
Submitted:
16 August 2023
Posted:
16 August 2023
You are already at the latest version
Abstract
Copepods are the most abundant metazoans on Earth, exhibit remarkable species diversity and global distribution. Accurate species identification of copepods presents a challenge due to their cryptic nature, necessitating meticulous examination and differentiation. To address this challenge, innovative approaches utilizing molecular markers have emerged, enabling more reliable and precise species identification. Mitochondrial genes, which are characterized by high variability, have proven invaluable in uncovering taxonomically significant variations among copepod species. In this study, we present the first complete mitochondrial genome analysis of the marine calanoid copepod Bestiolina similis (Sewell, 1914). Recent investigations employing whole mitochondrial genome-based phylogenetic analyses have generated considerable interest in exploring the deep branching patterns of arthropods. The complete mitochondrial genome of B. similis spans 23,704 base pairs and includes 13 protein-coding genes and 20 transfer RNA genes. The nucleotide composition of the mitochondrial genome is characterized by 43.42% adenine, 43.22% thymine, 6.48% guanine, and 6.88% cytosine. Maximum likelihood-based phylogenetic analysis of the mitochondrial genome were constructed of the available copepod species. Mitochondrial ge-nome-based phylogenies often reveal intriguing and distinctive topologies compared to traditional phylogenetic approaches. However, the sequencing of copepod mitochondrial genomes, particularly within the calanoid copepods remains limited. Therefore, this study aims to construct a phylogenetic tree based on the extracted mitochondrial genome, incorporating available data, to enhance our understanding of the evolutionary relationships among calanoid copepods.
Keywords:
Mitochondrial genome
; Calanoida
; Arthropod phylogeny
; Molecular taxonomy
; Chennai coast
1. Introduction
Copepods, the most abundant metazoans found on Earth, represent a diverse group of organisms consisting of thousands of species distributed worldwide [1,2]. Among them, the order Calanoida stands out as a particularly rich and significant component of marine ecosystems, encompassing a large number of species that exhibit subtle variations in their morphological characteristics [3,4]. Accurate identification of these species poses a challenge due to their cryptic nature, requiring careful examination and differentiation. To overcome this challenge, innovative approaches incorporating molecular markers have been developed, enabling more reliable and precise species identification [5].
Mitochondrial genes have proven to be highly variable and valuable tools in revealing taxonomically significant variation among copepod species. This variation enables clear discrimination between closely related species and facilitates the recognition of genetic divergence within conspecific groups, often associated with geographical isolation and cryptic speciation [6]. Consequently, mitochondrial (mt) genes have gained significant attention in recent years for investigating the taxonomic position of copepods. Among these genes, mitochondrial cytochrome oxidase I (mtCoI) has emerged as a common marker for rapid species identification of copepods [7]. However, while mtCoI is useful for species identification, it does not provide comprehensive insights into the phylogeny of copepod species, as it is known to be a fast-evolving gene [8]. In contrast, recent studies utilizing complete mitochondrial genome-based phylogenetic analyses have generated considerable interest in exploring the deep branching patterns of arthropods. Mitochondrial genome-based phylogenies have frequently revealed intriguing and sometimes striking topologies compared to traditional phylogenetic approaches [9,10].
The family Paracalanidae comprises dominant calanoid copepods in tropical oceans, playing a crucial role in the grazing behavior of ocean ecosystems [11]. Among them, Bestiolina similis is a prominent calanoid species found abundant in some parts of coast of India [12]. It belongs to the genus Bestolina (Andronov, 1991) within the Paracalanidae family, consisting of 11 species [13]. Bestiolina is a new genus which was previously considered along with genus Acrocalanus. However, Acrocalanus species are characterized by their absence of a fifth leg in female [14]. On the contrary, Bestiolina species has a small rudimentary fifth leg in female. In the past two decades, a significant number of Bestiolina species have been documented [15,16,17,18]. However, their small size (>1mm), inadequate sampling techniques (using mesh size above 200 µm) and misidentification with copepodite stages of other paracalanid species makes them challenging to study [19]. Due to the following reasons, accurate taxonomic identification can be challenging, prompting the use of molecular approaches for taxonomy. Therefore, the objective of this study is to extract and sequence the complete mitochondrial genome of the marine copepod B. similis for the first time and deposit it in GenBank for further investigations. Currently, only a limited number of copepod mitochondrial genomes have been sequenced, with only a few belonging to the calanoid group. Hence, this study will construct a phylogenetic tree based on the extracted mitochondrial genome, incorporating the available data, to enhance our understanding of the evolutionary relationships among calanoid copepods. This research contributes to the expanding knowledge of copepod biodiversity and provides valuable insights into their taxonomic and phylogenetic relationships in marine ecosystems.
2. Materials and Methods
2.1. Copepods collection and identification
Zooplankton samples were collected from the Marina beach (13°03'00'' N, 80°16'56.64'' E, India) using a standard Bolton silk zooplankton net with a mesh size of 150μ five nautical miles away from the coast during the month of August, 2022. The collected samples were preserved in 95% ethanol and transported to laboratory. Copepods were identified up to species level by carefully dissecting and studying the key characters [20,21].
2.2. DNA Extraction and quality Control
DNA from copepods was extracted using the Qiagen DNeasy Blood and Tissue Kit (Cat No.69506). The copepods were pelleted through centrifugation, and the supernatant was carefully removed, leaving behind the pellet, which was then air-dried. Subsequently, each microcentrifuge tube containing the pellet received ATL buffer and Proteinase K, followed by a brief vortexing and incubation at 56⁰C for 2 hours. RNase A treatment (MP Biomedicals; Cat. No.210107683) was carried out at 65⁰C for 20 minutes. The resulting lysate was thoroughly mixed with half the volume of absolute alcohol and loaded into a DNeasy mini spin column, placed within a 2 ml collection tube. The tubes were centrifuged at 8000 rpm for 1 minute, and the flow-through was discarded. The remaining column wash steps were conducted as per the manufacturer's instructions. Finally, DNA was eluted from the column using 10 mM Tris HCl, pH 8.0.
2.3. Library Preparation for mitochondrial genome
The DNA library preparation was carried out following the QIASeq FX DNA Library Preparation protocol (Cat#180475) in accordance with the manufacturer's instructions. The process involved enzymatic fragmentation, end-repair, and A-tailing of 1 ng to 10 ng of Qubit quantified DNA in a one-tube reaction using the FX Enzyme Mix provided in the QIASeq FX DNA kit. Subsequently, the end-repaired and adenylated fragments underwent adapter ligation, where an index-incorporated Illumina adapter was ligated to generate the sequencing library. The library was then subjected to 10 to 12 cycles of Indexing-PCR, including initial denaturation at 98˚C for 2 minutes, cycling (98˚C for 20 seconds, 60˚C for 30 seconds, 72˚C for 30 seconds), and final extension at 72˚C for 1 minute, to enrich the adapter-tagged fragments. Lastly, the amplified library was purified using Sera-MagTM Select beads (Cytiva, # 29343057), followed by a thorough quality control check of the library.
2.4. Illumina sequencing and data analysis
Illumina-compatible sequencing library was quantified by Qubit fluorometer (Thermo Fisher Scientific, MA, USA) and its fragment size distribution was analyzed on Agilent 2200 TapeStation. The libraries were subsequently subjected to paired-end sequencing on an Illumina NovoSeq 6000 sequencer for 150 cycles, following the manufacturer's instructions (Illumina, San Diego, USA). The sequencing generated approximately 26.8 to 34.3 million Illumina short reads data. To ensure data quality, the raw reads were trimmed using Trimgalore-v0.4.01 to remove adapter sequences and low-quality reads. The processed high-quality reads were then utilized for assembly, gene prediction, and annotation using the MitoZ-v3.4 tool, a specialized toolkit for animal mitochondrial genome assembly, annotation, and visualization.
2.5. Phylogenetic tree construction
The phylogenetic trees of mtDNA were constructed using the maximum likelihood (ML) method through MEGA-X software [22]. Existing mtDNA data from the NCBI database of relevant species were utilized for comparison and to produce the phylogenetic tree. Only data representing the genus level were considered for the analysis. Optimal trees were generated through heuristic searches with 1000 bootstraps [23]. The evolutionary history was inferred from the constructed trees using the Jukes-Cantor model [24]. A bootstrap consensus tree was obtained from 1000 replicates, representing the evolutionary history of the analyzed taxa [25]. Branches corresponding to partitions reproduced in less than 50% of bootstrap replicates were collapsed. The initial tree(s) for the heuristic search were automatically obtained using Neighbor-Joining and BioNJ algorithms applied to a matrix of pairwise distances estimated through the Maximum Composite Likelihood (MCL) approach. The topology with the most favorable log likelihood value was selected. Species distances were compared at the phylogenetic levels of calanoid copepods to assess their evolutionary relationships.
3. Results
3.1. Overview of the mitochondrial genome of B. similis
The complete mitochondrial genome sequence of B. similis found to be 23,704 base pair in length, including 13 PCGs and 20 tRNA genes (Figure 1)(Genbank: SUB12990372). It comprises a two longest non-coding region from the beginning of the genome (0 bp to ∼6.5k bp) to the end (∼21.9k bp to 23.7k bp). Other non-coding regions were below 1000 bp in length. Overall nucleotide composition of the complete mitochondrial genome is of 43.42% Adenine, 43.22% Thymine, 6.48% Guanine and 6.88% Cytosine.
3.2. Protein coding regions of the genome
The mtDNA of B. similis revealed 13 protein-coding genes. Table 1 shows the list of protein coding genes observed from the mt DNA. ND5, COX1, ND4 and CYB were observed to be the bigger genes in the genome with 1.713, 1572, 1299 and 1137 bp. ATP8 was observed to be the smallest gene with 156 bp. There was a big non-coding region with ∼1k bp observed between ATP8 and COX1.
3.3. Regions of tRNA genes
The mtDNA revealed 20 tRNAs coding genes each with ∼60 bp. There was ∼2k bp gap observed between the trnD(guc) and trnR(ucg) which was occupied by COX2 and ND4. Two gene coding for tRNA-Leu were observed - trnL(uaa) and trnL(uag). Table 2 gives an account of tRNA genes of the mtDNA of B. similis.
3.4. Phylogenetic tree based on mitochondrial genome of B. similis
The phylogenetic tree, constructed based on complete mtDNA, reveals a close relationship between the observed Bestiolina species and Paracyclopina and Calanus species (Figure 2). However, other calanoid species, such as Labidocera and Temora, appear to be distantly grouped from the Bestiolina and Calanus species. This finding indicates a substantial disorientation within the order Calanoida when considering the families based on complete mtDNA.
4. Discussion
The genus Bestiolina (Andronov, 1991), previously known as Bestiola [26], was distinguished from the genus Acrocalanus by the presence of 6 setae in P3 endopodite, which was 7 in the latter [27]. Since then, taxonomic and phylogenetic investigations on Bestiolina species have garnered considerable interest. Cornils and Blanco-Bercial [27] explored the evolution of Paracalanidae species through both morphological and molecular datasets, revealing that Bestiolina species displayed a close relationship with Acrocalanus species in molecular datasets. However, their relationship differed significantly in morphological datasets.
Species belonging to the genus Bestiolina are commonly found in tropical regions across different oceans, with some speculation about their origin in the Indo-Malayan region [14]. Previous studies by Sivakumar et al. [28], Nawaz et al. [4] Umer et al. [29], Dishad Begum [30], Shanthi and Ranamibai [31] and Rajthilak et al. [32] investigated the copepod diversity in and around the Chennai coast of India; however, none of them reported the occurrence of Bestiolina species. Therefore, this study marks the first report of Bestiolina species in Chennai waters. Nawaz et al. [10] observed a overall abundance of Paracalanidae copepods in Chennai waters. Despite the dominance of paracalanid copepods, Bestiolina species were not observed throughout the study. However, B. similis has been documented as dominant in other parts of the Indian coasts [11,33]. The occurrence of Bestiolina species in the present study suggests their potential migratory abilities.
The size range of metazoan mitochondrial (mt) genomes varies from 14 to 48 kb [34], encompassing genes encoding at least 13 proteins, 22 transfer RNAs, and 2 ribosomal RNAs (rRNA). Notably, the mitogenome typically includes a substantial non-coding region that plays a role in transcription initiation and gene replication [35]. In recent times, there has been an increasing trend in complete mitochondrial genome extraction for species identification and the study of phylogenetic and molecular evolution among closely related species [36]. The protein-coding genes (PCGs) of B. similis exhibit the same genes present in other calanoid mitochondrial genes, such as C. hyperbolus [37] and E. affinis [38]. Unlike Penella and Paracyclopina species, B. similis does possess the ATP8 gene [39], indicating a genomic structure similar to other calanoid species. However, the presence of rRNA genes was not observed in the present study. While vertebrate mitogenomes typically contain 2 rRNA genes, this feature is also common among most invertebrates. However, some cnidarins have been noted to exhibit reduced RNA genes in their genome [40], which may also apply to B. similis.
The phylogenetic tree constructed based on whole genome sequences revealed that E. affinis and L. rotunda were grouped together, forming a distant cluster from B. similis and C. simillimus. Consequently, B. similis exhibited a close proximity to P. nana, which belongs to the order Cyclopoida. This grouping indicates a lack of closely related species from both calanoid and cyclopoid orders in the phylogenetic construction. On the other hand, the positioning of Eutemora and Labidocera far from calanoid species suggests different evolutionary patterns within these calanoid species. Notably, only species from 4 genera of calanoid mitochondrial genomes have been extracted and studied thus far (including this study), which accounts for the displacement of these species in the tree. Furthermore, P. nana is the only cyclopoid species for which the complete mitochondrial genome has been extracted. The difficulty in extracting complete mitochondrial genomes is attributed to the challenges in long PCR amplification. B. similis was observed to possess <80% of A-T content, which may contribute to the possibility of long PCR amplification issues, as indicated by Ki et al [41]. . However, given the limited availability of mitogenomes, it is currently not feasible to draw conclusive phylogenetic relationships between the species. Therefore, future studies are needed to further investigate the molecular evolution of calanoid species based on whole mitogenomes.
Author Contributions
MAN and KS designed and executed the entire study. KS performed DNA extraction, quantification and library preparation. MAN, AS perform sample collection, analysis and DNA extraction. MAN, AS drafted the original manuscript. KS, AS Reviewed and approved the final manuscript.
Funding
The authors would like to thank the Ministry of Earth Sciences (MoES), Government of India for providing financial assistance for the completion of this work (MoES/36/OOIS/Extra/2018).
Data Availability Statement
Extracted genome is available in NCBI genome database with accession number
Acknowledgments
The authors MAN and KS would like to thank the Management, Principal, and Head of the Department of Biotechnology, KarpagaVinayaga College of Engineering and Technology, India for providing the necessary resources.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Huys, R.; Boxshall, G.A. Copepod evolution. The Ray society, London, 2004.
- Boxshall, G.A.; Defaye, D. Global diversity of copepods (Crustacea: Copepoda) in freshwater. Hydrobiologia 2008, 595, 195–207. [Google Scholar] [CrossRef]
- Frost, B.; Fleminger, A. A revision of the genus Clausocalanus (Copepoda:Calanoida) with remarks on distributional patterns in diagnostic characters. Berkeley: University of California Press, 1968; 1–235. [Google Scholar]
- Nawaz, M.A.; Sivakumar, K.; Baskar, G.; Vijayaraj, R. Diversity rhythm in pontellid copepods (Pontellidae: Copepoda) from the Covelong coast pre- and post-COVID-19 lockdown, Bay of Bengal. Turk. J. Zoo. 2023, 47, 71–80. [Google Scholar] [CrossRef]
- Blanco-Bercial, L.; Cornils, A.; Copley, N.; Bucklin, A. DNA barcoding of marine copepods: assessment of analytical approaches to species identification. PLoS Curr 2014, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Bucklin, A.; Frost, B.W.; Bradford-Grieve, J.; Allen, L.D.; Copley, N.J. Molecular systematic and phylogenetic assessment of 34 calanoid copepod species of the Calanidae and Clausocalanidae. Mar. Bio. 2003, 142, 333–343. [Google Scholar] [CrossRef]
- Elías-Gutiérrez, M.; Valdez-Moreno, M.; Topan, J.; Young, MR.; Cohuo-Colli, JA. Improved protocols to accelerate the assembly of DNA barcode reference libraries for freshwater zooplankton. Ecol Evol. 2018, 8, 3002–3018. [Google Scholar] [CrossRef]
- Lefébure, T.; Douady, C.J.; Gouy, M.; Gibert, J. Relationship between morphological taxonomy and molecular divergence within Crustacea: Proposal of a molecular threshold to help species delimitation. Mol. Phylo. Evo. 2006, 40, 435–447. [Google Scholar] [CrossRef]
- Hassanin, A. Phylogeny of Arthropoda inferred from mitochondrial sequences: strategies for limiting the misleading effects of multiple changes in pattern and rates of substitution. Mol. Phylo. Evo. 2006, 38, 100–116. [Google Scholar] [CrossRef]
- Lim, J.T.; Hwang, U.W. The complete mitochondrial genome of Pollicipes mitella (Crustacea, Maxillopoda, Cirripedia): non-monophylies of maxillopoda and crustacea. Mol.&cells 2006, 22, 314–322. [Google Scholar]
- Nawaz, M.A.; Sivakumar, K.; Baskar, G. Impact of variation in environmental parameters on the abundance of Paracalanidae (Calanoida: Copepoda) from the stressed tropical coast of India, Bay of Bengal. Russ. J. Mar. Bio, (Accepted for publication). 2023. [Google Scholar]
- Hani, P.M.; Jayalakshmi, K.J. Temporal variation in diversity, abundance, and size class structure of planktonic copepods from a tropical estuary. Aquat. Ecol. 2023, 57, 199–216. [Google Scholar] [CrossRef]
- World Register of Marine Species. Available from https://www.marinespecies.org at VLIZ. Accessed 2023-07-24.
- Kasturirangan, L.R. A Key for identification of The most common planktonic copepod of Indian coastal water (N.K. Panikkar, Ed.). Council of Scientific and Industrial Research, New Delhi, 1963.
- Ali, M.; Al-Yamani, F.; Prusova, I. Bestiolina arabica sp. nov. (Copepoda, Calanoida, Paracalanidae), a new species from the Northwestern Arabian Gulf. Crustaceana 2007, 80, 195–205. [Google Scholar] [CrossRef]
- Moon, S.Y.; Lee, W.; Soh, H.Y. A new species of Bestiolina (Crustacea: Copepoda: Calanoida) from the Yellow Sea, with notes on the zoogeography of the genus. Proceed. of Bio. Soc. of Washington 2010, 123, 32–46. [Google Scholar] [CrossRef]
- Mulyadi, M. Calanoid Copepods in Indonesian Waters. Research Center for Biology, Indonesia Institute of Sciences, Bogor, 2004.
- Suárez-Morales, E.; Almeyda-Artigas, R.J. A new species of Bestiolina (Copepoda: Calanoida: Paracalanidae) from the Northwestern Atlantic with comments on the distribution of the genus. Revista Mexicana de Biodiversidad 2016, 87, 301–310. [Google Scholar] [CrossRef]
- Dorado-Roncancio, J.; Gaviria, S.; Bernal-De La Torre, L.; Ahrens, M.J. A new species of Bestiolina (Crustacea, Copepoda, Calanoida, Paracalanidae) from coastal waters of the Colombian Pacific, including a worldwide key for the identification of the species. ZooKeys 2019, 2019 846, 1–18. [Google Scholar] [CrossRef]
- Razouls, C.; Desreumaux, N.; Kouwenberg, J.; Bovée, F. Biodiversity of Marine Planktonic Copepods (morphology, geographical distribution and biological data). Sorbonne University, CNRS, 2022. http://copepodes.obs-banyuls.fr/en.
- Boxshall, G.; Halsey, S. An introduction to copepod diversity. The Ray Society London, 2004.
- Kumar, S.; Tamura, K.; Nei, M. MEGA: Molecular Evolutionary Genetics Analysis. Pennsylvania State University, University Park, PA, 1993.
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution, 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Grauer, D.; Li, W.H. Fundamentals of molecular evolution, 2nd Edn. Sinauer Associates, 2000.
- Pattengale, N.D.; Alipour, M.; Bininda-Emonds, O.; Moret, B.; Stamatakis, A. How many bootstrap replicates are necessary? J. Comp. Bio. 2010, 17, 337–354. [Google Scholar] [CrossRef] [PubMed]
- Andronov, V.N. Veslonogie rachki Bestiola gen. n. (Copepoda, Paracalanidae). Zoologicheskii Zhurnal 1972, 51, 290–292. [Google Scholar]
- Cornils, A.; Blanco-Bercial, L. Phylogeny of the Paracalanidae Giesbrecht, 1888 (Crustacea: Copepoda: Calanoida). Mol. Phylo. Evo. 2013, 69, 861–872. [Google Scholar] [CrossRef]
- Sivakumar, K.; Nawaz, M.A.; Saboor, A. Population composition of calanoid copepods of the Chennai coast, Tamil Nadu. Ind. J.Geo-Mar. Sci. 2021, 50, 693–700. [Google Scholar]
- Umer, K.S.; Ebenezer, V.; Subramoniam, T. A short-term study on the effect of environmental factor variation on a zooplankton community. Ind. J.Geo-Mar. Sci 2020, 49, 1158–1164. [Google Scholar]
- Dilshad, B. I. Diversity and biology of calanoid copepods of Chennai coast India. Ph.D. Thesis, University of Madras, 2006.
- Santhi, M; Ramanibai, R. Studies on Copepods from Chennai Coast (Cooum and Adyar), Bay of Bengal-During the Cruise. Curr. Res. J. Bio. Sci. 2011, 3, 132–136. [Google Scholar]
- Rajthilak, C.; Perumal, P.; Santhanam, P.; Nandakumar, R.; Ananth, S. Spatial and temporal distributions of calanoid copepods (Crustacea; Arthropoda) along the Tamil Nadu coast (Southeast India). Ind. J.Geo-Mar. Sci. 2016, 45, 1578–1583. [Google Scholar]
- Bhattacharya, B.D.; Bhattacharya, A.; Rakshit, D.; Sarkar, S. Impact of the tropical cyclonic storm 'Aila' on the water quality characteristics and mesozooplankton community structure of Sundarban mangrove wetland, India. Ind. J.Geo-Mar. Sci. 2014, 43, 216–223. [Google Scholar]
- Crease, T.J. The complete sequence of the mitochondrial genome of Daphnia pulex (Cladocera: Crustacea). Gene 1999, 233, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Shadel, G.S.; Clayton, D.A. Mitochondrial DNA maintenance in vertebrates. Annual Review of Biochemistry 1997, 66, 409–435. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Peng, Y.; Zhang, K.; Liu, Y.; Li, J.; et al. Comparative analysis of the complete mitochondrial genomes of two species of Clupeiformes and the phylogenetic implications for Clupeiformes. J. Mar. Bio. Asso. of. Uni. King. 2022, 102, 445–456. [Google Scholar] [CrossRef]
- Kim, S. , Lim, B. J., Min, G. S., & Choi, H. G. The complete mitochondrial genome of Arctic Calanus hyperboreus (Copepoda, Calanoida) reveals characteristic patterns in calanoid mitochondrial genome. Gene, 2013, 520, 64–72. [Google Scholar]
- Choi, B. S.; Han, J.; Hwang, D. S.; Souissi, S.; Hagiwara, A.; Lee, J. S. Complete mitochondrial genome of the calanoid copepod Eurytemora affinis (Calanoida, Temoridae). Mito. DNA Part B 2019, 4, 2731–2733. [Google Scholar] [CrossRef]
- Liu, H.; Fu, Z.; Zhou, S.; Hu, J.; Yang, R.; Yu, G.; Ma, Z. The Complete Mitochondrial Genome of Pennella sp. Parasitizing Thunnus albacares. Front. Cell. and Infec. Microbio. 2022, 12, 945152. [Google Scholar] [CrossRef]
- Lavrov, D.V. Mitochondrial Genomes in Invertebrate Animals. In: Wells, R.D., Bond, J.S., Klinman, J., Masters, B.S.S. (eds) Molecular Life Sciences. Springer, New York, NY, 2018.
- Ki, J. S.; Park, H. G.; Lee, J. S. The complete mitochondrial genome of the cyclopoid copepod Paracyclopina nana: a highly divergent genome with novel gene order and atypical gene numbers. Gene 2009, 435, 13–22. [Google Scholar] [CrossRef]
Figure 1.
Complete mitochondrial genome of the calanoid copepod B. similis.
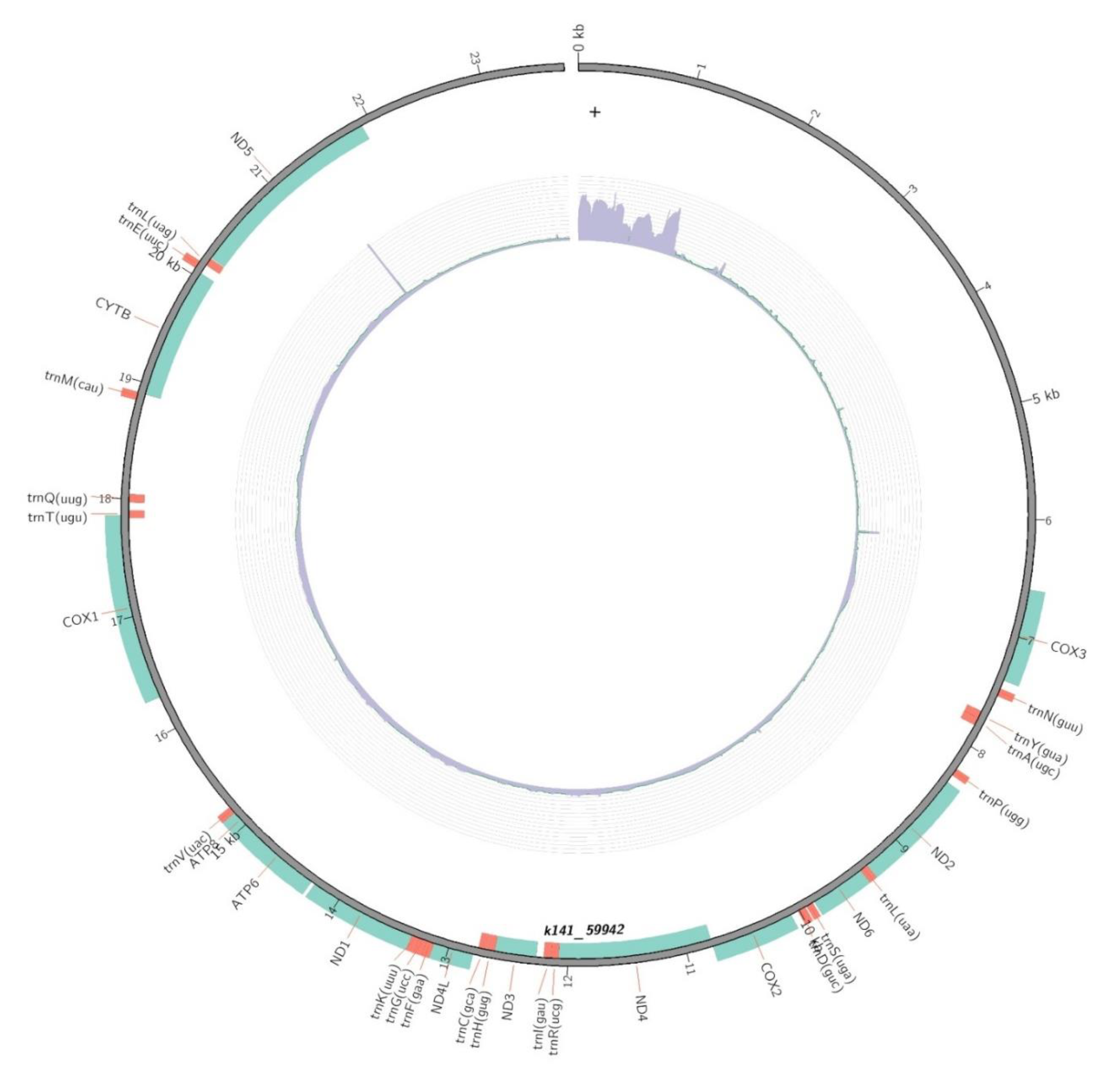
Figure 2.
Phyologenetic tree based on ML method using complete mtDNA.
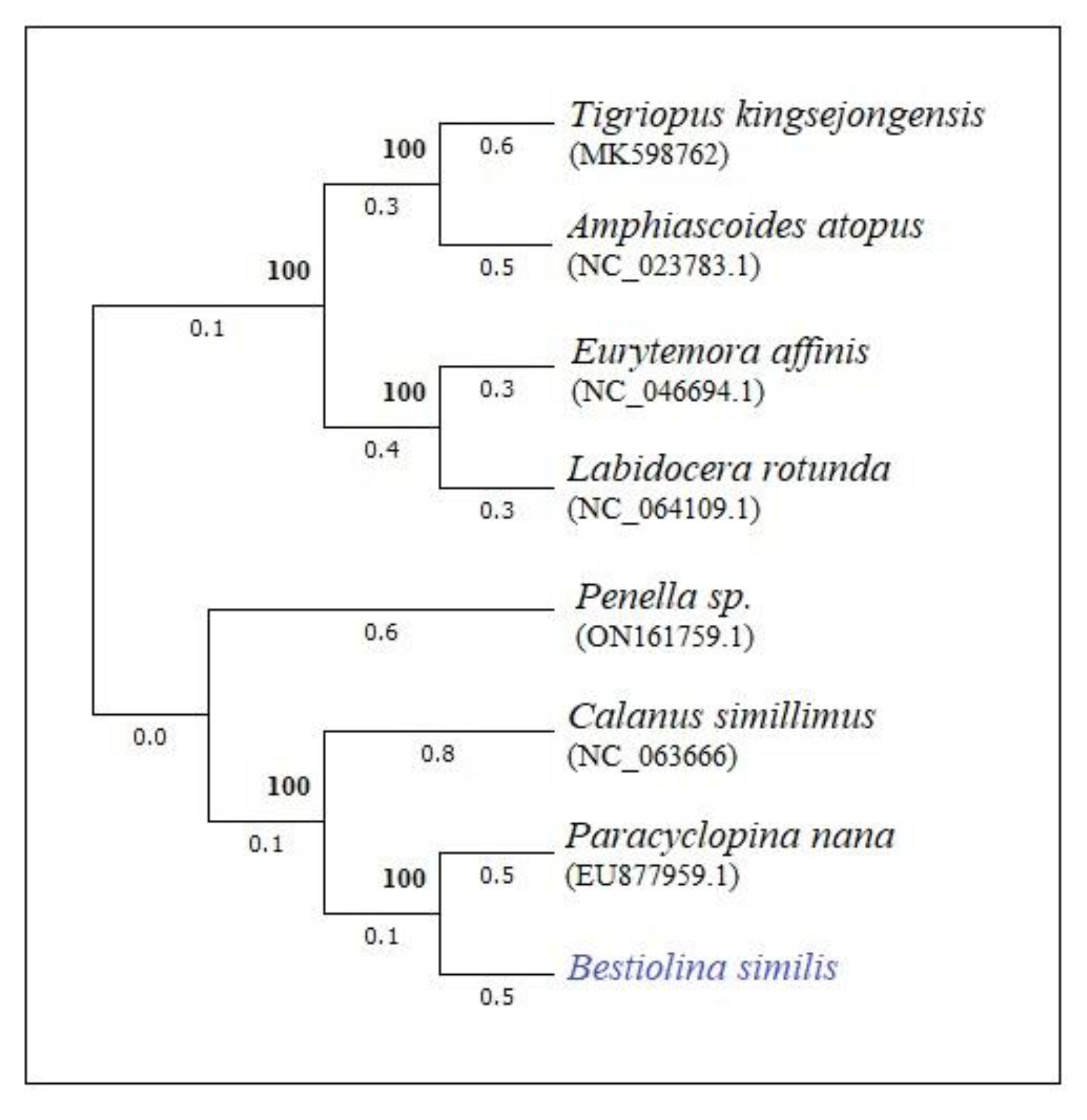

Table 1.
Protein coding regions of the mtDNA of B. similis.
| Start | End | Length(bp) | Gene name | Gene product |
|---|---|---|---|---|
| 6591 | 7382 | 792 | COX3 | Cytochrome c oxidase subunit III |
| 8357 | 9325 | 969 | ND2 | NADH dehydrogenase subunit 2 |
| 9394 | 9855 | 462 | ND6 | NADH dehydrogenase subunit 6 |
| 10079 | 10777 | 699 | COX2 | Cytochrome c oxidase subunit II |
| 10780 | 12078 | 1299 | ND4 | NADH dehydrogenase subunit 4 |
| 12267 | 12620 | 354 | ND3 | NADH dehydrogenase subunit 3 |
| 12794 | 13129 | 336 | ND4L | NADH dehydrogenase subunit 4L |
| 13327 | 14238 | 912 | ND1 | NADH dehydrogenase subunit 1 |
| 14271 | 14984 | 714 | ATP6 | ATP synthase F0 subunit 6 |
| 14981 | 15136 | 156 | ATP8 | ATP synthase F0 subunit 8 |
| 16292 | 17863 | 1572 | COX1 | Cytochrome c oxidase subunit I |
| 18909 | 20045 | 1137 | CYTB | Cytochrome b |
| 20204 | 21916 | 1713 | ND5 | NADH dehydrogenase subunit 5 |
Table 2.
tRNA genes of the mtDNA of B. similis.
| Start | End | Length(bp) | Gene name | Gene prodcut |
|---|---|---|---|---|
| 7457 | 7520 | 64 | trnN(guu) | tRNA-Asn |
| 7697 | 7759 | 63 | trnY(gua) | tRNA-Tyr |
| 7761 | 7823 | 63 | trnA(ugc) | tRNA-Ala |
| 8222 | 8284 | 63 | trnP(ugg) | tRNA-Pro |
| 9326 | 9393 | 68 | trnL(uaa) | tRNA-Leu |
| 9874 | 9930 | 57 | trnS(uga) | tRNA-Ser |
| 9947 | 10011 | 65 | trnD(guc) | tRNA-Asp |
| 12078 | 12139 | 62 | trnR(ucg) | tRNA-Arg |
| 12140 | 12202 | 63 | trnI(gau) | tRNA-Ile |
| 12623 | 12689 | 67 | trnH(gug) | tRNA-His |
| 12695 | 12757 | 63 | trnC(gca) | tRNA-Cys |
| 13130 | 13192 | 63 | trnF(gaa) | tRNA-Phe |
| 13195 | 13260 | 66 | trnG(ucc) | tRNA-Gly |
| 13262 | 13324 | 63 | trnK(uuu) | tRNA-Lys |
| 15137 | 15198 | 62 | trnV(uac) | tRNA-Val |
| 17840 | 17901 | 62 | trnT(ugu) | tRNA-Thr |
| 17974 | 18041 | 68 | trnQ(uug) | tRNA-Gln |
| 18845 | 18908 | 64 | trnM(cau) | tRNA-Met |
| 20062 | 20127 | 66 | trnE(uuc) | tRNA-Glu |
| 20130 | 20196 | 67 | trnL(uag) | tRNA-Leu |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



