
Submitted:
18 August 2023
Posted:
21 August 2023
You are already at the latest version
Abstract
Thimerosal (TmHg) has been used in thimerosal-containing vaccines (TCV) and various med-icines, although to our knowledge, there are no studies evaluating its effects on microglia. Microglia are a crucial glial component that have important functions in inflammatory diseases, particularly in the Central Nervous System (CNS). They are also considered a key player in neurotoxicity. Furthermore, microglia are responsible for the activation of immune responses and plays an essential role for the maintenance of brain homeostasis. Microglial response is important for the maintenance of CNS immune regulation. Moreover, microglia cells partici-pate in the removal of cellular debris and pathogens, playing an important role as neuroprotectants in several CNS diseases, including neurodegeneration. The objective of this study was to evaluate the effects of TmHg and its metabolite ethylmercury (EtHg) on microglial cells. Our novel findings demonstrated that microglia are more sensitive to TmHg than to EtHg with EC50 1.4 ± 0.4 μM and 2.3± 0.9 μM for 24h, respectively. The mercury compounds also induced the transcription of pro-inflammatory markers s such as IL-1β; iNOS, and to a lesser extent TNF-α expression. Likewise, the increase in Beclin-1 expression - a protein associated to autophagy - and the number of senescent cells indicate that microglia are activated by TmHg and EtHg. The effects of thimerosal may have different consequences depending on the health status and susceptibility of the populations.
Keywords:
microglia
; thimerosal
; ethylmercury
; inflammation
; neurotoxicity
1. Introduction
Mercury is a toxic heavy metal that is widely distributed in nature through processes such as degasification of earth´s crust or volcanic activity [1,2,3]. According to the World Health Organization (WHO), it is considered one of the top 10 chemicals of “major public health concern” and a ubiquitous pollutant [4]. Mercurial toxicity depends mainly on 3 factors: the chemical form of mercury, the dose, and the rate of exposure [3].
Exposure to mercury compounds can affect different organs such as the kidney, the liver, with major health effects on the Central Nervous System (CNS) [2], encompassing behavioural changes, emotional liability, somatosensory irritation, and alterations in mood scores [5] that mostly result from neuronopathies.
TmHg has an history of clinical use as antimicrobial and antifungal that ultimately led to its inclusion as a preservative in vaccines and medicines such as, formulations for ophthalmic application. There is a notorious scarcity of studies employing TmHg although the ones that were performed show the deleterious effect of this organomercurial compound [6,7,8,9,10]. Despite the lack of epidemiological evidence of significant neurotoxicity in humans, regulatory authorities, such as FDA, advised against its use in all vaccines routinely recommended for children 6 years of age or younger given the potential health effects of thimerosal [11]. The agency also recognizes the need to conduct additional studies to further evaluate safety.
Microglia, the tissue-resident macrophages of the brain represent 10% of the total glial cell population in adults [1], and are a crucial glial component of the CNS. Microglia serve several important functions in CNS, including, surveillance of baseline neuronal activity, rapid response to tissue damage, significant regulation of brain development, developmental engulfment of neural progenitor cells, maintenance of neuronal networks and promotion of neuronal survival [12]. Microglia are also considered the principal innate immune cells of CNS, being responsible for the maintenance of immune homeostasis [12]. Additionally, microglia have several important housekeeping functions like, ion regulation, metabolic support, extracellular matrix formation, neurotransmitter recycling and modulation of synaptic transmission [13,14]. Microglia play a key function is phagocytosis, a complex process that recognizes altered or non-self-cells, bind and digest particles [15]. Being a type of differentiated tissue macrophage, microglia cells are responsible for the elimination of pathogens, dead cells, redundant synapses, protein aggregates, and other antigens that may harm the CNS. However, these brain macrophages are different from the other tissue macrophages due to their unique homeostatic phenotype [16].
Microglia heterogeneity is characterized by changes in the morphology phenotype. Microglial reactive states are complex and the classification of M1 and M2 states and their subcategories is no longer sufficient to encompass the heterogeneous and dynamic nature of these cells. In spite of that, there has been some consensus that upon encountering Damage Associated Molecular Patterns (DAMPs) or other foreign material, microglia enter a ‘classically’ activated or ‘M1’ state, leading to a drastic increase in the production of inflammatory cytokines including IL-6 (Interleukin 6), IL-1β (Interleukin 1β), TNF-α (Tumour Necrosis Factor α), and IFN-γ (Interferon γ), as well as Nitric Oxide (NO) and Reactive Oxygen Species (ROS). The release of these molecules is important for the removal of unnecessary cells and pathogens, affording a neuroprotective role to the CNS [17,18]. It has been suggested that so-called ‘M1’ macrophages and microglia may be more vulnerable to ferroptotic or iron-induced cell death due to their being enriched in inducible Nitric Oxide Synthase (iNOS).
Microglia undergo senescence related to aging disease development (Disease-Associated Microglia, DAM) being characterized by a limited capacity for division. Senescent cells are still metabolically active, and capable of secreting molecules to modify the environment, the so-called Senescence-Associated Secretory Phenotype (SASP). SASP is associated with increased secretion of pro-inflammatory molecules, and also those involved in processes of matrix-degradation such as Tumour Necrosis Factor α (TNF-α) and Interleukin-6 (IL-6). Senescent microglia encompass features of both M1 and M2 microglia reactive states [19].
Activation of β-Galactosidase expression has also been used as a senescence marker. For instance, Martínez-Zamudio and coworkers (2021) [20] used a second-generation fluorogenic substrate for β-galactosidase and multi-parameter flow cytometry, to demonstrate that peripheral blood mononuclear cells (PBMCs) isolated from healthy humans increasingly display cells with high senescence-associated β-galactosidase (SA-βGal) activity with advancing donor age.This hydrolase enzyme resides in lysosomes and coverts β-galactosidase into monosaccharides under acidic pH conditions. Nevertheless, there are controversial results that observed its increase in hippocampal neurons from 3-months-old mice [20].
Beclin-1 is a conserved autophagy protein, that acts in the initiating phase of autophagy, forming an isolating membrane containing the cytoplasmic material to form the autophagosome[18]. Beclin 1 may not only function in autophagosome formation, but also in autophagosome/endosome maturation, and that temporally modulated or spatially modulated interactions between Beclin-1 and its positive regulators and negative regulators may govern these activities [21]. Decreases in Beclin 1 expression and/or functional activity have been linked to increased susceptibility to cancer, Alzheimer´s disease, Huntington´s disease, and desmin-related cardiomyopathy; alterations in microbial pathogenesis defects in apoptotic corpse clearance and development; and aging [21].
Autophagy is a fundamental cellular process that eliminates molecules and subcellular elements, including nucleic acids, proteins, lipids, and organelles, via lysosome-mediated degradation to promote homeostasis, differentiation, development and survival. While autophagy is intimately linked to health, the intricate relationship among autophagy, aging and disease remains unclear [22]. Autophagy is induced during and facilitates the senescence process, and the two pathways are functionally intertwined [23].
Overall, microglia play an important role in brain inflammatory diseases, thus being a focal point in the study of therapeutic targets for the development of new drugs [24,25]. Furthermore, other studies demonstrated that glial cells when activated by metal ions, such as mercury compounds, can release mediators of inflammatory and immune response [5,26]. Surprisingly, the effects of thimerosal over these cells remain unknown.
The aim of this study is to evaluate the effects of TmHg/EtHg on microglia to elucidate it the compounds affect these cells and their functions and what could be the consequences for susceptible populations.
2. Materials and Methods
2.1. Cell culture
The mouse microglia cell line, N9, was a kind gift from Prof. Dora Brites (Research Coordinator, Faculty of Pharmacy, Universidade de Lisboa and Group Leader of NeuroIn, iMed.ULisboa) it is a retroviral cell line, resulting from the spontaneous immortalization of microglia isolated from the cortex of C57BL/6 mouse embryos and has similar characteristics to primary microglia cultures such as phagocytose activity upon liposaccharide stimulation, TNF-α cytokine secretion upon β-amyloid exposure, migration, and inflammation-related features. These cells were grown in RPMI 1640 Medium containing Glutamax (Invitrogen) and supplemented with 10% of Fetal Bovine Serum (FBS) and 1% Penicillin/streptomycin mixture. Cultures were maintained in a humidified incubator at 37°C and 5% CO2. All cell manipulations were performed in a laminar flow cabinet with sterile reagents such as Versene (PBS (1x) +EDTA), Trypsin-EDTA 0.05% (10-15min, 37°C) and PBS 1x. These were preheated in water at 37°C during 20 minutes before contact with cell cultures. When cultures reached 70-80% confluence, the cells were subcultured or plated for experiments.
2.2. Mercury compounds
The mercury compounds used in the present study were TmHg (> 97%; Sigma) and its metabolite EtHg (Alfa Aeser). The work solutions were prepared to final concentration of 0.1M with sterile DMSO (Alfa Aeser). In each experimental step, fresh solutions of both compounds were prepared with PBS and brought to a final concentration of 1mM. The necessary volumes were added to cells medium to obtain a final concentration between 0.5 μM and 5 μM based on the results of previous studies (see Table 2).
2.3. Cell viability
Viability assays are used to determine the proportion of viable cells after a traumatic procedure like primary disaggregation or cell separation[27]. In the present study, the overall viability was determined with the MTT assay. The MTT assay is based on the conversion of [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (MTT) into insoluble purple formazan crystals by the action of mitochondrial reductase [28]. The effects of the mercury compounds on microglia were determined at 0.5 μM and 5 μM. Briefly, cells were incubated in 96 well-plates at a density of 5x103 cells/well. After 24h, EtHg at final concentration of 0.5, 1, 2, 3, 4, 5 μM and TmHg at final concentration of 0.5, 1, 2.5 and 5 μM were added to respective wells. Following 24h, 48h and 72h of incubation with the mercury compounds, the MTT solution was added to a final concentration of 400 mg/ml. After a 2h in a humified incubator at 37°C and 5% CO2, the medium was removed, and the formazan crystals formed were subsequently dissolved in 4:1 DMSO/ glycine pH 10.5 buffer. After 15 minutes shaking at room temperature, the absorbance was measured at 550 nm on a microplate reader (Zenyth 3100, Anthos Labtec Instruments). The concentration that causes 50% of cell proliferation (EC50) was calculated in comparison to the control group at each time point.
2.4. Determination of gene expression
The expression of TNF-α; iNOS, and 1L-1β was determined through quantitative Real Time-Polymerase Chain reaction (qRT-PCR) after RNA isolation kit (NZYTech, Portugal) and quantification in each sample and cDNA synthesis kit (NZYTech, Portugal), according to the manufacturer’s protocols.
2.4.1. RNA Isolation
The cells were placed in 6 well-plates (0.1x106cell/well) to achieve the 70%-80% of confluence. After 2 days, the medium was renewed and was added the mercury compounds, EtHg and TmHg, with the final concentration of 0.5; 1; 2 and 5 μM and Lipopolysaccharide (LPS) (300ng/mL). After 3h, 6h and 24h of exposure, cells were collected and RNA was extracted from with a NYZ total Isolation kit (NZYTech, Portugal), according to manufacturer instructions. First, the cells were collected and spun at 6,000g for 5 minutes at 4°C. Next, the supernatant was removed and at the pellet was added NR buffer from kit and finally, β-mercaptoethanol. Then, the samples were transferred to a homogenization column and after centrifuged at 11,000g for 1min, the flow-through was collected. In order to induce DNA precipitation, 70% of ethanol was joined to samples[29] Consequently, the lysates were load on a binding column and after NI buffer addition, digestion mixture (DNase I + Digestion buffer) was added and incubated at room temperature for 15 minutes. Following NWR1 and NWR2 buffer was added to lysates. In the final step, RNA was eluted with RNase free water and stored at -80°C until analysis.
2.4.2. RNA quantification and cDNA synthesis
The concentration of RNA in each sample was determined by nanodrop analysis in an OMEGA Tristar Spectrophotometer and the purity was analysed by A260/A280 and A260/A230 ratio. For A260/A280 the very pure RNA should be with ratio of ~2.1 and each value higher than 1.8 was considered acceptable purity. For cDNA analysis we used the NZY First-Strand cDNA Synthesis Kit (NZYTech, Portugal), according to manufacturer’s instructions. For this purpose, a sample volume of 10 pg and 5 µg of RNA were mixed with NZYRT Master Mix, NZYRT Enzyme mix and DEPC-treated H2O to a final volume of 20 μl. Next, samples were incubated at 25°C for 10 min, 50°C for 30 min, 85°C for 5 min to inactivate the reaction and then chilled at 4 °C. Subsequently, 1.5 μl of NZY RNase H from the kit was added to samples followed by incubation at 37°C for 20 min. The cDNA obtained was stored at -20°C until qRT-PCR experiment.
2.4.3. qRT-PCR
qRT-PCR was performed on an Applied Biosystems QuantStudio™ 7 Flex real-time PCR system (Applied Biosystems, Foster City, CA, USA). Preparation of each sample for each gene included mixing of 100 ng of cDNA template with a NZYqPCR GMM (2x) Rox plus, nuclease-free water and the respective forward and reverse primers (400nM). The amplification reaction included an initial 2 min stage at 50°C, followed by 10 min at 95°C and 40 cycles of 15 seconds at 95°C and 1 minute at 60°C. The results were determined according to 2-ΔΔCT method and expressed comparatively to the respective control. Besides the target genes iNOS, TNF-α and IL-1β, glyceraldehyde 3- phosphate dehydrogenase (GAPDH) was used as a reference gene. Primer sequences for each gene are reported on

Table 1.
Genomic sequence of GADPH, iNOS, TNF-α and IL-1β genes. The efficiency of primers was determined by evaluating the amplification Ct of serial dilutions (1x10-1 to 1x10-6) of the cDNA template and the values obtained varied between 104% and 108%.
Table 1.
Genomic sequence of GADPH, iNOS, TNF-α and IL-1β genes. The efficiency of primers was determined by evaluating the amplification Ct of serial dilutions (1x10-1 to 1x10-6) of the cDNA template and the values obtained varied between 104% and 108%.
| Gene | Forward sequence | Reverse sequence |
|---|---|---|
| GAPDH | 5´GGA GAG TGT TTC CTC GTC CC 3´ | 5´ATG AAG GGG TCG TTG ATG GC 3´ |
| iNOS | 5´GTT CTC AGC CCA ACA ATA CAA GA 3´ | 5´GTG GAC GGG TCG ATG TCA C 3´ |
| TNF-α | 5´TAG CCC ACG TCG TAG CAA AC 3´ | 5´GCA GCC TTG TCC CTT GAA GA 3´ |
| IL-1β | 5´TGC CAC CTT TTG ACA GTG ATG 3´ | 5´ATG TGC TGC TGC GAG ATT TG 3´ |
2.5. Determination of microglia autophagy
Autophagy is a “cellular function in which the cell degrades its own components in the lysosome” and in the N9 cell line it was measured by analysing Beclin-1 expression through Western-Blot, since this protein is an essential autophagy protein [30].
For western blot, firstly it was necessary to prepare cellular lysates. Briefly, cells were plated in 6 well-plates with 1x105 cells/ml per well. After reaching 70%-80% confluence, the medium was renovated, and cells were contaminated with positive control (LPS), EtHg/TmHg at 0.5; 1; 2 μM and incubated at 37°C with 5% of CO2 for 3h and 24h. After trypsinization, cells were centrifuged at 600 g for 5 minutes, the supernatant was removed, the pellet was washed with phosphate-buffered saline (PBS) and 35 μL of lysis buffer (25 mM Tris; 2.5 mM EDTA; 2.5mM EGTA; 20 mM NaF, 1 mM Na3NO4; 100mM NaCl; 20 mM C3H9Na2O6; 10 mM Na2H2P2O7; 0.5% Triton) was added. The total protein concentration of final lysates was quantified with the Bradford method in the soluble fraction after centrifugation of lysates at 12,000 g for 5 min. Briefly, samples were incubated with Coomassie dye (Bio-Rad; diluted 5 times) in 96-well plate and absorbance was measured at 595 nm in the microplate reader (Zenyth 3100, Anthos Labtec Instruments). Protein concentration was calculated from a calibration curve (1–12 μg of protein μL−1) with BSA. For the electrophoresis step, 40 μg of each sample were mixed with LDS, DTT 1M and water to a final volume of 20 μL. Subsequently, the samples were preheated at 70°C for 20 minutes to denature proteins and loaded on a 4-12% Bis-Tris Gel and the electrophoresis took place for 1h at 140 V. After electrophoresis, samples were transferred (transfer buffer: 25 mM Tris + 192 mM Glycine) to a nitrocellulose membrane (30 V, 2 h). Loading and transfer efficiency were evaluated by staining membranes with Ponceau S. Then the membrane was incubated with 5% milk solution during 1h at room temperature and afterwards incubated with anti-Beclin primary antibody (1:2000) overnight at 4°C. After five washes with PBST the nitrocellulose membrane was incubated with the secondary antibody anti-mouse (1:2000) during 2 h for further analysis of chemiluminescence.
2.6. Assessment of microglia Senescence
Senescence was evaluated by measuring beta-galactosidase (SA-β-galactosidase) activity. The presence of this biomarker is independent of DNA synthesis and distinguishes senescent cells from quiescent cells [31]. Beta-galactosidase activity was determined through the Cellular Senesce Assay Kit (Millipore) according to manufacturer’s instructions. Briefly, N9 cells (1x105 cells/well) were plated in 6-well plates, and upon reaching 70%-80% of confluence, either LPS (300 ng/ml), EtHg or TmHg (0.5; 1; 2 μM) were added to wells. According to kit instructions, a Fixing solution diluted in PBS (1:100) was added first to samples, followed by overnight incubation with the detection solution (SA-β-galactosidase) at 37°C without CO2 and protected from light. Next, the SA-β-galactosidase solution was removed, and the nuclei were counterstained with haematoxylin. The images for each sample were obtained using Zen 2012 (blue edition, Zeiss) and AxioCam 105 color (Zeiss) adapted to an AxioSkope HBO50 microscope (Zeiss). The percentage of blue staining cells, i.e. senescence cells, was determined relative to the total cell number for each image (original magnification: 40X).
2.7. Statistical analysis
The results represent the mean ± SEM of three to five independent experiments. Statistical analysis was performed using the GraphPad Prism software and consequence analysis of data was performed through a One-way-ANOVA. Multiple comparisons between exposure groups and control were analysed by the Turkey’s post hoc test. the p-values <0.05 were considered statistically significant.
3. Results
3.1. Cytotoxicity effects of TmHg and EtHg on N9 cell line
Both mercury compounds under study affected the viability of N9 cells in a concentration- and time-dependent manner (Figure 1). After 24 h, the EC50 is 2.3 ± 0.9 μM and 1.4 ± 0.4 μM for EtHg and TmHg, respectively. Moreover, the results showed that TmHg was more toxic to N9 cells than EtHg, at all studied time-points.
3.2. Effects of TmHg/EtHg at pro-inflammatory markers
Exposure to TmHg and EtHg elicited IL-1β expression, especially after 3 h (Figure 2A) with 5μM EtHg and 2μM TmHg.
iNOS expression significantly increased after 3h exposure to 5μM EtHg and 2μM TmHg (Figure 2B).
TNF-α expression in response to Hg compound exposures was indistinguishable from controls (p>0.05) (Figure 2C).
LPS was used as a positive control, that activates M1 microglia phenotype, promoting the release of inflammatory cytokines and therefore, it induces a high response in all genes studied [32].
3.3. Autophagy evaluation: Beclin-1 expression
Beclin-1 participates in the early stages of autophagy process and for that reason its determination was used to evaluate the consequences of microglia inflammation. As shown in Figure 3 B-C, after 3 h exposure to both Hg compounds, a concentration-dependent increase in the expression of the Beclin-1 protein was noted, and it was more pronounced in response to TmHg treatment (Figure 3 B-C).
3.4. Senescence evaluation
Senescence evaluation was carried out by determining SA-β-Galactosidase expression, counting the blue stained cells under light microscopy as noted in in the Materials and Methods section (Figure 4).
The expression of SA-β-Galactosidase showed a concentration-dependent increase, with exposure to both EtHg, but especially TmHg (Figure 5).
4. Discussion
In this novel work, we demonstrated that N9 are more sensitive to TmHg than to its metabolite, EtHg, with EC50 values of 1.4 μM (0.6 μg/mL) and 2.3 μM (0.5 μg/mL) respectively, after 24h exposure. TmHg and EtHg have been poorly tested yet considered less toxic than other compounds such as MeHg. However, based on results obtained by different research groups TmHg appears equally toxic to the N9 cell line (Table 2), consistent with a prominent effect of this compound on microglia, which could adversely affect both immunological and nervous systems.
Notably, compared to another glial cell type, astrocytes, microglia show higher susceptibility to mercury compounds [37]. Although analogous effects of MeHg have been seen in microglia, it is noteworthy that TmHg is more toxic than MeHg in SHSY5Y cells [36]. Additionally, our group has shown that Hg2+ exposure in N9 cells had an EC50 of 42.1 ± 3.7μM. Comparatively, TmHg and EtHg inhibited proliferation at concentrations significantly lower, being more toxic to N9 cell line [34].
Given the results of the viability assay, 2 µM TmHg was selected as the maximal concentration for subsequent assays with the longest exposure timepoint for 24h, since we were primarily interested in determining the effects on microglial differentiation, avoiding massive cell death.
Since IL-1β is responsible for the activation of microglia and iNOS is released as function of IL-1β, it is possible to conclude that microglia are activated in the presence of both mercury compounds (Figure 2 and Figure 6). In a previous study N9 exposure to Hg2+, also led to higher mRNA levels of pro-inflammatory cytokines [34], and high levels of inflammatory genes transcription were associated with the activation of the p38 pathway and ROS production [34].
Regarding TNF-α, , its mRNA levels were not significantly increased in comparison to IL-1β and iNOS. This result is corroborative with data published for microglia exposure upon MeHg exposure, which also failed to provoke significant changes in TNF-α mRNA expression[38].
The activation of the immune system in general can be beneficial in response to harmfull stimulii, such as pathogens, damaged cells contributing to maintain homeotasis [39]. Nevertheless, excessive activation and inflammation can lead to deleterious effects on the immune system. Accordingly, the autophagy process was evaluated [40]. The results of Beclin-1 expression (Figure 3 A-C) indicated this protein was induced by both EtHg and TmHg and at both timepoints tested, with the most significant increase noted with 2μM TmHg after 24h exposure. The high values of Beclin-1 indicate that activation of microglia by mercury compounds has the potential to interfere with cerebral homeostasis (Figure 6). On the other hand, Wang and coworkers (2012) [41] demonstrated a link between oncogenic signaling, the core autophagy machinery, and cytoskeletal proteins in the intermediate filament family. Akt signaling, intermediate filaments and proteins may be mechanistically linked to autophagy inhibition and tumorigenesis through regulation of the Beclin 1 complex. The higher expression of this protein with TmHg, indicates that it triggers more efficiently the autophagosome membrane formation initiating the autophagic process. The values of Beclin-1 expression were lower in LPS treated-cells, which is related to inhibition of autophagosome formation and autophagy by this endotoxin [42].
Senescent cells develop a non-proliferative state but can maintain their viability [43,44]. Senescence was only evaluated at 24h, assuming that it will occur after the autophagy process. Analogous to autophagy, the results demonstrated increased levels of senescent cells with TmHg exposure consistent with an inflammatory response (Figure 6). Notably, several studies have addressed the activation of microglia by mercurial at the gene transcription level, but this is the first time that autophagy and senescence were evaluated together.
5. Conclusions
Overall, the results show that TmHg and EtHg elicit microglial inflammation, which leads to microglial damage and loss of function. This inflammatory process is important for by two reasons. First of all, it supplies additional evidence to the ones that are against the exposure of newborns to TmHg even for a health protective reason as is the case of vaccine administration, since our results go in favor that mercury can lead to microglial damage and loss of function and therefore interfere with neurobehavioral development and function in children and also affects the immune system. Secondly, this activation pathway merits further attention with reference to malignant brain tumors as cancer cells with upregulated autophagy have a less aggressive behavior and are more responsive to chemotherapy. Glioblastoma that is one of the most recurrent and lethal tumors resistant to therapy, that encompasses a large number of microglia cells, therefore, the repurposing of TmHg [45,46] for adults’ treatment can be of great importance in the search for new therapeutic approaches.
Author Contributions
Conceptualization, C Carvalho and V Branco; Methodology, C Alfenim, V Branco and R Vaz; Software, I. Bramatti, C Alfenim and R Vaz; Validation, C Alfenim, I Bramatti and R Vaz; Formal analysis, C Alfenim, I Bramatti and R Vaz, C Carvalho, V Branco and M Aschner ; Investigation, C Alfenim, I Bramatti and R Vaz; Resources, C Carvalho; Data curation, C Alfenim, V Branco and C Carvalho; Writing—original draft preparation, C Carvalho and C Alfenim; Writing—C Carvalho, I Bramatti and M Aschner; Visualization, C Carvalho and M Aschner; Supervision, C Carvalho and V Branco; Project administration, C Carvalho; Funding acquisition, C Carvalho. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by FCT - Portugal through projects Target_Cancer (PTDC/MED-FAR/31136/2017) and by iMed.ULisboa UIB04138/2020 both from FCT. Vasco Branco is financed by national funds via FCT through Norma Transitória - DL57/2016/CP1376/CT002. MA was supported in part by a grant from the National Institute of Environmental Health Sciences (NIEHS) R01 ES07331.
Institutional Review Board Statement
Not applied.
Informed Consent Statement
Not applied.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Figueiredo, N.L.; Canário, J.; O’Driscoll, N.J.; Duarte, A.; Carvalho, C. Aerobic Mercury-resistant bacteria alter Mercury speciation and retention in the Tagus Estuary (Portugal). Ecotoxicol Environ Saf. 2016, 124, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Carocci, A.; Catalano, A.; Lauria, G.; Sinicropi, M.S.; Genchi, G. Brief History of the Development of the Transfusion Service, How to Recruit Voluntary Donors in the Third World? 2015, 238, 22–28. [Google Scholar] [CrossRef]
- Bernhoft, R.A. Mercury toxicity and treatment: A review of the literature. J Environ Public Health. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Caetano, T.; Branco, V.; Cavaco, A.; Carvalho, C. Risk assessment of methylmercury in pregnant women and newborns in the island of Madeira (Portugal) using exposure biomarkers and food-frequency questionnaires. Journal of Toxicology and Environmental Health - Part A: Current Issues. 2019, 82, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Watson, G.E.; Brewer, R.; Zareba, G.; Eto, K.; Takahashi, H.; Marumoto, M.; Love, T.; Gary, J.; Neurology, C.; City, M.; Section, P.; Disease, M.; City, M.; Biology, C. HHS Public Access, 2021, 88–98. [CrossRef]
- Sharpe, M.A.; Livingston, A.D.; Baskin, D.S. Thimerosal-derived ethylmercury is a mitochondrial toxin in human astrocytes: Possible role of fenton chemistry in the oxidation and breakage of mtDNA. J Toxicol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Cinca, I.; Dumitrescu, I.; Onaca, P.; Serbanescu, A.; Nestorescu, B. Accidental ethyl mercury poisoning with nervous system, skeletal muscle, and myocardium injury. J of Neurology, Neurosurgery, and Psychiatry 1979. [Google Scholar] [CrossRef]
- Migdal, C.; Tailhardat, M.; Courtellemont, P.; Haftek, M.; Serres, M. Responsiveness of human monocyte-derived dendritic cells to thimerosal and mercury derivatives. Toxicol Appl Pharmacol. 2010, 246, 66–73. [Google Scholar] [CrossRef]
- Migdal, C.; Foggia, L.; Tailhardat, M.; Courtellemont, P.; Haftek, M.; Serres, M. Sensitization effect of thimerosal is mediated in vitro via reactive oxygen species and calcium signaling. Toxicology. 2010, 274, 1–9. [Google Scholar] [CrossRef]
- Ida-Eto, M.; Oyabu, A.; Ohkawara, T.; Tashiro, Y.; Narita, N.; Narita, M. Prenatal exposure to organomercury, thimerosal, persistently impairs the serotonergic and dopaminergic systems in the rat brain: Implications for association with developmental disorders. Brain Dev. 2013, 35, 261–264. [Google Scholar] [CrossRef]
- FDA (Food and drug administration), Questions about vaccine safety. 2018. Available online: https://www.fda.gov/vaccines-blood-biologics/vaccines/thimerosal-vaccines-questions-and-answers (accessed on 16 August 2023).
- Norris, G.T.; Kipnis, J. Immune cells and CNS physiology: Microglia and beyond. Journal of Experimental Medicine. 2019, 216, 60–70. [Google Scholar] [CrossRef]
- Poon, C.C.; Sarkar, S.; Yong, V.W.; Kelly, J.J.P. Glioblastoma-associated microglia and macrophages: Targets for therapies to improve prognosis. Brain. 2017, 140, 1548–1560. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Soares, N.L.; Vieira, H.L.A. Microglia at the Centre of Brain Research: Accomplishments and Challenges for the Future. Neurochem Res. 2022, 47, 218–233. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Kan, L.K.; Williams, D.; Drummond, K.; O’Brien, T.; Monif, M. The role of microglia and P2X7 receptors in gliomas. J Neuroimmunol. 2019, 332, 138–146. [Google Scholar] [CrossRef]
- Wolf, S.A.; Boddeke, H.W.G.M.; Kettenmann, H. Microglia in Physiology and Disease. Annu Rev Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef]
- Greenwood, E.K.; Brown, D.R. Senescent microglia: The key to the ageing brain? Int J Mol Sci. 2021, 22. [Google Scholar] [CrossRef]
- Martínez-Zamudio, R.I.; Dewald, H.K.; Vasilopoulos, T.; Gittens-Williams, L.; Fitzgerald-Bocarsly, P.; Herbig, U. Senescence-associated β-galactosidase reveals the abundance of senescent CD8+ T cells in aging humans. Aging Cell. 2021, 20, 1–15. [Google Scholar] [CrossRef]
- He, C.; Levine, B. The Beclin 1 interactome. Curr Opin Cell Biol. 2010, 22, 140–149. [Google Scholar] [CrossRef]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; Rubinsztein, D.C.; Partridge, L.; Kroemer, G.; Labbadia, J.; Fang, E.F. Autophagy in healthy aging and disease. Nat Aging. 2021, 1, 634–650. [Google Scholar] [CrossRef]
- Young, A.R.J.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.J.; Tavaré, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; Narita, M. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Caldeira, C.; Cunha, C.; Vaz, A.R.; Falcão, A.S.; Barateiro, A.; Seixas, E.; Fernandes, A.; Brites, D. Key aging-associated alterations in primary microglia response to beta-amyloid stimulation. Front Aging Neurosci. 2017, 9, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Badie, B.; Schartner, J. Role of microglia in glioma biology. Microsc Res Tech. 2001, 54, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Watters, J.J.; Schartner, J.M.; Badie, B. Microglia function in brain tumors. J Neurosci Res. 2005, 81, 447–455. [Google Scholar] [CrossRef]
- Vaz, A.R.; Pinto, S.; Ezequiel, C.; Cunha, C.; Carvalho, L.A.; Moreira, R.; Brites, D. Phenotypic effects of wild-type and mutant SOD1 expression in n9 murine microglia at steady state, inflammatory and immunomodulatory conditions. Front Cell Neurosci. 2019, 13, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of cell viability by the MTT assay. Cold Spring Harb Protoc. 2018, 2018, 469–471. [Google Scholar] [CrossRef]
- Oda, Y.; Sadakane, K.; Yoshikawa, Y.; Imanaka, T.; Takiguchi, K.; Hayashi, M.; Kenmotsu, T.; Yoshikawa, K. Highly Concentrated Ethanol Solutions: Good Solvents for DNA as Revealed by Single-Molecule Observation. ChemPhysChem. 2016, 17, 471–473. [Google Scholar] [CrossRef]
- Renaud, J.; Martinoli, M.G. Development of an insert co-culture system of two cellular types in the absence of cell-cell contact. Journal of Visualized Experiments. 2016, 2016, 1–12. [Google Scholar] [CrossRef]
- Itahana, K.; Campisi, J.; Dimri, G.P. Methods to detect biomarkers of cellular senescence: The senescence-associated β-galactosidase assay. Methods in Molecular Biology. 2007, 371, 21–31. [Google Scholar] [CrossRef]
- Brites, D.; Vaz, A.R. Microglia centered pathogenesis in ALS: Insights in cell interconnectivity. Front Cell Neurosci. 2014, 8, 1–24. [Google Scholar] [CrossRef]
- Nishioku, T.; Takai, N.; Miyamoto, K.I.; Murao, K.; Hara, C.; Yamamoto, K.; Nakanishi, H. Involvement of caspase 3-like protease in methylmercury-induced apoptosis of primary cultured rat cerebral microglia. Brain Res. 2000, 871, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Branco, V.; Coppo, L.; Aschner, M.; Carvalho, C. N-Acetylcysteine or Sodium Selenite Prevent the p38-Mediated Production of Proinflammatory Cytokines by Microglia during Exposure to Mercury (II). Toxics. 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Aschner, M.; Syversen, T. Glutathione modulation influences methyl mercury induced neurotoxicity in primary cell cultures of neurons and astrocytes. Neurotoxicology. 2006, 27, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.; Branco, V.; Lu, J.; Holmgren, A.; Carvalho, C. Toxicological effects of thiomersal and ethylmercury: Inhibition of the thioredoxin system and NADP+-dependent dehydrogenases of the pentose phosphate pathway. Toxicol Appl Pharmacol. 2015, 286, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Li, X.; Yin, Z.; Sidoryk-Weogonekgrzynowicz, M.; Jiang, H.; Farina, M.; Rocha, J.B.T.; Syversen, T.; Aschner, M. Comparative study on the response of rat primary astrocytes and microglia to methylmercury toxicity. Glia. 2011, 59, 810–820. [Google Scholar] [CrossRef]
- Bassett, T.; Bach, P.; Chan, H.M. Effects of methylmercury on the secretion of pro-inflammatory cytokines from primary microglial cells and astrocytes. Neurotoxicology. 2012, 33, 229–234. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. 2018. Available online: www.impactjournals.com/oncotarget/.
- Su, P.; Zhang, J.; Wang, D.; Zhao, F.; Cao, Z.; Aschner, M.; Luo, W. The role of autophagy in modulation of neuroinflammation in microglia. Neuroscience. 2016, 319, 155–167. [Google Scholar] [CrossRef]
- Wang, R.C.; Wei, Y.; An, Z.; Zou, Z.; Xiao, G.; Bhagat, G.; White, M.; Reichelt, J.; Levine, B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science (1979) 2012, 338, 956–959. [Google Scholar] [CrossRef]
- Ye, X.; Zhu, M.; Che, X.; Wang, H.; Liang, X.J.; Wu, C.; Xue, X.; Yang, J. Lipopolysaccharide induces neuroinflammation in microglia by activating the MTOR pathway and downregulating Vps34 to inhibit autophagosome formation. J Neuroinflammation. 2020, 17, 1–17. [Google Scholar] [CrossRef]
- Rajendran, P.; Alzahrani, A.M.; Hanieh, H.N.; Kumar, S.A.; Ammar, R.B.; Rengarajan, T.; Alhoot, M.A. Autophagy and senescence: A new insight in selected human diseases. J Cell Physiol. 2019, 234, 21485–21492. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell. 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Pires, V.; Bramatti, I.; Aschner, M.; Branco, V.; Carvalho, C. Thioredoxin Reductase Inhibitors as Potential Antitumors: Mercury Compounds Efficacy in Glioma Cells. Front Mol Biosci. 2022, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bramatti, I.; Aschner, M.; Branco, V.; Carvalho, C. Co-exposure of human glioblastoma cells to Thimerosal and Temozolamide inhibits the thioredoxin system, HIF-1α, VEGF, and STAT3 phosphorylation. 2023. In Submission.
Figure 1.
Cytotoxicity effects of mercury compounds during cell growth- N9 cell line were exposed to EtHg and TmHg at different concentrations for 24h, 48h and 72h and the results were obtained with the MTT assay. Data are expressed as activity relative to control and the values are mean ± SEM of three independent experiments; *p≤0.05 **p ≤ 0.01 vs control.
Figure 1.
Cytotoxicity effects of mercury compounds during cell growth- N9 cell line were exposed to EtHg and TmHg at different concentrations for 24h, 48h and 72h and the results were obtained with the MTT assay. Data are expressed as activity relative to control and the values are mean ± SEM of three independent experiments; *p≤0.05 **p ≤ 0.01 vs control.
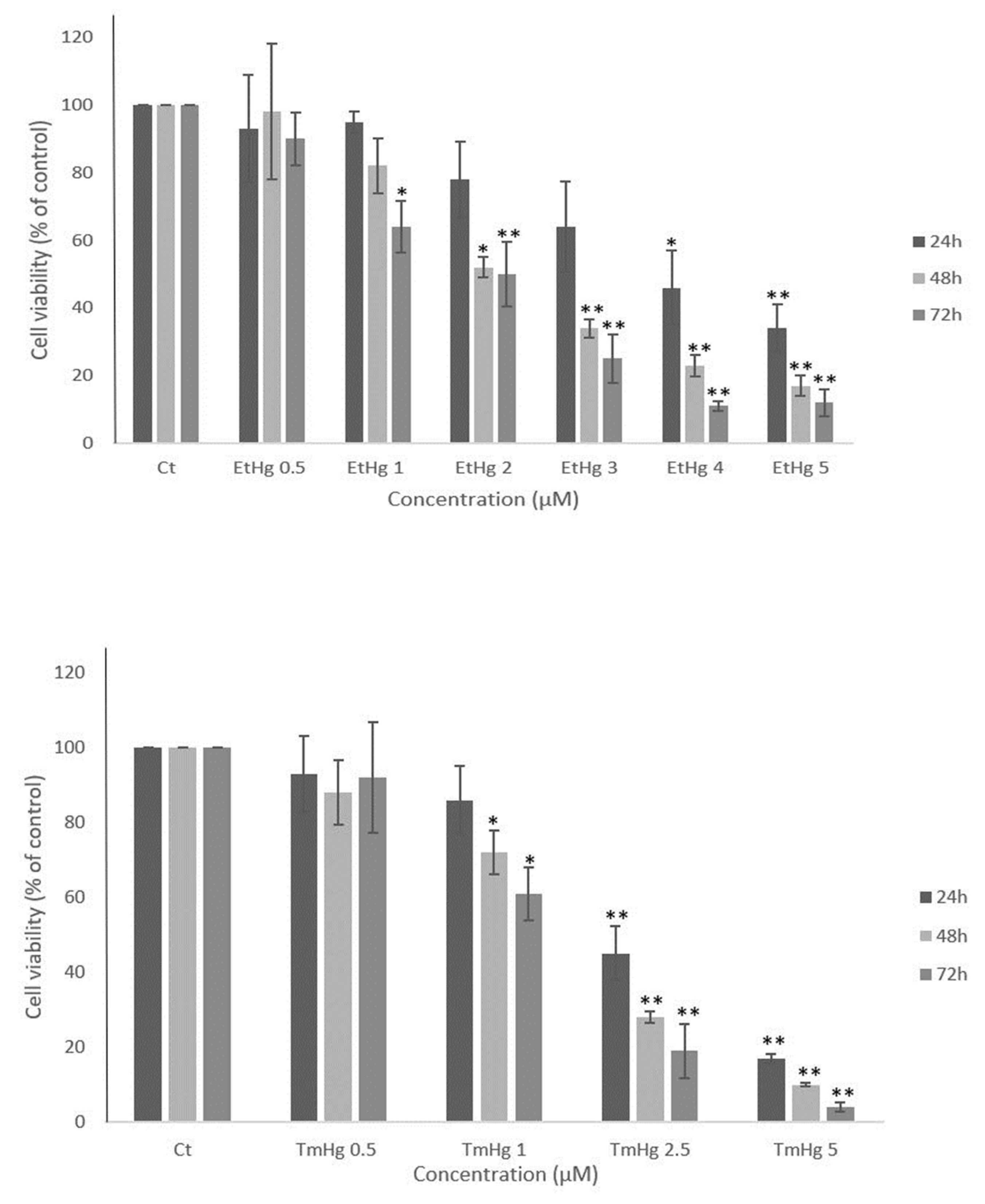
Figure 2.
Expression of inflammation mediators. N9 cell line was exposure to different concentrations of EtHg and TmHg for 3h; 6h and 24h. Expression of Inflammatory cytokines such as Interleukins, IL-1β (A); Inducible nitric oxide, iNOS (B) and tumour necrosis factor, TNF-α (C) was quantified by RT-PCR. Data are expressed as fold-change relative to control and the values are the mean ± SEM of five independent experiments; *p≤ 0.05 **p≤ 0.01 vs control.
Figure 2.
Expression of inflammation mediators. N9 cell line was exposure to different concentrations of EtHg and TmHg for 3h; 6h and 24h. Expression of Inflammatory cytokines such as Interleukins, IL-1β (A); Inducible nitric oxide, iNOS (B) and tumour necrosis factor, TNF-α (C) was quantified by RT-PCR. Data are expressed as fold-change relative to control and the values are the mean ± SEM of five independent experiments; *p≤ 0.05 **p≤ 0.01 vs control.
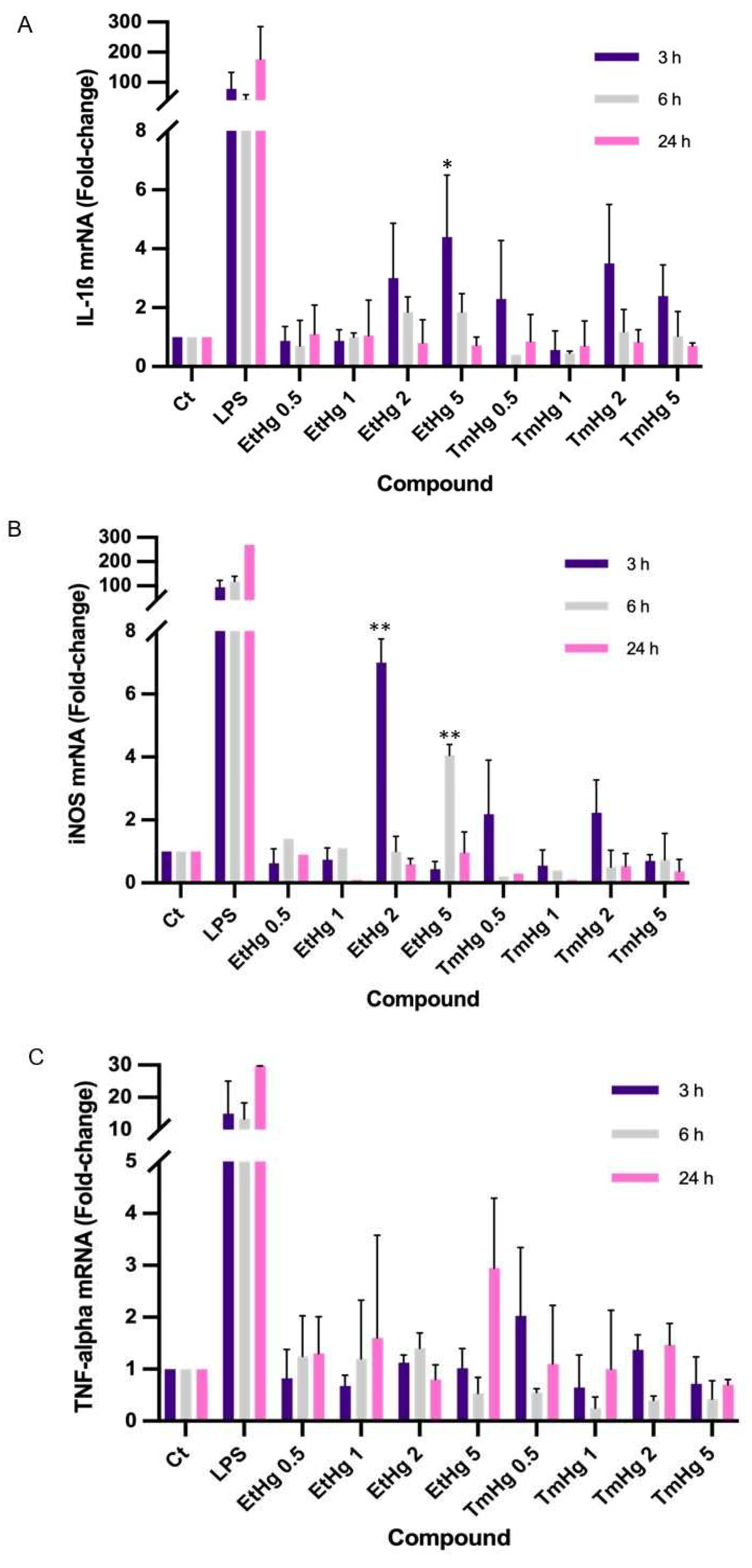
Figure 3.
Beclin-1 expression on N9 cell line. Cells were exposed to EtHg and TmHg for 3 and 24 h; A- Representative images of protein Beclin-1 expression (fold change). B, C- Results of densitometric analysis of Beclin-1 for EtHg and TmHg, respectively. Results are expressed relatively to control, and the values are mean ± SEM of four independent experiments; *p<0.05 vs control.
Figure 3.
Beclin-1 expression on N9 cell line. Cells were exposed to EtHg and TmHg for 3 and 24 h; A- Representative images of protein Beclin-1 expression (fold change). B, C- Results of densitometric analysis of Beclin-1 for EtHg and TmHg, respectively. Results are expressed relatively to control, and the values are mean ± SEM of four independent experiments; *p<0.05 vs control.
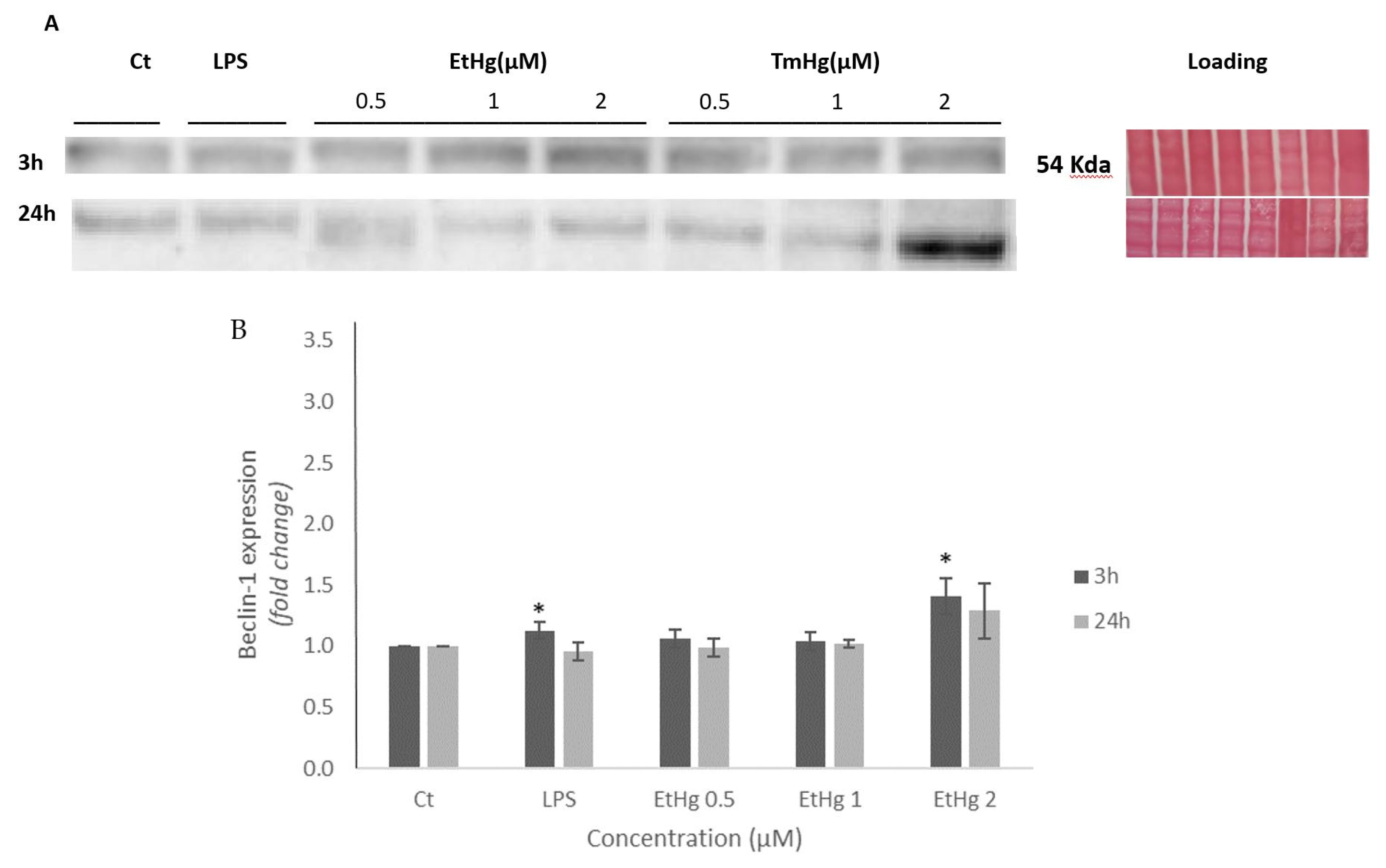
Figure 4.
Representative images of N9 senescent cells from five fields data sets obtained in each of three indepedent experiments- The images for each sample were obtained using Zen 2012 (blue edition, Zeiss) and AxioCam 105 color (Zeiss) adapted to an AxioSkope HBO50 microscope (Zeiss). The percentage of blue staining cells, i.e. senescence cells, was determined relative to the total cell number for each image (original magnification: 40x). Scale bar: 10mm.
Figure 4.
Representative images of N9 senescent cells from five fields data sets obtained in each of three indepedent experiments- The images for each sample were obtained using Zen 2012 (blue edition, Zeiss) and AxioCam 105 color (Zeiss) adapted to an AxioSkope HBO50 microscope (Zeiss). The percentage of blue staining cells, i.e. senescence cells, was determined relative to the total cell number for each image (original magnification: 40x). Scale bar: 10mm.
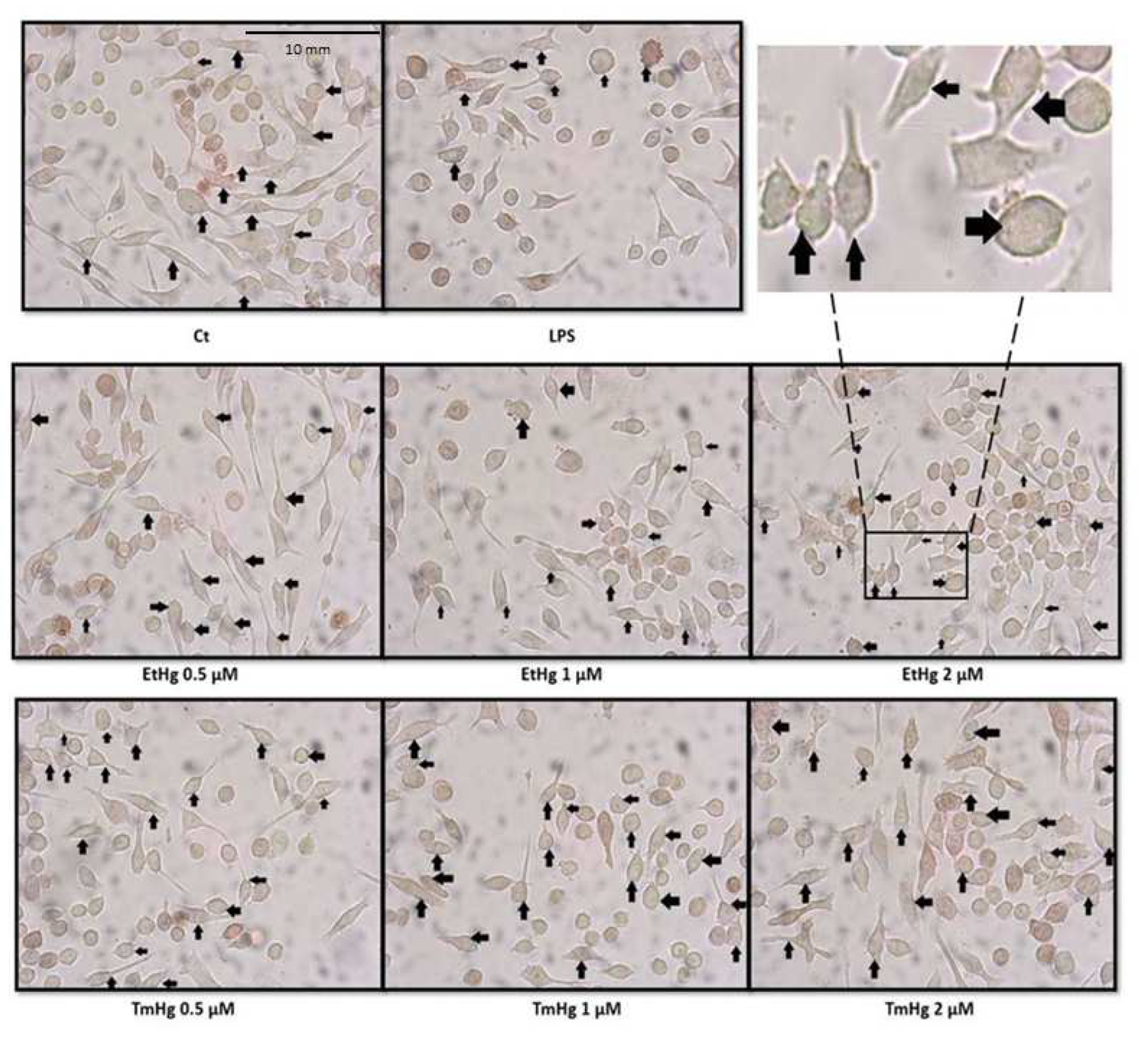
Figure 5.
N9 microglia cells were exposed to different concentrations of EtHg and TmHg during 24 h to evaluate changes on SA-β-galactosidase levels. Data are expressed relatively to control, and the values are mean ± SEM of three independent experiments **p<0.01 vs control.
Figure 5.
N9 microglia cells were exposed to different concentrations of EtHg and TmHg during 24 h to evaluate changes on SA-β-galactosidase levels. Data are expressed relatively to control, and the values are mean ± SEM of three independent experiments **p<0.01 vs control.
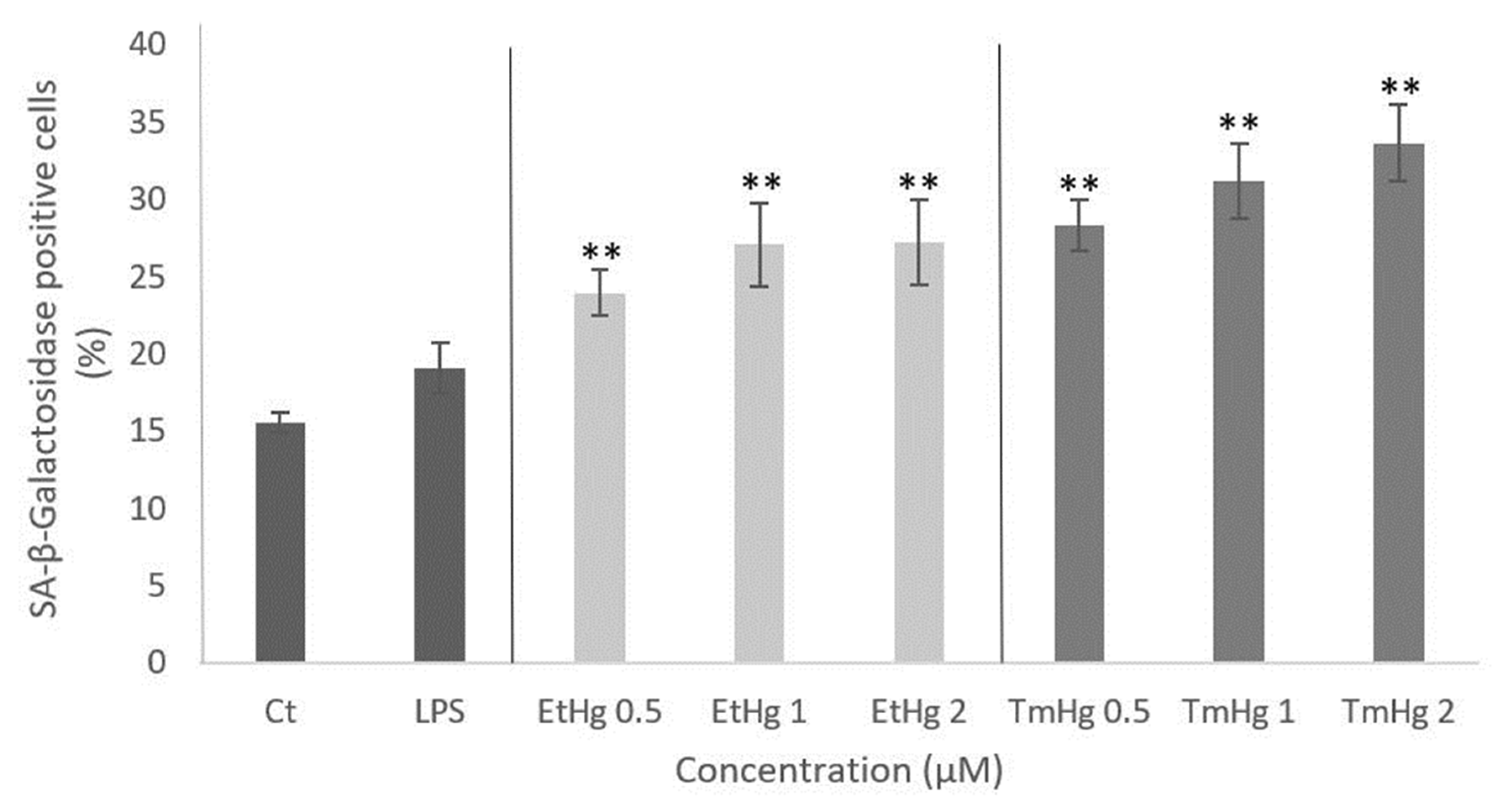
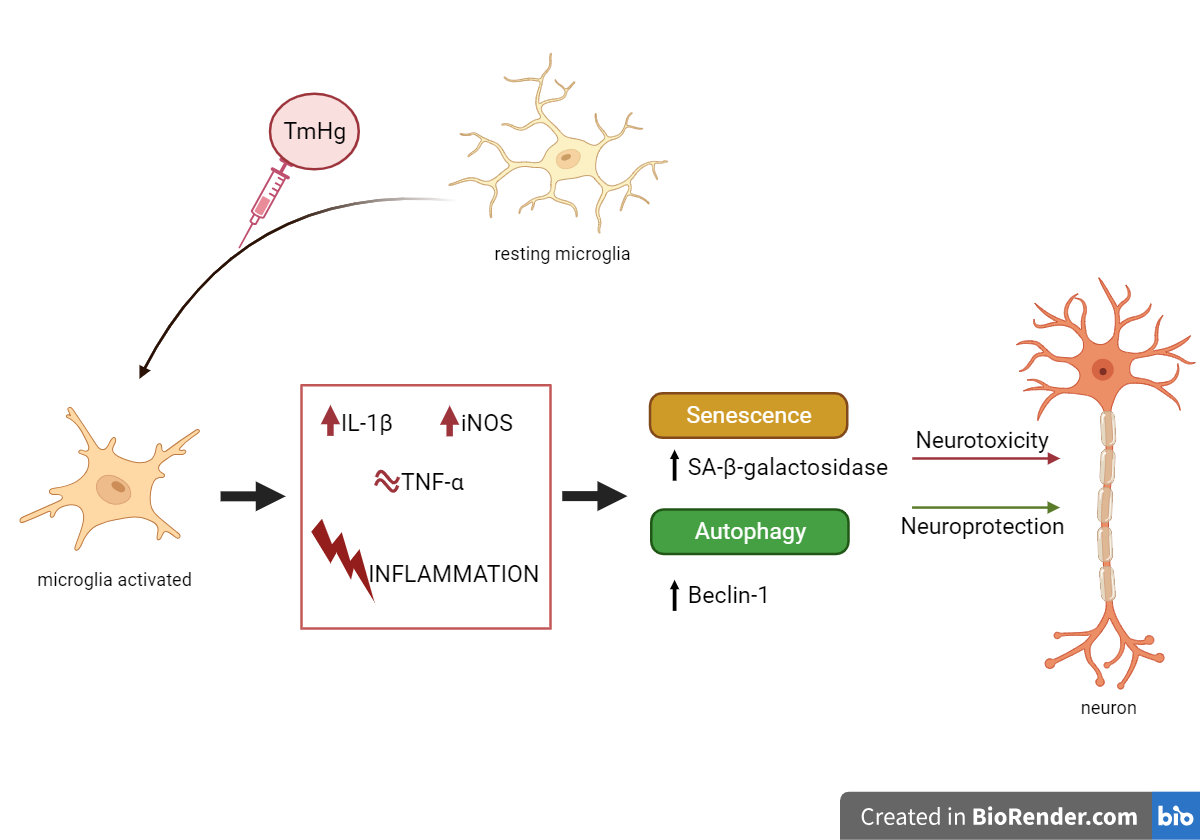
Figure 6.
Schematic representation of the results interpretation. N9 microglia cells exposed to different concentrations of EtHg or TmHg elicited the increase in markers of inflammation. This inflammatory response led to increased autophagy and senescence. The effects of thimerosal may have different consequences depending on the health status and susceptibility of the populations. This figure was created in BioRender.com.
Figure 6.
Schematic representation of the results interpretation. N9 microglia cells exposed to different concentrations of EtHg or TmHg elicited the increase in markers of inflammation. This inflammatory response led to increased autophagy and senescence. The effects of thimerosal may have different consequences depending on the health status and susceptibility of the populations. This figure was created in BioRender.com.
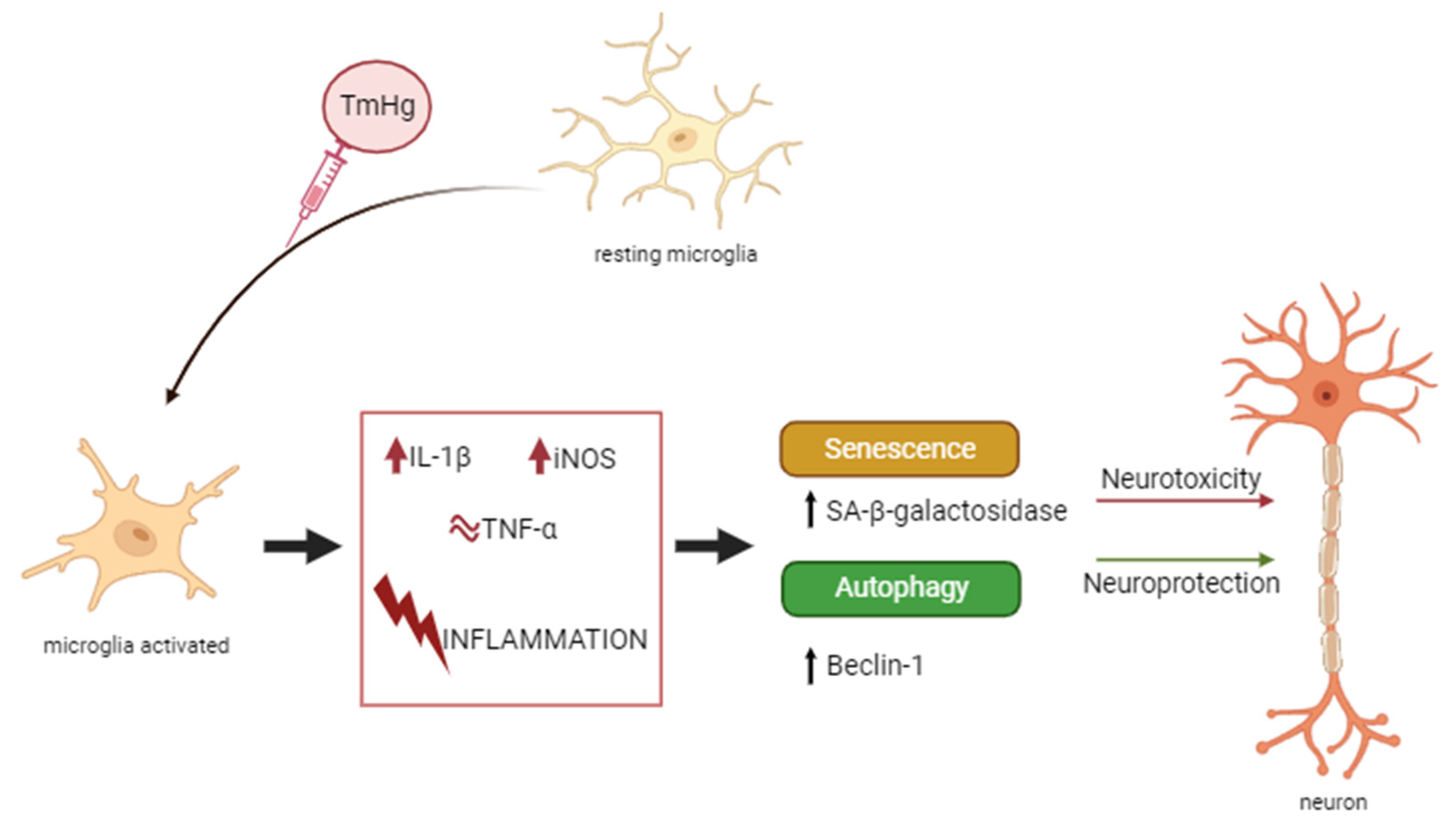
Table 2.
Comparative cytotoxicity from different neural cell lines and different mercury compounds.
| Performed Assays | Reference | |||||
| Cell line | Compound | EC50 (μM) | LC50 (μM) | |||
| 24h | 48h | 72h | 24h | |||
| N9 | EtHg | 2.3 ± 0.9 | 1.5 ± 0.2 | 1.4 ± 0.5 | --------- | Presentdengfeistudy |
| TmHg | 1.4 ± 0.4 | 1.2 ± 0.1 | 1.0 ± 0.2 | --------- | ||
| MeHg | ------ | ------ | ----- | 2.6 | [33] | |
| Hg2+ | 42.1 ± 3.7 | ------ | ------ | ------ | [34] | |
| Cerebellar astrocytes | MeHg | 1.32 | ------ | ------ | ------ | [35] |
| SHSY5Y | EtHg | 2.38 ± 0.28 | 1.49 ± 0.57 | 2.05 ± 0.64 | ------ | [36] |
| TmHg | 4.67 ± 0.14 | 4.21 ± 0.79 | 2.70 ± 0.34 | ------ | ||
------ No data; EC50: Concentration that reduces 50% the cell proliferation; Lc50: Concentration that kills 50% of cells.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



