
Submitted:
18 August 2023
Posted:
22 August 2023
You are already at the latest version
Abstract
Using the colloidal method, Au NP’s were deposited onto 6 different supports (TiO2, and -Al2O3, HFeO2, CeO2, and C), and their catalytic activity and selectivity were compared in the alkylation of aniline by benzyl alcohol. Correlations were made between the nature of the support, the Au NP size and H-binding energy.
Keywords:
Au-supported catalyst
; structure-activity-relationship
; H-borrowing catalysis
; Alkylation of amine
1. Introduction
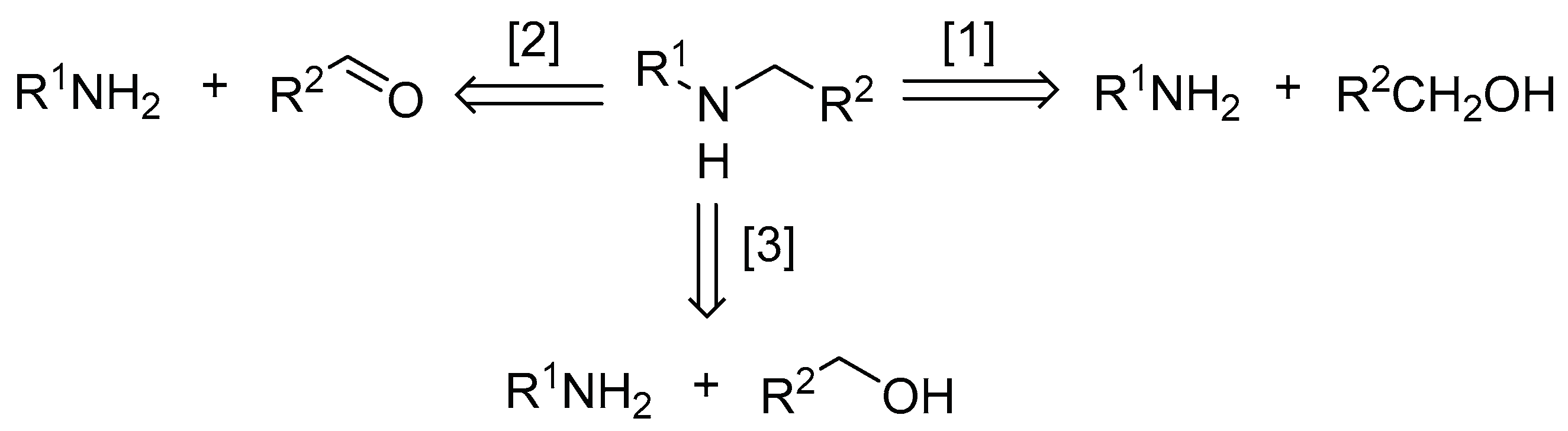
Secondary amines are an industrially important feedstock used in the synthesis of fine chemicals, surfactant, dyes, agrochemicals, functionalized materials, and biologically active compounds. Secondary amines are mostly derived from primary amines; either by the reaction with a reactive alkyl halide, or reductive amination with a carbonyl compound (Scheme 1, routes 1 and 2). These reactions utilize stoichiometric amounts of toxic alkyl halides or hazardous hydridic reductants, which generate stoichiometric amounts of by-products that require complex workup procedures for product purification.
In comparison, catalytic alkylation of amines using a primary alcohol (Scheme 1, route 3), also known as ‘H-auto transfer’ or ‘hydrogen-borrowing’ reactions, is considered to be a ‘greener’ method for the synthesis of secondary amines, as no extraneous reagents are needed, and only water is produced as the by-product.[1,2] The reaction broadly follows the following elementary steps (Scheme 2): (i) dehydrogenation/oxidation of alcohol to the corresponding aldehyde or ketone, (ii) condensation of carbonyl group with amine to form an imine intermediate, which is (iii) reduced by the metal-hydride formed in step (i) to the amine product.
A plethora of homogeneous and heterogenous metal catalysts had been reported for the H-auto transfer reaction, and the subject has been covered by a number of comprehensive reviews.[3,4,5] Often, the imine, R2CH=NR1 and the tertiary amine (R2CH2)2NR1 were observed as side products, necessitating the addition of a base to suppress these impurities. The inclusion of a base in such reactions can be problematic, as it is known that these type of reactions can, indeed, proceed under basic conditions without metal catalysts.[6,7]
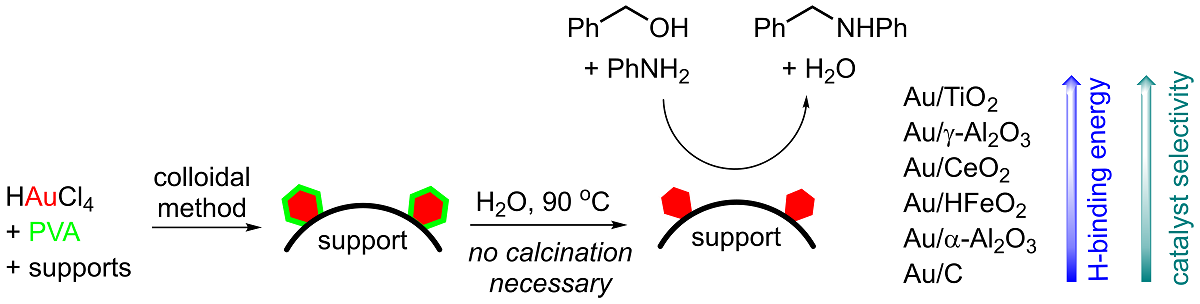
In earlier work, we demonstrated that the reaction can be performed with very high selectivity, in the absence of additives or base, using a commercially available Au/TiO2 catalyst (AUROliteTM) in a packed bed reactor. [8] Subsequently, we initiated a study to examine how metal support interactions (MSI) may affect the catalyst performance in these reactions. At a very basic level, MSI reduce the mobility of metal nanoparticles on the catalyst support and their tendency to agglomerate, enhancing catalytic activity by the maintenance of particle sizes during catalytic turnover.[9] However, MSI may also be attributed to the presence of special sites at the perimeter of metal particles, where chemical and electronic properties of both the metal and support atoms can influence the adsorbed species, thus bestowing added catalytic activity.[10] In an earlier study by Ishida et al.,[11] Au catalysts supported on 9 metal oxides were prepared by different methods (milling, co-precipitation and DP), and utilized in the reaction of aniline with benzyl alcohol. It was found that while basic and neutral supports promote catalytic activity (measured by alcohol conversion), the selectivity to secondary amine is attributed to the adsorption of aniline by hydrogen bonding with Lewis acidic sites on the surface of the metal oxides. However, the study did not appear to account for differences of Au nanoparticles, which can result from the different methods of operation.
In this work, we aim to prepare a series of Au NP’s supported on different supports, in an attempt to delineate the relationship(s) between the size of the NP and the nature of the support, with their catalytic activity in H-auto transfer reactions.
2. Results and discussion
2.1. Catalyst preparation
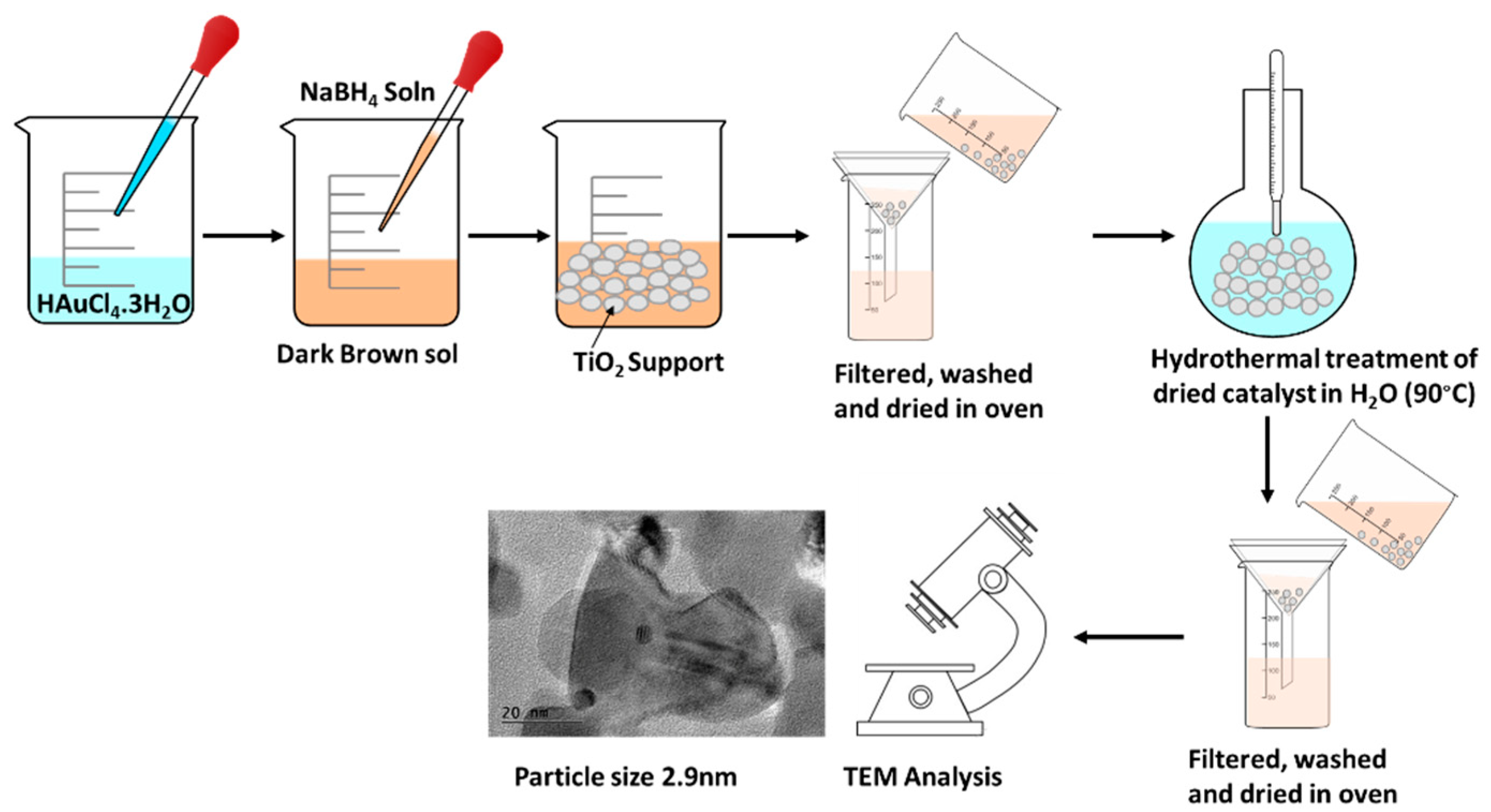
The sol immobilization method (colloidal synthesis) was chosen for the preparation of the Au catalysts. The procedure deposits pre-formed Au NP’s onto a solid support. As the metal nanoparticles are formed independently of the support, it can offer more consistent results in terms of control of particle sizes.[12] The colloidal method was deployed successfully by Hutchings and co-workers to produce 1 wt% Au/TiO2 catalysts using polyvinyl alcohol (PVA) as a stabilizer.[13] In this procedure, the resultant catalyst particles were then heated in water at 90 °C for 2 h to remove the water-soluble stabilizer (Figure 1). As the catalyst was not exposed to very high temperatures, the size of the Au nanoparticles can be preserved (2.9 nm). However, calcination of the catalyst at temperatures ≥300° can also lead to significant sintering of the metallic NP’s.
In the present work, we deployed the colloidal method to deposit Au NP’s onto seven different supports: titania (TiO2, P25), carbon (C), ferric oxyhydroxide (HFeO2), ceria (CeO2), g-alumina (g-Al2O3) and silica (SiO2). Following the treatment with water, the ‘as-prepared’ catalysts were air-dried in an oven at 100 °C for 24 h. The samples were subjected to analyses by ICP-OES (wt% Au), TEM (average particle size) and BET (surface area, Appendix A). To assess the removal of PVA stabilizer, the amount of residual C on the catalysts were also quantified by combustion analysis. The results are summarised in Table 1.
With the exception of SiO2 (entry 7) deposition of AuNP was achieved with varying degrees of success on the supports. The method successfully reproduced Au/TiO2 with very similar properties as that reported before, with close to 1 wt% loading of Au and average particle sizes of 2.9 nm (entry 1). The method also provided very small Au NP’s on g-Al2O3 (2.6 nm), with a lower 0.6 wt% catalyst loading (entry 2). In contrast, the deposition of Au NP’s on a-Al2O3 was poor, with very low loading (0.3 wt%) and very large particle sizes (entry 3). We attribute this to the small surface area afforded by this material, with limited availability of sites for the pre-formed Au NP’s to adhere to. The supported Au NP’s on the surface are also likely to be in close proximity, which can agglomerate, even under mild thermal treatment conditions, to form large NP’s with very wide size distributions (Figure 3, vide infra). The removal of PVA stabilizer from these three catalysts using the hydrothermal treatment was found to be successful, with <0.1 wt% of carbon remaining.
Preparation of Au/C by deposition-precipitation methods is known to be difficult, due to the hydrophobic nature of carbon and low density of surface OH groups.[9] Using the colloidal method, 0.9 wt% Au loading can be achieved (entry 5). The average particle size (3.4 nm) of the Au NP’s is slightly bigger than that deposited on TiO2 and g-Al2O3, but the particle size distribution is reasonably narrow (Figure 3). This shows that fairly good size control can be achieved by the method onto relatively unfunctionalized supports, as long as there is a large surface area for the NP to deposit onto. In comparison, the availability of surface hydroxyl groups in HFeO2 allowed the target 1 wt% of Au to be achieved (entry 5). Although the average particle size of 3.7 nm was obtained, a wide particle size distribution between 2-10 nm was found (Figure 3), suggesting substantial agglomeration has occurred during the drying process, likely to be also due to the poor distribution of NP on the small surface area. Preparation of Au/CeO2 using the colloidal method was previously described to produce Au NP’s of around 3 nm.[14] In this work, we were able to obtain a 0.8 wt% of Au on CeO2, but we were not able to establish the particle sizes due to poor contrast between Au and the dense support on the TEM grid. For the two catalysts supported on HFeO2 and CeO2, 0.4 and 0.5 wt% of residual carbon can be found, signifying that the hot water treatment was not effective for the removal of PVA from these catalysts. Subsequently, these catalysts were calcinated at 200 °C under 5% H2-N2 to remove the residual stabilizer. The other Au catalyst supported on a reducible metal oxide–Au/TiO2—was also subjected to the same thermal treatment to provide a comparison.
2.2. Catalyst activity
The catalytic activities of the Au catalysts for the H-auto transfer reactions were subsequently assessed. Using benzyl alcohol (1) and aniline (2) as model substrates, the evaluation was conducted in parallel under the same conditions (Scheme 3). Apart from the expected product 3, the reaction mixture may also contain reaction intermediates imine 4, and benzaldehyde 5. Competitive formation of toluene (6) as a side-product is also possible, but this can escape detection as it is often employed as a reaction solvent; in this case, the quantification of 6 can be achieved by using 2-methyl-2-butanol as a solvent.
The catalytic results are presented in Table 2. Turnover frequencies (TOF) were used to compare the catalyst activities to account for the different amount of Au deposited on each support. The performance of Au/TiO2 prepared by the colloidal method mirrors earlier results obtained with commercially available Au/TiO2,[8] with very good selectivity for the expected product 3 (entry 1). Au/g-Al2O3 also performed very well, with very similar outcomes (entry 2). In contrast, the corresponding catalyst supported on the a-allotrope was practically inactive (entry 3), which is perhaps unsurprising, given the much larger particle sizes.
On the other hand, while Au supported on activated carbon and HFeO2 both afforded similar average NP’s of 3.4 and 3.7 nm (Table 1, entries 4 and 5), the former is inactive, compared to the moderate turnover obtained with Au/HFeO2; even in the presence of residual PVA (Table 2, entries 4 and 5). The observation seems to validate the earlier report that the nature of the support has a significant effect on catalyst activity.[11] It is also interesting to see that the formation of the imine intermediate 4 was observed as the major product for the catalyst supported on activate carbon compared to the other metal oxides, which also suggests that the availability of surface hydroxyl groups are important for the H-transfer necessary to convert 4 to 3. To test this further, H2 temperature-programmed desorption (H-TPD) studies were performed with the different Au catalysts. By multiplying the peak integration value from H-TPD with the desorption temperature, the approximate binding energies can be calculated by multiplying the peak integration values from H-TPD analysis by the peak desorption temperatures. The values were subsequently plotted against the observed amine selectivity (Table 3 and Figure 2). This result reveals that a binding energy above 10 mV K-1 is required to maintain a high selectivity (>95%) for the secondary amine 3.
Thermal treatment of Au catalysts supported on reducible metal oxides (including TiO2, HFeO2 and CeO2) a reducing atmosphere is known to induce strong-metal-support interactions which can affect the catalyst activity, either in a positive or negative way.10 Conversely, thermal treatment of Au catalysts prepared by the colloidal method is also found to be very susceptible to agglomeration under thermal conditions, leading to reduced catalyst activity in CO oxidation reactions.[13] In the present study, calcination resulted in three different observations: The calcination of Au/TiO2 led to a decrease in catalyst activity, but did not affect the selectivity for amine 3 (Table 2, entries 1 vs 7), while the calcination of Au/FeO2 increased the catalytic activity but did not affect the selectivity (Table 2, entry 5 vs 8). Last but not least, the calcination of CeO2 led to deterioration of both the reactivity and selectivity (entries 6 and 9). These results show that calcinating these catalysts (with corresponding changes in particle size) does not lead to any improvements in the selectivity of the process.
Indeed, agglomeration of the Au NP’s may also occur during the catalytic turnover conditions. Following reactions at 180 ⁰C to 30 minutes, the catalysts were recovered and subject to TEM analyses (Table 4 and Figure 3). In all cases, Au particle sizes increased by between 16-29% for the metal oxide supports (Entries 1, 2, 3 and 5). In contrast, recovered Au/C catalyst was found to be 1.5 times bigger than the ‘as-prepared’ catalyst (entry 4), with a very dramatic changes in particle distribution (Figure 3). From this, we can surmise that one of the main roles of the (metal oxide) support is to maintain the particle size of the Au NP during the catalytic reaction.
3. Materials and Methods
3.1. General
Unless otherwise stated, all chemical precursors, solvents and standards employed in this work were procured commercially and used without further purification. TiO2 support (P25-Degussa) was employed in this work, containing 25% anatase and 75% rutile phases, and all other catalyst supports were provided by Johnson Matthey plc. The conversion of substrates to products was monitored using a HP6890 Gas Chromatograph, equipped with a H2 flame ionisation detector and an HP5 Agilent column (30 m × 320 μm × 0.25 μm). The percentage of conversion and selectivity was determined by comparison with known standards, using calibration plots and 4-tert-butylphenol (50 mM in methanol) as an external standard. TEM images were captured at the Harvey Flowers Electron Microscopy Suite at Imperial College London, using a JEOL 2010 TEM instrument operated at 200 kV, with a probe current of 108 µA, and a Gatan Orios camera. XRD analysis was conducted at Johnson Matthey plc using a Bruker AXS D8 diffractometer. ICP-OES analysis was conducted at Johnson Matthey plc using a Perkin Elmer Optima instrument. Chemisorption studies (H-TPD) were performed at Johnson Matthey plc using Altamira AMI-200 apparatus.
3.2. Preparation of Catalysts
Preparation of Au/support:[13] A colloidal solution of Au was prepared by addition of poly(vinyl)alcohol (PVA, Mw 9,000-10,000, 80% hydrolysed, 0.1 wt%) to HAuCl4.3H2O (0.50 mmol of Au) solution in 500 mL of H2O. A freshly prepared solution of aq. NaBH4 (13 mM) was then added to the mixture to form a dark brown sol. After 30 min, the colloidal solution was added to the requisite support (e.g. TiO2) with vigorous stirring (500 rpm). The resulting material was collected by filtration and exhaustively washed with deionised H2O. The catalyst was dried in an oven at 100 ̊C for 24 h, before it was transferred into a round bottom flask, and heated in deionised H2O at 90 ̊C with vigorous stirring (500 rpm) for 2 h to remove the PVA.13 The resulting solid was collected, washed thoroughly with deionised H2O and dried in an oven at 100 ̊C for 24 h.
Thermal treatments (calcination): The ‘as-prepared’ catalysts (above) were reduced under a flow of N2/H2 (200 mL/min), using a Carbolite STF tube furnace, respectively. A temperature ramp of 10 °C/min-1 up to 200 °C was applied, held for 2 h, before cooling to room temperature.
3.3. Catalyst screening
An Endeavor® catalyst screening system (Biotage) was employed in this part of the work. The reactor consists of 8 parallel reaction vessels with glass inserts (working volume 5 mL). Each reaction vessel was charged with a catalyst (0.9 mol% Au, average particle size 190 μm), and 2 mL stock solutions of aniline (0.5 M) and benzyl alcohol (0.5 M) in 2-methyl-2-butanol. The reaction vessels were sealed and purged with 3 cycles of N2, before pressurised to 15 bar. The biphasic mixtures were stirred using paddles (250 rpm) and heated to 180 °C for 30 min. Having cooled to room temperature, the reaction aliquots were extracted, diluted and analysed using a HP 6890 Gas Chromatograph equipped with an FID detector and an Agilent HP5 column (30 m × 320 μm × 0.25 μm). 1 μL of analyte solution was injected into the inlet, which was heated to 250 °C with a split ratio of 5:1. The system was operated under a constant pressure of 20 psi with an initial column temperature of 50 °C, held for 0.5 min, heated to 65 °C @2.5 °C/min, and finally to 200 °C @25 °C/min. The conversion of benzyl alcohol and selectivity of imines, amines and other intermediates were calculated using known standards and 4-tert-butylphenol (50 mM in methanol) as an external standard. The benzyl alcohol conversion and product selectivity were calculated as follows:
4. Conclusions
In this work, attempts were made to deposit pre-formed Au NP’s onto six different supports using the colloidal method, followed by a mild hydrothermal treatment to remove the PVA stabilizer and drying (temperatures of <100 ⁰C). The resultant material was found to contain varying amounts of Au (from 0-1 wt%) and the size and distribution of Au NP’s are largely dependent on the available surface area, which has a pronounced effect on the agglomeration of deposited NP’s, even under the mild conditions. The catalytic activity was found to be predominantly dominated by particle size—calcination of the reducible metal oxides did not lead to any significant enhancement in catalytic turnover. Agglomeration of Au during the catalytic turnover is highly dependent on the nature of the support, and this will also account for the different catalyst activity. Last but not least, a direct correlation between the H2 binding efficiency and amine selectivity can be observed, which can be used as an important selection criterion for future catalyst design and development.
Author Contributions
The research work was performed by Luka V. Hare (née Tallon) in partial fulfilment of her PhD degree. K.H., J.C., P.E. and K.K.H. participated in the design of experiments, provision of lab space, resources and interpretation of results. F.P. produced the first draft and produced additional figures for the paper, which was subsequently refined by K.H., J.C., P.E. and K.K.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by EPSRC and Johnson Matthey plc (EPSRC industrial CASE award, ref: 11220222). F.P. was funded by the EPSRC’s UK Catalysis Hub (“Catalysis for the Circular Economy and Sustainable Manufacturing”, EP/R027129/1).
Data Availability Statement
All the research data generated from this study are contained within the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
The surface area of Au/TiO2 was in good agreement with the reported value of 47 m2 g-1 of commercial Au/TiO2 (Strem Au AUROliteTM). The values for Au/Al2O3 (γ and α), Au/CeO2 and Au/HFeO2 were in agreement with the values reported in the literature.15-18 The surface area for Au/C is lower than expected for activated charcoal, which may be due to the residual PVA on the surface.
References
- Podyacheva, E.; Afanasyev, O. I.; Vasilyev, D. V.; Chusov, D. Borrowing Hydrogen Amination Reactions: A Complex Analysis of Trends and Correlations of the Various Reaction Parameters. ACS Catal. 2022, 12, 7142–7198. [Google Scholar] [CrossRef]
- Wang, X. Z.; Wang, H. L.; Shi, F. Alcohol Amination for N-Alkyl Amine Synthesis with Heterogeneous Catalysts. Prog. Chem. 2020, 32, 162–178. [Google Scholar] [CrossRef]
- Hameury, S.; Bensalem, H.; Vigier, K. D. Sustainable Amination of Bio-Based Alcohols by Hydrogen Borrowing Catalysis. Catalysts 2022, 12, 1306. [Google Scholar] [CrossRef]
- Corma, A.; Navas, J.; Sabater, M. J. Advances in One-Pot Synthesis through Borrowing Hydrogen Catalysis. Chem. Rev. 2018, 118, 1410–1459. [Google Scholar] [CrossRef] [PubMed]
- Reed-Berendt, B. G.; Polidano, K.; Morrill, L. C. Recent advances in homogeneous borrowing hydrogen catalysis using earth-abundant first row transition metals. Org. Biomol. Chem. 2019, 17, 1595–1607. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A. S.; Afanasyev, O. I.; Chusov, D. Borrowing hydrogen amination: Whether a catalyst is required? J. Catal. 2022, 413, 1070–1076. [Google Scholar] [CrossRef]
- Porcheddu, A.; Chelucci, G. Base-Mediated Transition-Metal-Free Dehydrative C-C and C-N Bond-Forming Reactions from Alcohols. Chem. Rec. 2019, 19, 2398–2435. [Google Scholar] [CrossRef] [PubMed]
- Zotova, N.; Roberts, F. J.; Kelsall, G. H.; Jessiman, A. S.; Hellgardt, K.; Hii, K. K. Catalysis in flow: Au-catalysed alkylation of amines by alcohols. Green Chem. 2012, 14(1), 226–232. [Google Scholar] [CrossRef]
- Corma, A.; Garcia, H. Supported Gold Nanoparticles as Catalysts for Organic Reactions. Chem. Soc. Rev. 2008, 37, 2096–2126. [Google Scholar] [CrossRef] [PubMed]
- Pu, T.; Zhang, W.; Zhu, M. Engineering Heterogeneous Catalysis with Strong Metal–Support Interactions: Characterization, Theory and Manipulation. Angew. Chem. Int. Ed. 2023, 62, e202212278. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Takamura, R.; Takei, T.; Akita, T.; Haruta, M. Support effects of metal oxides on gold-catalyzed one-pot N-alkylation of amine with alcohol. Appl. Catal. 2012, (413-414), 261-266. [CrossRef]
- Quinson, J.; Kunz, S.; Arenz, M. Surfactant-Free Colloidal Syntheses of Precious Metal Nanoparticles for Improved Catalysts. ACS Catal. 2023, 13, 4903–4937. [Google Scholar] [CrossRef]
- Lopez-Sanchez, J. A.; Dimitratos, N.; Hammond, C.; Brett, G. L.; Kesavan, L.; White, S.; Miedziak, P.; Tiruvalam, R.; Jenkins, R. L.; Carley, A. F. Facile removal of stabilizer-ligands from supported gold nanoparticles. Nat. Chem. 2011, 3, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Menegazzo, F.; Signoretto, M.; Fantinel, T.; Manzoli, M. Sol-immobilized vs deposited-precipitated Au nanoparticles supported on CeO2 for furfural oxidative esterification. J. Chem. Technol. Biotechnol. 2017, 92, 2196–2205. [Google Scholar] [CrossRef]
- Karpenko, A.; Leppelt, R.; Plzak, V.; Cai, J.; Chuvilin, A.; Schumacher, B.; Kaiser, U.; Behm, R. J. Influence of the catalyst surface area on the activity and stability of Au/CeO2 catalysts for the low-temperature water gas shift reaction. Top. Catal. 2007, 44, 183–198. [Google Scholar] [CrossRef]
- R. M. Cornell, U. Schwertmann, The Iron Oxides: Structure, Properties, Reactions, Occurences and Uses, Wiley, 2003. [CrossRef]
- Yalamaç, E.; Trapani, A.; Akkurt, S. Sintering and microstructural investigation of gamma–alpha alumina powders. Eng. Sci. Technol. Int. J. 2014, 17, 2–7. [Google Scholar] [CrossRef]
- Trueba, M.; Trasatti, S. P. γ-Alumina as a Support for Catalysts: A Review of Fundamental Aspects. Eur. J. Inorg. Chem. 2005, 2005, 3393–3403. [Google Scholar] [CrossRef]
- Nijkamp, M. G.; Raaymakers, J. E. M. J.; van Dillen, A. J.; de Jong, K. P. Hydrogen storage using physisorption—materials demands. Appl. Phys. A 2001, 72, 619–623. [Google Scholar] [CrossRef]
Scheme 1.
Common synthetic routes for transforming primary amines to secondary amines.
Scheme 2.
Alkylation of amine by an alcohol by the H-borrowing mechanism.
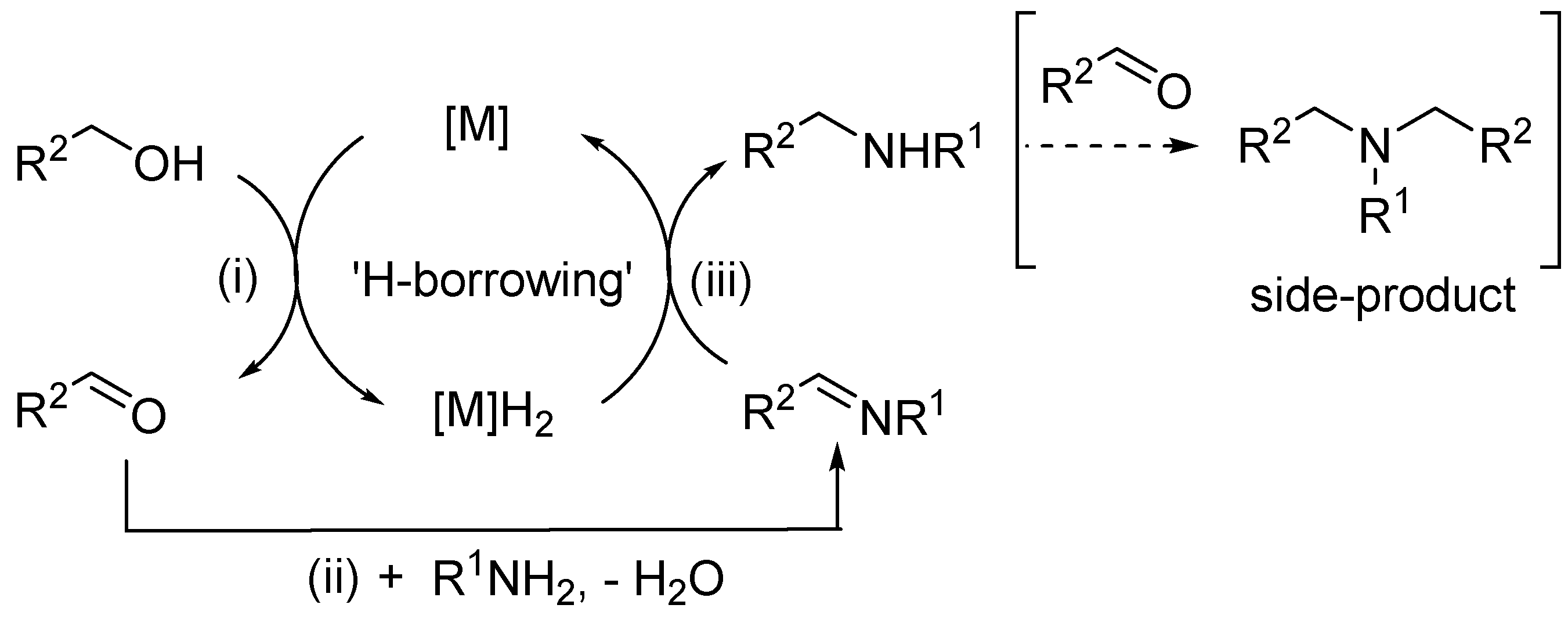
Figure 1.
Colloidal synthesis and hydrothermal treatment of Au/TiO2.
Scheme 3.
The reaction of benzyl alcohol with aniline under catalytic conditions.
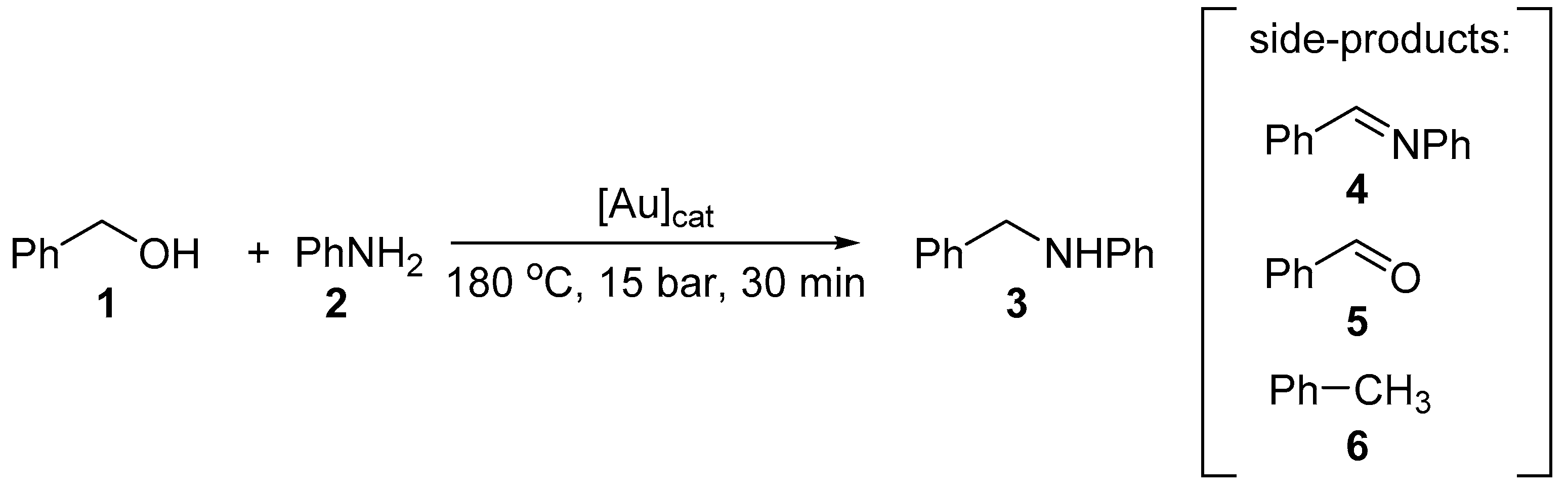
Figure 2.
Binding energy versus selectivity for amines for the ‘as prepared’ Au supported catalysts.
Figure 2.
Binding energy versus selectivity for amines for the ‘as prepared’ Au supported catalysts.
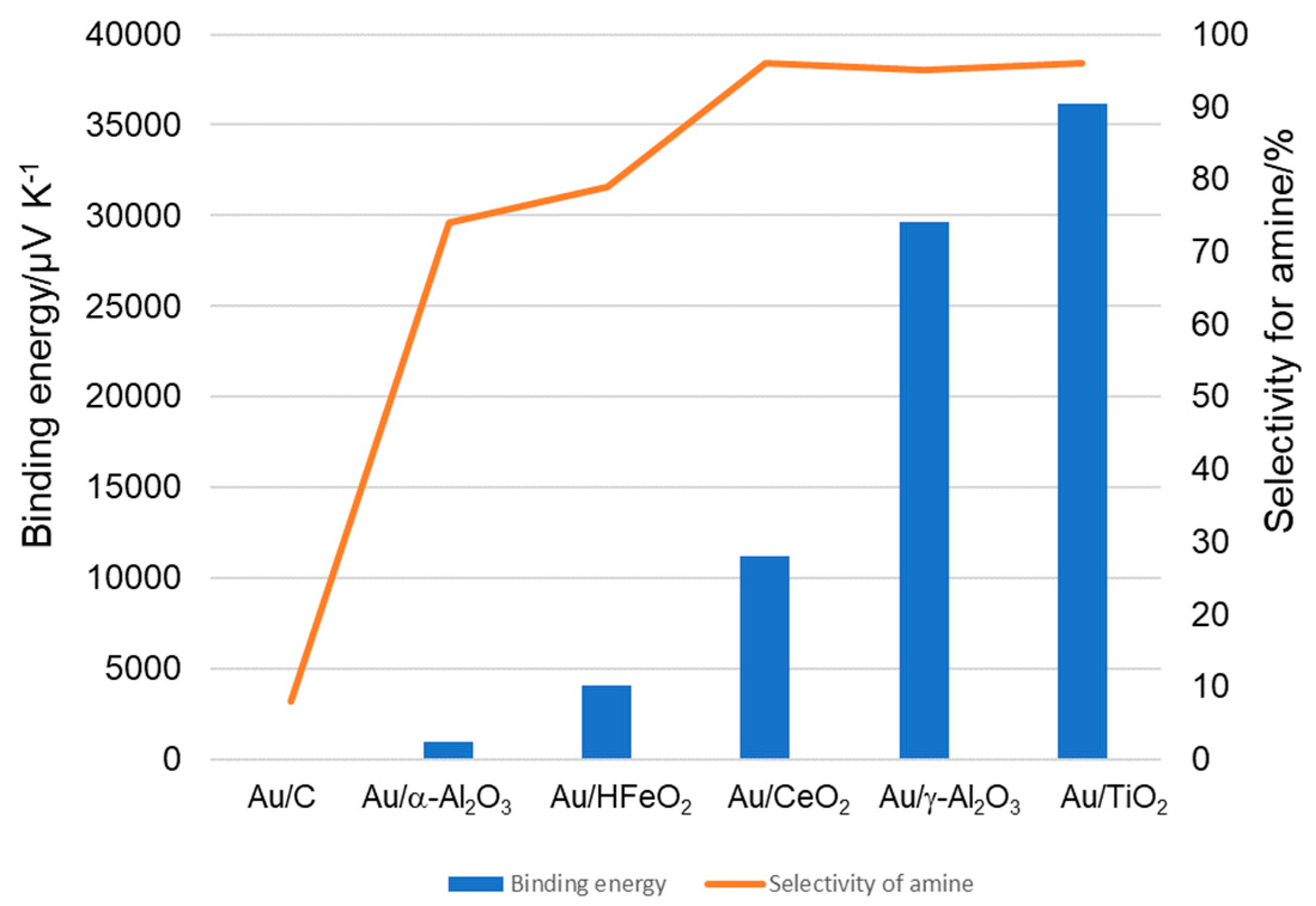
Figure 3.
Particle distribution graphs of Au/support before and after catalysis.
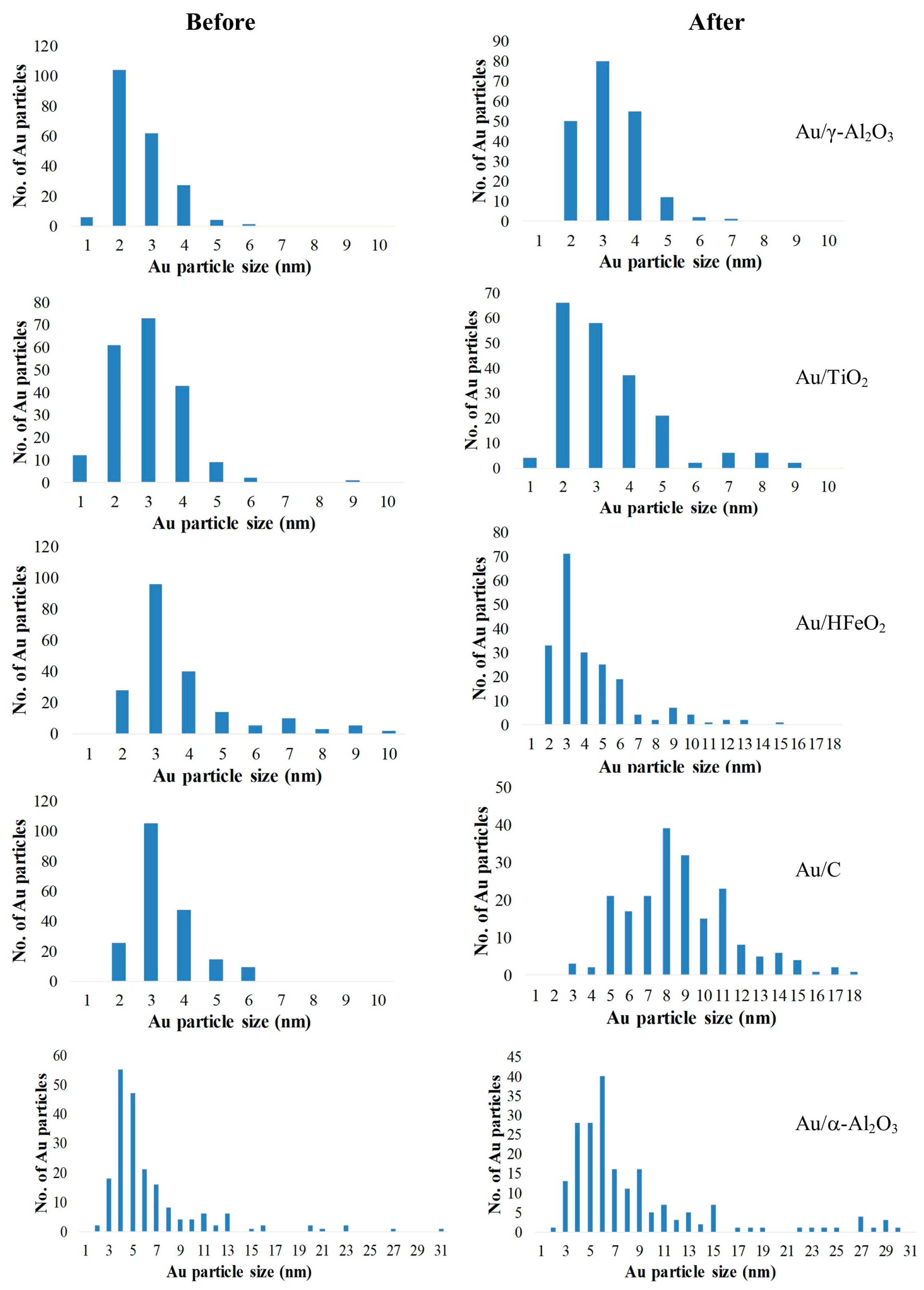

Table 1.
Physical properties of Au catalysts (‘as-prepared’).
| Entry | Catalyst | Au loading[a]/wt% |
Av. Part. size[b]/ /nm | Surface area[c] /m2g-1 | Carbon/wt% |
|---|---|---|---|---|---|
| 1 | Au/TiO2 | 0.9 | 2.9 | 55 (±0.20) | <0.1 |
| 2 | Au/g-Al2O3 | 0.6 | 2.6 | 141 (±0.20) | <0.1 |
| 3 | Au/a-Al2O3 | 0.3 | 6.5 | 13 (±0.01) | <0.1 |
| 4 | Au/C | 0.9 | 3.4 | 689 (±7.00) | n.d. |
| 5 | Au/HFeO2 | 1.0 | 3.7 | 16 (±0.03) | 0.4 |
| 6 | Au/CeO2 | 0.8 | n.d.[d] | 242 (±1.25) | 0.5 |
| 7 | Au/SiO2 | 0.0 | - | - | n.d. |
1 Determined by ICP-OES. bDetermined by TEM (average of 200 particles), particle distribution graphs are provided in FIgure 3. cDetermined by BET. [d]Not determined due to poor contrast.
Table 2.
Catalytic performance of Au catalysts in the alkylation of aniline by benzyl alcohol.
| Entry | Catalyst | Thermal treatment | %Conversion1 | TOF2/h-1 | Selectivity 3:4:5:6 (%) |
|---|---|---|---|---|---|
| 1 | 0.9% Au/TiO2 | ‘as-prepared’ | 80 | 172 | 96:3:0:1 |
| 2 | 0.6% Au/γ-Al2O3 | ‘as-prepared’ | 84 | 181 | 95:4:1:1 |
| 3 | 0.3% Au/α-Al2O3 | ‘as-prepared’ | 6 | 13 | 24:0:1:14 |
| 4 | 0.9% Au/C | ‘as-prepared’ | 1 | 2 | 8:64:1:14 |
| 5 | 1% Au/HFeO2 | ‘as-prepared’ | 413 | 88 | 79:16:2:3 |
| 6 | 0.8% Au/CeO2 | ‘as-prepared’ | 423 | 90 | 96:2:2:0 |
| 7 | 0.9% Au/TiO2 | 5% H2-N2, 200 °C | 66 | 142 | 95:3:0:1 |
| 8 | 1% Au/HFeO2 | 5% H2-N2, 200 °C | 54 | 116 | 81:12:0:6 |
| 9 | 0.8% Au/CeO2 | 5% H2-N2, 200 °C | 28 | 60 | 82:13:1:4 |
1Determined by GC using 4-tert-butylphenol as an external standard, based on benzyl alcohol. Results are an average of 2 runs (error ± 3%). 2TOF (h-1) (moles of benzyl alcohol converted/moles of Au). 3Residual organic material remaining on material (see Table 1). 4Poor mass balance (of products) observed due to the low conversion.
Table 3.
Comparison of binding energy versus selectivity for amine for the Au supported catalysts.
| Catalyst | Binding energy[a] (μV K-1) | Selectivity of amine/% |
|---|---|---|
| Au/C (COL) | 0 | 8 |
| Au/α-Al2O3 (COL) | 928 | 74 |
| Au/HFeO2 (COL) | 4108 | 79 |
| Au/CeO2 (COL) | 11231 | 96 |
| Au/γ-Al2O3 (COL) | 29610 | 95 |
| Au/TiO2 (COL) | 36136 | 96 |
[a] Binding energy = mmol H2 per gram cat * peak integration.
Table 4.
Average particle sizes before and after the reaction.
| Entry | Catalyst | ‘as-prepared’/nm | Recovered/nm | Change/% |
|---|---|---|---|---|
| 1 | 0.9% Au/TiO2 | 2.9 | 3.4 | +17 |
| 2 | 0.6% Au/γ-Al2O3 | 2.6 | 3.2 | +23 |
| 3 | 0.3% Au/α-Al2O3 | 6.5 | 8.4 | +29 |
| 4 | 0.9% Au/C | 3.4 | 8.7 | +155 |
| 5 | 1% Au/HFeO2 | 3.7 | 4.3 | +16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



