
Submitted:
19 August 2023
Posted:
21 August 2023
You are already at the latest version
Abstract
Growth hormone (GH) is a peptide hormone that plays a crucial role in controlling growth, development, and lifespan. Molecular regulation of GH is accomplished via the GH receptor (GHR) gene, which is the main factor influencing human development and is essential to optimal functioning of the GH/IGF-I axis. Two GHR isoforms have been studied, according to the presence (flGHR) or absence (d3GHR) of exon 3. The d3GHR variant, which has recently been related to longevity, is associated with enhanced signal transduction and higher receptor function. Many of these studies indicated that the growth response to GH treatment may be affected. Individuals carrying the d3GHR isoform have higher receptor activity, improved signal transduction, and alterations in the treatment response and efficacy compared with those carrying the WT isoform. Further, studies performed in patients with acromegaly, Prader-Willi syndrome, Turner syndrome, small for gestational age (SGA), and growth hormone deficiency (GHD) suggested that the d3GHR variant may have an impact on the relationship between GH and IGF-I levels, height, weight, BMI, and other variables. Other research, however, revealed inconsistent results, which might have been caused by confounding factors, including limited sample sizes and different experimental methods. In this review, we lay out the complexity of the GHR isoforms and provide an overview of the major pharmacogenetic research conducted on this ongoing and unresolved subject.
Keywords:
Growth hormone receptor (GHR)
; Human growth hormone
; Deletion of exon 3 (d3GHR)
; Polymorphism
; Growth and development
; Hormone deficiency
1. Introduction
Growth hormone (GH) is a peptide hormone that is pulsatilely released from the somatotroph cells of the anterior pituitary [1,2] and plays a crucial role in controlling growth, development, and lifespan [3]. Humans have two GHR isoforms that differ in exon 3 retention or exclusion during splicing: a full-length isoform and a d3GHR isoform that lacks exon 3 [4]. This isoform is characterized by increased receptor activity, enhanced signal transduction and altered hormone binding [5,6]. Several studies investigated how this polymorphism might affect growth [7,8,9,10]. Additionally, the clinical and biochemical phenotype of patients with GH abundance or deficiency, such as those with acromegaly, Turner syndrome, and small for gestational age (SGA), were studied, along with their responsiveness to particular treatments, and it was discovered that, depending on the syndrome under investigation, the presence or absence of the d3GHR polymorphism has a positive or negative impact, respectively, on GH concentrations following treatment [11,12,13,14]. In a study of extremely long-lived individuals, d3GHR carriers demonstrated an approximately 10-year lifespan increase and d3GHR homozygotes across ages were an inch taller than WT individuals [15], and they have a significantly higher rate of postnatal catch-up growth [7]. In addition, in acromegaly patients, the d3GHR isoform was associated with a better clinical response to pegvisomant therapy [13]. This review presents information about the functional characteristics of the GHR with a particular emphasis on the d3GHR genotypes and their impact in numerous syndromes related to an excess or lack of GH. It also provides an overview of the major pharmacogenetic studies conducted on these ongoing and unsolved issues.
2. GHR Function and Structure
Growth hormone (GH) is a peptide hormone that is pulsatilely released from the somatotroph cells of the anterior pituitary. Growth hormone-releasing hormone (GHRH; positive regulation) and somatostatin (negative regulation) are the main hypothalamic hormones that control the release of GH [1,2]. GH performs an essential function in regulating lifespan, growth, and development [3], the metabolic function of GH affects protein, carbohydrate, and lipid metabolism [16]. The GH receptor (GHR) is mediated by the growth hormone receptor (GHR) gene, which is located on the short arm of chromosome 5, spanning at least 87 kb on the 5p13.1-p12 chromosome, encoded by nine exons and an exon in the 5' untranslated region. Exon 2 codes for the signal peptide, exons 3–7 for the extracellular domain, exon 8 for the transmembrane domain, and exons 9–10 for the intracellular domain (Figure 1) [5,17].
The GHR gene initiates many intracellular signaling pathways that lead to glucose metabolism alterations, modulation of cell proliferation genes, and generation of insulin-like growth factor (IGF)-1 [16]. When GH binds to its receptor, JAK2 and the receptor are phosphorylated, which initiates signaling via multiple pathways as shown in Figure 2. Receptor dimerization triggers a series of subsequent protein phosphorylations. STAT1, STAT3, and STAT5 are among the STAT proteins that GH can activate. The JAK-STAT pathway is considered as the main pathway by which GH affects gene transcription [18]. Stats 5a and 5b are involved in the synthesis of several GH-sensitive genes, including insulin-like growth factor 1 (IGF-1) and the acid labile subunit (ALS), a vital part of the IGF binding protein complex (IGF1-IGFBP 3-ALS) [19]. Tyrosyl phosphorylation of IRS proteins would be triggered by JAK2 activation, which promotes the recruitment of PI3K, regulating glucose transport and probably other cellular responses [20]. Apoptosis and autophagy are inhibited by upregulation of Akt, which promotes the phosphorylation of target genes and proteins. This might be because PI3K/Akt triggering causes the NF-kB system to become overactive [21]. Increased expression of anti-apoptotic proteins such as Mcl-1 has been linked to capacity of the PI3K pathway to mediate immunotherapy resistance [22,23]. The SHC/MAP kinase pathway is an additional pathway. Mitogen-activated protein kinases (MAPK) and the phosphoinositide-3-kinase/Akt signaling pathways are typically activated by SHC proteins to carry out their actions [24,25]. The pathway consisting of Grb2-SOS-Ras-Raf-MEK-ERK1,2 is activated by SHC adapter proteins [20]. The GHR also activates the Src family kinase signaling pathway independently of JAK2 [26].
Growth hormone signaling is thought to be a key aging regulator [27,28]. Lack of GH signaling significantly can expands lifespan, slows the aging process, increases the health span, reduces body size, delays maturation, and increases life expectancy [28]. For instance, a study found that mice homozygous for the GHR disruption lived significantly longer than WT mice; the disruption resulted in a 55% and 38% lifespan increase in males and females, respectively [29].The loss of receptor activity is mainly due to mutations in the extracellular domain that result in receptor insensitivity or Laron syndrome. However, excessive receptor stimulation by growth hormone leads to gigantism, adult acromegaly, and cancer [30].
Growth hormone receptor deficiency (GHRD) in humans is brought on by GHR gene mutations that result in low levels of both insulin and insulin-like growth factor 1 (IGF-1) [31]; as a result, the circulating levels of IGF-1 affect brain structure and function during development and aging [32]. By focusing on people with GHRD, numerous studies sought to clarify and analyze the role of the GHR. A recent study of 13 people with GHRD and 12 unaffected relatives found that the GHRD group showed trends toward larger dentate gyrus and CA1 regions of the hippocampus as well as larger surface areas in several frontal and cingulate regions. Additionally, the GHRD group exhibited improved task-related activation and cognitive performance in the frontal, parietal and hippocampal regions when compared with the controls [33]. By contrast, a different study in the Ecuadorian population revealed that people with GHRD are not significantly different in terms of their intellectual ability, and the production of IGF-1 that is induced by GH is not necessary for postnatal intellectual development or for normal brain growth in utero [34]. Yet, a study that looked at 10 patients with the Laron syndrome revealed parenchymal loss to varying degrees and below-average intelligence [35]. Other studies showed that individuals with GHRD had no diabetes, and only one had a non-lethal malignancy, compared to 17% with cancer and 5% with diabetes in the controls [36]. In the light of the various results discussed above, more research is required to better understand and demonstrate how these conserved genes contribute to aging and longevity.
3. GHR isoforms
In 2004, Dos Santos et al. reported on two isoforms of a common GH receptor (GHR), the WT, which contains exon 3 (flGHR), and the mutated form, which lacks it (d3GHR) [4]. Exon 3 encodes a portion of the extracellular domain of the receptor, and its absence or presence results in differential GH-binding abilities [37,38]. Since then, a wide range of studies have examined how this GHR polymorphism affects people with various conditions. This article compares the effects of both isoforms in various disorders and unbiasedly assesses these studies.
4. Deletion of exon 3 in GHR (d3GHR)
One of the GHR isoforms is known as "d3GHR" and is encoded by a transcript variant that is missing exon 3. Exon 3 is coding for a portion of the extracellular domain, and its deletion results in a loss of 66 base pairs (bp) that code for 22 amino acids in the N-terminal domain (residues 7–28) [38,39]. The presence of the two isoforms (flGHR (WT) and d3GHR) could be due to alternative splicing of the immature mRNA [39]. Another possible explanation for the occurrence of these isoforms is that they are a result of an intrachromosomal recombination event between two related primate-specific retro-elements (DNA sequences derived from a retrovirus) flanking exon 3, which took place late in primate evolution [40]. These sequences underwent recombination during cell division, which resulted in a 2.7-kb genomic deletion [4,38,40,41]. The loss of exon 3 is a crucial element in growth hormone pharmacogenetics, as it alters receptor processing, transport, stability, and binding to other ligands and affects receptor expression or function and signal transduction [42]. In the GHR, the absence of exon 3 increases GH signal transduction by 30%. More specifically, the deletion of exon 3 increases the sensitivity of the intracellular JAK-STAT pathway to growth hormone, which in turn increases the transcription of GH target genes [4]. In addition, the d3GHR isoform facilitates significant changes in physiological function and hormone binding [43] and is positively correlated with increased GH responsiveness [5], although GH receptor binding is unaffected [44,45]. The effects of increased GH transduction signaling via the d3GHR include: decreased feedback on pituitary GH production, lower circulating GH levels, decreased binding affinity to d3GHR-equipped cells, and the restoration of a desirable level of GH action on pertinent targets [46] (Figure 3).
Many studies examined the distribution of the various GHR isoforms among healthy controls and found that on average, 50% of controls had full-length GHR (fl/fl), 30%–40% were heterozygous for the exon 3 deletion (fl/d3), and 10%–20% were homozygous for the exon 3 deletion (d3/d3) [4,40,47,48]. The exon 3 deletion had no impact on receptor binding affinity or internalization, and thus, the molecular mechanisms underlying the higher bioactivity of d3GHR have not yet been clarified [49,50].
5. flGHR and d3GHR genotyping
Genotyping of the d3GHR polymorphism can be achieved by several methods. One of the widely used and prevalent method is utilizing the polymerase chain reaction (PCR) using specific primers [5,10,15,17]. According to Pantel et al., the GHR genotype test can be easily carried out using a PCR assay and the PCR amplification results can be analyzed using agarose gel electrophoresis [17]. The full-length wild type (fl) allele is represented by a PCR fragment of 935 base pairs using the G1 and G3 primers, while the deletion (d3) allele, is represented by a PCR fragment of 532 base pairs using the G1 and G2 primers (GenBank accession number AF155912) [17,47,51].
Another common method for genotyping is using a single-nucleotide polymorphism (SNP) which is specific for exon 3 deletion. rs6873545 is one of several SNPs in the GHR gene that relates to the presence or absence of exon 3. The more prevalent allele encompassing exon 3 correlates with the rs6873545(T) allele, while the less prevalent rs6873545(C) allele correlates with the exon 3-deficient d3GHR allele. In order to identify exon 3 deletion in GHR, rs6873545 has been used as a tag in numerous studies [52,53,54,55]. In 183 adult GHD patients, Glad et al. tested the TaqMan SNP genotyping using the tagSNP rs6873545 as a marker for the d3/fl polymorphism, and reported that the tagSNP makes it easier to examine the impacts of the d3/fl polymorphism, especially in large cohorts [56].
6. Genetic editing of d3GHR
Numerous studies have been carried out to mimic the deletion of exon 3 in cell lines and animal models utilizing various genome-editing methods to better understand the function and impact of deletion of exon 3 of the GHR and its positive or negative effects. In cell lines, researchers transiently cotransfected HEK293 fibroblasts with vectors expressing full-length GHR, d3GHR, or both using the LHRE-luciferase reporter plasmid to investigate whether the growth hormone receptor is linked to increased responsiveness to growth hormone. When the cells were exposed to different growth hormone concentrations, it was found that d3GHR caused higher transcriptional activity of the reporter construct and responded more strongly to growth hormone stimulation than the full-length GHR [4]. Chiloiro et al., also found that in comparison to the flGHR isoform, the d3GHR isoform has a higher sensitivity to endogenous and recombinant GH [57].
A further possibility to mimic the deletion of exon 3 is in animal models. Exon 3 of the GHR can deleted utilizing CRISPR-CAS9 genome editing. Following several backcrosses, the researchers subjected WT and d3GHR mice to AL (ad libitum) feeding and 40% caloric restriction (CR). The findings showed that exon 3 deletion has a protective effect against severe malnutrition, and in male mouse livers under CR, the d3GHR causes the expression of female-like genes and the disappearance of sexual dimorphism in weight. The d3GHR males under CR were significantly lighter than the WT males, making them nearly identical in weight to the d3GHR females. In this study, the authors demonstrate how sex and environmental factors can influence the impact of genetic variants [54].
7. Effects of the flGHR and d3GHR isoforms in healthy people
Several studies have been conducted to explain the relationship between the d3GHR isoform and birth weight [58], life span [15], metabolic activity [33], and time of puberty onset [59]. Exon 3 deletion is linked to increased height, decreased serum IGF-1, increased GH sensitivity, and longer life expectancy in men. Men homozygous for the presence of exon 3 had an approximately 10-year lifespan increase and were an inch taller than WT isoform carrier individuals [15].
A study on 43 mother-child pairs with normal pregnancies demonstrated that the presence of the d3GHR polymorphism in the fetus is associated with a markedly reduced concentration of hGH-V and IGF-I in maternal serum, a reduced fetal growth rate in the third trimester, and lower birth weight compared to the wild type [60], which can illustrate its association with smaller placental and birth size [58,59]. However, in later developmental stages and in adults, the effect of the d3GHR isoform on organismal growth is reversed. People who carry the d3GHR isoform are more sensitive to GH [15], and there is early onset of puberty in boys when the d3/d3 genotype is compared with the fl/fl genotype [59]. In addition, the d3GHR homozygotes and heterozygotes showed a significantly higher rate of postnatal catch-up growth [7].
In a longitudinal study, 385 healthy individuals from birth to adulthood (ages 23–25) were divided into three groups based on birth size: small for gestational age (SGA n=130), appropriate for gestational age (AGA n=162), and large for gestational age (LGA n=93). The d3/d3 genotype increased in SGA and AGA individuals and decreased in LGA. fl/d3GHR was not related to plasma IGF1 levels in adults, and to height, weight, metabolic phenotype, or rising cardiovascular risk [61]. In another study, the d3GHR isoform is linked to a lower risk of pre-hypertension in boys but a higher risk of both pre- and hypertension in girls [62].
Yet, in a Saudi population investigation of 230 healthy adults, the GHR genotypes were distributed as follows: 39.1% for fl/fl, 44.8% for fl/d3, and 16.1% for d3/d3. They found no discernible differences between the GHR genotypes in height, weight, or body mass index [63]. Further, in healthy Turkish adults (n=477), 35% fl/fl, 39% fl/d3, and 26% d3/d3, the three GHR genotype groups did not differ in terms of height SDS (standard deviation score) or BMI SDS [64].
8. Effects of the flGHR and d3GHR isoforms on various GHR disorders
Numerous studies have shown that growth hormone receptor deficiencies are the source of a variety of disorders and syndromes, some of which appear in childhood, whereas others have their onset in adolescence and maturity. The responsiveness, timing and efficacy of treatments, GH and IGF-I levels, height, weight, BMI, and other variables were found to be significantly influenced by the d3GHR isoform [4,65,66,67]. Some of the studies found no correlation effects in the growth response and hGH therapy on d3GHR carriers [47], while others with a favorable correlation [4,67], which resulted in two meta-analyses that demonstrated moderate favorable dominant impacts of the d3GHR genotype in the response to GH in d3GHR carriers with short stature [68,69]. Yet, a pharmacogenetic investigation discovered no correlation between growth hormone response and the d3GHR polymorphism in patients with growth hormone deficiency, ISS, and SGA [65]. In conclusion, the results imply that the d3GHR, as examined among flGHR and d3GHR carriers, may be implicated in regulating the effectiveness of GH therapies.
In the following chapter, we will review how each syndrome is affected by the various GHR genotypes, notably exon 3 deletion.
8.1. Prader-Willi syndrome (PWS)
Prader–Willi syndrome (PWS) is a highly complex and prevalent genetic disorder that occurs in approximately 1 in 15,000–30,000 births. The syndrome is dictated by paternally imprinted loss of region 15 q11-q13 [51,70]. This genetic disorder affects multiple systems, the most consistent major manifestation of which is growth hormone insufficiency, leading to familial short stature, small hands and feet, decreased muscle mass, early childhood-onset hyperphagia, obesity, hypotonia with poor sucking, and poor weight gain in infancy [71]. Individuals with PWS frequently have lower muscle mass and strength as well as severe hypotonia that may increase the risk or tendency for scoliosis, which is determined by standing X-rays that show a skeletal back curve greater than 10° [44]. GH administration is used to treat PWS patients with lean body mass and increased stature [70,72].
Possession of the d3 allele confers a protective effect by increasing GH sensitivity in scoliosis over the life span and improving physiological parameters such as bone density and muscle mass; in fact, those with scoliosis who carried the d3 allele had more gradual increases in weight and height with age than those with the WT genotype [51]. In 2011, a study of 74 PWS patients and 100 healthy controls found that patients with at least one d3 allele had significantly higher height SDS and IGF-1 levels than those with flGHR before starting rhGH therapy [73].
In other study, d3GHR isoform carriers experienced a faster 1.5 cm/month increase in height while receiving GH treatment, compared to 0.87 cm/month for those with the WT isoform. Furthermore, in the absence of GH treatment, homozygotes for d3GHR grow faster than both fl/d3 and fl/fl; this result supports the theory that the d3 allele increases sensitivity to GH [74]. According to Schreiner et al., in preterm infants with very low birth weight, postnatal catch-up is significantly more common among those who carry at least one d3 allele than in those who are homozygous for the full-length isoform, defining the d3GHR genotype as a predictor of the post-natal growth pattern [7]. As a result, and in light of the aforementioned studied, the d3GHR isoform has a protective and beneficial effect on PWS patients.
8.2. Acromegaly
Acromegaly is a rare disorder caused by excess secretion of GH, usually due to a pituitary tumor [75]. These elevated GH levels lead to an increase in the production of insulin-like growth factor I (IGF-I), mainly in the liver but also in other tissues [76]. Screening for acromegaly is often performed by measuring random GH concentrations and IGF-1. In acromegaly, log GH concentrations are directly correlated with IGF-1 concentrations [77,78]. In active acromegaly patients, the relationship between serum IGF-I and serum GH is gender-specific: females have lower serum IGF-I concentrations than males with the same serum GH level [79]. The distribution of the GHR genotypes (fl/fl, fl/d3, and d3/d3) was examined in numerous studies involving acromegaly patients, and in most studies, roughly half of acromegaly patients carried at least one d3 allele. The prevalence of homozygote d3/d3GHR individuals is 5%–20% (Table 1).
Pegvisomant (PEGV) is a brand-new growth hormone receptor antagonist, which improves symptoms and maintains IGF-1 balance in acromegaly patients over the course of up to 12 weeks of treatment. As a result, PEGV is the best medication for controlling IGF-1 in acromegaly [80]. A study reported that exon 3 deletion predicted a better response to PEGV therapy [13], and for d3GHR carriers, Pegvisomant dosage and treatment time were reduced, and IGF-I levels were normalized faster [81]. Other research, however, found that the response to PEGV treatment or determination of the necessary PEGV dose does not differ between GHR genotypes [82].
Numerous studies have examined the connection between various GHR genotypes and several comorbidities linked to patients with active acromegaly, such as hypertension, obesity, T2DM, colonic polyps, heart disease, obstructive sleep apnea syndrome, vertebral fractures (VF), and heart disease, in addition to the effect of each genotype on GH, IGF-I, and BMI levels [37,78,83,84,85,86,87].
As shown by the studies cited above and in Table 1, the effects of the different GHR genotypes on individuals with acromegaly vary considerably. Further, in vivo and in vitro research is required to fully understand how d3GHR impacts the concentrations of GH and IGF-1 as well as other metabolic markers in acromegaly.
8.3. Turner syndrome
The description of female patients with Turner syndrome (TS) was published by Henry Turner in 1938 [92]. Nevertheless, in 1930, Dr. Ullrich reported a case of an 8-year-old girl with signs of TS; therefore, this syndrome is also called Ullrich-Turner syndrome [93]. This syndrome affects 1 in every 2500 females and is the most prevalent sex chromosome abnormality [94]. Short stature, which affects 98% of patients, is the most prevalent symptom [95]. Between 95% and 100% of TS patients experience growth failure and shorter adult height [96].
The FDA and other regulatory bodies around the world have approved rhGH therapy, which has been shown in numerous studies to increase adult stature in TS with varying effects on final adult height [97,98]. Growth hormone therapy works to increase final adult height, but the treatment benefit varies across studies and is dependent on a variety of factors, including the age when the therapy starts, GH dose, length of therapy, and bone age [11,99,100,101] in addition to the patient's GHR genotype, which we will discuss in this chapter. As a result, numerous studies have investigated how different GHR genotypes affect the way in which TS patients will respond to GH therapy.
In a study involving 48 TS patients (fl/fl=24, fl/d3=17, and d3/d3=7) the d3/d3 group showed a significantly higher total height gain than the fl/fl or the fl/d3 groups (final adult height as compared to their starting height). Additionally, d3GHR homozygous girls display a unique GH responsiveness that controls their weight and led to BMI reduction [11]. Another study found that girls with one or two copies of the d3GHR isoform displayed an increase in height velocity and outgrew their growth prediction during the first year of treatment, but gains in weight, IGFBP-3, and IGF-1 were not significantly different [101]
However, the concentrations of HV gain, BMI, IGF-1, and IGF binding protein 3 (IGFBP-3) between those with and without exon 3 were not significantly different [99], In addition, Turkish TS patients who receive GH treatment for one and two years, founded no impact of the GHR exon 3 polymorphism on growth [8].
8.4. Small for gestational age (SGA)
SGA is a clinical term for newborn infants whose length and/or weight is below normal for their gestational age and sex [102] and affects approximately 3%–10% of live births [103,104]. Compared with infants born with a normal length and weight for gestational age, SGA is linked to cardiovascular disease, neurodevelopmental impairment, T2DM, lower intelligence, increased risk for insulin resistance, and adult short stature [105,106]. Ninety percent of infants with SGA show catch-up growth, but 10% of them remain diminutive throughout childhood and adolescence [107]. Clinical research revealed that rhGH is a successful treatment for children of short stature who are born with SGA. As a result, children of short stature who are born SGA and who do not achieve catch-up growth by the ages of 2–4 are candidates for rhGH therapy. Children with SGA have been shown to respond differently to rhGH therapy depending on their GH status [103,108]. Numerous studies showed that GH treatment increases adult height in SGA people who did not exhibit catch-up growth [109].
In 536 healthy newborns subjects, SGA (n = 192), LGA (n = 200), and AGA (n = 144), the d3/d3 GHR genotype was found to be twice as common in the AGA and LGA cohorts compared to SGA. However, no significant differences in the frequency distribution of the GHR genotypes between LGA and AGA newborns were found. The information disqualifies the GHR exon 3 deletion polymorphism as a potential genetic cause of LGA pregnancies [110].
Table 2 summarizes the studies that have been published on the relationship between the GHR isoforms and SGA children. Numerous studies support the concept that the d3GHR isoform is linked to a significant increase in compensatory growth in SGA children [9,10] and a better growth response to hGH treatments [4,111]. However, other studies have also supported the lack of a connection between GHR genotypes and patients' responses to hGH therapy [12,112] (Table 2).
8.5. Growth hormone deficiency (GHD)
GHD is a condition in which GH production is reduced or nonexistent. Idiopathic isolated GHD is the most prevalent form of the illness, with an incidence rate of 1:4000 to 1:10,000 [113]. The most common cause of GH deficiency in adult humans is pituitary or peripituitary tumors [114]. In patients suspected of having adult GHD, a diagnosis must be made before replacement therapy with recombinant human GH (rhGH) can be considered [115].
According to numerous studies, children with GHD who lack exon 3 (d3GHR) respond to rhGH more favorably than those who do not [4,116]. GHD patients carrying at least one d3 allele had a noticeably better growth rate in the first year of hGH replacement and attained a taller adult height [67]. After the first two years of recombinant human GH treatment in 181 patients with severe isolated GH deficiency, subjects with the d3/d3GHR isoform significantly outgrew those with the full-length GHR in terms of both height gain and HV SDS score. For both HV SDS and height gain, a dose-dependent effect of the d3GHR isoform was observed [117]. According to a meta-analysis of 15 studies, in GHD, but not in non-GHD, d3GHR is associated with increased baseline height. Additionally, during the first year of rhGH therapy, d3GHR stimulates growth velocity by an additional effect of roughly 0.5 cm. This effect is stronger at lower rhGH doses and at older ages [69].
GH peak level (higher vs lower) and d3GHR (fl/fl vs d3 carriers) combined status was associated with height change over 3 years. d3GHR carriers with lower peak GH had lower growth than subjects with fl/fl. Conversely, d3GHR carriers with higher peak GH had better growth [118]. Whereas other results from various studies were inconsistent. Isolated GH deficiency (IGHD) patients who received replacement doses of GH for a year, showed that the d3GHR isoform does not affect the response to GH treatment or growth predictions [14]. Caucasian patients who have severe GH deficiency with the fl/d3 or d3/d3 genotype did not require less rhGH therapy to improve their quality of life than those with the fl/fl genotype [119].
9. Conclusion and Future Perspectives
According to numerous studies on GHR polymorphism that have been accumulating in recent years, one of the many factors that affect the phenotype of both children and adults with GHD or excess secretion of GH, as well as responsiveness to treatment, is the genotype of the GHR, "flGHR" or missing exon 3 "d3GHR". d3GHR, is the first genetic factor that controls how each individual responds to GH treatment. Numerous investigations on the GHR exon 3 genotype in people with various syndromes, including children receiving hGH therapy, girls with Turner syndrome, children born small for gestational age, and patients with acromegaly, corroborated the beneficial effect of the d3GHR isoform on GH actions. In healthy individuals, exon 3 deletion is associated with greater height, a longer life span and higher GH sensitivity [15]. However, some studies did not identify any significant differences in height, weight, or other characteristics among the GHR genotypes [61,63].
Several studies on PWS patients who had received GH treatment indicated that those with the d3 allele grew taller faster than those with the fl/fl subtype. This gene confers protection by increasing growth hormone sensitivity in scoliosis over the course of the person's life and improving physiological features, including bone density and muscle mass. It was also positively connected with spontaneous postnatal growth velocity. Acromegaly patients who receive the GHR antagonist pegvisomant and have the d3GHR isoform typically respond better to treatment than those with the fl/fl GHR isoform. Other research, however, revealed that d3GHR carriers had a higher risk of VF, as well as a high prevalence of obesity and hypertension. In a comparison of girls with d3GHR isoform-positive Turner syndrome and girls with fl/fl GHR, those positive for the d3GHR isoform showed a significantly greater total gain in height. Various studies indicated that d3GHR SGA children carriers grew more spontaneously and responded better to growth hormones, and patients with GHD who lack exon 3 (d3GHR) responded more favorably to rhGH and had higher baseline heights. The conflicting findings that have thus far been reported, however, warrant some thought and may be attributed to confounding elements such as small sample sizes, limitations in large prospective studies, variations in experimental design across studies, the individuals' growth parameters prior to the treatment, sex, and age. In addition, take into consideration that growth rate is a quantitative polygenic variable that is impacted by maternal factors both during pregnancy and afterward. We believe that additional studies, particularly sizable prospective studies, should be performed in order to better understand the role of the d3GHR isoform in the various GHR diseases and to evaluate the significance of this polymorphisms in GH pharmacogenetics. We suggest that CRISPR-CAS9 or other genome editing technologies to mimic GHR diseases in animal models, followed by the induction of the exon 3 deletion, can be utilized and assist in learning more about the function of the d3GHR isoform in life extension and treatment effectiveness.
Author Contributions
Conceptualization, G.F. and G.A.; methodology, G.F. and G.A.; writing—original draft preparation, G.F.; writing—review and editing, G.A. and L.S.; visualization, G.F.; supervision, G.A. All authors have read and agreed to the published version of the manuscript.
Funding Statement
This research received no external funding.
Acknowledgments
No funds, grants, or other support was received to assist with the preparation of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lu, M.; Flanagan, J.U.; Langley, R.J.; Hay, M.P.; Perry, J.K. Targeting growth hormone function: strategies and therapeutic applications. Signal Transduct. Target. Ther. 2019. [Google Scholar]
- Melmed, S. The pituitary: Fourth edition; 2017.
- Ashpole, N.M.; Sanders, J.E.; Hodges, E.L.; Yan, H.; Sonntag, W.E. Growth hormone, insulin-like growth factor-1 and the aging brain. Exp. Gerontol. 2015. [Google Scholar]
- Dos Santos, C.; Essioux, L.; Teinturier, C.; Tauber, M.; Goffin, V.; Bougnères, P. A common polymorphism of the growth hormone receptor is associated with increased responsiveness to growth hormone. Nat. Genet. 2004. [Google Scholar] [CrossRef] [PubMed]
- Palizban, A.A.; Radmansorry, M.; Bozorgzad, M. Exon 3-deleted and full-length growth hormone receptor polymorphism frequencies in an Iranian population. Res. Pharm. Sci. 2014. [Google Scholar]
- Filopanti, M.; Giavoli, C.; Grottoli, S.; Bianchi, A.; De Marinis, L.; Ghigo, E.; Spada, A. The exon 3-deleted growth hormone receptor: Molecular and functional characterization and impact on GH/IGF-I axis in physiological and pathological conditions. J. Endocrinol. Invest. 2011. [Google Scholar] [CrossRef]
- Schreiner, F.; Stutte, S.; Bartmann, P.; Gohlke, B.; Woelfle, J. Association of the growth hormone receptor d3-variant and catch-up growth of preterm infants with birth weight of less than 1500 grams. J. Clin. Endocrinol. Metab. 2007, 92. [Google Scholar] [CrossRef]
- Baş, F.; Darendeliler, F.; Aycan, Z.; Çetinkaya, E.; Berberoǧlu, M.; Şklar, Z.; Öcal, G.; Timirci, Ö.; Çetinkaya, S.; Darcan, Ş.; et al. The exon 3-deleted/full-length growth hormone receptor polymorphism and response to growth hormone therapy in growth hormone deficiency and turner syndrome: A multicenter study. Horm. Res. Paediatr. 2012, 77. [Google Scholar] [CrossRef]
- Wegmann, M.G.; Thankamony, A.; Roche, E.; Hoey, H.; Kirk, J.; Shaikh, G.; Ivarsson, S.A.; Söder, O.; Dunger, D.B.; Juul, A.; et al. The exon3-deleted growth hormone receptor gene polymorphism (d3-GHR) is associated with insulin and spontaneous growth in short SGA children (NESGAS). Growth Horm. IGF Res. 2017, 35. [Google Scholar] [CrossRef]
- Garrido, N.P.; Pujana, M.; Berger, M.; Ramírez, P.; Guercio, G.; Belgorosky, A.; Marino, R. Growth hormone receptor gene polymorphism. Spontaneous catch up growth in small for gestational age patients. Medicina (B. Aires). 2021, 81. [Google Scholar]
- Binder, G.; Trebar, B.; Baur, F.; Schweizer, R.; Ranke, M.B. Homozygosity of the d3-growth hormone receptor polymorphism is associated with a high total effect of GH on growth and a low BMI in girls with Turner syndrome. Clin. Endocrinol. (Oxf). 2008, 68. [Google Scholar] [CrossRef]
- Carrascosa, A.; Esteban, C.; Espadero, R.; Fernández-Cancio, M.; Andaluz, P.; Clemente, M.; Audí, L.; Wollmann, H.; Fryklund, L.; Parodi, L. The d3/fl-growth hormone (GH) receptor polymorphism does not influence the effect of GH treatment (66 μg/kg per day) or the spontaneous growth in short non-GH-deficient small-for-gestational-age children: Results from a two-year controlled prospective study in 170 Spanish patients. J. Clin. Endocrinol. Metab. 2006, 91. [Google Scholar] [CrossRef]
- Bernabeu, I.; Alvarez-Escolá, C.; Quinteiro, C.; Lucas, T.; Puig-Domingo, M.; Luque-Ramírez, M.; De Miguel-Novoa, P.; Fernandez-Rodriguez, E.; Halperin, I.; Loidi, L.; et al. The exon 3-deleted growth hormone receptor is associated with better response to pegvisomant therapy in acromegaly. J. Clin. Endocrinol. Metab. 2010, 95. [Google Scholar] [CrossRef] [PubMed]
- Blum, W.F.; Machinis, K.; Shavrikova, E.P.; Keller, A.; Stobbe, H.; Pfaeffle, R.W.; Amselem, S. The growth response to growth hormone (GH) treatment in children with isolated GH deficiency is independent of the presence of the exon 3-minus isoform of the GH receptor. J. Clin. Endocrinol. Metab. 2006, 91. [Google Scholar] [CrossRef] [PubMed]
- Ben-Avraham, D.; Govindaraju, D.R.; Budagov, T.; Fradin, D.; Durda, P.; Liu, B.; Ott, S.; Gutman, D.; Sharvit, L.; Kaplan, R.; et al. The GH receptor exon 3 deletion is a marker of male-specific exceptional longevity associated with increased GH sensitivity and taller stature. Sci. Adv. 2017. [Google Scholar] [CrossRef]
- Bonert, V.S.; Melmed, S. Growth Hormone. In The Pituitary: Fourth Edition; 2017; ISBN 9780128041697. [Google Scholar]
- Pantel, J.; Machinis, K.; Sobrier, M.-L.; Duquesnoy, P.; Goossens, M.; Amselem, S. Species-specific Alternative Splice Mimicry at the Growth Hormone Receptor Locus Revealed by the Lineage of Retroelements during Primate Evolution. J. Biol. Chem. 2000. [Google Scholar] [CrossRef]
- Postel-Vinay, M.C.; Kelly, P.A. Growth hormone receptor signalling. Baillieres. Clin. Endocrinol. Metab. 1996, 10. [Google Scholar] [CrossRef]
- Kurzer, J.H.; Carter-Su, C. Growth Hormone Induced Activation and Regulation of JAK2 and STAT Proteins. In Signal Transducers and Activators of Transcription (STATs); 2003.
- Carter-Su, C.; Schwartz, J.; Argetsinger, L.S. Growth hormone signaling pathways. Growth Horm. IGF Res. 2016, 28. [Google Scholar] [CrossRef]
- Rascio, F.; Spadaccino, F.; Rocchetti, M.T.; Castellano, G.; Stallone, G.; Netti, G.S.; Ranieri, E. The pathogenic role of PI3K/AKT pathway in cancer onset and drug resistance: an updated review. Cancers (Basel). 2021, 13. [Google Scholar] [CrossRef]
- Hähnel, P.S.; Thaler, S.; Antunes, E.; Huber, C.; Theobald, M.; Schuler, M. Targeting AKT signaling sensitizes cancer to cellular immunotherapy. Cancer Res. 2008, 68. [Google Scholar] [CrossRef]
- Noh, K.H.; Kang, T.H.; Kim, J.H.; Pai, S.I.; Lin, K.Y.; Hung, C.F.; Wu, T.C.; Kim, T.W. Activation of Akt as a mechanism for tumor immune evasion. Mol. Ther. 2009, 17. [Google Scholar] [CrossRef]
- Ravichandran, K.S. Signaling via Shc family adapter proteins. Oncogene 2001, 20. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.B.M.; Prigent, S.A. Insights into the Shc family of adaptor proteins. J. Mol. Signal. 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Dehkhoda, F.; Lee, C.M.M.; Medina, J.; Brooks, A.J. The growth hormone receptor: Mechanism of receptor activation, cell signaling, and physiological aspects. Front. Endocrinol. (Lausanne). 2018. [Google Scholar] [CrossRef] [PubMed]
- Aguiar-Oliveira, M.H.; Bartke, A. Growth hormone deficiency: Health and longevity. Endocr. Rev. 2019, 40. [Google Scholar] [CrossRef] [PubMed]
- Bartke, A. Growth hormone and aging. Rev. Endocr. Metab. Disord. 2021, 22, 71–80. [Google Scholar] [CrossRef]
- Coschigano, K.T.; Clemmons, D.; Bellush, L.L.; Kopchick, J.J. Assessment of growth parameters and life span of GHR/BP gene-disrupted mice. Endocrinology 2000, 141. [Google Scholar] [CrossRef]
- Brooks, A.J.; Waters, M.J. The growth hormone receptor: Mechanism of activation and clinical implications. Nat. Rev. Endocrinol. 2010, 6. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, B.C.; Maheshwari, H.G.; Li, H.; Reed, M.; Lozykowski, M.; Okada, S.; Catalog, L.; Coschigamo, K.; Wagner, T.E.; et al. A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse). Proc. Natl. Acad. Sci. U. S. A. 1997, 94. [Google Scholar] [CrossRef]
- Trejo, J.L.; LLorens-Martín, M. V.; Torres-Alemán, I. The effects of exercise on spatial learning and anxiety-like behavior are mediated by an IGF-I-dependent mechanism related to hippocampal neurogenesis. Mol. Cell. Neurosci. 2008, 37. [Google Scholar] [CrossRef]
- Nashiro, K.; Guevara-Aguirre, J.; Braskie, M.N.; Hafzalla, G.W.; Velasco, R.; Balasubramanian, P.; Wei, M.; Thompson, P.M.; Mather, M.; Nelson, M.D.; et al. Brain structure and function associated with younger adults in growth hormone receptor-deficient humans. J. Neurosci. 2017, 37. [Google Scholar] [CrossRef]
- Kranzler, J.H.; Rosenbloom, A.L.; Martinez, V.; Guevara-Aguirre, J. Normal intelligence with severe insulin-like growth factor I deficiency due to growth hormone receptor deficiency: A controlled study in a genetically homogeneous population. J. Clin. Endocrinol. Metab. 1998, 83. [Google Scholar] [CrossRef]
- Shevah, O.; Kornreich, L.; Galatzer, A.; Laron, Z. The intellectual capacity of patients with Laron syndrome (LS) differs with various molecular defects of the growth hormone receptor gene. Correlation with CNS abnormalities. Horm. Metab. Res. 2005, 37. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Aguirre, J.; Balasubramanian, P.; Guevara-Aguirre, M.; Wei, M.; Madia, F.; Cheng, C.W.; Hwang, D.; Martin-Montalvo, A.; Saavedra, J.; Ingles, S.; et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Mormando, M.; Nasto, L.A.; Bianchi, A.; Mazziotti, G.; Giampietro, A.; Pola, E.; Pontecorvi, A.; Giustina, A.; De Marinis, L. GH receptor isoforms and skeletal fragility in acromegaly. Eur. J. Endocrinol. 2014, 171. [Google Scholar] [CrossRef]
- Urbanek, M.; MacLeod, J.N.; Cooke, N.E.; Liebhaber, S.A. Expression of a human growth hormone (hGH) receptor isoform is predicted by tissue-specific alternative splicing of exon 3 of the hGH receptor gene transcript. Mol. Endocrinol. 1992, 6. [Google Scholar] [CrossRef]
- Mercado, M.; Davila, N.; Mc leod, J.F.; Baumann, G. Distribution of growth hormone receptor messenger ribonucleic acid containing and lacking exon 3 in human tissues. J. Clin. Endocrinol. Metab. 1994, 78. [Google Scholar] [CrossRef]
- Pantel, J.; Machinis, K.; Sobrier, M.L.; Duquesnoy, P.; Goossens, M.; Amselem, S. Species-specific alternative splice mimicry at the growth hormone receptor locus revealed by the lineage of retroelements during primate evolution. A novel mechanism accounting for protein diversity between and within species. J. Biol. Chem. 2000, 275. [Google Scholar] [CrossRef]
- Saenger, P.; Reiter, E. Genetic factors associated with small for gestational age birth and the use of human growth hormone in treating the disorder. Int. J. Pediatr. Endocrinol. 2012, 2012. [Google Scholar] [CrossRef]
- Kelly, P.A.; Djiane, J.; Postel-Vinay, M.C.; Edery, M. The prolactin/growth hormone receptor family. Endocr. Rev. 1991, 12. [Google Scholar] [CrossRef]
- Urbanek, M.; Russell, J.E.; Cooke, N.E.; Liebhaber, S.A. Functional characterization of the alternatively spliced, placental human growth hormone receptor. J. Biol. Chem. 1993, 268. [Google Scholar] [CrossRef]
- McQuillan, D.J.; Handley, C.J.; Campbell, M.A.; Bolis, S.; Milway, V.E.; Herington, A.C. Stimulation of proteoglycan biosynthesis by serum and insulin-like growth factor-I in cultured bovine articular cartilage. Biochem. J. 1986, 240. [Google Scholar] [CrossRef] [PubMed]
- Meulenbelt, I.; Bijkerk, C.; Miedema, H.S.; Breedveld, F.C.; Hofman, A.; Valkenburg, H.A.; Pols, H.A.P.; Slagboom, P.E.; Van Duijn, C.M. A genetic association study of the IGF-1 gene and radiological osteoarthritis in a population-based cohort study (the Rotterdam study). Ann. Rheum. Dis. 1998, 57. [Google Scholar] [CrossRef] [PubMed]
- Bougnères, P. The exon-3 deletion of the growth hormone receptor (GHR) gene still has a limited impact in clinical endocrinology. J. Clin. Endocrinol. Metab. 2010, 95. [Google Scholar] [CrossRef] [PubMed]
- Audí, L.; Esteban, C.; Carrascosa, A.; Espadero, R.; Pérez-Arroyo, A.; Arjona, R.; Clemente, M.; Wollmann, H.; Fryklund, L.; Parodi, L.A.; et al. Exon 3-deleted/full-length growth hormone receptor polymorphism genotype frequencies in Spanish short small-for-gestational-age (SGA) children and adolescents (n = 247) and in an adult control population (n = 289) show increased fl/fl in short SGA. J. Clin. Endocrinol. Metab. 2006, 91. [Google Scholar] [CrossRef] [PubMed]
- Montefusco, L.; Filopanti, M.; Ronchi, C.L.; Olgiati, L.; La-Porta, C.; Losa, M.; Epaminonda, P.; Coletti, F.; Beck-Peccoz, P.; Spada, A.; et al. D3-Growth hormone receptor polymorphism in acromegaly: Effects on metabolic phenotype. Clin. Endocrinol. (Oxf). 2010. [Google Scholar] [CrossRef]
- Tiulpakov, A.; Rubtsov, P.; Dedov, I.; Peterkova, V.; Bezlepkina, O.; Chrousos, G.P.; Hochberg, Z. A novel C-terminal Growth Hormone Receptor (GHR) mutation results in impaired GHR-STAT5 but normal STAT-3 signaling. J. Clin. Endocrinol. Metab. 2005, 90. [Google Scholar] [CrossRef]
- Sobrier, M.L.; Duquesnoy, P.; Duriez, B.; Amselem, S.; Goossens, M. Expression and binding properties of two isoforms of the human growth hormone receptor. FEBS Lett. 1993, 319. [Google Scholar] [CrossRef]
- Butler, M.G.; Hossain, W.; Hassan, M.; Manzardo, A.M. Growth hormone receptor (GHR) gene polymorphism and scoliosis in Prader-Willi syndrome. Growth Horm. IGF Res. 2018, 39. [Google Scholar] [CrossRef]
- Pelekanos, R.A.; Sardesai, V.S.; Dekker Nitert, M.; Callaway, L.K.; Fisk, N.M.; Jeffery, P.L. Rapid method for growth hormone receptor exon 3 delete (GHRd3) SNP genotyping from archival human placental samples. Endocrine 2015, 49. [Google Scholar] [CrossRef]
- Resendez, S.; Saitou, M.; Parisi, L.R.; Wu, F.; Nakagome, S.; Satta, Y.; Atilla-Gokcumen, G.E.; Mu, X.; Gokcumen, O. Sex-specific phenotypic effects and evolutionary history of an ancient deletion polymorphism of the human growth hormone receptor. bioRxiv 2019. [Google Scholar]
- Saitou, M.; Resendez, S.; Pradhan, A.J.; Wu, F.; Lie, N.C.; Hall, N.J.; Zhu, Q.; Reinholdt, L.; Satta, Y.; Speidel, L.; et al. Sex-specific phenotypic effects and evolutionary history of an ancient polymorphic deletion of the human growth hormone receptor. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Dias, C.; Giordano, M.; Frechette, R.; Bellone, S.; Polychronakos, C.; Legault, L.; Deal, C.L.; Goodyer, C.G. Genetic variations at the human growth hormone receptor (GHR) gene locus are associated with idiopathic short stature. J. Cell. Mol. Med. 2017, 21. [Google Scholar] [CrossRef] [PubMed]
- Glad, C.A.M.; Johannsson, G.; Carlsson, L.M.S.; Svensson, P.A. Rapid and high throughput genotyping of the growth hormone receptor exon 3 deleted/full-length polymorphism using a tagSNP. Growth Horm. IGF Res. 2010, 20. [Google Scholar] [CrossRef] [PubMed]
- Chiloiro, S.; Mirra, F.; Federico, D.; Giampietro, A.; Visconti, F.; Rossi, L.; Pontecorvi, A.; De Marinis, L.; Bianchi, A. The Role of Growth Hormone Receptor Isoforms and Their Effects in Bone Metabolism and Skeletal Fragility. Protein Pept. Lett. 2020, 27. [Google Scholar] [CrossRef]
- Padidela, R.; Bryan, S.M.; Abu-Amero, S.; Hudson-Davies, R.E.; Achermann, J.C.; Moore, G.E.; Hindmarsh, P.C. The growth hormone receptor gene deleted for exon three (GHRd3) polymorphism is associated with birth and placental weight. Clin. Endocrinol. (Oxf). 2012, 76. [Google Scholar] [CrossRef]
- Sørensen, K.; Aksglaede, L.; Petersen, J.H.; Leffers, H.; Juul, A. The exon 3 deleted growth hormone receptor gene is associated with small birth size and early pubertal onset in healthy boys. J. Clin. Endocrinol. Metab. 2010, 95. [Google Scholar] [CrossRef]
- Jensen, R.B.; Boas, M.; Nielsen, J.E.; Maroun, L.L.; Jørgensen, A.; Larsen, T.; Main, K.M.; Juul, A. A common deletion in the growth hormone receptor gene (d3-GHR) in the offspring is related to maternal placental GH levels during pregnancy. Growth Horm. IGF Res. 2020, 55. [Google Scholar] [CrossRef]
- Martins, C.S.; Fernandes-Rosa, F.L.; Espineira, A.R.; de Souza, R.M.; de Castro, M.; Barbieri, M.A.; Bettiol, H.; Jorge, A.L.; Antonini, S.R. The growth hormone receptor exon 3 polymorphism is not associated with height or metabolic traits in healthy young adults. Growth Horm. IGF Res. 2014, 24. [Google Scholar] [CrossRef]
- Chen, X.; Liu, C.; Yang, S.; Yang, Y.; Chen, Y.; Zhao, X.; Zhu, W.; Zhao, Q.; Ni, C.; Huang, X.; et al. Gender Specificity and Local Socioeconomic Influence on Association of GHR fl/d3 Polymorphism With Growth and Metabolism in Children and Adolescents. Front. Pediatr. 2022, 10. [Google Scholar] [CrossRef]
- Kaabi, Y.A. Frequency of the exon 3-deleted/full-length growth hormone receptor polymorphism in Saudi Arabian population. Saudi Med. J. 2017, 38. [Google Scholar] [CrossRef]
- Baş, F.; Keleşoǧlu, F.; Timirci, Ö.; Eryilmaz, S.K.; Bozkurt, N.; Aydin, B.K.; Bundak, R.; Isbir, T.; Darendeliler, F. The distribution of exon 3-deleted/full-length growth hormone receptor polymorphism in the Turkish population. JCRPE J. Clin. Res. Pediatr. Endocrinol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Dauber, A.; Meng, Y.; Audi, L.; Vedantam, S.; Weaver, B.; Carrascosa, A.; Albertsson-Wikland, K.; Ranke, M.B.; Jorge, A.A.L.; Cara, J.; et al. A genome-wide pharmacogenetic study of growth hormone responsiveness. J. Clin. Endocrinol. Metab. 2020, 105. [Google Scholar] [CrossRef] [PubMed]
- Van Der Klaauw, A.A.; Van Der Straaten, T.; Baak-Pablo, R.; Biermasz, N.R.; Guchelaar, H.J.; Pereira, A.M.; Smit, J.W.A.; Romijn, J.A. Influence of the d3-growth hormone (GH) receptor isoform on short-term and long-term treatment response to GH replacement in GH-deficient adults. J. Clin. Endocrinol. Metab. 2008, 93. [Google Scholar] [CrossRef] [PubMed]
- Jorge, A.A.L.; Marchisotti, F.G.; Montenegro, L.R.; Carvalho, L.R.; Mendonca, B.B.; Arnhold, I.J.P. Growth hormone (GH) pharmacogenetics: Influence of GH receptor exon 3 retention or deletion on first-year growth response and final height in patients with severe GH deficiency. J. Clin. Endocrinol. Metab. 2006, 91. [Google Scholar] [CrossRef] [PubMed]
- Renehan, A.G.; Solomon, M.; Zwahlen, M.; Morjaria, R.; Whatmore, A.; Audí, L.; Binder, G.; Blum, W.; Bougnres, P.; Santos, C. Dos; et al. Growth hormone receptor polymorphism and growth hormone therapy response in children: A bayesian meta-analysis. Am. J. Epidemiol. 2012, 175. [Google Scholar] [CrossRef]
- Wassenaar, M.J.E.; Dekkers, O.M.; Pereira, A.M.; Wit, J.M.; Smit, J.W.; Biermasz, N.R.; Romijn, J.A. Impact of the exon 3-deleted Growth Hormone (GH) receptor polymorphism on baseline height and the growth response to recombinant human GH therapy in GH-Deficient (GHD) and non-GHD children with short stature: A systematic review and meta-analysis. J. Clin. Endocrinol. Metab. 2009, 94. [Google Scholar] [CrossRef]
- Chung, M.S.; Langouët, M.; Chamberlain, S.J.; Carmichael, G.G. Prader-Willi syndrome: reflections on seminal studies and future therapies: Prader-Willi Syndrome. Open Biol. 2020, 10. [Google Scholar] [CrossRef]
- Angulo, M.A.; Butler, M.G.; Cataletto, M.E. Prader-Willi syndrome: A review of clinical, genetic, and endocrine findings. J. Endocrinol. Invest. 2015, 38. [Google Scholar] [CrossRef]
- Myers, S.E.; Carrel, A.L.; Whitman, B.Y.; Allen, D.B. Sustained benefit after 2 years of growth hormone on body composition, fat utilization, physical strength and agility, and growth in Prader-Willi syndrome. J. Pediatr. 2000, 137. [Google Scholar] [CrossRef]
- Park, S.W.; Lee, S.T.; Sohn, Y.B.; Kim, S.H.; Cho, S.Y.; Ko, A. ra; Ji, S.T.; Kwon, J.Y.; Yeau, S.; Paik, K.H.; et al. A polymorphism in the growth hormone receptor is associated with height in children with Prader-Willi syndrome. Am. J. Med. Genet. Part A 2011. [Google Scholar] [CrossRef]
- 74. Butler, Merlin G., Jennifer Roberts, Jena Hayes, Xiaoyu Tan, and A.M.M. Growth hormone receptor (GHR) gene polymorphism and prader–willi syndrome. Am. J. Med. Genet. Part A 2013, 161, 1647–1653. [CrossRef] [PubMed]
- Melmed, S. Acromegaly: Medical Progress. N. Engl. J. Med. 2006, 355. [Google Scholar] [CrossRef] [PubMed]
- Melmed, S. Acromegaly pathogenesis and treatment. J. Clin. Invest. 2009, 119. [Google Scholar] [CrossRef] [PubMed]
- Dobrashian, R.D.; O’Halloran, D.J.; Hunt, A.; Beardwell, C.G.; Shalet, S.M. Relationships between insulin-like growth factor-1 levels and growth hormone concentrations during diurnal profiles and following oral glucose in acromegaly. Clin. Endocrinol. (Oxf). 1993, 38. [Google Scholar] [CrossRef] [PubMed]
- Schmid, C.; Krayenbuehl, P.A.; Bernays, R.L.; Zwimpfer, C.; Maly, F.E.; Wiesli, P. Growth hormone (GH) receptor isoform in acromegaly: Lower concentrations of GH but not insulin-like growth factor-I in patients with a genomic deletion of exon 3 in the GH receptor gene. Clin. Chem. 2007, 53. [Google Scholar] [CrossRef]
- Parkinson, C.; Ryder, W.D.J.; Trainer, P.J. The relationship between serum GH and serum IGF-I in acromegaly is gender-specific. J. Clin. Endocrinol. Metab. 2001, 86. [Google Scholar] [CrossRef]
- Van Der Lely, A.J.; Hutson, R.K.; Trainer, P.J.; Besser, G.M.; Barkan, A.L.; Katznelson, L.; Klibanski, A.; Herman-Bonert, V.; Melmed, S.; Vance, M.L.; et al. Long-term treatment of acromegaly with pegvisomant, a growth hormone receptor antagonist. Lancet 2001, 358. [Google Scholar] [CrossRef]
- Bianchi, A.; Mazziotti, G.; Tilaro, L.; Cimino, V.; Veltri, F.; Gaetani, E.; Pecorini, G.; Pontecorvi, A.; Giustina, A.; De Marinis, L. Growth hormone receptor polymorphism and the effects of pegvisomant in acromegaly. Pituitary 2009, 12. [Google Scholar] [CrossRef]
- Franck, S.E.; Broer, L.; Van Der Lely, A.J.; Kamenicky, P.; Bernabéu, I.; Malchiodi, E.; Delhanty, P.J.D.; Rivadeneira, F.; Neggers, S.J.C.M.M. The Effect of the Exon-3-Deleted Growth Hormone Receptor on Pegvisomant-Treated Acromegaly: A Systematic Review and Meta-Analysis. Neuroendocrinology 2017, 105. [Google Scholar] [CrossRef]
- Colao, A.; Ferone, D.; Marzullo, P.; Lombardi, G. Systemic Complications of Acromegaly: Epidemiology, Pathogenesis, and Management. Endocr. Rev. 2004, 25. [Google Scholar] [CrossRef]
- Colao, A.; Pivonello, R.; Auriemma, R.S.; Galdiero, M.; Ferone, D.; Minuto, F.; Marzullo, P.; Lombardi, G. The association of fasting insulin concentrations and colonic neoplasms in acromegaly: A colonoscopy-based study in 210 patients. J. Clin. Endocrinol. Metab. 2007, 92. [Google Scholar] [CrossRef] [PubMed]
- Colao, A.; Spinelli, L.; Marzullo, P.; Pivonello, R.; Petretta, M.; Di Somma, C.; Vitale, G.; Bonaduce, D.; Lombardi, G. High prevalence of cardiac valve disease in acromegaly: An observational, analytical, case-control study. J. Clin. Endocrinol. Metab. 2003, 88. [Google Scholar] [CrossRef] [PubMed]
- Pontes, J.; Madeira, M.; Lima, C.H.A.; Ogino, L.L.; de Paula Paranhos Neto, F.; de Mendonça, L.M.C.; Farias, M.L.F.; Kasuki, L.; Gadelha, M.R. Exon 3-deleted growth hormone receptor isoform is not related to worse bone mineral density or microarchitecture or to increased fracture risk in acromegaly. J. Endocrinol. Invest. 2020, 43. [Google Scholar] [CrossRef] [PubMed]
- Park, H.Y.; Hwang, I.R.; Seo, J.B.; Kim, S.W.; Seo, H.A.; Lee, I.K.; Kim, J.G. Association between the growth hormone receptor exon 3 polymorphism and metabolic factors in Korean patients with acromegaly. Endocrinol. Metab. 2015, 30. [Google Scholar] [CrossRef] [PubMed]
- Kamenicky, P.; Dos Santos, C.; Espinosa, C.; Salenave, S.; Galland, F.; Le Bouc, Y.; Maison, P.; Bougnères, P.; Chanson, P. D3 GH receptor polymorphism is not associated with IGF1 levels in untreated acromegaly. Eur. J. Endocrinol. 2009, 161. [Google Scholar] [CrossRef] [PubMed]
- Cinar, N.; Dagdelen, S.; Yorgun, H.; Canpolat, U.; Kabakçı, G.; Erbas, T. The clinical and cardiometabolic effects of d3-growth hormone receptor polymorphism in acromegaly. Pituitary 2015, 18. [Google Scholar] [CrossRef]
- Mercado, M.; González, B.; Sandoval, C.; Esquenazi, Y.; Mier, F.; Vargas, G.; De Los Monteros, A.L.E.; Sosa, E. Clinical and biochemical impact of the d3 growth hormone receptor genotype in acromegaly. J. Clin. Endocrinol. Metab. 2008, 93. [Google Scholar] [CrossRef]
- Wassenaar, M.J.E.; Biermasz, N.R.; Pereira, A.M.; Van Der Klaauw, A.A.; Smit, J.W.A.; Roelfsema, F.; Van Der Straaten, T.; Cazemier, M.; Hommes, D.W.; Kroon, H.M.; et al. The exon-3 deleted growth hormone receptor polymorphism predisposes to long-term complications of acromegaly. J. Clin. Endocrinol. Metab. 2009, 94. [Google Scholar] [CrossRef]
- Classic pages in obstetrics and gynecology by Henry, H. Turner. A syndrome of infantilism, congenital webbed neck, and cubitus valgus. Endocrinology, vol. 23, pp. 566-574, 1938. Am. J. Obstet. Gynecol. 1972, 113. [Google Scholar] [CrossRef]
- Ullrich, O. Über typische Kombinationsbilder multipler Abartungen. Z. Kinderheilkd. 1930, 49. [Google Scholar] [CrossRef]
- Cui, X.; Cui, Y.; Shi, L.; Luan, J.; Zhou, X.; Han, J. A basic understanding of Turner syndrome: Incidence, complications, diagnosis, and treatment. Intractable Rare Dis. Res. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Elsheikh, M.; Dunger, D.B.; Conway, G.S.; Wass, J.A.H. Turner’s Syndrome in Adulthood. Endocr. Rev. 2002, 23. [Google Scholar] [CrossRef]
- Los, E.; Rosenfeld, R.G. Growth and growth hormone in Turner syndrome: Looking back, looking ahead. Am. J. Med. Genet. Part C Semin. Med. Genet. 2019, 181. [Google Scholar] [CrossRef] [PubMed]
- Van den Broeck, J.; Massa, G.G.; Attanasio, A.; Matranga, A.; Chaussain, J.L.; Price, D.A.; Aarskog, D.; Wit, J.M. Final height after long-term growth hormone treatment in Turner syndrome. J. Pediatr. 1995, 127. [Google Scholar] [CrossRef]
- Li, P.; Cheng, F.; Xiu, L. Height outcome of the recombinant human growth hormone treatment in turner syndrome: A meta-analysis. Endocr. Connect. 2018, 7. [Google Scholar] [CrossRef]
- Ko, J.M.; Kim, J.M.; Cheon, C.K.; Kim, D.H.; Lee, D.Y.; Cheong, W.Y.; Kim, E.Y.; Park, M.J.; Yoo, H.W. The common exon 3 polymorphism of the growth hormone receptor gene and the effect of growth hormone therapy on growth in Korean patients with Turner syndrome. Clin. Endocrinol. (Oxf). 2010, 72. [Google Scholar] [CrossRef]
- Chernausek, S.D.; Attie, K.M.; Cara, J.F.; Rosenfeld, R.G.; Frane, J. Growth Hormone Therapy of Turner Syndrome: The Impact of Age of Estrogen Replacement on Final Height 1. J. Clin. Endocrinol. Metab. 2000, 85. [Google Scholar] [CrossRef]
- Binder, G.; Baur, F.; Schweizer, R.; Ranke, M.B. The d3-growth hormone (GH) receptor polymorphism is associated with increased responsiveness to GH in turner syndrome and short small-for- gestational-age children. J. Clin. Endocrinol. Metab. 2006. [Google Scholar] [CrossRef]
- Zanelli, S.A.; Rogol, A.D. Short children born small for gestational age outcomes in the era of growth hormone therapy. Growth Horm. IGF Res. 2018, 38. [Google Scholar] [CrossRef]
- Hwang, I.T. Efficacy and safety of growth hormone treatment for children born small for gestational age. Korean J. Pediatr. 2014, 57. [Google Scholar] [CrossRef]
- López-Siguero, J.P.; Martínez-Aedo, M.J.; Bermúdez de la Vega, J.A.; Bosch-Muñoz, J.; Lechuga-Sancho, A.M.; Villalobos, T. Growth hormone treatment does not to lead to insulin resistance nor excessive rise in IGF-1 levels, while improving height in patients small for gestational age A long-term observational study. Clin. Endocrinol. (Oxf). 2022, 96. [Google Scholar] [CrossRef] [PubMed]
- Chernausek, S.D. Update: Consequences of abnormal fetal growth. J. Clin. Endocrinol. Metab. 2012, 97. [Google Scholar] [CrossRef] [PubMed]
- Saenger, P.; Czernichow, P.; Hughes, I.; Reiter, E.O. Small for gestational age: Short stature and beyond. Endocr. Rev. 2007, 28. [Google Scholar] [CrossRef] [PubMed]
- Pilling, E.L.; Elder, C.J.; Gibson, A.T. Growth patterns in the growth-retarded premature infant. Best Pract. Res. Clin. Endocrinol. Metab. 2008, 22. [Google Scholar] [CrossRef]
- Rapaport, R.; Lee, P.A.; Ross, J.L.; Saenger, P.; Ostrow, V.; Piccoli, G. Three years of growth hormone therapy in children born small for gestational age: Results from the answer program. Endocr. Connect. 2018, 7. [Google Scholar] [CrossRef]
- Clayton, P.E.; Cianfarani, S.; Czernichow, P.; Johannsson, G.; Rapaport, R.; Rogol, A.D. Consensus statement: Management of the child born small for gestational age through to adulthood: A consensus statement of the international societies of pediatric endocrinology and the growth hormone research society. J. Clin. Endocrinol. Metab. 2007, 92. [Google Scholar] [CrossRef]
- Vincenzi, M.; Ion Popa, F.; Corradi, M.; Gandini, A.; Teofoli, F.; Camilot, M.; Boner, A.; Cavarzere, P.; Gaudino, R.; Antoniazzi, F. Analysis of the d3-growth hormone receptor polymorphism in large cohorts of small, appropriate and large for gestational age newborns. Minerva Pediatr. 2016, 68. [Google Scholar]
- Dörr, H.G.; Bettendorf, M.; Hauffa, B.P.; Mehls, O.; Rohrer, T.; Stahnke, N.; Pfäffle, R.; Ranke, M.B. Different relationships between the first 2 years on growth hormone treatment and the d3-growth hormone receptor polymorphism in short small-for-gestational-age (SGA) children. Clin. Endocrinol. (Oxf). 2011, 75. [Google Scholar] [CrossRef]
- Carrascosa, A.; Audí, L.; Esteban, C.; Fernández-Cancio, M.; Andaluz, P.; Gussinyé, M.; Clemente, M.; Yeste, D.; Albisu, M.A. Growth hormone (GH) dose, but not exon 3-deleted/full-length GH receptor polymorphism genotypes, influences growth response to two-year GH therapy in short small-for-gestational-age children. J. Clin. Endocrinol. Metab. 2008, 93. [Google Scholar] [CrossRef]
- Esposito, S.; Leonardi, A.; Lanciotti, L.; Cofini, M.; Muzi, G.; Penta, L. Vitamin D and growth hormone in children: A review of the current scientific knowledge. J. Transl. Med. 2019, 17. [Google Scholar] [CrossRef]
- Sönksen, P.H. Replacement therapy in hypothalamo-pituitary insufficiency after childhood: Management in the adult. Horm. Res. Paediatr. 1990, 33. [Google Scholar] [CrossRef]
- Yuen, K.C.J.; Biller, B.M.K.; Radovick, S.; Carmichael, J.D.; Jasim, S.; Pantalone, K.M.; Hoffman, A.R. American Association of Clinical endocrinologists and American College of Endocrinology guidelines for management of growth hormone deficiency in adults and patients transitioning from pediatric to adult care. Endocr. Pract. 2019, 25. [Google Scholar] [CrossRef] [PubMed]
- Pilotta, A.; Mella, P.; Filisetti, M.; Felappi, B.; Prandi, E.; Parrinello, G.; Notarangelo, L.D.; Buzi, F. Common polymorphisms of the growth hormone (GH) receptor do not correlate with the growth response to exogenous recombinant human GH in GH-deficient children. J. Clin. Endocrinol. Metab. 2006, 91. [Google Scholar] [CrossRef] [PubMed]
- Räz, B.; Janner, M.; Petkovic, V.; Lochmatter, D.; Eblé, A.; Dattani, M.T.; Hindmarsh, P.C.; Flück, C.E.; Mullis, P.E. Influence of growth hormone (GH) receptor deletion of exon 3 and full-length isoforms on GH response and final height in patients with severe gh deficiency. J. Clin. Endocrinol. Metab. 2008, 93, 974–980. [Google Scholar] [CrossRef] [PubMed]
- Valsesia, A.; Chatelain, P.; Stevens, A.; Peterkova, V.; Belgorosky, A.; Maghnie, M.; Antoniazzi, F.; Koledova, E.; Wojcik, J.; Farmer, P.; et al. GH deficiency status combined with GH receptor polymorphism affects response to GH in children. Eur. J. Endocrinol. 2015, 173. [Google Scholar] [CrossRef]
- Adetunji, O.R.; Blair, J.C.; Javadpour, M.; Alfirevic, A.; Pirmohamed, M.; MacFarlane, I.A. Deletion of exon 3 in the growth hormone receptor gene in adults with growth hormone deficiency: Comparison of symptomatic and asymptomatic patients. Clin. Endocrinol. (Oxf). 2010, 72. [Google Scholar] [CrossRef]
Figure 1.
Genomic structure of the Growth Hormone Receptor gene.
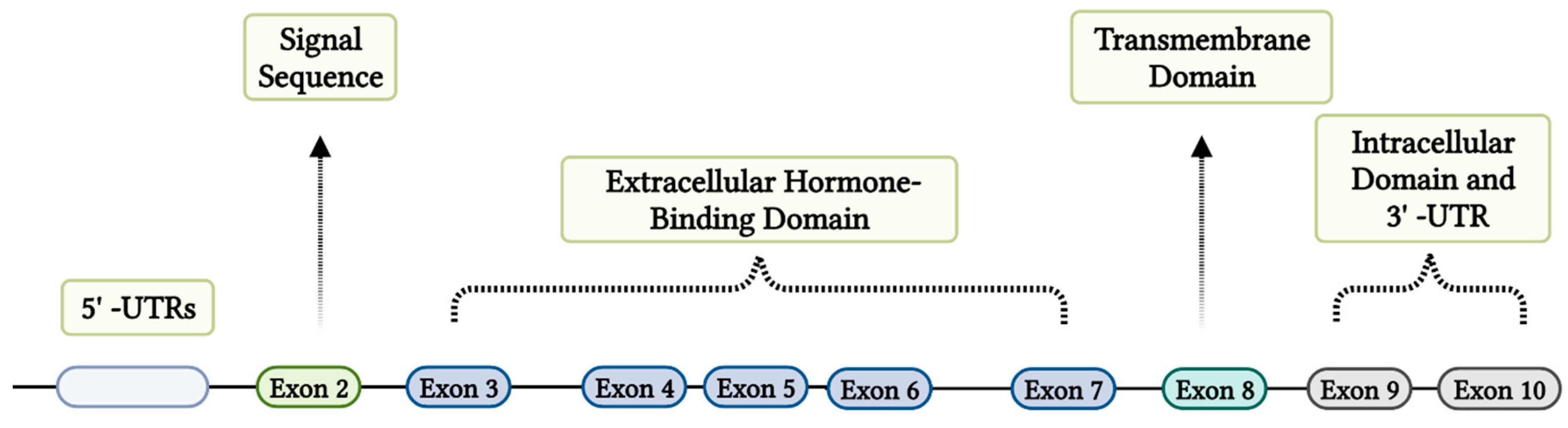
Figure 2.
GH receptor signaling pathways. JAK2 activation by GH initiates certain signaling pathways. SHC is phosphorylated by JAK2, which activates MAPK. JAK2 phosphorylates the transcription factor STAT as well. PI 3′-kinase activation via the phosphorylation of IRS proteins by JAK2 may be crucial for glucose metabolism. Abbreviations: GH, growth hormone; GHR, growth hormone receptor; JAK2, Janus kinase 2; IRS, insulin receptor substrates; MAPK, mitogen-activated protein kinase; STAT, signal transducers and activators of transcription; P, phosphate; PI 3′K, phosphatidylinositol 3-kinase.
Figure 2.
GH receptor signaling pathways. JAK2 activation by GH initiates certain signaling pathways. SHC is phosphorylated by JAK2, which activates MAPK. JAK2 phosphorylates the transcription factor STAT as well. PI 3′-kinase activation via the phosphorylation of IRS proteins by JAK2 may be crucial for glucose metabolism. Abbreviations: GH, growth hormone; GHR, growth hormone receptor; JAK2, Janus kinase 2; IRS, insulin receptor substrates; MAPK, mitogen-activated protein kinase; STAT, signal transducers and activators of transcription; P, phosphate; PI 3′K, phosphatidylinositol 3-kinase.
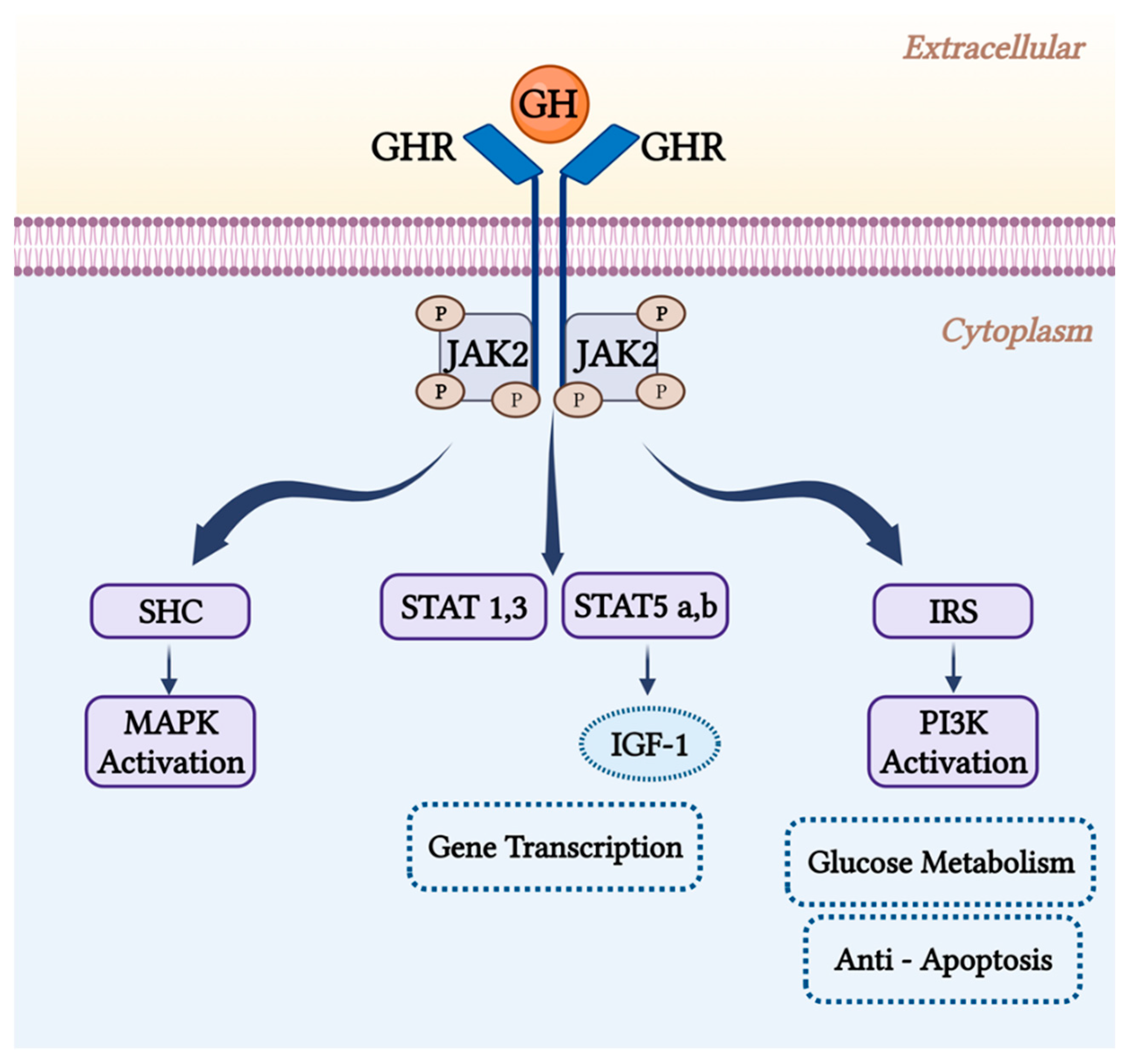
Figure 3.
The full-length GH receptor (flGHR) vs. deletion of exon 3 GH receptor (d3GHR) signaling pathways. The sensitivity of the intracellular JAK-STAT pathway to growth hormone is increased in the absence of exon 3, which raises the transcription of GH target genes in comparison to that of the flGHR isoform.
Figure 3.
The full-length GH receptor (flGHR) vs. deletion of exon 3 GH receptor (d3GHR) signaling pathways. The sensitivity of the intracellular JAK-STAT pathway to growth hormone is increased in the absence of exon 3, which raises the transcription of GH target genes in comparison to that of the flGHR isoform.
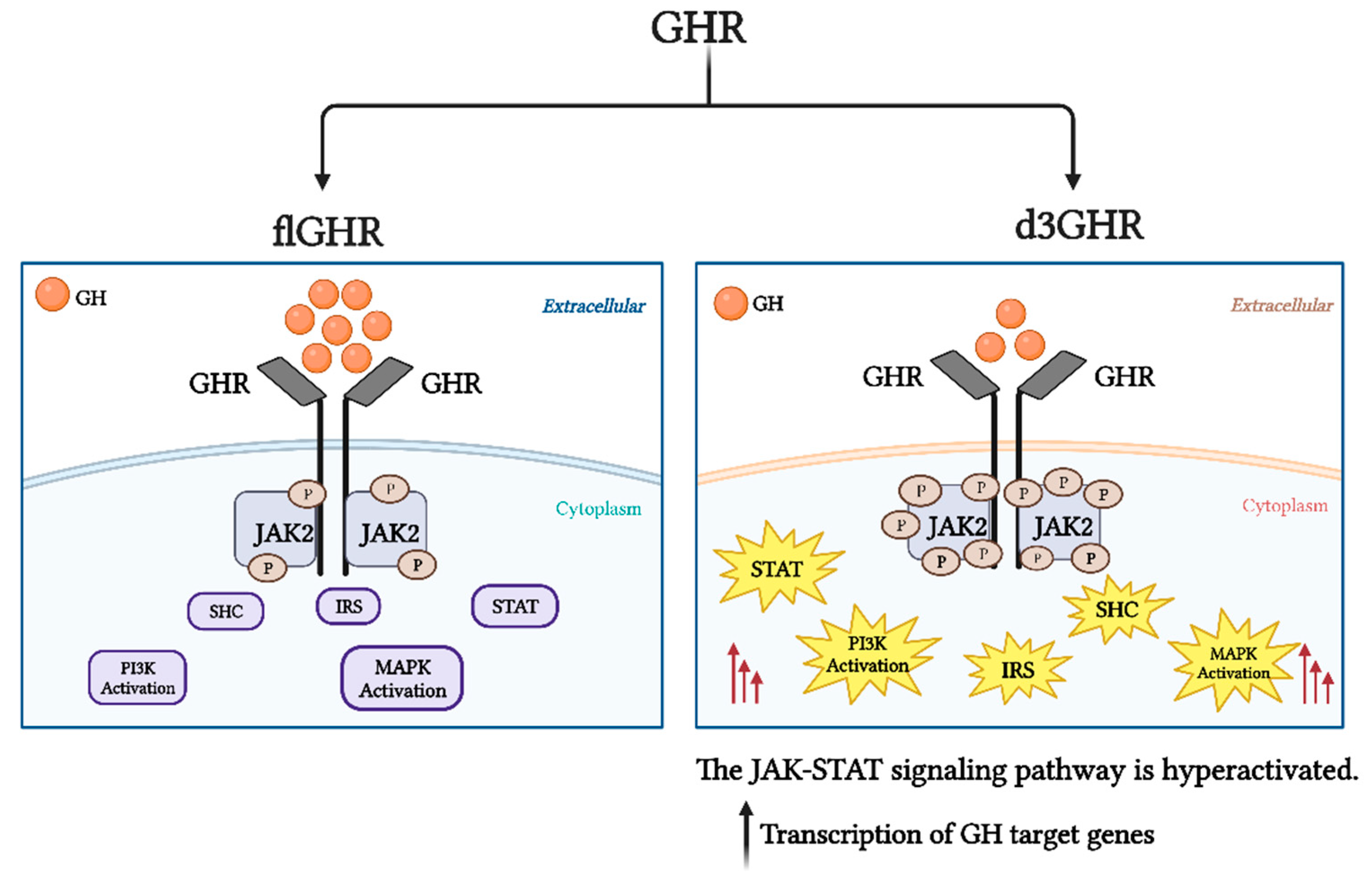

Table 1.
d3GHR polymorphism effects on growth parameters in several cohorts of acromegaly patients. Overview of main studies.
Table 1.
d3GHR polymorphism effects on growth parameters in several cohorts of acromegaly patients. Overview of main studies.
| Authors | No. of cases | fl/fl % |
fl/d3 % |
d3/d3 % |
Effects |
|---|---|---|---|---|---|
| Schmid, 2007 [78] | 44 | 50 | 50 | d3GHR carriers had lower GH concentrations. | |
| Kamenicky, 2009 [88] | 105 | 51 | 30 | 19 | No differences in the baseline levels of GH and IGF-1 concentrations. |
| Pontes, 2020 [86] | 88 | 40 | 60 | No correlation between the presence of d3GHR and increasing frequency of VF, worse BMD or bone microarchitecture. | |
| Cinar, 2015 [89] | 118 | 60.2 | 33.9 | 5.9 | No differences in the prevalence of hypertension, type 2 diabetes mellitus, or coronary artery disease. |
| Montefusco, 2010 [48] | 76 | 55.3 | 35.5 | 9.2 | d3GHR carriers had a lower BMI index. |
| Mercado, 2008 [90] | 148 | 45 | 32 | 22 | d3GHR carriers had higher post-treatment IGF-I concentrations and diabetes was more prevalent in them. |
| J E Wassenaar. 2009 [91] | 86 | 59 | 29 | 7 | d3GHR carriers had an increased prevalence of osteoarthritis, dolichocolon, and adenomatous colonic polyps. |
Table 2.
Effects of the flGHR and d3GHR isoforms on growth indicators in various cohorts of SGA children.
Table 2.
Effects of the flGHR and d3GHR isoforms on growth indicators in various cohorts of SGA children.
| Authors | No. of cases | Effects |
|---|---|---|
| Wegmann, 2017 [9] | 96 | d3GHR carriers had increased spontaneous growth, lower IS, and a compensatory increase in glucose, C-peptide, and insulin at baseline. |
| Garrido, 2021 [10] | 65 | d3GHR carriers have increased spontaneous growth. |
| Dos Santos, 2004 [4] | 76 | d3GHR carriers responded to hGH therapy 1.7 to 2 times better than those who had the flGHR isoform. |
| Dörr, 2011 [111] | 142 | d3GHR carriers exhibited a better growth response to growth hormone only in the first year but not in the second year. |
| Binder, 2006 [101] | 60 | d3GHR carriers grew significantly more quickly than anticipated. During the first year of rhGH treatment, the average height gain linked to d3GHR was roughly 0.75 cm. |
| Carrascosa, 2006 [12] | 170 | Spontaneous growth rate and responsiveness to 66 micro/k.d GH therapy are comparable in flGHR and d3GHR carriers. |
| Carrascosa, 2008 [112] | 60 | Both genotypes responded similarly to GH therapy after two years of response (32.1 ± 3.8 microg/kg.d). |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



