
Submitted:
23 August 2023
Posted:
25 August 2023
You are already at the latest version
Abstract
Head and neck squamous cell carcinoma (HNSCC) takes the sixth place among the most common cancers in the world. Abnormal methylation can be one of the reasons for this cancer. The aim of this study was to investigate the DNA promotor methylation status of cancer-associated genes (ATM, APC, CDO1, RB1, TP53, WIF1) in patients with HNSCC. Bisulfite Conversion and Methylation-Sensitive High-Resolution Melting was used for analysis of the DNA methylation level of normal and tumor tissues in 44 patients. There were significant differences in DNA methylation level between patient’s tumor and normal tissues for CDO1 and WIF1 genes in all subjects and subgroups (p<0.05). In T3 subgroup there was significant correlation between CDO1 gene methylation and age in the normal tissue. The same correlation was detected also for the WIF1 gene methylation in tumor tissue samples in the subgroup with T3 and in normal tissue samples in the subgroup with T4 (p<0,05). In all genes no significant differences were found between the subgroups (T2, T3, T4 stage, primary/recurrent lesion, non-keratinizing/keratinizing SCC, age before/ after 50, smokers/non-smokers) of the patients. Thus, changes in the expression of the CDO1 and WIF1 genes can affect mechanisms of the occurrence and development of HNSCC.
Keywords:
DNA methylation
; cancer associated genes
; head and neck squamous cell carcinoma
1. Introduction
Epigenetic changes of gene regulation (as DNA methylation, histone modification, chromatin remodeling, and non-coding RNAs) along with DNA mutation are the basis of molecular mechanisms of cancerogenesis and tumorigenesis. Therefore, the study of epigenetic markers for diagnosis, prognosis and prevention of cancer is of considerable interest for medicine. Detection of DNA methylation level has recently been used as biomarkers for early diagnosis, prognosis for identifying target genes for drug therapy.
The hypomethylation of single CpG dinucleotides has been one of the first changes found in tumor cells [1]. The large part of investigation has been carried out during DNA methylation in the promoter region of various genes [2,3]. There have been shown to be changes in the level of methylation of general and tissue specific cell genes in ontogenesis in utero and post-natal period.
Non-mutagenic etiology of cancer, such as disturbance of DNA methylation, is influenced by age, diet, transplacental, environmental and occupational exposure. Its dual nature of being in the same time reversible and transgenerational demands specific approach in interpretation of results. The age-related DNA methylation is associated with different diseases [4] and sex hormone status on DNA methylation in some cancer types [5]. There is a significant gap in understanding of association between the age expected DNA methylation level and cancer [6]. Especially, there is no data on pre- and postmenopausal differences in DNA methylation levels and its impact on cancer development.
Most intensive processes are in the genome of active proliferating cells, such as epithelial cells. Therefore, epithelial cells in various cancers can be a convenient model for studying epigenetic malformation. An example of onychopathology with epithelial tissue damage may be head and neck squamous cell carcinoma (HNSCC).
HNSCC corresponds to the majority of cases (~90%) of head and neck cancers (HNCs) [7]. HNCs are rated 6–7 in global prevalence among cancers, with 700,000 newly registered cases and 470,000 deaths a year [8]. The therapeutic options for HNCs include surgical treatment, radiotherapy and chemotherapy. The management tactics should be decided on personalized basis by a multidisciplinary board comprising an oncological surgeon, a chemotherapist, a radiotherapist, a psychologist, a rehabilitation specialist and a dentist [9,10].
Despite the advanced diagnostics, cancer screening and HPV vaccination programs, 60–70% of the cases are diagnosed as late as in stage III–IV, associated with low life expectancy and high risks of recurrence [11]. So, finding new potential targets for therapy is crucial.
The main risk factor for HNSCC is smoking. In some publications it was shown that smokers have a 10-fold higher risk of developing the disease than non-smokers; smoking in combination with frequent alcohol consumption increases the risk more than 35-fold [12,13,14]. Other risk factors include: ultraviolet and ionizing radiation, various toxic compounds, a weakened immune system, a diet low in vitamins A and B, age over 40, male, some viral infections [15].
DNA methylation also varies due to a number of factors including age and disease status. It was noted that silenced genes are often methylated while active genes remain largely unmethylated [16,17].
It should be noted that DNA methylation is the most studied epigenetic mechanism in HNSCC. It allows to observe changes in methylation patterns both in a whole genome and in individual genes, that can be used to identify new biomarkers of the disease [18]. Various types of biomaterials can be used for the study, including liquid biopsy samples, which provide a noninvasive alternative for early cancer detection. In particular, Zhou C. studied 27 aberrantly methylated genes with altered expression and showed that FAM135B methylation is a favorable independent prognostic marker for overall survival in patients with HNSCC [19].
We have previously investigated of DNA methylation status of some tumor associated genes (CDO1, MEST, RASSF1A, RASSF2, RASSF5 and WIF1) in patients with HNSCC [20]. There were significant differences in levels of DNA methylation between tumor and normal tissues in the CDO1 and WIF1 genes in all groups and subgroups of patients (larynx and other cancers, SCC keratinizing and non-keratinizing, primary and recurrent tumor, smokers and non-smokers) [21]. The methylation level in the CDO1 gene in tumor tissue was significantly increased in the T4 and T3 stage subgroups compared to T2 [22].
We did the investigation on another group of tumor and normal tissue of patients with HNSCC for further research. We chose other cancer associated genes: ATM, APC, RB1, TP53. We also took these two earlier studied genes (CDO1 and WIF1) as they had shown significant results in other patients (Table 1). The selected genes play a significant role in the regulation of cell proliferation, differentiation, and apoptosis, disruption of which can lead to oncopathology.
The aim of this study was to investigate the DNA promotor methylation status of cancer-associated genes (ATM, APC, CDO1, RB1, TP53, WIF1) in patients with HNSCC.
2. Materials and Methods
2.1. Patients characteristics
The study involved 44 patients (34 men and 10 women) with HNSCC treated at A. Tsyb Medical Radiology Center, Obninsk, and at P.A. Herzen Cancer Research Institute, Moscow. The study was approved by A. Tsyb MRC Ethics Committee (approval №634-17.11.2021).
For the first step of their therapy, all patients underwent surgery. The absence of other treatments before surgery was the main inclusion criteria.
The sampling time duration was 6 months. All patients were sampled before starting further therapies. Characteristics of the patients are presented in Table 2. 37 patients were diagnosed with primary HNSCC, and tumor recurrence after therapy was observed in 7 cases. Squamous cell carcinoma of the tongue was observed in 9, of the oral cavity in 6, of the floor of mouth in 3, maxillary sinus in 6 and larynx in 20 patients. The non-keratinizing type of tumor was observed in 28 patients, and the keratinizing type in 16 patients [21]. 26 out of 44 patients were smokers. The mean age of the men was 58 (30÷72) and that of the women was 58 (47÷81) years old. Distribution by age in all patients is shown in Figure 1: 4 patients of 30-40 years (9%), 8 patients of 41-50 years (18%), 9 patients of 52-59 years (20%), 20 patients of 61-70 years (44%) and 4 patients of 71-81 (9%). The patients are classified according to TNM system (Tumor-Node-Metastasis) [22], in which T1 - 2 (5%), T2 - 7 (16%), T3 - 20 (45%), T4 - 15 (34%).
Pre-operative contrast-enhanced CT image of three patients with various diagnoses is shown in Figure 2.
2.2. Sampling
Tumor and normal tissue samples from each patient were obtained during surgery and stored at -20oC.
2.3. DNA Extraction
DNA isolation from biomaterials was performed on microcolumns (K-SORB, № EX-514, Syntol, Russia) according to the manufacturer's instructions.
2.4. DNA methylation analysis.
Bisulfite conversion was performed with the EZ DNA Methylation-Lightning kit (ThermoFisher EpiJET Bisulfite Conversion Kit, K1461, ThermoFisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions.
Methylation of the promoter regions of the genes was performed with the Methylation-Sensitive High-Resolution Melting (MS-HRM) method using the CFX 96 Connect Real-time System (BioRad, USA).
The primers for the reaction were selected using Primer Blast software (Table 3). The ready-mix (PCR-Mix, M-428, Syntol, Russia) was used for two step PCR. Program of amplification was 95°С — 5 min; (95°С — 15 s, 60°С — 30 s, 72°С — 45 s) ×30 cycles; (95°С — 15 s, 50°С — 30 s, 72°С — 45 s) ×25 cycles [23]. Further, the intercalating dye EVA Green (Syntol, Russia) was added to the obtained products. Each sample was run in duplicate. Construction of the melting curve was performed according to the following program: 1st stage - 95° - 30 s; 2nd stage - 60° - 10 min, 3rd stage - melting analysis in the range 60°-90° with 0.2° step. MS-HRM was performed using Precision Melt Analysis Software, version 3 (BioRad, USA). A CFX96 amplifier (BioRad, USA) was used for PCR and MS-HRM. Methylation level was detected by fluorescence expressed in relative fluorescence units (RFU) [24].
2.5. Statistical analysis
Statistical analysis of the data was carried out using R language. The method used is chi-squared test. P values less than 0.05 were considered as statistically significant.
3. Results
The results average of methylation level of the DNA promoter is shown for all patient’s normal and tumor tissues and subgroups in Table 4. Significant differences in DNA methylation level between patient’s tumor and normal tissues were found for CDO1 and WIF1 genes in all persons (p<0.05). Significant differences are also observed for CDO1 and WIF1 genes in different subgroups of patient’s tumor and normal tissues (p<0.05). The exception is a subgroup of patients with T2 and non-smokers for the CDO1 gene. As for the WIF1 gene, the exceptions are subgroups of patients with T2, T3, patients after the age of 50 and non-smokers. Methylation level of the DNA promoter of ATM, APC, RB1, TP53 genes was not significant. Furthermore, no significant differences were found between the subgroups (T2, T3, T4 stage, with primary and recurrent lesion, non-keratinizing and keratinizing SCC, age before and after 50 years, smokers and non-smokers) of the examined patients.
Since then, the significant differences have been observed in the level of methylation between patient’s tumor and normal tissue only in the CDO1 and WIF1 genes. We analyzed the relationship between these data and age in samples of patients with different TNM system (Figure 3). There was a significant positive correlation in terms of the level of methylation in the gene CDO1 with age in normal tissue in patients with T3 (Age/Normal: y = -0,3102 + 0,0085*x; r = 0,66; p = 0,002; r2 = 0,43). The correlation between the level of methylation in the gene CDO1 with age in tumor tissue in patients with T3 was not significant but tend to be different (Age/Tumor3: y = -0,0641 + 0,0086*x; r = 0,42; p = 0,07; r2 = 0,17). There was a significant correlation between methylation level in the WIF1 gene and age in tumor tissue samples in the subgroup with T3 (Age/Tumor3: y = -0,4654 + 0,013*x; r = 0,59; p = 0,006; r2 = 0,35) and in normal tissue samples in the subgroup with T4 (Age/Normal: y = -0,0002 + 0,0019*x; r = 0,52; p = 0,05; r2 = 0,27).
4. Discussion
Epigenetic disorders in tumor tissue cells in various types of oncopathology are as important as mutations, according to abundant literature data from the last decade. Early-stage epigenetic alterations can be identified and exploited to diagnose tumors early and predict cancer predict the prognosis of cancer [25]. DNA methylation can be used as a molecular target for cancer treatment since it is reversible by pharmacological inhibition of DNA methyltransferase.
Our results from the evaluation of promoter methylation in normal and tumor tissues of HNSCC patients revealed significant differences for CDO1 and WIF1 genes in all patients and in the studied subgroups.
Many studies have shown an increased level of promoter methylation and, consequently, suppression of CDO1 gene in various tumor cells [26]. In primary breast cancer patients and those with prostate cancer, methylation level of the CDO1 gene's promoter region correlates strongly with tumor development and can be utilized as a reliable prognostic indicator [27]. Our findings are in line with the evidence from the literature since both the general group of patients with HNSCC and different subgroups had considerably greater levels of CDO1 gene promoter methylation in tumor tissue than in normal tissue. Moreover, significant differences were found between smokers and non-smokers. Gene regulation involves CDO1 promoter methylation. High levels of promoter CDO1 methylation have been seen in Barrett esophageal adenocarcinoma and prostate cancer patients, respectively [28, 29].
Non-small cell lung cancer frequently exhibits hypermethylation of the WIF1 (Wnt inhibitory factor-1) promoter region, despite the fact that patient characteristics such as age, sex, and smoking history are unrelated to the methylation status [30]. It has been established that Wnt signal transduction dysregulation can be one of the factors contributing to head and neck cancer. Inhibition of Wnt signaling induces apoptosis and inhibits tumor growth in many cancer types [31]. Methylation of WIF1 gene acting as one of the antagonists of this pathway is often associated with the development of this pathology [32]. The gene with the highest frequency of methylation in oral carcinomas was WIF1. WIF1 is methylated in 18% of patients with oral squamous cell carcinoma [33], 35% of patients with tongue carcinoma [34], and significantly methylated in nasopharyngeal tumors [35]. In this study substantial variations in the average WIF1 promoter methylation levels between tumor and normal tissues for both the entire patient population and the subgroups. It should be noted that there were significant differences for this parameter in the tumor tissue in the subgroups of patients with keratinizing and non-keratinizing HNSCC.
Mutations in the genes encoding proteins in the Wnt (Wingless/Int-1) pathway are rare in HNSCC, so this pathway is not believed to be significant for the pathogenesis of head and neck carcinomas [36]. It is worth mentioning that there are very few studies on methylation levels of the Wnt gene. Our data indicated no significant differences in normal and tumor tissues of patients in terms of the DNA methylation level of the Wnt gene.
It is well known that DNA methylation has association with cancer and ageing. Aberrant DNA methylation frequently coexists with ageing and illnesses, such as cancer. DNA methylation status can become disrupted according to age and certain disease stage. For example, promoter-specific hypermethylation and concurrent gene silence are linked to a wide range of malignancies [37]. We found a significant correlation between methylation level for gene CDO1 and WIF 1 in normal and tumor tissue and age only in subgroups with T3 and T4. These data indicate that there is some dependence of the level of methylation on the age of the patient. We may suppose that the individual epigenetic features of the patient’s genome play an important role in the results obtained. Perhaps the study should be repeated in a larger sample size and/or patients should be stratified according to other criteria.
Adenomatous Polyposis Coli (APC) is a tumor suppressor gene that, through Wnt/β-catenin signaling pathway signaling, inhibits cell proliferation. APC interacts with DNA repair proteins, DNA replication proteins, tubulin, and other components. It is not expressed in some types of cancer, in particular prostate, breast and colorectal cancer. Lack of expression is associated with reduced survival in cancer patients. Besides APC is involved in the regulation of response to chemotherapy in cancer cells [38]. The WNT/ β-catenin signaling pathway is activated by APC mutations in colorectal, endometrial, and prostate malignancies [39]. Hypermethylation of this gene has been detected in oral squamous cell carcinoma samples in some studies, but not in patients with head and neck cancers. Additionally, there were no differences in the methylation of the APC gene between HNSCC patients and healthy individuals. APC promoter was methylated in 7% of DNA samples taken from plasma of a population free of cancer. It was unrelated to a number of other putative risk factors, such as age, tobacco and alcohol use, family history of cancer, diet, and nutrition. Other research similarly failed to find a connection between clinical traits and outcomes, such as survival, and aberrant methylation [40,41]. Our results showed no significant differences in DNA methylation level for APC gene between patient’s tumor and normal tissues.
A well-known tumor suppressor, the Ataxia-telangiectasia-mutated (ATM) gene product, is essential for maintaining genomic stability. The mutated form of ATM gene is involved in cell cycle control, apoptosis, oxidative stress, and telomere maintenance, and its role as a risk factor for cancer development is well established. To ascertain how ATM gene mutations affect breast cancer risk, several investigations have been conducted [42,43]. In HNSCC, the ATM gene promoter is a target for abnormal hypermethylation. Reduced ATM function through epigenetic silencing is a likely mechanism contributing to HNSCC and, possibly, other tumor types, given the significant role that ATM plays in the maintenance of genome integrity and the causal role that genome instability plays in cancer onset and progression. It was found that hypermethylation of the ATM promoter is significantly correlated with a decrease in the average survival rate of patients. Furthermore, it was discovered that this link was unrelated to other possible predictive elements such tumor size, lymph node condition, clinical stage, and history of tobacco and alcohol use. Thus, this data seemingly indicates that hypermethylation of the ATM promoter can be used as a prognostic factor in HNSCC [44]. However, our data revealed no significant differences in DNA methylation level for ATM gene in patient’s tumor and normal tissues.
Functional properties caused by Tumor Protein P53 (TP53) gene mutations are involved in cancer development and progression [45,46]. One of the most prevalent changes in the TP53 gene in human tumors is somatic mutation, while the underlying cause of Li-Fraumeni syndrome, which predisposes to a variety of early-onset cancers, is germline mutation. Additionally, TP53 gene alterations are possible prognostic and predictive indicators, as well as prospective drug targets [47]. The majority of somatic genomic changes in HNSCC are mutations in the TP53 gene, according to a thorough integrative genomic study. Invasion, metastasis, genomic instability, and cancer cell proliferation are all facilitated by TP53 mutations, which can result in a loss of wild-type p53 activity or an increase in those activities. Interestingly, aggressiveness and worse survival following surgical treatment of HNSCC are related with disruptive TP53 mutations in tumor DNA [48]. Harris C.C. proposed that genetic analysis of the p53 gene in exfoliated cells detected in either body fluids or tissue biopsies may identify people at elevated cancer risk because mutations in the p53 gene can arise in precancerous lesions in the lung, breast, esophagus, and colon [49]. Hypermethylation of the promoter region of the tumor suppressor gene TP53 is often associated with a decrease in its activity, and can even lead to its silence, and, thus, to the loss of its function. This, in turn, contributes to the process of malignant transformation [50]. Unfortunately, our research showed no significant differences in DNA methylation level for TP53 gene between patient’s tumor and normal tissues.
The tumor suppressor gene retinoblastoma gene (Rb1) is essential for controlling the cell cycle by increasing G1/S arrest and growth limitation by blocking the E2F transcription factor. Even aneuploidy can result from RB1 loss, dramatically raising the chance of developing cancer. The RB1 gene is a part of a large gene family that includes RBL1 and RBL2, each of the three encoding structurally related proteins indicated as pRb, p107, and p130, respectively [51]. One frameshift and seven non-synonymous missense mutations were discovered in RB1 (pocket domain and spacer region) sequencing analysis. In the HNC tumor samples, RB1 promoter methylation study showed that 16% of its cytosines (3% in CpG) were methylated [52]. Sabir M. and colleagues indicate that both genetic and epigenetic RB1 changes may contribute to the pathogenesis of HNSCC among the Pakistani population [53]. However, our study showed no significant differences in DNA methylation level for Rb1 gene between patient’s tumor and normal tissues.
There were found no significant differences in all genes (T2, T3, T4 stage, with primary and recurrent lesion, non-keratinizing and keratinizing SCC, age before and after 50 years, smokers and non-smokers) of the patients. This may be due to the small number of persons examined.
Thus, we can conclude that the changes in the expression of the CDO1 and WIF1 genes can affect the mechanism of the occurrence and development of HNSCC and can be considered as prognostic and diagnostic markers for this pathology.
Future application of molecular-genetic and epigenetic studies on NHSCC can become the basis for creating target therapy and contribute in personalized medicine.
Author Contributions
Kurevlev S., molecular-genetic investigation, development of research design; Aghajanyan A. data analysis and interpretation, article writing, review of publications on the article topic, statistical analysis investigation; Tskhovrebova L. data analysis and interpretation, article writing, review of publications on the article topic; Gordon K. collection of biomaterials, clinical data analysis; Polyakov A. collection of biomaterials, clinical data analysis; Ratushny M. collection of biomaterials, clinical data analysis; Fatkhutdinov T., scientific editing supervision; All authors have read and agreed to the published version of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The study was done with the financial support of the Russian Federation represented by the Ministry of Education and Science of Russia; Agreement dated October 7, 2021 No. 075-15-2021-1356 (internal number of the Agreement 15.SIN.21.0011); (ID: RF 0951.61321X0012).
Institutional Review Board Statement
The study protocol was approved by the Biomedical Ethics Committee of the A.F. Tsyb Medical Radiology Research Center Branch of the Federal State Institution "National Medical Research Center of Radiology" of the Ministry of Health of the Russian Federation (protocol № 634 from 17.11.2021).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Esteller, M.; Corn, P.G.; Baylin, S.B.; Herman, J.G. A gene hypermethylation profile of human cancer. Cancer Res. 2001, 61, 3225–3229. [Google Scholar] [PubMed]
- Gupta, S.; Kumar, P.; Maini, J.; Kaur, H. Epigenetic Biomarkers in Head and Neck Cancer. J. Cancer Genet. Biomarkers 2018, 1, 41–50. [Google Scholar] [CrossRef]
- Castilho, R.M.; Squarize, C.H.; Almeida, L.O. Epigenetic Modifications and Head and Neck Cancer: Implications for Tumor Progression and Resistance to Therapy. Int. J. Mol. Sci. 2017, 18, 1506. [Google Scholar] [CrossRef]
- Salameh, Y.; Bejaoui, Y.; El Hajj, N. DNA Methylation Biomarkers in Aging and Age-Related Diseases. Front. Genet. 2020, 11, 171. [Google Scholar] [CrossRef]
- Johansson, A.; Palli, D.; Masala, G.; Grioni, S.; Agnoli, C.; Tumino, R.; Giurdanella, M.C.; Fasanelli, F.; Sacerdote, C.; Panico, S.; et al. Epigenome-wide association study for lifetime estrogen exposure identifies an epigenetic signature associated with breast cancer risk. Clin. Epigenetics 2019, 11, 66. [Google Scholar] [CrossRef]
- Chen, L.; Ganz, P.A.; Sehl, M.E. DNA Methylation, Aging, and Cancer Risk: A Mini-Review. Front. Bioinform. 2022, 2. [Google Scholar] [CrossRef]
- Rahman, Q.B.; Iocca, O.; Kufta, K.; Shanti, R.M. Global Burden of Head and Neck Cancer. Oral Maxillofac. Surg. Clin. North Am. 2020, 32, 367–375. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Hashim, D.; Genden, E.; Posner, M.; Hashibe, M.; Boffetta, P. Head and neck cancer prevention: from primary prevention to impact of clinicians on reducing burden. Ann. Oncol. 2019, 30, 744–756. [Google Scholar] [CrossRef]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef]
- Pfister, D.G.; Spencer, S.; Adelstein, D.; Adkins, D.; Anzai, Y.; Brizel, D.M.; Bruce, J.Y.; Busse, P.M.; Caudell, J.J.; Cmelak, A.J.; et al. Head and Neck Cancers, Version 2.2020, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2020, 18, 873–898. [Google Scholar] [CrossRef] [PubMed]
- Marcu, L.G.; Yeoh, E. A review of risk factors and genetic alterations in head and neck carcinogenesis and implications for current and future approaches to treatment. J. Cancer Res. Clin. Oncol. 2009, 135, 1303–1314. [Google Scholar] [CrossRef] [PubMed]
- WHO. int [Electronic resourse]: The world health report 2002: Reducing Risks, Promoting Healthy Life. – Available from: http://www.who.int/ whr/2002/en/whr02_en.pdf?ua=1. – Date of access: 20.06.2018. ISBN: 9241562072.
- WHO.int [Electronic resourse]: WHO global report on trends in tobacco smoking 2000–2025, 2015. – Available from: apps.who.int/iris/bitstre am/10665/156262/1/9789241564922_eng. – Date of access: 20.06.2018.
- Global Cancer Observatory. International Agency for Research on Cancer. World Health Organization. Available at: https://gco.iarc.fr/. Accessed on June 06, 2021.
- Demokan, S.; Dalay, N. Role of DNA methylation in head and neck cancer. Clin. Epigenetics 2011, 2, 123–150. [Google Scholar] [CrossRef] [PubMed]
- Maegawa, S.; Lu, Y.; Tahara, T.; Lee, J.T.; Madzo, J.; Liang, S.; Jelinek, J.; Colman, R.J.; Issa, J.-P.J. Caloric restriction delays age-related methylation drift. Nat. Commun. 2017, 8, 539. [Google Scholar] [CrossRef]
- Ovchinnikov, D.A.; Cooper, M.A.; Pandit, P.; Coman, W.B.; Cooper-White, J.J.; Keith, P.; Wolvetang, E.J.; Slowey, P.D.; Punyadeera, C. Tumor-suppressor Gene Promoter Hypermethylation in Saliva of Head and Neck Cancer Patients. Transl. Oncol. 2012, 5, 321–326. [Google Scholar] [CrossRef]
- Zhou, C.; Ye, M.; Ni, S.; Li, Q.; Ye, D.; Li, J.; Shen, Z.; Deng, H. DNA methylation biomarkers for head and neck squamous cell carcinoma. Epigenetics 2018, 13, 398–409. [Google Scholar] [CrossRef]
- Kurevlev, S.V.; Tskhovrebova, L.V.; Aghajanyan, A.V.; Fatkhudinov, T.K.; Gordon, K.B.; Azova, M.M. Methylation of the tumor associated genes in head and neck squamous cell carcinoma. Head Neck Tumors (HNT) 2023, 12, 61–70, (In Russ.). [Google Scholar] [CrossRef]
- Mete, O.; Wenig, B.M. Update from the 5th Edition of the World Health Organization Classification of Head and Neck Tumors: Overview of the 2022 WHO Classification of Head and Neck Neuroendocrine Neoplasms. Head Neck Pathol. 2022, 16, 123–142. [Google Scholar] [CrossRef]
- Amin MB, Edge S, Greene F, Byrd DR, Brookland RK, Washington MK, Gershenwald JE, Compton CC, Hess KR, et al. (Eds.). AJCC Cancer Staging Manual. 2017. (8th edition). Springer International Publishing: American Joint Commission on Cancer.
- Krasnyi, A.M.; Kurevlev, S.V.; Sadekova, A.А.; Sefihanov, T.G.; Kometova, V.V.; Rodionov, V.V. Primary tumor gene methylation profile in patients with luminal HER2-negative breast cancer when metastasizing to regional lymph nodes. (Russian). Biomedicinskaya himia. 2021, 67, 88–94. [Google Scholar]
- Słomka, M.; Sobalska-Kwapis, M.; Wachulec, M.; Bartosz, G.; Strapagiel, D. High Resolution Melting (HRM) for High-Throughput Genotyping—Limitations and Caveats in Practical Case Studies. Int. J. Mol. Sci. 2017, 18, 2316. [Google Scholar] [CrossRef]
- Yamashita, K.; Hosoda, K.; Nishizawa, N.; Katoh, H.; Watanabe, M. Epigenetic biomarkers of promoter DNA methylation in the new era of cancer treatment. Cancer Sci. 2018, 109, 3695–3706. [Google Scholar] [CrossRef] [PubMed]
- Brait, M.; Ling, S.; Nagpal, J.K.; Chang, X.; Park, H.L.; Lee, J.; Okamura, J.; Yamashita, K.; Sidransky, D.; Kim, M.S. Cysteine Dioxygenase 1 Is a Tumor Suppressor Gene Silenced by Promoter Methylation in Multiple Human Cancers. PLOS ONE 2012, 7, e44951. [Google Scholar] [CrossRef] [PubMed]
- Minatani, N.; Waraya, M.; Yamashita, K.; Kikuchi, M.; Ushiku, H.; Kojo, K.; Ema, A.; Nishimiya, H.; Kosaka, Y.; Katoh, H.; et al. Prognostic Significance of Promoter DNA Hypermethylation of cysteine dioxygenase 1 (CDO1) Gene in Primary Breast Cancer. PLOS ONE 2016, 11, e0144862–e0144862. [Google Scholar] [CrossRef] [PubMed]
- Meller, S.; Zipfel, L.; Gevensleben, H.; Dietrich, J.; Ellinger, J.; Majores, M.; Stein, J.; Sailer, V.; Jung, M.; Kristiansen, G.; et al. CDO1 promoter methylation is associated with gene silencing and is a prognostic biomarker for biochemical recurrence-free survival in prostate cancer patients. Epigenetics 2016, 11, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Kojima, K.; Yamashita, K.; Ushiku, H.; Katoh, H.; Ishii, S.; Tanaka, T.; Yokoi, K.; Suzuki, M.; Ooizumi, Y.; Igarashi, K.; et al. The clinical significance of cysteine dioxygenase type 1 methylation in Barrett esophagus adenocarcinoma. Dis. Esophagus 2017, 30, 1–9. [Google Scholar] [CrossRef]
- Yang, T.-M.; Leu, S.-W.; Li, J.-M.; Hung, M.-S.; Lin, C.-H.; Lin, Y.-C.; Huang, T.-J.; Tsai, Y.-H.; Yang, C.-T. WIF-1 promoter region hypermethylation as an adjuvant diagnostic marker for non-small cell lung cancer-related malignant pleural effusions. J. Cancer Res. Clin. Oncol. 2008, 135, 919–924. [Google Scholar] [CrossRef]
- He, B.; You, L.; Uematsu, K.; Xu, Z.; Lee, A.Y.; Matsangou, M.; McCormick, F.; Jablons, D.M. A Monoclonal Antibody against Wnt-1 Induces Apoptosis in Human Cancer Cells. Neoplasia 2004, 6, 7–14. [Google Scholar] [CrossRef]
- Paluszczak, J.; Sarbak, J.; Kostrzewska-Poczekaj, M.; Kiwerska, K.; Jarmuż-Szymczak, M.; Grenman, R.; Mielcarek-Kuchta, D.; Baer-Dubowska, W. The negative regulators of Wnt pathway—DACH1, DKK1, and WIF1 are methylated in oral and oropharyngeal cancer and WIF1 methylation predicts shorter survival. Tumor Biol. 2015, 36, 2855–2861. [Google Scholar] [CrossRef]
- Pannone, G.B.P.; Santoro, A.; Franco, R.; Aquino, G.; Longo, F.; Botti, G. WNT pathway in oral cancer: epigenetic inactivation of WNT-inhibitors. Oncol Rep. 2010, 24, 1035–1041. [Google Scholar]
- Supic, G.; Kozomara, R.; Jovic, N.; Zeljic, K.; Magic, Z. Hypermethylation of RUNX3 but not WIF1 gene and its association with stage and nodal status of tongue cancers. Oral Dis. 2011, 17, 794–800. [Google Scholar] [CrossRef]
- Chan, S.L.; Cui, Y.; van Hasselt, A.; Li, H.; Srivastava, G.; Jin, H.; Ng, K.M.; Wang, Y.; Lee, K.Y.; Tsao, G.S.W.; et al. The tumor suppressor Wnt inhibitory factor 1 is frequently methylated in nasopharyngeal and esophageal carcinomas. Lab. Investig. 2007, 87, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Yeh, K.-T.; Chang, J.-G.; Lin, T.-H.; Wang, Y.-F.; Shih, M.-C.; Lin, C.-C. Correlation between protein expression and epigenetic and mutation changes of Wnt pathway-related genes in oral cancer. Int. J. Oncol. 2003, 23, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.E.; Davies, T.J.; Mc Auley, M.T. The role of DNA methylation in ageing and cancer. Proc. Nutr. Soc. 2018, 77, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Stefanski, C.D.; Prosperi, J.R. Wnt-Independent and Wnt-Dependent Effects of APC Loss on the Chemotherapeutic Response. Int. J. Mol. Sci. 2020, 21, 7844. [Google Scholar] [CrossRef]
- Katoh, M.; Katoh, M. Molecular genetics and targeted therapy of WNT-related human diseases (Review). Int. J. Mol. Med. 2017, 40, 587–606. [Google Scholar] [CrossRef]
- Strzelczyk, J.K.; Krakowczyk. ; Owczarek, A.J. Aberrant DNA methylation of the p16, APC, MGMT, TIMP3 and CDH1 gene promoters in tumours and the surgical margins of patients with oral cavity cancer. J. Cancer 2018, 9, 1896–1904. [Google Scholar] [CrossRef]
- Ali, J.; Sabiha, B.; Jan, H.U.; Haider, S.A.; Khan, A.A.; Ali, S.S. Genetic etiology of oral cancer. Oral Oncol. 2017, 70, 23–28. [Google Scholar] [CrossRef]
- Stucci, L.S.; Internò, V.; Tucci, M.; Perrone, M.; Mannavola, F.; Palmirotta, R.; Porta, C. The ATM Gene in Breast Cancer: Its Relevance in Clinical Practice. Genes 2021, 12, 727. [Google Scholar] [CrossRef]
- Marabelli, M.; Cheng, S.-C.; Parmigiani, G. Penetrance ofATMGene Mutations in Breast Cancer: A Meta-Analysis of Different Measures of Risk. Genet. Epidemiology 2016, 40, 425–431. [Google Scholar] [CrossRef]
- Ai, L.; Vo, Q.N.; Zuo, C.; Li, L.; Ling, W.; Suen, J.Y.; Hanna, E.; Brown, K.D.; Fan, C.-Y. Ataxia-Telangiectasia-Mutated ( ATM ) Gene in Head and Neck Squamous Cell Carcinoma: Promoter Hypermethylation with Clinical Correlation in 100 Cases. Cancer Epidemiology Biomarkers Prev. 2004, 13, 150–156. [Google Scholar] [CrossRef]
- Lindemann, A.; Takahashi, H.; Patel, A.; Osman, A.; Myers, J. Targeting the DNA Damage Response in OSCC with TP53 Mutations. J. Dent. Res. 2018, 97, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Takahashi, S.; Ouchi, K.; Otsuki, Y.; Wakayama, S.; Ishioka, C. Different impacts of TP53 mutations on cell cycle-related gene expression among cancer types. Sci. Rep. 2023, 13, 4868. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 Mutations in Human Cancers: Origins, Consequences, and Clinical Use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008–a001008. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.; Khandelwal, A.R.; Wolf, G.T.; Rodrigo, J.P.; Mäkitie, A.A.; Saba, N.F.; Forastiere, A.A.; Bradford, C.R.; Ferlito, A. TP53 mutations in head and neck cancer. Mol. Carcinog. 2022, 61, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.C. p53 tumor suppressor gene: at the crossroads of molecular carcinogenesis, molecular epidemiology, and cancer risk assessment. Environ. Heal. Perspect. 1996, 104 (Suppl. 3), 435–439. [Google Scholar] [CrossRef]
- Golusinski, P.; Lamperska, K.; Pazdrowski, J.; Golusinski, W. Analiza występowania mutacji w obrębie genu TP53 u chorych na raka płaskonabłonkowego głowy i szyi [Analysis of mutations within the TP53 gene in patients with squamous cell carcinoma of the head and neck]. Otolaryngol. Polska 2011, 65, 114–121. [Google Scholar] [CrossRef]
- Di Fiore, R.; D'Anneo, A.; Tesoriere, G.; Vento, R. RB1 in cancer: Different mechanisms of RB1 inactivation and alterations of pRb pathway in tumorigenesis. J. Cell. Physiol. 2013, 228, 1676–1687. [Google Scholar] [CrossRef]
- Rusin, P.; Markiewicz, L.; Majsterek, I. Uwarunkowania genetyczne nowotworów głowy i szyi [Genetic predeterminations of head and neck cancer]. Postepy Hig Med Dosw (Online). Polish. 2008, 62, 490–501. [Google Scholar] [PubMed]
- Sabir, M.; Baig, R.M.; Ali, K.; Mahjabeen, I.; Saeed, M.; Kayani, M.A. Retinoblastoma (RB1) pocket domain mutations and promoter hyper-methylation in head and neck cancer. Cell. Oncol. 2014, 37, 203–213. [Google Scholar] [CrossRef]
Figure 1.
Distribution of all patients according to the years (58,68±11,05).
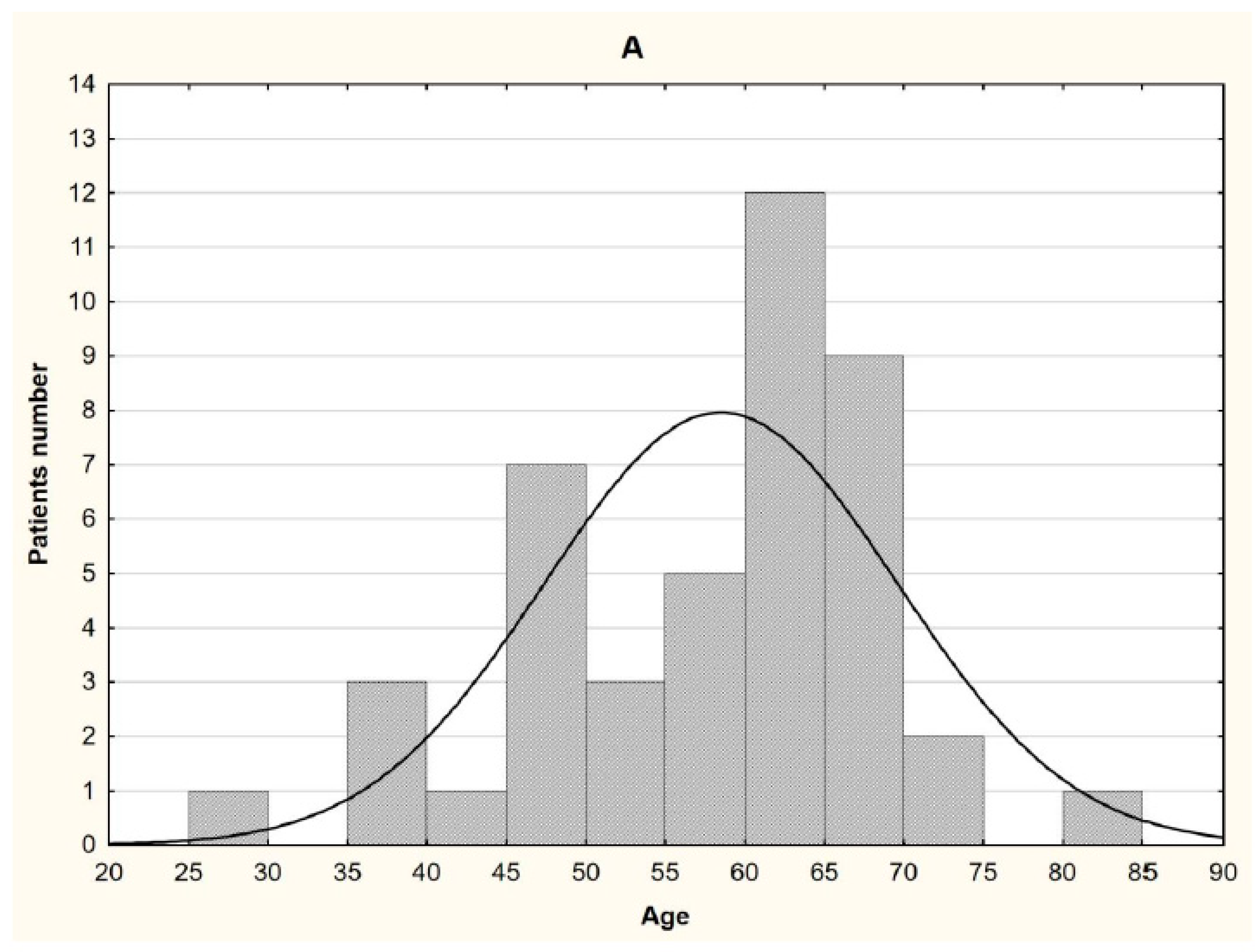
Figure 2.
Pre-operative contrast-enhanced CT images of three patients. A. Patient №37 - 49-year-old male diagnosed with locally advanced (T3N1M0) stage of laryngeal squamous cell non-keratinizing cancer. B. Patient №17 - 66-year-old female diagnosed with locally advanced (T3N1M0) stage squamous cell non-keratinizing cancer of mouth floor. C. Patient №41 - 72-year-old male diagnosed with locally advanced (T4aN0M0) stage laryngeal squamous cell keratinizing cancer.
Figure 2.
Pre-operative contrast-enhanced CT images of three patients. A. Patient №37 - 49-year-old male diagnosed with locally advanced (T3N1M0) stage of laryngeal squamous cell non-keratinizing cancer. B. Patient №17 - 66-year-old female diagnosed with locally advanced (T3N1M0) stage squamous cell non-keratinizing cancer of mouth floor. C. Patient №41 - 72-year-old male diagnosed with locally advanced (T4aN0M0) stage laryngeal squamous cell keratinizing cancer.
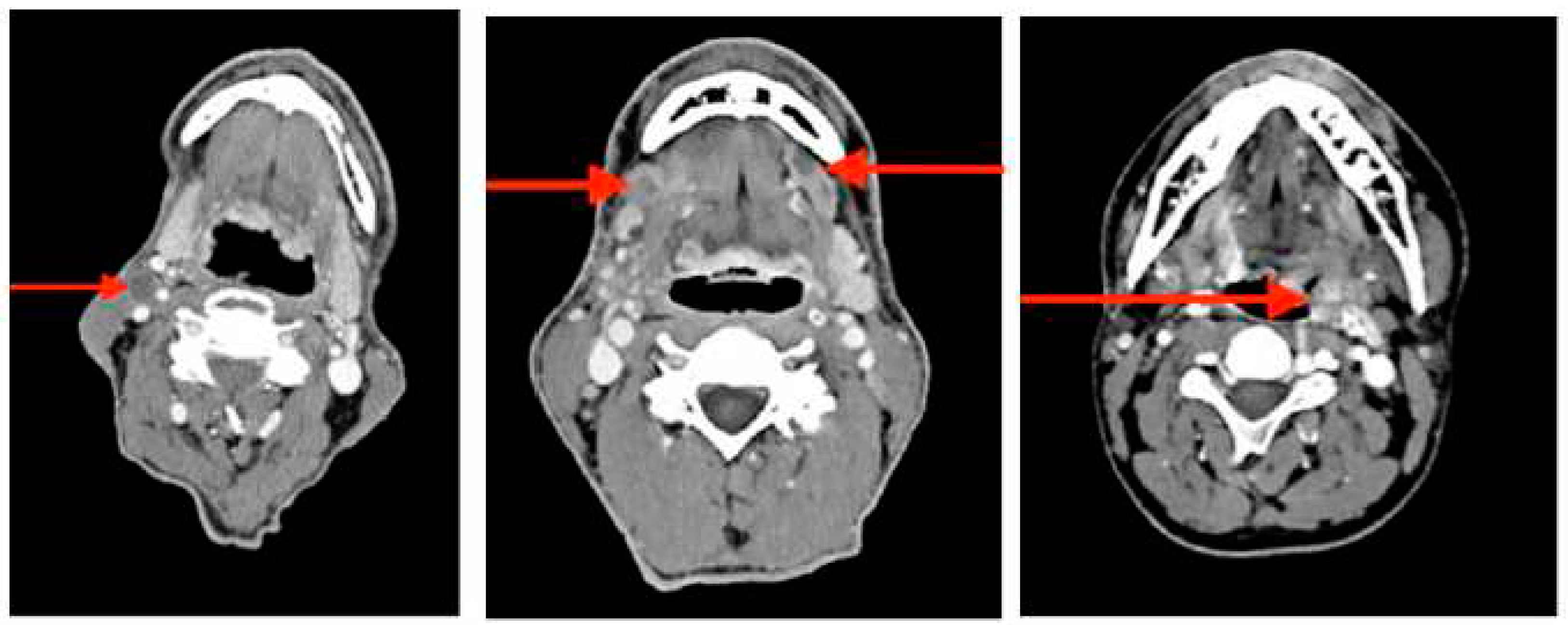
Figure 3.
The relationship between individual DNA methylation level in tumor and normal tissue in genes (CDO1, WIF1) and age in patients with different TNM classification. А. CDO1 gene, patients with T3; B. WIF1 gene, patients with T3; C. WIF1 gene, patients with T4.
Figure 3.
The relationship between individual DNA methylation level in tumor and normal tissue in genes (CDO1, WIF1) and age in patients with different TNM classification. А. CDO1 gene, patients with T3; B. WIF1 gene, patients with T3; C. WIF1 gene, patients with T4.
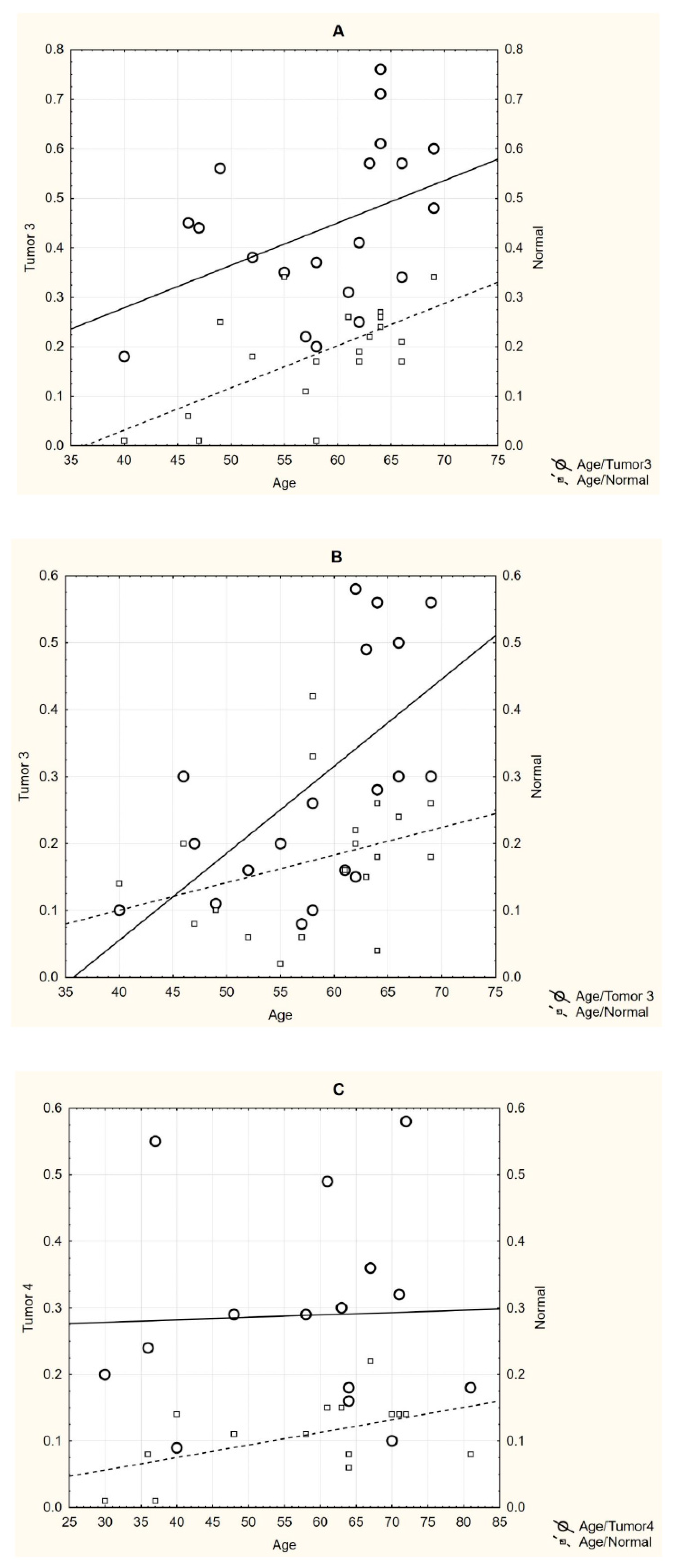

Table 1.
Characteristic of studied genes.
| Symbol | Gene name | Location | Exon count | Gene ID | Transcripts | MIM | Gene type | Gene function | References |
|---|---|---|---|---|---|---|---|---|---|
|
APC GS; DP2; DP3; BTPS2; DESMD; DP2.5; PPP1R46 |
Adenomatous Polyposis Coli | 5q22.2 | 20 | 5624 | NM_000038.6 | 611731 | protein-coding | tumor suppressor | 37; 39 |
|
ATM AT1; ATA; ATC; ATD; ATE; ATDC; TEL1; TELO1 |
Ataxia-telangiectasia-mutated | 11q22.3 | 67 | 472 | NM_000051.4 | 607585 | protein-coding | cell cycle regulation. | 42-44 |
|
CDO1 (CDO-I) |
Cysteine dioxygenase type 1 | 5q22.3 | 9 | 1036 | NM_001323565.2 | 603943 | protein-coding | tumor suppressor | 26-28 |
|
TP53 (P53; BCC7; LFS1; BMFS5; TRP53) |
Tumor Protein P53 | 17p13.1 | 11 | 7157 | NM_000546.6. | 191170 | protein-coding | tumor suppressor | 45-50 |
|
RB1 (RB; pRb; OSRC; pp110; p105-Rb; PPP1R130; p110-RB1) |
Retinoblastoma 1 | 13q14.2 | 27 | 5925 | NM_000321.3... | 614041 | protein-coding | cell cycle regulator, tumor suppressor |
51-53 |
|
WIF1 (WIF-1) |
WNT inhibitory factor 1 | 12q14.3 | 10 | 11197 | NM_007191.5 | 605186 | protein-coding | tumor suppressor | 19; 30-36 |
Table 2.
Characteristics of patient.
| Patient ID | Tumor origin | ICD-10 | TNM classification |
Type of lesion | Histological glade | Gender | Age | Smoker |
|---|---|---|---|---|---|---|---|---|
| 1 | Tongue n=9 | C02.0 | T1N0M0 | Prim | SCC non-keratinizing | М | 50 | y |
| 2 | C02.1 | T2N0M0 | Prim | SCC non-keratinizing | F | 59 | n | |
| 3 | C02.1 | T3N0M0 | Rec | SCC keratinizing | F | 66 | y | |
| 4 | C02.1 | T3N0M0 | Rec | SCC non-keratinizing | F | 47 | n | |
| 5 | C02.1 | T3N0M0 | Prim | SCC keratinizing | М | 63 | n | |
| 6 | C02.1 | T3N0M0 | Prim | SCC keratinizing | М | 40 | n | |
| 7 | C02.1 | T3N0M0 | Prim | SCC keratinizing | M | 46 | n | |
| 8 | C02.1 | T3N1M0 | Prim | SCC non-keratinizing | М | 52 | n | |
| 9 | C02.1 | T4N2M0 | Prim | SCC keratinizing | М | 36 | n | |
| 10 | Oral cavity n=6 | C03.0 | T2N0M0 | Prim | SCC non-keratinizing | F | 59 | n |
| 11 | C03.0 | T3N0M0 | Prim | SCC keratinizing | M | 69 | y | |
| 12 | C03.0 | T4N0M0 | Prim | SCC keratinizing | М | 37 | y | |
| 13 | C03.1 | T4N0M0 | Prim | SCC non-keratinizing | М | 30 | n | |
| 14 | C03.1 | T4N0M0 | Prim | SCC non-keratinizing | F | 63 | n | |
| 15 | C03.1 | T4N0M0 | Prim | SCC non-keratinizing | F | 48 | n | |
| 16 | Floor of mouth n=3 | C04.1 | T2N0M0 | Prim | SCC keratinizing | F | 67 | y |
| 17 | C04.1 | T3N1M0 | Prim | SCC non-keratinizing | F | 66 | y | |
| 18 | C04.1 | T4N0M0 | Prim | SCC keratinizing | F | 64 | y | |
| 19 | Maxillary sinus n=6 | C31.0 | T3N0M0 | Prim | SCC non-keratinizing | M | 58 | y |
| 20 | C31.1 | T3N0M0 | Prim | SCC non-keratinizing | M | 55 | y | |
| 21 | C31.8 | T3N0M0 | Rec | SCC non-keratinizing | M | 64 | y | |
| 22 | C31.0 | T4N0M0 | Rec | SCC non-keratinizing | M | 41 | n | |
| 23 | С31.8 | T4aN0M0 | Prim | SCC non-keratinizing | M | 61 | n | |
| 24 | C31.0 | T4aN0M0 | Prim | SCC non-keratinizing | M | 67 | n | |
| 25 | Larynx n=20 | C32.1 | T1N2M0 | Prim | SCC non-keratinizing | М | 48 | n |
| 26 | C32.0 | T2N0M0 | Prim | SCC non-keratinizing | F | 55 | n | |
| 27 | С32.8 | T2N0M0 | Prim | SCC non-keratinizing | M | 70 | n | |
| 28 | С32.0 | T2N0M0 | Prim | SCC non-keratinizing | M | 69 | y | |
| 29 | C32.0 | T2N0M0 | Prim | SCC non-keratinizing | М | 58 | y | |
| 30 | C32.0 | T3N0M0 | Prim | SCC keratinizing | M | 64 | y | |
| 31 | C32.0 | T3N0M0 | Prim | SCC non-keratinizing | M | 62 | n | |
| 32 | C32.0 | T3N0M0 | Prim | SCC keratinizing | М | 62 | n | |
| 33 | C32.8 | T3N0M0 | Rec | SCC non-keratinizing | М | 64 | y | |
| 34 | C32.8 | T3N0M0 | Prim | SCC non-keratinizing | М | 69 | y | |
| 35 | C32.8 | T3N0M0 | Prim | SCC non-keratinizing | М | 58 | y | |
| 36 | C32.9 | T3N0M0 | Prim | SCC keratinizing | M | 61 | n | |
| 37 | C32.8 | T3N1M0 | Prim | SCC non-keratinizing | M | 49 | y | |
| 38 | C32.8 | T3N0M0 | Rec | SCC non-keratinizing | М | 69 | n | |
| 39 | C32.0 | T4N2M0 | Prim | SCC non-keratinizing | М | 64 | n | |
| 40 | C32.0 | T4N0M0 | Prim | SCC keratinizing | М | 71 | y | |
| 41 | C32.8 | T4aN0M0 | Prim | SCC keratinizing | M | 72 | y | |
| 42 | C32.8 | T4aN0M0 | Prim | SCC non-keratinizing | M | 58 | n | |
| 43 | С32.8 | T4aN2aM0 | Prim | SCC non-keratinizing | M | 70 | y | |
| 44 | C32.8 | T4aN2bM0 | Rec | SCC non-keratinizing | F | 81 | n |
*M - male, F - female, SCC - squamous cell carcinoma, Prim - primary tumor, Rec - recurrent tumor, y - yes, n – no, ICD-10 - International Statistical Classification of Diseases and Related Health Problems.
Table 3.
Primer sequences used for Methylation-Specific PCR (MSP). * bp – base pair.
| Gene | Forward primer sequence (5’ → 3’) |
Reverse primer sequence (5’ → 3’) |
Product size (bp*) |
|---|---|---|---|
| ATM | GTTGGTTATTGGTGGATATGG | TAATTCCAAAACCCAAACTCTTAAC | 696 |
| APC | GTTGGTTATTGGTGGATATGG | AACCTACAAAACCAAAAACCAACTA | 600 |
| CDO1 | GGGAGGATGA ATTTTATAGATTTG |
TAAACTTCCATA ATAACCTACACCTC |
396 |
| RB1 | GATAGGGATGAGGTTTATAGTTATTTATTA | AAAATCCTATCACCATTCTACAAAC | 770 |
| TP53 | GGATTATTTGTTTTTATTTGTTATGG | CAAAACTCCACTCCTCTACCTAAAC | 495 |
| WIF1 | GAGTGATGTT TTAGGGGT |
CCTCAACCA AAACTATTCC |
464 |
Table 4.
Average level of promotor genes methylation in tumor and normal tissue in all patient and their subgroups. T-tumor, N-normal.
Table 4.
Average level of promotor genes methylation in tumor and normal tissue in all patient and their subgroups. T-tumor, N-normal.
| Patients (n) Genes |
Sample | All (n=44) |
TNM ** | Type of lesion | SCC | Age | Smokers | ||||||
| before 50 years (n=10) | after 50 years (n=34) |
Yes | No | ||||||||||
| T2 (n=7) |
T3 (n=20) |
T4 (n=15) |
Prim (n=37) |
Rec (n=7) | non-keratinizing (n=30) | keratinizing (n=14) | (n=20) |
(n=24) |
|||||
| M ± m, Range | |||||||||||||
| APC | T | 0.36±0.17 (0.01÷0.54) |
0.31±0.14 (0.05÷0.47) |
0.32±0.18 (0.01÷0.54) |
0.31±0.16 (0.01÷0.52) |
0.30±0.18 (0.01÷0.54) |
0.36±0.16 (0.01÷0.47) |
0.36±0.14 (0.01÷0.54) |
0.24±0.20 (0.01÷0.52) |
0.32±0.17 (0.01÷0.50) |
0.31±0.17 (0.01÷0.54) |
0.31±0.16 (0.01÷0.50) |
0.31±0.20 (0.01÷0.54) |
| N | 0.28±0.18 (0.01÷0.56) |
0.22±0.18 (0.07÷0.48) |
0.31±0.18 (0.04÷0.56) |
0.24±0.18 (0.01÷0.56) |
0.27±0.19 (0.01÷0.56) |
0.34±0.14 (0.01÷0.46) |
0.32±0.17 (0.01÷0.56) |
0.21±0.19 (0.01÷0.56) |
0.27±0.18 (0.04÷0.50) |
0.28±0.19 (0.01÷0.56) |
0.27±0.17 (0.01÷0.50) |
0.29±0.20 (0.01÷0.56) |
|
| ATM | T | 0.35±0.18 (0.01÷0.54) |
0.31±0.20 (0.04÷0.54) |
0.34±0.19 (0.01÷0.54) |
0.37±0.18 (0.06÷0.54) |
0.34±0.19 (0.01÷0.54) |
0.38±0.19 (0.01÷0.53) |
0.39±0.18 (0.01÷0.54) |
0.28±0.18 (0.01÷0.53) |
0.30±0.23 (0.01÷0.53) |
0.37±0.17 (0.04÷0.54) |
0.33±0.19 (0.01÷0.54) |
0.37±0.18 (0.04÷0.54) |
| N | 0.30±0.19 (0.01÷0.57) |
0.27±0.22 (0.06÷0.57) |
0.30±0.19 (0.01÷0.56) |
0.29±0.17 (0.07÷0.49) |
0.31±0.19 (0.02÷0.57) |
0.27±0.21 (0.01÷0.49) |
0.36±0.18 (0.01÷0.57) |
0.20±0.18 (0.01÷0.55) |
0.29±0.19 (0.02÷0.50) |
0.30±0.19 (0.01÷0.57) |
0.30±0.18 (0.02÷0.57) |
0.30±0.20 (0.01÷0.56) |
|
| CDO1 | T | 0.41±0.16* (0.18÷0.76) |
0.30±0.11 (0.18÷0.48) |
0.44±0.16* (0.18÷0.76) |
0.42±0.16* (0.18÷0.71) |
0.38±0.15* (0.18÷0.71) |
0.51±0.18* (0.26÷0.76) |
0.43±0.15* (0.18÷0.71) |
0.36±0.16* (0.18÷0.66) |
0.38±0.15* (0.18÷0.66) |
0.41±0.16* (0.18÷0.76) |
0.44*±0.16 (0.18÷0.76) |
0.36±0.16 (0.18÷0.71) |
| N | 0.20±0.11 (0.01÷0.53) |
0.20±0.10 (0.08÷0.34) |
0.19±0.10 (0.01÷0.34) |
0.19±0.11 (0.01÷0.32) |
0.19±0.11 (0.01÷0.34) |
0.23±0.11 (0.01÷0.34) |
0.21±0.10 (0.01÷0.34) |
0.17±0.10 (0.01÷0.32) |
0.17±0.13 (0.01÷0.32) |
0.21±0.09 (0.01÷0.34) |
0.20±0.11 (0.01÷0.34) |
0.20±0.10 (0.01÷0.34) |
|
| TP53 | T | 0.10±0.06 (0.01÷0.20) |
0.08±0.05 (0.01÷0.18) |
0.09±0.06 (0.01÷0.20) |
0.11±0.06 (0.01÷0.20) |
0.10±0.06 (0.01÷0.20) |
0.10±0.08 (0.01÷0.20) |
0.10±0.06 (0.01÷0.18) |
0.10±0.04 (0.01÷0.17) |
0.10±0.06 (0.01÷0.20) |
0.10±0.06 (0.01÷0.20) |
0.10±0.07 (0.01÷0.20) |
0.10±0.05 (0.01÷0.18) |
| N | 0.09±0.07 (0.01÷0.21) |
0.08±0.07 (0.01÷0.19) |
0.09±0.06 (0.01÷0.20) |
0.09±0.07 (0.01÷0.21) |
0.09±0.07 (0.01÷0.21) |
0.07±0.05 (0.01÷0.16) |
0.08±0.07 (0.01÷0.21) |
0.09±0.06 (0.01÷0.21) |
0.15±0.04 (0.09÷0.21) |
0.07±0.06 (0.01÷0.20) |
0.10±0.07 (0.01÷0.20) |
0.07±0.06 (0.01÷0.21) |
|
| RB1 | T | 0.26±0.12 (0.01÷0.40) |
0.21±0.12 (0.01÷0.36) |
0.27±0.13 (0.01÷0.40) |
0.27±0.12 (0.01÷0.38) |
0.26±0.13 (0.01÷0.40) |
0.27±0.13 (0.01÷0.36) |
0.28±0.11 (0.01÷0.40) |
0.24±0.15 (0.01÷0.38) |
0.27±0.10 (0.06÷0.39) |
0.25±0.14 (0.01÷0.40) |
0.26±0.12 (0.01÷0.39) |
0.27±0.14 (0.01÷0.40) |
| N | 0.25±0.11 (0.01÷0.40) |
0.26±0.11 (0.01÷0.38) |
0.26±0.11 (0.01÷0.40) |
0.23±0.10 (0.01÷0.36) |
0.26±0.10 (0.01÷0.40) |
0.23±0.14 (0.01÷0.36) |
0.26±0.10 (0.01÷0.40) |
0.23±0.12 (0.01÷0.36) |
0.22±0.10 (0.01÷0.34) |
0.24±0.12 (0.01÷0.40) |
0.25±0.10 (0.01÷0.38) |
0.26±0.12 (0.01÷0.40 |
|
| WIF1 | T | 0.30±0.16* (0.08÷0.58) |
0.33±0.15 (0.14÷0.58) |
0.30±0.17 (0.08÷0.58) |
0.29±0.15* (0.09÷0.58) |
0.29±0.15* (0.08÷0.58) |
0.34±0.16* (0.09÷0.56) |
0.26±0.13 (0.08÷0.58) |
0.37±0.18* (0.08÷0.59) |
0.23±0.13* (0.09÷0.55) |
0.32±0.15 (0.09÷0.58) |
0.29*±0.16 (0.08÷0.58) |
0.32±0.16 (0.08÷0.58) |
| N | 0.16±0.08 (0.01÷0.42) |
0.21±0.05 (0.15÷0.30) |
0.18±0.10 (0.01÷0.42) |
0.11±0.05 (0.01÷0.22) |
0.16±0.07 (0.01÷0.42) |
0.18±0.08 (0.08÷0.26) |
0.16±0.09 (0.01÷0.42) |
0.15±0.08 (0.01÷0.30) |
0.10±0.06 (0.01÷0.20) |
0.18±0.08 (0.01÷0.20) |
0.14±0.06 (0.01÷0.26) |
0.18±0.11 (0.01÷0.42) |
|
*Significant differences between tumor and normal tissues (p<0.05). ** Two patients had T1N0M0 and T1N2M0 stage with average methylation levels in ATM, APC, CDO1, RB1, TP53, WIF1 genes in patient’s tumor tissue equal to 0.26, 0.46, 0.33, 0.15, 0.35, 0.31 respectively, and in patient’s normal tissue equal to 0.46, 0.53, 0.31, 0.10, 0.30 and 0.23 respectively.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



