Characterization of closed-to-open Plg GII
In our earlier studies using X-ray crystallography, we revealed how the LBS of KRs interacts with lysine or arginine residues (3) on the surface of the neighbouring domains within the same molecule. These interactions lead to the formation of the closed Plg, as shown in Figures S1 and S2. However, the interactions between domains break up readily when LBSs form new interactions with external lysine and arginine residues on the surface of other molecules, such as receptors and fibrin, or L-lysine and analogues in solution. As a result, Plg transforms into an open form (3,12,20).
Figure 1 shows the result of an SEC-SAXS experiment (data collected immediately after size-exclusion chromatography) conducted on native human Plg GII. The closed and open Plg GII (Figure 1a, as shown in the normalized A280) are readily distinguishable on an SEC column. To generate the fully open Plg GII, 10 mM EACA was used, as described in the Materials and Methods. The two conformations have a comparable molecular mass (~90,000 Da), as reflected by the zero-angle scattering I(0). The radius of gyration (Rg) calculated via Guinier approximation is 31.7 ± 0.11 Å for the closed and 48.8 ± 0.9 Å for open conformations (Figure 1b). Such a significant difference in Rg between the two conformations shows that SAXS is indeed the method of choice to study the kinetics of Plg confirmational transition.
The SAXS data reveal that the closed Plg is a well-defined globular structure. Accordingly, the closed Plg GII showed a relatively shallow gradient at the low angle with a clear transition (Guinier knee) to the high angle (Figure 1b). This is further confirmed by the bell-shaped curve on the dimensionless Kratky plot (Figure 1c). The pairwise distribution function P(r) is a skewed bell curve, from which the maximum diameter (Dmax) is determined to be 105 Å (Figure 1d). In the Porod plot (Figure 1e), the curve plateaus around 0.08 Å-1, roughly approximating a spheroidal particle with a diameter of 78 Å.
On the other hand, the open Plg GII form is a disordered and flexible structure. Accordingly, the data reveal a steep slope at the low angle (Figure 1b) without a well-defined transition between the low and the higher angle. There is also a lack of distinct maxima in the dimensionless Kratky plot (Figure 1c). Meanwhile, the P(r) function reveals a Dmax of 170 Å (compared with 105 Å in the closed form (Figure 1d)) and without a plateau in the Porod plot (Figure 1e).
To further understand the population dynamics of Plg, we analyzed the SAXS data using the ensemble optimization method (EOM) (21,22) (Figure 1f) with the high-resolution crystal structure of individual domains of Plg (3). Here, an in-silico model was prepared in which flexible linkers of appropriate length joined each of the seven domains of plasminogen (PAp, five KRs and SP domains). Assuming no interaction between domains, a random pool of structures was generated that samples the total conformational space of Plg. A genetic algorithm was used to select subpopulations from this random pool that, when taken together, best fit the SAXS data. As shown in Figure 1f, the Rg distribution of the random pool consists of a broader peak with a large maximum dimension of 60 Å. The selected ensemble for the closed form is compact and relatively homogeneous, as evidenced by the narrow peak in the distribution plot. In contrast, the open form has a broader peak that is shifted to the right of the random ensemble pool, indicating that in the open form, Plg GII is more elongated than would be expected from purely random domain movement. This peak is significantly broader than the closed form, suggesting a more flexible structure.
Conformational transition studies using an HTP assay
An HTP assay to study the kinetics of Plg conversion from a closed to an open state was established for this work using Plg GII (see Materials and Methods and Figure 2). Figure 3 shows the scattering curves and Rg that resulted from the Guinier analysis of Plg GII treated with EACA concentrations ranging from 0-20 mM.
The data showed that the transition from closed to open is readily observable (Figure 3a); all intermediate Rg values can be interpreted as a linear composite of the open and closed forms. The eigenvalues plot (Figure 3b-c) from singular value decomposition showed that the minimum number of components is two, namely fully open and fully closed, without any stable intermediates. To analyze the transition profiles of Plg recorded from the fully closed to the fully open forms, we fitted the kinetic data as follows: the Rg of Plg GII in 0 mM EACA (32 Å) was defined as closed. The Rg of Plg in 20 mM EACA (49 Å) was defined as open. The best fit for the kinetic data is achieved using a multi-site binding model, which yields a Kopen of 0.35 mM. We use Kopen to define ligand efficacy; it refers to the concentration of a ligand at which 50% of all Plg molecules in a solution transformed from the closed to the open conformation. If the closed conformation is more stable or the ligand is weaker, there will be a higher Kopen value. The value of 0.35 mM found in this study is comparable to that of 0.45 mM previously reported on a mixed glycoform sample (9). The slope of the Hill plot is 1.5 (Figure 3d inset), suggesting that cooperative binding of at least two EACA molecules is required for the conformational change of Plg.
Plg conformational transition in the presence of L-lysine and peptides with a C-terminal lysine
We used the HTP assay detailed before to study the impact of free L-lysine residue alone and lysine-containing peptides derived from Plg-binding proteins, inhibitor α2AP and cell receptor Plg-RKT.
Plg-RKT is a 17 kDa transmembrane Plg receptor with a conserved lysine residue at the C-terminus (23,24). Previous studies showed that Plg/Plm binds to a synthetic peptide called CK10, corresponding to a C-terminal peptide of Plg-RKT (24).
α2-antiplasmin (α2AP) is a specific and efficacious plasmin inhibitor belonging to the SERPIN family. It inhibits free circulating plasmin in the system and regulates fibrinolysis (25). In addition to the conserved SERPIN core, it has two unique extensions, one at the N- and one at the C-terminus. Plg/Plm binds to the C-terminal extension, which contains five conserved lysine/arginine residues, one of which locates at the extreme C-terminus. MK12 and NK55, corresponding to the last 12 and 55 residues (Figure 4), were used in this work (26).
We determine the Kopen for Plg GII in the presence of L-lysine and these three peptides. All peptides have a C-terminal lysine, which is previously determined to be essential for Plg binding (24, 28). The results are shown in Figure 4; the Kopen for L-lysine is 7.47 ± 1.12 mM, and CK10 is 0.73 ± 0.24 mM. Surprisingly, we could not detect any conformational change for MK12, even at the highest concentration tested at 10 mM.
Our data revealed that the concentration of free L-lysine to induce a transition from closed to open of Plg GII is more than 20-fold of EACA and ~10-fold of CK10, confirming that L-lysine is a poor substrate. On the other hand, MK12 (441MEEDYPQFGSPK452) did not induce a conformational transition even at 10 mM, as mentioned previously, whereas CK10 (C139EQSKLFDSK147), which has only one internal lysine apart from the C-terminal lysine, is highly efficacious with a Kopen of 0.73 ± 0.24 mM. This data suggests that the internal lysine plays an indispensable role in CK10.
NK55 is also derived from the C-terminus of α2AP, but it has a total of seven internal lysine/arginine residues (398NPSAPRELKEQQDSPGNKDFLQSLKGFPRGDKLFGPDLKLVPPMEEDYPQFGSPK452) (25). The Kopen for NK55 is 0.08 ± 0.05 mM, which is significantly more efficacious than L-lysine (~100-fold) and CK10 (~10-fold). Although mapping the exact position of the internal lysine/arginine residues in CK10 and NK55 is beyond the scope of the current study, our findings strongly suggest that a minimum of one internal lysine residue is required for Plg GII to transit from its closed to an open form. We also proposed that in the case of NK55, the internal lysine/arginine residues bind to the Plg KRs in the same way as a zipper, first leading to a conformational change followed by stabilizing and constraining the open form.
Conformation stability of Plg GI and GII
We used SEC-SAXS to compare the conformation stability of Plg GI and GII (Figure 5), which were purified to homogeneity (Figure s3). Direct comparison of the scattering profile, Kratky plot, and P(r) functions of closed Plg GI and GII reveal that the Rg and Dmax (in Å) for GI are 34.1 ± 0.4 and 109.5; and for GII are 31.8 ± 0.6 and 109.3, respectively. Based on the data, there is a subtle difference between closed GI and GII.
The ab initio low-resolution envelope models of GI and GII (average NSD values of 0.541 and 0.626, respectively) agree with the X-ray crystal structures (Figure 5f,g). CRYSOL analysis indicates that the experimental SAXS data are highly consistent with GI and GII crystal structures (χ values of 0.78 and 1.19, respectively). Interestingly, our previous X-ray crystallography studies on the GI and GII revealed that the two glycoforms are similar except for the KR-3. In GI, KR-3 is N-linked glycosylated at Asn289, and it is not visible in the electron density (Figure 5e–g), suggesting it is a flexible domain. In the current study, KR-3 is also poorly defined in the solution model. This observation supports our previous conclusion that the N289-glycosylation makes the KR-3 domain highly flexible. Based on our current observation, highly flexible structure domains would not be accurately depicted in models generated from SAXS studies.
We proposed further that the relative flexibility of the KR-3 domain would significantly impact the overall conformational stability of Plg GI. Thus we also performed kinetics studies on GI and GII to verify this hypothesis using EACA and TXA in the HTP format (Figure S4, Table 1).
Our data confirmed that the closed GII is more stable; the Kopen of EACA and TXA for GII is ~1.75 fold higher than GI. Amongst these two ligands, EACA is weaker; the Kopen for both GI and GII is ~3 fold of TXA.
In comparison with L-lysine (Figures S5 and S6) based on the studies performed on Plg GII, the Kopen for TXA (0.12 ± 0.008 mM) and EACA 0.35 ± 0.02 mM) are 60 and 20-fold lower (Kopen for L-lysine is 7.47 ± 1.12 mM). These data, on the one hand, confirm the high efficacy of these anti-fibrinolytic therapeutics; they also permit a quantitative comparison of their performance. Importantly, our data support the notion that these lysine analogues can potentially outcompete the binding of Plg to its binding proteins Plg-RKT and α2AP (Figure S6) at a therapeutic dosage in vivo (plasma concentration up to ~1 mM TXA and ~10 mM EACA).
Discussion
Plg is a vital therapeutic target for thrombotic and hemostatic diseases. Functionally, the closed Plg must undergo a significant structural change to become an open form upon binding to a target; this conformational change is also essential for its activation by the host activators to occur. The mechanism and process of activation and inhibition have been under intense scrutiny for many decades (9,13,20,27). In this work, we used SEC-SAXS to fully characterise the closed and open conformations of native human Plg GII. This part of this work has provided crucial data to guide the setup of the HTP SAXS assays and validate the results obtained.
We used the HTP SAXS titration studies to show that the conformational change of Plg can be directly and readily measured. We also showed that Kopen correlates well, but reversely, with Kactivation by tPA (refer to Figure S4 for direct comparison). Relevant to this field, the generation of Plm enzyme activity (Kactivation) is often used as a readout for ligand-induced conformational change. Compared with Kopen, the Kactivation concentration is higher, >8-fold for EACA and >3.5-fold for TXA; accordingly, we proposed that Plg activation by tPA may occur only after it is fully open.
Using the HTP SAXS assay, we have confirmed the previous observations that the transition from closed to open conformation is a single-step process (20) . This conformational change is triggered by positively cooperative binding to at least two lysine or lysine analogues (4,20,27).
We have determined the Kopen values for EACA, TXA and L-lysine through ligand titration. This allows us to compare their ligand efficacy directly and accurately. Our findings indicate that TXA is the most efficacious ligand (Figure S6).
We also determined the Kopen of small peptide ligands derived from Plg-binding proteins, Plg-RKT receptor and Plg inhibitor α2AP. Previous studies revealed that both Plg-RKT and α2AP bind to Plg via the C-terminal lysine (23,28,29); here, we showed that the additional internal lysine(s) is essential. Specifically, MK12-MEEDYPQFGSPK from the last 12 residues of α2AP, which has a single C-terminal lysine residue, is not functional, whereas CK10-CEQSKLFSDK from the last 10 residues of Plg-RKT, which has an additional internal lysine residue, is highly efficacious. This observation also aligns with our observation that binding at least two lysine residues is required for a conformational change (Figure 3). NK55, which consists of eight lysine/arginine residues, confers a higher efficiency in inducing conformational change, most likely via forming a more stable complex. Our findings indicate that having only a C-terminal lysine in a peptide is insufficient for Plg conformational change. The exact location of the necessary internal lysine will require further investigation.
Further, Streptococcal Plg binding M-like protein (PAM) from Group A Streptococci has the highest reported binding affinity for Plg. In comparison, VEK35 (35 residues) and VEK75 (75 residues) peptides derived from PAM are extremely efficacious (Figure S6) (30). Using the same HTP method we reported here for the first time, we showed that the dimeric VEK75 has a much higher activity than its shorter and monomeric counterpart, VEK35. Intriguingly, neither of these peptides has a C-terminal lysine. The binding of these peptides to Plg, and presumably PAM (30), would involve a different mode to that of Plg-RKT and α2AP.
In this study, we also dissected, in full detail, the conformational stability of the two glycoforms. Our data revealed that the conformation of GII is more stable than GI. Previously, we hypothesized that the Asn289 N-glycan on GI enhances the mobility of KR3 and destabilizes its closed-form (3). Here, we generated the Kopen values for the direct comparison. We also showed that the superposed correlation between the X-ray crystal structures and the SAXS models is excellent; the disordered domain present in the crystal structure of GI (i.e. KR-3) is also poorly represented in the SAXS envelope. We proposed that, in solution, the mobile domain gives weak diffraction and therefore becomes difficult to resolve from the background. This study enables a direct evaluation of how post-translational modifications affect the stability of the conformation in Plg.
In summary, we have developed and validated an HTP SAXS assay which was used successfully to determine the kinetics and conformational changes of Plg in response to various ligand/receptor peptides.
Experimental Procedures
Preparation of Plg Glycoform I and II. Plg was isolated and purified from human plasma from Red Cross Blood Bank Australia via a three-step purification as previously described (3,6). Plg GI and GII were prepared with a TXA gradient (Figure S3). Closed Plg was prepared by exhaustive dialysis of purified protein into the assay buffer (100 mM sodium phosphate pH 7.4, 5% glycerol). Proteins were concentrated to 1 mg/ml in centrifugal filters (Merck Millipore, MA, USA). The final filtrate was kept as a solvent blank for SAXS experiments.
Preparation of effector ligands. EACA, TXA and L-lysine were purchased from Sigma-Aldrich (MO, USA), whereas peptides MK12, NK55 and CK10 were purchased from GL Biochem (Shanghai, China). Solutions of EACA, TXA and L-lysine (0.5-1.0 M) were prepared in ultrapure water. The peptide ligands are slightly acidic and therefore were dissolved in 0.1 M NH4OH with gentle sonication. MK12 and CK10 were prepared as 20 mM and NK55 as a 625 μM stock solution.
SAXSdata collection. Experiments were performed at the Australian Synchrotron SAXS/WAXS beamline using a fixed energy of 12 keV and a camera length of 2.6 m.
For the SEC-SAXS experiment, in-line size exclusion chromatography using a WTC-010S5 column (WYATT) with the co-flow setup was used. For the HTP assays, 1 mg/ml protein samples were mixed 1:1 (v/v) with effector solutions to a final volume of 50 μL in a 96-well plate and incubated for 15-30 minutes before data collection at 16 °C. The final protein concentration for these experiments was 0.5 mg/mL in the assay buffer unless otherwise specified.
For each sample, a matching buffer was used for background scatter subtraction. To minimize the effect of radiation damage, 50 μL samples were flowed past the X-ray beam in a 1.5 mm diameter quartz capillary at 4 μL.s-1. 15 to 20 2D scattering images were collected on a Pilatus 1M X-ray detector (Dectris) using 18x1-second exposures. The images were averaged, and the background was subtracted using the Scatterbrain software available at the Australian Synchrotron (31).
SAXS data analysis Averaged SAXS data was processed with ATSAS 2.7.2 software package (EMBL Hamburg, Germany). PRIMUS (32) was used for determining the radius of gyration (Rg) and protein molecular weight via Guinier approximation (33) and particle distance distribution function P(r) by indirect Fourier transform.
Ab initio models were generated with DAMMIN (34). Ten models were generated and averaged for each dataset using DAMAVER (35). The averaged model selected has a normalized spatial discrepancy (NSD) value equal to or less than 0.6. SUPCOMB was used to superimpose the low-resolution ab initio models onto the x-ray crystal structures of plasminogen (36).
Singular value decomposition of the titration series data was carried out using the SVDPLOT program in the ATSAS suite of software (37).
SAXS kinetic analysis. For each titration series, the Rg values were normalized as follows:
Normalized Rg values were plotted against ligand or peptide concentration ([L]) and analyzed with the following specific binding equation using GraphPad Prism 6 (GraphPad, CA, USA):
Where h is the Hill slope and the kinetic constant, K open , is the ligand concentration at which 50% of Plg is in the open conformation (i.e., normalized Rg is 0.5).
Plg activation assay. The generation of Plm was monitored in 96-well microtiter plates using chromogenic substrate S-2251 (Chromogenix, Milan, Italy) after 1-hour incubation of Plg with different concentrations of effector and tPA at 28oC. Each reaction mix contained 0.25 μM Plg, 0.01 μM tPA and the indicated amount of effector. After incubation, 0.2 mM of substrate S-2251 was added, and hydrolysis was monitored by continuous absorbance measurement at 405 nm (A405 nm). Initial rates of reaction (V0) were obtained by performing a linear regression of the first five minutes of the progress curve and were subsequently normalized by:
and plotted against the corresponding concentration of effectors ([L]). The resulting sigmoidal curves were fitted using the following:
where h is the Hill slope, and the kinetic constant Kactivation is the ligand concentration at which the V0 is 50% of the maximum.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
AQ prepared protein, performed experiments, analyzed data and wrote the paper; NC performed experiments, analyzed data and wrote the paper; TCD analyzed data and wrote the paper; PJC analyzed data and wrote the paper; JW conceived the idea, analyzed data and wrote the paper; RL conceived the idea, performed experiments, analyzed data, coordinated the study and wrote the paper. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
This work is supported by the National Health and Medical Research Council. SAXS data was collected on the SAXS/WAXS beamline at the Australian Synchrotron. The authors acknowledge the support of the SAXS beamline of Australian Synchrotron, particularly Dr Nigel Kirby. JW is an ARC Laureate Research Fellow.
Conflicts of Interest
The authors declare that they have no conflicts of interest with the contents of this article.
References
- Castellino, F.J.; Ploplis, V.A. Structure and function of the plasminogen/plasmin system. Thromb. Haemost. 2005, 93, 647–654. [Google Scholar] [PubMed]
- Rijken, D.C.; Lijnen, H.R. New insights into the molecular mechanisms of the fibrinolytic system. J. Thromb. Haemost. January 2009, 7, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Law, R.H.; Caradoc-Davies, T.; Cowieson, N.; Horvath, A.J.; Quek, A.J.; Encarnacao, J.A.; Steer, D.; Cowan, A.; Zhang, Q.; Lu, B.G.; et al. The X-ray crystal structure of full-length human plasminogen. Cell Rep. 2012, 1, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P.; Holland, S.K.; Cederholm-Williams, S.A.; Marshall, J.M.; Brown, A.J.; Spraggon, G.; Blake, C.C. The compact domain conformation of human Glu-plasminogen in solution. Biochim Biophys Acta. 1992, 1159, 155–161. [Google Scholar] [CrossRef]
- Lijnen, H.R. Elements of the Fibrinolytic System. 2001.
- Hayes, M.L. , and Castellino, F.J. Carbohydrate of the human plasminogen variants. II. Structure of the asparagine-linked oligosaccharide unit. J Biol Chem. 1979, 254, 8772–8776. [Google Scholar] [CrossRef] [PubMed]
- Hayes, M.L.; Castellino, F.J. Carbohydrate of the human plasminogen variants. III. Structure of the O-glycosidically linked oligosaccharide unit. J Biol Chem. 1979, 254, 8777–8780. [Google Scholar] [CrossRef]
- Pirie-Shepherd, S.R.; Stevens, R.D.; Andon, N.L.; Enghild, J.J.; Pizzo, S.V. Evidence for a novel O-linked sialylated trisaccharide on Ser-248 of human plasminogen 2. J Biol Chem. 1997, 272, 7408–7411. [Google Scholar] [CrossRef]
- Brockway, W.J.; Castellino, F.J. Measurement of the binding of antifibrinolytic amino acids to various plasminogens. Arch Biochem Biophys. 1972, 151, 194–199. [Google Scholar] [CrossRef]
- Molgaard, L.; Ponting, C.P.; Christensen, U. Glycosylation at Asn-289 facilitates the ligand-induced conformational changes of human Glu-plasminogen. FEBS Lett. 1997, 405, 363–368. [Google Scholar] [CrossRef]
- Takada, Y.; Makino, Y.; Takada, A. Glu-plasminogen I and II: their activation by urokinase and streptokinase in the presence of fibrin and fibrinogen. Thromb Res. 1985, 39, 289–296. [Google Scholar] [CrossRef]
- Marshall, J.M.; Brown, A.J.; Ponting, C.P. Conformational studies of human plasminogen and plasminogen fragments: evidence for a novel third conformation of plasminogen. Biochemistry. 1994, 33, 3599–3606. [Google Scholar] [CrossRef] [PubMed]
- Weisel, J.W.; Nagaswami, C.; Korsholm, B.; Petersen, L.C.; Suenson, E. Interactions of plasminogen with polymerizing fibrin and its derivatives, monitored with a photoaffinity cross-linker and electron microscopy. J Mol Biol. 1994, 235, 1117–1135. [Google Scholar] [CrossRef] [PubMed]
- Kornblatt, J.A.; Schuck, P. Influence of temperature on the conformation of canine plasminogen: an analytical ultracentrifugation and dynamic light scattering study. Biochemistry. 2005, 44, 13122–13131. [Google Scholar] [CrossRef]
- Tsutakawa, S.E.; Van Wynsberghe, A.W.; Freudenthal, B.D.; Weinacht, C.P.; Gakhar, L.; Washington, M.T.; Zhuang, Z.; Tainer, J.A.; Ivanov, I. Solution X-ray scattering combined with computational modeling reveals multiple conformations of covalently bound ubiquitin on PCNA. Proc Natl Acad Sci USA 2011, 108, 17672–17677. [Google Scholar] [CrossRef] [PubMed]
- Capp, J.A.; Hagarman, A.; Richardson, D.C.; Oas, T.G. The Statistical Conformation of a Highly Flexible Protein: Small-Angle X-Ray Scattering of S. aureus Protein A. Structure 2014, 29, 00186–00185. [Google Scholar]
- Dovega, R.; Tsutakawa, S.; Quistgaard, E.M.; Anandapadamanaban, M.; Low, C.; Nordlund, P. Structural and Biochemical Characterization of Human PR70 in Isolation and in Complex with the Scaffolding Subunit of Protein Phosphatase 2A. PLoS One. 2014, 9, e101846. [Google Scholar] [CrossRef]
- Shtykova, E.V.; Baratova, L.A.; Fedorova, N.V.; Radyukhin, V.A.; Ksenofontov, A.L.; Volkov, V.V.; Shishkov, A.V.; Dolgov, A.A.; Shilova, L.A.; Batishchev, O.V.; et al. Structural analysis of influenza A virus matrix protein M1 and its self-assemblies at low pH. PLoS One 2013, 8, e82431. [Google Scholar] [CrossRef]
- Sterckx, Y.G.; Volkov, A.N.; Vranken, W.F.; Kragelj, J.; Jensen, M.R.; Buts, L.; Garcia-Pino, A.; Jove, T.; Van Melderen, L.; Blackledge, M.; et al. Small-angle X-ray scattering- and nuclear magnetic resonance-derived conformational ensemble of the highly flexible antitoxin PaaA2. Structure 2014, 22, 854–865. [Google Scholar] [CrossRef]
- Mangel, W.F.; Lin, B.H.; Ramakrishnan, V. Characterization of an extremely large, ligand-induced conformational change in plasminogen. Science 1990, 248, 69–73. [Google Scholar] [CrossRef]
- Bernado, P.; Mylonas, E.; Petoukhov, M.V.; Blackledge, M.; Svergun, D.I. Structural characterization of flexible proteins using small-angle X-ray scattering. J Am Chem Soc. 2007, 129, 5656–5664. [Google Scholar] [CrossRef]
- Tria, G.; Mertens, H.D.; Kachala, M.; Svergun, D.I. Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ. 2015, 2, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Miles, L.A.; Lighvani, S.; Baik, N.; Andronicos, N.M.; Chen, E.I.; Parmer, C.M.; Khaldoyanidi, S.; Diggs, J.E.; Kiosses, W.B.; Kamps, M.P.; et al. The plasminogen receptor, Plg-R(KT), and macrophage function. J Biomed Biotechnol 2012, 2012, 250464. [Google Scholar] [CrossRef] [PubMed]
- Andronicos, N.M.; Chen, E.I.; Baik, N.; Bai, H.; Parmer, C.M.; Kiosses, W.B.; Kamps, M.P.; Yates, J.R., 3rd; Parmer, R.J.; Miles, L.A. Proteomics-based discovery of a novel, structurally unique, and developmentally regulated plasminogen receptor, Plg-RKT, a major regulator of cell surface plasminogen activation. Blood. 2010, 115, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, P.B. Antiplasmin: The forgotten serpin? FEBS J. 2005, 272, 4852–4857. [Google Scholar] [CrossRef]
- Frank, P.S.; Douglas, J.T.; Locher, M.; Llinás, M.; Schaller, J. Structural/functional characterization of the α2-plasmin inhibitor C-terminal peptide. Biochemistry 2003, 42, 1078–1085. [Google Scholar] [CrossRef]
- Christensen, U.; Molgaard, L. Positive co-operative binding at two weak lysine-binding sites governs the Glu-plasminogen conformational change. Biochem J. 1992, 285, 419–425. [Google Scholar] [CrossRef]
- Lu, B.G.; Sofian, T.; Law, R.H.; Coughlin, P.B.; Horvath, A.J. Contribution of conserved lysine residues in the alpha2-antiplasmin C terminus to plasmin binding and inhibition. J Biol Chem. 2011, 286, 24544–24552. [Google Scholar] [CrossRef]
- Hortin, G.L.; Gibson, B.L.; Fok, K.F. Alpha 2-antiplasmin’s carboxy-terminal lysine residue is a major site of interaction with plasmin. Biochem Biophys Res Commun. 1988, 155, 591–596. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Liang, Z.; Quek, A.J.; Ploplis, V.A.; Law, R.; Castellino, F.J. Dimerization is not a determining factor for functional high affinity human plasminogen binding by the group A streptococcal virulence factor PAM and is mediated by specific residues within the PAM a1a2 domain. J Biol Chem. 2014, 289, 21684–21693. [Google Scholar] [CrossRef]
- Kirby, N.; Cowieson, N.; Hawley, A.M.; Mudie, S.T.; McGillivray, D.J.; Kusel, M.; Samardzic-Boban, V.; Ryan, T.M. Improved radiation dose efficiency in solution SAXS using a sheath flow sample environment. Acta Crystallographica. Sect. D Struct. Biol. 2016, 72, 1254–1266. [Google Scholar] [CrossRef]
- Konarev, P.V. , Volkov, V.V., Sokolova, A.V., Koch, M.H.J., and Svergun, D.I. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003, 36, 1277–1282. [Google Scholar] [CrossRef]
- Guinier, A. La diffraction des rayons X aux tres petits angles; application a l’etude de phenomenes ultramicroscopiques. Ann. Phys. 1939, 12, 161–237. [Google Scholar] [CrossRef]
- Svergun, D.I. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J. 1999, 76, 2879–2886. [Google Scholar] [CrossRef] [PubMed]
- Volkov, V.V.; Svergun, D.I. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2003, 36, 860–864. [Google Scholar] [CrossRef]
- Kozin, M.B.; Svergun, D.I. Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 2001, 34, 33–41. [Google Scholar] [CrossRef]
- Franke, D.; Petoukhov, M.V.; Konarev, P.V.; Panjkovich, A.; Tuukkanen, A.; Mertens, H.D.T.; Kikhney, A.G.; Hajizadeh, N.R.; Franklin, J.M.; Jeffries, C.M.; et al. ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J Appl Crystallogr 2017, 50, 1212–1225. [Google Scholar] [CrossRef]
- Fischer, M.J.E. Amine Coupling Through EDC/NHS: A Practical Approach. In Surface Plasmon Resonance: Methods and Protocols; Mol, N.J., Fischer, M.J.E., Eds.; Humana Press: Totowa, NJ, USA, 2010; pp. 55–73. [Google Scholar]
Figure 1.
SAXS studies of closed and open Plg GII. Superposed of the closed (red) and open (blue) Plg GII: (a) SEC-SAXS profiles showing the I(0) (left Y-axis), normalized absorbance (A280) and Rg (right Y-axes); (b) SAXS profiles recorded; (c) Dimensionless Kratky plot showing the globular nature of the closed and the disordered nature of the open conformation; (d) P(r) analysis shows the distinctive difference in the dimensions of the two forms; (e) Porod-Debye plots showing the Porod plateau of the closed form; and (f) Ensemble Optimization Method (EOM) of closed and open Plg GII for analyzing the difference in Rg distribution. Distribution curves correspond to a random pool of 10,000 generated structures (grey) and the EOM-optimized ensemble of closed (blue) and open (red) plasminogen. The closed conformation is best represented by the compact distribution curve, and therefore it is a homogenous ensemble. The distribution curve for the open form has shifted to the right; with a broader curve, it represents a more heterogeneous ensemble.
Figure 1.
SAXS studies of closed and open Plg GII. Superposed of the closed (red) and open (blue) Plg GII: (a) SEC-SAXS profiles showing the I(0) (left Y-axis), normalized absorbance (A280) and Rg (right Y-axes); (b) SAXS profiles recorded; (c) Dimensionless Kratky plot showing the globular nature of the closed and the disordered nature of the open conformation; (d) P(r) analysis shows the distinctive difference in the dimensions of the two forms; (e) Porod-Debye plots showing the Porod plateau of the closed form; and (f) Ensemble Optimization Method (EOM) of closed and open Plg GII for analyzing the difference in Rg distribution. Distribution curves correspond to a random pool of 10,000 generated structures (grey) and the EOM-optimized ensemble of closed (blue) and open (red) plasminogen. The closed conformation is best represented by the compact distribution curve, and therefore it is a homogenous ensemble. The distribution curve for the open form has shifted to the right; with a broader curve, it represents a more heterogeneous ensemble.
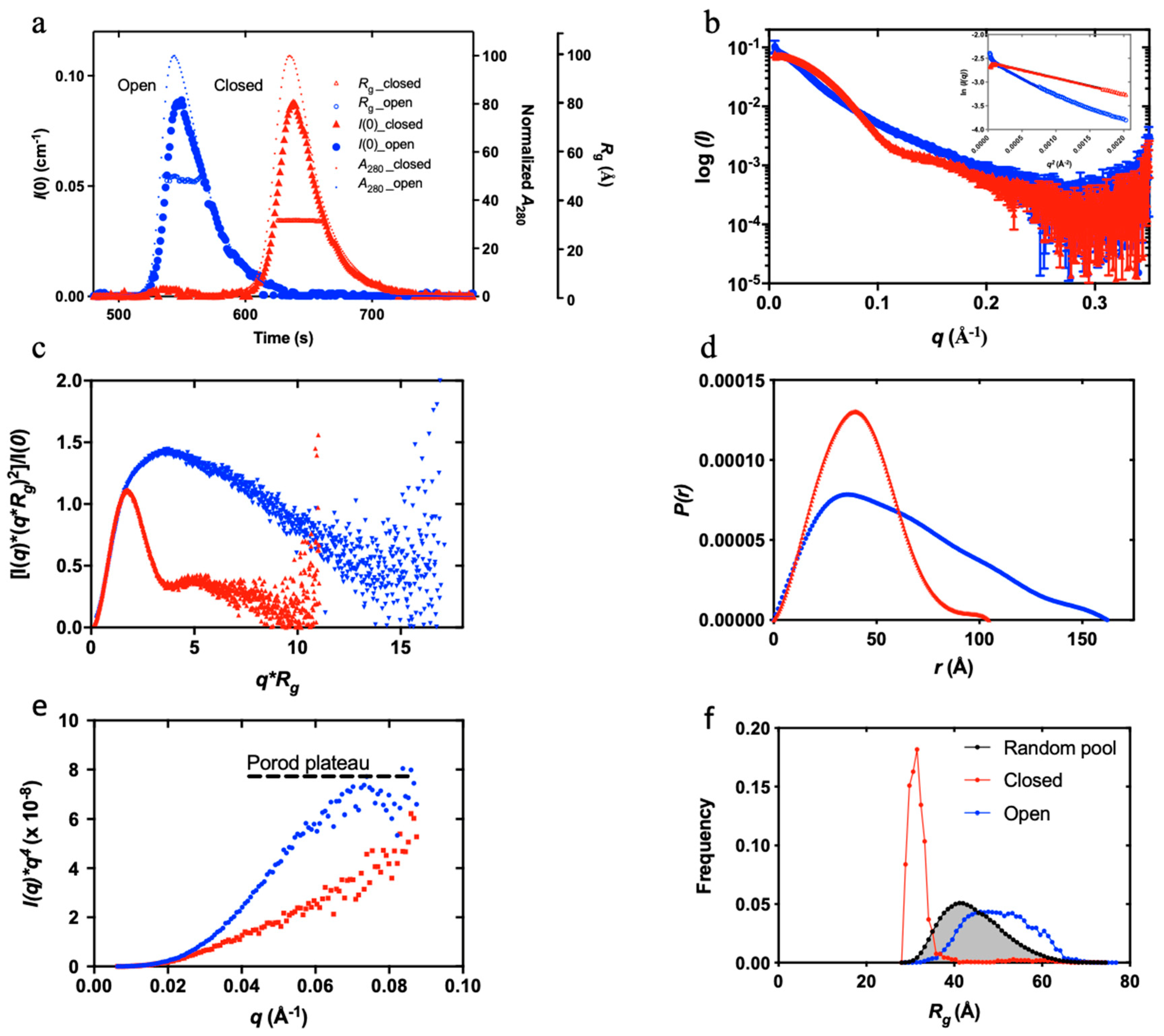
Figure 2.
Overview of the high-throughput kinetic studies of Plg conformational change. In this HTP method, ligand-induced Plg conformational change is set up in a 96-well plate format. (a) Ligands at the study concentration are mixed with Plg and incubated for 30 min. (b-c) The samples flowed past the X-ray beam in a quartz capillary, and scattering images were collected. (d) The resulting 2D scattering profile is averaged and buffer subtracted. (e) Rg values are derived by Guinier analysis (f) and then normalised before plotting against ligand concentration. The plot is fitted to a multi-site binding model, and Kopen, the ligand concentration required to induce 50% open Plg, is derived. Kopen is a kinetic parameter indicative of ligand efficacy or stability of closed conformation.
Figure 2.
Overview of the high-throughput kinetic studies of Plg conformational change. In this HTP method, ligand-induced Plg conformational change is set up in a 96-well plate format. (a) Ligands at the study concentration are mixed with Plg and incubated for 30 min. (b-c) The samples flowed past the X-ray beam in a quartz capillary, and scattering images were collected. (d) The resulting 2D scattering profile is averaged and buffer subtracted. (e) Rg values are derived by Guinier analysis (f) and then normalised before plotting against ligand concentration. The plot is fitted to a multi-site binding model, and Kopen, the ligand concentration required to induce 50% open Plg, is derived. Kopen is a kinetic parameter indicative of ligand efficacy or stability of closed conformation.
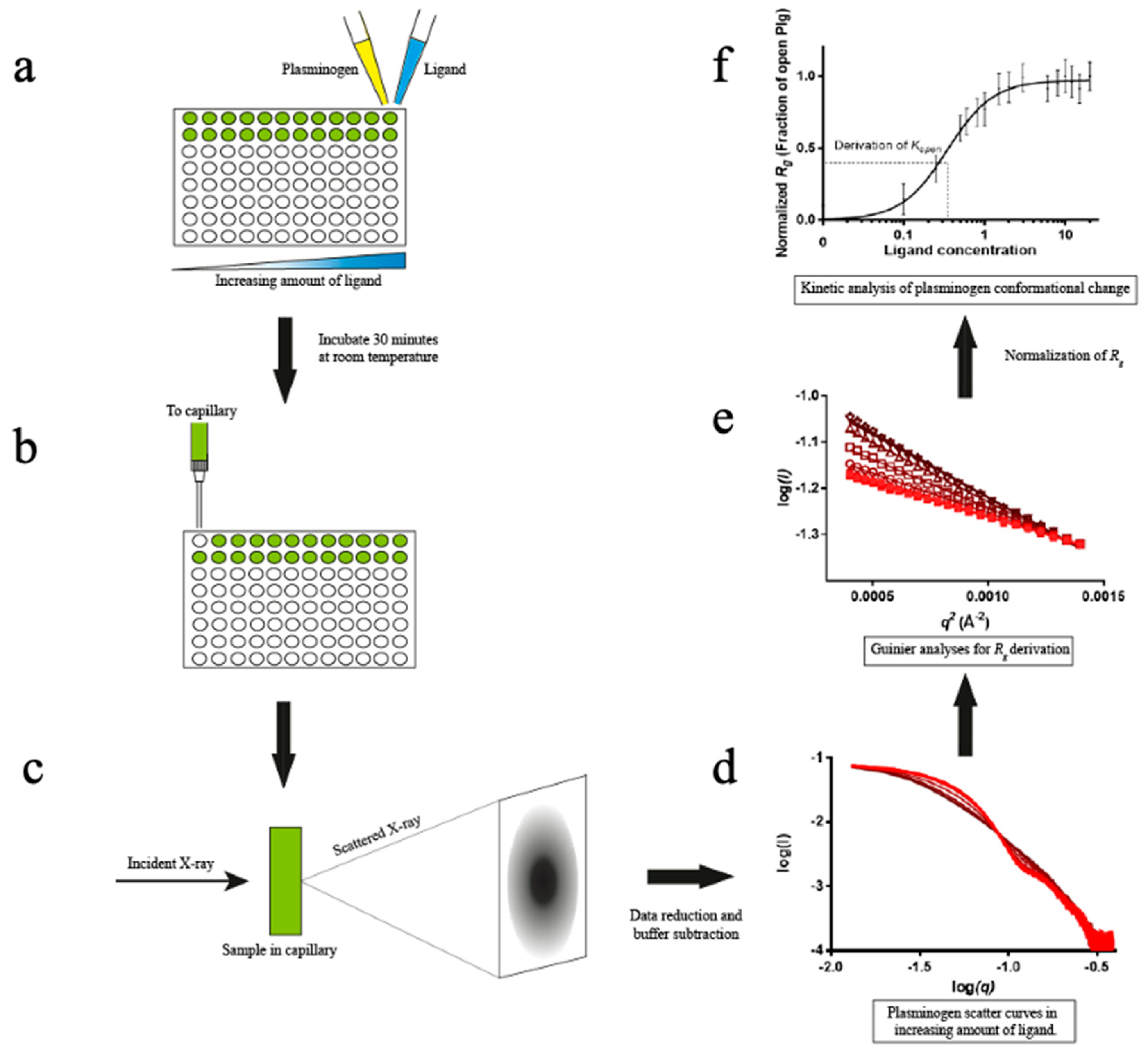
Figure 3.
Conformational change of Plg GII in the presence of EACA. (a) X-ray scattering curves from the EACA titration. The curves are represented in a red colour gradient, from closed (light red, solid arrow) to open form (dark red, blank arrow). (b) A plot of eigenvalues from singular value decomposition (SVD) analysis of scattering curves in the EACA titration series. The number of significant eigenvalues in the plot indicates the species contributing to the scattering data. (c) A plot of successive normalised eigenvectors from SVD analysis of scattering curves in (a). The first eigenvector is displayed at the top and the last at the bottom. (d) Fractions of Plg in the open form calculated from the SAXS scattering curves are plotted against EACA concentration. A multi-site cooperative kinetic curve is fitted and shown as a solid blue line. Inset: Hill plot showing positive cooperativity mechanism (hill slope= 1.5) of EACA binding.
Figure 3.
Conformational change of Plg GII in the presence of EACA. (a) X-ray scattering curves from the EACA titration. The curves are represented in a red colour gradient, from closed (light red, solid arrow) to open form (dark red, blank arrow). (b) A plot of eigenvalues from singular value decomposition (SVD) analysis of scattering curves in the EACA titration series. The number of significant eigenvalues in the plot indicates the species contributing to the scattering data. (c) A plot of successive normalised eigenvectors from SVD analysis of scattering curves in (a). The first eigenvector is displayed at the top and the last at the bottom. (d) Fractions of Plg in the open form calculated from the SAXS scattering curves are plotted against EACA concentration. A multi-site cooperative kinetic curve is fitted and shown as a solid blue line. Inset: Hill plot showing positive cooperativity mechanism (hill slope= 1.5) of EACA binding.
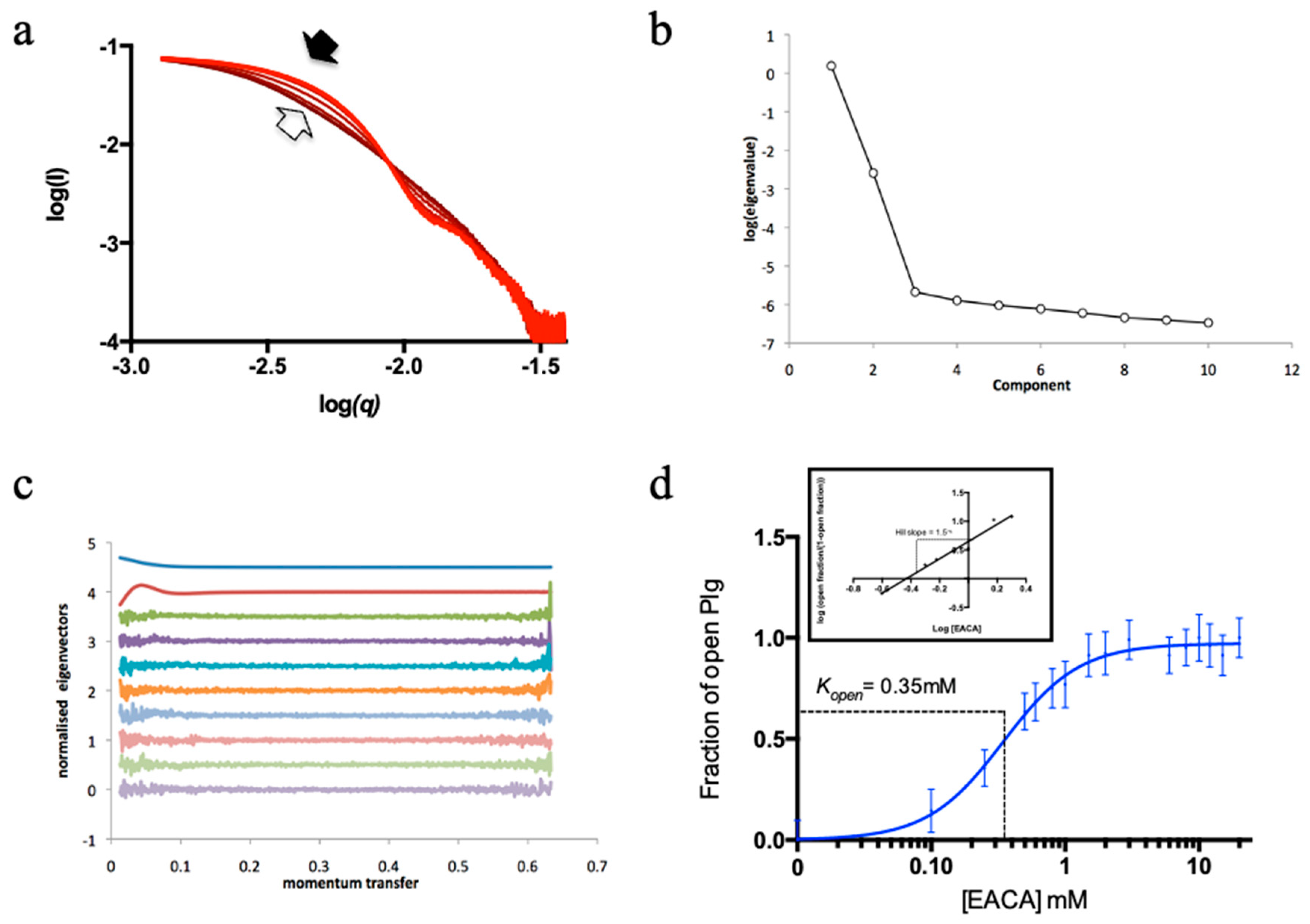
Figure 4.
Conformational change of Plg GII in the presence of L-lysine and synthetic peptides derived from Plg binding proteins. The graph shows the X-ray scattering titration curves of L-lysine (red) and synthetic peptides with a C-terminal lysine, namely CK10 (green) and NK55 (blue). MK12 was also tested, but at 10 mM, the highest concentration used, we did not observe any change in the SAXS profile of Plg GII. The Kopen derived from this study and information on the peptides regarding the corresponding residue number in the proteins and the residue sequence is shown below. Lysine and arginine residues are highlighted in bold font.
Figure 4.
Conformational change of Plg GII in the presence of L-lysine and synthetic peptides derived from Plg binding proteins. The graph shows the X-ray scattering titration curves of L-lysine (red) and synthetic peptides with a C-terminal lysine, namely CK10 (green) and NK55 (blue). MK12 was also tested, but at 10 mM, the highest concentration used, we did not observe any change in the SAXS profile of Plg GII. The Kopen derived from this study and information on the peptides regarding the corresponding residue number in the proteins and the residue sequence is shown below. Lysine and arginine residues are highlighted in bold font.
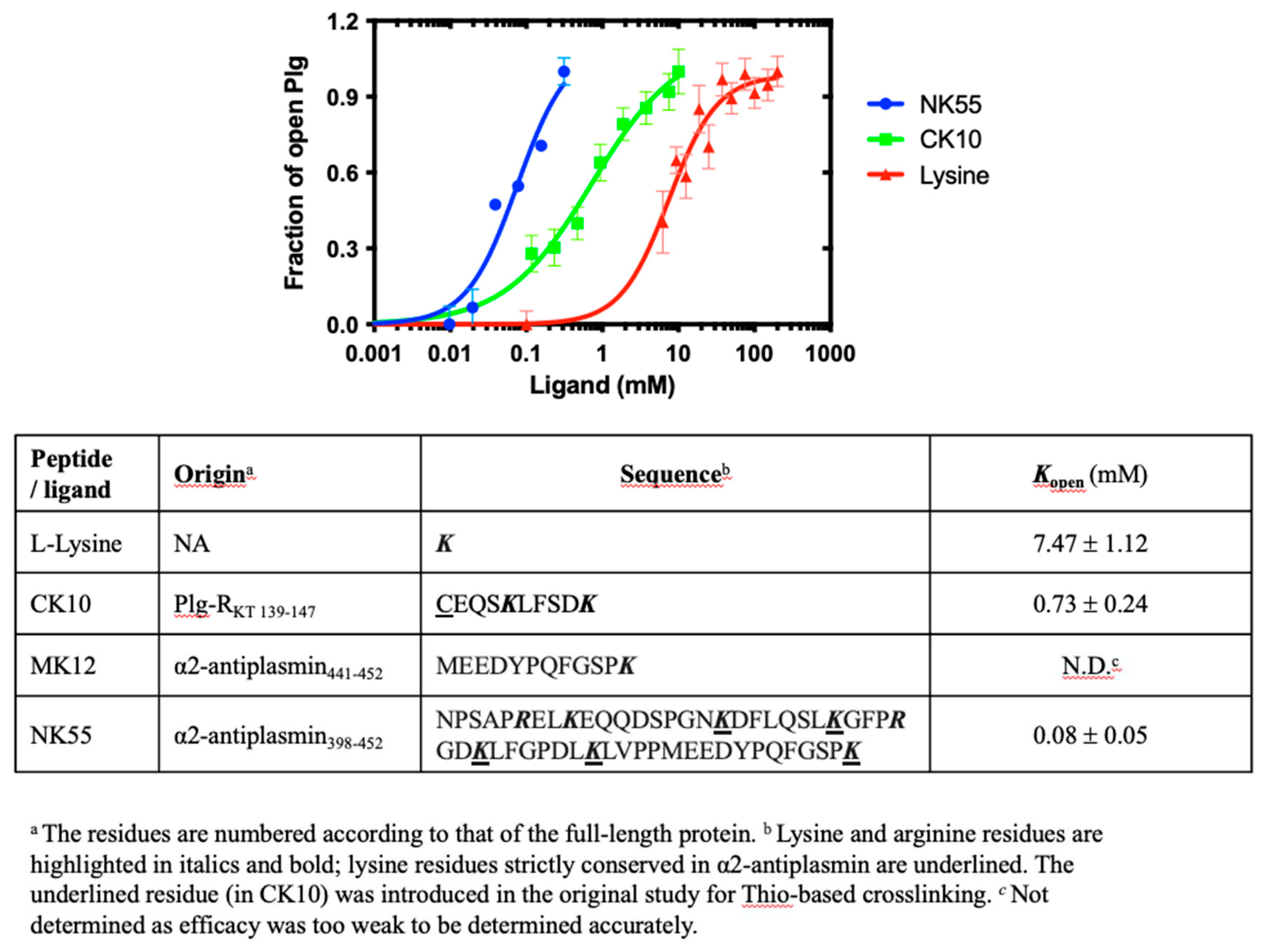
Figure 5.
Comparison of Plg GI and GII conformations. Superposed of GI (purple) and GII (green): (a) SEC-SAXS profiles showing the I(0) (left Y-axis), normalized absorbance (A280), and Rg (right Y-axes); (b) SAXS profiles recorded; (c) Dimensionless Kratky plot showing the globular nature and (d) P(r) analysis shows the similar dimensions of the two forms. (e) Superposition of Plg GI and GII crystal structures (PDB ID 4DUU and 4DUR, respectively, in the same colour scheme throughout). (f) Ab initio models of closed Plg GI and (g) GII; also shown are the correlation of scattering curves predicted from X-ray crystal structures using CRYSOL with the experimental data and the χ2 values.
Figure 5.
Comparison of Plg GI and GII conformations. Superposed of GI (purple) and GII (green): (a) SEC-SAXS profiles showing the I(0) (left Y-axis), normalized absorbance (A280), and Rg (right Y-axes); (b) SAXS profiles recorded; (c) Dimensionless Kratky plot showing the globular nature and (d) P(r) analysis shows the similar dimensions of the two forms. (e) Superposition of Plg GI and GII crystal structures (PDB ID 4DUU and 4DUR, respectively, in the same colour scheme throughout). (f) Ab initio models of closed Plg GI and (g) GII; also shown are the correlation of scattering curves predicted from X-ray crystal structures using CRYSOL with the experimental data and the χ2 values.
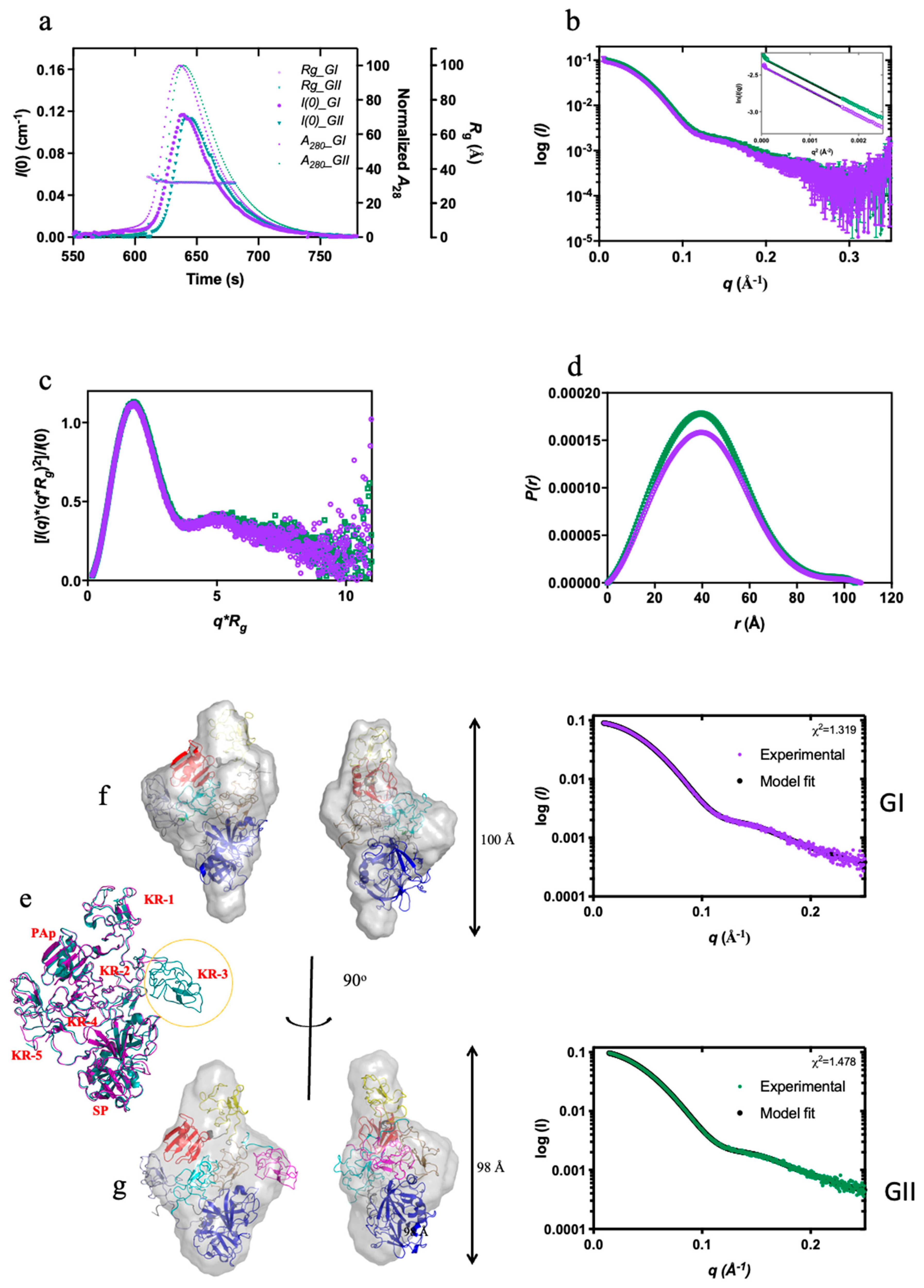

Table 1.
Kopen of Plg GI and GII in EACA (ε-aminocaproic acid) and TXA (Tranexamic acid). Kopen is the concentration of ligand required to induce the open conformation in 50% of the total Plg in solution (see Figure S4 for more info).
Table 1.
Kopen of Plg GI and GII in EACA (ε-aminocaproic acid) and TXA (Tranexamic acid). Kopen is the concentration of ligand required to induce the open conformation in 50% of the total Plg in solution (see Figure S4 for more info).
| Ligand | Kopen (mM) | ||
|---|---|---|---|
| Plg GI | Plg GII | Fold (GII/GI) | |
| EACA | 0.20 ± 0.01 | 0.35 ± 0.02 | 1.75 |
| TXA | 0.068 ± 0.002 | 0.120 ± 0.008 | 1.76 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



