
Submitted:
17 August 2023
Posted:
18 August 2023
You are already at the latest version
Abstract
Background: This study aimed to investigate the molecular profiles of stage III CRC patients from the international IDEA study. It also sought to correlate these profiles with Toll-like and vitamin D receptor polymorphisms, clinicopathological and epidemiological characteristics, and patient outcomes.
Methods: Whole Exome Sequencing and PCR-RFLP on surgical specimens and blood samples, respectively, were performed to identify molecular profiling and the presence of Toll-like and vitamin D polymorphisms. Bioinformatic analysis revealed mutational status.
Results: Among the enrolled patients, 63.7% were male, 66.7% had left-sided tumors, and 55.7% received CAPOX as adjuvant chemotherapy. Whole exome sequencing identified 59 mutated genes in 11 different signaling pathways from the Kyoto Encyclopedia of Genes and Genomes (KEGG) CRC panel. On average, patients had 8 mutated genes (range, 2-21 genes). Mutations in ARAF and MAPK10 emerged as independent prognostic factors for reduced DFS (p=0.027 and p<0.001, respectively), while RAC3 and RHOA genes emerged as independent prognostic factors for reduced OS (p=0.029 and p=0.006, respectively). Right-sided tumors were also identified as independent prognostic factors for reduced DFS (p=0.019) and OS (p=0.043). Additionally, patients with tumors in the transverse colon had mutations in genes related to apoptosis, PIK3-Akt, Wnt, and MAPK signaling pathways.
Conclusions: Molecular characterization of tumor cells can enhance our understanding of the disease course. Mutations may serve as promising prognostic biomarkers, offering improved treatment options. Confirming these findings with require larger patient cohorts and international collaborations to establish correlations between molecular profiling, clinicopathological and epidemiological characteristics and clinical outcomes.
Keywords:
colorectal cancer
; stage III
; molecular profiling
; IDEA study
; whole exome sequencing
; bioinfor-matics
1. Introduction
Colorectal cancer (CRC) is one of the most common malignancies and the second most common cause of death from cancer[1]. In 2020, 1.9 million new cases of CRC and approximately 935,000 deaths were reported[2]. By 2030, the global burden of CRC is predicted to be 60%, with more than 2.2 million new cases and 1.1 million deaths. By 2035, the total number of deaths from rectal and colon cancer is estimated to increase by 60% and 71.5%, respectively[2]. Depending on the stage of the disease at the time of diagnosis, both the treatment and prognosis differ. Patients with stage III CRC have an overall 5-year survival rate of 60%. Adjuvant chemotherapy aims to increase this rate and extend both the overall and disease-free survival[3]. Since 2004, folinic acid, fluorouracil, and oxaliplatin (FOLFOX) or capecitabine and oxaliplatin) for six months has been the standard treatment regimen[3,4]. However, oxaliplatin can lead to adverse effects, particularly peripheral sensory neuropathy[5].
Considering the increased incidence of the disease, toxicity of the treatment, cost, and efforts to reduce the duration of treatment for all the aforementioned reasons[5], the international IDEA study was designed to evaluate the hypothesis of non-inferiority of the 3-month vs. 6-month adjuvant chemotherapy with FOLFOX or CAPOX[6].
Although strong, AJCC/UICC-TNM staging (American Joint Committee on Cancer/Union Internationale Contre le Cancer—extent of primary tumor, regional lymph node involvement, presence of distant metastases) often fails to provide complete prognostic information because the outcome varies even among patients of the same stage[7]. Therefore, there is an urgent need to identify new tools that can contribute to CRC prognosis. The aim of the current study was to investigate the molecular profile of surgical specimens from stage III CRC patients enrolled in the international IDEA study, for whom paraffin-embedded cancer tissue was available. Following genetic profiling, correlations were performed with clinicopathological characteristics, as well as with patient outcomes. Additionally, the patients were tested for vitamin D receptor (VDR) and toll-like receptor (TLR) gene polymorphisms in peripheral blood, as our previous studies have demonstrated the role of such polymorphisms in tumor development and progression[8,9,10].
2. Patients and methods
2.1. Patient enrollment
The Hellenic Oncology Research Group (HORG) enrolled 708 patients with CRC in the international IDEA study (ClinicalTrials.gov Identifier: NCT01308086). Of these, 237 stage III patients with formalin-fixed paraffin-embedded (FFPE) tissues were included in the current study (Supplementary Table S1). All patients were aged > 18 years and received adjuvant chemotherapy with FOLFOX or CAPOX. None of the enrolled subjects had any other documented malignancies.
2.2. Formalin-Fixed Paraffin-embedded (FFPE) tissues
All surgical materials were evaluated by a specialized pathologist at the Department of Pathology of the University General Hospital of Heraklion, Crete, and the most representative and enriched tumor areas were selected for dissection. Healthy tissue was used as the control tissue. The specifications of the FFPE sections used for DNA extraction were as follows: tissue surface area, 25 mm2, section thickness, 10 μm; c. At least 10 sections with >50% cancer cells, d. Cancer cells were collected from areas rich in cancerous tissue and avoiding healthy tissue, adipose tissue, necrotic areas, and lymphocytes that decrease the content of cancer DNA. To facilitate the collection of appropriate cells from the 10 sections, an additional section was stained with hematoxylin-eosin to locate the cancerous areas.
2.3. TLR and VDR genotyping in blood samples
A total of 5 ml of peripheral blood in EDTA was collected from each patient, and DNA was extracted using the QIAamp DNA Blood Mini kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. The DNA concentration was determined using a NanoDrop ND-1000 v3.3 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA).
To determine the genetic variants of TLRs and VDRs, Polymerase chain reaction and restriction fragment length polymorphism (PCR-RFLP) were used to determine the genetic variants of TLRs and VDRs. For TLR2 196-to-174 Ins/Del genetic variants, PCR was used, while TLR4 (Asp299Gly and Thr399Ile) and TLR9 (T1237C and T1486C) genetic variants were determined using PCR-RFLP. The materials and conditions for each gene target have been previously described[8,9]. Similarly, for genotyping of VDR genetic variants at the TaqI, ApaI, FokI, and BsmI positions, PCR-RFLP was used. The reagents and PCR conditions have been previously described, in detail[8,9,10]. The patients were classified as wild-type, heterozygous, or homozygous for each single nucleotide polymorphism, based on the absence or presence of the restriction site in both alleles.
2.4. Whole Exome Sequencing (WES)
The Illumina DNA Prep with Enrichment kit (Illumina, San Diego, CA 92122) was used for library preparation and enrichment, and sequencing of both tumor and normal tissues was conducted using the NovaSeq 6000 system (Illumina). For each sample, at least 250 ng of high-quality DNA was quantified using a Qubit Fluorometer (Thermo Fisher Scientific, Paisley, UK), was used. Following the WES, two files (. fastq) containing sequencing data were extracted from each tissue (tumor and normal) using forward and reverse reads.
2.5. Bioinformatic analysis
After obtaining the raw data from the WES analysis, a bioinformatics pipeline was used to process the data and generate interpretations. This included alignment to the human genome, variant calling, variant filtering, and annotation of the variants. The processed data were analyzed to identify somatic variants and evaluate their functional significance, particularly those associated with CRC (Figure 1). Initially, the raw sequences were aligned to the human genome (version hg19/GRCh37)[11], and variant calling was conducted using the genome analysis toolkit (GATK) for SNPs and insertion/deletions (INDELs). This generates two variant call format files (VCF) for each patient, one for the tumor and one for the normal tissue, respectively[12]. The somatic variants were isolated by subtracting the germline variants from the tumor, and a custom gene panel composed of 62 genes associated with 11 signaling pathways was utilized based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database on CRC-correlated genes[13] (Table 1 and Table 2). Subsequently, ANNOVAR software was employed to annotate the SNPs and INDELs, providing functional information that can determine the biological significance of each variant and identify CRC-associated variants[14].
To identify clinically significant variants and the signaling pathways involved, a filtering strategy was employed based on the functional position of the variants. Variants located in exons and splice sites are isolated as they are more likely to cause diseases[15]. In addition, the variants located in exons were further filtered based on their functional consequences, excluding variants causing synonymous mutations. This step was considered synonymous mutations that do not affect the amino acid sequence of proteins and are therefore less likely to be clinically relevant. All analyses were run in the Anaconda Powershell Prompt (Anaconda3, Inc.) on Ubuntu 20.04.3 LTS.
2.6. Statistical analysis
After characterizing the patients’ molecular profiles, the clinical, pathological, and epidemiological characteristics were examined to determine their association with patient outcomes. Disease-free (DFS) and overall survival (OS) were calculated from the day of tumor excision until the first documented recurrence or death, respectively. Recurrence was defined as the presence of metastatic disease, local recurrence, or a second primary tumor. The possible associations between baseline characteristics, recurrence, and individual or concurrent mutations were compared using the 2-sided Fisher exact test for categorical variables. The association between risk factors and time-to-event endpoints was evaluated using the log-rank test, and the Kaplan-Meier method was used to generate DFS and OS curves. Univariate and multivariate Cox regression analyses were conducted to evaluate the correlation between the potential prognostic factors and DFS or OS. Statistical significance was defined as p ≤ 0.05, and the statistical tool used was SPSS v. 26.
3. Results
3.1. Patients
The current investigation enrolled 237 patients with stage III CRC, and their characteristics are displayed in Table 3 and Supplementary Table S1. Among these patients, 151 (63.7%) were male, 159 (67.1%) were <70 years old (median:64 years, range:18-84), 158 (66.7%) had tumor localization in the left colon, and 132 (55.7%) patients underwent CAPOX as adjuvant chemotherapy. Of the entire patient population, 116 (48.9%) were administered a 3-month treatment regimen, while 121 (51.1%) were administered a 6-month treatment regimen. Moreover, 84 patients were analyzed for VDR and TLR gene polymorphisms. Regarding VDRs, 10 (11.9%), 7 (8.2%), 5 (6.0%), and 17 (20.2%) patients presented TaqI, ApaI, FokI, and BsmI homozygous phenotypes, respectively. Moreover, regarding TLRs, 48 (57.1%), 38 (46.4%), 38 (46.4%), 37 (44.0%), and 37 (44.0%) patients presented TLR2 196-to-174, TLR4—Asp299Gly, TLR4—Thr399Ile), TLR9—T1237C and TLR9—T1486C homozygous phenotype, respectively.
3.2. Annotated variants for each position
The KEGG CRC panel detected 85,871 uniquely annotated variants. Of these, 602 were detected in exonic positions, including 25 splice variants. The remaining variants were non-coding, with 81,555 intronic variants being the most common, followed by 2,184 UTR variants, 788 downstream variants, and 742 upstream variants.
3.3. Identification of mutated genes
Mutated genes within the KEGG CRC gene panel for each patient were identified. On average, the patients exhibited eight mutated genes (range, 2–21 genes). The frequencies of mutations in each gene, specifically in exons and splicing sites, are presented in Table 4 and Figure 2.
From this analysis, it was observed that certain groups of patients had a higher mutation frequency in specific genes (Table 5). In brief, males had a significantly higher frequency of mutations in the JUN and MAPK3 genes than females (p=0.05, p=0.05, respectively). In terms of age groups, patients below 70 years of age had a higher frequency of mutations in TGFBR1 than those ≥70 years of age (p=0.012). Similarly, patients between 51-70 years old had more frequent mutations in BAD (p<0.001), RAC (p=0.016), AKT, AKT2, AKT3, APC, APPL1, AXIN1, AXIN2, BIRC5, DCC, GSK3B, KRAS, MAPK1, MAPK8, MAPK9, MAPK10, MLH1, MSH6, PΙK3CA, PIK3R1, PIK3R2, PIK3R5, RAF1, RALGDS, SMAD2, SMAD3, TCF7L2, TGFB1 and TGFB2 genes ( p=0.037). Moreover, mutation rates in ARAF, MAPK10, CASP, TCF7, and TGFΒ3 genes were significantly higher in patients relapsed after adjuvant treatment (p=0.027, p=0.044, p=0.003, and p=0.037, respectively).
Subsequently, it was demonstrated that patients with tumors located in the transverse colon, homozygous for mutated VDR alleles (TaqI, ApaI, FokI, BsmI) and homozygous for mutated TLR9 alleles (T1237C and T1486C), had mutations in genes that are mainly involved in the apoptosis, PIK3-AKT, Wnt and MAPK signaling pathways (Table 6).
3.4. Clinical outcome based on molecular profile and patients’ characteristics
Regarding disease-free survival (DFS), it was demonstrated that patients with ARAF mutations had a significantly shorter DFS (74 months, 95% CI:29.3 – 154.9 months) compared to those with wild-type ARAF mutations (133 months, 95% CI:101 – 138 months, p=0.017) (Figure 3A). Similarly, patients with MAPK10 mutations exhibit a significantly shorter DFS compared to those wild-type (12.5 months, 95% CI:0.0 – 29 months vs 108 months, 95% CI:101 – 114 months; p<0.001) (Figure 3B).
Similarly, it was demonstrated that patients with right-sided tumors experienced a significantly shorter overall survival (OS) compared to patients with left-sided tumors (92.1 months, 95% CI:82.5 – 122 months vs 111.3 months, 95% CI:104 – 135 months; p=0.011) (Figure 4A). Moreover, relapsed patients demonstrated a significantly shorter OS compared to those non-relapsed (81.1 months, 95% CI:70.4 – 118 months vs 104.3 months, 95% CI:93.9 – 130 months; p=0.008) (Figure 4Β). Moreover, patients with AKT1, APC2, ARAF, BAD, MAPK10, RAC3, RHOA, TGFB2 and TGFB3 mutations exhibited a significantly shorter OS compared to wild-type patients (68.9 vs 107.9 months, p=0.03; 89.7 vs 109.4 months, p=0.001; 43.3 vs 109.4 months, p<0.001; 34 vs 107.7 months, p=0.011; 19.3 vs 108.6, p>0.001; 55.6 vs 109, p<0.001; 45.2 vs 108.5 months, p=0.004; 69.5 vs 109.7 months, p=0.032 και 30.5 vs 108.1 months p<0.001, respectively) (Figures 4C-K). Finally, patients with MSH6 gene mutations had a significantly longer OS compared to those wild-type (120.7 months, 95% CI:111.3 – 130.1 months vs 100.9 months, 95% CI:93.7 – 108.1 108.1 months; p=0.008) (Figure 4L).
3.5. Univariate and multivariate Cox-regression analysis
Univariate analysis revealed that tumor localization in the right colon, and ARAF and MAPK10 mutations were associated with reduced DFS (Table 7). Multivariate analysis confirmed that tumor localization and ARAF and MAPK10 mutations were independent predictive factors of reduced DFS (HR=2.1; 95% CI:1.1-4.0; p=0.019; HR=3.9 ; 95% CI:1.2-13.1; p=0.027; HR=49; 95% CI:9.8-244.1; p<0.001 (Table 7).
Similarly, right-sided tumors and AKT1, APC2, ARAF, BAD, MAPK10, RAC3, RHOA, TGFB2, and TGFB3 were associated with an increased risk of shorter OS. In contrast, MSH6 mutations were demonstrated to be a good prognostic factor, as they were associated with a reduced risk for shorter OS. Multivariate analysis revealed that right-sided tumors and RAC3 and RHOA gene mutations emerged as independent predictors of reduced OS (HR=2.2; 95% CI:1.0-4.5; p=0.043; HR=3.5; 95% CI:1.1-10.7; p=0.029 and HR=9.5; 95% CI:1.9-47.7; p=0.006 (Table 7).
4. Discussion
Despite the potential benefits of presymptomatic screening and available treatments, CRC continues to be a significant public health concern[16]. Understanding the processes involved in CRC development and progression can help to identify new targets for treatment. Structural and functional changes in the DNA can offer vital insights into patient management[17,18]. As the normal colonic epithelium transforms into cancerous tissue, various mutations occur, leading to adenoma formation[19,20,21,22,23,24,25,26]. Extensive cancer cell proliferation through the RAS-RAF-MEK-ERK signaling pathway drives carcinogenesis, tumor invasion, and metastasis[27]. Moreover, the immune responses to cancer cells differ among patients with mutations[28,29,30,31,32,33,34].
The objective of this study was to analyze genetic changes in surgical samples from patients with stage III CRC. The study included 237 patients, and the WES and KEGG gene panel for CRC revealed 59 mutated genes belonging to 11 distinct signaling pathways. Of these, mutations in APC2, BRAF, MAPK10, MLH1, MSH6, RHOA, TGFβ, and TGFβ2 have been linked to a significant impact on patient survival. APC, TP53, KRAS, and MSH3 were the most commonly observed mutations in this study.
APC encodes an anti-tumor protein that competes with the Wnt signaling pathway and is involved in cell migration, adhesion, and apoptosis. APC mutations are responsible for familial adenomatous polyposis (FAP), an autosomal dominant precancerous disease that typically leads to malignancy. APC mutations are commonly observed in CRC cases[35]. Similarly, APC2 mutations, which are directly associated with APC’s tumor-suppressive function[36], have been linked to worse prognosis in CRC patients[37,38]. This study confirms that APC2 mutations in patients with stage III CRC are associated with lower overall survival but do not represent an independent prognostic factor. TP53 encodes an anti-tumor protein that regulates the expression of target genes, leading to cell cycle arrest, apoptosis, senescence, DNA repair, or metabolic changes. Similar to APC, TP53 are frequently observed in CRC cases[35]. Furthermore, mutations in the APC2 gene, which is directly linked to APC’s tumor-suppressive function[36], are also associated with worse prognosis in CRC patients[37,39]. This study confirms that while APC2 mutations in stage III CRC patients are linked to lower overall survival, they do not represent an independent prognostic factor. Various human cancers, including approximately 60% of CRC, are associated with mutations in the TP53 gene[40,41]. Prior studies have shown that mutations in TP53 resulting in the loss of its transcriptional activity, can lead to uncontrolled cellular proliferation in multiple organs, including the colon[42]. Similarly, KRAS mutations are the primary indicators of gastrointestinal cancers and are found in approximately 40% of patients with CRC (stage II-IV)[43]. They serve as negative prognostic factors for carcinogenesis and anti-EGFR therapy[44] because intracellular signal interruption leads to uncontrolled cellular proliferation and cancer. MSH3 mutations have been mainly linked to endometrial cancer, but there are reports of its relationship with inflammatory processes, such as ulcerative colitis and Crohn’s disease, which considerably increase the likelihood of CRC development[45,46]. MSH3-associated CRC seems to follow the classic APC pathway, as patients with adenomas and CRC carrying APC mutations showed MSH3 deficiency[47], as confirmed in this study. In addition to the common mutations detected in the patient group, mutations in AKT1, ARAF, BAD, MAPK10, RAC3, RHOA, TGFB2, and TGFB3 were associated with worse prognosis in this study. Furthermore, mutations in ARAF and MAPK10 were identified as independent prognostic factors for DFS, whereas mutations in RAC3 and RHOA were identified as independent prognostic factors for decreased OS.
This study sheds light on the association between mutations in genes involved in signaling pathways such as PI3K-Akt, MAPK, apoptosis, and CRC. To the best of our knowledge, this is the first report of its kind in the literature. The reactivation of embryonic self-renewal pathways, such as Hedgehog, Notch, and TGFβ/Stat3, is characteristic of most tumors, including CRC. The Wnt pathway is also essential in most CRC. Targeting embryonic pathways directly is likely to be more effective against stem and differentiated cancer cells [48,49,50]. Tumors that are addicted to increased regulated activity of the embryonic pathway, in combination with high tumor heterogeneity, may be more vulnerable to such therapies [51,52,53]. Patients with VDR polymorphisms had concurrent mutations in genes involved in cell cycle, apoptosis, PI3K-Akt, WNT, MAPK, ErbB, MSI, and RAS. Similarly, TLR9 polymorphisms were associated with mutations in genes involved in apoptotic signaling pathways, PI3K-Akt, and Wnt. Previous studies from our group have demonstrated that higher detection of TLR and VDR polymorphisms in CRC patients, especially advanced-stage patients, highlights the role of these polymorphisms in carcinogenesis, disease progression, and ultimately, patient survival[9,10,54]. Regarding DFS, tumors in the sigmoid or right colon and mutations in the ARAF and/or MAPK10 genes were associated with shorter DFS, a fact that has been confirmed in previous studies[55,56,57]. To our knowledge, this is the first study to highlight the role of ARAF and MAPK10 mutations as independent prognostic factors for decreased DFS.
The observation of statistically lower OS in patients with right colon tumors and gene mutations has been confirmed in the literature. Borakati et al. conducted A retrospective study and found that tumors in the right colon were independent prognostic factors for reduced OS after hepatic metastasectomy, regardless of the higher rates of liver metastases and larger metastases in the left colon[58]. Patients with mutations in genes, such as AKT1, APC2, ARAF, BAD, MAPK10, RAC3, RHOA, TGFB2, and TGFB3 had significantly reduced OS, as reported in other studies that also linked APC2, RHOA, and TGFB mutations to worse prognosis[37,38,59]. Conversely, studies have shown that mutations in MSH6 are associated with a lower risk of developing CRC, and patients with such mutations have a milder clinical presentation[60,61]. In the present study, patients with MSH6 mutations had a significantly longer OS, confirming MSH6 mutations are good prognostic factors. Concurrent mutations (co-mutations) are a significant factor that have been minimally investigated in CRC. Studies in patients with non-small cell lung cancer have shown distinct biological behavior and prognosis in KRAS/LKB1, KRAS/TP53, or KRAS/p16 mutated tumors[62]. Additionally, our group has previously reported the importance of evaluating the loss of LKB1 through immunohistochemistry in early stage CRC, particularly in BRAFV600E mutated tumors[63]. In the present study, several concurrent mutations were detected in patients, but no correlation was found with clinical/pathological characteristics or patient prognosis.
5. Conclusions
In conclusion, molecular characterization of cancer cells can enhance our understanding of the biological progression of this disease[64,65]. The findings of this study suggest that mutations are promising prognostic biomarkers. As personalized medicine has become the primary mode of therapy, knowledge of the precise mutation status of patients with CRC can lead to better therapeutic choices. However, further research is necessary with a larger patient cohort and international collaborations to confirm the correlation between patients’ molecular profiles, clinicopathological and epidemiological characteristics, and outcomes. Such research is expected to contribute to more precise clinical decision-making, personalized and improved care, and reduced toxicity of treatment, costs to patients, and burden on health systems.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Raw patient data.
Author Contributions
Conceptualization, I.M. and J.S.; methodology, I.M., E.P., K.V., M.S. and M.T.; software, I.M., E.P., P.T. and I.I.; validation, I.M., P.T. and M.T.; formal analysis, I.M.; data curation, I.M, D.M., J.T., N.G., M.T. and J.S..; writing—original draft preparation, I.M.; writing—review and editing, I.M., P.T., I.L. and J.S.; supervision, I.M., P.T. and J.S.; funding acquisition, I.M. and J.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was partially funded by the Hellenic Society of Medical Oncology (HeSMO) and the Gastrointestinal Cancer Study Group (GIC-SG).
Institutional Review Board Statement
This study was approved by the Ethics Committee/Institutional Review Board of the University Hospital of Heraklion (Number 7302/19-8-2009). All procedures were performed in accordance with the ethical standards of the institutional and/or national research committee and the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. All patients signed a written informed consent form for participation.
Data Availability Statement
All relevant data are within the paper and its Supporting Information files. Supplementary Table S1: Raw patient data
Acknowledgement
The authors would like to thank NATERA Inc. for providing whole exome sequencing analysis and sharing the raw data for further bioinformatic analysis.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Yu, H.; Hemminki, K. Genetic epidemiology of colorectal cancer and associated cancers. Mutagenesis 2020, 35, 207–219. [Google Scholar] [CrossRef]
- Sawicki, T.; Ruszkowska, M.; Danielewicz, A.; Niedzwiedzka, E.; Arlukowicz, T.; Przybylowicz, K.E. A Review of Colorectal Cancer in Terms of Epidemiology, Risk Factors, Development, Symptoms and Diagnosis. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef]
- Andre, T.; Boni, C.; Navarro, M.; Tabernero, J.; Hickish, T.; Topham, C.; Bonetti, A.; Clingan, P.; Bridgewater, J.; Rivera, F.; et al. Improved overall survival with oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment in stage II or III colon cancer in the MOSAIC trial. J Clin Oncol 2009, 27, 3109–3116. [Google Scholar] [CrossRef]
- Haller, D.G.; Tabernero, J.; Maroun, J.; de Braud, F.; Price, T.; Van Cutsem, E.; Hill, M.; Gilberg, F.; Rittweger, K.; Schmoll, H.J. Capecitabine plus oxaliplatin compared with fluorouracil and folinic acid as adjuvant therapy for stage III colon cancer. J Clin Oncol 2011, 29, 1465–1471. [Google Scholar] [CrossRef]
- Sobrero, A.; Grothey, A.; Iveson, T.; Labianca, R.; Yoshino, T.; Taieb, J.; Maughan, T.; Buyse, M.; Andre, T.; Meyerhardt, J.; et al. The hard road to data interpretation: 3 or 6 months of adjuvant chemotherapy for patients with stage III colon cancer? Ann Oncol 2018, 29, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Sobrero, A.F.; Shields, A.F.; Yoshino, T.; Paul, J.; Taieb, J.; Souglakos, J.; Shi, Q.; Kerr, R.; Labianca, R.; et al. Duration of Adjuvant Chemotherapy for Stage III Colon Cancer. N Engl J Med 2018, 378, 1177–1188. [Google Scholar] [CrossRef]
- Angell, H.K.; Bruni, D.; Barrett, J.C.; Herbst, R.; Galon, J. The Immunoscore: Colon Cancer and Beyond. Clin Cancer Res 2020, 26, 332–339. [Google Scholar] [CrossRef]
- Messaritakis, I.; Koulouridi, A.; Boukla, E.; Sfakianaki, M.; Vogiatzoglou, K.; Karagianni, M.; Gouvas, N.; Tsiaoussis, J.; Xynos, E.; Athanasakis, E.; et al. Investigation of Microbial Translocation, TLR and VDR Gene Polymorphisms, and Recurrence Risk in Stage III Colorectal Cancer Patients. Cancers (Basel) 2022, 14. [Google Scholar] [CrossRef]
- Messaritakis, I.; Vogiatzoglou, K.; Tsantaki, K.; Ntretaki, A.; Sfakianaki, M.; Koulouridi, A.; Tsiaoussis, J.; Mavroudis, D.; Souglakos, J. The Prognostic Value of the Detection of Microbial Translocation in the Blood of Colorectal Cancer Patients. Cancers (Basel) 2020, 12. [Google Scholar] [CrossRef]
- Messaritakis, I.; Koulouridi, A.; Sfakianaki, M.; Vogiatzoglou, K.; Gouvas, N.; Athanasakis, E.; Tsiaoussis, J.; Xynos, E.; Mavroudis, D.; Tzardi, M.; et al. The Role of Vitamin D Receptor Gene Polymorphisms in Colorectal Cancer Risk. Cancers (Basel) 2020, 12. [Google Scholar] [CrossRef]
- Available online: https://support.illumina.com/sequencing/sequencing_software/igenome.html.
- Geraldine A. Van der Auwera, B.D.O.C. Genomics in the cloud: Using docker, gatk, and wdl in terra, 1st ed.; 2020.
- Available online: https://www.genome.jp/pathway/hsa05210.
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010, 38, e164. [Google Scholar] [CrossRef]
- Deng, N.; Zhou, H.; Fan, H.; Yuan, Y. Single nucleotide polymorphisms and cancer susceptibility. Oncotarget 2017, 8. [Google Scholar]
- Brenner, H.; Chen, C. The colorectal cancer epidemic: challenges and opportunities for primary, secondary and tertiary prevention. Br J Cancer 2018, 119, 785–792. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Obuch, J.C.; Ahnen, D.J. Colorectal Cancer: Genetics is Changing Everything. Gastroenterol Clin North Am 2016, 45, 459–476. [Google Scholar] [CrossRef]
- Sullivan, B.A.; Noujaim, M.; Roper, J. Cause, Epidemiology, and Histology of Polyps and Pathways to Colorectal Cancer. Gastrointest Endosc Clin N Am 2022, 32, 177–194. [Google Scholar] [CrossRef]
- Uhlitz, F.; Bischoff, P.; Peidli, S.; Sieber, A.; Trinks, A.; Luthen, M.; Obermayer, B.; Blanc, E.; Ruchiy, Y.; Sell, T.; et al. Mitogen-activated protein kinase activity drives cell trajectories in colorectal cancer. EMBO Mol Med 2021, 13, e14123. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Chan, D.K.H.; Buczacki, S.J.A. Tumour heterogeneity and evolutionary dynamics in colorectal cancer. Oncogenesis 2021, 10, 53. [Google Scholar] [CrossRef]
- Fennell, L.; Kane, A.; Liu, C.; McKeone, D.; Hartel, G.; Su, C.; Bond, C.; Bettington, M.; Leggett, B.; Whitehall, V. Braf mutation induces rapid neoplastic transformation in the aged and aberrantly methylated intestinal epithelium. Gut 2022, 71, 1127–1140. [Google Scholar] [CrossRef]
- Chen, T.; Zeineldin, M.; Johnson, B.A.; Dong, Y.; Narkar, A.; Li, T.; Zhu, J.; Li, R.; Larman, T.C. Colonic epithelial adaptation to EGFR-independent growth induces chromosomal instability and is accelerated by prior injury. Neoplasia 2021, 23, 488–501. [Google Scholar] [CrossRef]
- Lee-Six, H.; Olafsson, S.; Ellis, P.; Osborne, R.J.; Sanders, M.A.; Moore, L.; Georgakopoulos, N.; Torrente, F.; Noorani, A.; Goddard, M.; et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature 2019, 574, 532–537. [Google Scholar] [CrossRef]
- Norgaard, K.; Muller, C.; Christensen, N.; Chiloeches, M.L.; Madsen, C.L.; Nielsen, S.S.; Thingholm, T.E.; Belcheva, A. Loss of mismatch repair signaling impairs the WNT-bone morphogenetic protein crosstalk and the colonic homeostasis. J Mol Cell Biol 2020, 12, 410–423. [Google Scholar] [CrossRef]
- Eklof, V.; Wikberg, M.L.; Edin, S.; Dahlin, A.M.; Jonsson, B.A.; Oberg, A.; Rutegard, J.; Palmqvist, R. The prognostic role of KRAS, BRAF, PIK3CA and PTEN in colorectal cancer. Br J Cancer 2013, 108, 2153–2163. [Google Scholar] [CrossRef]
- Fu, X.; Wang, X.; Duanmu, J.; Li, T.; Jiang, Q. KRAS mutations are negatively correlated with immunity in colon cancer. Aging (Albany NY) 2020, 13, 750–768. [Google Scholar] [CrossRef]
- Sun, J.; Yao, N.; Lu, P.; Wang, Y. Effects of mFOLFOX6 regimen combined with carrelizumab on immune function and prognosis in patients with microsatellite instability colorectal cancer. Cell Mol Biol (Noisy-le-grand) 2022, 67, 356–362. [Google Scholar] [CrossRef]
- Ratovomanana, T.; Cohen, R.; Svrcek, M.; Renaud, F.; Cervera, P.; Siret, A.; Letourneur, Q.; Buhard, O.; Bourgoin, P.; Guillerm, E.; et al. Performance of Next-Generation Sequencing for the Detection of Microsatellite Instability in Colorectal Cancer With Deficient DNA Mismatch Repair. Gastroenterology 2021, 161, 814–826. [Google Scholar] [CrossRef]
- Xiao, J.; Li, W.; Huang, Y.; Huang, M.; Li, S.; Zhai, X.; Zhao, J.; Gao, C.; Xie, W.; Qin, H.; et al. A next-generation sequencing-based strategy combining microsatellite instability and tumor mutation burden for comprehensive molecular diagnosis of advanced colorectal cancer. BMC Cancer 2021, 21, 282. [Google Scholar] [CrossRef]
- Huang, J.; Liu, H.; Zhao, Y.; Luo, T.; Liu, J.; Liu, J.; Pan, X.; Tang, W. MicroRNAs Expression Patterns Predict Tumor Mutational Burden in Colorectal Cancer. Front Oncol 2020, 10, 550986. [Google Scholar] [CrossRef]
- Bae, J.M.; Yoo, S.Y.; Kim, J.H.; Kang, G.H. Immune landscape and biomarkers for immuno-oncology in colorectal cancers. J Pathol Transl Med 2020, 54, 351–360. [Google Scholar] [CrossRef]
- Saller, J.; Qin, D.; Felder, S.; Coppola, D. Microsatellite Stable Colorectal Cancer With an Immunogenic Phenotype: Challenges in Diagnosis and Treatment. Clin Colorectal Cancer 2020, 19, 123–131. [Google Scholar] [CrossRef]
- Available online: https://www.ncbi.nlm.nih.gov/gene/324.
- Available online: https://www.ncbi.nlm.nih.gov/gene/10297.
- Sun, Y.; Tian, H.; Xu, X.; Wang, L. Low expression of adenomatous polyposis coli 2 correlates with aggressive features and poor prognosis in colorectal cancer. Bioengineered 2020, 11, 1027–1033. [Google Scholar] [CrossRef]
- Geng, Y.; Zheng, X.; Hu, W.; Wang, Q.; Xu, Y.; He, W.; Wu, C.; Zhu, D.; Wu, C.; Jiang, J. Hsa_circ_0009361 acts as the sponge of miR-582 to suppress colorectal cancer progression by regulating APC2 expression. Clin Sci (Lond) 2019, 133, 1197–1213. [Google Scholar] [CrossRef]
- Geng, Y.; Zheng, X.; Hu, W.; Wang, Q.; Xu, Y.; He, W.; Wu, C.; Zhu, D.; Jiang, J. Hsa_circ_0009361 acts as the sponge of miR-582 to suppress colorectal cancer progression by regulating APC2 expression. Clin Sci (Lond) 2019, 133, 1197–1213. [Google Scholar] [CrossRef]
- Marcel, V.; Perrier, S.; Aoubala, M.; Ageorges, S.; Groves, M.J.; Diot, A.; Fernandes, K.; Tauro, S.; Bourdon, J.C. Delta160p53 is a novel N-terminal p53 isoform encoded by Delta133p53 transcript. FEBS Lett 2010, 584, 4463–4468. [Google Scholar] [CrossRef]
- Yin, Y.; Stephen, C.W.; Luciani, M.G.; Fåhraeus, R. p53 stability and activity is regulated by Mdm2-mediated induction of alternative p53 translation products. Nature Cell Biology 2002, 4, 462–467. [Google Scholar] [CrossRef]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb Perspect Med 2016, 6, a026104. [Google Scholar] [CrossRef]
- Andreyev, H.J.; Norman, A.R.; Cunningham, D.; Oates, J.; Dix, B.R.; Iacopetta, B.J.; Young, J.; Walsh, T.; Ward, R.; Hawkins, N.; et al. Kirsten ras mutations in patients with colorectal cancer: the ‘RASCAL II’ study. Br J Cancer 2001, 85, 692–696. [Google Scholar] [CrossRef]
- De Roock, W.; Claes, B.; Bernasconi, D.; De Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 2010, 11, 753–762. [Google Scholar] [CrossRef]
- Biscaglia, G.; Latiano, A.; Castellana, S.; Fontana, R.; Gentile, A.; Latiano, T.; Corritore, G.; Panza, A.; Nardella, M.; Martino, G.; et al. Germline Alterations in Patients With IBD-associated Colorectal Cancer. Inflamm Bowel Dis 2022, 28, 447–454. [Google Scholar] [CrossRef]
- Xie, Z.; Ke, Y.; Chen, J.; Li, Z.; Wang, C.; Chen, Y.; Ding, H.; Cheng, L. Prevalence and Spectrum of Predisposition Genes With Germline Mutations Among Chinese Patients With Bowel Cancer. Front Genet 2021, 12, 755629. [Google Scholar] [CrossRef]
- Perne, C.; Peters, S.; Cartolano, M.; Horpaopan, S.; Grimm, C.; Altmuller, J.; Sommer, A.K.; Hillmer, A.M.; Thiele, H.; Odenthal, M.; et al. Variant profiling of colorectal adenomas from three patients of two families with MSH3-related adenomatous polyposis. PLoS One 2021, 16, e0259185. [Google Scholar] [CrossRef]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol 2011, 8, 97–106. [Google Scholar] [CrossRef]
- Medema, J.P. Cancer stem cells: the challenges ahead. Nat Cell Biol 2013, 15, 338–344. [Google Scholar] [CrossRef]
- Pattabiraman, D.R.; Weinberg, R.A. Tackling the cancer stem cells—what challenges do they pose? Nat Rev Drug Discov 2014, 13, 497–512. [Google Scholar] [CrossRef]
- Bienz, M.; Clevers, H. Linking colorectal cancer to Wnt signaling. Cell 2000, 103, 311–320. [Google Scholar] [CrossRef]
- Segditsas, S.; Tomlinson, I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene 2006, 25, 7531–7537. [Google Scholar] [CrossRef]
- Hernandez-Aya, L.F.; Gonzalez-Angulo, A.M. Targeting the phosphatidylinositol 3-kinase signaling pathway in breast cancer. Oncologist 2011, 16, 404–414. [Google Scholar] [CrossRef]
- Messaritakis, I.; Stogiannitsi, M.; Koulouridi, A.; Sfakianaki, M.; Voutsina, A.; Sotiriou, A.; Athanasakis, E.; Xynos, E.; Mavroudis, D.; Tzardi, M.; et al. Evaluation of the detection of Toll-like receptors (TLRs) in cancer development and progression in patients with colorectal cancer. PLoS One 2018, 13, e0197327. [Google Scholar] [CrossRef]
- Deng, J.; Zhou, S.; Wang, Z.; Huang, G.; Zeng, J.; Li, X. Comparison of Prognosis and Lymph Node Metastasis in T1-Stage Colonic and Rectal Carcinoma: A Retrospective Study. Int J Gen Med 2022, 15, 3651–3662. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kim, K.; Cho, S.H.; Chun, S.M.; Tak, E.; Hong, Y.S.; Kim, J.E.; Kim, T.W. Longitudinal change of genetic variations in cetuximab-treated metastatic colorectal cancer. Cancer Genet 2021, 258-259, 27–36. [Google Scholar] [CrossRef]
- Ohnami, S.; Maruyama, K.; Chen, K.; Takahashi, Y.; Hatakeyama, K.; Ohshima, K.; Shimoda, Y.; Sakai, A.; Kamada, F.; Nakatani, S.; et al. BMP4 and PHLDA1 are plausible drug-targetable candidate genes for KRAS G12A-, G12D-, and G12V-driven colorectal cancer. Mol Cell Biochem 2021, 476, 3469–3482. [Google Scholar] [CrossRef] [PubMed]
- Borakati, A.; Froghi, F.; Shetye, A.; Fusai, G.K.; Davidson, B.R.; Mirnezami, R. Assessing the Impact of Primary Tumour Location on Survival After Resection of Colorectal Liver Metastases: A Propensity Weighted Retrospective Cohort Study. World J Surg 2022, 46, 1734–1755. [Google Scholar] [CrossRef] [PubMed]
- Il, J.H.; Young, C.Y.; Jun, B.M.; Ho, B.S.; Byung, B.S.; Jun, J.D.; Yong, K.S.; Soo, L.M.; Sik, C.M.; Ho, K.C. Expression of RhoA in Colorectal Cancers and Its Clinicopathological Significance. J Korean Soc Coloproctol 2008, 24, 460–466. [Google Scholar] [CrossRef]
- Bonadona, V.; Bonaïti, B.; Olschwang, S.; Grandjouan, S.; Huiart, L.; Longy, M.; Guimbaud, R.; Buecher, B.; Bignon, Y.J.; Caron, O.; et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. Jama 2011, 305, 2304–2310. [Google Scholar] [CrossRef]
- Ramsoekh, D.; Wagner, A.; van Leerdam, M.E.; Dooijes, D.; Tops, C.M.; Steyerberg, E.W.; Kuipers, E.J. Cancer risk in MLH1, MSH2 and MSH6 mutation carriers; different risk profiles may influence clinical management. Hered Cancer Clin Pract 2009, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Heymach, J.V. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat Rev Cancer 2019, 19, 495–509. [Google Scholar] [CrossRef]
- Sfakianaki, M.; Papadaki, C.; Tzardi, M.; Trypaki, M.; Alam, S.; Lagoudaki, E.D.; Messaritakis, I.; Zoras, O.; Mavroudis, D.; Georgoulias, V.; et al. Loss of LKB1 Protein Expression Correlates with Increased Risk of Recurrence and Death in Patients with Resected, Stage II or III Colon Cancer. Cancer Res Treat 2019, 51, 1518–1526. [Google Scholar] [CrossRef]
- He, K.; Wang, Y.; Zhong, Y.; Pan, X.; Si, L.; Lu, J. KRAS Codon 12 Mutation is Associated with More Aggressive Invasiveness in Synchronous Metastatic Colorectal Cancer (mCRC): Retrospective Research. Onco Targets Ther 2020, 13, 12601–12613. [Google Scholar] [CrossRef]
- Scott, A.; Goffredo, P.; Ginader, T.; Hrabe, J.; Gribovskaja-Rupp, I.; Kapadia, M.R.; Weigel, R.J.; Hassan, I. The Impact of KRAS Mutation on the Presentation and Prognosis of Non-Metastatic Colon Cancer: an Analysis from the National Cancer Database. J Gastrointest Surg 2020, 24, 1402–1410. [Google Scholar] [CrossRef]
Figure 1.
Overview of the next generation sequencing (NGS) analysis pipeline.
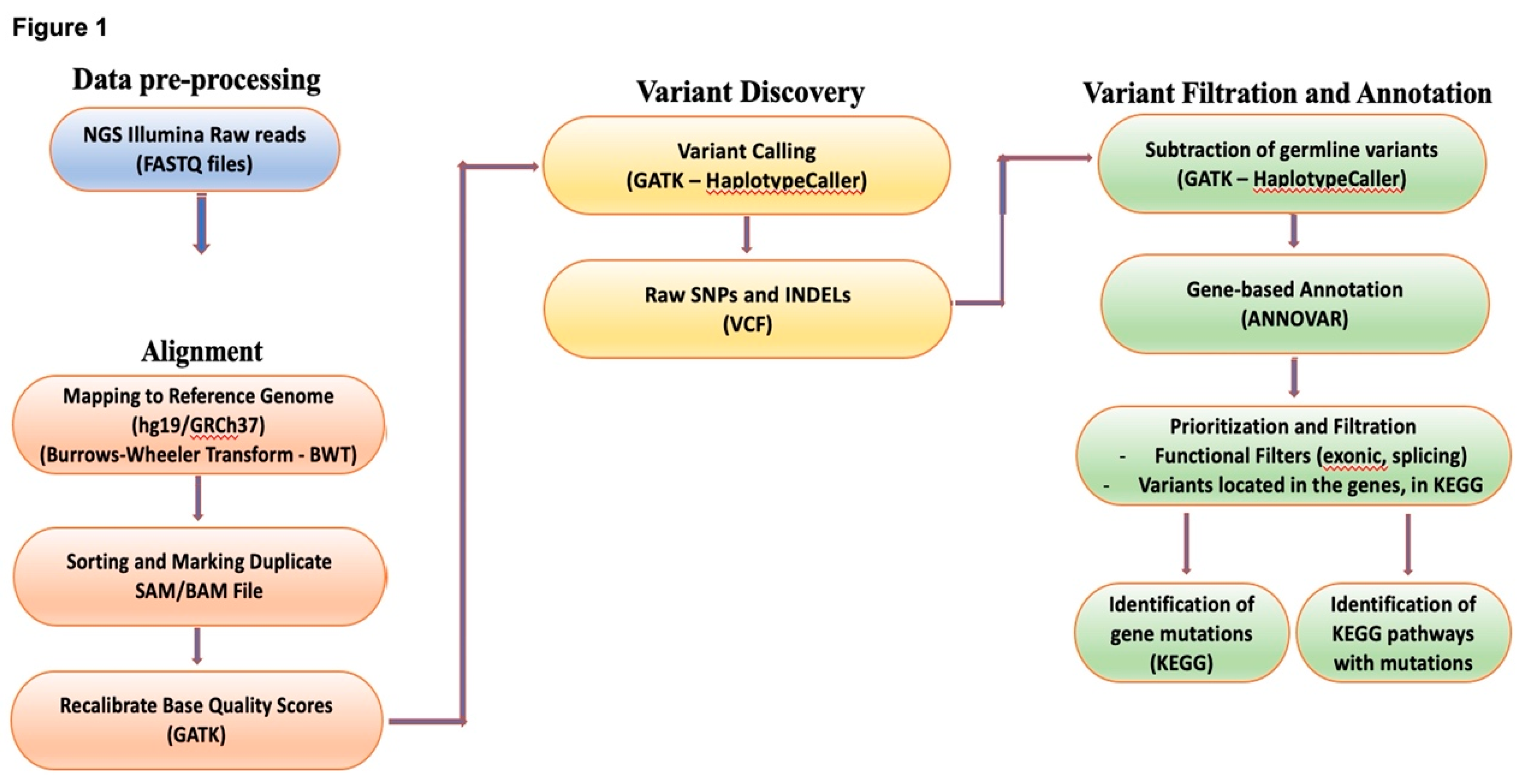
Figure 2.
Frequency of mutations in each gene located in exons and splicing sites.
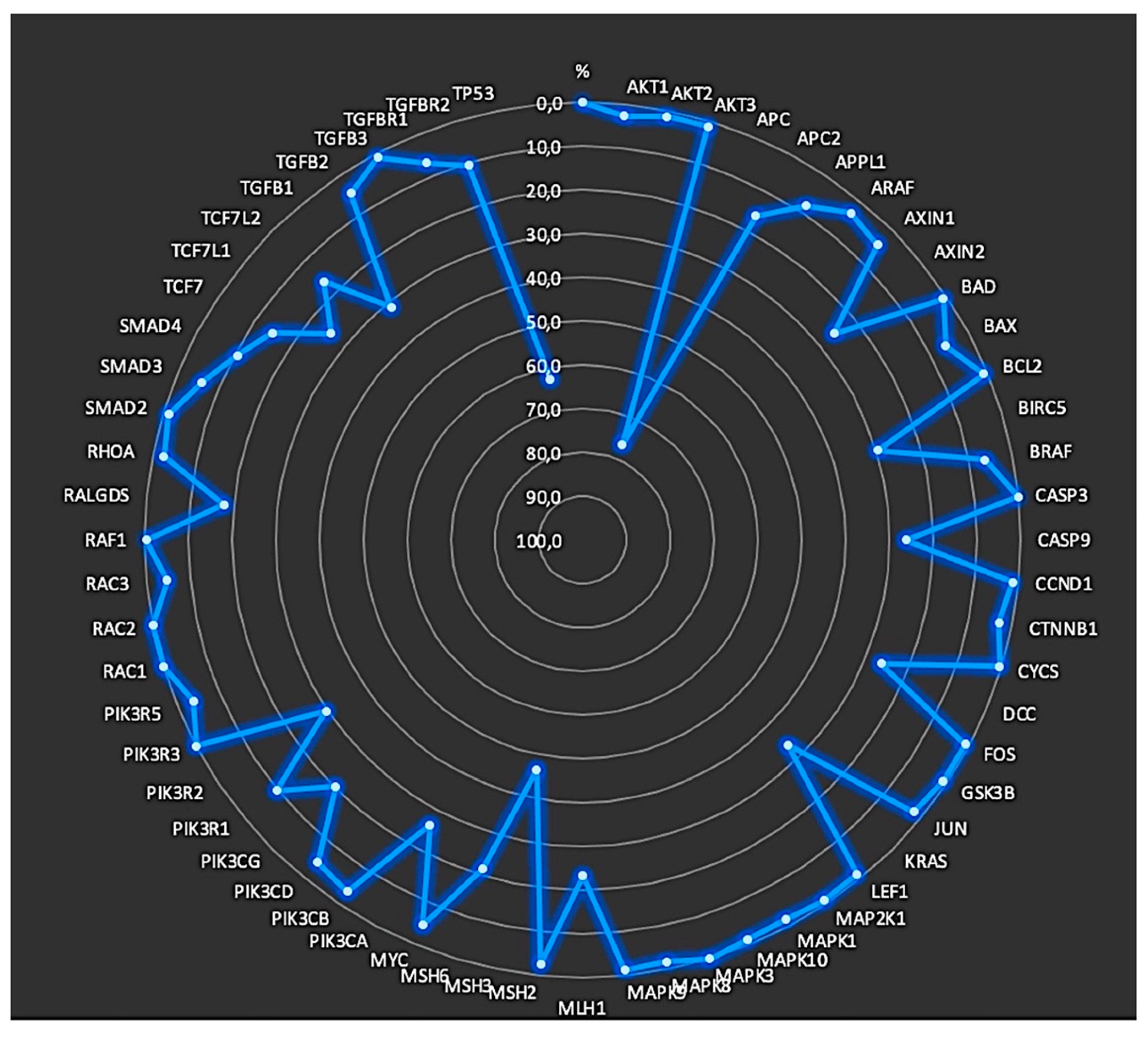
Figure 3.
Kaplan Meier curve for disease-free survival (DFS) according to (A) ARAF and (B) MAPK10 mutations.
Figure 3.
Kaplan Meier curve for disease-free survival (DFS) according to (A) ARAF and (B) MAPK10 mutations.
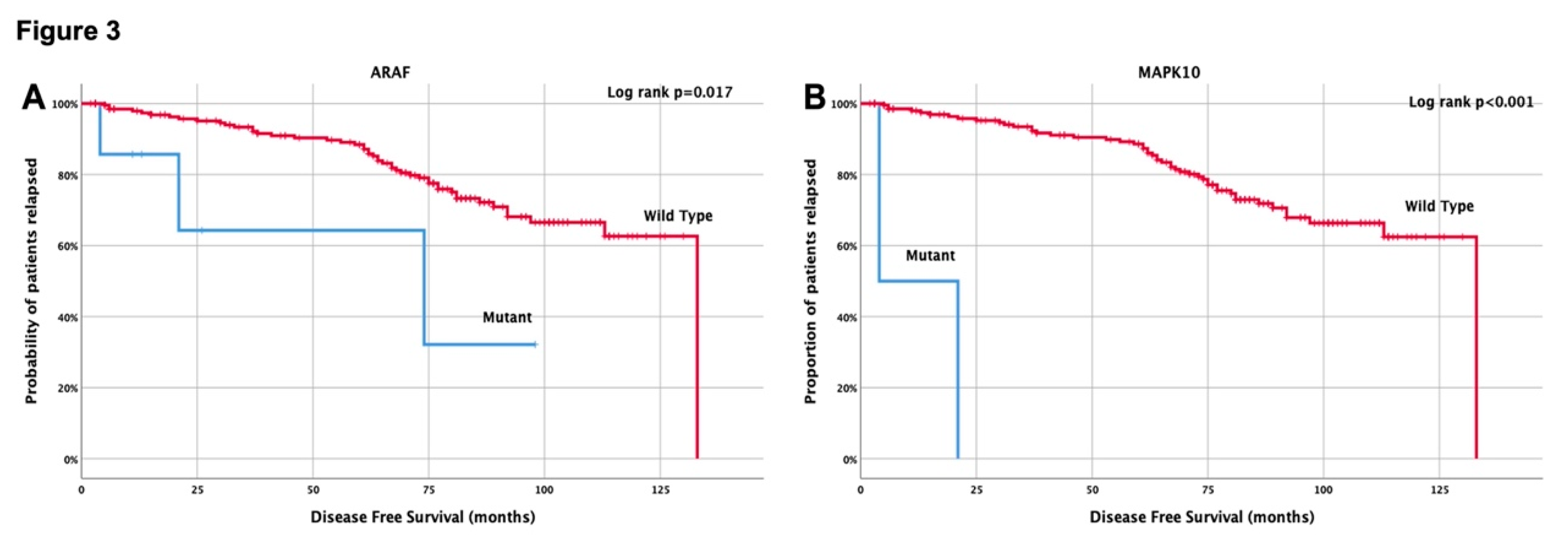
Figure 4.
Kaplan Meier curve for overall survival (OS) according to (A) tumor sidedness, (B) relapse status, (C–L) AKT1, APC2, ARAF, BAD, MAPK10, RAC3, RHOA, TGFB2, TGFB3 and MSH6 mutations.
Figure 4.
Kaplan Meier curve for overall survival (OS) according to (A) tumor sidedness, (B) relapse status, (C–L) AKT1, APC2, ARAF, BAD, MAPK10, RAC3, RHOA, TGFB2, TGFB3 and MSH6 mutations.
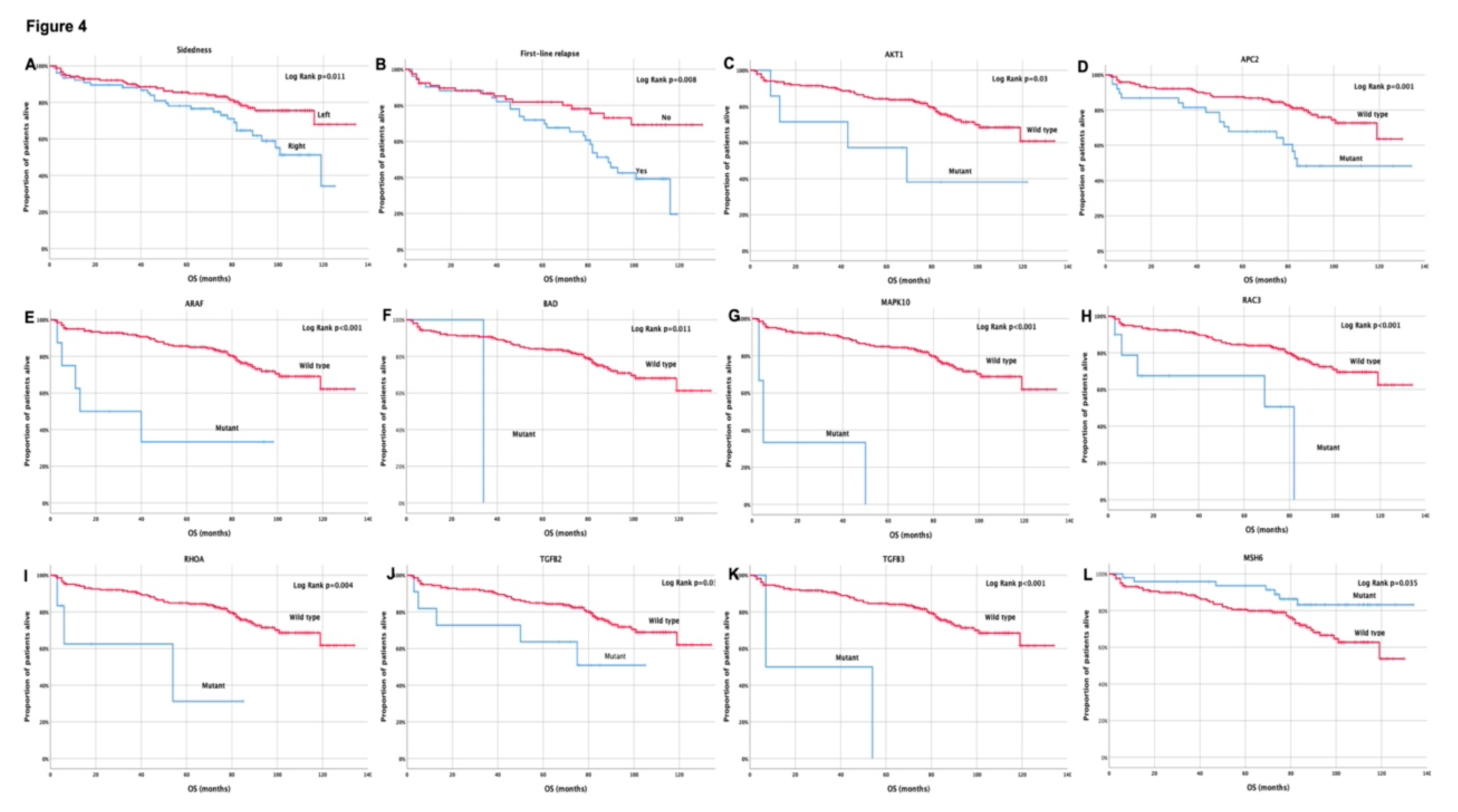

Table 1.
Gene panel for colorectal cancer (CRC) based on Kyoto Encyclopedia of Genes and Genomes (KEGG).
Table 1.
Gene panel for colorectal cancer (CRC) based on Kyoto Encyclopedia of Genes and Genomes (KEGG).
| Gene | Chromosome | Location based on GRCh37.p13 assembly | Source[https://www.ensembl.org/index.html] | |
|---|---|---|---|---|
| Start | End | |||
| AKT1 | 14 | 105235686 | 105262085 | Ensembl:ENSG00000142208 |
| AKT2 | 19 | 40736224 | 40791252 | Ensembl:ENSG00000105221 |
| AKT3 | 1 | 243651535 | 244014381 | Ensembl:ENSG00000117020 |
| APC | 5 | 112043195 | 112181936 | Ensembl:ENSG00000134982 |
| APC2 | 19 | 1450120 | 1473243 | Ensembl:ENSG00000115266 |
| APPL1 | 3 | 57261757 | 57307499 | Ensembl:ENSG00000157500 |
| ARAF | X | 47420604 | 47431307 | Ensembl:ENSG00000078061 |
| AXIN1 | 16 | 337440 | 402723 | Ensembl:ENSG00000103126 |
| AXIN2 | 17 | 63524681 | 63557766 | Ensembl:ENSG00000168646 |
| BAD | 11 | 64037300 | 64052176 | Ensembl:ENSG00000002330 |
| BAX | 19 | 49458132 | 49465055 | Ensembl:ENSG00000087088 |
| BCL2 | 18 | 60790579 | 60987002 | Ensembl:ENSG00000171791 |
| BIRC5 | 17 | 76210334 | 76221716 | Ensembl:ENSG00000089685 |
| BRAF | 7 | 140413128 | 140624729 | Ensembl:ENSG00000157764 |
| CASP3 | 4 | 185548850 | 185570601 | Ensembl:ENSG00000164305 |
| CASP9 | 1 | 15817896 | 15851285 | Ensembl:ENSG00000132906 |
| CCND1 | 11 | 69455924 | 69469242 | Ensembl:ENSG00000110092 |
| CTNNB1 | 3 | 41240996 | 41281934 | Ensembl:ENSG00000168036 |
| CYCS | 7 | 25158275 | 25164879 | Ensembl:ENSG00000172115 |
| DCC | 18 | 49866567 | 51062269 | Ensembl:ENSG00000187323 |
| FOS | 14 | 75745531 | 75748933 | Ensembl:ENSG00000170345 |
| GSK3B | 3 | 119540168 | 119813294 | Ensembl:ENSG00000082701 |
| JUN | 1 | 59246463 | 59249719 | Ensembl:ENSG00000177606 |
| KRAS | 12 | 25358180 | 25403863 | Ensembl:ENSG00000133703 |
| LEF1 | 4 | 108968704 | 109090088 | Ensembl:ENSG00000138795 |
| MAP2K1 | 15 | 66679250 | 66783882 | Ensembl:ENSG00000169032 |
| MAPK1 | 22 | 22113946 | 22221970 | Ensembl:ENSG00000100030 |
| MAPK10 | 4 | 86931558 | 87374348 | Ensembl:ENSG00000109339 |
| MAPK3 | 16 | 30125426 | 30134541 | Ensembl:ENSG00000102882 |
| MAPK8 | 10 | 49514720 | 49647403 | Ensembl:ENSG00000107643 |
| MAPK9 | 5 | 179660143 | 179719083 | Ensembl:ENSG00000050748 |
| MLH1 | 3 | 3703500 | 37092337 | Ensembl:ENSG00000076242 |
| MSH2 | 2 | 47630206 | 47710367 | Ensembl:ENSG00000095002 |
| MSH3 | 5 | 79950471 | 80172634 | Ensembl:ENSG00000113318 |
| MSH6 | 2 | 48010284 | 48034092 | Ensembl:ENSG00000116062 |
| MYC | 8 | 128747680 | 128755197 | Ensembl:ENSG00000136997 |
| PIK3CA | 3 | 178866145 | 178957881 | Ensembl:ENSG00000121879 |
| PIK3CB | 3 | 138371540 | 138553770 | Ensembl:ENSG00000051382 |
| PIK3CD | 1 | 9711789 | 9789172 | Ensembl:ENSG00000171608 |
| PIK3CG | 7 | 106505727 | 106549425 | Ensembl:ENSG00000105851 |
| PIK3R1 | 5 | 67511584 | 67597649 | Ensembl:ENSG00000145675 |
| PIK3R2 | 19 | 18263973 | 18281342 | Ensembl:ENSG00000105647 |
| PIK3R3 | 1 | 46505812 | 46640573 | Ensembl:ENSG00000117461 |
| PIK3R5 | 17 | 8782233 | 8869024 | Ensembl:ENSG00000141506 |
| RAC1 | 7 | 6414158 | 6443598 | Ensembl:ENSG00000136238 |
| RAC2 | 22 | 37621310 | 37640309 | Ensembl:ENSG00000128340 |
| RAC3 | 17 | 79989554 | 79992080 | Ensembl:ENSG00000169750 |
| RAF1 | 3 | 12625100 | 12705616 | Ensembl:ENSG00000132155 |
| RALGDS | 9 | 135973109 | 136024597 | Ensembl:ENSG00000160271 |
| RHOA | 3 | 49396578 | 49449409 | Ensembl:ENSG00000067560 |
| SMAD2 | 18 | 45335328 | 45457243 | Ensembl:ENSG00000175387 |
| SMAD3 | 15 | 67357940 | 67487507 | Ensembl:ENSG00000166949 |
| SMAD4 | 18 | 48556583 | 48611412 | Ensembl:ENSG00000141646 |
| TCF7 | 5 | 133450372 | 133483901 | Ensembl:ENSG00000081059 |
| TCF7L1 | 2 | 85360515 | 85537510 | Ensembl:ENSG00000152284 |
| TCF7L2 | 10 | 114710006 | 114927437 | Ensembl:ENSG00000148737 |
| TGFB1 | 19 | 41836228 | 41859827 | Ensembl:ENSG00000105329 |
| TGFB2 | 1 | 218518678 | 218617961 | Ensembl:ENSG00000092969 |
| TGFB3 | 14 | 76424440 | 76449354 | Ensembl:ENSG00000119699 |
| TGFBR1 | 9 | 101867395 | 101916474 | Ensembl:ENSG00000106799 |
| TGFBR2 | 3 | 30647994 | 30735634 | Ensembl:ENSG00000163513 |
| TP53 | 17 | 7571739 | 7590808 | Ensembl:ENSG00000141510 |
Table 2.
Signaling pathways and the associated genes for colorectal cancer (CRC) based on Kyoto Encyclopedia of Genes and Genomes (KEGG).
Table 2.
Signaling pathways and the associated genes for colorectal cancer (CRC) based on Kyoto Encyclopedia of Genes and Genomes (KEGG).
| Pathway | KEGG CRC genes | References [https://www.genome.jp] |
|---|---|---|
| Cell cycle | CCND1, GSK3B, MYC, SMAD2, SMAD3, SMAD4, TGFB1, TGFB2, TGFB3, TP53 | https://www.genome.jp/pathway/hsa04110 |
| p53 signaling pathway | BAX, BCL2, CASP3, CASP9, CCND1, CYCS, TP53 | https://www.genome.jp/pathway/hsa04115 |
| Apoptosis | AKT1, AKT2, AKT3, BAD, BAX, BCL2, BIRC5, CASP3, CASP9, CYCS, FOS, JUN, KRAS, MAP2K1, MAPK1, MAPK10, MAPK3, MAPK8, MAPK9, PIK3CA, PIK3CB, PIK3CD, PIK3R1, PIK3R2, PIK3R3, RAF1, TP53, DCC, APPL1 | https://www.genome.jp/pathway/hsa04210 |
| mTOR signaling pathway | BRAF, GSK3B, KRAS, MAP2K1, MAPK1, PIK3CA, PIK3CB, PIK3CD, PIK3R1, PIK3R2, PIK3R3, RAF1, RHOA | https://www.genome.jp/pathway/hsa04150 |
| PI3K-Akt signaling pathway | AKT1, AKT2, AKT3, BAD, BCL2, CASP9, GSK3B, KRAS, MAP2K1, MAPK1, MAPK3, MYC, PIK3CA, PIK3CB, PIK3CD, PIK3CG, PIK3R1, PIK3R2, PIK3R3, PIK3R5, RAC1, RAF1, TP53 | https://www.genome.jp/pathway/hsa04151 |
| Wnt signaling pathway | APC, APC2, AXIN1, AXIN2, CCND1,CTNNB1, GSK3B, JUN, LEF1, MAPK10, MAPK8, MAPK9, MYC, RAC1, RAC2, RAC3, RHOA, SMAD3, SMAD4, TCF7, TCF7L1, TCF7L2, TP53 | https://www.genome.jp/pathway/hsa04310 |
| TGF-beta signaling pathway | MAPK1, MAPK3, MYC, RHOA, SMAD2, SMAD3, SMAD4, TGFB1, TGFB2, TGFB3, TGFBR1, TGFBR2, | https://www.genome.jp/pathway/hsa04350 |
| MAPK signaling pathway | AKT1, AKT2, AKT3, ARAF, BRAF, CASP3, FOS, JUN, KRAS, LEF1, MAP2K1, MAPK1, MAPK10, MAPK3, MAPK8, MAPK9, MYC, RAC1, RAC2, RAC3, RAF1, TGFB1, TGFB2, TGFB3, TGFBR1, TGFBR2, TP53 | https://www.genome.jp/pathway/hsa04010 |
| ErbB signaling pathway | AKT1, AKT2, AKT3, ARAF, BAD, BRAF, GSK3B, JUN, KRAS, MAP2K1, MAPK1, MAPK10, MAPK3, MAPK8, MAPK9, MYC, PIK3CA, PIK3CB, PIK3CD, PIK3R1, PIK3R2, PIK3R3, RAF | https://www.genome.jp/pathway/hsa04012 |
| MSI pathway | APC, AXIN1, AXIN2, BAD, BAX, BCL2, GSK3B, MLH1, MSH2, MSH3, MSH6, TGFBR2 | https://www.genome.jp/dbget-bin/www_bget?path:map05210 |
| Ras signaling pathway | AKT1, AKT2, AKT3, BAD, KRAS, MAP2K1, MAPK1, MAPK10, MAPK3, MAPK8, MAPK9, PIK3CA, PIK3CB, PIK3CD, PIK3R1, PIK3R2, PIK3R3, RAC1, RAC2, RAC3, RAF1, RALGDS, RHOA | https://www.genome.jp/pathway/hsa04014 |
Table 3.
Patients characteristics.
| Characteristics | Number of patients (Ν=237) | % |
|---|---|---|
| Median age (range) | 64 (18–84) | |
| <70 | 159 | 67.1 |
| ≥70 | 78 | 32.9 |
| Gender | ||
| Males | 151 | 63.7 |
| Females | 86 | 36.3 |
| Tumor location | ||
| Cecum | 38 | 16.0 |
| Ascending | 42 | 17.7 |
| Transverse | 22 | 9.3 |
| Descending | 24 | 10.1 |
| Sigmoid | 111 | 46.8 |
| Sidedness | ||
| Left | 158 | 66.7 |
| Right | 79 | 33.3 |
| Performance status | ||
| 0-1 | 236 | 99.6 |
| >2 | 1 | .4 |
| Regimen | ||
| Folfox | 105 | 44.3 |
| Capox | 132 | 55.7 |
| Treatment duration | ||
| 3 months | 116 | 48.9 |
| 6 months | 121 | 51.1 |
Table 4.
Frequency of mutated patients in each gene.
| Gene | Mutant patients |
|---|---|
| AKT1 | 6 |
| AKT2 | 3 |
| AKT3 | 3 |
| APC | 181 |
| APC2 | 38 |
| APPL1 | 19 |
| ARAF | 8 |
| AXIN1 | 11 |
| AXIN2 | 61 |
| BAD | 2 |
| BAX | 14 |
| BCL2 | 2 |
| BIRC5 | 70 |
| BRAF | 15 |
| CASP3 | 0 |
| CASP9 | 62 |
| CCND1 | 3 |
| CTNNB1 | 7 |
| CYCS | 1 |
| DCC | 62 |
| FOS | 2 |
| GSK3B | 2 |
| JUN | 5 |
| KRAS | 80 |
| LEF1 | 3 |
| MAP2K1 | 2 |
| MAPK1 | 4 |
| MAPK10 | 3 |
| MAPK3 | 0 |
| MAPK8 | 4 |
| MAPK9 | 3 |
| MLH1 | 55 |
| MSH2 | 6 |
| MSH3 | 110 |
| MSH6 | 51 |
| MYC | 11 |
| PIK3CA | 62 |
| PIK3CB | 8 |
| PIK3CD | 11 |
| PIK3CG | 48 |
| PIK3R1 | 23 |
| PIK3R2 | 70 |
| PIK3R3 | 0 |
| PIK3R5 | 9 |
| RAC1 | 0 |
| RAC2 | 0 |
| RAC3 | 11 |
| RAF1 | 1 |
| RALGDS | 42 |
| RHOA | 6 |
| SMAD2 | 3 |
| SMAD3 | 14 |
| SMAD4 | 25 |
| TCF7 | 35 |
| TCF7L1 | 61 |
| TCF7L2 | 39 |
| TGFB1 | 74 |
| TGFB2 | 11 |
| TGFB3 | 2 |
| TGFBR1 | 16 |
| TGFBR2 | 25 |
| TP53 | 148 |
Table 5.
Frequency of mutations according to patients’ characteristics.
| Characteristics | Gene | No % (p value) |
|---|---|---|
| Gender | ||
| Male vs Female | JUN | 43.4% vs 25.0% (0.05) |
| MAPK3 | 57.5% vs 31.1% (0.05) | |
| Sideness | ||
| Left vs Right | MLH1 | 80% vs 20% (0.007) |
| MSH6 | 84.3% vs 15.7% (0.001) | |
| TCF7L1 | 77% vs 33% (0.019) | |
| Location | ||
| Colon vs Sigmoid | DCC | 90.3% vs 9.7% (0.04) |
| KRAS | 88.8% vs 11.2% (0.048) | |
| TGFBR2 | 96% vs 4% (0.043) | |
| Age | ||
| <70 vs ≥70 | TGFBR1 | 57.5% vs 33% (0.012) |
| 51-70 vs ≥70 vs <50 | BAD | 56.1% vs 30.7% vs 12.7% (<0.001) |
| 51-70 vs ≥70 vs <50 | RAC | 56.1% vs 30.7% vs 12.7% (0.016) |
| 51-70 vs ≥70 vs <50 | AKT, AKT2, AKT3, APC, APPL1, AXIN1, AXIN2, BIRC5, DCC, GSK3B, KRAS, MAPK1, MAPK8, MAPK9, MAPK10, MLH1, MSH6, PIK3CA, PIK3R1, PIK3R2, PIK3R3, PIK3R5, RAF1, RALGD5, SMAD2, SMAD3, TCF7L2, TGFB1, TGFB2 | 56.1% vs 30.7% vs 12.7% (0.037) |
| Relapse post Adj Chemotherapy | ||
| Yes vs No | ARAF, MAPK10 | 71.7% vs 20.5% (0.027) |
| CASP3 | 75.5% vs 22.6% (0.044) | |
| TCF7 | 75% vs 21.2% (0.003) | |
| TGFΒ3 | 74.5% vs 22.2% (0.037) |
Table 6.
Correlation of mutated signaling pathways and patients characteristics.
| Cell cycle | Apoptosis | PI3K-Akt | Wnt | MAPK | ErbB | MSI | RAS | |
|---|---|---|---|---|---|---|---|---|
| Transverse Colon | X | X | X | X | ||||
| TaqI Homozygous | X | X | X | X | X | X | X | |
| ApaI Homozygous | X | X | X | X | ||||
| FokI Homozygous | X | X | X | |||||
| BsmI Homozygous | X | X | X | |||||
| TLR9 -T1237C Homozygous | X | X | X | |||||
| TLR9–T1486C Homozygous | X | X | X |
Table 7.
Univariate and multivariate Cox regression analysis.
| Univariate | Multivariate | |||||||
|---|---|---|---|---|---|---|---|---|
| DFS | OS | DFS | OS | |||||
| Feature | HR (95%CI) | p-value | HR (95%CI) | p-value | HR (95%CI) | p-value | HR (95%CI) | p-value |
| Sidedness (Right vs Left) | 1.9 (1.2–3.2) | 0.012 | 2.1 (1.0–4.0) | 0.043 | 2.1 (1.1-4.0) | 0.019 | 2.2 (1.0-4.5) | 0.043 |
| AKT1 (Mutant vs Wild type) | 2.9 (1.1-8.2) | 0.039 | ||||||
| APC2 (Mutant vs Wild type) | 2.5 (1.4-4.5) | 0.002 | ||||||
| ARAF (Mutant vs Wild type) | 3.8 (1.2-12.1) | 0.027 | 5.0 (2.0-12.7) | 0.001 | 3.9 (1.2-13.1) | 0.027 | ||
| BAD (Mutant vs Wild type) | 8.6 (1.1-64.4) | 0.036 | ||||||
| MAPK10 (Mutant vs Wild type) | 43.0 (9.1-203.7) | <0.001 | 15.1 (4.6-50.3) | <0.001 | 49.0 (9.8-244.1) | <0.001 | ||
| MSH6 (Wild type vs Mutant) | 2.3 (1.0-5.1) | 0.041 | ||||||
| RAC3 (Mutant vs Wild type) | 4.7 (1.8-11.9) | 0.001 | 3.5 (1.1-10.7) | 0.029 | ||||
| RHOA (Mutant vs Wild type) | 4.8 (1.5-15.6) | 0.009 | 9.5 (1.9-47.7) | 0.006 | ||||
| TGFB2 (Mutant vs Wild type) | 2.6 (1.1-6.7) | 0.040 | ||||||
| TGFB3 (Mutant vs Wild type) | 9.0 (2.1-37.8) | 0.003 | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



