
Submitted:
22 August 2023
Posted:
23 August 2023
You are already at the latest version
Abstract
Background: The mechanisms underlying the effect of estradiol (E2) on low-density lipoprotein cholesterol (LDL-C) levels are not completely understood, although a role for proprotein con-vertase subtilisin/kexin type 9 (PCSK9) has been proposed. We aimed to investigate the association between levels of E2, PCSK9 and lipid parameters in premenopausal women undergoing in vitro fertilization (IVF). Methods: Healthy women undergoing IVF in the Department of Obstetrics and Gynecology of the University General Hospital of Ioannina were recruited. Levels of E2, pro-protein convertase subtilisin/kexin type 9 (PCSK9), total cholesterol (TCHOL), high-density lip-oprotein cholesterol (HDL-C), LDL-C, and triglycerides (TGs) were measured 10 days after ovarian depression (E2min) and 7 days after ovarian stimulation (E2max). Results: A total of 34 consecutive healthy women aged 38 (26-46) years old who underwent a full IVF cycle were included. E2 levels increased by 329.6% from E2min to E2max (108 [47-346] to 464 [241-2471] pg/mL, p<0.05). At the same time, serum PCSK9 levels significantly decreased by 30.8% (245 ± 80 to 170 ± 64 ng/mL, p<0.05). No significant changes in TCHOL, LDL-C, TGs and HDL-C were found (-0.4%, -3.8%, -2.2%, and +5.3%, p >0.05). Conclusions: The increase in endogenous E2 production during an IVF cycle was associated with a significant decrease in serum PCSK9 levels during a 7-day period.
Keywords:
estrogens
; proprotein convertase subtilisin/kexin type 9
; lipoprotein
; low-density lipoprotein cholesterol
; cholesterol
; lipids
; in vitro fertilization
1. Introduction
The epidemiology, pathophysiology, and outcomes of atherosclerotic cardiovascular disease (ASCVD) differ significantly between women and men [1]. This could be attributed to differences in gene expression and regulation of sex hormones resulting into different signaling pathways involved in atherosclerosis, including nitric oxide signaling, myocardial remodeling under stress, glucose regulation and lipid metabolism [1,2,3]. Indeed, premenopausal women have lower plasma low-density lipoprotein cholesterol (LDL-C) compared with post-menopausal ones and men [4]. LDL particles are removed from the circulation mainly through hepatic uptake via the LDL receptor (LDLR) [5]. The expression of LDLR is regulated at the transcriptional level by the sterol regulatory element binding protein 2 (SREBP-2), and at the post-translational level mainly by proprotein convertase subtilisin/kexin-type 9 (PCSK9), an inducible degrader of the LDLR [5]. Circulating PCSK9 binds to LDLR, leading to lysosomal degradation within cells, thus resulting in reduced LDLR expression on the cell membrane, decreased LDL catabolism, and increased plasma LDL-C levels [6].
Estrogens increase LDLR expression through estrogen receptor a (ERa), which inhibits the negative feedback regulation of cholesterol synthesis by the SREBP-2 pathway [7,8,9,10,11,12]. Studies in rats and humans have shown that high levels of endogenous estrogens reduce serum PCSK9, suggesting that estrogens also increase the number of LDLRs on hepatic cells through a post-transcriptional mechanism [13,14,15]. Indeed, PCSK9 levels are lower in women than in men, and higher in postmenopausal compared with premenopausal women [16]. In experimental studies, high-dose ethinylestradiol induced a 4-fold increased LDLR gene expression combined with a 50% suppression of hepatic PCSK9 gene expression [14].
We aimed to investigate the short-term effect of endogenous estradiol (E2) increase on serum PCSK9 and lipid levels. To this end, we established a human model to determine the relationship between changes of E2 levels with those of PCSK9 and lipids following ovarian suppression and stimulation during an in-vitro fertilization (IVF) cycle.
2. Materials and Methods
2.1. Study design
This was a prospective study of women who were scheduled for IVF at the Department of Assisted Reproduction at the University General Hospital of Ioannina, Ioannina, Greece. All subjects gave informed consent to participate, and the study protocol was approved by the Ethics Committee of the University General Hospital of Ioannina.
To induce controlled ovarian stimulation, endogenous estrogens were first suppressed using the gonadotropin releasing hormone agonist, leuprolide acetate. Leuprolide acetate was administered subcutaneously at a dosage of 1 mg/day starting on the 21st day of the menstrual cycle. Fourteen days later an E2 measurement (E2min) was carried out to verify suppression. Subsequently, leuprolide acetate dose was decreased to 0.5 mg/day and ovarian stimulation was initiated by subcutaneous administration of recombinant human follicle-stimulating hormone, follitropin beta, at a dosage of 250 IU/day. The response was followed up by an E2 measurement 7 days later (E2max) and by ultrasound scanning of the ovarian follicles 5, 8 and 10 days after the first follicle-stimulating hormone injection (Figure 1).
Serum levels of E2, PCSK9, total cholesterol (TC), high-density lipoprotein cholesterol (HDL-C), LDL-C and triglycerides (TGs) were measured at 2 time points: E2min and E2max.
2.2. Collection and analysis of blood samples
Blood samples were collected in EDTA tubes and centrifuged for 15 minutes at 1000 x g within 30 minutes of collection to separate the serum, which was stored at -80 °C. All laboratory determinations were carried out after a 12-h overnight fast.
Free E2 was measured using routine clinical assays (Estradiol Reagent Kit, Alinity, Abbott) (detection limit, 150 pg/mL). Apolipoprotein (ApoB)-depleted plasma was isolated after the sedimentation of all ApoB-containing lipoproteins with dextran sulfate-magnesium chloride as previously described and stored at -80 oC. TC and HDL-C were determined enzymatically in plasma and ApoB-depleted plasma, respectively, using commercially available kit (Human diagnostics, reference number 10028; Wiesbaden, Germany) according to the manufacturer’s instructions with slight modifications. Briefly, 5 μL total plasma samples or 10 μL ApoB-depleted plasma, and standards by varying volume (0-20 μL of 200 mg/dL cholesterol standard reagent) were added into each well of the microplate (Costar 3370, Corning, NY, USA) and mixed with working reagent (cholesterol enzyme and substrates mixture) in a 300 μL final volume. The reaction was performed for 10 min at 37 oC. The optical density of each well was measured at 510 nm using a microplate reader (Tecan infinite 200 PRO). Total plasma and ApoB-depleted plasma (HDL-rich content) cholesterol of the samples were calculated (mg/dL) using standard curve. Plasma TGs were determined using commercially available kit (Human diagnostics, reference number 10724; Wiesbaden, Germany) according to the manufacturer’s instructions with slight modifications. Briefly, 5 μL total plasma samples, and standards by varying the volume (0-20 μL of 200 mg/dL TGs standard reagent) were added into each well of the microplate (Costar 3370 Corning, NY, USA) and mixed with working reagent at a 300 μL final volume. The reaction was performed for 10 min at 37 oC. The optical density of each well was measured at 510 nm using a microplate reader (Tecan infinite 200 PRO). Plasma TGs (mg/dL) were calculated using standard curve. Samples and standards were assayed in duplicated. LDL-C was calculated by using the Friedewald formula (LDL-C=TC – TGs/5 – HDL-C) [if TGs were <400 mg/dL (4.52 mmol/L)]. The Human PCSK9 Quantikine ELISA kit (R&D Systems, cat. no. DPC900, Minneapolis, MN) was used to measure PCSK9 according to manufacturer’s instructions. Briefly, a monoclonal antibody specific for human PCSK9 was pre-coated on a microplate (ready to use). Fifty (50) µL of the standards and samples were pipetted into the wells and the plate was incubated for 2h at room temperature, where any PCSK9 present was bound to the immobilized antibody. Then, each well was aspirated and washed 3 times with cleaning buffer (400 µL/well). After washing away unbound conjugates, an enzyme-linked polyclonal antibody specific for human PCSK9 was added to the wells (200 µL per well) and the plate was incubated for 2h at room temperature. The plate was then washed to remove any unbound antibody-enzyme reagent and 200 µL of substrate solution was added to the wells and incubated for 30 min at room temperature. The color develops in proportion to the amount of bound PCSK9 at the initial stage. Color development was stopped (blue) by adding 50 µL stop solution and the intensity of the yellow color was measured using a microplate reader set at 450 nm. A standard curve was created by plotting the mean absorbance for each standard concentration on the ordinate against. Data could be linearized by plotting the log of human PCSK9 concentrations against the log of optical density (OD) and the best fit could be determined by regression analysis. The minimum detectable dose (MDD) of human PCSK9 ranges from 0.030-0.219 ng/mL and the mean MDD is 0.096 ng/mL. Intra- and inter-test CVs are 6.1 and 5.9%, respectively.
2.3. Statistical analysis
Continuous numeric variables are expressed as mean ± standard deviation (SD) or median (interquartile: IQR) if Gaussian or non-Gaussian distributed, respectively. Correlations between parameters were evaluated using Pearson’s and Spearman’s correlation coefficients. Paired sample t-test (parametric and non-parametric) was performed to investigate the change in numeric variables during follow-up. Two-tailed significance was defined as p<0.05. As this is a pilot study, no official power calculations were performed. The analyses were performed with the Statistical Package for Social Sciences (SPSS) v25.0 software (SPSS Statistics for Windows, Version 28.0. Armonk, New York, NY, USA: IBM Corp).
3. Results
Thirty-four consecutive healthy women aged 38 (26-46) years old with a median body-mass index 22.8 (19.1-26.7) kg/m2 were included in the present study. All women were non-smokers and not taking any medication. For all participants this was the first IVF cycle after a 12-month period of regular unprotected sexual intercourse not achieving pregnancy. All women were childless and had no miscarriages or abortions in the past.
Table 1 shows the changes in E2, PCSK9 and lipid levels between E2min and E2max time points.
As expected, E2 levels increased by almost 330% (p <0.05). PCSK9 levels were significantly reduced by 30.8% between these 2 time points (p<0.05). ΔPCSK9 was significantly correlated with ΔE2 (beta=-0.162, p=0.001). No significant changes were noted in subjects’ lipid levels; TCHOL, TGs and LDL-C levels decreased by 0.4%, 2.2% and 3.8%, respectively, (p >0.05), whereas HDL-C levels increased by 5.3% (p= >0.05). No significant correlations were found between changes in E2, and TC, LDL-C, TGs and HDL-C levels.
4. Discussion
In this study we demonstrated that short-term (7 days) elevation of endogenous E2 during an IVF cycle was associated with a significant reduction in PCSK9 levels and non-significant alterations in lipid profile.
Estrogen increases LDLR expression through nuclear receptor estrogen receptor-a (ERa) [7,8,9,10,11,12]. Li et al. by using HepG2 cells that express functional ERa and LDLR promoter constructs, have demonstrated that the complex of ERa with transcription factor Sp1 binds to the LDLR promoter, and induces LDLR transcription [12]. Emerging data suggest an additional effect of estrogens on lipid metabolism. Persson et al. showed that E2 decreases PCSK9 transcription, and thus contributes to elevated LDLR expression in rats [14]. It has also been shown that the G protein-coupled estrogen receptor (GPER) mediates the effect of estrogen on LDL-C via a non-genomic, post-transcriptional effect [17,18,19]. Of interest, estrogens upregulate LDLR levels mainly through GPER activation, which prevents PCSK9-dependent LDLR degradation in HepG2 cells [20]. In another study, Starr et al. showed that PCSK9 is required for post-transcriptional upregulation of LDLR by E2 in human hepatic HuH7 cells [21]. Furthermore, studies in rats and humans found that high levels of endogenous estrogens reduce serum PCSK9, suggesting that estrogens also increase the number of LDLRs on hepatic cells through a post-transcriptional mechanism [13,14,15]. Moreover, Persson et al. reported that high-dose treatment with ethinylestradiol reduced SREBP-2 mRNA levels by 55%, and PCSK9 expression by half [14].
A prolonged estrogen exposure may induce more pronounced effects οn lowering LDL-C levels, such as those demonstrated in studies with long-term estrogen replacement therapy (ERT) [22,23,24,25,26]. One study showed that a 2-month estrogen alone replacement therapy in postmenopausal women (n=184) was associated with a decrease in LDL-C by 8-9% and increase in HDL-C levels by 9% [27]. Another study of 90 hysterectomized and oophorectomized females, oral and transdermal ERT induced a decline in TC levels and LDL-C levels within 6 months, and an elevation in HDL-C and TGs [26].
In contrast to ours, a previous short-term study (6 days stimulation of estrogen synthesis) including 31 women who underwent IVF, showed that TCHOL, LDL-C and PCSK9 levels were significantly reduced by 14.0%, 20.2% and 14.0 %, respectively, while HDL-C levels remained unchanged [28]. This discrepancy could be due to the short duration and limited sample size of both studies. Of interest, E2 increase was lower in our study in comparison with that of Persson et al (+330% vs +3720%). This difference in endogenous E2 synthesis between two studies could be due to the use of gonadotropin nasal spray in Persson’s study instead of subcutaneous gonadotropin in our study [28].
Major limitations of this study are small sample, short-term duration and the lack of a control group.
5. Conclusions
Short-term increase of E2 during an IVF cycle is associated with a decrease of serum PCSK9 but no significant alteration in plasma lipids. Larger studies could elucidate this association and its clinical implication. T
Author Contributions
Conceptualization, K.Z. and E.L.; methodology, C.T.; validation, A.P., G.A. and F.B.; formal analysis, A.P and G.A.; writing—original draft preparation, A.P., G.A., F.B. and C.T.; writing—review and editing, K.Z. and E.L.; supervision, E.L. All authors have read and agreed to the published version of the manuscript.”
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Ethics Committee of the General Hospital of Ioannina, Greece (12985/24 May 2017).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data is unavailable due to privacy and local ethical restrictions.
Acknowledgments
We thank Dr Elefteria Deska MD, PhD and the Atherothrombosis Research Center/ Biochemistry Laboratory, Department of Chemistry, University of Ioannina.
Conflicts of Interest
A.P., C.T. and K.Z declare no conflict of interest. G.A. has received personal fees from Amgen and Novo Nordisk, outside the submitted work. FB has received honoraria and personal fees from Amgen, Novartis, Novo Nordisk and Viatris, outside the submitted work. E.L. reports personal fees and non-financial support from Amgen, personal fees from Servier, personal fees from Boehringer-Ingelheim, personal fees and non-financial support from AstraZeneca, personal fees from MSD, personal fees from Lilly, personal fees and non-financial support from Bayer, personal fees from Novartis, personal fees from Chiesi, outside the submitted work.
References
- Crea, F.; Battipaglia, I.; Andreotti, F. Sex differences in mechanisms, presentation and management of ischaemic heart disease. Atherosclerosis 2015, 241, 157–168. [Google Scholar] [CrossRef]
- Lu, C.; Donners, M.; Karel, J.; de Boer, H.; van Zonneveld, A.J.; den Ruijter, H.; Jukema, J.W.; Kraaijeveld, A.; Kuiper, J.; Pasterkamp, G.; et al. Sex-specific differences in cytokine signaling pathways in circulating monocytes of cardiovascular disease patients. Atherosclerosis 2023. [Google Scholar] [CrossRef] [PubMed]
- Regensteiner, J.G.; Reusch, J.E.B. Sex Differences in Cardiovascular Consequences of Hypertension, Obesity, and Diabetes: JACC Focus Seminar 4/7. J Am Coll Cardiol 2022, 79, 1492–1505. [Google Scholar] [CrossRef] [PubMed]
- Heiss, G.; Tamir, I.; Davis, C.E.; Tyroler, H.A.; Rifkand, B.M.; Schonfeld, G.; Jacobs, D.; Frantz, I.D., Jr. Lipoprotein-cholesterol distributions in selected North American populations: the lipid research clinics program prevalence study. Circulation 1980, 61, 302–315. [Google Scholar] [CrossRef]
- Gu, H.M.; Zhang, D.W. Hypercholesterolemia, low density lipoprotein receptor and proprotein convertase subtilisin/kexin-type 9. J Biomed Res 2015, 29, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Norata, G.D.; Tavori, H.; Pirillo, A.; Fazio, S.; Catapano, A.L. Biology of proprotein convertase subtilisin kexin 9: beyond low-density lipoprotein cholesterol lowering. Cardiovasc Res 2016, 112, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Ohlsson, C.; Hellberg, N.; Parini, P.; Vidal, O.; Bohlooly, Y.M.; Rudling, M.; Lindberg, M.K.; Warner, M.; Angelin, B.; Gustafsson, J.A. Obesity and disturbed lipoprotein profile in estrogen receptor-alpha-deficient male mice. Biochem Biophys Res Commun 2000, 278, 640–645. [Google Scholar] [CrossRef]
- Ribas, V.; Nguyen, M.T.; Henstridge, D.C.; Nguyen, A.K.; Beaven, S.W.; Watt, M.J.; Hevener, A.L. Impaired oxidative metabolism and inflammation are associated with insulin resistance in ERalpha-deficient mice. Am J Physiol Endocrinol Metab 2010, 298, E304–319. [Google Scholar] [CrossRef]
- Parini, P.; Angelin, B.; Rudling, M. Importance of estrogen receptors in hepatic LDL receptor regulation. Arterioscler Thromb Vasc Biol 1997, 17, 1800–1805. [Google Scholar] [CrossRef] [PubMed]
- Heine, P.A.; Taylor, J.A.; Iwamoto, G.A.; Lubahn, D.B.; Cooke, P.S. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc Natl Acad Sci U S A 2000, 97, 12729–12734. [Google Scholar] [CrossRef]
- Geary, N.; Asarian, L.; Korach, K.S.; Pfaff, D.W.; Ogawa, S. Deficits in E2-dependent control of feeding, weight gain, and cholecystokinin satiation in ER-alpha null mice. Endocrinology 2001, 142, 4751–4757. [Google Scholar] [CrossRef]
- Li, C.; Briggs, M.R.; Ahlborn, T.E.; Kraemer, F.B.; Liu, J. Requirement of Sp1 and estrogen receptor alpha interaction in 17beta-estradiol-mediated transcriptional activation of the low density lipoprotein receptor gene expression. Endocrinology 2001, 142, 1546–1553. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Galman, C.; Rudling, M.; Angelin, B. Influence of physiological changes in endogenous estrogen on circulating PCSK9 and LDL cholesterol. J Lipid Res 2015, 56, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Persson, L.; Galman, C.; Angelin, B.; Rudling, M. Importance of proprotein convertase subtilisin/kexin type 9 in the hormonal and dietary regulation of rat liver low-density lipoprotein receptors. Endocrinology 2009, 150, 1140–1146. [Google Scholar] [CrossRef]
- Seidah, N.G.; Prat, A. The biology and therapeutic targeting of the proprotein convertases. Nat Rev Drug Discov 2012, 11, 367–383. [Google Scholar] [CrossRef]
- Lakoski, S.G.; Lagace, T.A.; Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Genetic and metabolic determinants of plasma PCSK9 levels. J Clin Endocrinol Metab 2009, 94, 2537–2543. [Google Scholar] [CrossRef] [PubMed]
- Hussain, Y.; Ding, Q.; Connelly, P.W.; Brunt, J.H.; Ban, M.R.; McIntyre, A.D.; Huff, M.W.; Gros, R.; Hegele, R.A.; Feldman, R.D. G-protein estrogen receptor as a regulator of low-density lipoprotein cholesterol metabolism: cellular and population genetic studies. Arterioscler Thromb Vasc Biol 2015, 35, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.P.; Lee, J.Y.; Jeong, J.K.; Bae, S.W.; Lee, H.K.; Jo, I. Nongenomic stimulation of nitric oxide release by estrogen is mediated by estrogen receptor alpha localized in caveolae. Biochem Biophys Res Commun 1999, 263, 257–262. [Google Scholar] [CrossRef]
- Chambliss, K.L.; Shaul, P.W. Rapid activation of endothelial NO synthase by estrogen: evidence for a steroid receptor fast-action complex (SRFC) in caveolae. Steroids 2002, 67, 413–419. [Google Scholar] [CrossRef]
- Fu, W.; Gao, X.P.; Zhang, S.; Dai, Y.P.; Zou, W.J.; Yue, L.M. 17beta-Estradiol Inhibits PCSK9-Mediated LDLR Degradation Through GPER/PLC Activation in HepG2 Cells. Front Endocrinol (Lausanne) 2019, 10, 930. [Google Scholar] [CrossRef]
- Starr, A.E.; Lemieux, V.; Noad, J.; Moore, J.I.; Dewpura, T.; Raymond, A.; Chretien, M.; Figeys, D.; Mayne, J. beta-Estradiol results in a proprotein convertase subtilisin/kexin type 9-dependent increase in low-density lipoprotein receptor levels in human hepatic HuH7 cells. FEBS J 2015, 282, 2682–2696. [Google Scholar] [CrossRef]
- Barrett-Connor, E.; Wingard, D.L.; Criqui, M.H. Postmenopausal estrogen use and heart disease risk factors in the 1980s. Rancho Bernardo, Calif, revisited. JAMA 1989, 261, 2095–2100. [Google Scholar] [CrossRef]
- Bush, T.L.; Barrett-Connor, E.; Cowan, L.D.; Criqui, M.H.; Wallace, R.B.; Suchindran, C.M.; Tyroler, H.A.; Rifkind, B.M. Cardiovascular mortality and noncontraceptive use of estrogen in women: results from the Lipid Research Clinics Program Follow-up Study. Circulation 1987, 75, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Krauss, R.M.; Perlman, J.A.; Ray, R.; Petitti, D. Effects of estrogen dose and smoking on lipid and lipoprotein levels in postmenopausal women. Am J Obstet Gynecol 1988, 158, 1606–1611. [Google Scholar] [CrossRef] [PubMed]
- Metka, M.; Hanes, V.; Heytmanek, G. Hormone replacement therapy: lipid responses to continuous combined oestrogen and progestogen versus oestrogen monotherapy. Maturitas 1992, 15, 53–59. [Google Scholar] [CrossRef]
- Nanda, S.; Gupta, N.; Mehta, H.C.; Sangwan, K. Effect of oestrogen replacement therapy on serum lipid profile. Aust N Z J Obstet Gynaecol 2003, 43, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.J.; Jang, H.C.; Cho, D.H.; Min, Y.K. Effects of hormone replacement therapy on lipoprotein(a) and lipids in postmenopausal women. Arterioscler Thromb 1994, 14, 275–281. [Google Scholar] [CrossRef]
- Persson, L.; Henriksson, P.; Westerlund, E.; Hovatta, O.; Angelin, B.; Rudling, M. Endogenous estrogens lower plasma PCSK9 and LDL cholesterol but not Lp(a) or bile acid synthesis in women. Arterioscler Thromb Vasc Biol 2012, 32, 810–814. [Google Scholar] [CrossRef]
Figure 1.
Study design. Abbreviations: E2, estradiol; PCSK9, proprotein convertase subtilisin/kexin type 9; TC, total cholesterol; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; TGs, triglycerides.
Figure 1.
Study design. Abbreviations: E2, estradiol; PCSK9, proprotein convertase subtilisin/kexin type 9; TC, total cholesterol; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; TGs, triglycerides.
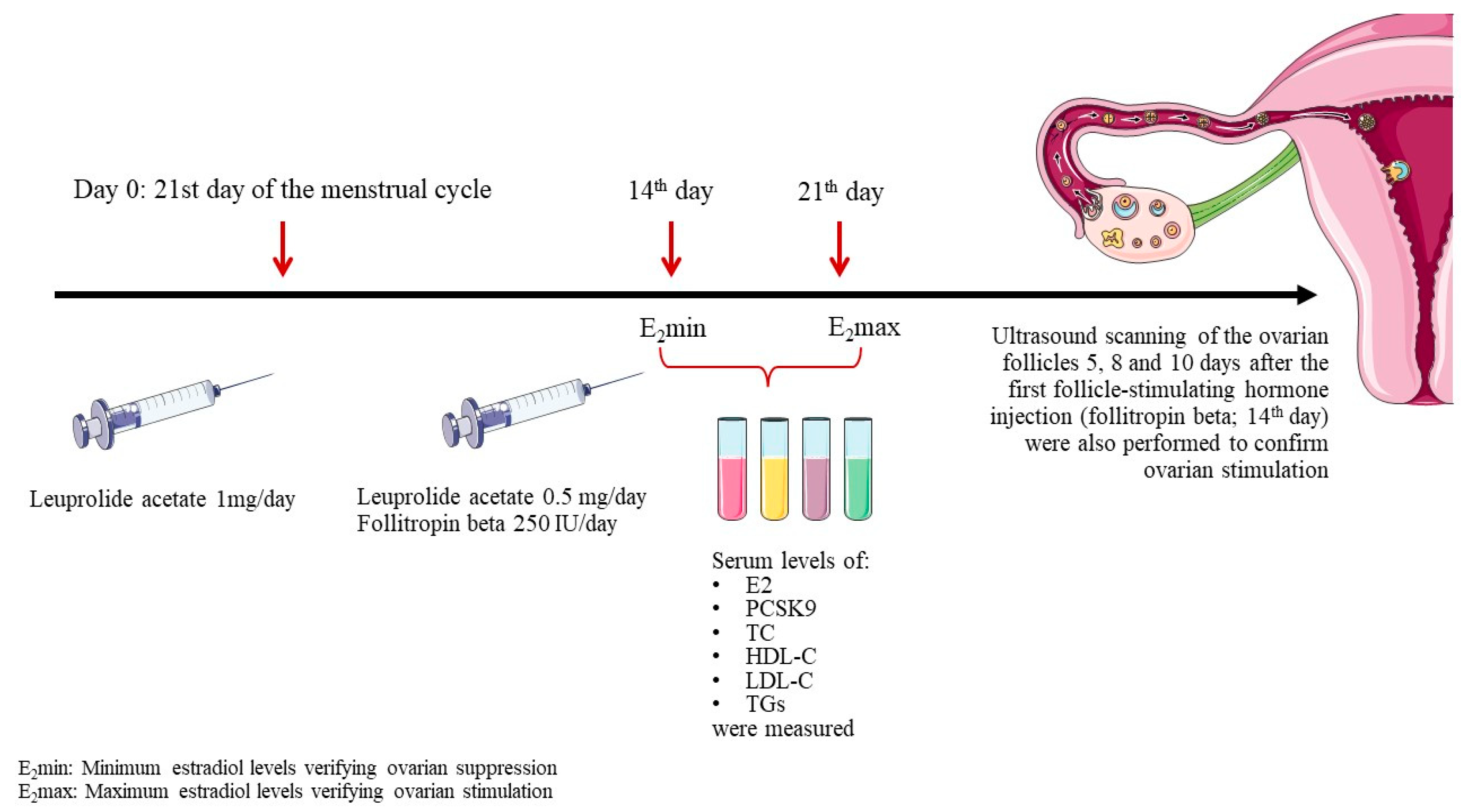

Table 1.
E2, PCSK9 and serum lipids levels of study participants (n=34) at ovarian depression (E2min) and ovarian stimulation (E2max).
Table 1.
E2, PCSK9 and serum lipids levels of study participants (n=34) at ovarian depression (E2min) and ovarian stimulation (E2max).
| E2min | E2max | Change, % | |
|---|---|---|---|
| E2, pg/mL | 108 (47-346) | 464 (241-2471) | + 329.6* |
| ΤC, mg/dL | 164 ± 44 | 163 ± 42 | - 0.4 |
| TGs, mg/dL | 69 ± 36 | 67 ± 24 | - 2.2 |
| LDL-C, mg/dL | 108 ± 43 | 103 ± 42 | - 3.8 |
| HDL-C, mg/dL | 42 ±10 | 45 ± 14 | + 5.3 |
| PCSK9, ng/mL | 245 ± 80 | 170 ± 64 | - 30.8* |
Abbreviations: E2; estradiol, TC; total cholesterol, TGs; triglycerides, LDL-C; low-density cholesterol, HDL-C; high-density cholesterol, PCSK9; Proprotein convertase subtilisin/kexin type 9. Values are expressed as median (interquartile range: IQR) or mean ± standard deviation (SD), *p <0.05 for comparison versus E2min group.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



