
Submitted:
23 August 2023
Posted:
24 August 2023
You are already at the latest version
Abstract
Precision medicine approaches often relies on complex and integrative analysis of multiple biomarkers from “omics” data to generate insights that can help either diagnostics, prognostics or therapeutical decisions. Such insights are often made using Machine learning (ML) models that make sample classification for a particular phenotype (yes/no). Building such models is a challenge and time-consuming, requiring advanced coding skills and mathematical modelling expertise. Artificial intelligence (AI) is a methodological solution that has the potential to facilitate, optimize and scale model development. In this work, we developed an AI-based, user-friendly and code-free platform (https://digitalphenomics.com) that fully automates the development of predictive models from quantitative “omics” data. Here, we show the application of this tool with the development of cancer survival prognostics models using real-life data from breast, lung and renal cancer transcriptomes. We report and compare their sensitivity, specificity, accuracy and Receiver Operating Characteristic (ROC) curve Area Under the Curve (AUC). Further, we report the associated sets of genes (biomarkers) and their expression pattern that are predictive of cancer survival. Moreover, we made our models available as online tools to generate prognostic predictions based on the gene expression of the biomarkers. In conclusion, we demonstrated that our tool is a robust user-friendly solution to develop bespoke predictive tools from “omics” data which facilitate precision medicine introduction to the point-of-care.
Keywords:
software tools
; bioinformatics
; cancer prognostics
; predictive modelling
1. Introduction
Transcriptomics, proteomics, metabolomics and lipidomics are examples of high-throughput “omics” methodologies often described as precision medicine approaches which enable a quantitative screening of multiple key biomarkers [1,2,3]. These methodologies have been often used to characterize human tissue variability and correlation with diseases such as cancer in attempt to find new biomarkers that predict disease outcomes and response to therapy [2,3,4]. Transcriptomics has particular importance as is an affordable and accurate gene expression quantification technique [1]. Often the identified gene expression biomarkers do not have enough predictive power on their own to provide robust insights to decision-making at the point-of-care [5,6]. This has been a persisting problem for cancer prognostics as the currently used biomarkers still have low predictive power, explaining only 25% to 75% of the cases [7].
Appling machine learning (ML) modelling frameworks to “omics” data have been considered a methodological solution for combining biomarkers and improving the predictive capacity of biomarkers [6,8,9,10]. Once applied ML on a “omics” dataset associated with a phenotype outcome, these frameworks find key features (biomarkers signatures) that compose a predictive model with a certain predictive power and performance (e.g. sensitivity, specificity and accuracy) [6,11]. Models, in turn, are able to score a new input of “omics” data with unknown phenotype/outcome and make a binary phenotype classification (yes/no) [6,8]. However, technical challenges and limitations associated with the implementation of ML have been preventing the full application of its potential to the point-of-care [12]. One critical limitation is associated to the complexity of developing and validating ML algorithms [10,13]. These are hard and time-consuming tasks that require advanced coding skills and mathematical modelling expertise to successfully implement and test supervised learning classification algorithms [12]. Another, is to choose the correct ML algorithms (e.g. random forests, neural networks, support vector machines and regression models) which is suitable to describe the data [8,9]. Besides, often the chosen ML algorithm has numerous tunning parameters for model refinement, which makes it almost humanly impossible to find the best possible model in reasonable timings without a systematic approach.
Automating ML-based model building and validation through artificial intelligence has been proven to be useful for optimal model generation and provides a much faster and more effective route to achieving better-performing models [14,15]. Genetic and Evolutionary inspired AI algorithms have been used in an attempt to optimize predictive models from “omics” datasets [16]. For example, TPOT (genetic) and EvA-3 (evolutionary) are two such algorithms that have been applied in the generation of optimal predictive models for early ovarian cancer detection, aneuploidies detection and cancer prognostics [14,17,18]. Although these approaches facilitate model development, they are not user-friendly and code-free. Further, currently ML algorithms have been rendering poor performance predictive models in cancer prognostics using transcriptomics [19]. In this work, we develop a novel AI-driven, user-friendly and code-free web platform for the automated generation of predictive models from “omics” datasets (https://digitalphenomics.com ). Here, we applied the tool for the generation of breast, lung and renal cancer survival prognostics models.
2. Materials and Methods
2.1. Tumour transcriptomics datasets
Tumour transcriptomics datasets were built using real-life biomedical data consisting of TCGA transcriptomics data of tumour biopsies of patients with breast, lung and renal cancers [20,21]. Transcriptomics data were collected from the 2021 updated records of the Human Protein Atlas database which contained mRNA expression (FPKM) of 200 genes from 1075 anonymized cancer patients [22,23]. We curated the collected data in the same way as previously done to make it comparable with previously generated model performances made by TPOT. Therefore, we selected the same 58 genes, considered key components of signalling pathways involved in the regulation of epithelial-to-mesenchymal transition, which has a role in cancer invasion and metastasis acquisition [24]. From the patient’s metadata collected, we selected transcriptomes associated with patients that have been reported to survive over 5 years after the diagnostics (good prognostics) or with a reported death lower than 2 years (poor prognostic). The sample numbers of the datasets are summarized in Table 1. A CSV dataset file for each cancer type was created with the gene IDs (first column) and the respective FPKM mRNA expression values of all patient samples (following columns). We also created a metadata CSV file that maps the survival phenotype of each patient sample with the expression data on the dataset. The datasets were made available in the digital phenomics platform (https://digitalphenomics.com ).
2.2. Platform development
The Digital Phenomics Platform version 1.0 was developed under a micro-services architecture design for scaling with robust performance on multiple servers. These micro-services included: Cybersecurity, Encrypted relational database, Encrypted models and datasets storage, Private and secure users’ environment, Containers systems for independent running microservices (Docker), Queueing system, AI-driven model building; FTP system, API management and supervision; And visualization tools. Multiple coding languages and frameworks were used for the development of the platform. These included Javascript, Python, HTML, PHP, bash and Nodejs.
2.3. Model Generation
Predictive models were generated on the digital phenomics platform (https://digitalphenomics.com ) version 1.0. Model generation used the AI software O2P-Mgen version 1.0 developed by the Bioenhancer Systems LTD. This AI was programmed to conduct all model training, optimization, refinement and validation automatedly. Using this tool, the data for model training is selected by the AI with a proportion always lower than 50% of the dataset, leaving the remaining data for testing. The AI performs supervised ML to develop models using an evolution-inspired algorithm that finds the best combination of biomarkers under a multi-objective fitness function for optimal sensitivity and specificity (EvA-3 algorithm version 2.0). To build models, the AI was programmed to search for biomarkers characteristics that reflect up-regulations, down-regulations, gene activations or gene expression inhibitions (e.g. gene knockouts) in the model training groups (positives vs negatives). For the cancer datasets, we set as positives the good survival prognostics and negatives the poor survival prognostics. By default, the AI only selects biomarkers up-regulations and down-regulations on the training data when the p-values are below 0.05 to ensure enough statistical significance. In the case of gene activations and inhibitions, the AI was programmed to look for a binary expression behaviour on data, considering a residual degree of tolerance. Models were constructed by the AI using the generic scoring function (Score), where: Pi is the absolute distance between the median level of the biomarker i on the group of positives for the phenotype and the sample value; Ni is the absolute distance between the median level of the biomarker i on the group of positives for the phenotype and the sample value; Wi is the enrichment of the biomarker i on the group positive for the phenotype; and n the total number of the biomarker in the model.
3. Results
3.1. Digital Phenomics Platform
We developed a novel user-friendly platform, Digital Phenomics Platform, tailored for the generation of predictive models from “omics” data. We made this platform available online (http://digitalphenomics.com ). The platform is organized into modules that address a particular functionality (Figure 1). The GENERATOR module enabled us to build datasets (drag and drop) and build predictive models using the uploaded datasets. Building models was straightforward, only requires to press the add button (+) or edit icon and type/modify; models’ name, description, AI learning time and maximum false positive rate allowed. Upon saving the request, the AI initializes the model building which may take minutes to hours depending on the amount of learning time requested.
Once a model is generated, its characteristics and performance can be analysed by clicking on the bar icon. This generates a table with their; predictive biomarkers, median levels (positive prediction), type of predictive regulation (e.g. up-regulation, down-regulation) and the associated p-value. A dynamic ROC curve and a heatmap of the predictions is generated, which optionally can be download. The heatmap shows the biomarkers scoring, overall predictive score, and outcomes on all the data used to build the model. With this heatmap, users can easily identify the false positives and negatives. To generate predictions from unknown samples using the models, we implemented the PREDICT module (Figure 2). In this tool, it is required to insert the values of the model biomarkers and submit them for prediction. Upon submission, the results are shown instantaneously on the platform in a visually and intuitive manner.
3.2. Testing Model Generation with Transcriptomics data
We tested the model generation potential of the Digital Phenomics Platform with real-life tumour transcriptomics datasets (table 1). Using these datasets, we request the generation of 100 models for cancer survival prognostics with AI learning times ranging from 1 to 120 min with a maximum of false positive rate of 25% and repeated 5 times. All models were successfully generated with specificities > 75% fulfilling the user setup requirement, indicating that the tool is robust. The accuracy of the generated prediction tools was also checked by recapitulating the datasets outcomes and prediction scores, where we manually checked the 3 models and calculated the sensitivities and specificities. The generated models’ ROC’s AUC shows an increase of predictive power with learning time, reaching a plateau between 30-60 min (Figure 3). The results also indicate a performance variability in model building independent of the learning time. On the other hand, these results show that the predictive power is also dependent on the dataset. In contrast, the number of predictive biomarkers identified by the AI negatively correlated with the overall models’ performance, indicating that the AI was struggling to make models from the renal and lung transcriptomics datasets. Further, the model generation was observed to be approximately 2.4 times the learning time. This is because the AI uses the user-defined learning time to model refinement and requires to dedicate time to process the data for finding the synergic effect of biomarkers on model performance.
3.3. Cancer Prognostic Models
We developed breast, lung and renal cancer survival prognostic models using the transcriptomics datasets (Table 1) and the digital phenomics platform. The models are made available for the generation of predictions on https://digitalphenomics.com. The models performance, the number of predicted biomarkers that compose the model and their scoring cut-off are presented in Table 2. The obtained model’s ROC’s curve (Figure 4) and their area under the curve values (Table 2) indicates these models have good predictive power [25]. The sensitivity, specificity and Accuracy of the Breast cancer prognostic model were superior to 85% (Table 2), indicating that this model has a good performance, suitable for making predictions on new cancer transcriptomics data [25]. Lung and renal cancer models had lower performances (Table 2) but yet with sensitivities, specificities and accuracies always superior to 70%, indicating that these are reasonable models to generate predictions on new data [25]. The biomarkers of the cancer prognostic models and their associated predicted behaviour is represented in Figure 5. We found mainly up-regulations and some down-regulations of gene expression in cancer survival phenotypes. Interestingly, these results show both distinct and conservative gene expression patterns between breast, lung and renal cancer. We identified 8 genes (PI3K, β-catenin, MET, EGF, TCF, LEF, Delta1 and Frizzled) that have up-regulated expression and 1 (SNAIL1) with down-regulation, conservative across the 3 cancers types.
4. Discussion
Current “omics” based precision medicine frameworks still rely on experienced bioinformaticians with expertise in data science and modelling to generate ML based predictive [13]. Although this is an ideal scenario for the academy, it is not efficient to implement such frameworks to the point-of-care as it is required to scale it to the population level. Our developed solution using AI replaces the role of the specialized bioinformatician modeller and made it possible unspecialized laboratory personnel to generate and apply predictive models as online tools to the point-of-care. This is mainly because our tool is user-friendly, and doesn’t require users to have any coding and advanced mathematical modelling skills. Furthermore, the tool was designed for scaling to large datasets and multiple users working in parallel. This makes it possible for many academic or industry related laboratories to apply ML on “omics” data without investing in specialized human resources and computational resources, which can be an economic burden above £100,000 per year. Thus, we believe that our solution may have an impact on opening new possibilities to academic labs, start-ups and diagnostic companies that want to focus on precision medicine approaches.
Although our platform solution showed robust results, we highlight some limitations and disadvantages in comparison to other solutions. One is the fact that the digital phenomics platform relies only on a scoring-based evolutionary algorithm for model generation, whereas other AI-driven auto ML frameworks such as TPOT use an exclusive library of algorithms that include random forests, support vector machines and neural networks [14]. Another limitation is that it can only develop supervised learning classifiers (yes/no) which require a list of categorical features (biomarkers) associated with numerical values (quantitative data). This brings some uncertainty in the generated predictions when it’s near the scoring cut-off of the phenotype yes/no decision. The observed variability associated with the performance of generated models in each attempt and the dependency of the dataset can be also considered as a limitation of this technology. This implies trial-and-error attempts from the users to get the best-performing model. Furthermore, once new data comes, a model does not update automatedly by the AI. It is required a user intervention to conduct another model development attempt. A future version of the AI algorithm should take into account these limitations towards improvement that minimizes the impact of these limitations.
Importantly, the Digital Phenomics Platform have rendered promising breast, lung and renal cancer survival prognostic models from tumour transcriptomics data (Table 2). Our models rendered much higher predictive power (86% >AUCs > 77%) in comparison to the ones generated using TPOT on the same datasets (70% >AUCs > 48%) [19]. This suggests that our AI-driven modelling framework outcompetes the capacity of the compendium of ML algorithms implemented in TPOT for transcriptomics data. In comparison to other published ML models, our model for breast cancer prognostics performed with a superior sensitivity (86%) in comparison to the reported 35-64%, whereas the specificity was inferior (85%) to the 97-99% [26]. Besides, the obtained AUC for the breast cancer prognostic model (86%) was comparable to the 80-92% reported for other models [26]. For lung and renal cancer prognostic models, we obtained in this work slightly superior (up to 10%) in comparison to the ones published using other modelling approaches [27,28]. This suggests that our cancer prognostic models are competitive alternatives to the ones already published.
Interestingly, the obtained conservative patterns of gene expression among cancer types are compatible with the main markers of epithelial phenotype (β-catenin) and incompatible with the markers of the mesenchymal phenotype (SNAIL1) [24,29]. This may explain partially the survival prognostics as the mesenchymal phenotype and the overexpression of SNAIL1 is often correlated with cancer invasion, whereas the epithelial phenotype often correlates with benign cancers [30,31]. However, the other biomarkers identified are considered to be involved in epithelial-to-mesenchymal transitions, known to be highjacked during cancer invasion [32,33]. According to a regulatory network model of epithelial-to-mesenchymal transitions, the identified biomarkers are more compatible with mesenchymal than a more invasive hybrid phenotype [24,34]. Thus, our results agrees with this idea, but also highlights the complexity and heterogenicity of cancer deregulations and its correlation with survival prognostics [7,26,28,35].
5. Conclusions
In this work, we developed a novel AI-driven platform for the generation of predictive models from ‘omics’ data. Here, we demonstrated that the platform is a user-friendly, coding-free, robust and scalable solution suitable to be applied as a precision medicine tool in the point-of-care. This was illustrated with the application of the platform for the generation of breast, lung and renal cancer prognostics from transcriptomics data. Importantly, with this work, we enabled the usage of competitive and novel cancer prognostic models which can be accessed online for the generation of predictions through the digital phenomics platform.
Supplementary Materials
No supplementary material is supplied.
Author Contributions
Conceptualization, R.P. and U.F.; methodology, R.P. and U.F.; software, v; validation, T.P; formal analysis, T.P.; data curation, T.P.; writing—original draft preparation, U.F.; writing—review and editing, R.P.; supervision, R.P. All authors have read and agreed to the published version of the manuscript.”
Funding
Please add: This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data in this research is available at https://digitalphenomics.com.
Acknowledgments
We acknowledge Bioenhancer Systems LTD and URA Informatics LTD for supporting the resources necessary to conduct the analysis and maintain the tools online.
Conflicts of Interest
U. Filho. and R. Pais. declare a potential conflict of interest as they are directors of Bioenhancer Systems LTD and URA Informatics, respectively. T. Pais declares no conflict of interest.
References
- Van Allen, E.M.; Robinson, D.; Morrissey, C.; Pritchard, C.; Imamovic, A.; Carter, S.; Rosenberg, M.; McKenna, A.; Wu, Y.M.; Cao, X.; et al. A Comparative Assessment of Clinical Whole Exome and Transcriptome Profiling across Sequencing Centers: Implications for Precision Cancer Medicine. Oncotarget 2016, 7, 52888–52899. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science (80-. ). 2015, 347, 1260419–1260419. [Google Scholar] [CrossRef]
- Kholodenko, B.; Yaffe, M.B.; Kolch, W. Computational Approaches for Analyzing Information Flow in Biological Networks. Sci. Signal. 2012, 5, 1–14. [Google Scholar] [CrossRef]
- Ao Kong, A.; Gupta, C.; Ferrari, M.; Agostini, M.; Bedin, C.; Bouamrani, A.; Tasciotti, E.; Azencott, R. Biomarker Signature Discovery from Mass Spectrometry Data. IEEE/ACM Trans. Comput. Biol. Bioinforma. 2014, 11, 766–772. [Google Scholar] [CrossRef]
- Kim, H.; Kwon, H.J.; Kim, E.S.; Kwon, S.; Suh, K.J.; Kim, S.H.; Kim, Y.J.; Lee, J.S.; Chung, J.-H. Comparison of the Predictive Power of a Combination versus Individual Biomarker Testing in Non–Small Cell Lung Cancer Patients Treated with Immune Checkpoint Inhibitors. Cancer Res. Treat. 2022, 54, 424–433. [Google Scholar] [CrossRef]
- Mann, M.; Kumar, C.; Zeng, W.F.; Strauss, M.T. Artificial Intelligence for Proteomics and Biomarker Discovery. Cell Syst. 2021, 12, 759–770. [Google Scholar] [CrossRef]
- Šutić, M.; Vukić, A.; Baranašić, J.; Försti, A.; Džubur, F.; Samaržija, M.; Jakopović, M.; Brčić, L.; Knežević, J. Diagnostic, Predictive, and Prognostic Biomarkers in Non-Small Cell Lung Cancer (NSCLC) Management. J. Pers. Med. 2021, 11, 1102. [Google Scholar] [CrossRef]
- De Ridder, D.; De Ridder, J.; Reinders, M.J.T. Pattern Recognition in Bioinformatics. Brief. Bioinform. 2013, 14, 633–647. [Google Scholar] [CrossRef]
- 9. Ricardo Jorge Pais Bioinformatics and Predictive Modelling as Tools for Clinical Diagnostics. Insights Omnia-Health 2020, 30–34.
- Swan, A.L.; Mobasheri, A.; Allaway, D.; Liddell, S.; Bacardit, J. Application of Machine Learning to Proteomics Data: Classification and Biomarker Identification in Postgenomics Biology. Omi. A J. Integr. Biol. 2013, 17, 595–610. [Google Scholar] [CrossRef]
- Rahman, J.; Rahman, S. The Utility of Phenomics in Diagnosis of Inherited Metabolic Disorders. Clin. Med. J. R. Coll. Physicians London 2019. [CrossRef] [PubMed]
- Pais, R.J. Predictive Modelling in Clinical Bioinformatics: Key Concepts for Startups. BioTech 2022, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pais, R.J. Predictive Modelling in Clinical Bioinformatics: Key Concepts for Startups. BioTech 2022, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Le, T.T.; Fu, W.; Moore, J.H. Scaling Tree-Based Automated Machine Learning to Biomedical Big Data with a Feature Set Selector. Bioinformatics 2020, 36, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Olson, R.S.; Urbanowicz, R.J.; Andrews, P.C.; Lavender, N.A.; Kidd, L.C.; Moore, J.H. Automating Biomedical Data Science Through Tree-Based Pipeline Optimization. In Lecture Notes in Computer Science (including subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics); Springer, Cham, 2016; Vol. 9597, pp. 123–137 ISBN 9783319312033.
- Telikani, A.; Gandomi, A.H.; Tahmassebi, A.; Banzhaf, W. Evolutionary Machine Learning: A Survey. ACM Comput. Surv 2021, 54, 1–35. [Google Scholar] [CrossRef]
- Pais, R.J.; Iles, R.K.; Zmuidinaite, R. MALDI-ToF Mass Spectra Phenomic Analysis for Human Disease Diagnosis Enabled by Cutting-Edge Data Processing Pipelines and Bioinformatic Tools. Curr. Med. Chem. 2020, 27. [Google Scholar] [CrossRef] [PubMed]
- Pais, R.J.; Zmuidinaite, R.; Lacey, J.C.; Jardine, C.S.; Iles, R.K. A Rapid and Affordable Screening Tool for Early-Stage Ovarian Cancer Detection Based on MALDI-ToF MS of Blood Serum. Appl. Sci. 2022, 12, 3030. [Google Scholar] [CrossRef]
- Pais, R.J.; Lopes, F.; Parreira, I.; Silva, M.; Silva, M.; Moutinho, M.G. Predicting Cancer Prognostics from Tumour Transcriptomics Using an Auto Machine Learning Approach. In Proceedings of the Med. Sci Forum; MDPI: Basel Switzerland, August 8, 2023; p. 6. [Google Scholar]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Mills Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Publ. Gr. 2013. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A Pathology Atlas of the Human Cancer Transcriptome. Science (80-.). 2017, 357. [Google Scholar] [CrossRef]
- Pontén, F.; Jirström, K.; Uhlen, M. The Human Protein Atlas-a Tool for Pathology. J. Pathol. J Pathol 2008, 216, 387–393. [Google Scholar] [CrossRef]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a Knowledge-Based Human Protein Atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef] [PubMed]
- Pais, R.J. Simulation of Multiple Microenvironments Shows a Pivot Role of RPTPs on the Control of Epithelial-to-Mesenchymal Transition. Biosystems. 2020, 198. [Google Scholar] [CrossRef] [PubMed]
- Dankers, F.J.W.M.; Traverso, A.; Wee, L.; van Kuijk, S.M.J. Prediction Modeling Methodology. In Fundamentals of Clinical Data Science; Springer International Publishing: Cham, 2019; pp. 101–120. [Google Scholar]
- Boeri, C.; Chiappa, C.; Galli, F.; De Berardinis, V.; Bardelli, L.; Carcano, G.; Rovera, F. Machine Learning Techniques in Breast Cancer Prognosis Prediction: A Primary Evaluation. Cancer Med. 2020, 9, 3234–3243. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Ma, X.; Song, P. A Prognostic Model of Non Small Cell Lung Cancer Based on TCGA and ImmPort Databases. Sci. Rep. 2022, 12, 437. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Xu, Y.; Han, C.; Wang, Z.; Li, J.; Wang, Q.; Che, X. Identification of a Prognostic Risk Signature of Kidney Renal Clear Cell Carcinoma Based on Regulating the Immune Response Pathway Exploration. J. Oncol. 2020, 2020, 1–8. [Google Scholar] [CrossRef]
- Zeisberg, M.; Neilson, E.G. Biomarkers for Epithelial-Mesenchymal Transitions. J. Clin. Invest. 2009, 119, 1429–1437. [Google Scholar] [CrossRef]
- Zhao, W.; Zhou, Y.; Xu, H.; Cheng, Y.; Kong, B. Snail Family Proteins in Cervical Squamous Carcinoma: Expression and Significance. Clin. Invest. Med. 2013, 36, E223–E233. [Google Scholar] [CrossRef]
- Howard, S.; Deroo, T.; Fujita, Y.; Itasaki, N. A Positive Role of Cadherin in Wnt/β-Catenin Signalling during Epithelial-Mesenchymal Transition. PLoS One 2011, 6, e23899. [Google Scholar] [CrossRef]
- Krakhmal, N. V; Zavyalova, M. V; Denisov, E. V; Vtorushin, S. V; Perelmuter, V.M. Cancer Invasion: Patterns and Mechanisms. Acta Naturae 2015, 7, 17–28. [Google Scholar] [CrossRef]
- Savagner, P. Epithelial-Mesenchymal Transitions: From Cell Plasticity to Concept Elasticity. Curr. Top. Dev. Biol. 2015, 112, 273–300. [Google Scholar] [CrossRef]
- Steinway, S.N.; Zanudo, J.G.T.; Ding, W.; Rountree, C.B.; Feith, D.J.; Loughran, T.P.; Albert, R. Network Modeling of TGFβ Signaling in Hepatocellular Carcinoma Epithelial-to-Mesenchymal Transition Reveals Joint Sonic Hedgehog and Wnt Pathway Activation. Cancer Res. 2014, 74, 5963–5977. [Google Scholar] [CrossRef] [PubMed]
- van de Wouw, A. .; Janssen-Heijnen, M.L..; Coebergh, J.W..; Hillen, H.F..; Hemminki, K.; Fiorentini, G.; D’Aprile, M.; Giorgi, F.; Parziale, A.; Contu, A. Comparison of Survival of Patients with Metastases from Known versus Unknown Primaries: Survival in Metastatic Cancer. BMC Cancer 2013, 13–36. [Google Scholar] [CrossRef]
Figure 1.
Digital Phenomics Platform under the generation of models. On the right, there is an actionable link for the GENERATOR and PREDICTOR modules. Models link on GENERATOR returns a table that shows all models developed and enables editing (pencil icon) or creating a new model (+ icon). .
Figure 1.
Digital Phenomics Platform under the generation of models. On the right, there is an actionable link for the GENERATOR and PREDICTOR modules. Models link on GENERATOR returns a table that shows all models developed and enables editing (pencil icon) or creating a new model (+ icon). .
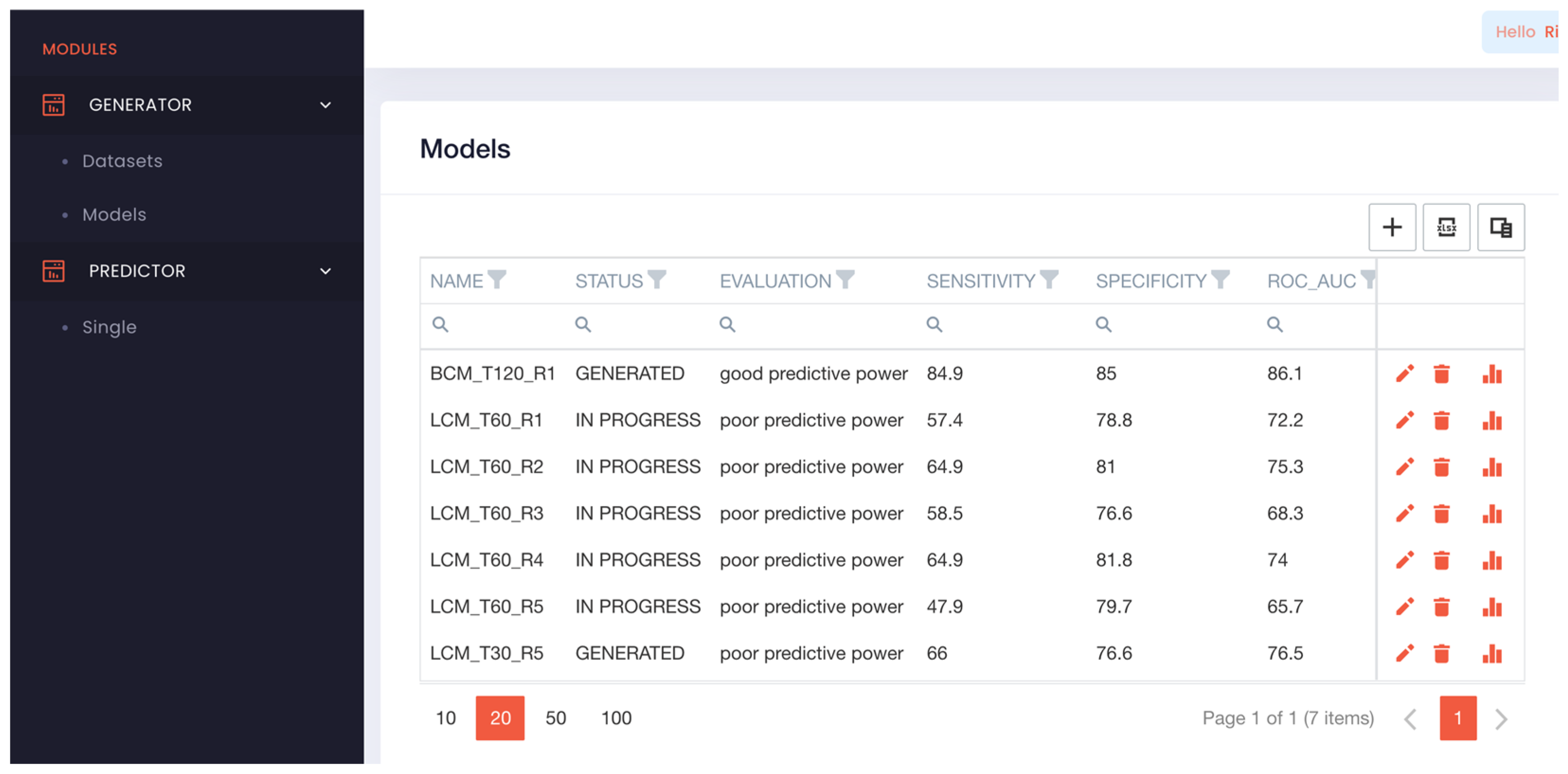
Figure 2.
The PREDICTOR module tool User interface on the digital phenomics platform. (a) Model selection and biomarker input values. (b) Unknown Sample prediction example using the PREDICT tool.
Figure 2.
The PREDICTOR module tool User interface on the digital phenomics platform. (a) Model selection and biomarker input values. (b) Unknown Sample prediction example using the PREDICT tool.

Figure 3.
Generated cancer survival prognostic models attempt for breast, lung and renal tumour transcriptomics datasets. (a) ROC-AUC as a function of AI learning time. The AUC of the ROC is considered here as the main index of predictive power (b) Number of predictive biomarkers in each model as a function of the AI learning time. All models built were conducted at a 25% maximum false positive rate allowed. .
Figure 3.
Generated cancer survival prognostic models attempt for breast, lung and renal tumour transcriptomics datasets. (a) ROC-AUC as a function of AI learning time. The AUC of the ROC is considered here as the main index of predictive power (b) Number of predictive biomarkers in each model as a function of the AI learning time. All models built were conducted at a 25% maximum false positive rate allowed. .
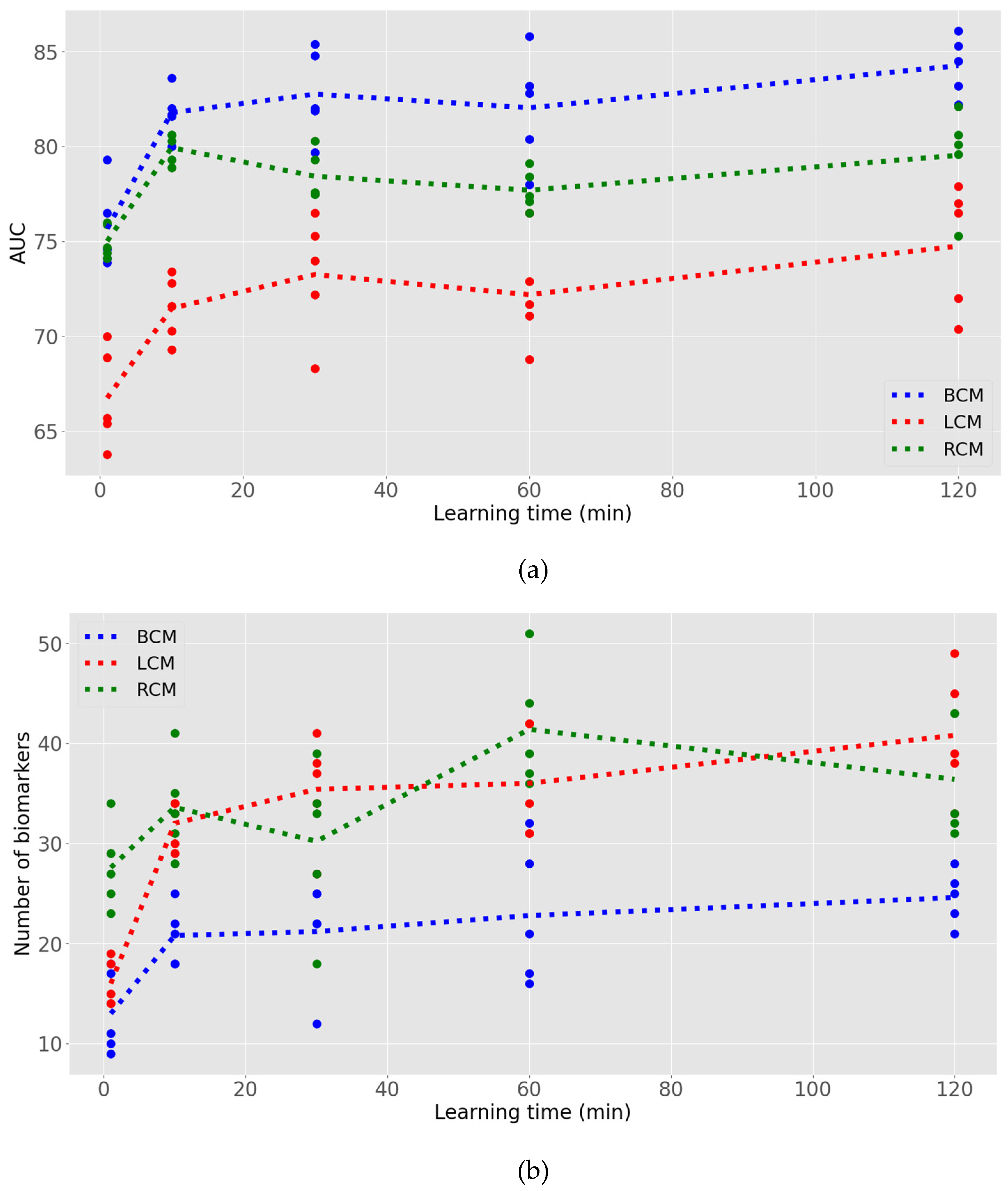
Figure 4.
Receiver Operating Characteristics (ROC) curves of the selected cancer survival prognostics models (Table 2). ROC’s adapted from the ones downloaded from the digital phenomics platform.
Figure 4.
Receiver Operating Characteristics (ROC) curves of the selected cancer survival prognostics models (Table 2). ROC’s adapted from the ones downloaded from the digital phenomics platform.
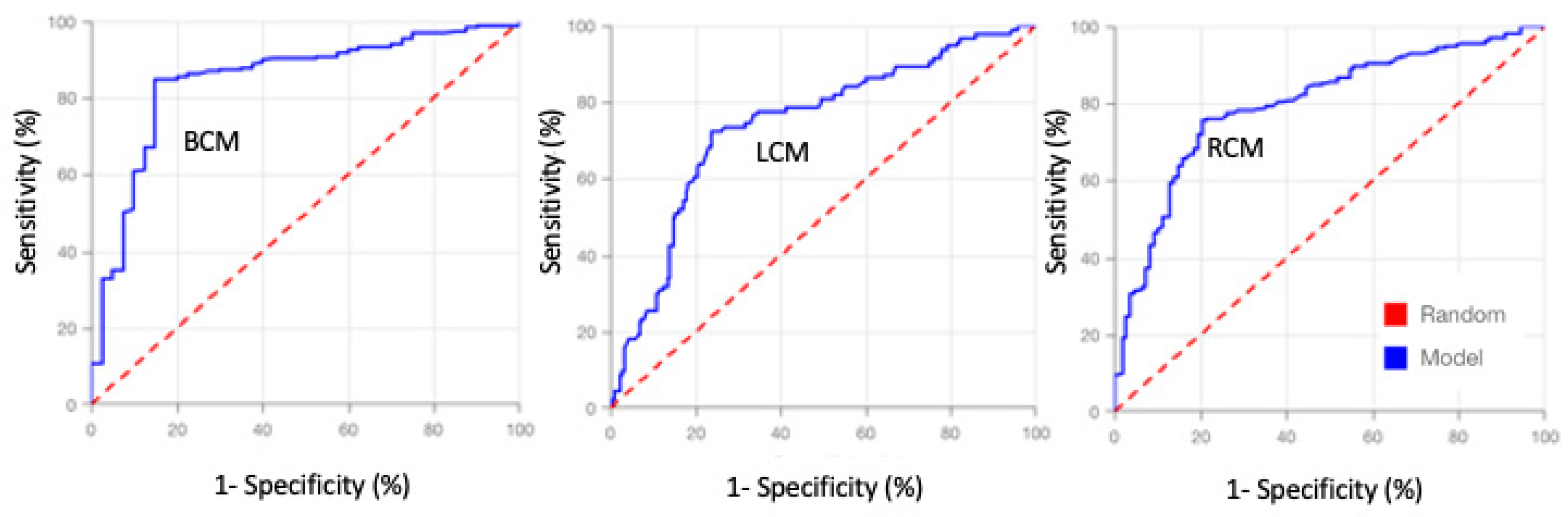
Figure 5.
The identified regulatory pattern of gene expression of the predictive biomarkers of each prognostic model. Models are indicated by BCM (breast cancer model), LCM (lung cancer model), and RCM (renal cancer model). Red indicates up-regulation, blue indicates down-regulation and grey indicates no predictive effect. All 58 Genes in the datasets are depicted in the y-axis, and gene product names are shown instead of the Ensembl gene IDs.
Figure 5.
The identified regulatory pattern of gene expression of the predictive biomarkers of each prognostic model. Models are indicated by BCM (breast cancer model), LCM (lung cancer model), and RCM (renal cancer model). Red indicates up-regulation, blue indicates down-regulation and grey indicates no predictive effect. All 58 Genes in the datasets are depicted in the y-axis, and gene product names are shown instead of the Ensembl gene IDs.
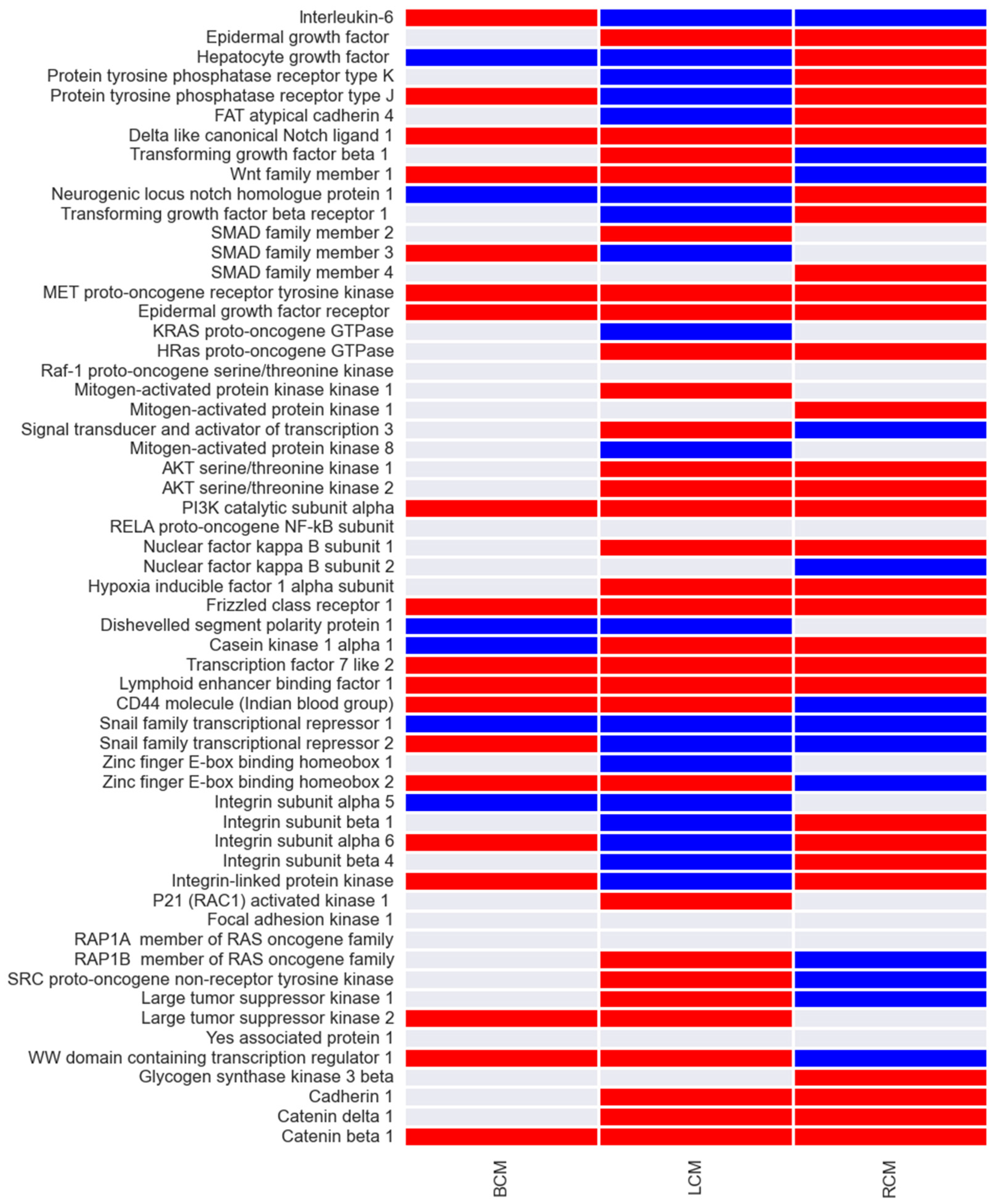

Table 1.
Cancer transcriptomics datasets and their sampling numbers.
| TCGA refs | N poor prognostics |
N good prognostics |
N Total |
Tumour Tissue | Dataset ID |
|---|---|---|---|---|---|
| BRCA | 40 | 199 | 239 | Breast | TCGA BCSD |
| LUSC, LUAD | 231 | 94 | 325 | Lung | TCGA LCSD |
| KICH, KIRC, KIRP | 108 | 210 | 318 | Renal | TCGA RCSD |
Table 2.
Performances of the best cancer survival prognostic models generated by the digital phenomics platform.
Table 2.
Performances of the best cancer survival prognostic models generated by the digital phenomics platform.
| Cut-off | AUC | Accuracy | Specificity | Sensitivity | N Biomarkers |
Cancer | Model |
|---|---|---|---|---|---|---|---|
| 220.7 | 86.1% | 84.9% | 85.0% | 84.9% | 25 | Breast | BCM |
| 690.0 | 77.0% | 75.1% | 76.2% | 72.3% | 49 | Lung | LCM |
| 530.2 | 82.1% | 77.0% | 79.6% | 75.7% | 43 | Renal | RCM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



