
Submitted:
25 August 2023
Posted:
25 August 2023
You are already at the latest version
Abstract
The king ratsnake (Elaphe carinata) of the genus Elaphe is a common large non-venomous snake that is widely distributed in Southeast and East Asia, and is an economically important farmed snake species. As a non-venomous snake, the king snake that is predatory on venomous snakes such as cobras and pit vipers. The immune mechanisms of which has been unclear. Despite their economic and research importance, genomic resources which will benefit studies in toxicology, phylogeography and immunogenetics are lacking. In this study, we use single-tube long fragment read (stLFR) sequencing to display the first complete genome of a King ratsnake from Huangshan City, Anhui province in China. The genome size is 1.56GB with a scaffold N50 of 6.53M, the total length of the genome is approximately 621Mb, and the repeat content is 38.90%. Additionally, we predicted 22,339 protein-coding genes, of which 22,065 had functional annotations. Our genome is a potentially useful addition to those currently available for snakes.
Keywords:
Genetics and Genomics
; Zoology
; Animal Genetics
Data Description
The king ratsnake (Elaphe carinate) of family Colubridae and genus Elaphe is a large oviparous snake[1] that is found in many provinces in South-eastern China, the southern edge of the distribution area can reach northern Guangdong, Guangxi and Taiwan, while the northern edge is located in the Beijing-Tianjin area (Figure 1). Also distributed in northern Vietnam and several islands (Ryukyu Islands, including the Senkaku Islands) in Japan [2,3]. E. carinata mainly inhabit mountainous and hilly areas and generally feed on rodents, birds, and eggs. Its juveniles differ greatly from adults, and when threatened, can use its anal glands to secrete a foul-smelling fluid [3]. King ratsnakes are farmed in many countries as an important food source as they provide a large amount of protein[4]. According to the China Red Data Book of Endangered Animals [5] (Zhao, 1998), the king snake is listed as a vulnerable species. The common name of "king ratsnake" refers to its habit of eating other snakes, according to reports, due to a special protein in the blood, the non-venomous king snake has a strong antagonistic effect on the venom of some poisonous snakes whose toxins are mainly blood-circulating poisons, such as bamboo leaf green and sharp-nosed viper (Deinagkistrodon acutus) snakes. However, the exact immune mechanism for this protection is unknown. As snake antivenom is the only treatment that is effective in preventing or reversing the effects of snake venom[6], the genome of the king ratsnake may provide new insight into antivenoms.
In the present study, we assembled the first highly contiguous E. carinate genome by using stLFR sequencing data and combined with next-generation sequencing data for correction. The resulting genome, which is comparable in genome size to the previously sequenced corn snake Pantherophis guttatus [7] but more contiguous, is valuable for further studies, such as snake evolution and venom immunity.
Main Content
Context
As a snake with a long history of captive breeding, the reproduction and virus carrying of the king ratsnake has been well studied[8,9], but there is insufficient research on its immune resistance and a general lack of genomic resources. Here we demonstrate the de novo assembly of a highly contiguous king ratsnake genome with a genome size of 1.56 Gb based on stLFR sequencing data (Table 1). The maximal length of scaffold is 49.75M and the N50 length is 6.53M. The GC content of E. carinate is 40.25%. Based on the characteristics of the published snake genome sequences, the assembled genomes were shown to be highly available and contiguous. Here, we present the draft genome sequence of E. carinata. It will be an invaluable resource for understanding snake venom resistance.
Methods
Samples and Ethics Statement
An adult E.carinata (NCBI:txid74364) individual from Huangshan City in Anhui province, which was collected for DNA sequencing and RNA sequencing. After the individual died naturally, the samples were transferred to dry ice and quickly frozen, then kept at -80℃ until further use. We used four tissues and organs of liver, stomach, kidney and muscle for RNA sequencing. In addition, single-tube long fragment read (stLFR) sequencing only used muscle samples. Sample collection and experimental studies were both approved by the Institutional Review Board of BGI (BGI-IRB E22017) . All procedures are carried out in accordance with the guidelines of the BGI-IRB.
Nucleic Acid Isolation, Library Preparation, and Sequencing
We extracted DNA according to the method of Wang et al[10]. A stLFR co-barcoded DNA library was constructed using the MGIEasy stLFR Library Prep Kit (MGI, China). Sequencing was performed using a BGISEQ-500 sequencer. The genomic DNA kit (AxyPrep, USA ) was used to isolate DNA for WGS sequencing in the meantime. Total RNA was extracted according to manufacturer 's instructions by using TRlzol reagent (Invitrogen, USA ). Integrity and concentration of DNA and RNA were assessed using Qubit 3.0 Fluorometer (Life Technologies, USA) and Agilent 2100 Bioanalyzer System (Agilent, USA). Use 200–400 bp RNA fragments for reverse transcription of cDNA libraries.
Genome Assembly, Annotation and Assessment
The stLFR sequencing data were assembled using Supernova software (v2.1.1)[10]. Based on the WGS data, the assembly was gap filled and redundant removed using GapCloser (v1.12-r6)[11] and redundans (v0.14a)[12], respectively.
We first identified de novo repeats using Repeat Finder (TRF) [13] (v. 4.09), LTR finder (v1.0.6) [14] and RepeatModeler [15] (v1.0.8). These repeats were then used together with RepBase in RepeatMasker[16] (v. 3.3.0) as known elements for identifying transposable elements, and known repeat elements were searched using RepeatProteinMask[17] (v. 3.3.0) in genome sequences. For protein-coding gene prediction, we first use Augustus[18] (v3.0.3) for de novo prediction. Based on the RNA-seq data filtered clean by Trimmomatic[19] (v0.30), the transcripts were assembled using Trinity[20] (v2.13.2), and compared with the king ratsnake genome through Programto Assemble Spliced Alignments (PASA)[21] (v2.0.2) to obtain the gene structure. For homology-based prediction, we used Blastall[22] (v2.2.26) with an E-value cut-off of 1e-5 to map the protein sequences by comparing four sets of high-quality data of Crotalus tigris, Pseudonaja textilis, Notechis scutatus and Thamnophis elegans from the UniProt database (release-2020_05) with the king ratsnake genome. GeneWise[23] (v2.4.1) was used to analyze alignment results to predict gene models. We used the MAKER pipeline[24] (v3.01.03) to generate final gene set representing RNA-seq, homology, and de novo predicted genes.
Functional annotation was completed by using SwissProt[25], TrEMBL[25], and (KEGG)[26] databases to perform BLAST comparison on structurally annotated gene sets, and the E value cut-off value was 1e-5. InterProScan[27] (v5.52-86.0) was used to count and visualize structural domain information, and Gene Ontology (GO) terms were used for gene enrichment.
The genome integrity was evaluated by Benchmarking Universal Single-Copy Orthologs (BUSCO v5.2.2), with parameters set to genome mode and dataset input set to vertebrata_odb10[28].
We used OrthoFinderv2.3.7 (RRID:SCR_017118)[29] to search for single-copy orthologs in the protein sequences of Rana temporaria(GCA_905171775.1), Gopherus evgoodei (GCA_007399415.1), Podarcis muralis(GCA_004329235.1), Pseudonaja textilis(GCA_900518735.1), Thamnophis elegans(GCA_009769535.1) Pantherophis guttatus(GCA_001185365.2), and to construct phylogenetic trees by orthogroups. A total of 1307 single-copy loci were found.
Results
Usually, genome-wide repetitive elements are important for eukaryotic evolution[30]. In E. carinata, the content of repetitive elements in the genome accounted for 38.90%, and the total length reached 621Mb (Table 2 and Table 3). Among all repetitive elements, LINE accounted for 38.41%, DNA accounted for 17.11% and unknown types of repetitive elements accounted for 31.93% (Figure 3). This indicates that the content and quantity of repeating elements is one of the sources of species differences.
A total of 22,065 functional genes were annotated, and the annotations associated with the TrEMBL database accounted for the largest proportion, reaching 97.92%(Table 4). In addition, all genes were annotated with KEGG, which showed the highest number in pathways such as Human Diseases, Organismal Systems and Metabolism, and the highest number of Signal Transduction genes in Environmental Information Processing. In Additionally, GO gene enrichment for E. carinata revealed that, among 25 biological process pathways, 251 genes were related to immune system processes, and 2 genes were related to detoxification (Figure 4).
Data Validation and Quality Control
When assessing the quality of the genome, we performed a completeness assessment of the assembly with BUSCO v3.1.0 (RRID:SCR_015008) [31]using the vertebrata_odb10 dataset [31]. This assembly was able to match 83.2% of the complete BUSCOs. (Figure 5) .
By screening closely related species, Rana temporaria, Gopherus evgoodei, Podarcis muralis, Pseudonaja textilis, Thamnophis elegans, Pantherophis guttatus were filtered to construct a phylogenetic tree. Consistent with previous studies[32], our data can construct a phylogenetic trees and cluster closely related species. (Figure 6)
Reuse Potential
King ratsnake has both nutritive and medicinal value, and the growth and development of individuals and snake eggs has been widely studied[33]. However, there are insufficient studies and genomics data on its immune system. Only Sun et al. have done relevant research on the development of the immune system in the embryonic stage of the king snake[34].
Our data can be combined with other snake genome data for phylogenetic studies to construct the developmental evolutionary history of snakes and other reptiles. In addition, the genomic data can provide new insights into the study of the immune system, snake venom resistance genes and their mechanisms of action.
Data Availability
Competing Interests
The authors declare no conflict financial interests.
Consent for Publication
Not applicable.
Author Contributions
Song Huang, He Wang and Tianming Lan designed and initiated the project. Yi Zhang, Tierui Zhang, Zhihao Jiang and Jing Yu collected the samples. Xinge Wang, Zicheng Su and performed the DNA extraction, Diancheng Yang, Yanan Gong and Zhangbo Cui performed genome assembly. Jiale Fan and Ruyi Huang performed data analysis and wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgments
Our project was financially supported by the Doctoral Research Starting Foundation of Anhui Normal University (752017), National Natural Science Foundation of China (NSFC 31471968) and Guangdong Provincial Key Laboratory of Genome Read and Write (grant no. 2017B030301011). This work was also supported by China National GeneBank (CNGB).
References
- Zhao, E.; Huang, M.; Zong, Y. 中国动物志: 爬行纲. 第三卷, 有鳞目 蛇亚目 [Fauna Sinica. Reptilia Vol. 2. Squamata. Ophidia]. Science Press: Beijing, China, 1999.
- Xiang, J.; Pingyue, S.; Xuefeng, X.; Weiguo, D. Relationships among body size, clutch size, and egg size in five species of oviparous colubrid snakes from Zhoushan Islands, Zhejiang, China. Dong Wu Xue Bao [Acta Zool. Sin. ] 2000, 46, 138–145. [Google Scholar]
- Chao, L.-L.; Hsieh, C.-K.; Shih, C.-M. First report of Amblyomma helvolum (Acari: Ixodidae) from the Taiwan stink snake, Elaphe carinata (Reptilia: Colubridae), collected in southern Taiwan. Ticks Tick-Borne Dis. 2013, 4, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; He, J.; Deng, S.; Zhang, H.; Liu, G.; Li, S.; et al. Integrated analysis of mRNA and miRNA expression profiles reveals muscle growth differences between fast-and slow-growing king ratsnakes (Elaphe carinata). Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2020, 248, 110482. [Google Scholar] [CrossRef] [PubMed]
- Zhao, E.; Wang, S. China red data book of endangered animals: Amphibia and Reptilia; Science Press: 1998.
- Suryamohan, K.; Krishnankutty, S.P.; Guillory, J.; Jevit, M.; Schröder, M.S.; Wu, M.; et al. The Indian cobra reference genome and transcriptome enables comprehensive identification of venom toxins. Nat. Genet. 2020, 52, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Ullate-Agote, A.; Milinkovitch, M.C.; Tzika, A.C. The genome sequence of the corn snake (Pantherophis guttatus), a valuable resource for EvoDevo studies in squamates. Int. J. Dev. Biol. 2015, 58, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.-F.; Li, H.; Gao, J.-F.; Ji, X. Geographical variation in reproductive traits and trade-offs between size and number of eggs in the king ratsnake, Elaphe carinata. Biol. J. Linn. Soc. 2011, 104, 701–709. [Google Scholar] [CrossRef]
- Wu, Q.; Xu, X.; Chen, Q.; Ji, J.; Kan, Y.; Yao, L.; et al. Genetic analysis of avian gyrovirus 2 variant-related Gyrovirus detected in farmed king ratsnake (Elaphe carinata): The first report from China. Pathogens 2019, 8, 185. [Google Scholar] [CrossRef]
- Weisenfeld, N.I.; Kumar, V.; Shah, P.; Church, D.M.; Jaffe, D.B. Direct determination of diploid genome sequences. Genome Res. 2017, 27, 757–767. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 2047–2217X. [Google Scholar] [CrossRef]
- Pryszcz, L.P.; Gabaldón, T. Redundans: An assembly pipeline for highly heterozygous genomes. Nucleic Acids Res. 2016, 44, e113. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Hao, W. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, 265–268. [Google Scholar]
- Smit, A.; Hubley, R.; Green, P. RepeatModeler Open-1.0; Institute for Systems Biology: Seattle, USA, 2015. [Google Scholar]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Current protocols in bioinformatics / editoral board, Andreas D Baxevanis [et al]. 2009.
- Tempel, S. Using and understanding RepeatMasker. In Mobile Genetic Elements; Springer: Berlin/Heidelberg, Germany, 2012; pp. 29–51. [Google Scholar]
- Stanke, M.; Steinkamp, R.; Waack, S.; Morgenstern, B. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res. 2004, 32, W309–W312. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef]
- Mount, D.W. Using the Basic Local Alignment Search Tool (BLAST). CSH Protocols 2007, 2007. [Google Scholar] [CrossRef]
- Birney, E.; Clamp, M.; Durbin, R. GeneWise and Genomewise. Genome Res. 2004, 14, 988–995. [Google Scholar] [CrossRef]
- Campbell, M.S.; Holt, C.; Moore, B.; Yandell, M. Genome Annotation and Curation Using MAKER and MAKER-P. Curr. Protoc. Bioinform. 2014, 48. [Google Scholar] [CrossRef]
- Amos, B.; Rolf, A. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 1, 45. [Google Scholar]
- Pitk, E. KEGG database. Novartis Found. Symp. 2006, 247, 91–103. [Google Scholar]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Holt, K.E. Benchmarking of long-read assemblers for prokaryote whole genome sequencing. F1000Research 2019, 8, 2138. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Fang, D.; Li, H.; Sahu, S.K.; Wang, Q.; Yuan, H.; et al. Chromosome-Scale Genome of Masked Palm Civet (Paguma larvata) Shows Genomic Signatures of Its Biological Characteristics and Evolution. Front. Genet. 2022, 12. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Vidal, N.; Hedges, S.B. The molecular evolutionary tree of lizards, snakes, and amphisbaenians. Comptes Rendus Biol. 2009, 332, 129–139. [Google Scholar] [CrossRef]
- Ji, X.; Du, W.-G.; Li, H.; Lin, L.-H. Experimentally reducing clutch size reveals a fixed upper limit to egg size in snakes, evidence from the king ratsnake, Elaphe carinata. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2006, 144, 474–478. [Google Scholar] [CrossRef]
- Sun, J.; Gao, H.; Lian, L.; Zhang, Z. Variation pattern and adaptive significance of different subtypes of leukocytes in the king ratsnakes (Elaphe carinata) from birth to 30 days of postembryonal period. Chin. J. Ecol. 2017, 36, 2246. [Google Scholar]
- Guo, X.; Chen, F.; Gao, F.; Li, L.; Liu, K.; You, L.; et al. CNSA: A data repository for archiving omics data. Database 2020, 2020, baaa055. [Google Scholar] [CrossRef]
- Feng, Z.C.; Li, J.Y.; Fan, Y.; Li, N.W.; Xiao, F.W. CNGBdb: China National GeneBank DataBase. Hereditas 2020, 42, 799–809. [Google Scholar]
Figure 1.
An E. carinate individual photographed by Diancheng Yang.

Figure 2.
A protocols.io collection of the protocols for sequencing snake genomes [9].
Figure 2.
A protocols.io collection of the protocols for sequencing snake genomes [9].

Figure 3.
Distribution of transposable elements (TEs) such as DNA transposons (DNA) and RNA transposons in the E. carinata genome. RNA transposons include DNAs, LINEs, LTRs, and SINEs. (a) Distribution of divergence rates for De novo sequences. (b) Distribution of divergence rates for known sequences. (c) Proportion and distribution of repeating elements.
Figure 3.
Distribution of transposable elements (TEs) such as DNA transposons (DNA) and RNA transposons in the E. carinata genome. RNA transposons include DNAs, LINEs, LTRs, and SINEs. (a) Distribution of divergence rates for De novo sequences. (b) Distribution of divergence rates for known sequences. (c) Proportion and distribution of repeating elements.
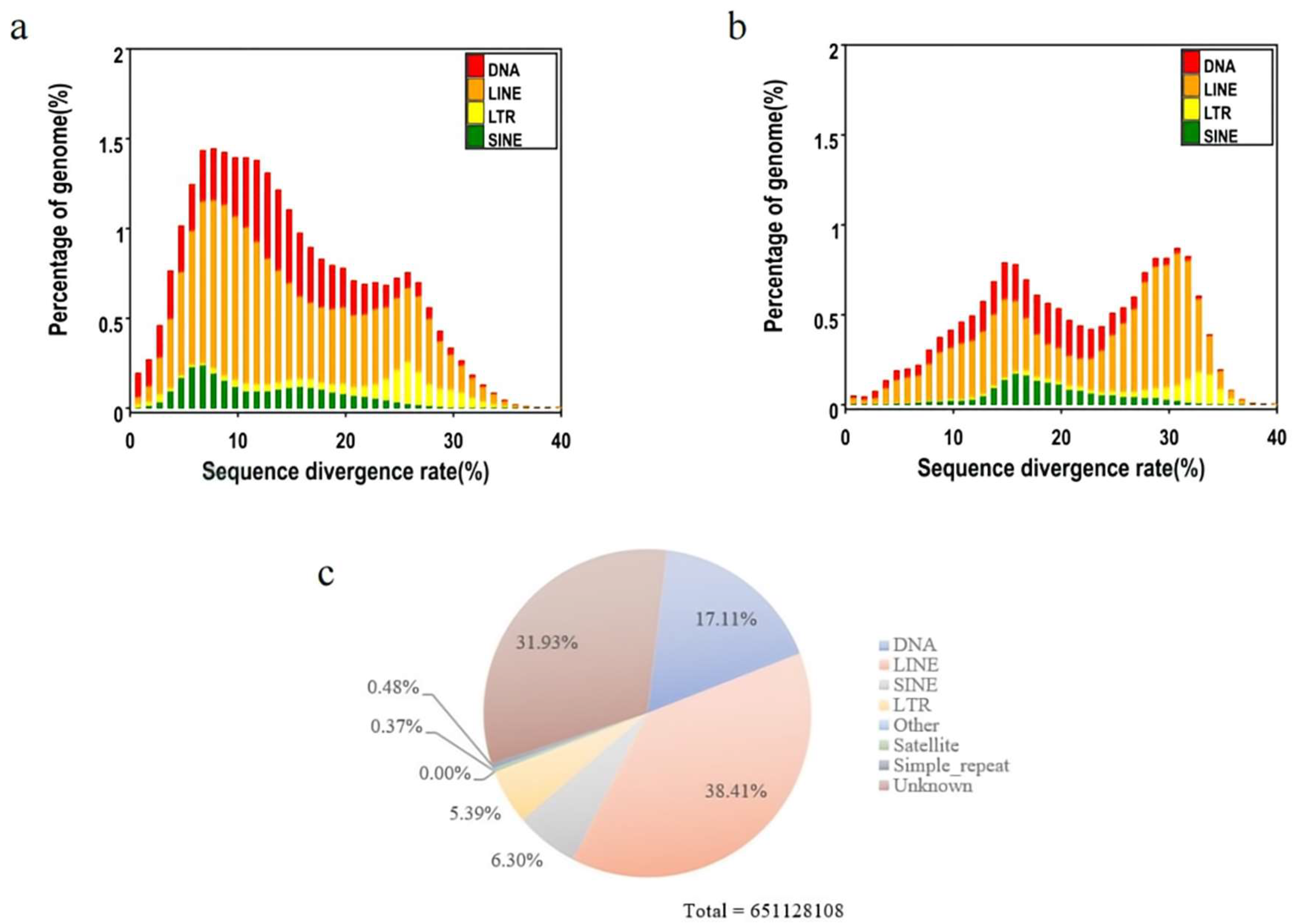
Figure 4.
Gene annotation results for E.carinata. (a) KEGG enrichment of E.carinata. (b) GO enrichment of E .carinata.
Figure 4.
Gene annotation results for E.carinata. (a) KEGG enrichment of E.carinata. (b) GO enrichment of E .carinata.
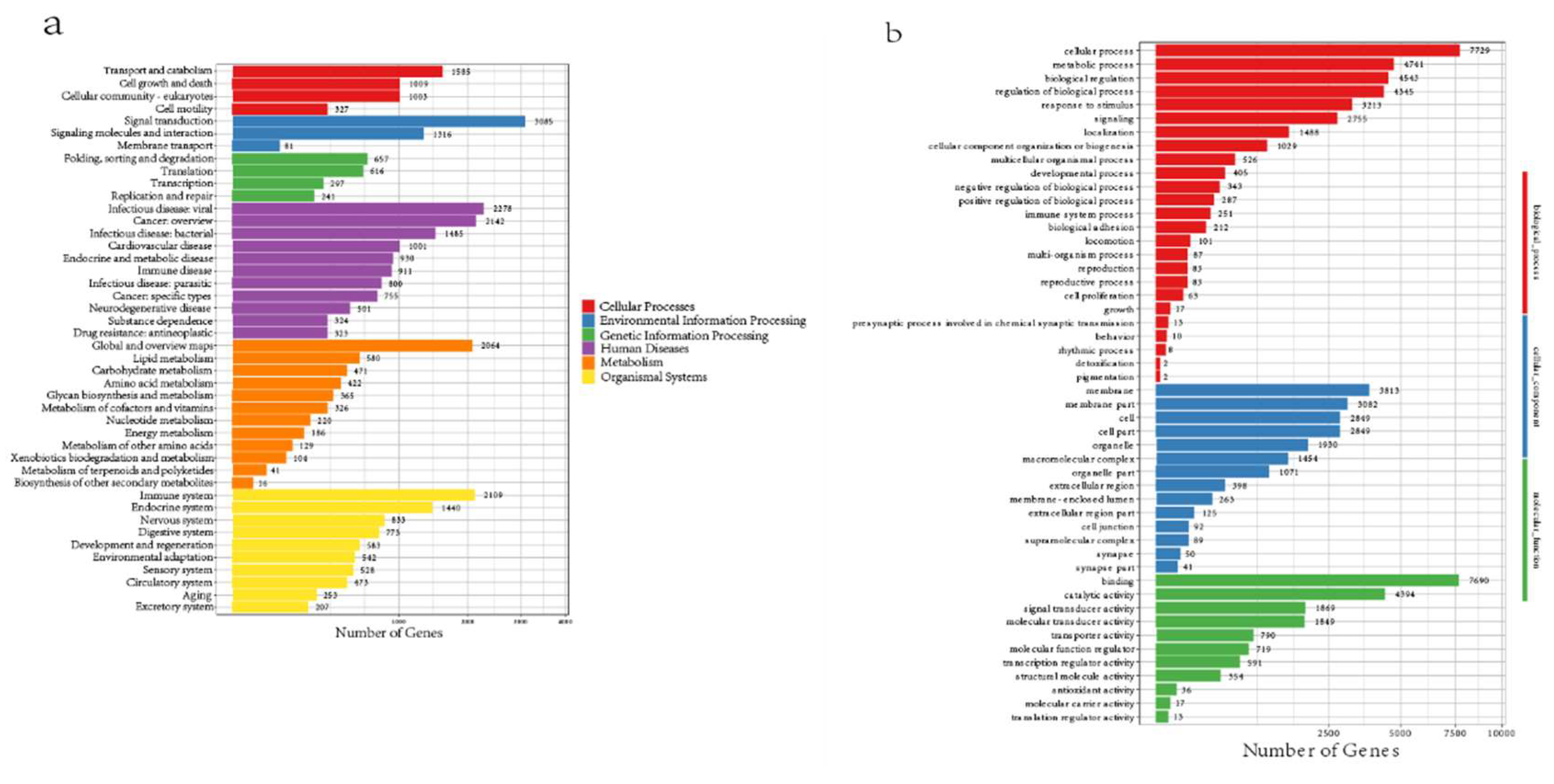
Figure 5.
BUSCO Assessment result of the E.carinata genome.
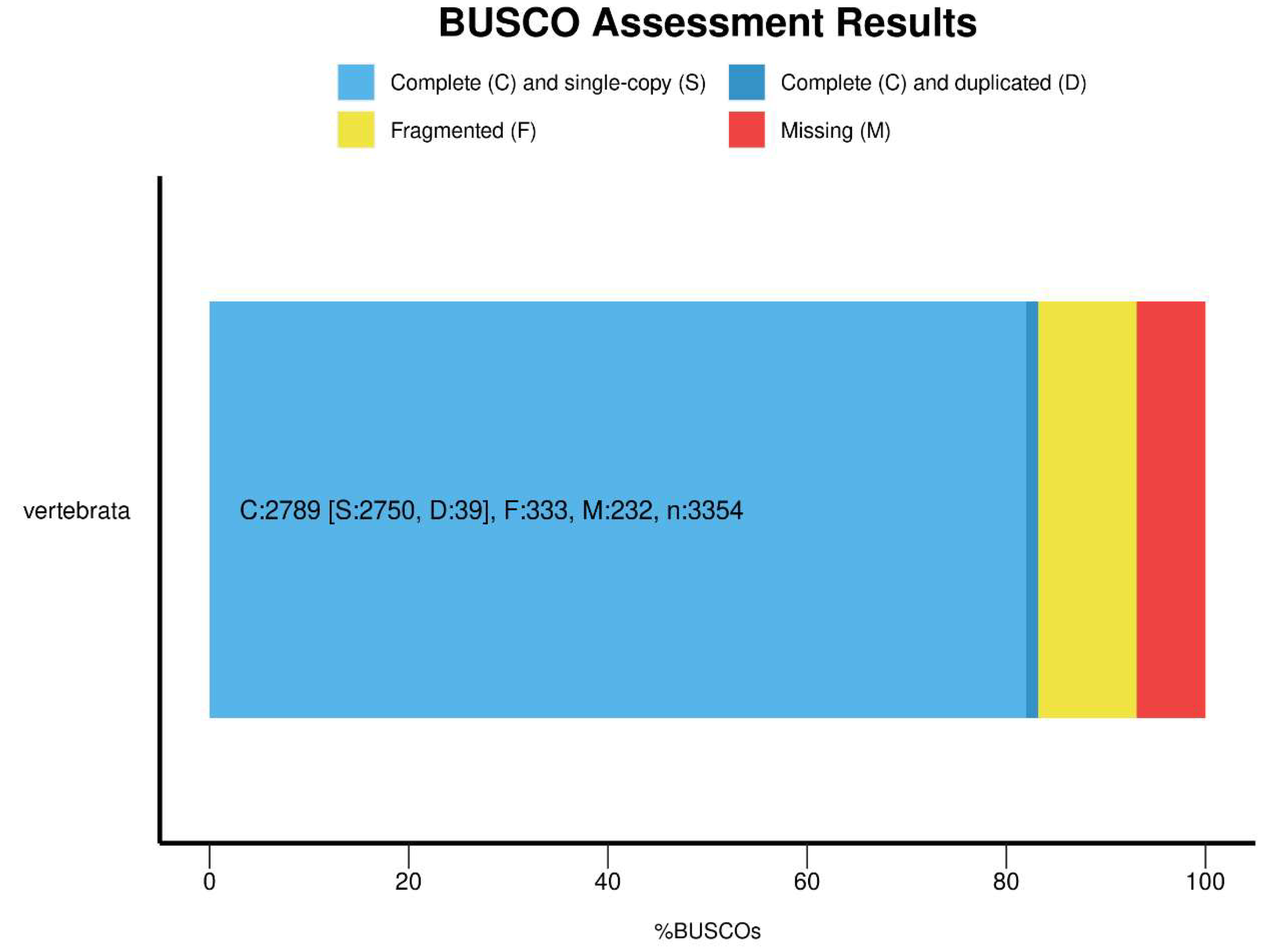
Figure 6.
Phylogenetic tree reconstructed using nuclear genome single-copy genes.
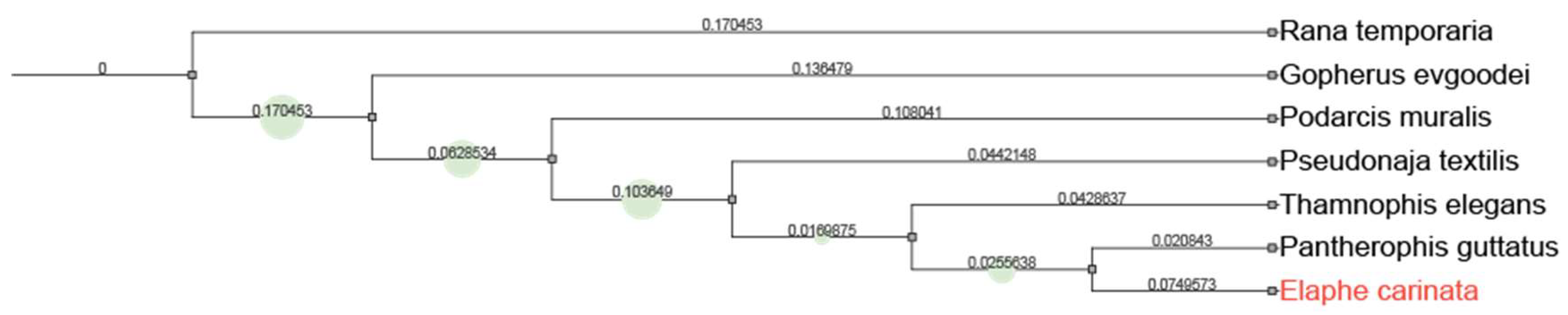

Table 1.
Summary of the features of the E.carinata genome.
| Contig | Scaffold | |
|---|---|---|
| Maximal length(bp) | 657733 | 52164798 |
| N90(bp) | 3039 | 4090 |
| N50(bp) | 45108 | 6847971 |
| number>=500bp | 187253 | 134573 |
| Ratio of Ns | 0.059 | 0.059 |
| GC content(%) | 40.25 | 40.25 |
| Genome size(bp) | 1574091846 | 1674021862 |
Table 2.
Content of various repeat sequences in the E. carinata genome.
| Type | Length(Bp) | % in genome |
|---|---|---|
| DNA | 114900759 | 6.863755 |
| LINE | 257937611 | 15.408258 |
| SINE | 42327923 | 2.528517 |
| LTR | 36199886 | 2.16245 |
| Other | 0 | 0 |
| Satellite | 2487376 | 0.148587 |
| Simple_repeat | 3251656 | 0.194242 |
| Unknown | 214450953 | 12.810523 |
| Total | 651128108 | 38.896034 |
Table 3.
Summary of transposable elements (TEs)in the E. carinata genome.
| Repbase TEs | TE protiens | De novo | Combined TEs | |||||
|---|---|---|---|---|---|---|---|---|
| Type | Length(Bp) | % in genome | Length(Bp) | % in genome | Length(Bp) | % in genome | Length(Bp) | % in genome |
| DNA | 44586593 | 2.663442 | 3037369 | 0.181441 | 114900759 | 6.863755 | 137315177 | 8.202711 |
| LINE | 172974640 | 10.332878 | 142896461 | 8.536117 | 257937611 | 15.408258 | 287262246 | 17.160006 |
| SINE | 27330057 | 1.632599 | 0 | 0 | 42327923 | 2.528517 | 52336172 | 3.126373 |
| LTR | 20332067 | 1.214564 | 26146398 | 1.561891 | 36199886 | 2.16245 | 48061022 | 2.870991 |
| Other | 28331 | 0.001692 | 291 | 0.000017 | 0 | 0 | 28622 | 0.00171 |
| Unknown | 0 | 0 | 0 | 0 | 214450953 | 12.810523 | 214450953 | 12.810523 |
| Total | 252872307 | 15.105675 | 171980912 | 10.273516 | 645389076 | 38.553205 | 685733449 | 40.963231 |
Table 4.
Summary of annotation results in the E. carinata genome.
| Values | Total | Swissprot-Annotated | KEGG-Annotated | TrEMBL-Annotated | Interpro-Annotated | GO-Annotated | Overall |
|---|---|---|---|---|---|---|---|
| Number | 22,339 | 20,796 | 19,836 | 21,874 | 21,604 | 15,169 | 22,065 |
| Percentage | 100% | 93.09% | 88.80% | 97.92% | 96.71% | 67.90% | 98.77% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



