Submitted:
18 August 2023
Posted:
21 August 2023
You are already at the latest version
Abstract
In the context of type 2 diabetes (T2DM), fasting and postprandial hyperglucagonemia have long been linked to the disease's development and progression. However, recent studies have brought to light the positive impact of glucagon-agonists on lipid metabolism and energy homeostasis. This review delves into the intricate relationship and underlying mechanisms between glucagon and nutrient composition, which may hold promise in devising novel therapeutic approaches for T2DM management.
Keywords:
gucagon
; glucose
; lipid
; amino acid
; hyperglucagonemia
1. Introduction
Glucagon, a 29-amino acid peptide, was discovered in 1921 (1), and was described as hyperglycemia substance due to contaminants in pancreatic extracts (2) in 1923. In 1948 it was established that glucagon is released from pancreatic α cells and later to a lesser extent, from brainstem neurons (3, 4). It is widely recognized that fasting and postprandial hyperglucagonemia play a crucial role in both the development and progression of type 2 diabetes (T2DM). However, researchers recently reshaped the role of glucagon in metabolism confirming that the biology of glucagon is more comprehensive and extends beyond hepatic hyperglycemic actions to exert effects on glucose, amino acids (AA) and lipid metabolism.
The hyperglycemic effect in T2DM is undisputedly present as demonstrated by glucagon receptor antagonists in humans which, however, induced hepatic side effects (5-7). The positive effects of glucagon-agonists on lipid metabolism, energy homeostasis and reduction of liver fat have been emphasized with the development of glucagon/GLP-1 co-agonists as well as GLP-1/GIP/glucagon triple agonists which are currently in clinical development/trials (8-10).
Nutrients, especially amino acids (AA), and non-nutrient components, stimulate glucagon secretion directly through sensory transporters and receptors or indirectly through their effects on cellular metabolism. Indeed, increasing protein intake, and thereby glucagon release, has shown positive effects in studies with orally treated T2DM patients (11, 12). We therefore review studies addressing the role of glucagon regulation by food intake. We describe the complex interplay of glucagon with glucose, protein/amino acid and lipid/fatty acid metabolism as well as secretion of insulin and other hormones to provide a mechanistic background. Our hypothesis is that the powerful stimulation of glucagon by AA and other food components may be exploited in the treatment of T2DM.
2. Glucagon Actions and Regulation
Glucagon was viewed primarily as the counter-regulatory hormone of insulin in the control of glucose homeostasis in the decades after its discovery (13, 14). Nowadays, glucagon is considered as a pleiotropic hormone metabolic actions of which include insulin secretion (15), regulation of lipid and AA metabolism, an increase of energy expenditure, modulation of food intake and satiety, and weight loss in both animals and humans (16-18). Furthermore, glucagon has been demonstrated to play a role in the regulation of bile acid metabolism, encompassing processes such as bile acid synthesis, uptake, and efflux (19).
The secretion of glucagon by α-cells is primarily associated with occurrences of hypoglycemia, acting as a safeguard against low blood sugar levels. Additionally, it is also stimulated by elevated circulating levels of AAs and fatty acids, as well as in response to adrenergic stimulation and circulating incretins (20). It is well established that β-cell derived secretory products including insulin, zinc and gamma-aminobutyric acid (GABA) GABA inhibit glucagon secretion(21). Meanwhile, it is potently suppressed by somatostatin (22), GLP-1, amylin, leptin, fatty acids and ketone bodies and stimulated by GIP and vagal stimulation. Meier et al. demonstrated that GLP-2 also stimulates glucagon secretion in healthy subjects (23) (Figure 1). And some medications such as furosemide or acetylsalicylic acid may influence prostaglandin (PG) synthesis, mainly PGE, which in turn control glucagon release. In this regard, details of glucagon receptor function are helpful to understand their linkage to metabolic effects of glucagon.
Glucagon receptors (GCGRs) belong to the class B of seven transmembrane (7TM) protein receptors known to activate adenylyl cyclase through Gαs-coupled proteins, which is accompanied by an increase in cellular cyclic AMP levels and activation of protein kinase A (PKA) (24, 25). Recently, GCGRs were also shown to activate the IP3 -pathway via Gq and activation of INSP3R1 in liver cells (26). GCGRs are highly expressed in the liver as well as multiple extrahepatic tissues (Figure 1), which play an essential role in glucose, AA, lipid and energy metabolism (21, 27, 28).
How Are GCGRs Regulated?
In 1996 research in broilers demonstrated that reduced glucagon binding was closely associated with reduced lipolysis, implying that downregulation of cell surface GCGRs is the mechanism by which glucagon induced desensitization of its ability to stimulate lipolysis in adipocytes(29). It has been reported that dihydroxy bile acids like chenodeoxycholic acid (CDCA) induce GCGR desensitization(30). A study in 2006 demonstrated that CDCA stimulated phosphorylation and heterologous desensitization of the GCGRs through protein kinase C (PKC) activation (31). Given that glucagon is one of several peptide hormones that increase glucose levels, Leibiger et al. investigated the broader implications of autocrine secretion-biosynthesis coupling, using glucagon as an example. Their study demonstrated that glucagon, through signaling via GCGR, PKC, and PKA, upregulates the expression of its own gene, providing evidence for a more widespread applicability of positive autocrine feedback (32).
Downstream signaling pathways of GCGRs involve activation of Gs and Gq, leading to the formation of intracellular cAMP and inositol 1,4,5-trisphosphate (IP3) with subsequent release of intracellular Ca2+. Activation of either Gs-induced protein kinase A (PKA) or Gq’s effect on Ca2+/calmodulin-dependent protein kinase can lead to the phosphorylation of cAMP response element binding protein (CREB). The metabolic effects of glucagon depend on its concentration, spatial features (mitochondrial vs. cytosolic effects), and substrate dependency. Thus, for ureagenesis and gluconeogenesis plasma concentrations of substrates are important, i.e. AAs and free fatty acids, respectively. It was described in previous studies that inositol triphosphate receptor 1 (INSP3R1) is the isoform primarily responsible for mitochondrial calcium signaling in hepatocytes and knocking down InsP3R1 reduced glucose production in isolated hepatocytes (33, 34). Perry et al. (26) implied that InsP3R1 activation and Ca2+/calmodulin-dependent protein kinase II (CAMKII) activity are important for the acute effect of glucagon in stimulating hepatic glucose production (HGP).
3. Glucagon and Glucose Metabolism
Glucose inhibits glucagon release upon oral or i.v. administration while hypoglycemia increases the secretion of glucagon to elevate hepatic glucose output by stimulation of glycogenolysis and gluconeogenesis, and additionally inhibits glycogenesis and glycolysis and induces ketone production through multiple mechanisms and thereby protects against hypoglycemia (24, 35, 36). Glucagon acts in concert with cortisol, growth hormone and the adrenergic hormones which also increase hepatic glucose output in hypoglycemia. Remarkably, the blockade of GCGRs does not impair the counter-regulation against hypoglycemia (7, 37, 38).
Although the level of glucagon rises rapidly in the early stage of fasting, circulating glucagon concentrations drop to postprandial levels upon prolonged fasting (see below), with persistently decreasing glycaemia due to glycogen depletion (39). Glucagon physiologically regulates the early phases of fasting in non-diabetic animals and humans which is relevant in real life since prolonged fasting is an unusual state in industrialized societies.
The inhibition of glucagon by increases of blood glucose is important to control blood glucose and is therefore finely tuned with insulin release. However, with the development of impaired glucose tolerance and T2DM, glucagon continues to induce glucose production during hyperglycemia in fasted and pre-prandial conditions (40-42). Both, fasting and postprandial hyperglucagonemia were proposed to trigger metabolic disturbances in obese and/or prediabetic subjects (43, 44). In 1970s, Unger et al. proposed that T2DM may not only the consequence of relative or absolute insulin deficiency, but also of glucagon excess in dogs (45). Reaven et al. later showed that hyperglucagonemia persists throughout the day in both obese and non-obese patients with T2DM, despite significant elevations in plasma insulin and glucose levels. These factors are traditionally expected to inhibit glucagon secretion under normal conditions (46). Recent studies have confirmed that most patients with T2DM exhibit abnormally high fasting plasma glucagon levels that do not appropriately decrease, and in some cases, may even increase after oral glucose tolerance test (OGTT) or carbohydrate intake (40, 42, 47-49). This failure to suppress glucagon secretion significantly contributes to post-prandial hyperglycemia by increasing hepatic glucose production in T2DM patients. Some researchers suggest that the difficulty in controlling glucagon could signal an initial problem with α-cells in the pancreas before insulin deficiency develops. This might involve reduced α-cell response to glucose and/or a lack of insulin’s suppressive effect within the pancreatic islets (21, 43, 50, 51). However, i.v. administration of glucose was reported to inhibit glucagon release similarly in people with or without diabetes. Since manifest diabetes is associated with increased fasting plasma glucose in the presence of increased plasma glucagon, the inhibition of glucagon release is partially lost. However, as further increases of glucose by i.v. infusion still inhibit glucagon release, the response appears to be maintained but right shifted(52).
What Are the Explanations for This Difference (oral vs i.v Glucose)?
A careful comparison of oral or i.v. glucose in healthy people showed greater suppression of glucagon by i.v. than by oral glucose (53). Since the i.v. glucose does not stimulate incretins, this cannot be explained by suppression of glucagon by GLP-1. However, as GIP was stimulated by oral but not i.v. glucose, one might postulate that GIP stimulated glucagon and thereby attenuated the effect of glucose, and possibly due to the glucagonotropic action of GLP-2 (53). In people with T2DM, i.v. glucose dose dependently suppressed glucagon even at elevated basal plasma glucose while oral glucose caused an initial stimulation of glucagon which was not explained by the levels of incretins hormones (54, 55). On the other hand, studies in isolated α-cells and pancreatic islets also provided evidences. Notably, glucose unexpectedly stimulates glucagon release in isolated α-cells via mechanisms which involve KATP-channels and Ca2+-induced depolarization (56) which pointed to indirect and possibly paracrine mechanisms of inhibition of glucagon release in vivo. In addition, glucose inhibits glucagon release in intact mouse and human islets apparently by paracrine mechanisms involving somatostatin release which lost their function in islets from diabetes patients (57-59). This would suggest that deficient stimulation of somatostatin release by glucose or insulin from delta-cells in islets accounts for the hyperglucagonemia in diabetic conditions. Meanwhile, somatostatin independent mechanisms have also been proposed (59-61). Recently, hyperglycaemia-induced Na+-dependent reduction of ATP production and thus mitochondrial impairment was proposed as a primary mechanism (62, 63).
An important observation is that hyperglucagonemia in the fasting state and in response to arginine disappears upon prolonged infusion of insulin in insulin-dependent patients. This is compatible with a dysregulation of α-cells due to hyperglycemia and/or insulin deficiency and not a primary defect of α-cell function (62, 64). This observation would suggest that glucose-induced dysregulation of energy metabolism in islets drives hyperglucagonemia. This raises the question, whether α -cell dysfunction also involves an exaggerated response to AA or fat, which may also involve mitochondrial mechanisms.
How Does Hypoglycemia Stimulate Glucagon Secretion?
Normally, hypoglycemia triggers a counter-regulatory response in the α-cells which does not happen in many T1DM and some T2DM patients. The comprehensive mechanisms how glucose regulates glucagon secretion remain unclear. It has been claimed that CNS and hepatoportal sensors (i.e. hypoglycemia activated gastrointestinal neurons in the brainstem and in several hypothalamic nuclei) contribute to the control of glucagon(65, 66). Recent studies also questioned whether the primary role of glucagon is to elevate glucose concentrations(67). While a fast’s beginning is marked by an initial surge in glucagon levels along with a decrease in blood glucose, an extended fast exceeding three days triggers a gradual reduction in circulating glucagon levels. Surprisingly, these levels eventually normalize to what is typically seen after a meal, even in the presence of consistently low blood glucose(39). Furthermore, the administration of glucagon in hypoglycemic individuals who have been fasting for more than 3 days does not produce any significant changes in glycaemia, possibly due to depleted glycogen stores(68).
Two recent studies claimed that Irak4 (Interleukin-1 receptor associated kinase-4) controls hypoglycemia-induced glucagon secretion by modulating hypothalamic Il-1β signaling, and Agpat5 activation in AgRP (agouti-related protein) neurons leads to hypoglycemia-induced glucagon secretion (69, 70). It is established that sodium-glucose co-transporter 1 (SGLT-1) and the generation of reactive oxygen species (ROS) released from β-cells, are involved in increasing α-cell proliferation and glucagon secretion (71). The role of glucagon in contributing to hyperglycemia in diabetes was reinforced by observing lower glucose levels in mice with knockout of the glucagon receptors (GCGRs) (72), as well as improved glucose and HbA1c in humans treated with acute and prolonged pharmacological receptor antagonisms (GRAs) in clinical and preclinical trials, although accompanied with hepatic side effects such as increases in transaminases, liver fat accumulation and dyslipidemia in addition to α-cell hyperplasia (73-75).
The reason why counter-regulation fails in diabetic patients is not fully understood. Interestingly, inhibiting mitochondrial ATP production or pharmacologically acti-vating KATP channels with diazoxide mimics the dysregulation of glucagon secre-tion (63). These observations collectively suggest that the glucagon secretion defect in diabetic patients may stem from disrupted mitochondrial metabolism, though the exact mechanisms remain unclear.
It has been proposed that glucagon increases with the onset of obesity and fatty liver as a consequence of hepatic glucagon resistance (76) and insulin resistance due to inappropriate regulation of glucagon by fasting and a static glucagon/insulin ratio (77). Normally fasting induces an increase glucagon/insulin ratio which leads a predominant glucagon signaling to increase hepatic intracellular concentrations of the second messenger cAMP and downstream PKA and CREB signaling pathways, increasing hepatic glucose output (78). One study highlighted the role of nutrient signaling via mTOR complex 1 (mTORC1) regulation to control glucagon secretion and α-cell mass (79). However, one recent study in mice demonstrated that chronic hyperglucagonemia can improve glucose homeostasis by downregulating hepatic GCGR expression, inducing hepatic “glucagon resistance”, and enhancing insulin secretion (80).
4. Glucagon and Amino Acid Metabolism
In addition to its established glucose-regulatory effects, glucagon powerfully regulates hepatic AA turnover by increasing activities of necessary transporters and enzymes in the urea cycle through cAMP-PKA-CREB protein (81, 82). In fact, there is evidence to suggest that the impact of glucagon signaling may vary between fasting and post-prandial conditions (83). Glucagon activates the transcription of AA transporters located on the hepatocyte membrane, thus allowing increased AA uptake and substrate availability for ureagenesis (84). In turn, AAs enhance glucagon secretion from α cells (85). This generates a glucagon and AA feedback loop, referred to as “liver-α-cell axis”, which might be as important for metabolism as the glucagon-glucose loop (86-90). In line with this, genetic, GCGR-antibody or pharmacological inhibition of glucagon signaling leads to α-cell hypersecretion and hyperplasia as well as a decrease of hepatic AA transporters and gene expression involved in AA metabolism, resulting in dramatically increased plasma concentrations of some but not AAs (86, 89, 91). Recently, decreased AAs levels were reported to reduce target of rapamycin (mTOR) signaling in α-cells and suppress α-cell proliferation (92). At the hormonal level, protein-rich meals acutely increase glucagon secretion depending on the kinetics of AA plasma levels (93, 94). If excess AA, i.e. more than can be utilized for protein synthesis, are taken up with meals or liberated by proteolysis upon fasting, they are used as energy substrates (95). Since muscle and other organs cannot handle the amino groups, they delaminate the AA and take the carbon moiety for Krebs cycle and use primarily alanine to shuttle the amino groups to the liver. Alanine therefore predominates within 15 glucogenic AA (96) and is preferentially taken up by the liver in the presence of elevated glucagon for glucose production (known as glucose–alanine cycle or Cahill cycle) (97-99). This glucose–alanine cycle is dysregulated in dysglycaemia in humans with obesity and T2DM, as exemplified by heightened splanchnic (that is, viscera and liver) alanine uptake. One recent study demonstrated that alanine transport and aminotransferase (ALT) isoform expression (ALT and ALT2) were remarkably higher in obese, prediabetes and overt diabetic mouse models and in individuals with metabolic diseases. In addition, a GCGR antagonist reduced hyperglycemia accompanied by blunting of increased ALT/ALT2 activity in mice (98).
Glucagon regulates not only ureagenesis, but also affects renal nitrogen excretion (93). Glucagon affects fluid and solute transport in the distal tubule and collecting duct by increasing hepatic cAMP secretion, which, in turn, influences proximal tubule reabsorption of urea. This interaction increases the fractional excretion of urea, sodium, potassium and phosphates. After oral protein loading, there was a significant correlation between GFR and urinary urea nitrogen excretion rate (93). Furthermore, branched chain amino acids (BCAA) do not induce an increase in renal hemodynamics (100). A post-prandial increase in plasma glucagon could potentially counteract amino acid and insulin-stimulated mTORC1 activation, leading to the suppression of protein synthesis in the liver. After ingesting a protein-rich diet, the liver shows increased rates of translation initiation and protein synthesis compared to fasted animals. A hypothesis suggests that glucagon resistance, a molecular phenomenon affecting the physiological effects of glucagon on glucose, AA, and lipid metabolism, may contribute to the development of T2DM and metabolic diseases (94). In the healthy liver, glucagon binds to the hepatic GCGR and increases AA metabolism through urea production. Glucagon may influence several steps in this process, including various biochemical shunts in the carbamoyl phosphate and urea cycles. In subjects with liver diseases, such as NAFLD, GCGR resistance may affect this liver-α cell axis, as reduced hepatic GCGR expression or impaired GCGR signaling leads to decreased urea production, which in turn leads to hyperaminoacidemia and subsequent compensatory hyperglucagonemia. Furthermore, hepatic steatosis may impair glucagon-dependent enhancement of AA catabolism in mice and humans with NAFLD (101). A recent study reported that elevated plasma levels of total AAs associate with hyperglucagonemia in NAFLD independently of glycemic control (102). This hypothesis may explain the hyperglucagonemia frequently observed in obesity, fatty liver and T2DM. However, the relative contributions of alpha to beta-cell cross talk and diminished inhibition of glucagon secretion by insulin resistance or deficiency and increased AA stimulation is presently unclear. Both, fatty liver and insulin resistance correlate well with glucagon levels in plasma (103).
Most circulating AAs have been shown to potently stimulate both glucagon and insulin secretion in animals and humans, although not all AAs are identical in their glucagontropic effects (104, 105). The purpose of this increase in glucagon release is believed to prevent hypoglycemia after protein intake, as AAs also stimulate insulin secretion. As early as the 1970s, Unger had found that alanine infusion induced an increase in glucagon secretion accompanied with very little stimulatory effect on insulin secretion in dogs. Lysine contributed to a lesser extent to α-cell secretion, while BCAAs had no effects on glucagon secretion whereas they elicit a significant insulin response (104, 106-108). Nevertheless, However, other studies have reported that BCAAs stimulate the secretion of both insulin and glucagon, particularly with oral administration resulting in greater and more prolonged secretion of both hormones (109, 110). Later in 1974 arginine was proven to enhance both insulin and glucagon secretion which support separate glucose and arginine receptors on both α and β cells in rodents, or via direct plasma membrane depolarization and Ca2+ influx in the α-cell (111, 112). In mice, leucine intake indeed improved glycemic control while some studies observed negative effects on glucose homeostasis for methionine and BCAAs (113-115). Furthermore, studies in animal and human isolated α cells indicated L-glutamine is a positive modulator of glucagon release and could regulate α cell proliferation and mass via mTOR-dependent nutrient sensing (116, 117). In the following decades, clinical studies often utilized specific AAs, although the effects of individual AAs on glucagon secretion remain an area of controversy. Primarily arginine, is established as an α-cell secretagogue, which induces significant increases in circulating glucagon and insulin regardless of ambient glycaemia (9). Recent research demonstrated that BCAAs directly raised intracellular Ca2+ levels in α cells, leading to increased plasma glucagon levels in diabetic mice. This suggests that disordered BCAA catabolism in pancreatic islet cells contributes to postprandial hypersecretion of glucagon in diabetes (118). Additionally, a separate study showed that glycine ingestion resulted in a slight decrease in serum glucose and increased insulin and glucagon concentrations in healthy human subjects (119, 120). Galsgaard et al. proposed that defective glucagon signaling to the liver results in hyper-aminoacidemia, which further stimulates the secretion of glucagon, possibly resulting in hyperplasia of α cells (121). However, the precise number of amino acids involved in the glucagon and amino acid feedback loop remains unclear, with glutamine suggested as a potential candidate (116). Additionally, which AAs are capable of stimulating glucagon secretion directly from pancreatic α-cells or via increasing GCGR signaling remains mysterious. The Holst group also found that alanine, arginine, cysteine, and proline are involved in the acute regulation of the liver-α-cell axis through administering AA mixtures in vivo in mice(105).Ingested whey protein in healthy participants induced hyperglucagonemia while suppressing free fatty acids, and showed that physiological hyperglucagonemia can override the hepatic actions of insulin, and postprandial hyperglucagonemia evolved to concurrently and synergistically collaborate with insulin to regulate glucose, AA, and nitrogen metabolism(122). Epidemiological evidence indicates that plant protein-based nutrition, with lower methionine and BCAAs and higher arginine content, is inversely associated with mortality and T2DM (123). There are studies indicating that soy protein normalized fasting hyperglucagonemia and improved glucagon resistance in obese Zucker (fa/fa) rats through inducing increase GCGR recycling to the membrane of adipocytes and its ligand-binding and G-protein selectively(123-126). Our recent study showed that plant protein, with lower methionine and BCAAs but higher arginine content, leads to greater postprandial increases in glucagon compared to animal protein. As a result, it requires higher insulin levels to control glucose metabolism, which seems to be associated with the rate of amino acid appearance in patients with T2DM(127).
5. Glucagon and Lipid Metabolism
Glucagon is recognized for its potent hypolipidemic effects. In humans, intravenous glucagon administration reduces plasma cholesterol, total esterified fatty acids, hepatic synthesis of triglycerides, and apolipoproteins by stimulating β-oxidation and lipolysis in the liver (128, 129). Furthermore, glucagon also reduces hepatic lipid accumulation and decrease hepatic lipid secretion through the inhibition of lipogenesis in the liver (130). In addition, glucagon stimulates lipolysis in the white adipose tissue, increased ketone-body production and fatty acid oxidation in humans and rats (24) (131, 132).
It is established that increased GCGR signaling has been linked to improved lipid metabolism. In 1979, studies explored glucagon’s role in the direct short-term regulation of hepatic free fatty acid (FFA) metabolism. They found that physiological concentrations of glucagon increased ketogenesis and reduced triglyceride synthesis from palmitate in hepatocytes from fed rats at FFA concentrations of 1.0 mM or lower (133). Glucagon could modulate FFA metabolism by both intrahepatic and extrahepatic mechanisms, i.e. glucagon reduces de novo fatty acid synthesis by inhibiting malonyl-CoA formation. This occurs after an increase in intracellular cAMP activates PKA, which phosphorylates and inactivates acetyl-CoA carboxylase (ACC) (134).
As GCGR are expressed on β-cells and may stimulate insulin through both GLP-1R and GCGR, one may speculate that intra-islet regulation of insulin by glucagon might contribute to its effect on lipid metabolism. As discussed above, GCGR antagonists (e.g., LY2409021, Volagidemab) have been considered as glucose-lowering therapy in T2DM patients but resulted in lipid disorders, whereas glucagon/GLP-1 receptors co-agonism improved dyslipidemia and reduced hepatic steatosis which brought up discussions regarding to the relationship between glucagon signaling and lipid metabolism (5, 135, 136).
It remains unclear how glucagon promotes hepatic mitochondrial fat oxidation and to what extent glucagon influences lipolysis in adipose tissue, especially in humans. Previous study confirmed that InsP3R-I is essential due to reduced glucose production with knockdown InsP3R-I in isolated hepatocytes (34). The recent discovery from Perry and co-workers(26) is quite impressive, which reported glucagon stimulates intrahepatic lipolysis through InsP3R-I/CAMKII-dependent activation with increased hepatic acetyl-CoA. In addition, glucagon stimulates hepatic mitochondrial oxidation through InsP3R-I-mediated calcium signaling. The INSP3R1–ATGL pathway appears to play a central role in the regulation of hepatic lipid metabolism in response to glucagon. This is supported by the fact that plasma non-esterified fatty acid levels did not change significantly in both INSP3R1-LKO mice and the control mice during glucagon infusion (26). Those results explain many of these actions since the cAMP/PKA mediated effects were transcriptional and did not explain the acute metabolic actions of glucagon.
In turn, the capability of FFAs to regulate glucagon secretion remains in debate although they are insulin secretagogues under some circumstances, and increased FFAs levels might be correlated with T2DM (21, 137-139). Early research in 1974 had shown that elevation of plasma FFA suppressed glucagon levels in people, which are supported by the following clinical studies (138, 140-142). Experiments on isolated rodent islets, an α-cell line, and human islets have shown that FFAs (oleate or palmitate) stimulate glucagon secretion. This occurs through signaling via fatty acid G-protein coupled receptors, β-oxidation of fatty acids, and activation of L-type Ca2+ channels. Additionally, it involves relieving the inhibitory paracrine action of somatostatin secreted from δ-cells (143-145). Some studies observed that palmitate stimulates glucagon secretion in a glucose-dependent manner. This means glucagon is secreted when the glucose concentration is below 10mM but not at 16.7mM (144, 146). Wang et al. reported long-chain FFA (linoleic acid) acutely stimulated glucagon secretion by activation of G protein coupled receptor 40 (GPR40) and phospholipase C to increase Ca2+ release and associated Ca2+ influx through Ca2+ channels in primary cultured rat pancreatic islets (147) (Figure 2). Similar effects have been observed in rat islets with oleic acid (148). The Danish group reported that short-term exposure to FFAs directly increases glucagon release from α-cells. The stimulatory action depends on the chain length, degree of unsaturation, and spatial configuration of FAs in isolated mouse islets and alpha TC1-6 cells. Saturated fatty acids (SFA) were found to be more effective than unsaturated fatty acids (USFA) in stimulating glucagon secretion (144, 149), and later new data indicated that prolonged exposure (up to 3 days) to palmitate and oleate leads to excessive lipid accumulation and induces time- and dose-dependent hyperglucagonemia in both isolated islets and alpha TC1-6 cells through oxidation (150, 151). Long-term culture of a clonal α-cell line with palmitate increased glucagon release and expression, likely through activation of the AMPK pathway (152). Conversely, insulin’s inhibitory effect on glucagon release was impaired after prolonged exposure to free fatty acids (FFAs) due to palmitate-induced insulin resistance, involving defects in the IRS-1/PI3K/Akt pathway (152). In rat islets, chronic exposure to fatty acids resulted in increased glucagon release but decreased glucagon content without altering glucagon gene expression (153, 154). Glucolipotoxicity conditions, such as combining palmitate with high glucose levels, can induce apoptosis in rodent α-cells (155). In clinical studies, intravenous or oral administration of a lipid emulsion did not alter glucagon secretion, but only oral lipid ingestion elicited a clear insulin response with increased GIP and GLP-1 concentrations(156). In other studies, no difference in glucagon secretion was observed after a high-fat or low-fat diet intake in both never-obese and post-obese women (157). However, an alternative study noted a slight but higher glucagon response to C4-dietary oil compared to C18-olive oil in overweight subjects with T2DM (158). Furthermore, a sharp increase of plasma glucagon concentrations was observed in healthy men after the ingestion of long-chain fatty acids (olive oil and C8 fatty acids) while no increase with short-chain fatty acid (C4) (159). Another study reported 6 months mono-unsaturated fatty acids (MUFA) intake contributes to larger post-lunch glucagon responses compared to a control meal in healthy young subjects (160). Interestingly, a recent study revealed that palmitate can cause a switch to a glucagon-secreting phenotype in intestinal GLP-1 secreting cells, suggesting the potential of fatty acids to induce extra-pancreatic glucagon (161). Moreover, both long- and short-chain FFA increase GIP concentrations which might be one potential reason to stimulate glucagon release in human(162, 163). Overall, the data from animals and humans support the hypothesis that chronic elevation of fatty acids may contribute to α-cell deregulation in T2DM.
Conclusions
In summary, glucagon plays a crucial role in glucose, AA and lipid metabolism, with its secretion tightly regulated by glucose levels and other factors. In patients with impaired glucose tolerance and T2DM, dysregulation of glucagon secretion leads to elevated fasting plasma glucagon concentrations even during hyperglycemia. The underlying mechanisms involve factors like incretins, somatostatin release, α cell dysfunction, hyperglycemia, and insulin deficiency. The exact mechanisms driving these dysregulations are still under investigation, presenting potential avenues for therapeutic intervention in diabetes management.
Furthermore, glucagon’s impact extends beyond glucose regulation, influencing hepatic amino acid turnover and lipid metabolism. The “liver-α-cell axis” links amino acids to glucagon secretion, contributing to metabolic imbalances in conditions like obesity and T2DM. Specific AAs, such as arginine and branched-chain amino acids, directly influence plasma glucagon levels. Moreover, glucagon’s potent hypolipidemic effects are attributed to its role in hepatic β-oxidation, lipolysis, and inhibition of lipogenesis. However, the complex interplay between glucagon and fatty acids requires further elucidation to understand its implications in metabolic diseases.
Overall, investigating the intricate mechanisms governing glucagon’s diverse functions is vital to unraveling its role in metabolic disorders like T2DM. Advancing our understanding of glucagon’s regulatory pathways may offer new opportunities for developing targeted therapies to restore metabolic balance and improve patient outcomes.
Author Contributions
Conceptualization, A F.H Pfeiffer.; software, Y Zheng.; writing—original draft preparation, JD. Zhang. and L Martens; writing—review and editing, Y Zheng. And A F.H Pfeiffer.; supervision, A F.H Pfeiffer. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
We express sincere gratitude to China Scholarship Council (CSC) for their financial support to JD.Zhang (No. 202008080164).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Glucagon-Molecular Physiology, Clinical and therapeutic.pdf.
- Murlin, J.R.; Clough, H.D.; Gibbs, C.B.F.; Stokes, A.M. Aqueous Extracts of Pancreas. J. Biol. Chem. 1923, 56, 253–296. [Google Scholar] [CrossRef]
- Sutherland, E.W.; de Duve, C. Origin and Distribution of the Hyperglycemic-Glycogenolytic Factor of the Pancreas. J. Biol. Chem. 1948, 175, 663–674. [Google Scholar] [CrossRef]
- Sasaki, H.; Ebitani, I.; Tominaga, M.; Yamatani, K.; Yawata, Y.; Hara, M. Glucagon-like substance in the canine brain. Endocrinol. Jpn. 1980, 27 (Suppl. 1), 135–140. [Google Scholar] [CrossRef]
- Guzman, C.B.; Zhang, X.M.; Liu, R.; Regev, A.; Shankar, S.; Garhyan, P.; et al. Treatment with LY2409021, a glucagon receptor antagonist, increases liver fat in patients with type 2 diabetes. Diabetes Obes. Metab. 2017, 19, 1521–1528. [Google Scholar]
- Pettus, J.H.; D’Alessio, D.; Frias, J.P.; Vajda, E.G.; Pipkin, J.D.; Rosenstock, J.; et al. Efficacy and Safety of the Glucagon Receptor Antagonist RVT-1502 in Type 2 Diabetes Uncontrolled on Metformin Monotherapy: A 12-Week Dose-Ranging Study. Diabetes Care. 2020, 43, 161–168. [Google Scholar]
- Kazierad, D.J.; Chidsey, K.; Somayaji, V.R.; Bergman, A.J.; Calle, R.A. Efficacy and safety of the glucagon receptor antagonist PF-06291874: A 12-week, randomized, dose-response study in patients with type 2 diabetes mellitus on background metformin therapy. Diabetes Obes. Metab. 2018, 20, 2608–2616. [Google Scholar]
- Bossart, M.; Wagner, M.; Elvert, R.; Evers, A.; Hubschle, T.; Kloeckener, T.; et al. Effects on weight loss and glycemic control with SAR441255, a potent unimolecular peptide GLP-1/GIP/GCG receptor triagonist. Cell Metab. 2022, 34, 59–74. [Google Scholar]
- Finan, B.; Capozzi, M.E.; Campbell, J.E. Repositioning Glucagon Action in the Physiology and Pharmacology of Diabetes. Diabetes. 2020, 69, 532–541. [Google Scholar]
- Rosenstock, J.; Wysham, C.; Frias, J.P.; Kaneko, S.; Lee, C.J.; Fernandez Lando, L.; et al. Efficacy and safety of a novel dual GIP and GLP-1 receptor agonist tirzepatide in patients with type 2 diabetes (SURPASS-1): A double-blind, randomised, phase 3 trial. Lancet 2021, 398, 143–155. [Google Scholar]
- Sucher, S.; Markova, M.; Hornemann, S.; Pivovarova, O.; Rudovich, N.; Thomann, R.; et al. Comparison of the effects of diets high in animal or plant protein on metabolic and cardiovascular markers in type 2 diabetes: A randomized clinical trial. Diabetes Obes. Metab. 2017, 19, 944–952. [Google Scholar]
- Markova, M.; Pivovarova, O.; Hornemann, S.; Sucher, S.; Frahnow, T.; Wegner, K.; et al. Isocaloric Diets High in Animal or Plant Protein Reduce Liver Fat and Inflammation in Individuals With Type 2 Diabetes. Gastroenterology 2017, 152, 571–585. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H.; Orci, L. Paracrinology of islets and the paracrinopathy of diabetes. Proc. Natl. Acad. Sci. USA 2010, 107, 16009–16012. [Google Scholar] [CrossRef] [PubMed]
- Sherwin, R.; Felig, P. Glucagon physiology in health and disease. Int. Rev. Physiol. 1977, 16, 151–171. [Google Scholar] [PubMed]
- Capozzi, M.E.; Wait, J.B.; Koech, J.; Gordon, A.N.; Coch, R.W.; Svendsen, B.; et al. Glucagon lowers glycemia when beta-cells are active. JCI Insight. 2019, 4, e129954. [Google Scholar] [CrossRef] [PubMed]
- Schulman, J.L.; Carleton, J.L.; Whitney, G.; Whitehorn, J.C. Effect of glucagon on food intake and body weight in man. J. Appl. Physiol. 1957, 11, 419–421. [Google Scholar] [CrossRef] [PubMed]
- Billington, D.C. Angiogenesis and its inhibition: Potential new therapies in oncology and non-neoplastic diseases. Drug Des. Discov. 1991, 8, 3–35. [Google Scholar]
- <Individual, but not simultaneous, glucagon and cholecystokinin infusions inhibit feeding in men.pdf>.
- Bouscarel B, Kroll SD, and Fromm, H. Signal transduction and hepatocellular bile acid transport: Cross talk between bile acids and second messengers. Gastroenterology 1999, 117, 433–452. [Google Scholar] [CrossRef]
- 20. Holst JJ, Knop FK, Vilsboll T, Krarup T, and Madsbad, S. Loss of incretin effect is a specific, important, and early characteristic of type 2 diabetes. Diabetes Care 2011, 34 (Suppl. 2), S251–S257. [CrossRef]
- Gromada, J.; Franklin, I.; Wollheim, C.B. Alpha-cells of the endocrine pancreas: 35 years of research but the enigma remains. Endocr. Rev. 2007, 28, 84–116. [Google Scholar] [CrossRef]
- <Somatostatin and diabetes.pdf>.
- Meier, J.J.; Nauck, M.A.; Pott, A.; Heinze, K.; Goetze, O.; Bulut, K.; et al. Glucagon-like peptide 2 stimulates glucagon secretion, enhances lipid absorption, and inhibits gastric acid secretion in humans. Gastroenterology 2006, 130, 44–54. [Google Scholar] [CrossRef]
- Habegger, K.M.; Heppner, K.M.; Geary, N.; Bartness, T.J.; DiMarchi, R.; Tschop, M.H. The metabolic actions of glucagon revisited. Nat. Rev. Endocrinol. 2010, 6, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Ramnanan, C.J.; Edgerton, D.S.; Kraft, G.; Cherrington, A.D. Physiologic action of glucagon on liver glucose metabolism. Diabetes Obes. Metab. 2011, 13 (Suppl. 1), 118–125. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Zhang, D.; Guerra, M.T.; Brill, A.L.; Goedeke, L.; Nasiri, A.R.; et al. Glucagon stimulates gluconeogenesis by INSP3R1-mediated hepatic lipolysis. Nature 2020, 579, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.E.; Drucker, D.J. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 2013, 17, 819–837. [Google Scholar] [CrossRef] [PubMed]
- Bansal, P.; Wang, Q. Insulin as a physiological modulator of glucagon secretion. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E751–E761. [Google Scholar] [CrossRef] [PubMed]
- Oscar, T.P. Down-regulation of glucagon receptors on the surface of broiler adipocytes. Poult. Sci. 1996, 75, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Kohout, T.A.; Lefkowitz, R.J. Regulation of G protein-coupled receptor kinases and arrestins during receptor desensitization. Mol. Pharmacol. 2003, 63, 9–18. [Google Scholar] [CrossRef]
- Ikegami, T.; Krilov, L.; Meng, J.; Patel, B.; Chapin-Kennedy, K.; Bouscarel, B. Decreased glucagon responsiveness by bile acids: A role for protein kinase Calpha and glucagon receptor phosphorylation. Endocrinology 2006, 147, 5294–5302. [Google Scholar] [CrossRef]
- Leibiger, B.; Moede, T.; Muhandiramlage, T.P.; Kaiser, D.; Vaca Sanchez, P.; Leibiger, I.B.; et al. Glucagon regulates its own synthesis by autocrine signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 20925–20930. [Google Scholar] [CrossRef]
- Feriod, C.N.; Oliveira, A.G.; Guerra, M.T.; Nguyen, L.; Richards, K.M.; Jurczak, M.J.; et al. Hepatic Inositol 1,4,5 Trisphosphate Receptor Type 1 Mediates Fatty Liver. Hepatol. Commun. 2017, 1, 23–35. [Google Scholar] [CrossRef]
- Wang, Y.; Li, G.; Goode, J.; Paz, J.C.; Ouyang, K.; Screaton, R.; et al. Inositol-1,4,5-trisphosphate receptor regulates hepatic gluconeogenesis in fasting and diabetes. Nature 2012, 485, 128–132. [Google Scholar] [CrossRef]
- Jiang, G.; Zhang, B.B. Glucagon and regulation of glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E671–E678. [Google Scholar] [CrossRef] [PubMed]
- Sokal, J.E. Effect of glucagon on gluconeogenesis by the isolated perfused rat liver. Endocrinology 1966, 78, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Gerich, J.E.; Lorenzi, M.; Bier, D.M.; Tsalikian, E.; Schneider, V.; Karam, J.H.; et al. Effects of physiologic levels of glucagon and growth hormone on human carbohydrate and lipid metabolism. Studies involving administration of exogenous hormone during suppression of endogenous hormone secretion with somatostatin. J. Clin. Investig. 1976, 57, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Lecavalier, L.; Bolli, G.; Gerich, J. Glucagon-cortisol interactions on glucose turnover and lactate gluconeogenesis in normal humans. Am. J. Physiol. 1990, 258, E569–E575. [Google Scholar] [CrossRef]
- Marliss, E.B.; Aoki, T.T.; Unger, R.H.; Soeldner, J.S.; Cahill, G.F., Jr. Glucagon levels and metabolic effects in fasting man. J. Clin. Invest. 1970, 49, 2256–2270. [Google Scholar] [CrossRef]
- Rohrer, S.; Menge, B.A.; Gruber, L.; Deacon, C.F.; Schmidt, W.E.; Veldhuis, J.D.; et al. Impaired crosstalk between pulsatile insulin and glucagon secretion in prediabetic individuals. J. Clin. Endocrinol. Metab. 2012, 97, E791–E795. [Google Scholar] [CrossRef]
- Menge, B.A.; Gruber, L.; Jorgensen, S.M.; Deacon, C.F.; Schmidt, W.E.; Veldhuis, J.D.; et al. Loss of inverse relationship between pulsatile insulin and glucagon secretion in patients with type 2 diabetes. Diabetes 2011, 60, 2160–2168. [Google Scholar] [CrossRef]
- Meier, J.J.; Kjems, L.L.; Veldhuis, J.D.; Lefebvre, P.; Butler, P.C. Postprandial suppression of glucagon secretion depends on intact pulsatile insulin secretion: Further evidence for the intraislet insulin hypothesis. Diabetes 2006, 55, 1051–1056. [Google Scholar] [CrossRef]
- Demant, M.; Bagger, J.I.; Suppli, M.P.; Lund, A.; Gyldenlove, M.; Hansen, K.B.; et al. Determinants of Fasting Hyperglucagonemia in Patients with Type 2 Diabetes and Nondiabetic Control Subjects. Metab. Syndr. Relat. Disord. 2018, 16, 530–536. [Google Scholar] [CrossRef]
- Lee, Y.H.; Wang, M.-Y.; Yu, X.-X.; Unger, R.H. Glucagon is the key factor in the development of diabetes. Diabetologia. 2016, 59, 1372–1375. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H.; Orci, L. The essential role of glucagon in the pathogenesis of diabetes mellitus. Lancet. 1975, 1, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Reaven, G.M.; Chen, Y.D.; Golay, A.; Swislocki, A.L.; Jaspan, J.B. Documentation of hyperglucagonemia throughout the day in nonobese and obese patients with noninsulin-dependent diabetes mellitus. J. Clin. Endocrinol. Metab. 1987, 64, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Raskin, P.; Unger, R.H. Hyperglucagonemia and its suppression. Importance in the metabolic control of diabetes. N. Engl. J. Med. 1978, 299, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Mitrakou, A.; Kelley, D.; Mokan, M.; Veneman, T.; Pangburn, T.; Reilly, J.; et al. Role of reduced suppression of glucose production and diminished early insulin release in impaired glucose tolerance. N. Engl. J. Med. 1992, 326, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Ipp, E. Impaired glucose tolerance: The irrepressible alpha-cell? Diabetes Care 2000, 23, 569–570. [Google Scholar] [CrossRef]
- Sonne, D.P.; Rehfeld, J.F.; Holst, J.J.; Vilsboll, T.; Knop, F.K. Postprandial gallbladder emptying in patients with type 2 diabetes: Potential implications for bile-induced secretion of glucagon-like peptide 1. Eur. J. Endocrinol. 2014, 171, 407–419. [Google Scholar] [CrossRef]
- Shah, P.; Vella, A.; Basu, A.; Basu, R.; Schwenk, W.F.; Rizza, R.A. Lack of suppression of glucagon contributes to postprandial hyperglycemia in subjects with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2000, 85, 4053–4059. [Google Scholar] [CrossRef]
- Knop, F.K.; Vilsboll, T.; Madsbad, S.; Holst, J.J.; Krarup, T. Inappropriate suppression of glucagon during OGTT but not during isoglycaemic i.v. glucose infusion contributes to the reduced incretin effect in type 2 diabetes mellitus. Diabetologia 2007, 50, 797–805. [Google Scholar] [CrossRef]
- Meier, J.J.; Deacon, C.F.; Schmidt, W.E.; Holst, J.J.; Nauck, M.A. Suppression of glucagon secretion is lower after oral glucose administration than during intravenous glucose administration in human subjects. Diabetologia 2007, 50, 806–813. [Google Scholar] [CrossRef]
- Bagger, J.I.; Knop, F.K.; Lund, A.; Holst, J.J.; Vilsboll, T. Glucagon responses to increasing oral loads of glucose and corresponding isoglycaemic intravenous glucose infusions in patients with type 2 diabetes and healthy individuals. Diabetologia 2014, 57, 1720–1725. [Google Scholar] [CrossRef] [PubMed]
- Bagger, J.I.; Knop, F.K.; Lund, A.; Vestergaard, H.; Holst, J.J.; Vilsboll, T. Impaired regulation of the incretin effect in patients with type 2 diabetes. J. Clin. Endocrinol. Metab. 2011, 96, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Olsen, H.L.; Theander, S.; Bokvist, K.; Buschard, K.; Wollheim, C.B.; Gromada, J. Glucose stimulates glucagon release in single rat alpha-cells by mechanisms that mirror the stimulus-secretion coupling in beta-cells. Endocrinology 2005, 146, 4861–4870. [Google Scholar] [CrossRef] [PubMed]
- Omar-Hmeadi, M.; Lund, P.E.; Gandasi, N.R.; Tengholm, A.; Barg, S. Paracrine control of alpha-cell glucagon exocytosis is compromised in human type-2 diabetes. Nat. Commun. 2020, 11, 1896. [Google Scholar] [CrossRef]
- Kellard, J.A.; Rorsman, N.J.G.; Hill, T.G.; Armour, S.L.; van de Bunt, M.; Rorsman, P.; et al. Reduced somatostatin signalling leads to hypersecretion of glucagon in mice fed a high-fat diet. Mol. Metab. 2020, 40, 101021. [Google Scholar] [CrossRef]
- Vergari, E.; Knudsen, J.G.; Ramracheya, R.; Salehi, A.; Zhang, Q.; Adam, J.; et al. Insulin inhibits glucagon release by SGLT2-induced stimulation of somatostatin secretion. Nat. Commun. 2019, 10, 139. [Google Scholar] [CrossRef]
- Yu, Q.; Shuai, H.; Ahooghalandari, P.; Gylfe, E.; Tengholm, A. Glucose controls glucagon secretion by directly modulating cAMP in alpha cells. Diabetologia 2019, 62, 1212–1224. [Google Scholar] [CrossRef]
- Almaca, J.; Molina, J.; Menegaz, D.; Pronin, A.N.; Tamayo, A.; Slepak, V.; et al. Human Beta Cells Produce and Release Serotonin to Inhibit Glucagon Secretion from Alpha Cells. Cell Rep. 2016, 17, 3281–3291. [Google Scholar] [CrossRef]
- Knudsen, J.G.; Hamilton, A.; Ramracheya, R.; Tarasov, A.I.; Brereton, M.; Haythorne, E.; et al. Dysregulation of Glucagon Secretion by Hyperglycemia-Induced Sodium-Dependent Reduction of ATP Production. Cell Metab. 2019, 29, 430–442. [Google Scholar] [CrossRef]
- Zhang, Q.; Ramracheya, R.; Lahmann, C.; Tarasov, A.; Bengtsson, M.; Braha, O.; et al. Role of KATP channels in glucose-regulated glucagon secretion and impaired counterregulation in type 2 diabetes. Cell Metab. 2013, 18, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Gerich, J.E.; Tsalikian, E.; Lorenzi, M.; Schneider, V.; Bohannon, N.V.; Gustafson, G.; et al. Normalization of fasting hyperglucagonemia and excessive glucagon responses to intravenous arginine in human diabetes mellitus by prolonged infusion of insulin. J. Clin. Endocrinol. Metab. 1975, 41, 1178–1180. [Google Scholar] [CrossRef] [PubMed]
- Steinbusch, L.; Labouebe, G.; Thorens, B. Brain glucose sensing in homeostatic and hedonic regulation. Trends Endocrinol. Metab. 2015, 26, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Stanley, S.; Moheet, A.; Seaquist, E.R. Central Mechanisms of Glucose Sensing and Counterregulation in Defense of Hypoglycemia. Endocr. Rev. 2019, 40, 768–788. [Google Scholar] [CrossRef] [PubMed]
- Nauck, M.A.; Meier, J.J. The incretin effect in healthy individuals and those with type 2 diabetes: Physiology, pathophysiology, and response to therapeutic interventions. Lancet Diabetes Endocrinol. 2016, 4, 525–536. [Google Scholar] [CrossRef]
- Fisher, M.; Sherwin, R.S.; Hendler, R.; Felig, P. Kinetics of glucagon in man: Effects of starvation. Proc. Natl. Acad. Sci. USA 1976, 73, 1735–1739. [Google Scholar] [CrossRef]
- Picard, A.; Berney, X.; Castillo-Armengol, J.; Tarussio, D.; Jan, M.; Sanchez-Archidona, A.R.; et al. Hypothalamic Irak4 is a genetically controlled regulator of hypoglycemia-induced glucagon secretion. Mol. Metab. 2022, 61, 101479. [Google Scholar] [CrossRef]
- Strembitska, A.; Labouebe, G.; Picard, A.; Berney, X.P.; Tarussio, D.; Jan, M.; et al. Lipid biosynthesis enzyme Agpat5 in AgRP-neurons is required for insulin-induced hypoglycemia sensing and glucagon secretion. Nat. Commun. 2022, 13, 5761. [Google Scholar] [CrossRef]
- Robson-Doucette, C.A.; Sultan, S.; Allister, E.M.; Wikstrom, J.D.; Koshkin, V.; Bhattacharjee, A.; et al. Beta-cell uncoupling protein 2 regulates reactive oxygen species production, which influences both insulin and glucagon secretion. Diabetes 2011, 60, 2710–2719. [Google Scholar] [CrossRef]
- Johnson, D.G.; Goebel, C.U.; Hruby, V.J.; Bregman, M.D.; Trivedi, D. Hyperglycemia of diabetic rats decreased by a glucagon receptor antagonist. Science 1982, 215, 1115–1116. [Google Scholar] [CrossRef]
- Spolitu, S.; Okamoto, H.; Dai, W.; Zadroga, J.A.; Wittchen, E.S.; Gromada, J.; et al. Hepatic Glucagon Signaling Regulates PCSK9 and Low-Density Lipoprotein Cholesterol. Circ. Res. 2019, 124, 38–51. [Google Scholar] [CrossRef]
- Okamoto, H.; Cavino, K.; Na, E.; Krumm, E.; Kim, S.Y.; Cheng, X.; et al. Glucagon receptor inhibition normalizes blood glucose in severe insulin-resistant mice. Proc. Natl. Acad. Sci. USA 2017, 114, 2753–2758. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.; Kim, J.; Aglione, J.; Lee, J.; Cavino, K.; Na, E.; et al. Glucagon Receptor Blockade With a Human Antibody Normalizes Blood Glucose in Diabetic Mice and Monkeys. Endocrinology 2015, 156, 2781–2794. [Google Scholar] [CrossRef] [PubMed]
- Wewer Albrechtsen, N.J.; Pedersen, J.; Galsgaard, K.D.; Winther-Sorensen, M.; Suppli, M.P.; Janah, L.; et al. The Liver-alpha-Cell Axis and Type 2 Diabetes. Endocr. Rev. 2019, 40, 1353–1366. [Google Scholar] [CrossRef] [PubMed]
- Stern, J.H.; Smith, G.I.; Chen, S.; Unger, R.H.; Klein, S.; Scherer, P.E. Obesity dysregulates fasting-induced changes in glucagon secretion. J. Endocrinol. 2019, 243, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Mutel, E.; Gautier-Stein, A.; Abdul-Wahed, A.; Amigo-Correig, M.; Zitoun, C.; Stefanutti, A.; et al. Control of blood glucose in the absence of hepatic glucose production during prolonged fasting in mice: Induction of renal and intestinal gluconeogenesis by glucagon. Diabetes 2011, 60, 3121–3131. [Google Scholar] [CrossRef]
- Bozadjieva, N.; Blandino-Rosano, M.; Chase, J.; Dai, X.Q.; Cummings, K.; Gimeno, J.; et al. Loss of mTORC1 signaling alters pancreatic alpha cell mass and impairs glucagon secretion. J. Clin. Invest. 2017, 127, 4379–4393. [Google Scholar] [CrossRef]
- Bozadjieva Kramer, N.; Lubaczeuski, C.; Blandino-Rosano, M.; Barker, G.; Gittes, G.K.; Caicedo, A.; et al. Glucagon Resistance and Decreased Susceptibility to Diabetes in a Model of Chronic Hyperglucagonemia. Diabetes 2021, 70, 477–491. [Google Scholar] [CrossRef]
- Snodgrass, P.J.; Lin, R.C.; Muller, W.A.; Aoki, T.T. Induction of urea cycle enzymes of rat liver by glucagon. J. Biol. Chem. 1978, 253, 2748–2753. [Google Scholar] [CrossRef]
- Hamberg, O.; Vilstrup, H. Regulation of urea synthesis by glucose and glucagon in normal man. Clin. Nutr. 1994, 13, 183–191. [Google Scholar] [CrossRef]
- Kazda, C.M.; Ding, Y.; Kelly, R.P.; Garhyan, P.; Shi, C.; Lim, C.N.; et al. Evaluation of Efficacy and Safety of the Glucagon Receptor Antagonist LY2409021 in Patients With Type 2 Diabetes: 12- and 24-Week Phase 2 Studies. Diabetes Care 2016, 39, 1241–1249. [Google Scholar] [CrossRef]
- Kilberg, M.S.; Barber, E.F.; Handlogten, M.E. Characteristics and hormonal regulation of amino acid transport system A in isolated rat hepatocytes. Curr. Top. Cell Regul. 1985, 25, 133–163. [Google Scholar] [PubMed]
- Ohneda, A.; Parada, E.; Eisentraut, A.M.; Unger, R.H. Characterization of response of circulating glucagon to intraduodenal and intravenous administration of amino acids. J. Clin. Invest. 1968, 47, 2305–2322. [Google Scholar] [CrossRef] [PubMed]
- Dean, E.D. A Primary Role for α-Cells as Amino Acid Sensors. Diabetes 2020, 69, 542–549. [Google Scholar] [CrossRef]
- Dean, E.D.; Unger, R.H.; Holland, W.L. Glucagon antagonism in islet cell proliferation. Proc. Natl. Acad. Sci. USA 2017, 114, 3006–3008. [Google Scholar] [CrossRef] [PubMed]
- Flakoll, P.J.; Borel, M.J.; Wentzel, L.S.; Williams, P.E.; Lacy, D.B.; Abumrad, N.N. The role of glucagon in the control of protein and amino acid metabolism in vivo. Metabolism 1994, 43, 1509–1516. [Google Scholar] [CrossRef]
- Boden G, Rezvani I, and Owen OE. Effects of glucagon on plasma amino acids. J. Clin. Investig. 1984, 73, 785–793. [Google Scholar] [CrossRef]
- Holst, J.J.; Wewer Albrechtsen, N.J.; Pedersen, J.; Knop, F.K. Glucagon and Amino Acids Are Linked in a Mutual Feedback Cycle: The Liver-alpha-Cell Axis. Diabetes 2017, 66, 235–240. [Google Scholar] [CrossRef]
- Miller, R.A.; Birnbaum, M.J. Glucagon: Acute actions on hepatic metabolism. Diabetologia 2016, 59, 1376–1381. [Google Scholar] [CrossRef]
- Solloway, M.J.; Madjidi, A.; Gu, C.; Eastham-Anderson, J.; Clarke, H.J.; Kljavin, N.; et al. Glucagon Couples Hepatic Amino Acid Catabolism to mTOR-Dependent Regulation of alpha-Cell Mass. Cell Rep. 2015, 12, 495–510. [Google Scholar] [CrossRef]
- Bankir, L.; Bouby, N.; Speth, R.C.; Velho, G.; Crambert, G. Glucagon revisited: Coordinated actions on the liver and kidney. Diabetes Res. Clin. Pract. 2018, 146, 119–129. [Google Scholar] [CrossRef]
- Janah, L.; Kjeldsen, S.; Galsgaard, K.D.; Winther-Sorensen, M.; Stojanovska, E.; Pedersen, J.; et al. Glucagon Receptor Signaling and Glucagon Resistance. Int. J. Mol. Sci. 2019, 20, 3314. [Google Scholar] [CrossRef]
- Koopman, R.; Walrand, S.; Beelen, M.; Gijsen, A.P.; Kies, A.K.; Boirie, Y.; et al. Dietary protein digestion and absorption rates and the subsequent postprandial muscle protein synthetic response do not differ between young and elderly men. J. Nutr. 2009, 139, 1707–1713. [Google Scholar] [CrossRef]
- Wahren, J.; Ekberg, K. Splanchnic regulation of glucose production. Annu. Rev. Nutr. 2007, 27, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Felig, P. The glucose-alanine cycle. Metabolism 1973, 22, 179–207. [Google Scholar] [CrossRef] [PubMed]
- Okun, J.G.; Rusu, P.M.; Chan, A.Y.; Wu, Y.; Yap, Y.W.; Sharkie, T.; et al. Liver alanine catabolism promotes skeletal muscle atrophy and hyperglycaemia in type 2 diabetes. Nat. Metab. 2021, 3, 394–409. [Google Scholar] [CrossRef] [PubMed]
- Snell, K.; Duff, D.A. The hepato-muscular metabolic axis and gluconeogenesis. Prog. Clin. Biol. Res. 1982, 102 Pt C, 279–291. [Google Scholar]
- Claris-Appiani, A.; Assael, B.M.; Tirelli, A.S.; Marra, G.; Cavanna, G. Lack of glomerular hemodynamic stimulation after infusion of branched-chain amino acids. Kidney Int. 1988, 33, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Winther-Sorensen, M.; Galsgaard, K.D.; Santos, A.; Trammell, S.A.J.; Sulek, K.; Kuhre, R.E.; et al. Glucagon acutely regulates hepatic amino acid catabolism and the effect may be disturbed by steatosis. Mol. Metab. 2020, 42, 101080. [Google Scholar] [CrossRef]
- Wewer Albrechtsen, N.J.; Junker, A.E.; Christensen, M.; Haedersdal, S.; Wibrand, F.; Lund, A.M.; et al. Hyperglucagonemia correlates with plasma levels of non-branched-chain amino acids in patients with liver disease independent of type 2 diabetes. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G91–G6. [Google Scholar] [CrossRef]
- Zhang, J.; Pivovarova-Ramich, O.; Kabisch, S.; Markova, M.; Hornemann, S.; Sucher, S.; et al. High Protein Diets Improve Liver Fat and Insulin Sensitivity by Prandial but Not Fasting Glucagon Secretion in Type 2 Diabetes. Front. Nutr. 2022, 9, 808346. [Google Scholar] [CrossRef]
- Rocha, D.M.; Faloona, G.R.; Unger, R.H. Glucagon-stimulating activity of 20 amino acids in dogs. J. Clin. Invest. 1972, 51, 2346–2351. [Google Scholar] [CrossRef] [PubMed]
- Galsgaard KD, Jepsen SL, Kjeldsen SAS, Pedersen J, Wewer Albrechtsen NJ, and Holst JJ. Alanine, arginine, cysteine, and proline, but not glutamine, are substrates for, and acute mediators of, the liver-alpha-cell axis in female mice. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E920–E9. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, A.; Kosaka, K. Effects of leucine and isoleucine infused intrapancreatically on glucagon and insulin secretion. Endocrinology 1972, 91, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.A.; Faloona, G.R.; Unger, R.H. The effect of alanine on glucagon secretion. J. Clin. Invest. 1971, 50, 2215–2218. [Google Scholar] [CrossRef]
- Kuhara, T.; Ikeda, S.; Ohneda, A.; Sasaki, Y. Effects of intravenous infusion of 17 amino acids on the secretion of GH, glucagon, and insulin in sheep. Am. J. Physiol. 1991, 260, E21–E26. [Google Scholar] [CrossRef] [PubMed]
- Gar, C.; Rottenkolber, M.; Prehn, C.; Adamski, J.; Seissler, J.; Lechner, A. Serum and plasma amino acids as markers of prediabetes, insulin resistance, and incident diabetes. Crit. Rev. Clin. Lab. Sci. 2018, 55, 21–32. [Google Scholar] [CrossRef]
- Adeva-Andany, M.M.; Lopez-Maside, L.; Donapetry-Garcia, C.; Fernandez-Fernandez, C.; Sixto-Leal, C. Enzymes involved in branched-chain amino acid metabolism in humans. Amino Acids. 2017, 49, 1005–1028. [Google Scholar] [CrossRef]
- Gerich, J.E.; Charles, M.A.; Grodsky, G.M. Characterization of the effects of arginine and glucose on glucagon and insulin release from the perfused rat pancreas. J. Clin. Invest. 1974, 54, 833–841. [Google Scholar] [CrossRef]
- Assan, R.; Attali, J.R.; Ballerio, G.; Boillot, J.; Girard, J.R. Glucagon secretion induced by natural and artificial amino acids in the perfused rat pancreas. Diabetes 1977, 26, 300–307. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, K.; LeBlanc, R.E.; Loh, D.; Schwartz, G.J.; Yu, Y.H. Increasing dietary leucine intake reduces diet-induced obesity and improves glucose and cholesterol metabolism in mice via multimechanisms. Diabetes 2007, 56, 1647–1654. [Google Scholar] [CrossRef]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef]
- Stone, K.P.; Wanders, D.; Orgeron, M.; Cortez, C.C.; Gettys, T.W. Mechanisms of increased in vivo insulin sensitivity by dietary methionine restriction in mice. Diabetes 2014, 63, 3721–3733. [Google Scholar] [CrossRef] [PubMed]
- Dean, E.D.; Li, M.; Prasad, N.; Wisniewski, S.N.; Von Deylen, A.; Spaeth, J.; et al. Interrupted Glucagon Signaling Reveals Hepatic alpha Cell Axis and Role for L-Glutamine in alpha Cell Proliferation. Cell Metab. 2017, 25, 1362–1373. [Google Scholar] [CrossRef] [PubMed]
- Ostenson, C.G.; Grebing, C. Evidence for metabolic regulation of pancreatic glucagon secretion by L-glutamine. Acta Endocrinol. 1985, 108, 386–391. [Google Scholar] [CrossRef]
- Wada, E.; Kobayashi, M.; Kohno, D.; Kikuchi, O.; Suga, T.; Matsui, S.; et al. Disordered branched chain amino acid catabolism in pancreatic islets is associated with postprandial hypersecretion of glucagon in diabetic mice. J. Nutr. Biochem. 2021, 97, 108811. [Google Scholar] [CrossRef]
- Gannon, M.C.; Nuttall, J.A.; Nuttall, F.Q. The metabolic response to ingested glycine. Am. J. Clin. Nutr. 2002, 76, 1302–1307. [Google Scholar] [CrossRef] [PubMed]
- Kalogeropoulou, D.; LaFave, L.; Schweim, K.; Gannon, M.C.; Nuttall, F.Q. Lysine ingestion markedly attenuates the glucose response to ingested glucose without a change in insulin response. Am. J. Clin. Nutr. 2009, 90, 314–320. [Google Scholar] [CrossRef]
- Galsgaard, K.D.; Winther-Sørensen, M.; Ørskov, C.; Kissow, H.; Poulsen, S.S.; Vilstrup, H.; et al. Disruption of glucagon receptor signaling causes hyperaminoacidemia exposing a possible liver-alpha-cell axis. Am. J. Physiol. -Endocrinol. Metabolism. 2018, 314, E93–E103. [Google Scholar] [CrossRef]
- Ang, T.; Bruce, C.R.; Kowalski, G.M. Postprandial Aminogenic Insulin and Glucagon Secretion Can Stimulate Glucose Flux in Humans. Diabetes 2019, 68, 939–946. [Google Scholar] [CrossRef]
- Song, M.; Fung, T.T.; Hu, F.B.; Willett, W.C.; Longo, V.D.; Chan, A.T.; et al. Association of Animal and Plant Protein Intake With All-Cause and Cause-Specific Mortality. JAMA Intern. Med. 2016, 176, 1453–1463. [Google Scholar] [CrossRef]
- Diaz-Villasenor A, Granados O, Gonzalez-Palacios B, Tovar-Palacio C, Torre-Villalvazo I, Olivares-Garcia V; et al. Differential modulation of the functionality of white adipose tissue of obese Zucker (fa/fa) rats by the type of protein and the amount and type of fat. J. Nutr. Biochem. 2013, 24, 1798–1809. [Google Scholar] [CrossRef]
- Tonstad, S.; Stewart, K.; Oda, K.; Batech, M.; Herring, R.P.; Fraser, G.E. Vegetarian diets and incidence of diabetes in the Adventist Health Study-2. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Pan, A.; Sun, Q.; Bernstein, A.M.; Manson, J.E.; Willett, W.C.; Hu, F.B. Changes in red meat consumption and subsequent risk of type 2 diabetes mellitus: Three cohorts of US men and women. JAMA Intern. Med. 2013, 173, 1328–1335. [Google Scholar] [CrossRef] [PubMed]
- Markova, M.; Hornemann, S.; Sucher, S.; Wegner, K.; Pivovarova, O.; Rudovich, N.; et al. Rate of appearance of amino acids after a meal regulates insulin and glucagon secretion in patients with type 2 diabetes: A randomized clinical trial. Am. J. Clin. Nutr. 2018, 108, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Pegorier, J.P.; Garcia-Garcia, M.V.; Prip-Buus, C.; Duee, P.H.; Kohl, C.; Girard, J. Induction of ketogenesis and fatty acid oxidation by glucagon and cyclic AMP in cultured hepatocytes from rabbit fetuses. Evidence for a decreased sensitivity of carnitine palmitoyltransferase I to malonyl-CoA inhibition after glucagon or cyclic AMP treatment. Biochem. J. 1989, 264, 93–100. [Google Scholar]
- Guettet, C.; Mathe, D.; Riottot, M.; Lutton, C. Effects of chronic glucagon administration on cholesterol and bile acid metabolism. Biochim. Biophys. Acta 1988, 963, 215–223. [Google Scholar] [CrossRef]
- Galsgaard, K.D.; Pedersen, J.; Knop, F.K.; Holst, J.J.; Wewer Albrechtsen, N.J. Glucagon Receptor Signaling and Lipid Metabolism. Front. Physiol. 2019, 10, 413. [Google Scholar] [CrossRef]
- Ahren, B. Glucagon--Early breakthroughs and recent discoveries. Peptides 2015, 67, 74–81. [Google Scholar] [CrossRef]
- Charron, M.J.; Vuguin, P.M. Lack of glucagon receptor signaling and its implications beyond glucose homeostasis. J. Endocrinol. 2015, 224, R123–R130. [Google Scholar] [CrossRef]
- Witters, L.A.; Trasko, C.S. Regulation of hepatic free fatty acid metabolism by glucagon and insulin. Am. J. Physiol. 1979, 237, E23–E29. [Google Scholar] [CrossRef]
- Watkins, P.A.; Tarlow, D.M.; Lane, M.D. Mechanism for acute control of fatty acid synthesis by glucagon and 3’:5’-cyclic AMP in the liver cell. Proc. Natl. Acad. Sci. USA 1977, 74, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Day, J.W.; Ottaway, N.; Patterson, J.T.; Gelfanov, V.; Smiley, D.; Gidda, J.; et al. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat. Chem. Biol. 2009, 5, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Pocai, A.; Carrington, P.E.; Adams, J.R.; Wright, M.; Eiermann, G.; Zhu, L.; et al. Glucagon-like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes 2009, 58, 2258–2266. [Google Scholar] [CrossRef] [PubMed]
- Boden, G.; Carnell, L.H. Nutritional effects of fat on carbohydrate metabolism. Best. Pr. Res. Clin. Endocrinol. Metab. 2003, 17, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Gerich, J.E.; Langlois, M.; Schneider, V.; Karam, J.H.; Noacco, C. Effects of alternations of plasma free fatty acid levels on pancreatic glucagon secretion in man. J. Clin. Invest. 1974, 53, 1284–1289. [Google Scholar] [CrossRef] [PubMed]
- Boden, G.; Shulman, G.I. Free fatty acids in obesity and type 2 diabetes: Defining their role in the development of insulin resistance and beta-cell dysfunction. Eur. J. Clin. Invest. 2002, 32 (Suppl. 3), 14–23. [Google Scholar] [CrossRef]
- Madison, L.L.; Seyffert, W.A., Jr.; Unger, R.H.; Barker, B. Effect on plasma free fatty acids on plasma glucagon and serum insulin concentrations. Metabolism 1968, 17, 301–304. [Google Scholar] [CrossRef]
- Edwards, J.C.; Taylor, K.W. Fatty acids and the release of glucagon from isolated guinea-pig islets of Langerhans incubated in vitro. Biochim. Biophys. Acta 1970, 215, 310–315. [Google Scholar] [CrossRef]
- <Effects of Alterations of Plasma Free Fatty Acid 1974.pdf>.
- Collins, S.C.; Salehi, A.; Eliasson, L.; Olofsson, C.S.; Rorsman, P. Long-term exposure of mouse pancreatic islets to oleate or palmitate results in reduced glucose-induced somatostatin and oversecretion of glucagon. Diabetologia 2008, 51, 1689–1693. [Google Scholar] [CrossRef]
- Olofsson, C.S.; Salehi, A.; Gopel, S.O.; Holm, C.; Rorsman, P. Palmitate stimulation of glucagon secretion in mouse pancreatic alpha-cells results from activation of L-type calcium channels and elevation of cytoplasmic calcium. Diabetes 2004, 53, 2836–2843. [Google Scholar] [CrossRef]
- Kristinsson, H.; Sargsyan, E.; Manell, H.; Smith, D.M.; Gopel, S.O.; Bergsten, P. Basal hypersecretion of glucagon and insulin from palmitate-exposed human islets depends on FFAR1 but not decreased somatostatin secretion. Sci. Rep. 2017, 7, 4657. [Google Scholar] [CrossRef] [PubMed]
- Bollheimer, L.C.; Landauer, H.C.; Troll, S.; Schweimer, J.; Wrede, C.E.; Scholmerich, J.; et al. Stimulatory short-term effects of free fatty acids on glucagon secretion at low to normal glucose concentrations. Metabolism 2004, 53, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhao, Y.; Gui, B.; Fu, R.; Ma, F.; Yu, J.; et al. Acute stimulation of glucagon secretion by linoleic acid results from GPR40 activation and [Ca2+]i increase in pancreatic islet {alpha}-cells. J. Endocrinol. 2011, 210, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K.; Maekawa, F.; Dezaki, K.; Nakata, M.; Yashiro, T.; Yada, T. Oleic acid glucose-independently stimulates glucagon secretion by increasing cytoplasmic Ca2+ via endoplasmic reticulum Ca2+ release and Ca2+ influx in the rat islet alpha-cells. Endocrinology 2007, 148, 2496–2504. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Abudula, R.; Chen, J.; Jeppesen, P.B.; Dyrskog, S.E.; Xiao, J.; et al. The short-term effect of fatty acids on glucagon secretion is influenced by their chain length, spatial configuration, and degree of unsaturation: Studies in vitro. Metabolism 2005, 54, 1329–1336. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Chen, L.; Jeppesen, P.B.; Nordentoft, I.; Hermansen, K. Stevioside counteracts the alpha-cell hypersecretion caused by long-term palmitate exposure. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E416–E422. [Google Scholar] [CrossRef]
- Hong, J.; Jeppesen, P.B.; Nordentoft, I.; Hermansen, K. Fatty acid-induced effect on glucagon secretion is mediated via fatty acid oxidation. Diabetes Metab. Res. Rev. 2007, 23, 202–210. [Google Scholar] [CrossRef]
- Piro, S.; Maniscalchi, E.T.; Monello, A.; Pandini, G.; Mascali, L.G.; Rabuazzo, A.M.; et al. Palmitate affects insulin receptor phosphorylation and intracellular insulin signal in a pancreatic alpha-cell line. Endocrinology 2010, 151, 4197–4206. [Google Scholar] [CrossRef]
- Dumonteil, E.; Magnan, C.; Ritz-Laser, B.; Ktorza, A.; Meda, P.; Philippe, J. Glucose regulates proinsulin and prosomatostatin but not proglucagon messenger ribonucleic acid levels in rat pancreatic islets. Endocrinology 2000, 141, 174–180. [Google Scholar] [CrossRef]
- Gremlich, S.; Bonny, C.; Waeber, G.; Thorens, B. Fatty acids decrease IDX-1 expression in rat pancreatic islets and reduce GLUT2, glucokinase, insulin, and somatostatin levels. J. Biol. Chem. 1997, 272, 30261–30269. [Google Scholar] [CrossRef]
- Ellingsgaard, H.; Ehses, J.A.; Hammar, E.B.; Van Lommel, L.; Quintens, R.; Martens, G.; et al. Interleukin-6 regulates pancreatic alpha-cell mass expansion. Proc. Natl. Acad. Sci. USA 2008, 105, 13163–13168. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, O.; Carr, R.D.; Deacon, C.F.; Holst, J.J.; Pacini, G.; Mari, A.; et al. Incretin hormone and insulin responses to oral versus intravenous lipid administration in humans. J. Clin. Endocrinol. Metab. 2011, 96, 2519–2524. [Google Scholar] [CrossRef] [PubMed]
- Raben, A.; Holst, J.J.; Madsen, J.; Astrup, A. Diurnal metabolic profiles after 14 d of an ad libitum high-starch, high-sucrose, or high-fat diet in normal-weight never-obese and postobese women. Am. J. Clin. Nutr. 2001, 73, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Mandoe, M.J.; Hansen, K.B.; Windelov, J.A.; Knop, F.K.; Rehfeld, J.F.; Rosenkilde, M.M.; et al. Comparing olive oil and C4-dietary oil, a prodrug for the GPR119 agonist, 2-oleoyl glycerol, less energy intake of the latter is needed to stimulate incretin hormone secretion in overweight subjects with type 2 diabetes. Nutr. Diabetes. 2018, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Mandoe, M.J.; Hansen, K.B.; Hartmann, B.; Rehfeld, J.F.; Holst, J.J.; Hansen, H.S. The 2-monoacylglycerol moiety of dietary fat appears to be responsible for the fat-induced release of GLP-1 in humans. Am. J. Clin. Nutr. 2015, 102, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Sloth, B.; Due, A.; Larsen, T.M.; Holst, J.J.; Heding, A.; Astrup, A. The effect of a high-MUFA, low-glycaemic index diet and a low-fat diet on appetite and glucose metabolism during a 6-month weight maintenance period. Br. J. Nutr. 2009, 101, 1846–1858. [Google Scholar] [CrossRef]
- Filippello, A.; Urbano, F.; Di Mauro, S.; Scamporrino, A.; Di Pino, A.; Scicali, R.; et al. Chronic Exposure to Palmitate Impairs Insulin Signaling in an Intestinal L-cell Line: A Possible Shift from GLP-1 to Glucagon Production. Int. J. Mol. Sci. 2018, 19, 3791. [Google Scholar] [CrossRef]
- Mandoe, M.J.; Hansen, K.B.; Hartmann, B.; Rehfeld, J.F.; Holst, J.J.; Hansen, H.S. The 2-monoacylglycerol moiety of dietary fat appears to be responsible for the fat-induced release of GLP-1 in humans. Am. J. Clin. Nutrition. 2015, 102, 548–555. [Google Scholar] [CrossRef]
- Freeland, K.R.; Wilson, C.; Wolever, T.M. Adaptation of colonic fermentation and glucagon-like peptide-1 secretion with increased wheat fibre intake for 1 year in hyperinsulinaemic human subjects. Br. J. Nutr. 2010, 103, 82–90. [Google Scholar] [CrossRef]
Figure 1.
Glucagon receptors on multiple organs and the stimulation/inhibition of glucagon release (this graph is generated with www.biorender.de).
Figure 1.
Glucagon receptors on multiple organs and the stimulation/inhibition of glucagon release (this graph is generated with www.biorender.de).
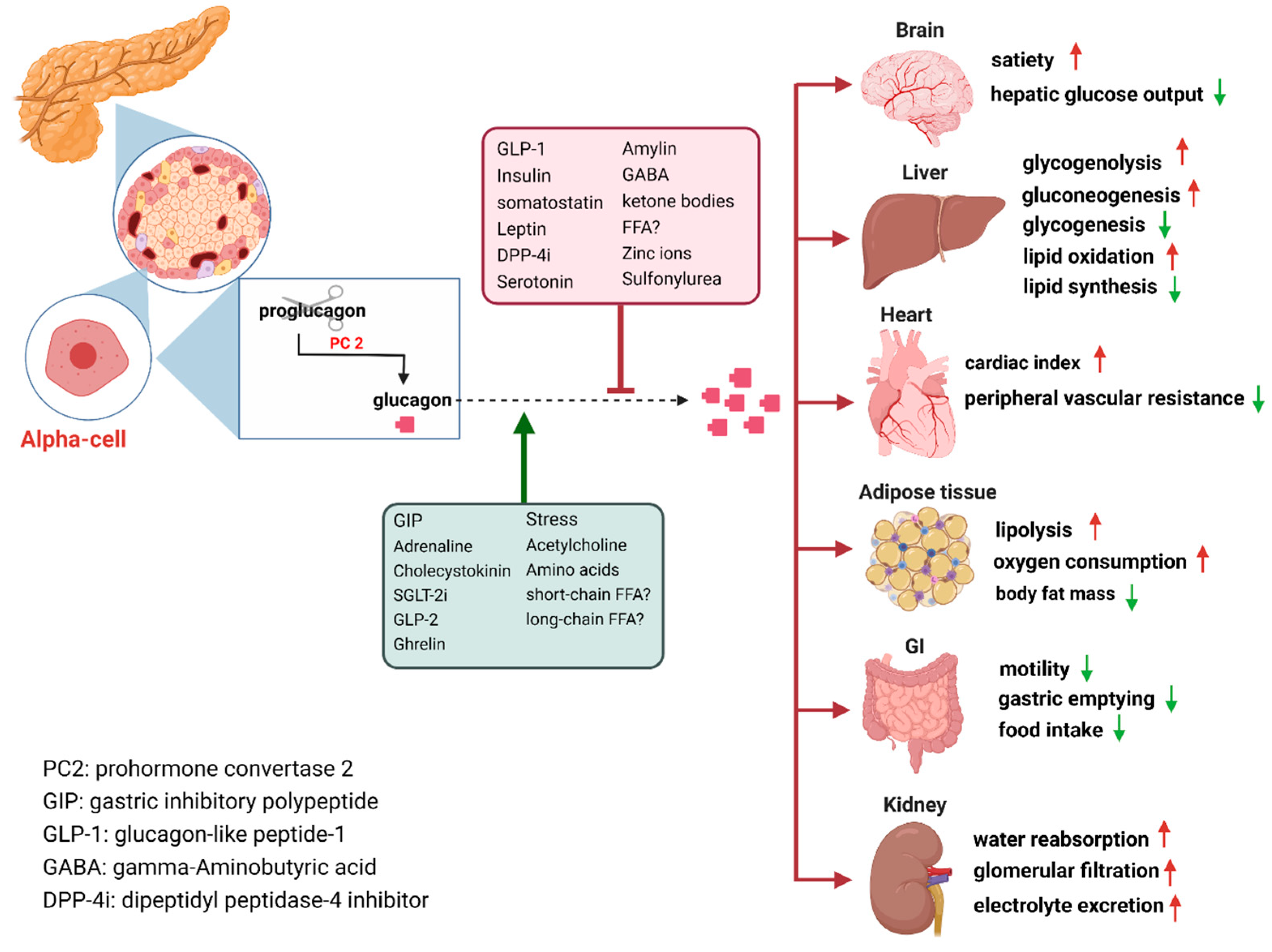
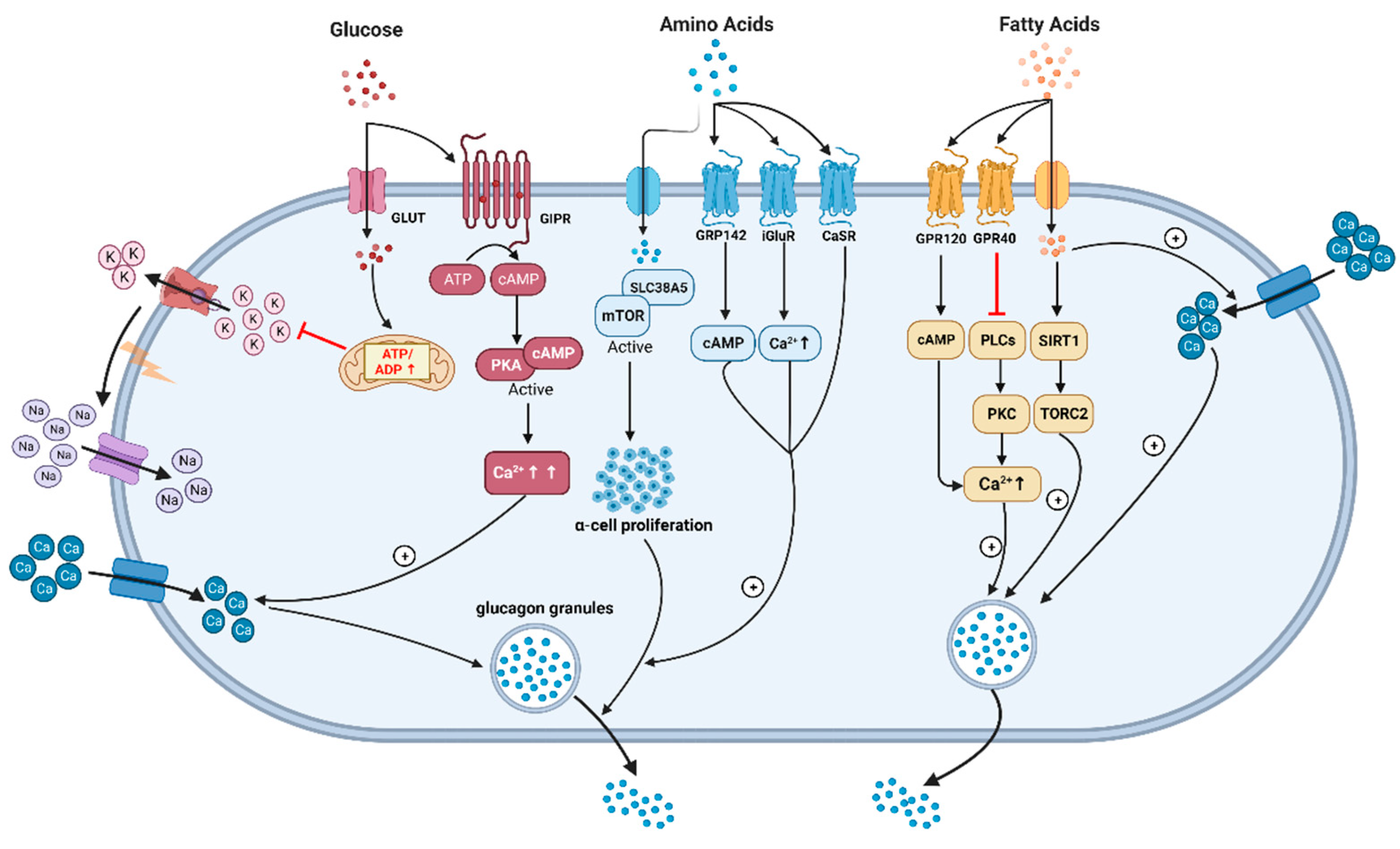
Figure 2.
The underlying mechanisms of glucagon stimulation to glucose, amino acids, and fatty acids (this graph is generated with www.biorender.de).
Figure 2.
The underlying mechanisms of glucagon stimulation to glucose, amino acids, and fatty acids (this graph is generated with www.biorender.de).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



