
Submitted:
22 August 2023
Posted:
23 August 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
To map Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV 2) spreading and evaluate immune responses variations against this virus, it was essential to locally set up efficient serological tests. The SARS-CoV2 immunogenic proteins were very expensive and not affordable mainly for Low and Middle Income Countries (LMICs). For this purpose, the commonly used antigen, Re-ceptor-Binding Domain (RBD) of Spike S1 protein (S1RBD), was produced using the the Baculovirus Expression Vector System (BEVS) . During the current study, the expression of S1RBD was monitored using western blot under different culture conditions. Different parameters were studied: Multiplicity Of Infection (MOI), cell density at infection and harvest time. Hence, optimal conditions for efficient S1RBD production were identified: MOI 3; cell density at infection 2-3x 106 cells/mL and time post-infection (tpi or harvest time) of 72h and 72-96 h successively for expression in shake-flasks and 7L-bioreactor. A high production yield of S1RBD varying between 4mg and 70 mg per liter of crude cell culture supernatant was achieved, respectively, in shake-flasks and in 7L-bioreactor. Moreover, the produced S1RBD showed an excellent antigenicity potential against COVID-19 patient sera evaluated by western blot. Thus, additional serological assays, such as ELISA, were developed using the purified S1RDB (published by other research groups).
Keywords:
Multiplicity of infection
; cell density at infection
; SARS-CoV-2
; Spike S1 glycoprotein
; Receptor binding domain
; Sf9 cells
1. Introduction
SARS-CoV-2 responsible for coronavirus disease 2019 (COVID-19), is an enveloped virus belonging to the betacoronavirus group of the Coronaviridae family. Its large single positively stranded RNA, of 29891 nucleotides and 9860 amino acids, encodes four major structural proteins: the spike (S), envelope (E), membrane (M) and the nucleocapsid (N) proteins [1]. To date, there is no specific treatment for COVID-19. Despite the newly commercialized vaccines relatively quickly developed, due to the emergency that the situation imposes, the virus continues its propagation globally. The strategies used for all commercial vaccines were based on the immunogenic potential of the trimeric spike glycoprotein (S), composed of two subunits S1 and S2 [2]. The S1 subunit contains the receptor-binding domain (RBD), essential for binding to the peptidase domain of angiotensin converting enzyme 2 (ACE2). The latter is widely expressed in various human organs including lungs, heart, brain and kidney. Thereby, it plays an essential role in virus attachment, fusion and entry into the host cell [3]. On the other hand, the S2 subunit is involved in the fusion of the virus and the host cell membranes [4,5,6,7]. Thanks to genome sequencing, scientists discovered the emergence of new variants even more contagious due to different mutations at the Spike protein level [89]. Indeed, the RBD region, within the spike S1 subunit, is the cornerstone of the virus for its spread through infected cells containing the ACE2 receptor [10]. Moreover, RBD has been reported to be highly immunogenic and mediate protective responses. Therefore, RBD-based vaccines and diagnostic approaches have been extensively developed [3,11,12]. For example, four versions of the spike (S1) protein of SARS-CoV-2 were expressed in Sf9 insect cells, including the RBD. They showed excellent antigenicity againist convalescent COVID-19 patient sera through ELISA and a high neutralization titer in mice [12]. This prompted us to further study the BEVS process parameters, for optimized RBD production. Some influent parameters such as the time of post-infection (tPI), cell density at infection and multiplicity of infection (MOI) were concerned. Sf9 insect cells grown on a commercial animal component free medium (SF900II) were used. In fact, BEVS, first described in the early 1980s, was successfully used for the production of various recombinant proteins [13]. During this study, we investigated the expression of the RBD (His-tagged) of SARS-CoV2 S1 subunit (S1RBD) under different culture conditions. The upstream and downstream process parameters were optimized. This may be of utmost interest for the production of several recombinant proteins with therapeutic and diagnostic potential.
2. Materials and Methods
2.1. Upstream Processing
2.1.1. Construction of the recombinant pFastBac-S1RBD and bacmid generation
The RBD cDNA sequence (residues 319-541) of the SARS-CoV-2 spike S1 subunit (GenBank accession number: QHD43416.1) was cloned into the baculovirus shuttle pFastBac™/ CT-TOPO™ vector (Invitrogen), downstream to the gp67 signal sequence and an His-tag, in the C-terminal extremity. In fact, the RBD constructs were fused with an N-terminal gp67 signal peptide and a C-terminal His6 tag to facilitate extracellular secretion and permit affinity purification, respectively (Figure S1). The recombinant pFASTBac-S1RBD vector was kindly provided by Chris KP Mok from HKU-Pasteur Research Pole, Li Ka Shing Faculty of Medicine, The University of Hong Kong, China (Perera et al., 2020). The generation of recombianant bacmid DNA, containing the RBD sequence, was performed using the Bac-to-BacTM C-His TOPOTM expression system kit following the manual user guidelines (Invitrogen). Briefly, DH10Bac competent cells (Invitrogen) were transformed with the recombinant pFastBac-RBD plasmid to generate the recombinant bacmid DNA, by homologous recombination. Then, twelve white colonies (on LB agar plates containing: 50µg/mL Kanamicin, 7µg/mL gentamicin, 10µg/mL tetracycline, 100µg/mL X-Gal and 40µg/mL IPTG) were screened by PCR. pUC/M13 Forward: 5'-CCCAGTCACGACGTTGTAAAACG-3' and pUC/M13 Reverse: 5'-AGCGGATAACAATTTCACACAGG-3' primers were used. The PCR product was visualized on a 1.5% agarose gel stained with ethidium bromide. Finally, the recombinant bacmid was isolated using the PureLink HiPure Plasmid DNA Miniprep Kit (Invitrogen).
2.1.2. Insect cell culture
Sf9 insect cell line, a cell line derived from the pupal ovary tissue of the Fall Army worm (Spodoptera frugiperda) was used in this study. Sf9 cells were grown in suspension, on SF900II serum free medium (Gibco, catalogue number 11496-015) without antibiotics addition in polycarbonate Erlenmyer flasks (Corning) at 27ºC and 115-120 rpm. The seeding cell density was 0.5 x106 cells/mL and subcultivation frequency was twice a week.
2.1.3. Transfection
Sf9 cells were maintained in exponential growth phase, at a cell density of 1.5-2.5 x 106 cells/mL and viability higher than 95%, in 25cm2 culture flasks containing a total of 2 x 106 cells at a final volume of 5mL. Twenty µL of ExpiFectamineTM Sf transfection reagent were diluted in 500µL medium, in a sterile Eppendorf tube. Then, 2 µg of the recombinant bacmid DNA were added. The mixture was inverted 10 times and incubated for 5 minutes at room temperature to form a DNA-lipid complex. Transfected cells were incubated at 27°C for 4-5 days to generate the recombinant baculoviruses (Figure S2). Untransfected cells cultivated under the same conditions served as a control.
2.1.4. Amplification of baculovirus stocks
When the viability falled to 70-80%, transfected cells were harversted by centrifugation at 3500xg for 10 minutes and the supernatant containing the recombinant baculoviruses (P0 stock) was stored at 4°C ( stable for 6 months) or at -80°C (long term storage). The recombinant P0 baculovirus stock was amplified to obtain a high titer of recombinant virus by adding 0.5 mL of P0 to 20 mL of Sf9 cells (2 x 106 cells/mL) and incubation for 2-4 days until reaching a viability between 70 and 80%. The cells were harvested and the culture supernatant, containing the recombinant baculoviruses (P1 stock), was stored at 4°C for further baculovirus amplification as described in Figure S3.
2.1.5. Baculovirus titration using a cell viability assay (TCID50)
Tissue culture infective dose (TCID50) method was improved by Mena et al., (2003) [14]. In this method, viral titration was performed by measuring cell viability based on the cleavage of the tetrazolium ring of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) by a mitochondrial dehydrogenase in viable cells [15]. A magenta coloration results from that reaction and allows the quantification of viable cells via spectrophotometry. Sf9 cells were grown in Sf900 II medium to reach a cell density of 3x 105 cells/mL. Gentamicin was added to the cell culture at a concentration of 0.05% (vol/vol) to avoid any contamination. Then, 50µL per well of cell culture were added. The virus was diluted from 10-1 to 10-10 in another 96-well plate containing 90µL of medium per well. In fact, serial dilutions (10µL virus in 90µL medium) were performed from line 2 to 11. The plate was incubated at 27°C for 6 days. After that, 10µL of MTT solution (5g/L) were added to each well and the plate was incubated at 27°C for 2 hours under gentle shaking. Afterwards,the plates were centrifuged at 2000xg for 10 min and the supernatant was discarded with a multi-channel pipet. After solubilisation of the magenta salt crystals with Dim2thylsulfoxyde (DMSO), the absorbance was measured at 570 nm with a microplate reader. To calculate the titer (plaque forming units: pfu /mL), data were analyzed using SigmaPlot 12.0 software.
2.1.6. Production of S1RBD in Shake-Flasks
Recombinant baculovirus, P1 stock, was used to infect Sf9 cells and protein expression was monitored, by western blot analysis, at different time points post-infection (0h, 24, 72, 96 and 120h) using two different multiplicities of infection (MOI 1 and 5). Furthermore, protein expression was analysed at additional MOI values (0.01, 0.5, 1, 3, 5, 7 and 10) in Sf9 cells using a cell density at infection of 2 × 106 cells/ml and a viability higher than 95% at 27°C for 72 h post-infection with shaking at 110 rpm. S1RBD protein expression was also analysed using different cell densities at infection (1 x 106, 2 x 106, 3 x 106, 4 x 106, 5 x 106 and 7 x 106 cells/mL) at an MOI of 3.
2.1.7. Production of S1RBD in 7L-controlled bioreactors
Large scale production of S1RBD was carried out in a 7-L bioreactor (BioBraun, Melsungen, Germany) as described by Trabelsi et al. (2006) [16]. It is equipped with a marine impeller and a spin filter (pore size: 22 µm) fixed to the axis. The following conditions were maintained during both cell proliferation and infection phases: no pH regulation by CO2 sparging or addition of NaHCO3 at 88 g/L, dissolved oxygen was adjusted to 50% air saturation by continuous surface aeration. The temperature was maintained at 27 °C and the agitation rate at 125 rpm. Batch culture mode was started with a seeding cell density of 5 × 105 cells/mL. When cell density reached 4x106 cells/mL, the cells were infected with recombinant baculovirus (titer 1.085x109 pfu/mL) at an MOI of 3. After cell infection, samples were taken daily to determine cell density, cell viability, osmolarity and S1RBD expression level.
2.2. Downstream Processing
2.2.1. Tangential flow filtration (TFF)
The infected Sf9 culture supernatants (from erlenmeyer flasks or bioreactor) were harvested by centrifugation at 4500 rpm for 20 min at 4°C. Culture supernatants were then pre-treated by filtration at a flow rate of 20 mL/min through MF-Millipore membrane filter, 8 µm pore size (Milliopre, Catalogue number SCWP14250) . Afterwards, the clarified supernatants were concentrated and buffer exchanged by TFF using cogent microscale System (Merck Millipore, MA, USA) as follows: A MasterFlex peristaltic pump fed the supernatant to a 10 KDa molecular weight cutoff cassette. Two references were tested respectively for small scale (Maximum volume 500 mL) and medium scale (volumes between 500 mL and 5L) experiments: Pellicon XL 50cm2 (Biomax Media; catalogue number PXP010A50; Millipore) and Pellicon 3/ 0.11 2 (ultracel membrane; catalogue number P3C010C01; Millipore). The pre-treated supernatant was first concentrated to 50% of the initial volume and then buffer exchanged with 4 to 5 volumes of 20mM PBS, 0.5M NaCl, 20mM imidazole, pH 7.4. Subsequently, the concentration to the desired level was performed. Finally, the TFF cassette was rinced , regenerated and stored according to manufacturer instructions.
2.2.2. Protein purification
Recombinant His-tagged RBD Spike S1 (S1RBD) protein was purified from TFF retentate based on immobilized metal ion affinity chromatography using the HisTrap FF, 5mL column (catalogue number 17524801; GE Healthcare) following the manufacturer’s procedure by AKTÄ purifier system (GE Healthcare Life Sciences, Uppsala, Sweden). Prior to loading, the samples should be filtrated through 0.22 µm low protein binding filters. We used Sarto Scale 25 (Sartorius, Catalogue number 5235307HV-LX-C) and Sartopore Platinium Capsules (single use, Sartorius, Catalogue number 5491307H4-SO-B) respectively for small scale (maximum 200 mL) and medium scale (200 mL to 1L) assays. The ultra low binding filters were kindly offered by Sartorius, Stedim Biotech, GmbH. The column was first equilibrated with at least 5 column volumes of the binding buffer (20mM PBS, 0.5M NaCl, 20mM imidazole, pH 7.4). Then, the TFF-treated culture supernatant, was applied to the column at a flow rate of 1mL/mL. The flow rate used at all other purification steps was 5mL/min. Afterwards, the column was washed with a minimum of 10 CV binding buffer. The elution of the S1RBD was carried out using the elution buffer (20mM PBS, 0.5M NaCl, 500mM imidazole, pH 7.4). Elution fraction containing the purified S1RBD were pooled, concentrated and buffer exchanged with Vivaspin device 10KDa (Sartorius, ). PD10 desalting colums (GE healthcare) were also tested for buffer exchange (imidazole elimination). A second purification step, using size exclusion chromatography, was performed on a Superdex® 200 Increase 10/300 GL column (GE Healthcare, catalogue number 28990944) according to manufacturer's instructions. The equilibration and elution buffer was 20mM PBS containing 0.5M NaCl pH 7.4. A 0.5 mL protein sample was injected through a 2mL sample loop. Separation was performed at a flow rate of 0.75 mL/min. The eluate was collected into 1 mL fractions using a fraction collector. Protein concentrations of the purified and non-purified samples were determined according to the Bradford microassay protocol [17]with bovine serum albumin (BSA) as standard, using the Quick Start™ Bradford reagent (Bio-Rad, Hercules, USA).
2.2.3. SDS-PAGE
Protein fractions were analyzed on SDS-PAGE 4-12%, Bis-Tris, 0.75 mm, 12-well mini protein gel (Biorad) under reduced (+5% β-mercaptoethanol) and unreduced (without β-mercaptoethanol) conditions to monitor protein size, purity, and dimerization. The gels were visualized by silver staining method using ProteoSilverTM Stain Kit (Sigma-Aldrich) according to the manufacture’s instructions. Visualization using Coomassie Brilliant Blue G-250 (BioRad) was also performed.
2.2.4. Western blot analysis
Samples of 10µg purified S1RBD were analyzed on a 12% SDS-polyacrylamide gel using the Bio-Rad Mini Trans-Blot® cell system. Proteins were transferred to a nitrocellulose membrane (GE Healthcare). The membrane was first saturated using blocking buffer (Phosphate-Buffered Saline: 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4 + 0.05% Tween-20, v/v + 5% skimmed, dry milk, w/v) during 1h at room temperature. Then, the membrane was washed twice with 20 ml PBST for 5 min under gentle agitation. After that, it was incubated with the anti-His(C-term)-HRP antibody (Invitrogen, Life Technologies) diluted 1:5000. For the Western blot immunoassay [adapted from 18; 19], a supplementary addition of convalescent sera from Tunisian patients infected with SARS-Cov2, diluted 1:200 in PBS-T + 2.5% skimmed dry milk (w/v) overnight at 4°C under gentle agitation is also needed. Following, three washing steps with PBS-T were carried out. For detection of His-tagged S1RBD, a signal was detected directly using the Enhanced Chemiluminescence (ECL) kit (GE Healthcare). For the immunoblotting detection of anti-IgG SARS-Cov2 antibodies, the membrane was incubated with the secondary peroxidase-conjugated anti- human IgG (HRP) (Invitrogen) (1:10000) for 1 hour at room temperature. The signal was then detected using the ECL kit.
The current work was carried out within national and international projects. The study protocol was approved by the national ethical committee placed in Institut Pasteur of Tunis (references of ethical approval: 2021/02/I/LR16IPT and 2020/21/I/LR16IPT).
3. Results and discussion
3.1. Small scale productionof the His-tagged S1RBD Spike protein in Sf9 cells
3.1.1. Preliminary assay
A Cell density and cell viability respectively of, 6.18x106 cells/mL and 88% were reached 72h post-infection using a cell density at infection of 2x106 cells/mL. The assay was carried out at an MOI of 5, in a-250mL buffled shake-flask with a working volume of 20mL (Figure 1). We noted that best infection efficiency was obtained with cells in the active (better the centre) exponential growth phase which confirmed the litterarture. For example, Carinhas et al. (2008), explained that a new culture inoculated with cells from the middle exponential phase can achieve a slightly higher cell concentration and protein expression yields than one from the late exponential phase. That could be linked to the higher percentage of active cells (in phase S and G1) in the inoculum [20].
To further optimise the His-tagged S1RBD Spike protein expression in Sf9 cells, we the effect of MOI, cell density at infection and harvest time were studied.
3.1.1. MOI assay
The effect of MOI on the recombinant His-tagged S1RBD expression (detection by western blot analysis using anti-Histidine antibodies) was studied in 250mL buffled shake-flasks at different MOIs ( we started with: 1and 5). As shown in Figure 2, S1RBD protein was detected, in the culture supernatant, only after 48h PI and 24 h PI respectively at MOI 1 and MOI 5, to 96h of post-infection and no signal was detected at120h of post-infection (This was also confirmed in other assays such as Figure S4).
In addition, any significant difference (related to S1RBD expression level at 72h of post-infection) was detected during the increase of working volume from 20 to 500 mL (Figure 2b’ only; this sample was only analyzed by western blot ). Such observation can be considered as a good indicator for a successful scaling up. Morover, better protein expression was reached with an MOI of 3. This refers to the band intensities of the corresponding western blot results. A comparison was carried out with the protein (S1RBD) band intensity obteined at MOIs 1 and 5 and a fixed cell density at infection of 2 x 106 cells/mL (Figure 2). As known for BEVS expression system, MOI’s higher than unity should always be used to maximise protein expression yield [22].
It is of utmost importance here to mention that a low MOI, inferior to 1, is mainly used for virus amplification as it allows a small subset of cells to be infected initially and to produce budded virus. Thus, the infection of Sf9 cells at a low MOI is considered as a key factor in successful virus amplification [23]. .
This prompted us to analyse S1RBD expression, at 72h post-infection, using additional MOI values: 0.01; 0.1; 0.5; 1; 3; 5; 7 and 10. As presented in Figure 3a, higher viral titer (3,08x109 pfu/mL) was observed at low MOI values (0.01; 0.1; 0.5 and 1). The eperiments were carried out similtaneously in shake flasks at a fixed cell density at infection (2x106 cells/mL).
S1RBD was efficiently expressed (72 h PI), in Sf9 cells, at all tested MOI levels (MOI 0.01 was only analyzed by virus titration) with slightly better expression at MOI 3 (Figure 3c). After purification, the protein yield of 4.3 mg and and 1.4 mg S1RBD per liter of crude cell culture supernatant was achieved (Table 1 in supplementary data).
In fact, optimal MOI depends on the cell line, the baculovirus, the medium, the mode of operation, and the physiological state of the cells. MOI affects the protein productivity, the production of defective particles and the time of exposure to proteases [13].
Based on protein yield after purification (line 8, Figure 3b and other data not shown), the MOI 3 was selected for the scale-up and further studies.
3.1.2. Cell density at infection assay
Cell density at infection is also an important parameter to consider while expressing proteins on BEVS system.
Shake flask experiments were carried out using different cell densities at infection. (1 x 106, 2 x 106, 3 x 106, 4 x 106, 5 x 106 and 7 x 106 cells/mL) at a fixed MOI of 3. As shown in Figure 4 , better S1RBD expression levels were observed at cell dencities of 2 x 106 and 3 x 106 cells/mL at 72h post-infection.
Zhang et al., reported that MOI and cells density at infection (CDI) are correlated. They explained that higher productivities were obtained in cultures infected at higher CDI and MOI. Indeed, studies that have addressed the combined effects of CDI and MOI on recombinant protein titers demonstrate that for each CDI there is an optimum MOI for maximum protein production [24].
For scaling-up experiments (bioreactor cultures), we planned to use a minimum CDI of 3x106 cells/mL and an MOI of 3.
3.2. Scale-up of the optimised process
Enhancement of SARS-CoV-2 spike RBD protein production has been extensively studied recently, e.g. in HEK293 cells, due to its high-sensitivity in serological assays [3; 25]. In the current work, we investigated the scalability of the process by producing S1RBD protein in larger working volumes of 500mL and 5L, respectively, in 2L buffled shake-flasks and a 7L stirred bioreactor.
Shake-flask cultures were infected at MOI 3 and a cell density at infection of 2 x 106 cells/mL.
On the other hand, The bioreactor culture was initiated at a starting cell density of 0.5 x 106 cells/mL. After 96h (exponential cell growth phase), the cells reached 4x106 cells/Ml. They were infected at an MOI of 3. In the bioreactor experiments, cell density, cell viability, medium osmolarity, pH and S1RBD expression level were monitored until 120h post-infection.
The highest cell density was 5.55 x 106 cells/mL at 24h post-infection (Figure 5 a). Cell viability decreased, to approximately 92-95% by 96h post-infection and then dropped to 51% at 120h post-infection (Figure 5b). Interestingly, increased expression of S1RBD was observed in the bioreactor at 96h and 120h post-infection, compared to shake-flask experiments where respectively low and no expression levels were detected 96 and 120h PI(Figure 5c).
As described by Thompson et al. [26], the time of harvest may depend on the MOI.
3.3. Downstream process
Purification of S1RBD started with cell harvest. The crude culture supernatant was then clarified by centrifucation (for low volumes) and 8µm or depth filters (for large volumes). Then, TFF was performed (buffer exchange and concentration as described in the Materials and Methods section). The culture supernatants were then concentrated at least 10 -fold to reach a final volume of 150mL and 30mL corresponding to a total protein concentration of 0.32mg/mL and 1.11 mg/mL, respectively (Table 1). Afterwards, The S1RBD protein, tagged at its C-terminus with hexa-histidine, was purified, in a two -step purification, using HisTrap FF column with a high purity of more than 95% estimated on silver staining SDS-PAGE 12%, to allow a more sensitive detection of impurities. Finally, polishing and imidazole salt elimination were performed with Superdex Peptide or PD10 columns for small volumes (Figure 6a, line E6). One step-purification followed by Vivaspin buffer exchange was also successful.
Eluted fractions were then pooled and protein levels were quantified using a colorimetric assay (Bradford). The one-step purification protocol results on an average yield of 4.45+0.15 mg per liter of crude cell supernatant (Table 1), which is about at least 2 fold higher than the yield obtained in HEK293SF cells using non-viral and viral production approaches [3]. The purified S1RBD was analyzed by western blot (Figure 6b). A lower purification yield of S1RBD protein was also obtained using the one-step HisTrap FF column purification (Figure 6c, lanes E3, E4 and E5). Therefore, the elution fractions from E3 to E10 were pooled, concentrated (MWCO 10 kDa) using Vivaspin centrifugal concentrators and quantified. A volume of 0.5mL, with a protein concentration of 1,55mg/mL, was injected into the AKTA protein purification system for a second purification step by size exclusion chromatography. Thus, a higher purification efficiency was observed after analysis on silver stain SDS-PAGE 12% (Figure 6d, lane F18). The two-step purification protocol leads to an average yield of 1.4 mg per liter of cell culture (Table 1), which is approximately 1.5 times higher than the yield obtained in HEK293SF cells [3]. The lower yield obtained with the two-step purification protocol could be explained by the protein loss during the procedure.
Interestingly, for clarified (8µm membrane) 7L-culture harvests, the single-step purification method, results in a yield of at least 70 mg S1RBD per liter of crude supernatant. The corresponding band (27 kDa) was detected by SDS-PAGE 12% and confirmed by Western Blot (Table 1; Figure 4d). The obtained production yield was about 50 times higher than the yield obtained using a 2L shake flasks. This proves to the effectiveness of the upstream and downstream process.
Using different cell expression hosts, Expi293F™, higher yields were obtained for RBD (approx. 90 mg/L) 3 days post transfection. Such yield was only achieved by HEK293-E6 cells when the culture was extended two extra days (harvesting at Day 5 post-transfection) [27].
Dimerization of the purified S1RBD was verified by non-reducing SDS-PAGE analysis and as expected, two bands appeared in the line in which the protein sample was not reduced with 2-mercaptoethanol; a dominant band with a Molecular weight of 27 kDa and a less intense band with a Mw of about 54 kDa (Figure 7).
In the control lines where the samples were reduced in the presence of 2-mercaptoethanol, a distinct single band was observed. These results corroborate those reported by Tee et al. (2020) [28] who showed two bands of the same molecular weight for the unreduced RBD protein produced in HEK cells.The S1RBD sequence (residues 319-541) has four disulfide bonds (C336-C361, C379-C432, C391-C525 and C480-C488) and a free C538 that may be involved in intermolecular dimerization [7].
3.4. Immunoassays of the His-tagged S1RBD Spike protein
To demonstrate the antigenicity of S1RBD against anti-IgG-SARS-CoV2 antibodies, a Western blot was developed using Tunisian convalescent COVID-19 patient sera. Indeed, the anti-IgG antibodies recognized the purified S1RBD protein, and Wetern blot analysis clearly showed a band with a molecular weight of 30KDa corresponding to S1RBD in both sera 5 and 6 of the convalescent COVID-19 patients, although the band was more pronounced inpatient 5, which could be explained by a higher IgG anti-Sars-CoV2 anibody titer (Figure 8).
Recently, recombinant SRBD protein produced in Sf9 insect cells was reported to have excellent antigenicity with convalescent COVID-19 patient sera [28; 12]. We also observed the presence of a second band with a higher molecular weight of about 52KDa, which could correspond to a dimer of S1RBD, despite the reduced conditions used and the slight difference in molecular weight compared to S1RBD dimer (~60KDa) tested under non-reduced conditions (Figure 7) [29]. BEVS expression vector system expressed S1RBD produced in SF9 cells has excellent antigenic potential as reported by Li et al. [12].
It has also been shown that RBD dimerization induced high levels of RBD-binding and SARS-CoV-2 neutralizing antibody in both mice and non-human primate [30].
The purified S-RBD protein was used by colleagues to perform national and international ELISA tests and it’s activity was successfully assessed.The ELISA data were recently published [31-33].
While this coronavirus outbreak continues to spread, many studies focused on conserved epitopes that may allow structure-based design not only of a SARS-CoV-2 vaccine but also of cross-protective antibody responses against future coronavirus epidemics and pandemics [34].
4. Conclusions
In this study, the entire upstream and downstream process for the production of the RBD spike S1 protein of Sars-CoV2 was characterized using Sf9 insect cells using the baculovirus vector expression system (BEVS). A purification yield higher than 4 and 70 mg S1RBD per liter of crude culture supernatant was achieved respectively in shake flasks and stirred bioreactor. We demonstrated that this expression system was efficient for the overproduction of immunologically active S1RBD, towards IgG SARS-CoV2 antibody, with a high potential for use in serological assays such as ELISA. These optimized production processes, using the baculovirus-insect cell system, will be of interest for other studies aimed at the overproduction of recombinant proteins for therapeutic or diagnostic purposes.
The current results, protein production using insect cells-BEVS, were obtained in bioreactor operated in batch mode. The MOI and CDI were selected after small-scale testing in shake flasks. The BEVS is widely used for the manufacture of biologics, including vaccines for humans and animals, therapeutics, gene therapy vectors, and biopesticides.
This work was directed by national priorities during COVID-19 outbreak. The choose of BEVS expression system was based on availability of know-how and cost effectiveness.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Figure S1: Map of the recombinant pFASTBac-S1RBD vector.; Figure S2: Schematic representation of the preparation of the recombinant bacmid (a) and transfection of Sf9 cells (b); Figure S3: Schematic representation of the amplification process of recombianant baculovirus (a) and S1RBD protein (b); Figure S4: S1RBD expression in Sf9 cells. Total proteins from the culture supernatant were analysed at different time points post infection (tPI) with MOI 1 (a) and MOI 5 (b). The assays were analysed by Coomassie-stained SDS-PAGE 12% and Western blot. The cell density at infecton was 2 x 106 cells/mL in buffled shake-flask. Equal amounts of total proteins (10 μg) were loaded onto the SDS-PAGE gel and Table 1: Supplementary data related to downstream process.
Author Contributions
M.B., A.C., M.B.Z., I.A.,HA, S.S, KT and S.R. carried out the experiments; S.R and M.B. performed the data analysis and wrote the manuscript; Conceptualization, K.T. and SR; methodology, SR; validation, SR and KT; formal analysis, SR; investigation, SR, KT and MB; resources, M.B.A. and C.BA.; data analysis, M.B., A.C., KT and SR; writing original draft preparation, M.B. and SR; Review & editing, S.R.; visualization, S.R., C.B.; supervision, S.R.. Project administration, S.R., C.B. and M.B.A.; funding acquisition, C.B., KT and M.B.A.
Funding
This study was funded, next to Institut Pasteur of Tunis (IPT) resources, by: 1. The « URGENCE COVID-19 » fundraising campaign of Institut Pasteur (special thanks to Pr Hechmi Louzir and Dr Sinda Zarrouk). 2. TheTunisian ministry of higher education and scientific research; federated research projects (PRF: Projets de Recherche Fédérés); COVID-19 R&I program; Fight, Predict, Act: Federated COVID-19 Strategy; PRFCOVID19-D5P1” registered under the reference 2020/21/I/LR16IPT. 3. Institut Pasteur International Network (IPIN); project “EASI”: ELISA Assays development for SARS-COV2 within Institut Pasteur International Network (IPIN). This collaborative project is based on the tripartite agreement signed between Institut Pasteur de Tunis: IPT; University of Hong Kong - Pasteur Research PoleHong Kong: HKU and Institut Pasteur of Paris: IP (We are involved in Work package 2). 4. The French Ministry for Europe and Foreign Affairs via the project “REPAIR; International Pasteurian Research in Response to Coronavirus in Africa” registered under the reference 2021/02/I/LR16IPT.
Acknowledgments
The authors sincerely thank Drs Nicholas Wu (University of Illinois at Urbana) and Chris Ka Pun Mok From the HKU-Pasteur Research Pole, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong SAR, China, for providing us the recombinant pFastBac-S1RBD plasmid used in this study. They are also grateful for Prs Hechmi Louzir (General Director of IPT) and Mohamed Koussay Dellagi, director for scientific foresight of the international network of pasteur institutes for securing funds and for their encouragement.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. The Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Herrera, N.G.; Morano, N.C.; Celikgil, A.; Georgiev, G.I.; Malonis, R.J.; Lee, J.H.; Tong, K.; Vergnolle, O.; Massimi, A.B.; Yen, L.Y.; et al. Characterization of the SARS-CoV-2 S Protein: Biophysical, Biochemical, Structural, and Antigenic Analysis. ACS Omega 2021, 6, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Farnós, O.; Venereo-Sánchez, A.; Xu, X.; Chan, C.; Dash, S.; Chaabane, H.; Sauvageau, J.; Brahimi, F.; Saragovi, U.; Leclerc, D.; et al. Rapid High-Yield Production of Functional SARS-CoV-2 Receptor Binding Domain by Viral and Non-Viral Transient Expression for Pre-Clinical Evaluation. Vaccines 2020, 8, 654. [Google Scholar] [CrossRef]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Tortorici, M.A.; Snijder, J.; Xiong, X.; Bosch, B.-J.; Rey, F.A.; Veesler, D. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. PNAS 2017, 114, 11157–11162. [Google Scholar] [CrossRef] [PubMed]
- Millet, J.K.; Whittaker, G.R. Physiological and molecular triggers for SARS-CoV membrane fusion and entry into host cells. Virology 2018, 517, 3–8. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; Qin, H.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, 1284–1294.e9. [Google Scholar] [CrossRef]
- Rees-Spear, C.; Muir, L.; Griffith, S.A.; Heaney, J.; Aldon, Y.; Snitselaar, J.L.; Thomas, P.; Graham, C.; Seow, J.; Lee, N.; et al. The impact of Spike mutations on SARS-CoV-2 neutralization. bioRxiv 2021.01.15.42 6849. [CrossRef]
- Xie, Y.; Karki, C.B.; Du, D.; Li, H.; Wang, J.; Sobitan, A.; Teng, S.; Tang, Q.; Li, L. Spike Proteins of SARS-CoV and SARS-CoV-2 Utilize Different Mechanisms to Bind With Human ACE2. Front. Mol. Biosci. 2020, 7. [Google Scholar] [CrossRef]
- Liu, Z.; Xu, W.; Xia, S.; Gu, C.; Wang, X.; Wang, Q.; Zhou, J.; Wu, Y.; Cai, X.; Qu, D.; et al. RBD-Fc-based COVID-19 vaccine candidate induces highly potent SARS-CoV-2 neutralizing antibody response. Signal Transduction and Targeted Therapy 2020, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zheng, Q.; Yu, H.; Wu, D.; Xue, W.; Xiong, H.; Huang, X.; Nie, M.; Yue, M.; Rong, R.; et al. SARS-CoV-2 spike produced in insect cells elicits high neutralization titres in non-human primates. Emerg Microbes Infect 2020, 9, 2076–2090. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Gómez, A.; Sánchez-Mirón, A.; García-Camacho, F.; Molina-Grima, E.; Chisti, Y. Protein production using the baculovirus-insect cell expression system. Biotechnol Prog 2014, 30, 1–18. [Google Scholar] [CrossRef]
- Mena, J.A.; Ramírez, O.T.; Palomares, L.A. Titration of non-occluded baculovirus using a cell viability assay. Biotechniques 2003, 34, 260–262. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Trabelsi, K.; Rourou, S.; Loukil, H.; Majoul, S.; Kallel, H. Optimization of virus yield as a strategy to improve rabies vaccine production by Vero cells in a bioreactor. J Biotechnol 2006, 121, 261–271. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Liang, F.-Y.; Lin, L.-C.; Ying, T.-H.; Yao, C.-W.; Tang, T.-K.; Chen, Y.-W.; Hou, M.-H. Immunoreactivity characterisation of the three structural regions of the human coronavirus OC43 nucleocapsid protein by Western blot: Implications for the diagnosis of coronavirus infection. J Virol Methods 2013, 187, 413–420. [Google Scholar] [CrossRef]
- Faburay, B.; Wilson, W.; McVey, D.S.; Drolet, B.S.; Weingartl, H.; Madden, D.; Young, A.; Ma, W.; Richt, J.A. Rift Valley Fever Virus Structural and Nonstructural Proteins: Recombinant Protein Expression and Immunoreactivity Against Antisera from Sheep. Vector Borne Zoonotic Dis 2013, 13, 619–629. [Google Scholar] [CrossRef]
- Carinhas, N.; Bernal, V.; Yokomizo, A.Y.; Carrondo, M.J.T.; Oliveira, R.; Alves, P.M. Baculovirus production for gene therapy: the role of cell density, multiplicity of infection and medium exchange. Appl Microbiol Biotechnol 2009, 81, 1041–1049. [Google Scholar] [CrossRef]
- Radford, K.M.; Cavegn, C.; Bertrand, M.; Bernard, A.R.; Reid, S.; Greenfield, P.F. The indirect effects of multiplicity of infection on baculovirus expressed proteins in insect cells: secreted and non-secreted products. Cytotechnology 1997, 24, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Irons, S.L.; Chambers, A.C.; Lissina, O.; King, L.A.; Possee, R.D. Protein Production Using the Baculovirus Expression System. Curr Protoc Protein Sci 2018, 91, 5–5. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.T.; Peter, C.H.; Greenfield, P.F.; Reid, S.; Nielsen, L.K. Low multiplicity infection of insect cells with a recombinant baculovirus: The cell yield concept. Biotechnol Bioeng 1996, 49, 659–666. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Enden, G.; Merchuk, J.C. Insect cells-Baculovirus system: Factors affecting growth and low MOI infection. Biochemical Engineering Journal 2005, 27, 8–16. [Google Scholar] [CrossRef]
- Mehalko, J.; Drew, M.; Snead, K.; Denson, J.-P.; Wall, V.; Taylor, T.; Sadtler, K.; Messing, S.; Gillette, W.; Esposito, D. Improved production of SARS-CoV-2 spike receptor-binding domain (RBD) for serology assays. Protein Expression and Purification 2021, 179, 105802. [Google Scholar] [CrossRef]
- Thompson, C.M.; Montes, J.; Aucoin, M.G.; Kamen, A.A. Recombinant Protein Production in Large-Scale Agitated Bioreactors Using the Baculovirus Expression Vector System. Methods Mol Biol 2016, 1350, 241–261. [Google Scholar] [CrossRef]
- Castro, R.; Nobre, L.S.; Eleutério, R.P.; Thomaz, M.; Pires, A.; Monteiro, S.M.; Mendes, S.; Gomes, R.A.; Clemente, J.J.; Sousa, M.F.Q.; et al. Production of high-quality SARS-CoV-2 antigens: Impact of bioprocess and storage on glycosylation, biophysical attributes, and ELISA serologic tests performance. Biotechnology and Bioengineering. 2021, 118, 2202–2219. [Google Scholar] [CrossRef] [PubMed]
- Tee, K.L.; Jackson, P.J.; Scarrott, J.M.; Jaffe, S.R.P.; Johnson, A.O.; Johari, Y.; Pohle, T.H.; Mozzanino, T.; Price, J.; Grinham, J.; et al. . Purification of recombinant SARS-CoV-2 spike, its receptor binding domain, and CR3022 mAb for serological assay. bioRxiv 2020. [Google Scholar] [CrossRef]
- Perera, R.A.; Mok, C.K.; Tsang, O.T.; Lv, H.; Ko, R.L.; Wu, N.C.; Yuan, M.; Leung, W.S.; Chan, J.M.; Chik, T.S.; et al. Serological assays for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), March 2020. Eurosurveillance 2020, 25, 2000421. [Google Scholar] [CrossRef]
- An, Y.; Li, S.; Jin, X.; Han, J.B.; Xu, K.; Xu, S.; Han, Y.; Liu, C.; Zheng, T.; Liu, M.; et al. A tandem-repeat dimeric RBD protein-based covid-19 vaccine zf2001 protects mice and nonhuman primates. Emerg Microbes Infect. 2022, 11, 1058–1071. [Google Scholar] [CrossRef]
- Kharroubi, G.; Cherif, I.; Ghawar, W.; Dhaouadi, N.; Yazidi, R.; Chaabane, S.; Snoussi, M.-A.; Salem, S.; Ben Hammouda, W.; Ben Hammouda, S.; et al. Incidence and risk factors of SARS-CoV-2 infection among workers in a public health laboratory in Tunisia. Arch Virol 2023, 168, 69. [Google Scholar] [CrossRef] [PubMed]
- Cherif, I.; Kharroubi, G.; Chaabane, S.; Yazidi, R.; Dellagi, M.; Snoussi, M.A.; Salem, S.; Marzouki, S.; Kammoun Rebai, W.; Rourou, S.; et al. COVID-19 in Tunisia (North Africa): Seroprevalence of SARS-CoV2 in the General Population of the Capital City Tunis. Diagnostics 2022, 12, 971. [Google Scholar] [CrossRef] [PubMed]
- Benabdessalem, C.; Ben Hamouda, W.; Marzouki, S.; Faye, R.; Mbow, A.-A.; Diouf, B.; Ndiaye, O.; Dia, N.; Faye, O.; Sall, A.-A.; et al. Development and comparative evaluation of SARS-CoV-2 S-RBD and N based ELISA tests in various African endemic settings. Diagnostic Microbiology and Infectious Disease (2023). 115903. [CrossRef] [PubMed]
- Yuan, M.; Wu, N.C.; Zhu, X.; Lee, C.-C.D.; So, R.T.Y.; Lv, H.; Mok, C.K.P.; Wilson, I.A. A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science 2020, 368, 630–633. [Google Scholar] [CrossRef]
Figure 1.
Monitoring of Cell density (a) and viability (b) during 72 hours post-infection (PI). The preliminary assay was carried out in a 125mL shake buffled shake-flask with a working volume of 20mL, a cell density at infection of 2 x 106 cells/mL and an MOI of 5. The dark bar refers to infected flask and the light bar to blank (non infected flask). The assay was repeated 3 times and the bar errors indicate the mean standard deviation.
Figure 1.
Monitoring of Cell density (a) and viability (b) during 72 hours post-infection (PI). The preliminary assay was carried out in a 125mL shake buffled shake-flask with a working volume of 20mL, a cell density at infection of 2 x 106 cells/mL and an MOI of 5. The dark bar refers to infected flask and the light bar to blank (non infected flask). The assay was repeated 3 times and the bar errors indicate the mean standard deviation.
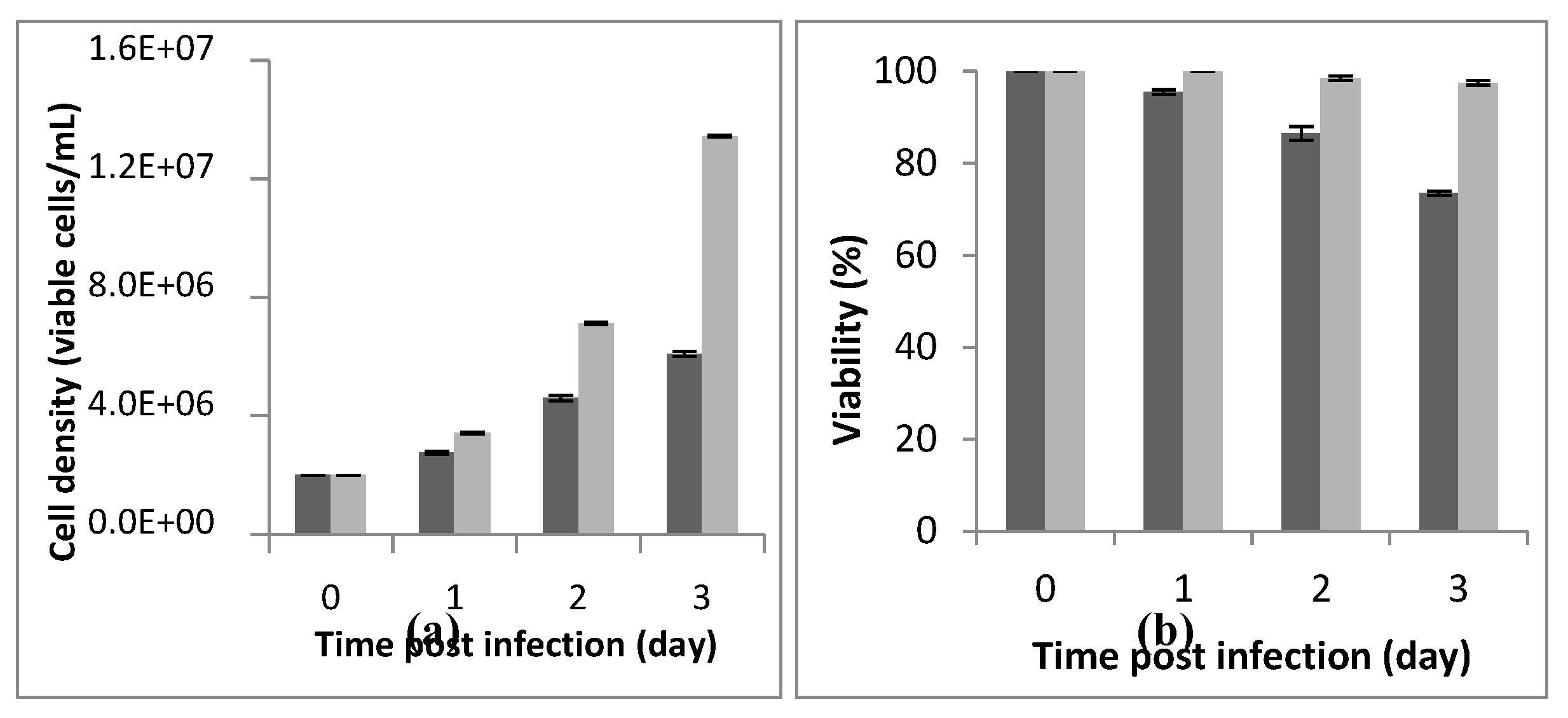
Figure 2.
Monitoring S1RBD protein expression on Sf9 cells grown in erlenmeyer flasfs. The cells were infected at 2x106 cells/mL at MOI 1 (a) and MOI 5 (b). Supernatents harvested at different times post infection (tPI) were analysed by Coomassie staining 12% SDS-PAGE and Western blot. Working volume increase (from 20 to 500 mL) effect was also assessed in buffled shake-flasks. Equal amounts of total proteins (10 μg) were loaded onto the SDS-PAGE gel.
Figure 2.
Monitoring S1RBD protein expression on Sf9 cells grown in erlenmeyer flasfs. The cells were infected at 2x106 cells/mL at MOI 1 (a) and MOI 5 (b). Supernatents harvested at different times post infection (tPI) were analysed by Coomassie staining 12% SDS-PAGE and Western blot. Working volume increase (from 20 to 500 mL) effect was also assessed in buffled shake-flasks. Equal amounts of total proteins (10 μg) were loaded onto the SDS-PAGE gel.
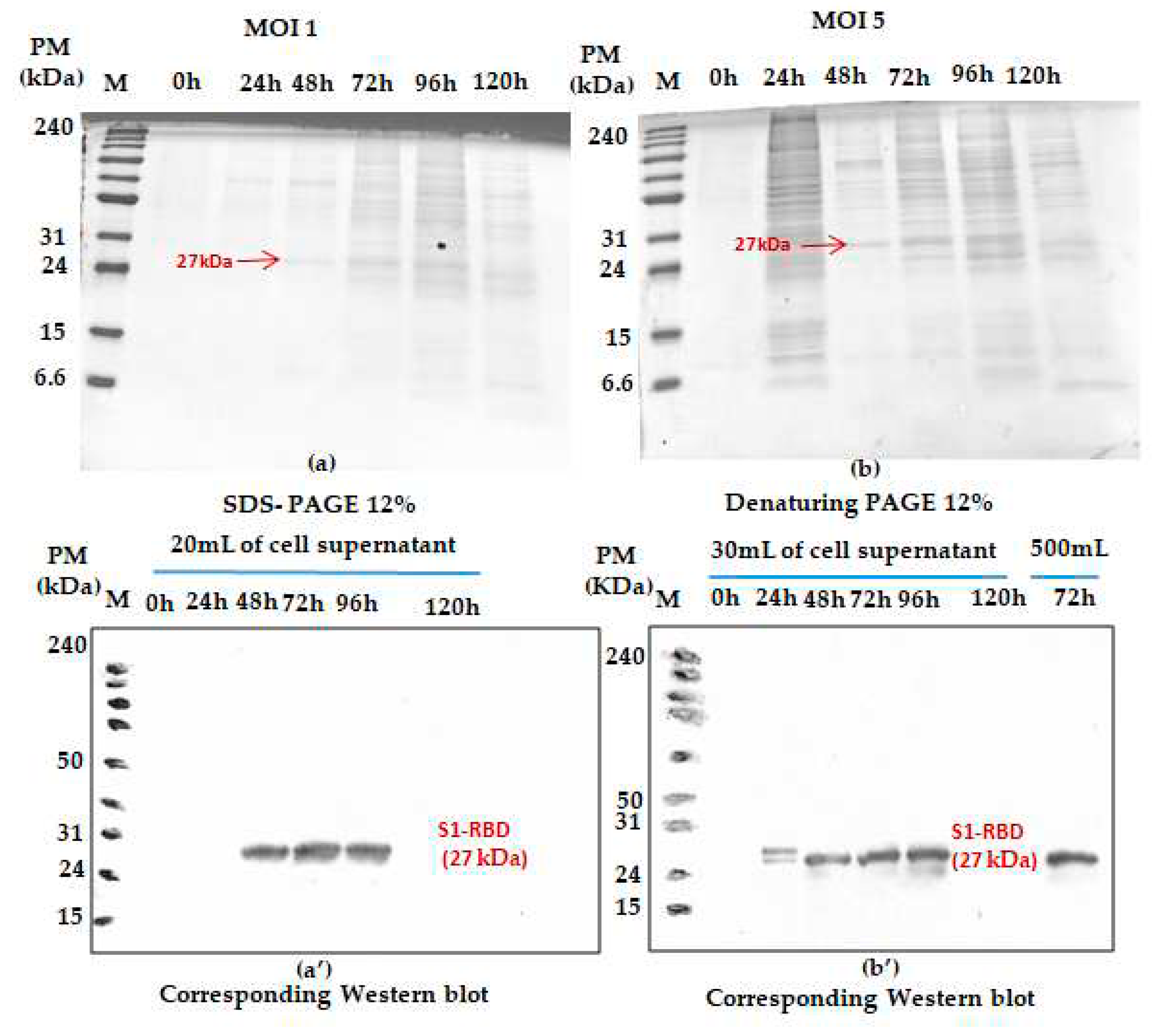
Figure 3.
Recombiant baculovirus titer and S1RBD expression at different MOIs. The virus titer (a) as well as S1RBD expression profile 72h post-infection using Coomassie-stained SDS-PAGE 12% (b) and western blot (c).
Figure 3.
Recombiant baculovirus titer and S1RBD expression at different MOIs. The virus titer (a) as well as S1RBD expression profile 72h post-infection using Coomassie-stained SDS-PAGE 12% (b) and western blot (c).
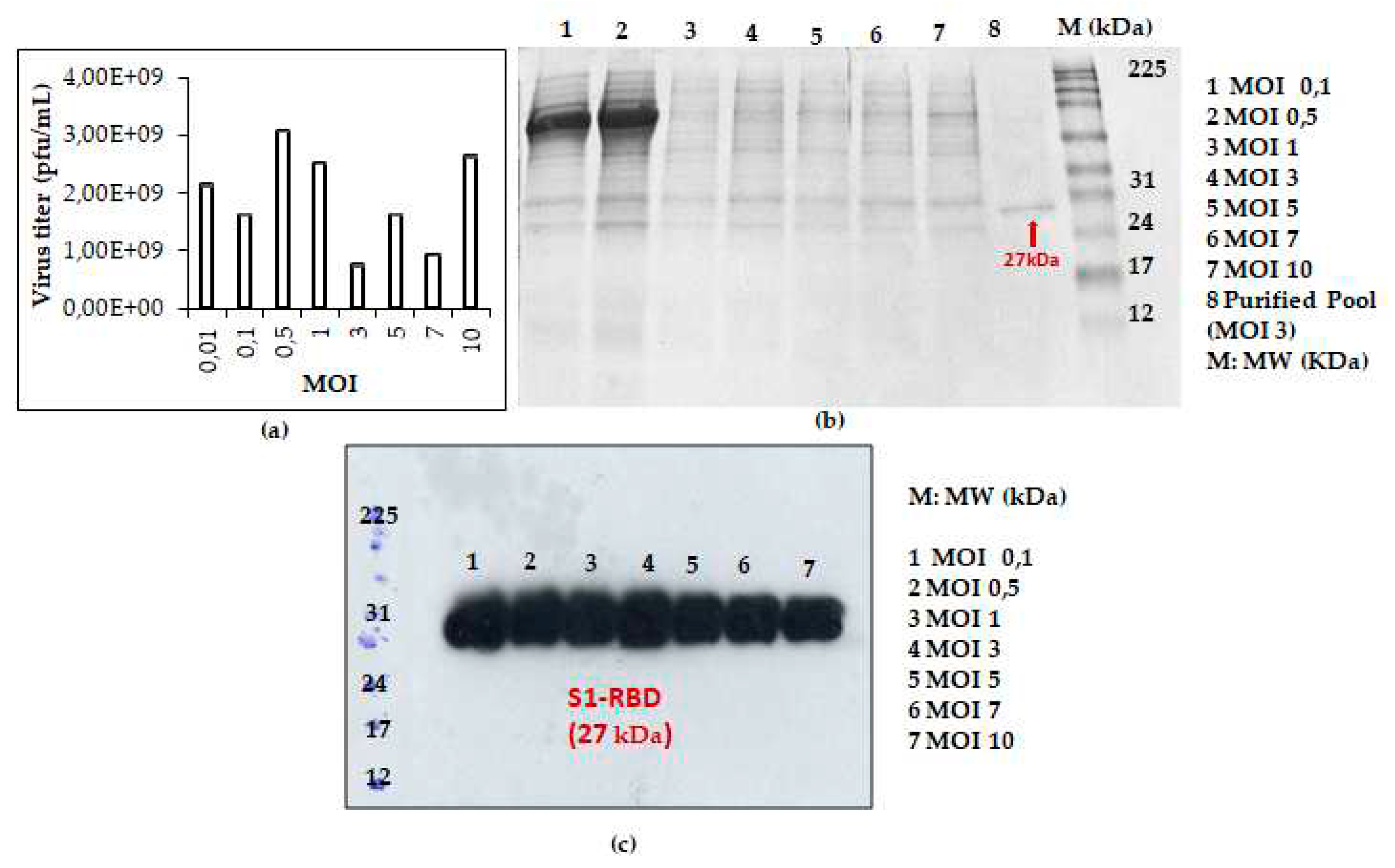
Figure 4.
S1RBD expression profiles at different cell densities at infection (1 x 106 and 2 x 106 cells/mL (a); 3 x 106 and 4 x 106 cells/mL (b); 5 x 106 and 7 x 106 cells/mL (c)) and a fixed MOI of 3 in shake-flasks. Equal amounts of total proteins (10 μg) were loaded onto the SDS-PAGE gel.
Figure 4.
S1RBD expression profiles at different cell densities at infection (1 x 106 and 2 x 106 cells/mL (a); 3 x 106 and 4 x 106 cells/mL (b); 5 x 106 and 7 x 106 cells/mL (c)) and a fixed MOI of 3 in shake-flasks. Equal amounts of total proteins (10 μg) were loaded onto the SDS-PAGE gel.
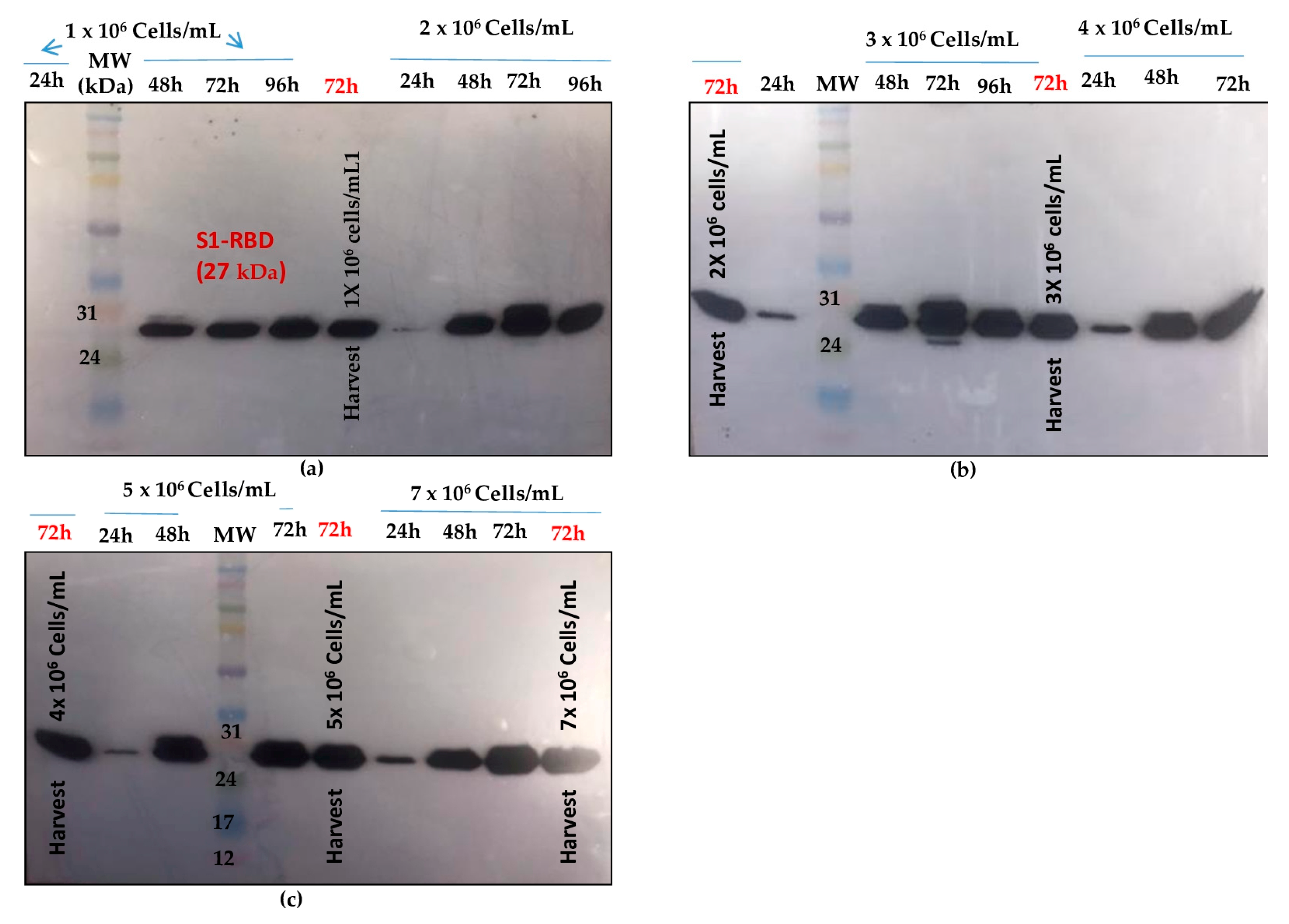
Figure 5.
Growth curve of infected Sf9 cells and production of S1RBD in a 7L bioreactor. Cell density and viability (a) (The arrow indicates the point of infection), and time course analysis of S1RBD expression in 7L stirred bioreactor (b). One step purification of S1RBD by affinity chromatography followed by Coomassie staining SDS-PAGE 12% (c).
Figure 5.
Growth curve of infected Sf9 cells and production of S1RBD in a 7L bioreactor. Cell density and viability (a) (The arrow indicates the point of infection), and time course analysis of S1RBD expression in 7L stirred bioreactor (b). One step purification of S1RBD by affinity chromatography followed by Coomassie staining SDS-PAGE 12% (c).
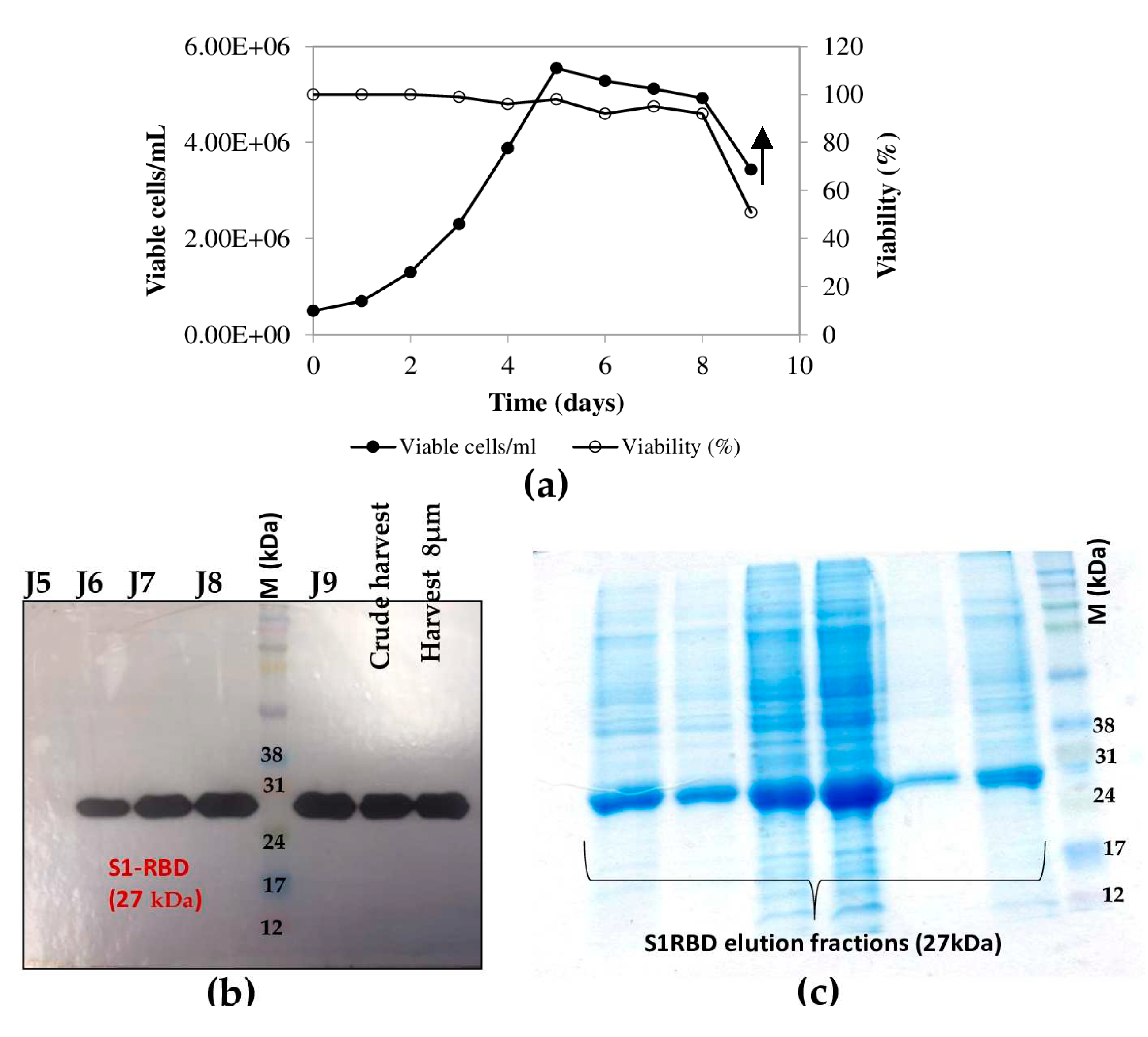
Figure 6.
Purification of S1RBD and its detection by western blot. (a) Single step purification of S1RBD by affinity column followed by Silver staining SDS/PAGE 12%: E5, E6 and E7 correspond to the eluted S1RBD fractions with the expected apparent molecular weight of 30 KDa. (b) Western blot analysis of the purified fractions. Two-step purification of S1RBD by affinity column (c) followed by size exclusion chromatography (d). Analysis was performed using silver-stained SDS-PAGE 12%. MW indicates the molecular weight of the protein marker.
Figure 6.
Purification of S1RBD and its detection by western blot. (a) Single step purification of S1RBD by affinity column followed by Silver staining SDS/PAGE 12%: E5, E6 and E7 correspond to the eluted S1RBD fractions with the expected apparent molecular weight of 30 KDa. (b) Western blot analysis of the purified fractions. Two-step purification of S1RBD by affinity column (c) followed by size exclusion chromatography (d). Analysis was performed using silver-stained SDS-PAGE 12%. MW indicates the molecular weight of the protein marker.
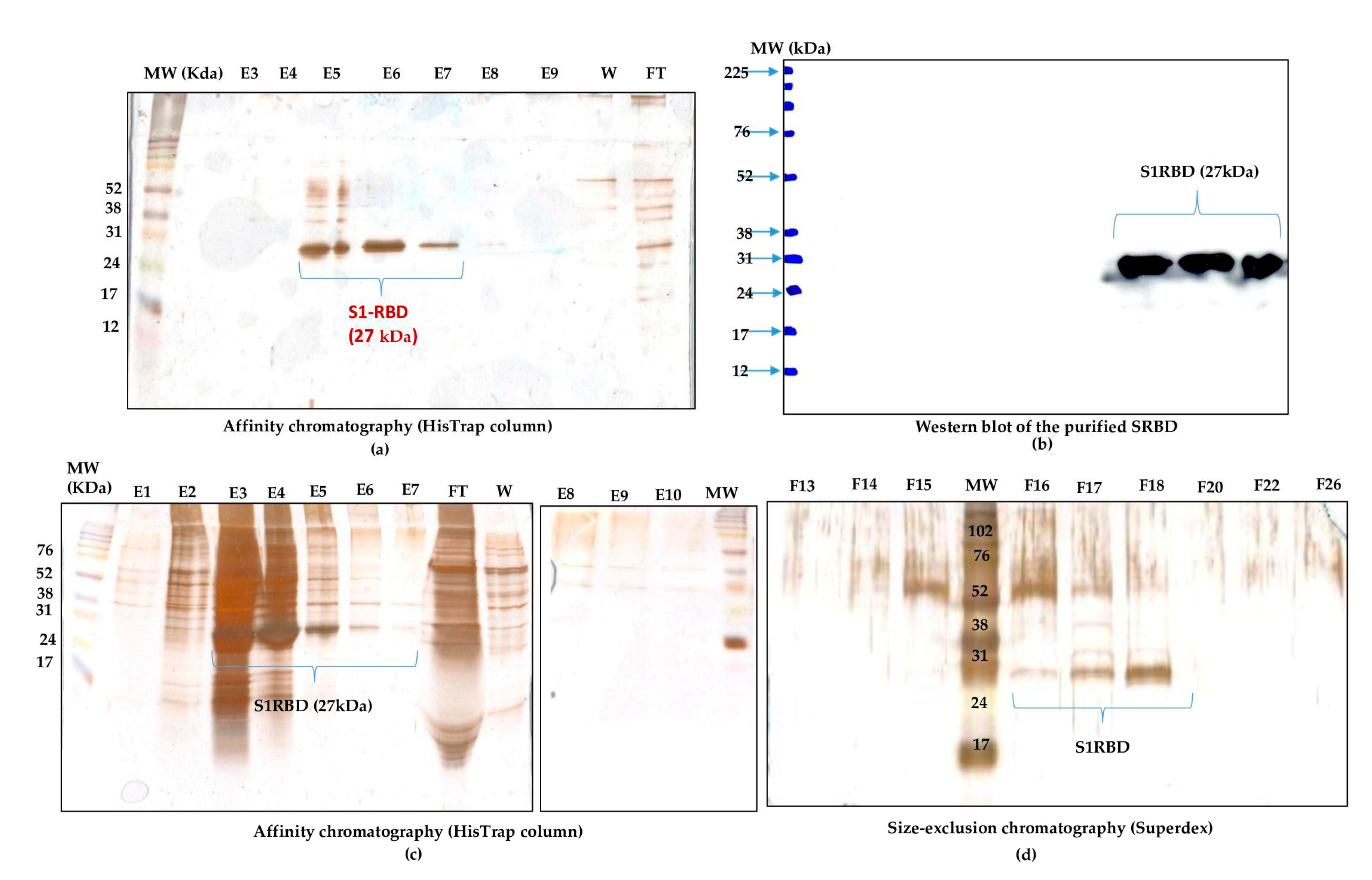
Figure 7.
Dimerization analysis of purified S1RBD on SDS-PAGE 12% under non-reduced (left) and reduced (right) conditions.
Figure 7.
Dimerization analysis of purified S1RBD on SDS-PAGE 12% under non-reduced (left) and reduced (right) conditions.
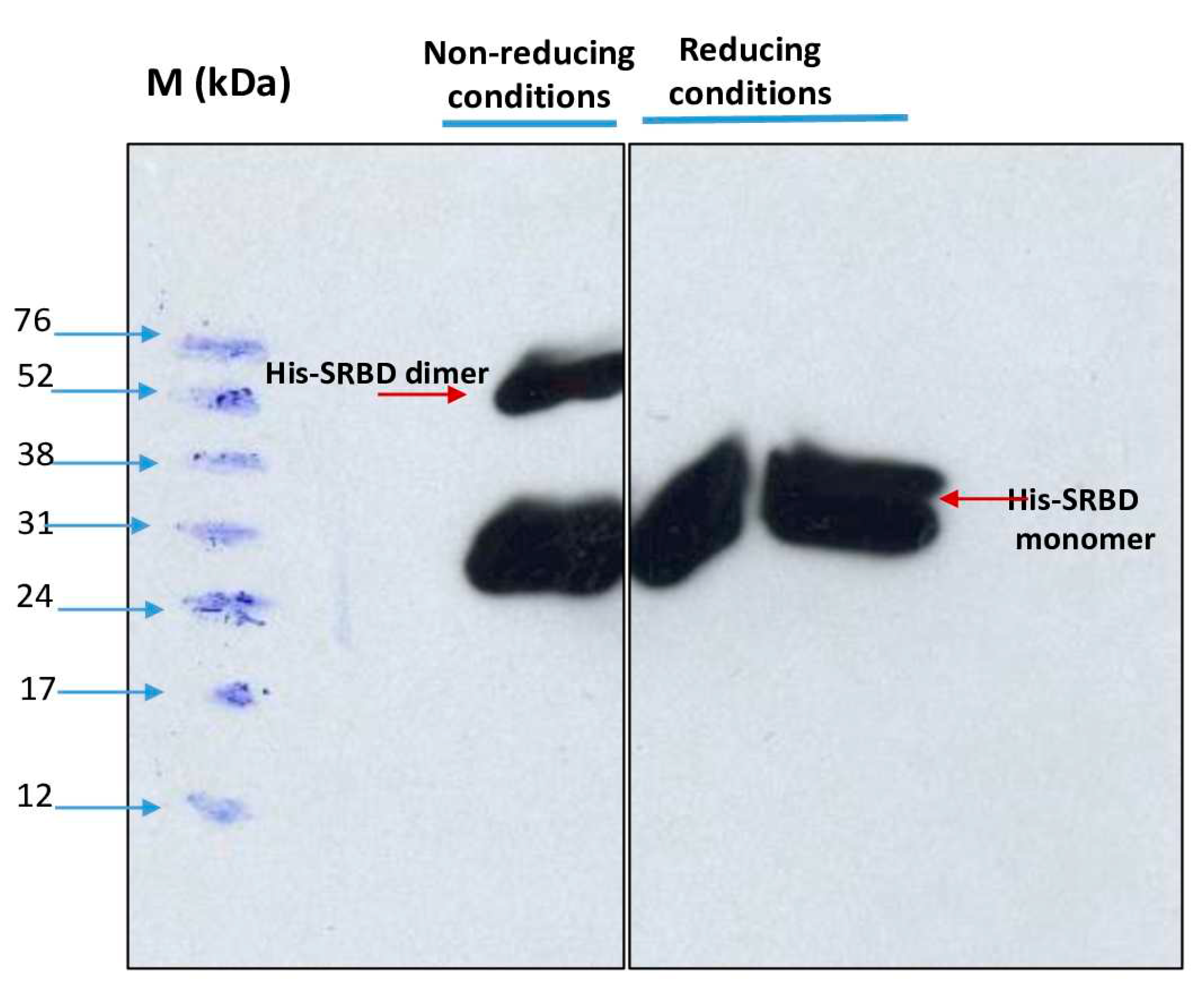
Figure 8.
Development of a Western blot assay to detect sero-positivity for SARS-CoV-2 infection. A negative control and 3 sera were used in duplicate.
Figure 8.
Development of a Western blot assay to detect sero-positivity for SARS-CoV-2 infection. A negative control and 3 sera were used in duplicate.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



