Submitted:
15 August 2023
Posted:
16 August 2023
Read the latest preprint version here
Abstract
Neurodegeneration is becoming one of the leading causes of death worldwide as the population expands and grows older. There is a growing desire to understand the mechanisms behind prion proteins as well as the prion-like proteins that make up neurodegenerative diseases (NDs) including Alzheimer’s Disease (AD). Both amyloid β (Aβ) and hyperphosphorylated tau (p-tau) proteins behave in ways similar to that of the infectious form of the prion protein, PrPSc, such as aggregating, seeding, and replicating under not yet fully understood mechanisms, thus the designation of prion-like. This review aims to highlight the shared mechanisms between prion-like proteins and prion proteins in the structural variations associated with aggregation and disease development. These mechanisms are largely focusing on the dysregulation of protein homeostasis, self-replication, and protein aggregation, and how this knowledge could contribute to diagnoses and treatments for the given NDs.
Keywords:
Prions
; Neurodegeneration
; Alzheimer’s Disease
; Protein Homeostasis
; Aggregation
; Prion-Like Proteins
; Amyloid Beta
; Hyperphosphorylated Tau
; Self Replication
1. Introduction
Prions are best understood as a misfolding of proteins within the brain that are not exclusive to just one species and can be transmitted between both humans and animals. The associated prion diseases are also called transmissible spongiform encephalopathies (TSEs). The term prion was coined in 1982 by Stanley B. Prusiner, as a shortened form of proteinaceous infectious particle, while studying Syrian hamsters [1]. This infectious, misfolded protein then becomes self-propagating in a not yet fully understood manner. The presence of prions always exists in animals in the forms of scrapie in sheep, chronic wasting disease (CWD) in cervids, and bovine spongiform encephalopathy (BSE) in cows being the most notable. Within humans, the most notable prion is PrPSc, which occurs when the protein PrPC is stimulated. The presentation of prions in humans can be seen in diseases such as Kuru, all variants of Creutzfeldt–Jakob Disease (Iatrogenic, Variant, Familial, and Sporadic), Gerstmann–Straüssler–Scheinker Syndrome (GSS), and both forms of Fatal Familial Insomnia (FFI) (Familial and Sporadic) [2]. However, there are differences in the pathogenesis of each of these diseases, as some may result from mutations in the Prion Protein (Prnp) gene, infection, or sporadic mutation.
The function of the PrPC protein is of interest due to its possibility of helping to shed light on the prion process, with some of the processes being in the development of nervous system cells and cell signaling [3]. Also, of growing interest regarding the PrPC protein is its ability to shift phases, and how these phase shifts may then interact with both prion-like and non-prion-like proteins.
With progress in the study of neurodegeneration, the definition of prions has evolved to be broader and widely encompassing, with the understanding that “prions are composed of host-encoded proteins that adopt alternative conformations, which are self-propagating” [4]. This broadening of what a prion is considered as effectively allows for proteins that exhibit similar behaviors to be considered prion-like, with this argument for more proteins to be considered prion-like being largely attributed to Adriano Aguzzi in 2009 due to his paper analyzing amyloid proteins' similarities to prion protein [5], with arguments being made that they should just be regarded as prions. Of significant consideration for rebranding are the two prion-like proteins, p-tau and Aꞵ, due to their presence in non-prion neurodegeneration, such as in AD. It is also worth noting that the interaction between PrPC and the amyloid β oligomer (AβO) present in AD can trigger a phase shift in their previously established relationship [6].
Despite the progress in understanding NDs, the contribution of prions and prion-like proteins in relation to neurodegeneration continues to pose many unresolved problems. Arguably the largest issue is the understanding of the mechanism that regulates ND development and progression. This mechanism can then be broken down into three subsections: protein homeostasis (proteostasis), self-replication, and aggregation. As seen in Figure 1 below, these subsections may be interpreted as having both potential promoters (to “aid” in continued neurodegeneration) and inhibitors (to slow/prevent continuation). Therefore, this review aims to mainly summarize the recent advances of prions and prion-like proteins through structural variations, underlying regulation mechanisms, and the impact these have on disease-associated protein aggregation.
2. Primary Structural Features of Prions and Prion-like Proteins
The three ND-related proteins that this review focuses on are PrPSc, Aβ, and p-tau. While in their biological forms, these three proteins each have their own roles that aid in maintaining neuronal homeostasis, neuronal growth, neuronal repair, stabilizing neuronal MT, and so on. The “infectious” forms become detrimental once a sizable amount has accumulated, allowing for symptoms to be observed. Recently, however, studies have begun to show that there may be interplay between astrocytes or glial cells that can be beneficial for reducing the infectious forms, while simultaneously there is a possibility that these sames things may be helping spread the infectious proteins throughout the central nervous system (CNS) [7]. As of now, there are no tests that can be performed on a still-living individual that can conclusively diagnose an individual with an ND, but there are tests that can be performed that have a high level of confidence regarding the given diagnosis, such as MRI, PET, CT, SPECT, spinal tap to sample spinal fluids, and electroencephalograms [8]; however, the only truly definitive way to diagnose NDs is post-mortem during a brain autopsy (neuropathology autopsy).
2.1. Prnp, PrPC, and PrPSc
The 253 amino acids long prion protein, PrP is encoded by the Prnp gene, which is expressed in the brain and other tissues. The initial non-infectious structural conformation of the prion protein, PrPC, is of alpha helices that are attached to the cell membrane at the C-terminal by glycophosphatidylinositol (GPI) [9]. Meanwhile, the infectious form of PrP is a β sheet-rich isoform and is highly insoluble, making it different to do a complete structural analysis of them with current methods. There are two main hypotheses as to how the conformational change from the normal, noninfectious PrPC form to the infectious PrPSc form occurs. The first is that the change is due to the N-terminal of the PrPC protein, which is disordered and thus has a flexible tail. The second is the change that occurs at the C-terminal of the protein, which is speculated to be a highly ordered globular domain containing two antiparallel β-sheets and three ɑ-helices, in contrast. These distinct differences leave room for speculation as to which terminal may be more in control of form conversion [10,11]. Furthermore, there are distinct strains of the PrPSc form, which can be categorized by histology, clinical signs, and most commonly, incubation period – where incubation period refers to the time elapsed between inoculation and onset of disease in animal models [12]. However, even though there are strains that are distinct biologically, not all can be distinguished biochemically [12].
2.2. Prion-Like Proteins: Amyloid ꞵ and Hyperphosphorylated Tau
The proteins present in AD-associated neurodegeneration are now deemed to be prion-like proteins. This designation results from the replication using a so-called self-template, similar to what is seen in prion proteins. The complete mechanisms behind the self-replication are still not understood, for both the prion-like proteins and prion proteins themselves, but there are many proposed mechanisms that have begun to be widely accepted.
The amyloid precursor protein (APP) is a type-I transmembrane protein whose proteolysis gives rise to amyloid ꞵ peptides. APP processing can undergo one of two pathways, one being the non-amyloidogenic pathway and the other being the amyloidogenic pathway. This second pathway contains what eventually leads to the aggregation of form flexible, soluble oligomers that cause neuropathy. Further within the realm of Aβ protein-related disease progression, of interest is how exactly the protein causes memory deficits alongside neurodegeneration. As such, Lesné et al, found that an extracellular 56-kDa soluble Aβ assembly, which was then termed Aβ*56 (Aβ star 56) is likely responsible for memory deficits by induction of transient physiological brain alterations as opposed to permanent neuropathological alterations [13].
Aꞵ can occur either sporadic or due to genetic factors. However, a recent study demonstrated that human Aβ entering the mice circulation can induce the occurrence of Aβ plaque in the brain. It is yet to be determined whether Aβ derived from blood can enter the brain [14]. Aꞵ pathology can be introduced to mice without human Aβ (hAꞵ) by parabiosis. These mice can then develop AD-related pathologies, as well as experience phosphorylation of tau. However, what remains to be fully seen in this model is if there is a notable cognitive decline [14].
Tau, as has been mentioned, is an important factor in microtubule (MT) generation and degeneration. However, as tau undergoes phosphorylation, the tau binding to the MTs weakens until it dissociates and then will begin to aggregate together. These disordered aggregates of p-tau are known as neurofibrillary tangles (NFTs) and neuropil threads and are one of the hallmarks of AD. As tau undergoes conformation changes, the ability to undergo phosphorylation changes, along with the ability of a kinase to access a given site on the tau protein [15]. There are strong indications that the tau in AD is resistant to being dephosphorylated due to the presence of filaments within the NFTs [15].
3. Mechanistic Insights into Structural Changes Driving Protein Aggregation and Neurodegeneration
3.1. Dysregulation of Protein Homeostasis Associated with Protein Aggregation
Homeostasis of proteins (proteostasis) is becoming a critical field in ND, as the maintained regulation of protein accumulation can prove crucial for negating or increasing the likelihood of developing an ND. When the proteostasis undergoes dysregulation, the given protein system is ultimately disrupted. It is this dysregulation that then gives rise to the blockade of proteasomal/lysosomal dependent protein degradation and accumulation of aberrant proteins as there is no longer a well-maintained level of newly synthesized proteins and clearance of old or damaged proteins.
Impaired proteasomal degradation has been strongly associated with the accumulation of misfolded and aggregated proteins in neurodegenerative diseases such as Alzheimer’s Disease and prion diseases [16,17]. In Alzheimer's Disease, the proteasomal system is compromised, leading to the accumulation of amyloid-beta plaques and tau tangles, which are hallmark protein aggregates [18,19,20]. Similarly, in prion diseases, the abnormal conversion of prion proteins into a misfolded and aggregated form overwhelms the proteasomal machinery, resulting in the accumulation of infectious prion particles [21,22]. Therefore, impaired proteasomal degradation plays a crucial role in the pathogenesis of both Alzheimer's Disease and prion diseases by contributing to the formation and persistence of protein aggregates [20].
Within proteostasis, there are many facets to consider for the clearance of overabundant proteins as well as defective proteins that can accumulate to cause pathological issues. As such, the mitochondrial quality of cells within the CNS may play a crucial role in controlling the neuropathology caused by PrPSc, Aβ, and p-tau, within these proteins' respective associated NDs. A recent study has found that in cells infected with Scrapie, there was an observed excessive induction of mitophagosome and mitophagolysosome formation, and the PrPSc protein was enriched in these areas, alluding to the possibility that Scrapie infection promotes mitophagy [23].
Impaired brain clearance mechanisms responsible for protein accumulation in NDs provide new diagnostic and therapeutic opportunities to delay or prevent clinical symptoms. The glymphatic system is one such, rather newly discovered, system that may be of significance in regards to the clearance of parenchymal proteins, such as Aβ and p-tau seen in AD. As a pathway for exchanging cerebrospinal fluid (CSF) and interstitial fluid (ISF), the glymphatic system is facilitated by aquaporin 4 (AQP4). Harrison et al. found impaired CSF-ISF exchange and AQP4 polarization in a mouse model of tauopathy. A novel AQP4 inhibitor, TGN-020, the glymphatic system disrupted glymphatic CSF-ISF exchange and tau protein clearance, suggesting that the glymphatic system could be a druggable target for AD treatment and possibly other NDs as well [24].
Liquid-liquid phase separation (LLPS) is a dynamic and reversible process that assembles biomolecular condensates as well as separates homogenous solutions into different phases: dilute and dense phases [25]. LLPS within proteins is part of a complex system that helps to control cellular functions, and abnormalities within this can lead to disease development [26]. As such, the PrP protein is not unique in the manner that it undergoes LLPS, having similar characteristics of other proteins that undergo LLPS, such as binding nucleic acids (NA) and being highly disordered [27], therefore an additional mechanism in maintaining protein homeostasis is that of protein phase separation. Understanding the conversion from the non-infectious to infectious forms of proteins is of great importance, as doing so creates pathways for potential treatments and diagnostics routes.
Thus far, the phase separations of the highly disordered PrP (and other ND-causing proteins) can be manipulated by NA aptamers to create aggregates of the bound NA:protein in similar ways to NA-free protein aggregates [27]. As protein concentration increases, the ratios of NA to protein becomes increasingly important to the likelihood of droplet formation and eventual aggregation. The addition of various NA aptamers impacts the protein structure and the ability to aggregate and maintain an aggregated state. Furthermore, there are studies showing that tau also undergoes phase separation, and can form complexes with RNA in varying salt concentrations [26]. Therefore, by elucidating the mechanisms in which differing RNAs/NAs cause conformation changes and aggregation of prion and prion-like proteins into condensates to occur, methods to prevent aggregation may be developed by preventing phase separation dysregulation.
Based on the high affinity interaction of PrPC with amyloid-β oligomers (AβO) and the fact that PrPC undergoes an LLPS in which α-helical Thr becomes unfolded, Kostyley et al. found the interaction between PrPC Lys residues and AβO created a hydrogel containing immobile AβO and relatively mobile PrPC. NMR studies of hydrogel PrPC revealed a specific α-helical conformation for natively unfolded amino-terminal Gly and Ala residues. Recombinant PrPC could extract endogenous AβO from human Alzheimer’s brain lysates into hydrogel, and a PrPC antagonist could release AβO from endogenous brain hydrogel [4]. The findings suggest that Aβ species from AD can drive coupled phase and conformational transitions of PrPC.
3.2. PTM Associated Structural Anomalies and Protein Aggregation
Alterations in primary gene and/or protein sequences or new structural domains, including peptide cleavage and mutations are essential as the structural basis of protein aggregation. Therefore, the differences in the conformation of a given protein can be the deciding factor for if an infection can fully take root in a brain. As such, all three proteins being looked at in this review are subject to structural changes that can then result in a distinct strain, depending on the causation of the structural change such as gene mutations or anomaly cleavage of proteins, among which post-translational modification (PTM) is a major contributing factor.
Accounting for impacting factors such as glycoforms within a structural model may give rise to representations of the various strains that are present within prion diseases. Since how different strains formed by the same protein elicit different clinical phenotypes remain poorly understood. Consideration of structural diversity may be the clue to understanding why these varying strains occur, based on ratios of un-, mono-, and di-glycosylated glycoforms and sialoglycoforms [28].
Regarding the PrPC protein, differences in the glycosylation will determine the likelihood of the formation of newly generated PrPSc proteins and their associated various strains, and the ability and patterns of these proteins to deposit within the CNS [29]. Beyond just the glycosylation of the PrPC protein being a source of new strains, patterns of carbohydrate epitopes on N-glycans, sialyation, and charge of glycans may also have an impact on strain formation and chances of occurring [30]. Tau is also subject to many different strains based on if and where it experiences phosphorylation, acetylation, and ubiquitination; which then dictates if the tau will be able to participate in AD fibrils [30].
While tau proteins occur in many different manners of phosphorylation in the brain, phosphorylation by CaM kinase is what is deemed responsible for the tau in the NFTs witnessed in AD [31]. It is not fully understood what mechanisms cause tau to become hyperphosphorylated, but one speculation is that it occurs due to an impairment of glucose levels, causing decreased tau O-GlcNAcylation which then causes hyperphosphorylation of tau that eventually leads to NFTs [32]. The NFTs that form eventually accumulate as insoluble forms. In a healthy brain, tau exists intrinsically disordered and can become ordered by having a binding partner. However, tau in an unhealthy brain is argued to be “misdisordered” and misordered, and this has been detected by the use of the antibody DC11 [33]. When DC11 is able to bind to tau, tau has taken on a new conformation, and this new confirmation-tau will then go on to form the NFTs and filaments that are seen in AD [33]. These findings demonstrate the impact of post-translational modification on the transition of structural anomaly.
3.3. Regulation of Protein Structures by Environmental Factors: Chaperones, RNA, and Ions
Present in prion diseases, as well as AD, is the development of amyloids, which eventually progresses into amyloidogenesis, as neurodegeneration progresses [34,35,36]. There may also be a macroenvironmental dependence that dictates the amyloid formation and infection chances of prion and prion-like proteins [37] as well as dictating the strain formation of the prion protein [38]. Continuing the focus on the environmental impact present within these proteins is also the microenvironment in regards to the determination structure, replication, and toxicity [39]. Along with the environmental dependence, concomitant is the hydrophobicity that may be present within a species due to alterations [40]. Metal ions, such as copper, may also have a role in defining protein conformations. Depending on the region of the protein and the type of ion being considered, the protein may experience a looser or tighter conformational change, as well as affect the likelihood of susceptibility of conversion from the noninfectious form to the infectious form [41].
Chaperones of the proteins can serve to either protect from infectious form conversions, but can also be a catalyst for these infectious forms to occur depending on abundance. The different chaperones introduced to a protein may also have different efficiency rates in protection and infection, this can be seen in studies focusing on yeast prions and their varying mutability [42,43]. The presence of RNA in an environment may also impact the conformational changes that a protein may undergo. Rai et al found that in heterotropic conditions and when RNA is present, the proteins of interest are able to undergo transformations until they eventually form a solid-like aggregation [27]. RNA aptamers also serve as a potential way to control the conformational change or structure that may occur but there is still much to be investigated regarding the use of these aptamers regarding structural regulation.
4. Disease-Related Self-Replication and Aggregation Model
4.1. Self-Replication of Prions and Prion-Like Proteins
Over the years, in attempts to elucidate self-replication of the prion or prion-like proteins, many models have been created, but seemingly none have well fitted to the disease models perfectly. The way prion and prion-like proteins replicate is arguably a key to understanding why these aggregates occur and how to potentially treat and prevent them. The self-replication of these proteins is based on a conformational change of the protein, essentially a refolding of the native protein to a structurally changed molecule. This is seen within the PrPC to PrPSc conversion, as the protein structure changes from being largely composed of alpha helixes to largely composed of beta sheets. It has been found that self-replication leading to aggregation is a default state of disease-causing proteins [44]. The difficulty of fully understanding this remains, however, due to the differences that may result from in vitro versus in vivo studies.
The protein aggregation seen displayed in AD is of interest due to the impact of two prion-like proteins at work. The aggregation associated with the Aꞵ plaques is still yet to be fully understood, as it can occur in different structural forms, such as roughly spherical or wispy, irregular deposits [45]. The tau protein aggregated is seen as neurofibrillary tangles (NFTs), groupings of insoluble p-tau, that eventually spread throughout the brain, in a manner similar to PrPSc [5]. The spread of infectious proteins via aggregation may also be influenced by the presence of chaperones, as inhibition of chaperone proteins may enhance aggregation, while enhancement may be a potential way to prevent the spread of infectious protein aggregates in a homeostasis-maintaining manner [46,47]. Such as the chaperone proSAAS that is neuronally expressed and its function in blocking Aβ undergoing fibrillation [47] or the overexpression of Rnq1 being toxic to [RNQ+] prions toxicity, thus preventing the conversion of other proteins into amyloid species [42].
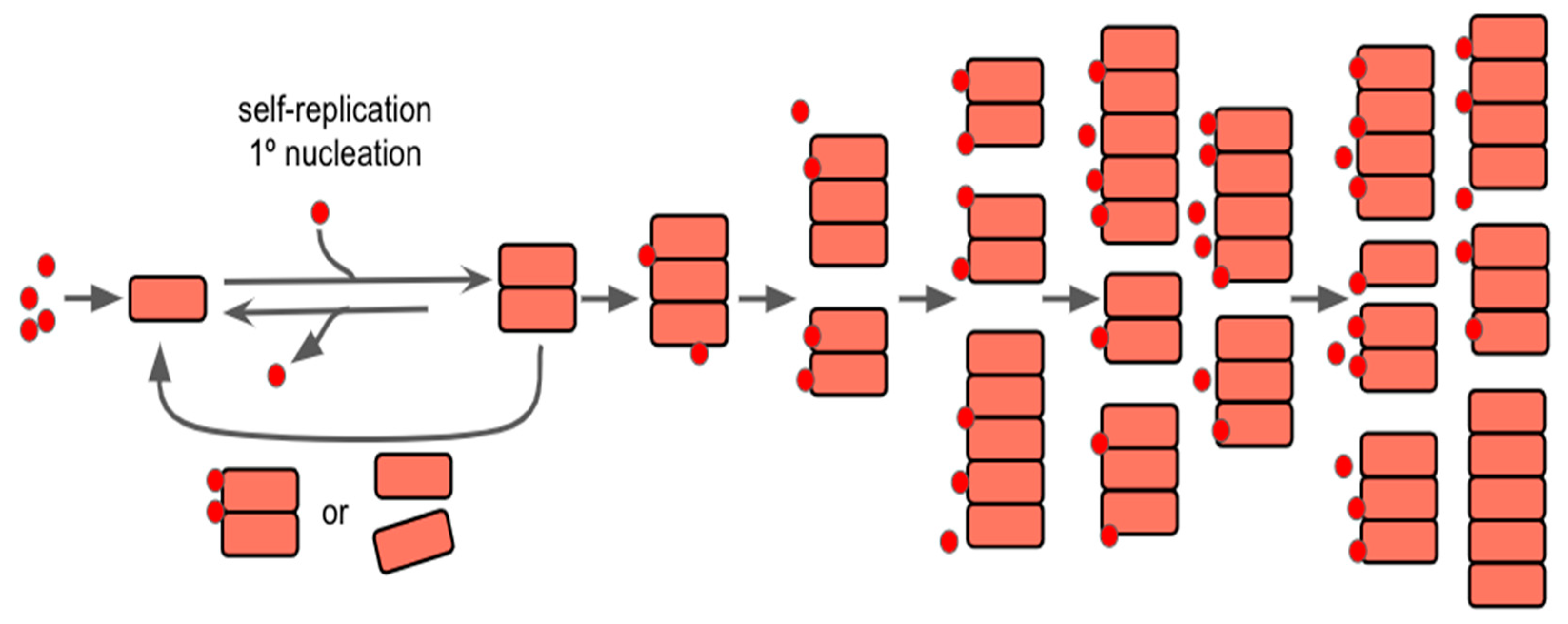
The action of aggregation replication of proteins within prion diseases and AD may also be thought of as being the default state, as it may have initially had a benefit that has yet to be evolved out of modern humans, assuming that the protein(s) in question have initially formed filamentous aggregates; as filamentous aggregate formation may be largely dependent on a secondary nucleation point, as described by Meisl et al in 2022 [44]. It is this secondary nucleation that causes the divide between normal growth and self-replication, as the secondary process is what allows the system to become autocatalytic and begin to grow exponentially. As seen within Figure 2, the introduction of new protein seeds on the surface of the aggregates allows for continued growth/lengthening and formation of new aggregates as time continues. As such, understanding the full function of the protein(s) role in the CNS may serve as a way to begin to focus studies in regards to preventing aggregation or preventing these secondary processes from occurring.
4.2. Neurological Inflammation
Alongside the protein self-replication and aggregation mechanism, another factor that may influence the mechanistic progressions of NDs is that of neurological inflammation. Initially, neuroinflammation may serve a protective role within the CNS, but if it is prolonged or under dysregulation, it can change its function into that of aiding the progression of diseases [48]. There is also growing evidence that neurological inflammation may serve as a way to spread the infectious proteins throughout the CNS via astrocytes and microglia [7,49].
Of growing interest regarding neurodegeneration caused by both prion and prion-like diseases is the potential role that astrocytes may play in the spreading of these misfolded proteins. It has been noted that animals have noticeable aggregate formation in astrocytes [50], as well as the possibility of astrocytes playing a role in prion infection [51]. As animal-based prions act rather similarly in infection models, it is possible to speculate that the human astrocyte would also be playing a crucial role in both prion and prion-like infections. In a PS19 mouse model as early as 3-4 months, the microglia are activated in the brain and spine; and at 6 months old, there are reactive astrocytes in the CA3 region of the brain in a study focusing on tauopathy [52]. This hypothesis has been further studied in animal models, as well as the role of astrocytes in neuroinflammation in not only prion disease but prion-like protein diseases, such as AD. Worth noting, however, is that astrocyte-associated infection, and by extension, the microglia-associated infection, spreading may be limited to a specific type of infection, due to there being three categories: Genetic, Sporadic, and Acquired, which occur at different rates.
5. Perspectives in Prevention, Diagnosis, and Therapy
All present methods of treating NDs are aimed at halting the progression of symptoms while also trying to prevent any new symptoms from developing for as long as possible. As of now, there is no cure for any ND, but there is considerable interest in developing increasingly improved treatments until a cure can be developed.
Both pharmaceutical-based and therapeutic-based treatments have a large number of aspects to target, such as 1) the prevention of Aβ protein from forming, accumulating, and spreading within the CNS; 2) delaying and preventing cognitive decline from occurring and treating it when/if it does occur; 3) treating neuroinflammation and any resulting apoptosis while trying to enhance neuroprotection. Treatments targeted towards the p-tau protein need to focus on the reduction of hyperphosphorylation from occurring and a reduction of the accumulation and spread of p-tau and NFTs in the CNS; as well as similar focus as 2) and 3) in the treatment of the Aβ proteins. Similar to treatments for Aβ protein and p-tau protein, the treatments for the PrPSc protein will also need to focus on prevention of formation, accumulation, and spread, as well as the previously listed focuses of 2) and 3), but also needs a focus on preventing infection from being able to potentially spread to other individuals and species.
5.1. Pharmaceutical-Based and Therapeutic-Based Treatment Methods
Pharmaceutical-based treatments for neurodegeneration have largely focused on immunosuppression in order to prevent the large spread and continual occurrence of misfolded proteins. Currently, a large focus of pharmaceutical treatment is focusing on researching the repurposing of already existing medication in order to speed up the development of a widespread treatment option [53].
Therapeutic treatments for neurodegeneration are largely focused on individuals that have a genetic predisposition to develop an ND. This focus aims to delay the onset of noticeable systems for as long as possible and tends to rely on pharmacological approaches in order to stave off the accumulation of the infectious proteins isoforms.
As the brain continues to be studied and systems within it are uncovered and researched, more potential treatment options for NDs become available. One such is the usage of the glymphatic system in AD in order to clear out Aβ and p-tau as the proteins accumulate. When the system was first described in 2012 by Maiken Nedergaard and colleagues, the dependency on astroglial cells was once again highlighted [54]. While most of the research on the glymphatic system has focused on the Aβ protein, such as in Peng et al., there is a growing number of studies focusing on the MT-associated tau protein, such as in Harrison et al. [24,55].
The AppNL-G-F/NL-G-F mouse model developed by Sakakibara and colleagues poses the potential to serve as a good base for developing therapeutic treatments for AD-related Aβ plaque formations [56]. Beyond just developing new animal models with better human accuracy, there needs to be a way to trace the presence of the proteins within humans, such as developing ways to flag the proteins, such as what Hosokawa-Muto, J. et al. did in order to perform FRET analysis on the protein, albeit this method was used in an attempt to elucidate structural information [57].
5.2. Notable Advancements in Treatments in Recent Years
PrPC can serve in a neuroprotective capacity when interacting with AβO in astrocyte cells to prevent oxidative damage that can eventually contribute to neurodegeneration [35]. However, PrPC can also interact with subsets of AβO in AD to further cognitive impairment as the disease progresses [58]. Furthermore, differing biomarkers that are used in the diagnosis and monitoring of AD are also a field of increasing focus. Such as monitoring the accumulation of neurotoxic protein aggregates and the resulting neurodegeneration through blood [59] or the potential for diagnosis and monitoring disease progression through the retina [60].
As reviewed by Murakami, K. et al., aptamers have become a popular topic in regard to the potential for treating amyloidogenic proteins, such as PrPSc and Aβ. The use of aptamers for treatment is a prosperous but difficult possibility [61]. This is due to the existence of DNA and RNA aptamers, as well as the many different options that currently exist to target a given part of a protein. CRISPR/Cas9 gene editing may also serve as a way to treat prions and prion-like proteins, especially for those with a genetic predisposition for a given disease. One study by Castle et al. in 2022 attempted to introduce a null Prnp allele or edited Prnp gene into mice, which while overall unsuccessful, showed promising aspects for a basis to further develop this possibility from reworking the issues that arose within the study, such as focusing on developing a complete germline [62].
Autophagy stimulation is another treatment method that has been of popular interest in regard to treating prion infection due to the ability to manipulate autophagy at different points in the infection stages. This, along with other treatments, poses difficulties due to the fear of the likelihood of prions developing resistance to treatment. As such, one group has recently tried a combination of autophagy stimulation and cellulose ethers as both are known to show positives results in regards to prion reduction, but this combination did not show as good results as compared to individual treatments leaving still more to investigate for possible combination treatments [63].
6. Conclusions
The significance of this review will lie in the improvement of understanding the mechanisms of structural changes and protein aggregation in prions and prion-like proteins that are seen in AD and the neurodegeneration associated with each respective protein. As more interest regarding prions and infectious proteins grows, the exploration of parallels and structural information will hopefully aid in developing increasingly successful treatments for preventing neurodegeneration before it starts. Along with helping to prevent continued ongoing neurodegeneration from occurring, and listing possibilities of infection occurrence in common materials, such as wood, concrete, and surgical equipment, as thus far there is no method that exists to successfully prevent transmission of prions.
Therefore, as more information regarding the structure of the infectious prion protein is achieved and understood, it will hopefully lead to better treatments for diseases associated with prions and prion-like proteins, such as AD, as explored throughout this review. Recent research on prions has begun to link novel structures and mutations, such as amino acid replacements, protein seeding, environmental changes, and hydrophobic cores, in prions and prion-like proteins to dysregulated homeostasis, leading to misfolding, propagation, and aggregation of these proteins. By summarizing recent advances in causal relations of structural anomaly and the potential of aggregation model, we disclose underlying mechanisms in the onset of neurodegeneration from the perspective of prion diseases and AD and the possible interlinking of the proteins associated with the two.
Author Contributions
Conceptualization, H.J.L., C.S.C., D. Y., and W.L.; writing—original draft preparation, C.S.C. and H.J.L.; writing—review and editing, H.J.L., D. Y., and W.L.; supervision, H.J.L.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
This research is supported by internal funding from New York University Shanghai.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Prusiner, S. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef]
- Colby, D.W.; Prusiner, S.B. Prions. Cold Spring Harb. Perspect. Biol. 2011, 3, a006833–a006833. [Google Scholar] [CrossRef] [PubMed]
- Legname, G. Elucidating the function of the prion protein. PLOS Pathog. 2017, 13, e1006458. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, A.; Condello, C.; Stöhr, J.; Yue, W.; Rivera, B.M.; Lee, J.C.; Woerman, A.L.; Halliday, G.; van Duinen, S.; Ingelsson, M.; et al. Aβ and tau prion-like activities decline with longevity in the Alzheimer’s disease human brain. Sci. Transl. Med. 2019, 11, eaat8462. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A. Beyond the prion principle. Nature 2009, 459, 924–925. [Google Scholar] [CrossRef]
- Kostylev, M.A.; Tuttle, M.D.; Lee, S.; Klein, L.E.; Takahashi, H.; Cox, T.O.; Gunther, E.C.; Zilm, K.W.; Strittmatter, S.M. Liquid and Hydrogel Phases of PrPC Linked to Conformation Shifts and Triggered by Alzheimer’s Amyloid-β Oligomers. Mol. Cell 2018, 72, 426–443. [Google Scholar] [CrossRef]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte Crosstalk in CNS Inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef]
- Valotassiou, V.; Malamitsi, J.; Papatriantafyllou, J.; Dardiotis, E.; Tsougos, I.; Psimadas, D.; Alexiou, S.; Hadjigeorgiou, G.; Georgoulias, P. SPECT and PET imaging in Alzheimer’s disease. Ann. Nucl. Med. 2018, 32, 583–593. [Google Scholar] [CrossRef]
- Hetz, C.; Maundrell, K.; Soto, C. Is loss of function of the prion protein the cause of prion disorders? Trends Mol. Med. 2003, 9, 237–243. [Google Scholar] [CrossRef]
- Lee, J.-H.; Bae, S.-E.; Jung, S.; Ahn, I.; Son, H.S. Discriminant analysis of prion sequences for prediction of susceptibility. Exp. Mol. Med. 2013, 45, e48–e48. [Google Scholar] [CrossRef]
- Yamamoto, N. Hot Spot of Structural Ambivalence in Prion Protein Revealed by Secondary Structure Principal Component Analysis. J. Phys. Chem. B 2014, 118, 9826–9833. [Google Scholar] [CrossRef]
- Angers, R.C.; Kang, H.-E.; Napier, D.; Browning, S.; Seward, T.; Mathiason, C.; Balachandran, A.; McKenzie, D.; Castilla, J.; Soto, C.; et al. Prion Strain Mutation Determined by Prion Protein Conformational Compatibility and Primary Structure. Science 2010, 328, 1154–1158. [Google Scholar] [CrossRef] [PubMed]
- Lesné, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-β protein assembly in the brain impairs memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Bu, X.-L.; Xiang, Y.; Jin, W.-S.; Wang, J.; Shen, L.-L.; Huang, Z.-L.; Zhang, K.; Liu, Y.-H.; Zeng, F.; Liu, J.-H.; et al. Blood-derived amyloid-β protein induces Alzheimer’s disease pathologies. Mol. Psychiatry 2017, 23, 1948–1956. [Google Scholar] [CrossRef]
- Hasegawa, M. Structure of NFT: Biochemical Approach. Adv. Exp. Med. Biol. 2019, 1184, 23–34. [Google Scholar] [CrossRef]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer's disease and Down's syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 1984, 122, 1131–1135. [Google Scholar] [CrossRef]
- Goate, A.; Chartier-Harlin, M.-C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M., Wischik, C.M., Crowther, R.A., Walker, J.E., Klug, A. Cloning and sequencing of the cDHA-encoding a core protein of the paired helical filament of Alzheimer’s disease: Identification of the microtubule-associated protein tau. Proc Natl Acad Sci USA, 1988, 85: 4051-4055.
- Goedert, M.; Clavaguera, F.; Tolnay, M. The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci. 2010, 33, 317–325. [Google Scholar] [CrossRef]
- McKinnon, C.; Goold, R.; Andre, R.; Devoy, A.; Ortega, Z.; Moonga, J.; Linehan, J.M.; Brandner, S.; Lucas, J.J.; Collinge, J.; et al. Prion-mediated neurodegeneration is associated with early impairment of the ubiquitin–proteasome system. Acta Neuropathol. 2015, 131, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Tian, C.; Wang, S.-B.; Xie, W.-L.; Guo, Y.; Zhang, J.; Shi, Q.; Chen, C.; Dong, X.-P. Activation of the macroautophagic system in scrapie-infected experimental animals and human genetic prion diseases. Autophagy 2012, 8, 1604–1620. [Google Scholar] [CrossRef]
- Kim, M.J.; Kim, H.J.; Jang, B.; Kim, H.J.; Mostafa, M.N.; Park, S.J.; Kim, Y.S.; Choi, E.K. Impairment of Neuronal Mitochondrial Quality Control in Prion-Induced Neurodegeneration. Cells 2022, 11, 2744. [Google Scholar] [CrossRef]
- Harrison, I.F.; Ismail, O.; Machhada, A.; Colgan, N.; Ohene, Y.; Nahavandi, P.; Ahmed, Z.; Fisher, A.; Meftah, S.; Murray, T.K.; et al. Impaired glymphatic function and clearance of tau in an Alzheimer’s disease model. Brain 2020, 143, 2576–2593. [Google Scholar] [CrossRef]
- Feng, Z.; Chen, X.; Wu, X.; Zhang, M. Formation of biological condensates via phase separation: Characteristics, analytical methods, and physiological implications. PEDIATRICS 2019, 294, 14823–14835. [Google Scholar] [CrossRef]
- Rai, S.K.; Khanna, R.; Avni, A.; Mukhopadhyay, S. Heterotypic electrostatic interactions control complex phase separation of tau and prion into multiphasic condensates and co-aggregates. 2023, 120. [CrossRef]
- Matos, C.O.; Passos, Y.M.; do Amaral, M.J.; Macedo, B.; Tempone, M.H.; Bezerra, O.C.L.; Moraes, M.O.; Almeida, M.S.; Weber, G.; Missailidis, S.; et al. Liquid-liquid phase separation and fibrillation of the prion protein modulated by a high-affinity DNA aptamer. FASEB J. 2020, 34, 365–385. [Google Scholar] [CrossRef]
- Baskakov, I.V.; Katorcha, E.; Makarava, N. Prion Strain-Specific Structure and Pathology: A View from the Perspective of Glycobiology. Viruses 2018, 10, 723. [Google Scholar] [CrossRef]
- DeArmond, S.J.; Sánchez, H.; Yehiely, F.; Qiu, Y.; Ninchak-Casey, A.; Daggett, V.; Camerino, A.P.; Cayetano, J.; Rogers, M.; Groth, D.; et al. Selective Neuronal Targeting in Prion Disease. Neuron 1997, 19, 1337–1348. [Google Scholar] [CrossRef]
- Baskakov, I.V. From Posttranslational Modifications to Disease Phenotype: A Substrate Selection Hypothesis in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 901. [Google Scholar] [CrossRef]
- Steiner, B.; Mandelkow, E.M.; Biernat, J.; Gustke, N.; Meyer, H.E.; Schmidt, B.; Mieskes, G.; Söling, H.D.; Drechsel, D.; Kirschner, M.W. Phosphorylation of microtubule-associated protein tau: identification of the site for Ca2(+)-calmodulin dependent kinase and relationship with tau phosphorylation in Alzheimer tangles. EMBO J. 1990, 9, 3539–3544. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.-X.; Iqbal, K. Hyperphosphorylation of Microtubule-Associated Protein Tau: A Promising Therapeutic Target for Alzheimer Disease. Curr. Med. Chem. 2008, 15, 2321–2328. [Google Scholar] [CrossRef] [PubMed]
- Kovacech, B.; Skrabana, R.; Novak, M. Transition of Tau Protein from Disordered to Misordered in Alzheimer’s Disease. Neurodegener. Dis. 2010, 7, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Ghetti, B.; Piccardo, P.; Frangione, B.; Bugiani, O.; Giaccone, G.; Young, K.; Prelli, F.; Farlow, M.R.; Dlouhy, S.R.; Tagliavini, F. Prion Protein Amyloidosis. Brain Pathol. 1996, 6, 127–145. [Google Scholar] [CrossRef]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common Structure of Soluble Amyloid Oligomers Implies Common Mechanism of Pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef]
- Marques, C.M.S.; Gomes, R.N.; Pedron, T.; Batista, B.L.; Cerchiaro, G. Cellular prion protein offers neuroprotection in astrocytes submitted to amyloid β oligomer toxicity. Mol. Cell. Biochem. 2022, 478, 1847–1865. [Google Scholar] [CrossRef]
- Pritzkow, S.; Morales, R.; Lyon, A.; Concha-Marambio, L.; Urayama, A.; Soto, C. Efficient prion disease transmission through common environmental materials. J. Biol. Chem. 2018, 293, 3363–3373. [Google Scholar] [CrossRef]
- Katorcha, E.; Gonzalez-Montalban, N.; Makarava, N.; Kovacs, G.G.; Baskakov, I.V. Prion replication environment defines the fate of prion strain adaptation. PLOS Pathog. 2018, 14, e1007093. [Google Scholar] [CrossRef]
- Siddiqi, M.K.; Kim, C.; Haldiman, T.; Kacirova, M.; Wang, B.; Bohon, J.; Chance, M.R.; Kiselar, J.; Safar, J.G. Structurally distinct external solvent-exposed domains drive replication of major human prions. PLOS Pathog. 2021, 17, e1009642. [Google Scholar] [CrossRef]
- Roterman, I.; Stapor, K.; Gądek, K.; Gubała, T.; Nowakowski, P.; Fabian, P.; Konieczny, L. On the Dependence of Prion and Amyloid Structure on the Folding Environment. Int. J. Mol. Sci. 2021, 22, 13494. [Google Scholar] [CrossRef]
- Salzano, G.; Brennich, M.; Mancini, G.; Tran, T.H.; Legname, G.; D’angelo, P.; Giachin, G. Deciphering Copper Coordination in the Mammalian Prion Protein Amyloidogenic Domain. Biophys. J. 2020, 118, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Douglas, P.M.; Treusch, S.; Ren, H.-Y.; Halfmann, R.; Duennwald, M.L.; Lindquist, S.; Cyr, D.M. Chaperone-dependent amyloid assembly protects cells from prion toxicity. Proc. Natl. Acad. Sci. 2008, 105, 7206–7211. [Google Scholar] [CrossRef] [PubMed]
- King, C.-Y. The Mutability of Yeast Prions. Viruses 2022, 14, 2337. [Google Scholar] [CrossRef]
- Meisl, G.; Xu, C.K.; Taylor, J.D.; Michaels, T.C.T.; Levin, A.; Otzen, D.; Klenerman, D.; Matthews, S.; Linse, S.; Andreasen, M.; et al. Uncovering the universality of self-replication in protein aggregation and its link to disease. Sci. Adv. 2022, 8, eabn6831. [Google Scholar] [CrossRef] [PubMed]
- Games, D.; Adams, D.; Alessandrini, R.; Barbour, R.; Borthelette, P.; Blackwell, C.; Carr, T.; Clemens, J.; Donaldson, T.; Gillespie, F.; et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature 1995, 373, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Sui, C.; Liu, F.; Chen, T.; Zhang, L.; Zheng, Y.; Liu, B.; Gao, C. The protein arginine methyltransferase PRMT9 attenuates MAVS activation through arginine methylation. Nat. Commun. 2022, 13, 1–16. [Google Scholar] [CrossRef]
- Peinado, J.R.; Chaplot, K.; Jarvela, T.S.; Barbieri, E.M.; Shorter, J.; Lindberg, I. Sequestration of TDP-43216-414 Aggregates by Cytoplasmic Expression of the proSAAS Chaperone. ACS Chem. Neurosci. 2022, 13, 1651–1665. [Google Scholar] [CrossRef]
- Li, B.; Chen, M.; Zhu, C. Neuroinflammation in Prion Disease. Int. J. Mol. Sci. 2021, 22, 2196. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 1–12. [Google Scholar] [CrossRef]
- Diedrich, J.F.; E Bendheim, P.; Kim, Y.S.; I Carp, R.; Haase, A.T. Scrapie-associated prion protein accumulates in astrocytes during scrapie infection. Proc. Natl. Acad. Sci. 1991, 88, 375–379. [Google Scholar] [CrossRef]
- Raeber, A.J.; Race, R.E.; Brandner, S.; Priola, S.A.; Sailer, A.; Bessen, R.A.; Mucke, L.; Manson, J.; Aguzzi, A.; Oldstone, M.B.; et al. Astrocyte-specific expression of hamster prion protein (PrP) renders PrP knockout mice susceptible to hamster scrapie. EMBO J. 1997, 16, 6057–6065. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.-M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.-Y. Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef]
- Durães, F.; Pinto, M.; Sousa, E. Old Drugs as New Treatments for Neurodegenerative Diseases. Pharmaceuticals 2018, 11, 44. [Google Scholar] [CrossRef] [PubMed]
- Lopes, D.M.; Llewellyn, S.K.; Harrison, I.F. Propagation of tau and α-synuclein in the brain: therapeutic potential of the glymphatic system. Transl. Neurodegener. 2022, 11, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Achariyar, T.M.; Li, B.; Liao, Y.; Mestre, H.; Hitomi, E.; Regan, S.; Kasper, T.; Peng, S.; Ding, F.; et al. Suppression of glymphatic fluid transport in a mouse model of Alzheimer's disease. Neurobiol. Dis. 2016, 93, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, Y.; Sekiya, M.; Saito, T.; Saido, T.C.; Iijima, K.M. Amyloid-β plaque formation and reactive gliosis are required for induction of cognitive deficits in App knock-in mouse models of Alzheimer’s disease. BMC Neurosci. 2019, 20, 13. [Google Scholar] [CrossRef]
- Hosokawa-Muto, J.; Yamaguchi, K.-I.; O Kamatari, Y.; Kuwata, K. Synthesis of double-fluorescent labeled prion protein for FRET analysis. Biosci. Biotechnol. Biochem. 2015, 79, 1802–1809. [Google Scholar] [CrossRef] [PubMed]
- Kostylev, M.A.; Kaufman, A.C.; Nygaard, H.B.; Patel, P.; Haas, L.T.; Gunther, E.C.; Vortmeyer, A.; Strittmatter, S.M. Prion-Protein-interacting Amyloid-β Oligomers of High Molecular Weight Are Tightly Correlated with Memory Impairment in Multiple Alzheimer Mouse Models. J. Biol. Chem. 2015, 290, 17415–17438. [Google Scholar] [CrossRef] [PubMed]
- Youn, Y.C.; Kang, S.; Suh, J.; Park, Y.H.; Kang, M.J.; Pyun, J.-M.; Choi, S.H.; Jeong, J.H.; Park, K.W.; Lee, H.-W.; et al. Blood amyloid-β oligomerization associated with neurodegeneration of Alzheimer’s disease. Alzheimer's Res. Ther. 2019, 11, 40. [Google Scholar] [CrossRef]
- Koronyo, Y.; Rentsendorj, A.; Mirzaei, N.; Regis, G.C.; Sheyn, J.; Shi, H.; Barron, E.; Cook-Wiens, G.; Rodriguez, A.R.; Medeiros, R.; et al. Retinal pathological features and proteome signatures of Alzheimer’s disease. Acta Neuropathol. 2023, 145, 409–438. [Google Scholar] [CrossRef]
- Murakami, K.; Izuo, N.; Bitan, G. Aptamers targeting amyloidogenic proteins and their emerging role in neurodegenerative diseases. J. Biol. Chem. 2022, 298, 101478. [Google Scholar] [CrossRef] [PubMed]
- Castle, A.R.; Wohlgemuth, S.; Arce, L.; Westaway, D. Investigating CRISPR/Cas9 gene drive for production of disease-preventing prion gene alleles. PLOS ONE 2022, 17, e0269342. [Google Scholar] [CrossRef] [PubMed]
- Abdulrahman, B.A.; Tahir, W.; Doh-Ura, K.; Gilch, S.; Schatzl, H.M. Combining autophagy stimulators and cellulose ethers for therapy against prion disease. Prion 2019, 13, 185–196. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Simplified Roadmap of Infection Progression and Potential Promoters and Inhibitors of Infection.
Figure 1.
Simplified Roadmap of Infection Progression and Potential Promoters and Inhibitors of Infection.
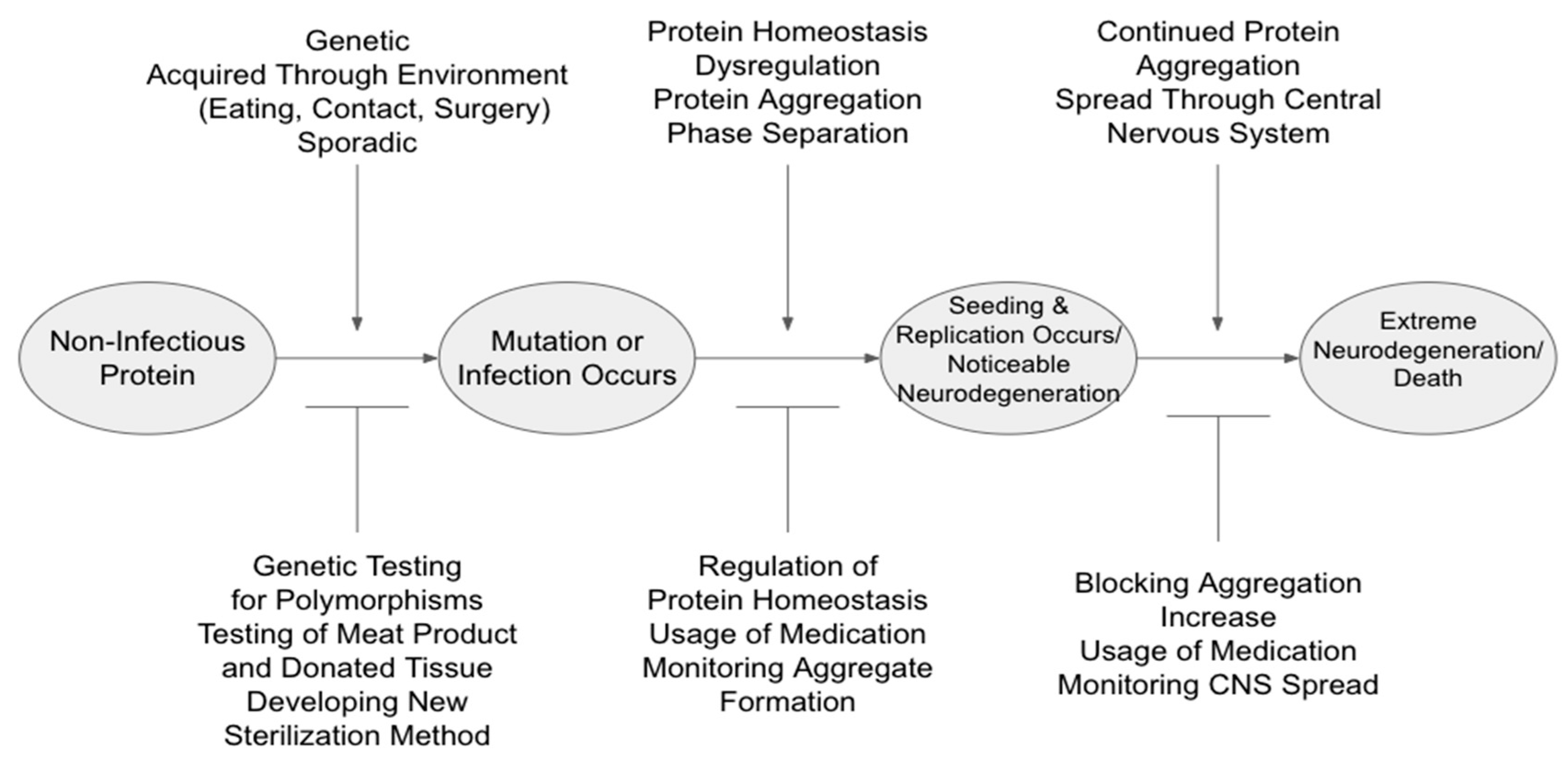
Figure 2.
Self-Replication and Nucleation Model of Aggregation Reaction and Concentration Over Time. The red dots are representative of seeds for nucleation and the pink squares are showing the aggregation of proteins over time. As time progresses, seeding continually happens, and the aggregates can continue to grow in length and/or split to create new ones. Modified from [44].
Figure 2.
Self-Replication and Nucleation Model of Aggregation Reaction and Concentration Over Time. The red dots are representative of seeds for nucleation and the pink squares are showing the aggregation of proteins over time. As time progresses, seeding continually happens, and the aggregates can continue to grow in length and/or split to create new ones. Modified from [44].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



