
Submitted:
16 August 2023
Posted:
17 August 2023
Read the latest preprint version here
Abstract
Meningiomas are the most prevalent primary intracranial tumors. The majority are benign but can undergo dedifferentiation in grades classified from I to III. Meningiomas tremendous variability in tumor behavior and slow growth rates complicate their diagnosis and treatment. A deeper comprehension of the molecular pathways and cellular microenvironment factors implicated in meningioma survival and pathology is needed. This review summarizes the known genetic and epigenetic aberrations involved in meningioma, with a focus on Neurofibromatosis type 2 (NF2) and non-NF2 mutations. Novel potential biomarkers for meningioma diagnosis and prognosis are also discussed, including epigenetic-, RNA-, and protein-based markers. Finally, the landscape of available meningioma-specific animal models is overviewed. Use of these animal models can enable planning of adjuvant treatment, potentially assisting in preoperative and postoperative decision-making. Discovery of novel biomarkers will allow more precise meningioma grading, including meningioma identification, subtype determination, and prediction of metastasis, recurrence, and response to therapy. Moreover, these biomarkers may be exploited in the development of personalized targeted therapies that can distinguish between the 15 diverse meningioma subtypes.
Keywords:
Meningioma
; NF2 mutations
; biomarker
; miRNA
; proteomics
1. Introduction
Meningiomas are the most prevalent primary intracranial tumors. Meningiomas have an incidence of 7.86 cases per 100,000 persons per year, accounting for around 36% of all central nervous system (CNS) tumors and 53% of nonmalignant CNS tumors [1,2]. Risk factors of meningiomas include radiation therapy, diabetes, genetic susceptibility, arterial hypertension, estrogen use in women, and potentially smoking [3,4]. Nonmalignant meningiomas are more common in women than in men. Meningiomas are also more prevalent in older people and are largely prevalent in US black population [5]. Arachnoid cap cells, which are found in the thin spider-web-like meningeal membrane that surrounds the brain and spinal cord, are the origin of meningiomas. Most meningiomas are benign and are frequently discovered incidentally [1]. Nearly 80-90 % of meningiomas arise intracranially, while the remaining 10-20 % arise in the spinal cord [2]. Former and current editions of the World Health Organization (WHO) categorization of tumors of the CNS describe 15 unique meningioma subtypes with heterogeneous physical characteristics, which include histo- and cyto-morphological differences. WHO classification of CNS malignancies, divides the 15 meningioma subtypes into three groups: nine types are classified as WHO grade I (benign, low-grade, 80% of all meningiomas), three as grade II (intermediate, high-grade, atypical, 5-15 % of all meningiomas, higher chance of recurrence following gross total resection), and three as grade III (malignant, high-grade, anaplastic, 1–3% of all meningiomas, very poor clinical outcomes and higher possibility of recurrence and metastasis) [6-8]. Indeed, there is a huge divergence in individual clinical behaviors of atypical and malignant meningiomas (Grade II vs Grade III). The current WHO grading system, which depends mainly on histopathological features, is often inaccurate and unable to predict outcomes such as recurrence and patient survival. Therefore, the discovery of reliable meningioma biomarkers is an urgent priority for the prediction of treatment options and a better prognosis of this disease [9].
Meningioma was one of the first malignancies in which cytogenetic abnormalities were discovered. Recent genomic analyses of meningiomas revealed significant molecular variability. In fact, 60%–80% of meningiomas have a loss of one copy of 22q, which harbors the neurofibromatosis type 2 (NF2) gene, and this loss is usually coupled with alterations of the remaining NF2 allele [10-12]. In fact, up to 60% of sporadic meningiomas have biallelic inactivation of NF2 due to chromosome 22 monosomy combined with NF2 point mutations [13,14]. Studies conducted afterwards revealed that the probability of recurrence and malignancy are both correlated with an accumulation of other chromosomal abnormalities, most typically losses of 1p, 10, and 14q [15,16]. In addition to NF2 mutations, somatic mutations of tumor necrosis factor receptor-associated factor 7 (TRAF7), DNA-directed RNA polymerase II subunit RPB1 (POLR2A), Protein Kinase A Type 1a Regulatory Subunit (PRKAR1A), Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha (PIK3CA), Kruppel-Like Factor 4 (KLF4), AKT Serine/Threonine Kinase 1/ Protein Kinase B (AKT1), Smoothened Frizzled Class Receptor (SMO), Suppressor Of Fused Homolog (SUFU), and genes of the Transforming growth factor beta pathway (TGF) among others have been detected in meningiomas. Some of these mutations may co-occur with NF2 mutations while others occur independently of NF2 mutations. Interestingly, some of these mutations are implicated in certain meningiomas like those that appear in distinct locations or are of distinct histological subtypes or severity [17-22]; Figure 1 demonstrates the relation between genetic alterations, grades of meningioma, and anatomical location of the tumor in the CNS. However, these somatic driver mutations cannot inform treatment stratification for intracranial tumors [23], and there is an urgent need to understand how these genomic changes are linked to disease outcomes such as tumor recurrence following resection, response to radiotherapy, and overall survival [9].
While genomics markers of meningiomas, like NF2 mutations, have been explored, the search for other classes of biomarkers is in progress. For example, different WHO grades of meningiomas show differential protein profiles, paving the way for the discovery of protein-based biomarkers [24]. Along the same lines, epigenetic and mRNA biomarkers are currently under investigation in meningioma. There is evidence that defects in epigenetic regulation are essential for tumorigenesis and that genomic mutations can only partially explain the early stages of tumorigenesis. Indeed, epigenetic alterations of trimethylation of lysine 27 on histone 3 (H3K27me3) repress gene expression and have been implicated in the pathogenesis of intracranial tumors, and loss of H3K27me3 alterations has been associated with meningioma recurrence in retrospective clinical studies [25,26]. In addition, hypermethylation of TIMP3, Cyclin Dependent Kinase Inhibitor 2A (CDKN2A), and TP73 has been correlated with meningioma grade [27,28]. Ultimately, a panel of meningioma biomarkers combining epigenetics, transcriptomics, proteomics, and genomics biomarkers will be needed to predict behaviors of aggressive meningiomas with a high risk of progression or recurrence [29]. In this review, we aim to evaluate genetic and other molecular alterations involved in meningioma and how to exploit them for new biomarker discovery for diagnosis and prognosis including meningioma identification, grading and subtype determination, and risk of metastasis and recurrence.
2. Grading of Meningiomas
The majority of meningiomas (more than 80%) are WHO Grade I, with Grade I age-adjusted incidence rates of 3.68/100,000 and 8.56/100,000 in the male and female populations, respectively [6]. WHO grade II meningiomas have an age-adjusted incidence rate of 0.26 per 100,000 males and 0.30 per 100,000 females. WHO grade III meningiomas are a rare disease with age-adjusted incidence rates of 0.08 per 100,000 males and 0.09 per 100,000 females [30]. Diagnosis of meningioma is made through imaging, and a biopsy is not necessary if imaging strongly suggests a meningioma [31]. Asymptomatic meningiomas grow linearly at a rate of 2-4 millimeters per year, however, there can be instances where there is no growth in volume [32]. This aspect highlights the significance of surveillance in untreated patients with asymptomatic meningioma. Grade II and III meningiomas are usually symptomatic or have a high tendency for growth and undergo gross total resection [33]. Occasionally, not all the tumor is accessible for resection leading to recurrence since it has been observed that the extent of resection affects recurrence rates [34]. The estimated 10-year overall survival for benign meningiomas is 81.4%, compared to 57.1% for malignant ones. Grade II tumors 10-year overall survival rate is around 53%, while grade III tumors sadly have this rate as 0% [2]. Meningiomas with distant metastasis are rare and have only been documented in few case reports or brief case series [35-38]. The lungs, bones, spinal cord, and liver are the most common secondary metastasis sites of meningiomas [35]. Only 6% of metastases are discovered at the time of diagnosis, while 93% of metastatic meningiomas are discovered after the main tumor has been diagnosed and removed [35].
Recent developments in genomics have led to further stratification of meningioma subtypes based on alterations in somatic gene copy numbers and genome-wide profiling of DNA methylation [20,39,40]. Patel et al. combined whole genome sequencing and transcriptome analysis and suggested another classification of meningiomas into 3 major types: type A have missense mutations in TRAF7, KLF4, and AKT1 and have minimal chromosomal alterations [41], similar to previous findings in benign meningiomas [39]; type B are NF2-deficient non-aggressive meningiomas; and type C are more aggressive meningiomas which have a significant chromosomal instability and chromosomal gains and losses, most commonly loss of both chr22q and chr1p [41]. Using these molecular principles, Tsitsikov et al. compared transcriptional profiles of four of the most common benign types of meningiomas: 1) NF2 loss versus meningiomas with TRAF7 missense mutations, 2) NF2 tumors with or without additional loss of chr1p, 3). TRAF7 meningiomas with additional missense mutations in AKT1 or KLF4. Their analysis showed distinct transcriptional programs specific for each meningioma genotype [39]. Other studies have integrated multiple parameters, including DNA methylation, RNA-seq, and cytogenetic profiling to enhance the grading of meningiomas [42,43]. The significant differences of the molecular profiles between the different meningioma grades led to the recognition of certain high-risk molecular signatures in the WHO 2021 classification of CNS tumors [8]. In this WHO classification, loss of H3K27me3 is indicative of aggressive meningioma behavior and recurrence, and homozygous deletions of CDKN2A/B and mutations of TERT promoter (pTERT) are criteria for grade 3 meningioma, since they are linked to increased risk of recurrence [8,44,45]. However, these added molecular markers can specify only a subtype of meningiomas that that are at high risk of recurrence. This further underscores the need to include more molecular markers for meningioma identification and that meningioma grading should not depend on histopathology only [45].
3. Genomic Alterations and Epigenetic Modifications in Meningiomas
Advances in technology over the last few decades have led to an ongoing rapid growth in the understanding of the oncogenesis and genomic profiles of meningiomas. One outcome of such advances was the association between meningioma formation and NF2 gene inactivation. Later, genomics studies have identified numerous meningioma genetic alterations many of which were not in the NF2 gene [46]. NF2 is named after neurofibromatosis type 2 which is a genetic condition in which benign tumors grow along the nerves responsible for hearing and balance; mutations in the NF2 gene were found to cause the disease. NF2 gene is located on chromosome 22q12.2 and codes for a 69 kDa protein, Merlin [47]. Merlin protein can be found in a variety of adult and embryonic human tissues, specifically in Schwann, meningeal, lens, and nerve cells. Merlin is a cytoskeletal protein that functions in crosslinking membrane proteins with the cytoskeleton [47]. Loss of Merlin protein interrupts normal cell growth by creating gaps in adherens junctions [48]. Merlin is known to act as a tumor suppressor by inhibiting cell growth through contact inhibition and activation of multiple signaling pathways [49], and genetic inactivation of NF2 prevents the production of Merlin, leading to meningioma formation [28]. Figure 2 illustrates NF2/Merlin signaling pathways in a normal arachnoid cap cell in comparison to an NF2-deficient meningioma cell.
NF2 is the most recurrently mutated gene in sporadic and radiation-induced meningiomas [50]. Merlin inactivation, due to mutations in NF2, is involved in about half of sporadic meningiomas [49]. In fact, 60% of meningiomas have been characterized by an NF2 gene deficiency caused by promoter methylation, epigenetic inactivation, monosomy of chromosome 22, or a somatic mutation [51]. Low expression of Merlin was associated with tumor recurrence, and worse overall survival and progression-free survival (PFS) in large patient studies [52,53]. These studies suggested that the mutation status NF2 can act as a biomarker of the survival, prognosis, and risk of tumor recurrence in meningioma patients.
The presence of NF2 mutations is the basis for the classification of meningioma into a subtype that has NF2 gene alterations and a subtype associated with non-NF2 somatic mutations [46]. Meningiomas with mutations in non-NF2 genes are less common, more heterogeneous, and often result in different tumor phenotypes [22,54]. Indeed, missense mutations in TRAF7, KLF4, and AKT1 exist in 30%, 14%, and 12% of non-NF2 meningiomas respectively [53,55]. Studies have identified driver TRAF7 somatic mutations in meningioma tumorigenesis [22,54]. These are the most common non-NF2 mutations and are detected in over 30% of non-NF2, grade I tumors, whereas grade III tumors were less likely to result from these mutations. TRAF7 mutations are exclusive of NF2 mutations, suggesting that the two genes act along the same pathway. Additionally, TRAF7 mutations instigate meningioma growth by acting in combination with one of various co-mutations such as KLF4 and AKT1. KLF4 and AKT1 mutations co-exist with TRAF7 mutations, but not with each other [22,54,55]. KLF4 is a transcription factor that regulates differentiation in a variety of cell types and its expression is essential to reprogram adult cells into pluripotent adult stem cells while AKT1 is involved in proliferation signaling and is a well-characterized oncogene [56].
In addition, there are rare germline mutations in meningioma including Switch/Sucrose non-Fermentable Family (SWI/SNF) Related, Matrix Associated, Actin Dependent Regulator of Chromatin, Subfamily B, Member 1 (SMARC B1), SMARC E1, BAP1, and SUFU genes. SMARC B1 and SMARC E1 mutations are frequently reported in familial syndromes with multiple meningiomas [46]. Mammalian SWI/SNF complex is a multi-subunit chromatin remodeling complex that uses the energy of ATP hydrolysis to remodel nucleosomes and regulate DNA accessibility in fundamental cellular processes, such as transcription, and DNA replication and repair. Mutations of components of SWI/SNF complex are frequently observed in numerous human cancers; however, the underlying mechanisms by which SWI/SNF components contribute to tumorigenesis or drug sensitivity warrant further investigation. It also remains unknown whether and how SWI/SNF mutations or defects could be exploited for therapeutic purposes [57].
Epigenetic modifications are major regulators of gene expression and there is evidence that abnormalities in epigenetic regulation are a critical part of the process of tumorigenesis. Modification of DNA methylation profiles is one of the best-characterized epigenetic alterations implicated in carcinogenesis. Cancer cells usually undergo a global hypomethylation of their genomes, with only selected regions around specific gene promoter regions undergoing DNA hypermethylation. The altered DNA methylation profiles cause alterations in gene expression [46,58]. Conserved CpG islands next to gene regulatory elements in cancer cells exhibit DNA hypermethylation and gene silencing, which correlate with tumor aggressiveness and recurrence. These abnormal changes in DNA methylation are usually unique and can be exploited to characterize a cancer type [46,59,60]. Indeed, methylation profiles of specific genes were shown to correlate with a shorter time to meningioma recurrence [61]. These results can be used to predict prognosis and guide the selection of therapeutic options. As mentioned, loss of H3K27me3 modifications has been associated with meningioma recurrence in retrospective clinical studies [25,26], and hypermethylation of TIMP3, CDKN2A, and TP73 has been correlated with meningioma grade [27,28]. Hypo- and hyper-methylation of numerous other genes have been correlated with the severity, recurrence, and metastasis of meningiomas, as has been reviewed in [59,62] and Table 1. Changes in DNA methylation patterns can be combined with the existing molecular biomarkers to further classify meningiomas into subtypes of different severity and potential for recurrence or metastasis [59]. Choudhury et al. have developed a tool, Meningioma Methylation Classifier (https://william-c-chen.shinyapps.io/MeninMethylClassApp/), that classifies meningiomas according to their DNA methylation status [63]. As proposed by Singh et al. genome-wide DNA methylation profiling represents a paradigm shift in meningioma classification, prognostic prediction, and treatment strategy [62].
4. NF2/ Merlin Signaling Pathways in Meningioma
Merlin is known to interrupt cellular growth by signaling through several cellular signaling pathways (Figure 2) such as inhibition of the RAS/RAF/MEK/ERK mitogen-activated protein kinase (MAPK) signaling pathway, which is relevant to organism growth and development and survival of cells [64]. In normal meningeal cells, Merlin forms a complex with the receptor tyrosine kinase human epidermal growth factor (ERB B2, HER2) and integrin β1 at the cell membrane. This complex inhibits protein kinase B (AKT) and extracellular signal-regulated kinase (ERK) MAPK by preventing the accumulation of ERB B2 and ERB B3 (HER3), two members of the epidermal growth factor receptor (EGFR) family (Figure 2). Merlin can also act upstream of the RAS/RAF/MEK/ERK pathway by inhibiting activation of RAS and RAC following growth factor stimulation (Figure 2) [65]. Merlin deficiency due to NF2 mutations often results in the overactivation of the RAS/ERK pathway, therefore leading to tumor development (Figure 2) [49]. It is common for RAS expression to be elevated in patients with meningiomas. Furthermore, the extent of the RAS increase could serve as an index for determining the degree of malignancy and grade of the meningioma [64]. A study analyzing the expression of various signaling proteins in 70 primary meningiomas indicated strong immunoexpression of RAS and RAF in almost all grade I meningiomas. However, the expression of RAS and RAF was decreased in grade II and III meningiomas, suggesting that these tumors might have other dysregulated pathways than that of RAS/ERK. Additionally, the same study found that RAF was associated with meningioma recurrence, thus highlighting the importance of the RAS/RAF/MEK/ERK pathway activation for meningioma growth [66]. Furthermore, animal models have shown that inhibition of Ras activity suppresses proliferation and induces apoptosis of meningioma cells [64], suggesting that Ras might be an ideal target in meningioma treatment. However, further research is needed on the dysregulation of the RAS/ERK pathway in meningioma.
Merlin has also been reported to signal through the Hippo tumor suppression pathway (Figure 2), the main pathway of cellular growth and regulation of organ/tissue size. The mechanism by which Merlin regulates upstream signals of this pathway are not fully understood yet. However, it is known that loss of Merlin lipid binding ability severely compromises Hippo pathway. NF2 mutants that result in a Merlin protein deficient in phosphoinositide binding prevent osmotic stress-induced activation of the Hippo pathway [67]. Experiments in Drosophila and mice as well as in vitro using human cells have shown that NF2 acts through this pathway to keep tissue growth in check. Deletion of NF2 in human cells was sufficient to completely abolish the Hippo pathway response to glucose starvation, actin disruption, or serum deprivation [68]. Inactivating mutations of the NF2 gene, inactivate the Hippo pathway allowing the transcription factors Yes-associated protein (YAP)/Transcriptional coactivator with PDZ-binding motif (TAZ) to move into the nucleus and form a complex with TEADS (TEA domain) transcription factor, thus promoting cell proliferation and preventing apoptosis by activating the transcription of genes such as AXL Receptor Tyrosine Kinase (AXL), Cysteine-Rich Angiogenic Inducer 61 (CYR61), and Connective Tissue Growth Factor (CTGF). A study involving the analysis of 57 meningiomas demonstrated a significant elevation of expression of these Hippo pathway-associated genes, in tumors involving NF2 mutations, but without any correlation with the grade of meningioma [67]. Indeed, high levels of YAP1 were found to have nuclear localization in meningiomas, and targeting YAP1 activity was shown to be a potential treatment option in meningioma [69]. In addition, YAP is frequently altered, usually by fusion with other proteins, in meningiomas and other tumor types associated with neurofibromatosis type 2. There are also data suggesting the exclusivity of YAP fusion events with NF2 mutations [70]. These provide evidence that Hippo pathway dysregulation is a common driver of oncogenesis in meningiomas and other rare cancer types of the CNS [70]. TEAD palmitoylation inhibitors prevented the growth of NF2-null schwannoma and NF2-null meningioma cells in vitro and a mouse model [71]. Similarly, constitutive activation of YAP1 or the presence of YAP1-MAML2, a fusion protein that was identified in several meningioma patients, can drive the formation of tumors that resemble NF2 mutant meningiomas [70]. Nevertheless, further research is needed to understand the oncogenic functions of the Hippo pathway in meningioma in order to exploit these functions in diagnosis and the discovery of specific therapeutic targets for treatment of meningioma and other tumors.
Merlin interacts with the phosphoinositide 3-kinase /AKT/mammalian target of rapamycin (PI3K/AKT/mTOR) signaling axis (Figure 2), which contributes to the regulation of cell growth and proliferation [72]. Activation of PI3K by a growth factor for example, will cause phosphorylation and activation of AKT which can activate mTOR complex (mTORC), allowing the translation of mTOR target proteins [72]. Merlin inhibits the activation of PI3K by binding phosphatidylinositol 3-kinase enhancer-L [73]. The PI3K/AKT/mTOR axis is overactive in meningioma [74]. Activating mutations of AKT were identified in a subtype of meningiomas [74], and high-grade meningiomas have higher expression levels of AKT, which support a role for PI3K/AKT in meningioma [66,74]. High levels of active phosphorylated mTOR were associated with shorter PFS and increased recurrence in atypical meningiomas [75]. Merlin negatively regulates mTORC whereas Merlin-deficient meningioma cell lines and tumors show constitutive activation of mTORC1 [76]. Merlin-mediated inhibition of mTROC is PI3K/AKT- and ERK MAPK-independent implying the existence of a non-canonical mechanism of mTORC1 inactivation by Merlin [74]. This mechanism remains unexplored and requires further research. Inhibitors of mTORC1 were tested using in vitro, in vivo in animal meningioma models, and in patients and were shown to significantly reduce the proliferation of meningioma cell lines and animal models [74]. Moreover, the combined inhibition of mTORC and angiogenesis increased overall progression-free survival to 22 months in 17 patients with progressive or refractory symptomatic meningiomas [74,77]. Similarly, mTORC inhibition was safe and extended the PFS of 28 patients with recurrent or progressive grade II-III meningiomas in a Phase 2 trial [78].
Merlin also acts as a negative regulator of the forkhead box M1 (FOXM1)/WNT signaling pathway (Figure 2). The WNT signaling pathway is essential during embryogenesis and CNS development and is known to be associated with cancer cell growth and rapid tumor development [51,79]. Components of WNT signaling regulate multiple aspects of brain development in vertebrate embryos. WNT signaling leads to the accumulation of the transcription factor β-catenin in the cytoplasm and its subsequent translocation to the nucleus. Relatedly, mutations in the β-catenin gene have been reported in a variety of human tumors [79]. A study by Lau et al. illustrated a relationship between Merlin and WNT signaling in human glioma cells where re-expression of Merlin reduced WNT signaling. The levels of WNT receptor Frizzled-1 (FZD1) were reduced and the expression of molecules that inhibit WNT signaling, Dickkopf-1 (DKK1) and Dickkopf-2 (DKK2) were increased [80]. Additionally, hypermethylation and inhibition of polycomb repressive complex (PRC) that cause NF2 mutations have been shown to potentiate WNT signaling. Mutated NF2 serves as a functional switch for FOXM1 transcription. Overexpression of FOXM1 due to the lack of regulation by Merlin promotes meningioma cell proliferation and viability. FOXM1 interacts with β-catenin to increase WNT signaling [51].
Overall, signaling pathways that are affected by Merlin loss of function continue to emerge as possible targets for therapy [65]. However, a lot remains unknown in regards to the exact mechanisms by which these pathways influence meningioma grading, pathology, and prognosis.
5. Biomarkers of Meningiomas
5.1. Current Diagnosis and Prognosis
Current approaches for diagnosis of meningiomas rely on patient medical history, physical examination, and use of radiological techniques like computed tomography (CT) scans and magnetic resonance imaging (MRI). MRI is the gold standard for radiologic diagnosis and is also used for long-term follow-up as there is no exposure to radiation [33]. However, in cases where MRI is counter-indicated such as in patients with pacemakers, contrast-enhanced CT scans are used [81]. The challenge in using radiology to diagnose meningiomas is the similarity of meningiomas to other intracranial lesions in MRI and CT scans, complicating diagnosis. Figure 1 depicts the grades of meningioma and their anatomical locations in the CNS, where other CNS tumors may also arise further complicating diagnosis. For example, in the diagnostic process, whenever a suspected meningioma is encountered, the possibility of it being a hemangiopericytoma is also considered. Meningiomas originate from meningothelial cells (arachnoid cap cells), while hemangiopericytomas arise from pericytes, which are cells found in close proximity in the blood vessels. Furthermore, meningiomas present in the cerebral hemispheres can be challenging to distinguish from dural (pachymeningeal) metastases, particularly metastases of prostate, lung, kidney, or breast cancers, primary glial tumors that extend into the subarachnoid space, and hematopoietic neoplasms like extra-axial non-Hodgkin lymphoma [82-85]. Moreover, meningiomas at the base of the skull, particularly at the cerebello-pontine angle, must be distinguished from vestibular and trigeminal schwannomas and neoplastic meningitis. In order for imaging modalities to detect meningiomas, the tumor must grow to a certain size. This becomes another major limiting factor of diagnosis since meningiomas are slow-growing tumors, so the patient remains undiagnosed for early-stage tumors for a long period. For example, fibrous meningiomas and meningothelial meningiomas take an average of 26.3 years and 17.8 years, respectively, until a tumor mass is discovered after the initial cellular change [86]. In meningioma diagnosis, the challenge is not only to confirm the diagnosis of meningioma but also to identify its subtype and grading. MRI can help in the diagnosis of meningiomas, but it may not be able to distinguish between different meningioma subtypes. Studies have also shown that patient movement during the MRI examination can introduce motion artifacts, compromising image quality and diagnostic accuracy [87,88]. All these challenges involving imaging can be avoided by the use of histopathological assessment, which is becoming the new criterion for the diagnosis of meningiomas [31]. Histological techniques provide static snapshots of tissue morphology, lacking real-time or dynamic information about cellular processes or molecular interactions. However, this involves obtaining a tissue biopsy, which not only is an invasive procedure but also may not be a widely available option. The quality of the biopsy sample, which might occasionally be constrained by tumor location, size, or level of vascularity, can also impact the accuracy of diagnosis [87,88]. Differentiation between different CNS tumor types and meningioma, and meningioma subtype determination and grading requires the discovery of new meningioma-specific biomarkers. Collectively, the limitations of MRI and histological techniques highlight the need for new biomarker discovery to enhance diagnostic accuracy, improve early disease detection, and enable non-invasive monitoring of disease progression.
5.2. The Need for a Profile of Biomarkers of Different Types
The need for new meningioma biomarker discovery is underscored by the complex WHO histological diagnostic criteria and the varied morphological characteristics of meningioma subtypes. The complexity is most prominent in WHO grade II tumors, where inter-observer discrepancy can reach 12.2%, as opposed to 7% in grade I and 6.4% in grade III tumors [89,90]. Grade II tumors can behave biologically similarly to grade I or III tumors with unexpected clinical outcomes due to their very diverse histological characteristics [26,91]. Furthermore, grade I meningiomas that are clinically aggressive can also have clinical outcomes resembling those of grade II tumors [92]. These uncertainties make it clear that imaging and classical histological techniques alone cannot be used to predict the prognosis and clinical course of meningiomas and further highlight the need for the discovery of novel meningioma biomarkers. These novel biomarkers can assist in the diagnosis, management, and prognosis of meningiomas given the growing emphasis on an integrated molecular approach to diagnosing CNS tumors [93,94]. Currently, there is a lack of non-invasive meningioma diagnostic or prognostic biomarkers. These biomarkers may have an impact on the early detection of meningiomas, patient management, and clinical outcomes [95,96] .
Proteomics, metabolomics, epigenomics, RNA sequencing (RNA-seq), and single cell RNA-seq (scRNA-seq) are emerging approaches that have aided in the discovery of new biomarkers for several diseases and ailments. These biomarkers include specific molecules, genetic variations, or imaging characteristics that are associated with the presence, severity, or progression of diseases. They may offer an opportunity to develop more accurate diagnostic tests, predict treatment responses, identify therapeutic targets, and monitor disease progression in a non-invasive manner. Marastoni and Barresi have most recently reviewed the potential of these emerging technologies in comparison to histopathological markers and WHO grading. They compared meningioma grading based on meningioma methylation status in several studies and concluded that DNA methylation profiles are more accurate predictors of meningioma prognosis than the WHO grading system [45]. In this regard, Kishida et al. first reported that recurrent meningiomas have a greater number of methylated genes in comparison with nonrecurrent meningiomas, indicating the prognostic potential of DNA methylation profiles in meningioma grading [97]. Later, Olar et al. reported that among a training cohort of 89 tumors and a validation set of 51 tumors, prognostically unfavorable high grade meningiomas have more methylated genes, chromosomal CNVs, and shorter recurrence-free survival than prognostically favorable low grade meningiomas [98]. Sahm et al. generated genome-wide DNA methylation profiles of 497 meningioma samples and concluded that DNA methylation profiling could distinguish six different clinically relevant methylation classes that also showed difference in mutational, cytogenetic, and gene expression patterns. They also indicated that classification according to these 6 methylation classes was more accurate than 2016 WHO grading at defining WHO Grade I meningiomas at high risk of progression, and WHO grade II meningiomas at lower risk of recurrence [99]. Nevertheless, the higher prognostic values of DNA methylation profiles has not been applied in routine diagnosis, due to high cost and the requirement of complex technologies [45].
To build on the success of meningioma grading using a combination of DNA methylation patterns and genetic alterations, an integrated molecular–morphological grading approach for meningioma grading was employed [45]. Maas et al. developed an integrated meningioma grading system based on following determinants: 2016 WHO grade, combined classes of DNA methylation patterns, genetic mutations, and chromosomal copy number changes of chromosomes 1p, 6q, and 14q. A score was given to each of the determinant. The minimal score of all determinants was 0 and the maximal score was 9 and a score of 0–2 indicated low risk, a score of 3–5 indicated intermediate risk, and a score of 6–9 indicated high risk meningiomas. The integrated grading system was superior at predicting recurrence risk of meningiomas than 2016 WHO grading, combined methylation classes, or chromosomal copy number changes, when validated in a set of 471 meningiomas [100]. Relatedly, Driver et al. designed another integrated grading scheme incorporating mitotic count and loss of chromosome 1p, 3p, 4, 6, 10, 14q, 18, 19, or CDKN2A was also shown to to more accurately identify meningiomas PFS and risk for recurrence, relative to WHO grading [101].
More recent studies have demonstrated that the best approach distinguish between three biologically distinct categories of meningiomas is to use an integrated molecular grading scheme by combining data from different kinds of biomarkers including somatic DNA point mutations, DNA methylation classes, transcriptomics, RNA-seq, and chromosomal instability (CIN)/cytogenetics [41-43,61]. Patel et al. studied 160 meningiomas covering the spectrum of the three WHO categories were subtyped using whole-exome sequencing (WES), RNA-seq, and cytogenetics [41]. Three types were delineated: Type A rarely recurring malignancies that carry mutations in TRAF7, AKT1, or KLF4 but do not exhibit chromosomal deletions; type B meningiomas that lack the chromatin-modifying enzyme PRC2 and are deficient in the NF2/Merlin protein; and type C, which is both NF2-deficient and marked by CIN, notably loss of chromosome 1p, and this type has worse recurrence rates [41,43]. Additionally, Nassiri et al. identified integrative molecular groupings using a multi-omics method by incorporating an investigation of somatic DNA point mutations, DNA methylation, mRNA levels, and somatic chromosomal copy-number aberrations [42,59]. Interestingly, they discovered four molecular clusters that, in contrast to WHO grading, independently correlated with recurrence-free survival and offered more accurate predictions of time to recurrence than WHO grading [42,59]. In confirmation, Choudhury et al. profiled 565 meningiomas and combined DNA methylation patterns with genetic, transcriptomic, biochemical, proteomic, and single-cell analyses and obtained similar results, showing that meningiomas exhibit three DNA methylation classes with different clinical outcomes, biological drivers and therapeutic vulnerabilities [61]. In this study, meningiomas segregated into Merlin-intact meningiomas (34%, best clinical outcomes and response to cytotoxic drugs, owing to the apoptotic function of the intact Merlin protein), immune-enriched meningiomas (38%, have intermediate prognosis, are distinguished by immune cell infiltration, HLA expression and lymphatic vessels, and have 22q loss and inactivation of NF2), and hypermitotic meningiomas (28%, have the worst prognosis, high aneuploidy with frequent chromosomal losses, loss of CDKN2A/B, hypermethylation, and resistance to cytotoxic drugs) [61]. Comparative genome hybridization was also used for the identification of chromosome 1p loss in radiation-induced meningioma, a less prevalent late danger of cranial irradiation which has a higher recurrence rate and pathologically malignant characteristics than sporadic meningioma [102]. A study of 31 meningioma cases, using exome, epigenome, and RNA-seq analyses, revealed the presence of NF2 rearrangements in radiation-induced meningioma, and this may be utilized to differentiate this type of meningioma from sporadic ones [103]. One study developed a meningioma progression score (MPscore) to quantify the likelihood of progression in meningioma and generalize this discriminative ability [104]. Accordingly, the MPscore served as a reliable surrogate for subtype III meningioma advancement, conveying that MPscore of subtype III was considerably higher than the MPscores of other subtypes [104]; hence, the meningioma recurrence-free survival rate and MPscore were highly correlated. It may be possible to create significant phenotypic meningioma profiles using non-invasive analysis to forecast tumor genetics and behavior. These profiles can then be used to guide non-invasive treatment and management decisions. Wang et al. pioneered the use of scRNA-seq analysis to study immune and non-immune cell types in tissues from non-tumor-associated dura versus primary meningioma tumor tissues of patients, revealing that the human dura has a complex immune microenvironment that is transcriptionally different from that of meningioma [105]. One pilot study integrated machine learning methods with bioinformatics techniques to categorize glioblastoma (GBM) subtypes associated with bevacizumab responsiveness based on existing miRNA profiling datasets [106]. This lays out new strategies that may be applied in meningioma biomarker identification to help classify, monitor, and provide therapeutic decisions in meningioma tumors. A newer emerging non-invasive methodology employed a zinc oxide nanowire-based device that can be used to extract a substantially higher diversity and quantity of miRNAs from urine, suggesting that urinary miRNA profiles are suitable for noninvasive CNS tumor mass screening since urinary miRNA expression has been correlated with the incidence of certain tumors [107].
Ongoing research in meningioma biomarker identification aims to integrate all these emerging molecular approaches to define an integrative set of new biomarkers that can non-invasively diagnose meningioma and stratify the different subtypes of meningioma. This can serve for a better prognosis of meningioma and the discovery of new therapeutic targets. Overall, the new integrated molecular approaches [41-43,61] have higher accuracy in predicting prognosis and risk of recurrence than 2016 or 2021 WHO grading systems or methylation-based classifications [45]. Based on these new integrated meningioma grading approaches, Marastoni and Barresi conclude their review by defining three meningioma classes which can complement WHO grading for the prediction of prognosis. Group 1 meningiomas have the best prognosis, are free of NF-2 mutations and chromosomal instability; may include AKT1, TRAF7, or KLF4 mutations, and are predicted good responses to cytotoxic therapies. Group 2 meningiomas have intermediate prognosis, NF-2 inactivation, and are free of chromosomal instabilities and enriched in immune cells. Group 3 meningiomas have worst prognosis and high chromosomal instability and proliferation indices, show resistance to cytotoxic therapies, and may have pTERT mutations and/or CDKN2A/B deletion. Although these new classifications were not part of the 2021 WHO meningioma grading, they are expected to guide meningioma grading in the near future. Application of these new grading schemes in clinical practice may face difficulties, but new proteomic studies have indicated that meningiomas may be classified may be stratified using specific immunostaining targets that can replace the need for sophisticated methods like profiling of DNA methylation or RNA-Seq [45].
5.3. Exploring Protein Biomarkers as Meningioma Biomarkers
A panel of meningioma biomarkers incorporating proteomics may be able to predict aggressive meningiomas with a high risk of metastasis or recurrence. However, challenges of identifying proteomics-based predictive, prognostic, and monitoring biomarkers go beyond detection of the prevalence of the disease and must in addition consider the type of targeted therapy, response rates to therapy, and time to event analysis, including progression free survival and mortality [108]. For future research, overcoming these biological and technical difficulties is essential, and should be considered throughout the design phase of discovery, during biomarker development, and should be confirmed using distinct validation cohorts [108]. Interestingly, protein-based diagnostic biomarkers may be used as theranostic biomarkers where the protein biomarker is combined with therapeutic agents, such as radioactive compounds [109]. For example, somatostatin receptor subtype 2 (SSTR2) mRNA is overexpressed by all subtypes of meningiomas; therefore, somatostatin peptide analogues (SSTa) have been labeled by different radionuclides for the detection of meningioma using positron emission tomography (PET) imaging as well as therapy, that has been termed targeted peptide receptor radionuclide therapy (PRRT). Using PRRT with SSTa, Saglues et al. were able to prolong the 6-month progression-free survival of progressive refractory WHO grade I and II meningiomas, but not aggressive WHO grade II tumors [110]. Another study reported that prostate-specific membrane antigen (PSA) protein expression increases as meningiomas progress in grade or as a result of recurrence and that 98.9% of 91 included meningioma samples express PSA in endothelial cells. The study proposed PSA as a potential theranostic marker of meningioma [111]. Large-scale randomized trials are needed for the transformation of potential theranostic biomarkers into clinical practice guidelines.
a. Serum Protein Biomarkers
There are no blood biomarkers that currently exist for meningioma, and the discovery of non-invasive protein biomarkers in the serum of patients is a major area of interest in meningioma diagnosis. A serum biomarker can be any substance that changes measurably in the serum as a tumor develops [112], hence it should be able to detect the presence of meningiomas and determine their grades and subtypes. Typically, these biomarkers should be highly expressed on the surface of circulating malignant cells, or shed into the blood stream by tumor cells [112]. Using an immunoassay-based detection, it was shown that a panel of seven serum proteins (caspase-3, CD69, prolactin, epidermal growth factor (EGF), chemokine (C-C motif) ligand 24 (CCL24), amphiregulin (AREG), and heparin-binding EGF (HB-EGF)) were strongly expressed in grade I meningioma samples, with caspase-3 emerging as the highest differentially expressed protein [113]; however, vascular endothelial growth factor D (VEGFD), transforming growth factor (TGF-α), E-Selectin, B-cell activating factor (BAFF), interleukin-12 (IL-12), chemokine CCL9, and growth hormone (GH) levels were downregulated [113]. This coincides with the results of a previous study that reported elevated caspase3 immunoreactivity in grade II and grade III meningioma tissues and proposed caspase 3 as an independent unique predictor of early recurrence [114]. Meningiomas have been linked to the activation of complement cascades by increasing the expression of a few complement (C) components, including C5, C8 beta chain, C6, and C4-B [115]. Particularly, C3, a key protein in tumorigenesis of meningiomas, was found to be down regulated in grade II Meningioma when compared to grade I [116]. Moreover, elevated levels of proteins involved in blood coagulation and hemostasis, such as antithrombin-III, alpha-2-antiplasmin, vitamin K-dependent protein S, fibrinogen alpha chain, plasminogen, alpha-2-macroglobulin, and coagulation factor XII, were associated with different grades of meningioma [115].
Hypoxia markers in serum can be potentially used in the diagnosis of meningioma. Hypoxia is a common feature of many malignant neoplasms. In hypoxia, the transcription factor hypoxia-inducible factor 1 (HIF-1) binds to hypoxia response elements (HREs) and regulates the expression of hypoxia-responsive genes, thereby coordinating many of the responses to hypoxic stress. HIF-1 target genes include the angiogenic factor VEGF, erythropoietin (EPO), glucose transporter-1 (GLUT1), and several glycolytic enzymes which contain HREs in their promoter or enhancer regions [117]. In a study by El-Benhawy et al., serum levels of hypoxia markers HIF-1α, VEGF, and lactate dehydrogenase (LDH) were considerably decreased after radiotherapy in meningioma patients [118]. Previous studies have demonstrated that acidic pH increases angiogenesis and migration of glioma stem cells by activating glioma stem cell markers [119]. This opens the question of whether elevated LDH levels and acidic pH could also be related to meningioma progression. According to another study, the expression of the endogenous hypoxia marker carbonic anhydrase 9 was highly expressed in more than 50% (29 of 62) of the included meningioma patients, had an expression that was substantially related with higher-grade histology and was prevalent in recurrent tumors [120].
Endocan is another potential serum biomarker of meningioma. Endocan serum levels were found to vary in relation to meningioma grade; the higher the meningioma grade, the higher the endocan serum levels [121]. These results confirm results of a previous study that tested the levels of endocan in glioma and meningioma brain tumors and concluded that the levels of endocan are increased in tumors of glioma and meningioma patients and the amount of increase correlated with the degree of malignancy [122].
b. Cerebrospinal Fluid Protein Biomarkers
The blood-brain barrier prevents brain tumor-specific molecules from being released into blood circulation, and this limits the number of biomarkers in serum of CNS tumors [123]. As a result, cerebral spinal fluid (CSF) has been investigated for its potential use in the diagnosis of brain tumors [123]. Indeed, oncologists clinically use CSF protein biomarkers because of their utility not only in diagnosis but also in the treatment and evaluation of recurrent malignancies [124]. Brain ventricles are filled with CSF, which also encircles the brain and bone marrow in the subarachnoid space [124], so it is directly in contact with the extracellular environment of the CNS. Hence, CSF cytology is amenable to collection, and lumbar puncture is a non-invasive way of collecting CSF [124]. In one investigation, two dimensional (2D) gel electrophoresis and mass spectrometry (MS) analysis of CSF samples allowed the identification of upregulated meningioma-specific CSF proteins. The upregulated proteins included apolipoprotein E (APO-E), alpha-1-antitrypsin, and prostaglandin synthases (Table 1) [124,125]. APO-E is present in normal human tissue as well as intracranial neoplasms, and APO-J has anti-amyloidogenic function, acting as a prominent carrier protein of soluble circulating amyloids in bodily fluids. Both APO-E and APO-J are considered as potential CSF biomarkers for detection of meningiomas [124]. Notably, a recent study measured the level of three APOE peptides (SELEEQLTPVAEETR, LGPLVEQGR, and AATVGSLAGQPLQER) in meningioma CSF samples, and the results indicated a 2.21-fold increase of APO-E in grade II as compared to grade I meningioma [116]. On the other hand, ApoA-I, a multifunctional protein involved in regulating immune responses as well as cholesterol transport [126], was downregulated in meningioma grade II tissue compared to meningioma grade I [116]. Additionally, prostaglandin H2 D-isomerase (PTGDS) has been proposed as a potential biomarker of meningioma. Kim et al. reported that CSF of meningioma patients had reduced PTGDS expression [124], and a recent study validated that PTGDS had considerably higher expression in grade I meningioma than in grade II [116]. In the CSF of children with medulloblastoma, another CNS tumor, total prostaglandin D2 synthase levels were reduced by six times, most likely as a result of the host reaction to the presence of the tumor [127]. This sheds light on CSF prostaglandin D2 synthase that could be tested as a potential biomarker of meningioma.
EGF-containing fibulin-like extracellular matrix protein 1 (EFEMP1) levels in CSF of meningioma patients were considerably higher compared to controls (Table 1) [128]. Similarly, CSF levels of carcinoembryonic antigen (CEA), a protein tumor marker that is frequently elevated in a number of human malignancies, can be used for diagnosing of primary and metastatic brain tumors including meningeal carcinomas (Table 1) [123]. A previous investigation reported on the concentrations of three tumor markers CEA, cytokeratin 19 fragments (CYFRA 21-1), and neuron-specific enolase (NSE) in CSF of 35 lung cancer patients with meningeal carcinomatosis of lung cancer and 35 patients with benign brain tumors [129]. The three markers were significantly higher in serum and CSF of the meningeal carcinomatosis than in the group with benign disease [129].
5.4. LncRNA and miRNA in Diagnosis and Prognosis of Meningiomas
microRNAs (miRNAs) are short non-coding RNAs that suppress the translation of proteins, typically by binding to the 3′ untranslated regions (3’UTR) of target mRNAs [130]. Their transcription is deregulated in several malignancies and many miRNAs have been recognized as disease biomarkers [130]. Meningiomas exihibit increased expression levels miR-451, miR-711, and miR-935 (Table 1) [128]. Circulating miRNAs have been identified in CSF [131]. Zhi et al. compared miRNA expression profiles of 200 miRNAs between 110 meningioma tumors and 35 “normal” adjacent tissue samples [132]. Three novel miRNAs- miR-29c-3p, miR-219-5p, and miR-190a- were proposed as potential prognostic meningioma indicators (Table 1). Advanced clinical stages of meningioma were associated with downregulation of miR-29c-3p and miR-219-5p and an upregulation of miR-190a. These miRNAs were also strongly linked with elevated meningioma recurrence rates, suggesting the utility of these miRNAs in predicting recurrence [132]. In a different study, down regulation of miR-331-3p combined with partial resection of meningioma were found to be the most significant predictive biomarkers. Indeed, miR-331-3p predictive power superseded that of miR-15a-5p (P=0.038), miR-146a-5p (P=0.053), and miR-331-3p (P=0.09), in an enlarged patient cohort [133]. Moreover, Zhi et al. examined the expression of 200 microRNAs in meningioma cells and discovered that miR-17-5p, miR-199a, miR-190a, miR-186-5p, miR-155-5p, miR-22-3p, miR-24-3p, miR- 26b-5p, mmiR-27a-3p, miR-27b-3p, miR-96-5p, and miR-146a-5p were significantly upregulated in meningioma cells and acted as oncogenic factors, while miR-29c-3p and miR-219-5p were significantly downregulated in meningioma cells [134]. Particularly, miR-21 [135], as well as miR-219-5p [136], enable the distinction of the primary meningioma histological types, with their expression positively correlated with the clinical stages of meningioma [135,136]. Similarly, the serum levels of miRNA in meningioma patients was examined and miR-106a-5p, miR-219-5p, miR-375, and miR-409-3p were significantly increased, whereas the serum levels of miR-197 and miR-224 were markedly decreased [136]. In a study on tissue samples from 55 patients with atypical meningiomas (43 from a radiosensitive group and 12 from a radioresistant group), there were seven significantly upregulated miRNAs (miR-4286, miR-4695-5p, miR-6732-5p, miR-6855-5p, miR-7977, miR-6765-3p, miR-6787-5p); while seven miRNAs were significantly downregulated (miR-1275, miR-30c-1-3p, miR-4449, miR-4539, miR-4684-3p, miR-6129, miR-6891-5p) in patients resistant to radiotherapy [135]. In a different study, miR-181d expression was found to be higher in meningiomas, and this increase in expression was more pronounced in correlation with the advancement of tumor grade [137]. On the other hand miR-200a, exhibited much lower expression levels in recurrent meningiomas than in initially diagnosed ones [138].
Extracellular vesicles (EVs) are nano-sized, lipid bilayer-enclosed structures released by all living cells. EVs cargo includes bioactive molecules, like nucleic acids, proteins, lipids, and metabolites. EVs mediate cell-cell communication and have been shown to have physiologically essential functions as well as pathology-related processes such as in cancer and during viral infection [139]. EVs cargoes have been proposed as biomarkers of different diseases, including CNS tumors [128]. EVs were also shown to exist in serum as well as CSF [128,140]. The transcription factor GATA-4 was reported to be overexpressed in malignant meningiomas, where it negatively regulates the expression of miR-497-195 cluster and maintains cell viability [135,141]. miR-497 levels were found to be reduced in serum EVs derived from patients with high-grade compared to benign meningioma, due to overexpression of GATA-4 in these tumors [141]. Future research is needed to examine the clinical implications of EVs miR-497 in the resistance to treatment exihibited by high-grade meningioma. These studies also suggest the possibility of using transcription factors and their target miRNAs as new tissue-specific biomarkers for higher-grade meningiomas. Finally, future research should investigate CSF as well as serum EVs and their cargoes as non-invasive biomarkers of meningioma. In this regard, Ricklefs et al. have recently demonstrated the diagnostic potential of plasma EVs and indicated that DNA carried by EVs reflects the methylation profiles, mutations, and copy number variations of the meningioma .cells from which they asre derived [142].
Malignant meningiomas have been shown to be significantly regulated by long non-coding RNAs (lncRNAs). LncRNAs are non-coding genes whose transcripts are more than 200 nucleotides [143]. LncRNAs can bind chromatin, attract protein complexes to modify chromatin states, and subsequently control gene expression [144]. In one instance, LncRNAs can control miRNA function by acting as endogenous miRNA sponges to inhibit miRNA function and consequently block the silencing of miRNA target genes [145]. Differential profiling of patients with different meningioma grades and recurrence revealed that mRNA levels of Immunoglobulin superfamily containing leucine rich repeat 2 (ISLR2), anti-mullerian hormone (AMH), and LncRNA-GOLGA6A-1 exhibited the highest prognostic power to predict meningioma recurrence (Table 1) [146]. Interestingly, ISLR2, AMH, and LncRNA-GOLGA6A-1 transcription is controlled by several transcription factors including KLF4 which is linked to activating mutations of meningiomas [146]. Invasive meningioma associated transcript 1 (IMAT1) is a LncRNA which was shown to be expressed more strongly in invasive than non-invasive meningiomas [145]. IMAT1 overexpression significantly increased proliferation and invasion of human meningioma cells expressing KLF4. On the other hand, IMAT1 knockout had the opposite effect, suggesting that IMAT1 lncRNA can severely reduce KLF4 anti-tumor effects [145]. Li et al. found that, in malignant meningioma, lncRNA-LINC00702 can operate as an oncogene by controlling the miR-4652-3p/ZEB1 axis and activating the WNT/-CATENIN signaling pathway [147]. Further research was conducted by Xing et al. [148] who discovered that lncRNA-LINC00460 was highly expressed in meningiomas, and increased meningioma metastasis and progression via binding to microRNA-539/MMP-9. Additionally, other findings showed that maternally expressed gene 3 (MEG3), a well-known lncRNA, was significantly down-regulated in meningioma tissues and cells, acting as a tumor suppressor and decreasing the expression of A-kinase anchor protein 12 (AKAP12) by targeting miR-29c to suppress cell-cycle, migration, invasion, and proliferation in vitro [149]. Other LncRNAs such as LncRNA-NUP210, LncRNA-SPIRE2, LncRNA-SLC7A1, and LncRNA-DMTN were upregulated in meningioma [134].
6. Animal Models for Discovery of Meningioma Biomarkers
The development of several mouse models of meningioma has immensely benefited the field of meningioma research. Such in vivo models have provided a better understanding of the underlying biological mechanisms of meningioma pathology. Relatedly, these models were employed as tools for the discovery of various biomarkers that are altered in meningiomas. The first mouse model to be developed for meningioma research was the heterotopic xenograft mouse model [150]. In this model, human immortalized cell lines or patient-derived tumor cells (glioblastoma or meningioma) are injected subcutaneously into mice. Mixing a basement membrane protein mixture (Matrigel) with meningioma cells prior to injection has proven to increase the success rates of tumor development in mice [151]. The resulting tumors exhibit both immunohistochemical and histological features which are consistent with meningiomas. However, they lack the key components of the meningioma-specific microenvironment including the CSF, bone, arachnoid, and the brain. The orthotopic xenograft model overcomes this limitation through injection of the meningioma cells intracranially into immunocompromised mice. McCutcheon et al. established the first meningioma orthotopic xenograft model, using the IOMM-Lee meningioma cell line and first passage primary cell cultures [152]. Previous studies have described the usage of a wide variety of injection sites and volumes as well as different cell types and numbers during xenografting [153]. The utilization of atypical and malignant meningioma cell lines resulted in very high tumor take rates with almost all immunocompromised mice developing tumors post-injection [153]. Immortalized benign meningioma cell lines produced more heterogenous results, with tumor takes that ranged between 55% and 100% [154]. Closely monitoring tumor take and growth is done in a simpler manner with heterotopic mice models as compared to orthotopic models. Currently, imaging using small-animal MRI [155] and bioluminescence-based methods [156] are the two main techniques being utilized for tumor monitoring in orthotopic models. It is noteworthy that small-animal MRI is expensive and lacks ready availability.
The anatomy, histology, and genetic driver events in an animal model of a tumor should ideally closely mimic the human tumor. Additionally, the ability to manipulate tumor initiation from different temporal and spatial perspectives is key for the successful establishment of a tumor model. Genetically engineered mouse models (GEMMs) facilitate these features by allowing researchers to extensively edit and manipulate genes [157]. Using the Cre-loxP system in GEMMs allows site-specific DNA modifications such as insertions, deletions, and translocations and has been extensively used in meningiomas research following advances in the molecular analysis of human meningiomas [153]. Second generation GEMMs used for meningioma research introduced modifications to promoter of the prostaglandin-D2-synthase (PGDS) gene to establish meningiomas in mice [158]. In the CNS, PGDS is responsible for prostaglandin D2 biosynthesis and was identified as a marker of meningeal cells in rats, mice, and humans [159-161]. Another system used to establish GEMMs is the RCAS-TVA gene delivery system, which is popular for modeling human cancer [162]. Overexpression of the platelet-derived growth factor (PDGF) in arachnoïdal cells using the RCAS-TVA system leads to meningioma development independently of Nf2 mutations [66]. PDGF overexpression combined with the presence of Nf2 mutations and the additional loss of Cdkn2ab was shown to induce malignant progression in this model [158]. New GEMMs are needed to improve our understanding of the biological mechanisms involved in meningioma tumorigenesis. This will facilitate preclinical drug evaluation as well as the discovery of new specific meningeal markers. The advantages and limitations of the mentioned meningioma pre-clinical models which can be utilized for biomarkers research are presented in Table 2.
GEMMs were utilized by Kalamarides et al. to demonstrate that the excision of Nf2 exon 2 in arachnoïdal cells is rate-limiting for meningioma development in mice where 30% of mice developed meningiomas [163]. Meningiomas appeared in mice at four months of age and were histologically similar to human ones. It was also reported that Nf2 and p53 mutations do not synergize in meningeal tumorigenesis, since disease frequency or progression were not affected with additional p53 hemizygosity. In a follow-up study, the same authors reported that meningothelial proliferation and meningioma frequency were increased, without variations in the tumor grade, in mice nullizygous for the tumor suppressor p16 (Ink4a), revealing a synergy between Nf2 and p16 inactivation in meningioma development [164]. Another genetic study revealed that meningioma progression in mice was facilitated by a cooperation between Nf2 and cdkn2ab [165]. Deleting cdKn2ab was associated with shorter latency and an elevated frequency of meningiomas in mice [165].
Interestingly, Mandara et al. investigated steroid receptors in canine and feline meningiomas and revealed that among nine meningiomas from dogs and five from cats that were examined utilizing immunohistochemistry, meningiomas with a high proliferation index exhibited the lowest levels of progesterone receptor (PR) expression [166]. Alterations in estrogen receptor expression were not significant in the investigated samples [166]. In a xenograft mouse model, it was found that PR expression was dependent on the cell-line utilized for injection [167]. In both heterotopic and orthotopic approaches, transplantation of low-passage patient-derived tumor cells formed meningiomas positive for PR and vimentin. However, subcutaneous injection of high-passage cells yielded PR-negative and vimentin-positive tumors, consistent with high-grade meningiomas [167]. An in vivo study utilizing a heterotopic xenograft mouse model demonstrated that FoxM1 is a key transcription factor and oncogenic driver in meningioma progression [168]. The authors injected OMM-Lee cells in control nude mice and in nude mice pre-treated with siomycin A, a FoxM1 inhibitor. Inhibition of FoxM1 resulted in the formation of significantly smaller tumors. Moreover, the knock down of FOXM1 in meningiomas decreased the number of β CATENIN-expressing and Ki67-positive proliferating tumor cells [168]. However, overexpressing FOXM1 in transplanted benign meningioma cell-lines failed to produce tumors in mice, suggesting that FOXM1 alone was insufficient to drive meningioma growth in vivo [168]. The heterotopic xenograft mouse model was also used to explore the role of miR-200a in meningioma tumors growth [169]. Subcutaneous injection of meningioma cell line SF4433-Fluc overexpressing miR-200a into athymic mice resulted in an increase in caspase 3/7 activity and apoptosis of the injected cells. Almost all mice that received cells transfected with miR-200a developed tumors that failed to grow or that exhibited a marked reduction in size, indicating that miR-200a blunted the ability of meningioma cells to form tumors [169]. Tuchen et al. employed an orthotopic xenograft mouse model to assess the role of receptor tyrosine kinases (RTKs) in meningioma progression [170]. Using sorafenib and regorafenib RTKs inhibitors which target the phosphorylation of p44/42 ERK through the downregulation of the PDGFR. Monitoring tumor growth using small-animal MRI revealed that inhibition of RTKs inhibited growth and invasion of meningioma cells [170]. The availability of several mouse meningioma models represents a tool that can be exploited for further advances in meningioma biomarker discovery. Despite several limitations (Table 2), these pre-clinical models are continuously being optimized to enhance meningioma research. For example, the CRISPR-Cas 9 technology seems promising for next-generation mouse models of meningioma. Moreover, it is relevant to develop new GEMMs that explore targetable somatic mutations found in human meningiomas such as TRAF7, AKT1, and PIK3CA among others.
7. Conclusions
Meningiomas are the most prevalent primary intracranial tumors, accounting for 36% of all CNS tumors. There are various types and subtypes of meningiomas, bestowing them with a wide heterogeneity and complicating diagnosis. In addition, there are overlapping characteristics between benign and malignant subtypes, necessitating the presence of fast and effective diagnostic biomarkers.
Currently, there are no viable indicators of diagnosis, prognosis, or management of these tumors. Radiological imaging, mainly CT and MRI, is the main method of meningioma diagnosis. Unfortunately, imaging is not always suitable since it requires a big tumor size, presents overlapping findings with other CNS tumors, and needs continuous radiation exposure for follow-up. Imaging cannot anticipate the clinical behavior of meningiomas. All these limitations in current methods of diagnosis and prognosis necessitate the development of new meningioma biomarkers.
Currently, WHO classification of meningiomas is based on histopathology; however, due to the heterogeneity of meningiomas, likely, the future diagnosis, prognosis, and therapy of meningiomas will likely be based on a multi-omics approach by combining genomics, proteomics, and epigenetic landscapes.
NF2 gene mutations have been used as potential meningioma biomarkers, but proteomic-based biomarkers are better suited to accommodate meningioma diversity. Several prospective biomarkers are currently being researched such as serum protein expression patterns, CSF proteins, miRNA, and lncRNA. Furthermore, the use of the available meningioma animal models will facilitate the discovery of new tumor meningioma biomarkers. The ultimate diagnosis of meningioma may require a panel of biomarkers of different types to cope with the heterogeneity of this disease. Such biomarkers when available will lead to fast and accurate stratification and grading of the different meningioma subtypes, and enhance pre-operative and post-operative decision-making. Importantly, these biomarkers may offer new targets for the development of new meningioma therapies including theranostic meningioma therapies.
Funding
This research is funded by a grant from Morehouse School of Medicine to Firas Kobeissy and a grant from Qatar University to Abdullah A. Shaito.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gittleman, H.R.; Ostrom, Q.T.; Rouse, C.D.; Dowling, J.A.; de Blank, P.M.; Kruchko, C.A.; Elder, J.B.; Rosenfeld, S.S.; Selman, W.R.; Sloan, A.E.; et al. Trends in central nervous system tumor incidence relative to other common cancers in adults, adolescents, and children in the United States, 2000 to 2010. Cancer 2015, 121, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Vecchione-Koval, T.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro-Oncology 2017, 19, v1–v88. [Google Scholar] [CrossRef] [PubMed]
- Flint-Richter, P.; Mandelzweig, L.; Oberman, B.; Sadetzki, S. Possible interaction between ionizing radiation, smoking, and gender in the causation of meningioma. Neuro-Oncology 2011, 13, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.; Pülhorn, H.; Röhrig, B.; Rainov, N.G. Predisposing conditions and risk factors for development of symptomatic meningioma in adults. Cancer Detection and Prevention 2005, 29, 440–447. [Google Scholar] [CrossRef]
- Achey, R.L.; Gittleman, H.; Schroer, J.; Khanna, V.; Kruchko, C.; Barnholtz-Sloan, J.S. Nonmalignant and malignant meningioma incidence and survival in the elderly, 2005–2015, using the Central Brain Tumor Registry of the United States. Neuro-Oncology 2019, 21, 380–391. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathologica 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Yang, S.Y.; Park, C.K.; Park, S.H.; Kim, D.G.; Chung, Y.S.; Jung, H.W. Atypical and anaplastic meningiomas: prognostic implications of clinicopathological features. J Neurol Neurosurg Psychiatry 2008, 79, 574–580. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Loewenstern, J.; Rutland, J.; Gill, C.; Arib, H.; Pain, M.; Umphlett, M.; Kinoshita, Y.; McBride, R.; Donovan, M.; Sebra, R.; et al. Comparative genomic analysis of driver mutations in matched primary and recurrent meningiomas. Oncotarget 2019, 10, 3506–3517. [Google Scholar] [CrossRef]
- Dumanski, J.P.; Carlbom, E.; Collins, V.P.; Nordenskjöld, M. Deletion mapping of a locus on human chromosome 22 involved in the oncogenesis of meningioma. Proceedings of the National Academy of Sciences 1987, 84, 9275–9279. [Google Scholar] [CrossRef]
- Schneider, G.; Lutz, S.; Henn, W.; Zang, K.D.; Blin, N. Search for putative suppressor genes in meningioma: significance of chromosome 22. Human Genetics 1992, 88, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Wellenreuther, R.; Kraus, J.A.; Lenartz, D.; Menon, A.G.; Schramm, J.; Louis, D.N.; Ramesh, V.; Gusella, J.F.; Wiestler, O.D.; von Deimling, A. Analysis of the neurofibromatosis 2 gene reveals molecular variants of meningioma. The American journal of pathology 1995, 146, 827–832. [Google Scholar] [PubMed]
- Gutmann, D.H.; Giordano, M.J.; Fishback, A.S.; Guha, A. Loss of merlin expression in sporadic meningiomas, ependymomas and schwannomas. Neurology 1997, 49, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Ruttledge, M.H.; Sarrazin, J.; Rangaratnam, S.; Phelan, C.M.; Twist, E.; Merel, P.; Delattre, O.; Thomas, G.; Nordenskjöld, M.; Collins, V.P.; et al. Evidence for the complete inactivation of the NF2 gene in the majority of sporadic meningiomas. Nat Genet 1994, 6, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, M.D.; Espinosa, A.B.; Maíllo, A.; Sayagués, J.M.; Alguero, M.d.C.; Lumbreras, E.; Díaz, P.; Gonçalves, J.M.; Onzain, I.; Merino, M.; et al. Characterization of chromosome 14 abnormalities by interphase in situ hybridization and comparative genomic hybridization in 124 meningiomas: correlation with clinical, histopathologic, and prognostic features. American journal of clinical pathology 2005, 123, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Sayagués, J.M.; Tabernero, M.D.; Maíllo, A.; Espinosa, A.; Rasillo, A.; Díaz, P.; Ciudad, J.; López, A.; Merino, M.; Gonçalves, J.M.; et al. Intratumoral Patterns of Clonal Evolution in Meningiomas as Defined by Multicolor Interphase Fluorescence in Situ Hybridization (FISH). The Journal of Molecular Diagnostics 2004, 6, 316–325. [Google Scholar] [CrossRef]
- Strickland, M.R.; Gill, C.M.; Nayyar, N.; D'Andrea, M.R.; Thiede, C.; Juratli, T.A.; Schackert, G.; Borger, D.R.; Santagata, S.; Frosch, M.P.; et al. Targeted sequencing of SMO and AKT1 in anterior skull base meningiomas. Journal of Neurosurgery 2017, 127, 438–444. [Google Scholar] [CrossRef]
- Clark, V.E.; Harmancı, A.S.; Bai, H.; Youngblood, M.W.; Lee, T.I.; Baranoski, J.F.; Ercan-Sencicek, A.G.; Abraham, B.J.; Weintraub, A.S.; Hnisz, D.; et al. Recurrent somatic mutations in POLR2A define a distinct subset of meningiomas. Nature Genetics 2016, 48, 1253–1259. [Google Scholar] [CrossRef]
- Brastianos, P.K.; Horowitz, P.M.; Santagata, S.; Jones, R.T.; McKenna, A.; Getz, G.; Ligon, K.L.; Palescandolo, E.; Van Hummelen, P.; Ducar, M.D.; et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nature Genetics 2013, 45, 285–289. [Google Scholar] [CrossRef]
- Sahm, F.; Bissel, J.; Koelsche, C.; Schweizer, L.; Capper, D.; Reuss, D.; Böhmer, K.; Lass, U.; Göck, T.; Kalis, K.; et al. AKT1E17K mutations cluster with meningothelial and transitional meningiomas and can be detected by SFRP1 immunohistochemistry. Acta Neuropathologica 2013, 126, 757–762. [Google Scholar] [CrossRef]
- Reuss, D.E.; Piro, R.M.; Jones, D.T.W.; Simon, M.; Ketter, R.; Kool, M.; Becker, A.; Sahm, F.; Pusch, S.; Meyer, J.; et al. Secretory meningiomas are defined by combined KLF4 K409Q and TRAF7 mutations. Acta Neuropathologica 2013, 125, 351–358. [Google Scholar] [CrossRef]
- Clark, V.E.; Erson-Omay, E.Z.; Serin, A.; Yin, J.; Cotney, J.; Ozduman, K.; Avsar, T.; Li, J.; Murray, P.B.; Henegariu, O.; et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science 2013, 339, 1077–1080. [Google Scholar] [CrossRef]
- Aldape, K.; Nejad, R.; Louis, D.N.; Zadeh, G. Integrating molecular markers into the World Health Organization classification of CNS tumors: a survey of the neuro-oncology community. Neuro Oncol 2017, 19, 336–344. [Google Scholar] [CrossRef]
- Papaioannou, M.-D.; Djuric, U.; Kao, J.; Karimi, S.; Zadeh, G.; Aldape, K.; Diamandis, P. Proteomic analysis of meningiomas reveals clinically distinct molecular patterns. Neuro-Oncology 2019, 21, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Nassiri, F.; Wang, J.Z.; Singh, O.; Karimi, S.; Dalcourt, T.; Ijad, N.; Pirouzmand, N.; Ng, H.K.; Saladino, A.; Pollo, B.; et al. Loss of H3K27me3 in meningiomas. Neuro Oncol 2021, 23, 1282–1291. [Google Scholar] [CrossRef]
- Katz, L.M.; Hielscher, T.; Liechty, B.; Silverman, J.; Zagzag, D.; Sen, R.; Wu, P.; Golfinos, J.G.; Reuss, D.; Neidert, M.C.; et al. Loss of histone H3K27me3 identifies a subset of meningiomas with increased risk of recurrence. Acta Neuropathologica 2018, 135, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Bello, M.J.; Amiñoso, C.; Lopez-Marin, I.; Arjona, D.; Gonzalez-Gomez, P.; Alonso, M.E.; Lomas, J.; de Campos, J.M.; Kusak, M.E.; Vaquero, J.; et al. DNA methylation of multiple promoter-associated CpG islands in meningiomas: relationship with the allelic status at 1p and 22q. Acta Neuropathol 2004, 108, 413–421. [Google Scholar] [CrossRef]
- Barski, D.; Wolter, M.; Reifenberger, G.; Riemenschneider, M.J. Hypermethylation and transcriptional downregulation of the TIMP3 gene is associated with allelic loss on 22q12.3 and malignancy in meningiomas. Brain Pathol 2010, 20, 623–631. [Google Scholar] [CrossRef]
- Nazem, A.A.; Ruzevick, J.; Ferreira, M.J. Advances in meningioma genomics, proteomics, and epigenetics: insights into biomarker identification and targeted therapies. Oncotarget 2020, 11, 4544–4553. [Google Scholar] [CrossRef]
- Kshettry, V.R.; Ostrom, Q.T.; Kruchko, C.; Al-Mefty, O.; Barnett, G.H.; Barnholtz-Sloan, J.S. Descriptive epidemiology of World Health Organization grades II and III intracranial meningiomas in the United States. Neuro-Oncology 2015, 17, 1166–1173. [Google Scholar] [CrossRef]
- Goldbrunner, R.; Minniti, G.; Preusser, M.; Jenkinson, M.D.; Sallabanda, K.; Houdart, E.; von Deimling, A.; Stavrinou, P.; Lefranc, F.; Lund-Johansen, M.; et al. EANO guidelines for the diagnosis and treatment of meningiomas. The Lancet Oncology 2016, 17, e383–e391. [Google Scholar] [CrossRef] [PubMed]
- Hashiba, T.; Hashimoto, N.; Izumoto, S.; Suzuki, T.; Kagawa, N.; Maruno, M.; Kato, A.; Yoshimine, T. Serial volumetric assessment of the natural history and growth pattern of incidentally discovered meningiomas. Journal of Neurosurgery 2009, 110, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Buerki, R.A.; Horbinski, C.M.; Kruser, T.; Horowitz, P.M.; James, C.D.; Lukas, R.V. An overview of meningiomas. Future Oncology 2018, 14, 2161–2177. [Google Scholar] [CrossRef] [PubMed]
- Chotai, S.; Schwartz, T.H. The Simpson Grading: Is It Still Valid? Cancers (Basel) 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Surov, A.; Gottschling, S.; Bolz, J.; Kornhuber, M.; Alfieri, A.; Holzhausen, H.-J.; Abbas, J.; Kösling, S. Distant metastases in meningioma: an underestimated problem. Journal of Neuro-Oncology 2013, 112, 323–327. [Google Scholar] [CrossRef]
- Enomoto, T.; Aoki, M.; Kouzaki, Y.; Abe, H.; Imamura, N.; Iwasaki, A.; Inoue, T.; Nabeshima, K. WHO Grade I Meningioma Metastasis to the Lung 26 Years after Initial Surgery: A Case Report and Literature Review. NMC Case Report Journal 2019, 6, 125–129. [Google Scholar] [CrossRef]
- Paix, A.; Waissi, W.; Antoni, D.; Adeduntan, R.; Noël, G. Visceral and bone metastases of a WHO grade 2 meningioma: A case report and review of the literature. Cancer/Radiothérapie 2017, 21, 55–59. [Google Scholar] [CrossRef]
- Liu, Y.; Li, J.; Duan, Y.; Ye, Y.; Xiao, L.; Mao, R. Subcutaneous Metastasis of Atypical Meningioma: Case Report and Literature Review. World Neurosurgery 2020, 138, 182–186. [Google Scholar] [CrossRef]
- Tsitsikov, E.N.; Hameed, S.; Tavakol, S.A.; Stephens, T.M.; Tsytsykova, A.V.; Garman, L.; Bi, W.L.; Dunn, I.F. Specific gene expression signatures of low grade meningiomas. Front Oncol 2023, 13, 1126550. [Google Scholar] [CrossRef]
- Harmancı, A.S.; Youngblood, M.W.; Clark, V.E.; Coşkun, S.; Henegariu, O.; Duran, D.; Erson-Omay, E.Z.; Kaulen, L.D.; Lee, T.I.; Abraham, B.J.; et al. Integrated genomic analyses of de novo pathways underlying atypical meningiomas. Nat Commun 2017, 8, 14433. [Google Scholar] [CrossRef]
- Patel, A.J.; Wan, Y.W.; Al-Ouran, R.; Revelli, J.P.; Cardenas, M.F.; Oneissi, M.; Xi, L.; Jalali, A.; Magnotti, J.F.; Muzny, D.M.; et al. Molecular profiling predicts meningioma recurrence and reveals loss of DREAM complex repression in aggressive tumors. Proc Natl Acad Sci U S A 2019, 116, 21715–21726. [Google Scholar] [CrossRef] [PubMed]
- Nassiri, F.; Liu, J.; Patil, V.; Mamatjan, Y.; Wang, J.Z.; Hugh-White, R.; Macklin, A.M.; Khan, S.; Singh, O.; Karimi, S.; et al. A clinically applicable integrative molecular classification of meningiomas. Nature 2021, 597, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Bayley, J.C.t.; Hadley, C.C.; Harmanci, A.O.; Harmanci, A.S.; Klisch, T.J.; Patel, A.J. Multiple approaches converge on three biological subtypes of meningioma and extract new insights from published studies. Sci Adv 2022, 8, eabm6247. [Google Scholar] [CrossRef] [PubMed]
- Torp, S.H.; Solheim, O.; Skjulsvik, A.J. The WHO 2021 Classification of Central Nervous System tumours: a practical update on what neurosurgeons need to know-a minireview. Acta Neurochir (Wien) 2022, 164, 2453–2464. [Google Scholar] [CrossRef]
- Marastoni, E.; Barresi, V. Meningioma Grading beyond Histopathology: Relevance of Epigenetic and Genetic Features to Predict Clinical Outcome. Cancers (Basel) 2023, 15. [Google Scholar] [CrossRef]
- Pawloski, J.A.; Fadel, H.A.; Huang, Y.W.; Lee, I.Y. Genomic Biomarkers of Meningioma: A Focused Review. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Ruttledge, M.H.; Xie, Y.G.; Han, F.Y.; Giovannini, M.; Janson, M.; Fransson, I.; Werelius, B.; Delattre, O.; Thomas, G.; Evans, G.; et al. Physical mapping of the NF2/meningioma region on human chromosome 22q12. Genomics 1994, 19, 52–59. [Google Scholar] [CrossRef]
- Ichimura, K.; Yuasa, Y. [Molecular biological analysis of neurofibromatosis type 2 gene]. Nihon Rinsho 1993, 51, 2462–2466. [Google Scholar]
- Cui, Y.; Ma, L.; Schacke, S.; Yin, J.C.; Hsueh, Y.P.; Jin, H.; Morrison, H. Merlin cooperates with neurofibromin and Spred1 to suppress the Ras-Erk pathway. Hum Mol Genet 2021, 29, 3793–3806. [Google Scholar] [CrossRef]
- Maggio, I.; Franceschi, E.; Tosoni, A.; Nunno, V.D.; Gatto, L.; Lodi, R.; Brandes, A.A. Meningioma: not always a benign tumor. A review of advances in the treatment of meningiomas. CNS Oncol 2021, 10, Cns72. [Google Scholar] [CrossRef]
- Moussalem, C.; Massaad, E.; Minassian, G.B.; Ftouni, L.; Bsat, S.; Houshiemy, M.N.E.; Alomari, S.; Sarieddine, R.; Kobeissy, F.; Omeis, I. Meningioma genomics: a therapeutic challenge for clinicians. J Integr Neurosci 2021, 20, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Hua, L.; Han, T.; Tian, M.; Wang, D.; Tang, H.; Sun, S.; Chen, H.; Cheng, H.; Zhang, T.; et al. The CREB-binding protein inhibitor ICG-001: a promising therapeutic strategy in sporadic meningioma with NF2 mutations. Neurooncol Adv 2020, 2, vdz055. [Google Scholar] [CrossRef] [PubMed]
- Youngblood, M.W.; Miyagishima, D.F.; Jin, L.; Gupte, T.; Li, C.; Duran, D.; Montejo, J.D.; Zhao, A.; Sheth, A.; Tyrtova, E.; et al. Associations of meningioma molecular subgroup and tumor recurrence. Neuro Oncol 2021, 23, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Abedalthagafi, M.; Bi, W.L.; Aizer, A.A.; Merrill, P.H.; Brewster, R.; Agarwalla, P.K.; Listewnik, M.L.; Dias-Santagata, D.; Thorner, A.R.; Van Hummelen, P.; et al. Oncogenic PI3K mutations are as common as AKT1 and SMO mutations in meningioma. Neuro Oncol 2016, 18, 649–655. [Google Scholar] [CrossRef]
- Bi, W.L.; Zhang, M.; Wu, W.W.; Mei, Y.; Dunn, I.F. Meningioma Genomics: Diagnostic, Prognostic, and Therapeutic Applications. Front Surg 2016, 3, 40. [Google Scholar] [CrossRef]
- Zotti, T.; Scudiero, I.; Vito, P.; Stilo, R. The Emerging Role of TRAF7 in Tumor Development. J Cell Physiol 2017, 232, 1233–1238. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Yuan, J.; Deng, Y.; Fan, X.; Shen, J. Emerging role of SWI/SNF complex deficiency as a target of immune checkpoint blockade in human cancers. Oncogenesis 2021, 10, 3. [Google Scholar] [CrossRef]
- Ross, J.P.; Rand, K.N.; Molloy, P.L. Hypomethylation of repeated DNA sequences in cancer. Epigenomics 2010, 2, 245–269. [Google Scholar] [CrossRef]
- Robert, S.M.; Vetsa, S.; Nadar, A.; Vasandani, S.; Youngblood, M.W.; Gorelick, E.; Jin, L.; Marianayagam, N.; Erson-Omay, E.Z.; Günel, M.; et al. The integrated multiomic diagnosis of sporadic meningiomas: a review of its clinical implications. J Neurooncol 2022, 156, 205–214. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, K. Pan-cancer analysis of frequent DNA co-methylation patterns reveals consistent epigenetic landscape changes in multiple cancers. BMC Genomics 2017, 18, 1045. [Google Scholar] [CrossRef]
- Nassiri, F.; Mamatjan, Y.; Suppiah, S.; Badhiwala, J.H.; Mansouri, S.; Karimi, S.; Saarela, O.; Poisson, L.; Gepfner-Tuma, I.; Schittenhelm, J.; et al. DNA methylation profiling to predict recurrence risk in meningioma: development and validation of a nomogram to optimize clinical management. Neuro Oncol 2019, 21, 901–910. [Google Scholar] [CrossRef]
- Singh, J.; Sharma, R.; Shukla, N.; Narwal, P.; Katiyar, A.; Mahajan, S.; Sahu, S.; Garg, A.; Sharma, M.C.; Suri, A.; et al. DNA methylation profiling of meningiomas highlights clinically distinct molecular subgroups. J Neurooncol 2023, 161, 339–356. [Google Scholar] [CrossRef]
- Choudhury, A.; Magill, S.T.; Eaton, C.D.; Prager, B.C.; Chen, W.C.; Cady, M.A.; Seo, K.; Lucas, C.G.; Casey-Clyde, T.J.; Vasudevan, H.N.; et al. Meningioma DNA methylation groups identify biological drivers and therapeutic vulnerabilities. Nat Genet 2022, 54, 649–659. [Google Scholar] [CrossRef]
- Jiang, C.; Song, T.; Li, J.; Ao, F.; Gong, X.; Lu, Y.; Zhang, C.; Chen, L.; Liu, Y.; He, H.; et al. RAS Promotes Proliferation and Resistances to Apoptosis in Meningioma. Mol Neurobiol 2017, 54, 779–787. [Google Scholar] [CrossRef]
- Pecina-Slaus, N. Merlin, the NF2 gene product. Pathol Oncol Res 2013, 19, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Mawrin, C.; Sasse, T.; Kirches, E.; Kropf, S.; Schneider, T.; Grimm, C.; Pambor, C.; Vorwerk, C.K.; Firsching, R.; Lendeckel, U.; et al. Different activation of mitogen-activated protein kinase and Akt signaling is associated with aggressive phenotype of human meningiomas. Clin Cancer Res 2005, 11, 4074–4082. [Google Scholar] [CrossRef] [PubMed]
- G Mougel, G.M. , R Appay, A Querdray, C Roche, A Jijon, I Konstantinova, A Soude, T Graillon, A Barlier. A Hippo signaling pathway is strongly involved in meningioma tumorigenesis Neuro-Oncology 2022, 24. [Google Scholar] [CrossRef]
- Hong, A.W.; Meng, Z.; Plouffe, S.W.; Lin, Z.; Zhang, M.; Guan, K.L. Critical roles of phosphoinositides and NF2 in Hippo pathway regulation. Genes Dev 2020, 34, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Baia, G.S.; Caballero, O.L.; Riggins, G.J. The Hippo signaling pathway and translational opportunities for brain cancers. CNS Oncol 2012, 1, 113–115. [Google Scholar] [CrossRef] [PubMed]
- Sievers, P.; Chiang, J.; Schrimpf, D.; Stichel, D.; Paramasivam, N.; Sill, M.; Gayden, T.; Casalini, B.; Reuss, D.E.; Dalton, J.; et al. YAP1-fusions in pediatric NF2-wildtype meningioma. Acta Neuropathol 2020, 139, 215–218. [Google Scholar] [CrossRef]
- Laraba, L.; Hillson, L.; de Guibert, J.G.; Hewitt, A.; Jaques, M.R.; Tang, T.T.; Post, L.; Ercolano, E.; Rai, G.; Yang, S.M.; et al. Inhibition of YAP/TAZ-driven TEAD activity prevents growth of NF2-null schwannoma and meningioma. Brain 2022. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Wang, Y.; Zhou, C.; Mei, W.; Zeng, C. PI3K/Akt/mTOR Pathway and Its Role in Cancer Therapeutics: Are We Making Headway? Front Oncol 2022, 12, 819128. [Google Scholar] [CrossRef] [PubMed]
- Rong, R.; Tang, X.; Gutmann, D.H.; Ye, K. Neurofibromatosis 2 (NF2) tumor suppressor merlin inhibits phosphatidylinositol 3-kinase through binding to PIKE-L. Proc Natl Acad Sci U S A 2004, 101, 18200–18205. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Karas, P.J.; Hadley, C.C.; Bayley, V.J.; Khan, A.B.; Jalali, A.; Sweeney, A.D.; Klisch, T.J.; Patel, A.J. The Role of Merlin/NF2 Loss in Meningioma Biology. Cancers (Basel) 2019, 11. [Google Scholar] [CrossRef]
- Barresi, V.; Lionti, S.; La Rocca, L.; Caliri, S.; Caffo, M. High p-mTOR expression is associated with recurrence and shorter disease-free survival in atypical meningiomas. Neuropathology 2019, 39, 22–29. [Google Scholar] [CrossRef]
- James, M.F.; Han, S.; Polizzano, C.; Plotkin, S.R.; Manning, B.D.; Stemmer-Rachamimov, A.O.; Gusella, J.F.; Ramesh, V. NF2/merlin is a novel negative regulator of mTOR complex 1, and activation of mTORC1 is associated with meningioma and schwannoma growth. Mol Cell Biol 2009, 29, 4250–4261. [Google Scholar] [CrossRef]
- Shih, K.C.; Chowdhary, S.; Rosenblatt, P.; Weir, A.B., 3rd; Shepard, G.C.; Williams, J.T.; Shastry, M.; Burris, H.A., 3rd; Hainsworth, J.D. A phase II trial of bevacizumab and everolimus as treatment for patients with refractory, progressive intracranial meningioma. J Neurooncol 2016, 129, 281–288. [Google Scholar] [CrossRef]
- Plotkin, S.R.; Kumthekar, P.; Wen, P.Y.; Barker, F.G.; Stemmer-Rachamimov, A.; Beauchamp, R.L.; Jordan, J.T.; Muzikansky, A.; Ramesh, V. Multi-center, single arm phase II study of the dual mTORC1/mTORC2 inhibitor vistusertib for patients with recurrent or progressive grade II-III meningiomas. Journal of Clinical Oncology 2021, 39, 2024–2024. [Google Scholar] [CrossRef]
- Pecina-Slaus, N.; Kafka, A.; Lechpammer, M. Molecular Genetics of Intracranial Meningiomas with Emphasis on Canonical Wnt Signalling. Cancers (Basel) 2016, 8. [Google Scholar] [CrossRef]
- Lau, Y.K.; Murray, L.B.; Houshmandi, S.S.; Xu, Y.; Gutmann, D.H.; Yu, Q. Merlin is a potent inhibitor of glioma growth. Cancer Res 2008, 68, 5733–5742. [Google Scholar] [CrossRef]
- Nowosielski, M.; Galldiks, N.; Iglseder, S.; Kickingereder, P.; von Deimling, A.; Bendszus, M.; Wick, W.; Sahm, F. Diagnostic challenges in meningioma. Neuro-Oncology 2017, 19, 1588–1598. [Google Scholar] [CrossRef] [PubMed]
- Tagle, P.; Villanueva, P.; Torrealba, G.; Huete, I. Intracranial metastasis or meningioma? Surgical Neurology 2002, 58, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Bendszus, M.; Warmuth-Metz, M.; Burger, R.; Klein, R.; Tonn, J.C.; Solymosi, L. Diagnosing dural metastases: the value of 1 H magnetic resonance spectroscopy. Neuroradiology 2001, 43, 285–289. [Google Scholar] [CrossRef]
- Bourekas, E.C.; Wildenhain, P.; Lewin, J.S.; Tarr, R.W.; Dastur, K.J.; Raji, M.R.; Lanzieri, C.F. The dural tail sign revisited. AJNR. American journal of neuroradiology 1995, 16, 1514–1516. [Google Scholar]
- Wilms, G.; Lammens, M.; Marchal, G.; Demaerel, P.; Verplancke, J.; Van Calenbergh, F.; Goffin, J.; Plets, C.; Baert, A.L. Prominent dural enhancement adjacent to nonmeningiomatous malignant lesions on contrast-enhanced MR images. AJNR. American journal of neuroradiology 12.
- Huttner, H.B.; Bergmann, O.; Salehpour, M.; El Cheikh, R.; Nakamura, M.; Tortora, A.; Heinke, P.; Coras, R.; Englund, E.; Eyüpoglu, I.Y.; et al. Meningioma growth dynamics assessed by radiocarbon retrospective birth dating. EBioMedicine 2018, 27, 176–181. [Google Scholar] [CrossRef]
- Narai, A.; Hermann, P.; Auer, T.; Kemenczky, P.; Szalma, J.; Homolya, I.; Somogyi, E.; Vakli, P.; Weiss, B.; Vidnyanszky, Z. Movement-related artefacts (MR-ART) dataset of matched motion-corrupted and clean structural MRI brain scans. Sci Data 2022, 9, 630. [Google Scholar] [CrossRef]
- Usman, M.; Latif, S.; Asim, M.; Lee, B.-D.; Qadir, J. Retrospective Motion Correction in Multishot MRI using Generative Adversarial Network. Scientific Reports 2020, 10, 4786. [Google Scholar] [CrossRef] [PubMed]
- Harter, P.N.; Braun, Y.; Plate, K.H. Classification of meningiomas—advances and controversies. Chinese Clinical Oncology 2017, 6, S2–S2. [Google Scholar] [CrossRef]
- Rogers, C.L.; Perry, A.; Pugh, S.; Vogelbaum, M.A.; Brachman, D.; McMillan, W.; Jenrette, J.; Barani, I.; Shrieve, D.; Sloan, A.; et al. Pathology concordance levels for meningioma classification and grading in NRG Oncology RTOG Trial 0539. Neuro-Oncology 2016, 18, 565–574. [Google Scholar] [CrossRef]
- Zhang, Q.; Jia, G.-J.; Zhang, G.-B.; Wang, L.; Wu, Z.; Jia, W.; Hao, S.-Y.; Ni, M.; Li, D.; Wang, K.; et al. A Logistic Regression Model for Detecting the Presence of Malignant Progression in Atypical Meningiomas. World Neurosurgery 2019, 126, e392–e401. [Google Scholar] [CrossRef]
- Parada, C.A.; Osbun, J.W.; Busald, T.; Karasozen, Y.; Kaur, S.; Shi, M.; Barber, J.; Adidharma, W.; Cimino, P.J.; Pan, C.; et al. Phosphoproteomic and Kinomic Signature of Clinically Aggressive Grade I (1.5) Meningiomas Reveals RB1 Signaling as a Novel Mediator and Biomarker. Clinical Cancer Research 2020, 26, 193–205. [Google Scholar] [CrossRef]
- Deng, J.; Hua, L.; Bian, L.; Chen, H.; Chen, L.; Cheng, H.; Dou, C.; Geng, D.; Hong, T.; Ji, H.; et al. Molecular diagnosis and treatment of meningiomas: an expert consensus (2022). Chinese Medical Journal 2022, 135, 1894–1912. [Google Scholar] [CrossRef]
- Gritsch, S.; Batchelor, T.T.; Gonzalez Castro, L.N. Diagnostic, therapeutic, and prognostic implications of the 2021 World Health Organization classification of tumors of the central nervous system. Cancer 2022, 128, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Al-Rashed, M.; Foshay, K.; Abedalthagafi, M. Recent Advances in Meningioma Immunogenetics. Front Oncol 2019, 9, 1472. [Google Scholar] [CrossRef]
- Sofela, A.A.; McGavin, L.; Whitfield, P.C.; Hanemann, C.O. Biomarkers for differentiating grade II meningiomas from grade I: a systematic review. Br J Neurosurg 2021, 35, 696–702. [Google Scholar] [CrossRef]
- Kishida, Y.; Natsume, A.; Kondo, Y.; Takeuchi, I.; An, B.; Okamoto, Y.; Shinjo, K.; Saito, K.; Ando, H.; Ohka, F.; et al. Epigenetic subclassification of meningiomas based on genome-wide DNA methylation analyses. Carcinogenesis 2012, 33, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Olar, A.; Wani, K.M.; Wilson, C.D.; Zadeh, G.; DeMonte, F.; Jones, D.T.; Pfister, S.M.; Sulman, E.P.; Aldape, K.D. Global epigenetic profiling identifies methylation subgroups associated with recurrence-free survival in meningioma. Acta Neuropathol 2017, 133, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Sahm, F.; Schrimpf, D.; Stichel, D.; Jones, D.T.W.; Hielscher, T.; Schefzyk, S.; Okonechnikov, K.; Koelsche, C.; Reuss, D.E.; Capper, D.; et al. DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol 2017, 18, 682–694. [Google Scholar] [CrossRef] [PubMed]
- Maas, S.L.N.; Stichel, D.; Hielscher, T.; Sievers, P.; Berghoff, A.S.; Schrimpf, D.; Sill, M.; Euskirchen, P.; Blume, C.; Patel, A.; et al. Integrated Molecular-Morphologic Meningioma Classification: A Multicenter Retrospective Analysis, Retrospectively and Prospectively Validated. J Clin Oncol 2021, 39, 3839–3852. [Google Scholar] [CrossRef]
- Driver, J.; Hoffman, S.E.; Tavakol, S.; Woodward, E.; Maury, E.A.; Bhave, V.; Greenwald, N.F.; Nassiri, F.; Aldape, K.; Zadeh, G.; et al. A molecularly integrated grade for meningioma. Neuro Oncol 2022, 24, 796–808. [Google Scholar] [CrossRef]
- Liu, J.; Xia, C.; Wang, G. Multi-Omics Analysis in Initiation and Progression of Meningiomas: From Pathogenesis to Diagnosis. Frontiers in Oncology 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, S.; Suppiah, S.; Tonge, P.D.; Jalali, S.; Danesh, A.; Bruce, J.P.; Mamatjan, Y.; Klironomos, G.; Gonen, L.; Au, K.; et al. Therapeutic radiation for childhood cancer drives structural aberrations of NF2 in meningiomas. Nat Commun 2017, 8, 186. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Qian, J.; Ma, C. MPscore: A Novel Predictive and Prognostic Scoring for Progressive Meningioma. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef]
- Wang, A.Z.; Bowman-Kirigin, J.A.; Desai, R.; Kang, L.-I.; Patel, P.R.; Patel, B.; Khan, S.M.; Bender, D.; Marlin, M.C.; Liu, J.; et al. Single-cell profiling of human dura and meningioma reveals cellular meningeal landscape and insights into meningioma immune response. Genome Medicine 2022, 14, 49. [Google Scholar] [CrossRef]
- Shi, J. Machine learning and bioinformatics approaches for classification and clinical detection of bevacizumab responsive glioblastoma subtypes based on miRNA expression. Sci Rep 2022, 12, 8685. [Google Scholar] [CrossRef]
- Kitano, Y.; Aoki, K.; Ohka, F.; Yamazaki, S.; Motomura, K.; Tanahashi, K.; Hirano, M.; Naganawa, T.; Iida, M.; Shiraki, Y.; et al. Urinary MicroRNA-Based Diagnostic Model for Central Nervous System Tumors Using Nanowire Scaffolds. ACS Applied Materials & Interfaces 2021, 13, 17316–17329. [Google Scholar] [CrossRef]
- Bhawal, R.; Oberg, A.L.; Zhang, S.; Kohli, M. Challenges and Opportunities in Clinical Applications of Blood-Based Proteomics in Cancer. Cancers 2020, 12, 2428. [Google Scholar] [CrossRef] [PubMed]
- Gomes Marin, J.F.; Nunes, R.F.; Coutinho, A.M.; Zaniboni, E.C.; Costa, L.B.; Barbosa, F.G.; Queiroz, M.A.; Cerri, G.G.; Buchpiguel, C.A. Theranostics in Nuclear Medicine: Emerging and Re-emerging Integrated Imaging and Therapies in the Era of Precision Oncology. Radiographics 2020, 40, 1715–1740. [Google Scholar] [CrossRef]
- Salgues, B.; Graillon, T.; Horowitz, T.; Chinot, O.; Padovani, L.; Taïeb, D.; Guedj, E. Somatostatin Receptor Theranostics for Refractory Meningiomas. Curr Oncol 2022, 29, 5550–5565. [Google Scholar] [CrossRef]
- Tubre, T.; Hacking, S.; Alexander, A.; Brickman, A.; Delalle, I.; Elinzano, H.; Donahue, J.E. Prostate-Specific Membrane Antigen Expression in Meningioma: A Promising Theranostic Target. J Neuropathol Exp Neurol 2022. [Google Scholar] [CrossRef]
- Zhang, D.; Ye, S.; Pan, T. The role of serum and urinary biomarkers in the diagnosis of early diabetic nephropathy in patients with type 2 diabetes. PeerJ 2019, 7, e7079. [Google Scholar] [CrossRef]
- Erkan, E.P.; Ströbel, T.; Dorfer, C.; Sonntagbauer, M.; Weinhäusel, A.; Saydam, N.; Saydam, O. Circulating Tumor Biomarkers in Meningiomas Reveal a Signature of Equilibrium Between Tumor Growth and Immune Modulation. Frontiers in Oncology 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidou, A.E.; Givalos, N.; Gakiopoulou, H.; Korkolopoulou, P.; Kotsiakis, X.; Boviatsis, E.; Agrogiannis, G.; Mahera, H.; Patsouris, E. Caspase-3 immunohistochemical expression is a marker of apoptosis, increased grade and early recurrence in intracranial meningiomas. Apoptosis 2007, 12, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Abbritti, R.V.; Polito, F.; Cucinotta, M.; Lo Giudice, C.; Caffo, M.; Tomasello, C.; Germanò, A.; Aguennouz, M. Meningiomas and Proteomics: Focus on New Potential Biomarkers and Molecular Pathways. Cancer Genomics Proteomics 2016, 13, 369–379. [Google Scholar] [PubMed]
- Ghantasala, S.; Pai, M.G.J.; Biswas, D.; Gahoi, N.; Mukherjee, S.; Kp, M.; Nissa, M.U.; Srivastava, A.; Epari, S.; Shetty, P.; et al. Multiple Reaction Monitoring-Based Targeted Assays for the Validation of Protein Biomarkers in Brain Tumors. Front Oncol 2021, 11, 548243. [Google Scholar] [CrossRef] [PubMed]
- Abou Khouzam, R.; Brodaczewska, K.; Filipiak, A.; Zeinelabdin, N.A.; Buart, S.; Szczylik, C.; Kieda, C.; Chouaib, S. Tumor Hypoxia Regulates Immune Escape/Invasion: Influence on Angiogenesis and Potential Impact of Hypoxic Biomarkers on Cancer Therapies. Front Immunol 2020, 11, 613114. [Google Scholar] [CrossRef]
- El-Benhawy, S.A.; Sakr, O.A.; Fahmy, E.I.; Ali, R.A.; Hussein, M.S.; Nassar, E.M.; Salem, S.M.; Abu-Samra, N.; Elzawawy, S. Assessment of Serum Hypoxia Biomarkers Pre- and Post-radiotherapy in Patients with Brain Tumors. J Mol Neurosci 2022. [Google Scholar] [CrossRef]
- Valvona, C.J.; Fillmore, H.L.; Nunn, P.B.; Pilkington, G.J. The Regulation and Function of Lactate Dehydrogenase A: Therapeutic Potential in Brain Tumor. Brain Pathol 2016, 26, 3–17. [Google Scholar] [CrossRef]
- Yoo, H.; Baia, G.S.; Smith, J.S.; McDermott, M.W.; Bollen, A.W.; VandenBerg, S.R.; Lamborn, K.R.; Lal, A. Expression of the Hypoxia Marker Carbonic Anhydrase 9 Is Associated with Anaplastic Phenotypes in Meningiomas. Clinical Cancer Research 2007, 13, 68–75. [Google Scholar] [CrossRef]
- Fattahi, M.J.; Sedaghat, F.; Malekzadeh, M.; Nejat, A.A.; Poostkar, M.; Saberi, Y.; Taghipour, M.; Ghaderi, A. Endocan serum levels in patients with low- and high-grade meningiomas: does this biomarker have an indicative role? The Egyptian Journal of Neurology, Psychiatry and Neurosurgery 2021, 57, 92. [Google Scholar] [CrossRef]
- Atukeren, P.; Kunbaz, A.; Turk, O.; Kemerdere, R.; Ulu, M.O.; Turkmen Inanir, N.; Tanriverdi, T. Expressions of Endocan in Patients with Meningiomas and Gliomas. Dis Markers 2016, 2016, 7157039. [Google Scholar] [CrossRef] [PubMed]
- Shalaby, T.; Achini, F.; Grotzer, M.A. Targeting cerebrospinal fluid for discovery of brain cancer biomarkers. Journal of Cancer Metastasis and Treatment 2016, 2, 176–187. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, S.K.; Yoo, Y.C.; Park, N.H.; Park, D.B.; Yoo, J.S.; An, H.J.; Park, Y.M.; Cho, K.G. Proteome analysis of human cerebrospinal fluid as a diagnostic biomarker in patients with meningioma. Med Sci Monit 2012, 18, Br450–Br460. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, R.; Elabbas, A.; Vyas, A.; Osborne, D.; Chigurupati, H.D.; Abbas, L.F.; Kampa, P.; M, H.F.; Sarwar, H.; Alfonso, M. Utilization of Cerebrospinal Fluid Proteome Analysis in the Diagnosis of Meningioma: A Systematic Review. Cureus 2021, 13, e20707. [Google Scholar] [CrossRef]
- Georgila, K.; Vyrla, D.; Drakos, E. Apolipoprotein A-I (ApoA-I), Immunity, Inflammation and Cancer. Cancers 2019, 11, 1097. [Google Scholar] [CrossRef]
- Rajagopal, M.U.; Hathout, Y.; MacDonald, T.J.; Kieran, M.W.; Gururangan, S.; Blaney, S.M.; Phillips, P.; Packer, R.; Gordish-Dressman, H.; Rood, B.R. Proteomic profiling of cerebrospinal fluid identifies prostaglandin D2 synthase as a putative biomarker for pediatric medulloblastoma: A pediatric brain tumor consortium study. Proteomics 2011, 11, 935–943. [Google Scholar] [CrossRef]
- Xiao, F.; Lv, S.; Zong, Z.; Wu, L.; Tang, X.; Kuang, W.; Zhang, P.; Li, X.; Fu, J.; Xiao, M.; et al. Cerebrospinal fluid biomarkers for brain tumor detection: clinical roles and current progress. Am J Transl Res 2020, 12, 1379–1396. [Google Scholar]
- Wang, P.; Piao, Y.; Zhang, X.; Li, W.; Hao, X. The concentration of CYFRA 21-1, NSE and CEA in cerebro-spinal fluid can be useful indicators for diagnosis of meningeal carcinomatosis of lung cancer. Cancer Biomark 2013, 13, 123–130. [Google Scholar] [CrossRef]
- O'Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front Endocrinol (Lausanne) 2018, 9, 402. [Google Scholar] [CrossRef]
- Kopkova, A.; Sana, J.; Fadrus, P.; Slaby, O. Cerebrospinal fluid microRNAs as diagnostic biomarkers in brain tumors. Clin Chem Lab Med 2018, 56, 869–879. [Google Scholar] [CrossRef]
- Zhi, F.; Zhou, G.; Wang, S.; Shi, Y.; Peng, Y.; Shao, N.; Guan, W.; Qu, H.; Zhang, Y.; Wang, Q.; et al. A microRNA expression signature predicts meningioma recurrence. Int J Cancer 2013, 132, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Slavik, H.; Balik, V.; Vrbkova, J.; Rehulkova, A.; Vaverka, M.; Hrabalek, L.; Ehrmann, J.; Vidlarova, M.; Gurska, S.; Hajduch, M.; et al. Identification of Meningioma Patients at High Risk of Tumor Recurrence Using MicroRNA Profiling. Neurosurgery 2020, 87, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Eraky, A.M. Non-coding RNAs as Genetic Biomarkers for the Diagnosis, Prognosis, Radiosensitivity, and Histopathologic Grade of Meningioma. Cureus 2023, 15, e34593. [Google Scholar] [CrossRef] [PubMed]
- Gareev, I.; Beylerli, O.; Liang, Y.; Xiang, H.; Liu, C.; Xu, X.; Yuan, C.; Ahmad, A.; Yang, G. The Role of MicroRNAs in Therapeutic Resistance of Malignant Primary Brain Tumors. Frontiers in Cell and Developmental Biology 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhi, F.; Shao, N.; Li, B.; Xue, L.; Deng, D.; Xu, Y.; Lan, Q.; Peng, Y.; Yang, Y. A serum 6-miRNA panel as a novel non-invasive biomarker for meningioma. Scientific Reports 2016, 6, 32067. [Google Scholar] [CrossRef]
- Carneiro, V.; Cirino, M.; Panepucci, R.; Peria, F.; Tirapelli, D.; Colli, B.; Carlotti Jr, C.G. The Role of MicroRNA 181d as a Possible Biomarker Associated With Tumor Progression in Meningiomas. Cureus 2021, 13, e19158. [Google Scholar] [CrossRef]
- Urbschat, S.; Landau, B.; Bewersdorf, N.C.; Schuster, C.; Wagenpfeil, G.; Schulz-Schaeffer, W.J.; Oertel, J.; Ketter, R. MicroRNA 200a as a histologically independent marker for meningioma recurrence: Results of a four microRNA panel analysis in meningiomas. Cancer Med 2022. [Google Scholar] [CrossRef]
- Gurunathan, S.; Kang, M.H.; Kim, J.H. Diverse Effects of Exosomes on COVID-19: A Perspective of Progress From Transmission to Therapeutic Developments. Front Immunol 2021, 12, 716407. [Google Scholar] [CrossRef]
- Saugstad, J.A.; Lusardi, T.A.; Van Keuren-Jensen, K.R.; Phillips, J.I.; Lind, B.; Harrington, C.A.; McFarland, T.J.; Courtright, A.L.; Reiman, R.A.; Yeri, A.S.; et al. Analysis of extracellular RNA in cerebrospinal fluid. J Extracell Vesicles 2017, 6, 1317577. [Google Scholar] [CrossRef]
- Negroni, C.; Hilton, D.A.; Ercolano, E.; Adams, C.L.; Kurian, K.M.; Baiz, D.; Hanemann, C.O. GATA-4, a potential novel therapeutic target for high-grade meningioma, regulates miR-497, a potential novel circulating biomarker for high-grade meningioma. EBioMedicine 2020, 59, 102941. [Google Scholar] [CrossRef]
- Ricklefs, F.L.; Maire, C.L.; Wollmann, K.; Dührsen, L.; Fita, K.D.; Sahm, F.; Herold-Mende, C.; von Deimling, A.; Kolbe, K.; Holz, M.; et al. Diagnostic potential of extracellular vesicles in meningioma patients. Neuro Oncol 2022, 24, 2078–2090. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J.S.; Amaral, P.P.; Carninci, P.; Carpenter, S.; Chang, H.Y.; Chen, L.-L.; Chen, R.; Dean, C.; Dinger, M.E.; Fitzgerald, K.A.; et al. Long non-coding RNAs: definitions, functions, challenges and recommendations. Nature Reviews Molecular Cell Biology 2023. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Lan, Y.; Xie, A.; Shi, J.; Zhao, H.; Xu, L.; Zhu, S.; Luo, T.; Zhao, T.; Xiao, Y.; et al. Comprehensive analysis of long noncoding RNA (lncRNA)-chromatin interactions reveals lncRNA functions dependent on binding diverse regulatory elements. J Biol Chem 2019, 294, 15613–15622. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Ge, Y.; Wang, D.; Liu, Q.; Sun, S.; Hua, L.; Deng, J.; Luan, S.; Cheng, H.; Xie, Q.; et al. LncRNA-IMAT1 Promotes Invasion of Meningiomas by Suppressing KLF4/hsa-miR22-3p/Snai1 Pathway. Mol Cells 2022, 45, 388–402. [Google Scholar] [CrossRef] [PubMed]
- Slavik, H.; Balik, V.; Kokas, F.Z.; Slavkovsky, R.; Vrbkova, J.; Rehulkova, A.; Lausova, T.; Ehrmann, J.; Gurska, S.; Uberall, I.; et al. Transcriptomic Profiling Revealed Lnc-GOLGA6A-1 as a Novel Prognostic Biomarker of Meningioma Recurrence. Neurosurgery 2022, 91, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Ren, J.; Ma, J.; Wu, J.; Zhang, R.; Yuan, H.; Han, X. LINC00702/miR-4652-3p/ZEB1 axis promotes the progression of malignant meningioma through activating Wnt/beta-catenin pathway. Biomed Pharmacother 2019, 113, 108718. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.; Wang, S.; Li, Q.; Ma, Y.; Sun, P. Long noncoding RNA LINC00460 targets miR-539/MMP-9 to promote meningioma progression and metastasis. Biomed Pharmacother 2018, 105, 677–682. [Google Scholar] [CrossRef]
- Ding, C.; Yi, X.; Xu, J.; Huang, Z.; Bu, X.; Wang, D.; Ge, H.; Zhang, G.; Gu, J.; Kang, D.; et al. Long Non-Coding RNA MEG3 Modifies Cell-Cycle, Migration, Invasion, and Proliferation Through AKAP12 by Sponging miR-29c in Meningioma Cells. Frontiers in Oncology 2020, 10. [Google Scholar] [CrossRef]
- Rana, M.W.; Pinkerton, H.; Thornton, H.; Nagy, D. Heterotransplantation of human glioblastoma multiforme and meningioma to nude mice. Proc Soc Exp Biol Med 1977, 155, 85–88. [Google Scholar] [CrossRef]
- Jensen, R.L.; Leppla, D.; Rokosz, N.; Wurster, R.D. Matrigel augments xenograft transplantation of meningioma cells into athymic mice. Neurosurgery 1998, 42, 130–135. [Google Scholar] [CrossRef]
- McCutcheon, I.E.; Friend, K.E.; Gerdes, T.M.; Zhang, B.M.; Wildrick, D.M.; Fuller, G.N. Intracranial injection of human meningioma cells in athymic mice: an orthotopic model for meningioma growth. J Neurosurg 2000, 92, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Boetto, J.; Peyre, M.; Kalamarides, M. Mouse Models in Meningioma Research: A Systematic Review. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Püttmann, S.; Senner, V.; Braune, S.; Hillmann, B.; Exeler, R.; Rickert, C.H.; Paulus, W. Establishment of a benign meningioma cell line by hTERT-mediated immortalization. Lab Invest 2005, 85, 1163–1171. [Google Scholar] [CrossRef]
- van Furth, W.R.; Laughlin, S.; Taylor, M.D.; Salhia, B.; Mainprize, T.; Henkelman, M.; Cusimano, M.D.; Ackerley, C.; Rutka, J.T. Imaging of murine brain tumors using a 1.5 Tesla clinical MRI system. Can J Neurol Sci 2003, 30, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Baia, G.S.; Dinca, E.B.; Ozawa, T.; Kimura, E.T.; McDermott, M.W.; James, C.D.; VandenBerg, S.R.; Lal, A. An orthotopic skull base model of malignant meningioma. Brain Pathol 2008, 18, 172–179. [Google Scholar] [CrossRef]
- Castle, K.D.; Chen, M.; Wisdom, A.J.; Kirsch, D.G. Genetically engineered mouse models for studying radiation biology. Transl Cancer Res 2017, 6, S900–s913. [Google Scholar] [CrossRef]
- Peyre, M.; Salaud, C.; Clermont-Taranchon, E.; Niwa-Kawakita, M.; Goutagny, S.; Mawrin, C.; Giovannini, M.; Kalamarides, M. PDGF activation in PGDS-positive arachnoid cells induces meningioma formation in mice promoting tumor progression in combination with Nf2 and Cdkn2ab loss. Oncotarget 2015, 6, 32713–32722. [Google Scholar] [CrossRef]
- Kawashima, M.; Suzuki, S.O.; Yamashima, T.; Fukui, M.; Iwaki, T. Prostaglandin D synthase (beta-trace) in meningeal hemangiopericytoma. Mod Pathol 2001, 14, 197–201. [Google Scholar] [CrossRef]
- Urade, Y.; Kitahama, K.; Ohishi, H.; Kaneko, T.; Mizuno, N.; Hayaishi, O. Dominant expression of mRNA for prostaglandin D synthase in leptomeninges, choroid plexus, and oligodendrocytes of the adult rat brain. Proc Natl Acad Sci U S A 1993, 90, 9070–9074. [Google Scholar] [CrossRef]
- Yamashima, T.; Sakuda, K.; Tohma, Y.; Yamashita, J.; Oda, H.; Irikura, D.; Eguchi, N.; Beuckmann, C.T.; Kanaoka, Y.; Urade, Y.; et al. Prostaglandin D synthase (beta-trace) in human arachnoid and meningioma cells: roles as a cell marker or in cerebrospinal fluid absorption, tumorigenesis, and calcification process. J Neurosci 1997, 17, 2376–2382. [Google Scholar] [CrossRef]
- von Werder, A.; Seidler, B.; Schmid, R.M.; Schneider, G.; Saur, D. Production of avian retroviruses and tissue-specific somatic retroviral gene transfer in vivo using the RCAS/TVA system. Nat Protoc 2012, 7, 1167–1183. [Google Scholar] [CrossRef] [PubMed]
- Kalamarides, M.; Niwa-Kawakita, M.; Leblois, H.; Abramowski, V.; Perricaudet, M.; Janin, A.; Thomas, G.; Gutmann, D.H.; Giovannini, M. Nf2 gene inactivation in arachnoidal cells is rate-limiting for meningioma development in the mouse. Genes Dev 2002, 16, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Kalamarides, M.; Stemmer-Rachamimov, A.O.; Takahashi, M.; Han, Z.Y.; Chareyre, F.; Niwa-Kawakita, M.; Black, P.M.; Carroll, R.S.; Giovannini, M. Natural history of meningioma development in mice reveals: a synergy of Nf2 and p16(Ink4a) mutations. Brain Pathol 2008, 18, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Peyre, M.; Stemmer-Rachamimov, A.; Clermont-Taranchon, E.; Quentin, S.; El-Taraya, N.; Walczak, C.; Volk, A.; Niwa-Kawakita, M.; Karboul, N.; Giovannini, M.; et al. Meningioma progression in mice triggered by Nf2 and Cdkn2ab inactivation. Oncogene 2013, 32, 4264–4272. [Google Scholar] [CrossRef] [PubMed]
- Mandara, M.T.; Ricci, G.; Rinaldi, L.; Sarli, G.; Vitellozzi, G. Immunohistochemical identification and image analysis quantification of oestrogen and progesterone receptors in canine and feline meningioma. J Comp Pathol 2002, 127, 214–218. [Google Scholar] [CrossRef]
- Michelhaugh, S.K.; Guastella, A.R.; Varadarajan, K.; Klinger, N.V.; Parajuli, P.; Ahmad, A.; Sethi, S.; Aboukameel, A.; Kiousis, S.; Zitron, I.M.; et al. Development of patient-derived xenograft models from a spontaneously immortal low-grade meningioma cell line, KCI-MENG1. J Transl Med 2015, 13, 227. [Google Scholar] [CrossRef]
- Kim, H.; Park, K.J.; Ryu, B.K.; Park, D.H.; Kong, D.S.; Chong, K.; Chae, Y.S.; Chung, Y.G.; Park, S.I.; Kang, S.H. Forkhead box M1 (FOXM1) transcription factor is a key oncogenic driver of aggressive human meningioma progression. Neuropathol Appl Neurobiol 2020, 46, 125–141. [Google Scholar] [CrossRef]
- Saydam, O.; Shen, Y.; Würdinger, T.; Senol, O.; Boke, E.; James, M.F.; Tannous, B.A.; Stemmer-Rachamimov, A.O.; Yi, M.; Stephens, R.M.; et al. Downregulated microRNA-200a in meningiomas promotes tumor growth by reducing E-cadherin and activating the Wnt/beta-catenin signaling pathway. Mol Cell Biol 2009, 29, 5923–5940. [Google Scholar] [CrossRef]
- Tuchen, M.; Wilisch-Neumann, A.; Daniel, E.A.; Baldauf, L.; Pachow, D.; Scholz, J.; Angenstein, F.; Stork, O.; Kirches, E.; Mawrin, C. Receptor tyrosine kinase inhibition by regorafenib/sorafenib inhibits growth and invasion of meningioma cells. Eur J Cancer 2017, 73, 9–21. [Google Scholar] [CrossRef]
- Murnyák, B.; Bognár, L.; Klekner, Á.; Hortobágyi, T. Epigenetics of Meningiomas. Biomed Res Int 2015, 2015, 532451. [Google Scholar] [CrossRef]
- Di Bonaventura, R.; Martini, M.; Cenci, T.; Caccavella, V.M.; Barresi, V.; Gessi, M.; Albanese, A.; Lauretti, L.; Pallini, R.; D'Alessandris, Q.G.; et al. Dissecting Stemness in Aggressive Intracranial Meningiomas: Prognostic Role of SOX2 Expression. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Anis, S.E.; Lotfalla, M.; Zain, M.; Kamel, N.N.; Soliman, A.A. Value of SSTR2A and Claudin - 1 in Differentiating Meningioma from Schwannoma and Hemangiopericytoma. Open Access Maced J Med Sci 2018, 6, 248–253. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Title: Association between genetic/cytogenetic alteration, grade of meningioma, and anatomical location of the tumor. Meningioma arises in the meningeal layers of the brain or spinal cord. They are commonly seen in the parasagittal, brain convexity, posterior fossa, and spine. Grade I (benign) meningiomas are found in the parasagittal and posterior fossa with alterations in chromosome 22 and variation of the second allele of neurofibromatosis 2 (NF2). Genetic alterations in AKT1, PIK3CA, SMO, TRAF7, KLF4, SMARCB1, and PTERT also take place in Grade I meningiomas, in the presence or absence of NF2 mutations depending on the gene. Grade Il (atypical) meningiomas tend to invade the brain convexity & spine and can have a loss of a copy of chromosomes 1,10, or 14. They display genetic alterations in NF2, PTERT, & SMARCEl. Grade III (malignant or anaplastic) meningiomas tend to invade the parts of the brain that are closest to the tumor with an absence of chromosome 9p and genetic alterations of NF2, BAP1, LDH229, and PTERT.
Figure 1.
Title: Association between genetic/cytogenetic alteration, grade of meningioma, and anatomical location of the tumor. Meningioma arises in the meningeal layers of the brain or spinal cord. They are commonly seen in the parasagittal, brain convexity, posterior fossa, and spine. Grade I (benign) meningiomas are found in the parasagittal and posterior fossa with alterations in chromosome 22 and variation of the second allele of neurofibromatosis 2 (NF2). Genetic alterations in AKT1, PIK3CA, SMO, TRAF7, KLF4, SMARCB1, and PTERT also take place in Grade I meningiomas, in the presence or absence of NF2 mutations depending on the gene. Grade Il (atypical) meningiomas tend to invade the brain convexity & spine and can have a loss of a copy of chromosomes 1,10, or 14. They display genetic alterations in NF2, PTERT, & SMARCEl. Grade III (malignant or anaplastic) meningiomas tend to invade the parts of the brain that are closest to the tumor with an absence of chromosome 9p and genetic alterations of NF2, BAP1, LDH229, and PTERT.

Figure 2.
NF2/Merlin signaling in a normal meningeal cell vs Merlin-deficient meningioma cell. Merlin is an effective inhibitor of major signaling pathways that lead to cell proliferation, protein synthesis, and angiogenesis. In a normal meningeal arachnoid cap cell, the NF2 gene encodes for Merlin. Merlin is a cytoskeletal protein that interacts and complexes with integrin 3, receptor tyrosine kinases (RTK), and -catenin to inhibit mTOR signaling pathway, MAPK pathway, and WNT pathway among others. Merlin inhibits downstream effectors of these pathways including RAS, PI3K, AKT, mTOR, and -CATENIN. Additionally, Merlin interferes with the translocation of -CATENIN into the nucleus, inhibiting canonical WNT signaling. Merlin also inhibits transcription factors YAP/TAZ and TEA by interacting with components of the Hippo pathway. Loss of Merlin function to NF2 mutations, such as in meningioma activates these pathways (indicated by arrows) and leads to cell proliferation, protein synthesis, and angiogenesis, contributing to meningioma incidence and progression.
Figure 2.
NF2/Merlin signaling in a normal meningeal cell vs Merlin-deficient meningioma cell. Merlin is an effective inhibitor of major signaling pathways that lead to cell proliferation, protein synthesis, and angiogenesis. In a normal meningeal arachnoid cap cell, the NF2 gene encodes for Merlin. Merlin is a cytoskeletal protein that interacts and complexes with integrin 3, receptor tyrosine kinases (RTK), and -catenin to inhibit mTOR signaling pathway, MAPK pathway, and WNT pathway among others. Merlin inhibits downstream effectors of these pathways including RAS, PI3K, AKT, mTOR, and -CATENIN. Additionally, Merlin interferes with the translocation of -CATENIN into the nucleus, inhibiting canonical WNT signaling. Merlin also inhibits transcription factors YAP/TAZ and TEA by interacting with components of the Hippo pathway. Loss of Merlin function to NF2 mutations, such as in meningioma activates these pathways (indicated by arrows) and leads to cell proliferation, protein synthesis, and angiogenesis, contributing to meningioma incidence and progression.
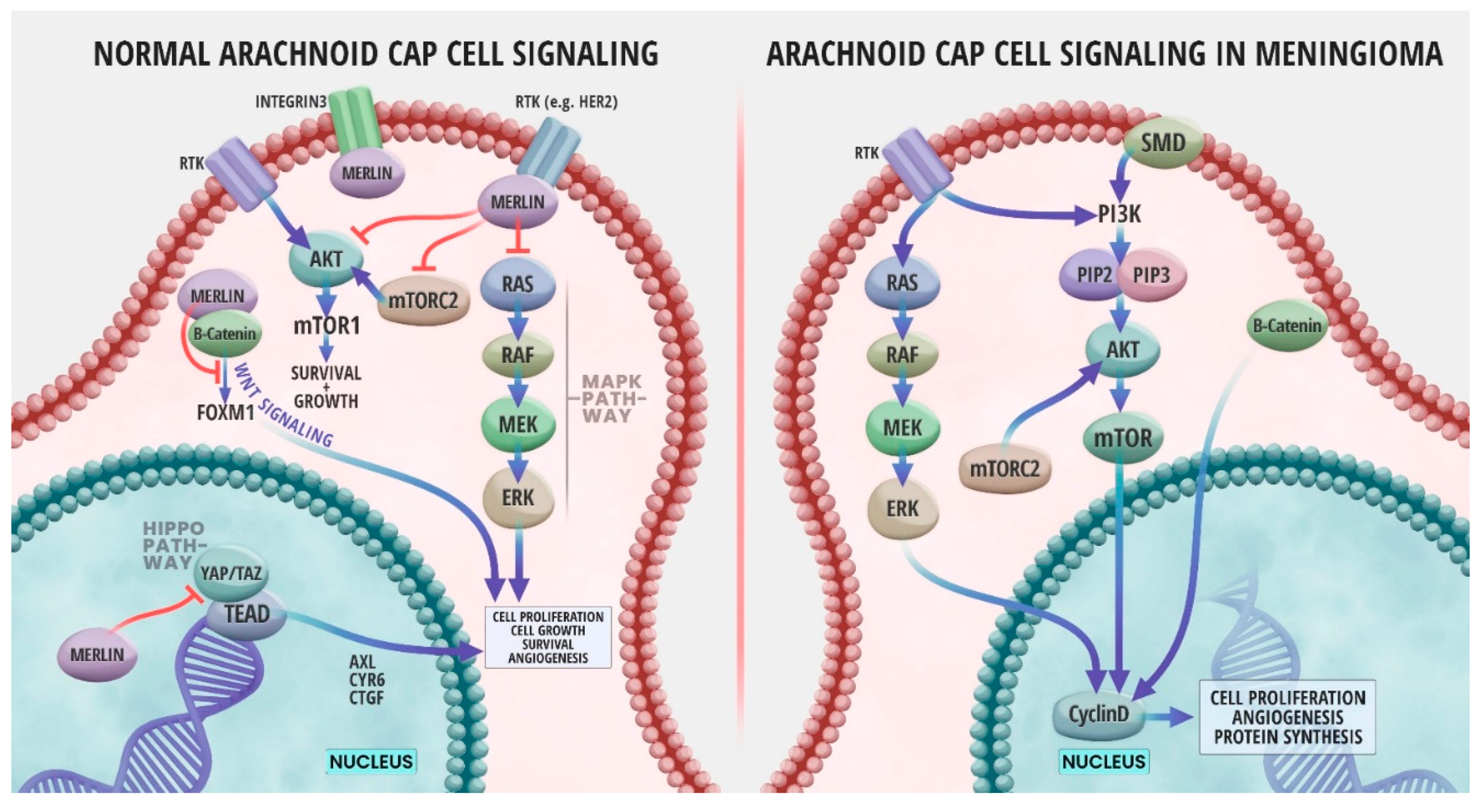

Table 1.
Measures of Association of Known Biomarkers of Meningioma.
| Biomarker Type | Known Biomarkers | Study design | Clinical Use | Reference | Publication Date |
|---|---|---|---|---|---|
| Genomic | NF2, TRAF7,AKT1, KLF4, SMO PIK3CA | Human studies (Review) | Diagnosis | [29] | 2020 |
| miRNA | miR-451, miR-711, miR-190a miR-17-5p, miR-199a, miR-190a, miR-186-5p, miR-155-5p, miR-22-3p, miR-24-3p, miR- 26b-5p, miR-27a-3p, miR-27b-3p, miR-96-5p,miR-146a-5p miR-106a-5p, miR-219-5p, miR-375, and miR-409-3p miR-4695-5p, miR-4286, miR-6732-5p, miR-6855-5p, miR-7977, miR-6765-3p, miR-6787-5p miR-181d |
Human clinical trial In-vivo human clinical trial Clinical Study In-vivo human Cohort study Clinical Human study Human clinical study on tissues and plasma |
Diagnosis/therapy response Prognosis Diagnosis,prognosis, histological grade & radiosensitivity Non-invasive diagnostic/prognostic Prognosis Diagnosis and prognosis |
[128] [132] [134] [136] [135] [137] |
2020 2013 2023 2016 2021 2021 |
| LncRNA | lnc-GOLGA6A-1 lncRNA-LINC00460 LncRNA- NUP210, LncRNA-SPIRE2, LncRNA-SLC7A1, and LncRNA-DMTN |
Human Clinical independent cohort study In-vitro human clinical study Clinical study |
Prognosis and pathogenesis Diagnosis Diagnosis,prognosis, histological grade & radiosensitivity |
[146] [149] [134] |
2022 2020 2023 |
| Epigenetic | TIMP3, HOXA7,HOXA9, HOXA10, RASSF1A SOX2 |
- Human clinical trial |
prognosis prognosis |
[171] [172] |
2015 2022 |
| Proteomic | APO-E, APO-J, PTGDS Caspase-3 EFEMP1 CEA |
Clinical study Screening Cohort Study Human clinical trial Review |
Diagnosis Non-invasive diagnosis and prognosis Diagnosis/therapy response Diagnosis |
[124] [113] [128] [123] |
2012 2019 2020 2016 |
| Histological | SSTR2A, Claudin-1 CA9 |
Human Clinical case Clinical study |
Diagnosis and sub-type determination Poor prognosis |
[173] [120] |
2018 2007 |
Table 2.
Advantages and Limitations of Xenograft and Genetically Engineered Mouse Models of Meningiomas.
Table 2.
Advantages and Limitations of Xenograft and Genetically Engineered Mouse Models of Meningiomas.
| Mouse Model | Advantages | Limitations |
|---|---|---|
| Heterotopic xenograft model | Very reliable in term of tumor take rates. | Lacks the key components of the meningiomas specific microenvironment. |
| Orthotopic xenograft model |
|
|
| Genetically Engineered Mouse Models (GEMM) |
|
|
* These Limitations are in common with the heterotopic xenograft model.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



