
Submitted:
25 August 2023
Posted:
25 August 2023
You are already at the latest version
Abstract
Advanced genomics, transcriptomics and epigenomics techniques are providing unprecedented insights into the understanding of the molecular underpinnings of the central nervous system, including the neuro-sensory cochlea of the inner ear. Here, we report for the first time a comprehensive and updated overview of the most advanced omics techniques for the study of nucleic acids, and their applications in cochlear research. We describe the available in vitro and in vivo models for hearing research, the principles of genomics, transcriptomics and epigenomics, alongside their most advanced technologies (like single cell omics and spatial omics), which allows to investigate the molecular events that occur at single cell resolution retaining the spatial information.
Keywords:
Omics
; Cochlea
; Single cell omics
; Spatial omics
; Epigenomics
; Transcriptomics
; Genomics
; Organ of Corti.
1. Introduction
According to the World Health Organization (WHO) reports, 432 million adults and 32 million children are affected by disabling hearing loss and it is estimated that this number would increase to 700 million by 2050[1]. In particular, sensorineural hearing loss (SNHL) is characterized by the deterioration of the neuro-sensory structure of the inner ear - the cochlea - and leads to irreversible hearing loss that affects communication, speech and cognition, with a clear impact on the quality of life and severe socio-economic consequences. SNHL can be caused by either congenital or acquired factors (noise exposure, ototoxic drugs, ageing, strial or metabolic dysfunctions)[2]. The severity of the aetiology can range from synaptic disconnectivity of the sensory epithelium[3] - composed of inner (IHCs)/outer hair cells (OHCs) and supporting cells (SCs) - to critical cases of the loss of hair cells (HCs). The latter process is often followed by the degeneration of the downstream spiral ganglion neurons (SGNs)[4], whose axons form the auditory nerve. Although cochlear implants and hearing aids exhibit some beneficial outcomes in deaf patients, they cannot entirely replace the cochlea’s functionality[5]. Thus, management-based approaches must give way to disease-modifying interventions. This strategy needs a more thorough and in-depth understanding of the molecular events that could eventually become novel therapeutic targets and/or diagnostic biomarkers of SNHL, to be exploited also in cochlear regeneration strategies. Thanks to the technological advancements in the field of molecular biology, recent progress has been made in identifying and characterizing novel genes involved in hearing loss[6], as well as new molecular mechanisms of cochlear development[7], degeneration and regeneration[8]. In this review, we aim to present cutting-edge molecular methodologies that have been used to investigate the genome, epigenome and transcriptome in cochlear research, as well as methods that could be employed in the next future to expand our understanding in the field, such as the latest sophisticated single cell and spatial genomics, transcriptomics and epigenomics.
2. Experimental models in inner ear research
Modelling inner ear disorders is important to understand the molecular bases of hearing, as well as the mechanisms of deafness in humans. Currently, the only possibility to study human inner ear disorders is from cadavers[9], since sampling tissues from alive subjects would cause irreversible damage to the intricate inner ear structures. Hence, this is possible only in cases of inner ear tumors[10,11]. Moreover, non-invasive techniques, such as magnetic resonance imaging (MRI) and computerized tomography (CT), cannot lead to a detailed understanding of the inner ear pathogenesis[12]. Therefore, most of the models for studies on the cochlea are based on cell cultures from animals or on animal models. Figure 1 summarizes the experimental models that are currently available and used in cochlear research.
2.1. In vitro and ex vivo models: cochlear cell lines, organotypic cultures and organoids
Establishment of cochlear cell cultures has been challenging for a long time due to the paucity of the tissue and the poor accessibility of the inner ear. With the advent of the Immortomouse™, a transgenic mouse model carrying the temperature-sensitive tsA58 variant of the SV40 T-antigen, it became possible to develop immortalized cell lines from the inner ear[13]. Therefore, to date most of the available cell lines are derived from ImmunomortomouseTM, including the ImmunomortomouseTM otocysts E9.5 (IMOs), HCs cultures (like UB/OC-1, UB/OC-2, HEI-OC1), and cells of the organ of Corti which include either HCs or SCs (e.g., OC-k1to 4). The only human inner ear cell line developed so far is the immortalized endolymphatic sac (ES) cell line[14], while none has been developed yet for the human organ of Corti. In addition to cell cultures, cochlear explants (or organotypic cultures) are another efficient experimental tool to identify and characterize molecular and genetic pathways which play a role in specification and patterning of cells in their natural environment[15]. A recent improvement of the organotypic cochlear models is based on the use of microfluidic chambers for organ-on-chip culture, which allows to reproduce a more controlled microenvironment[16] . However, cochlear explants require to use a large number of animals, and there are technical issues for the isolation of the intact tissue to be cultured. Inner ear organoids, derived from induced pluripotent stem cells (iPSCs) or embryonic stem cells (ESCs), represent a relevant alternative to recapitulate the physiological dynamics of the cochlea in terms of cell type yield and functionality, particularly for HCs. However, it is only claimed that a small portion of the organoid cultures contain functional sensory HCs, and the reason why this happens has not yet been fully understood[17]. It should be stressed that optimization of inner ear organoid culture could lead to better drug screening programs and disease modelling opportunities.
2.2. In vivo models
Animal models used in hearing research are based on multiple species (rats, guinea pigs, mice, chinchillas, gerbils, birds and zebrafish), that differ in the physiological and anatomical characteristics of the auditory system, and then offer different view angles to study the inner ear[18]. These animals include genetic models of spontaneous or inherited hearing loss, carrying mutations for specific genes associated with hearing impairment in humans. For instance, mutations in the genes encoding for collagen and non-collagen proteins (like α-tectorin) that are important for the structure of the basilar membrane (BM) and of the tectorial membrane (TM) in the organ of Corti, cause SNHL. These mutations successfully recapitulate cochlear degeneration in mice, for instance disproportionate micromelia (Dmm) mice, spondyloepiphyseal dysplasia congenita (sedc) mice, Col2a1G574S mice, Col9a1 KO mice, chondrodysplasia (Cho) mice, all carrying mutations in collagen genes; Tecta∆ENT/∆ENT and TectaY1870C/Y1870C, both carrying mutation in the gene encoding for α-tectorin, Tecta [19,20]. Also, mutations in genes encoding for myosin and other proteins important for HCs function and mechanotransduction, as well as genes encoding for endolymph proteins, are associated with SNHL, and multiple genetic models have been successfully developed: Beethoven mice (mutations in TMC1), Usher syndrome models, Lcc and Ysb mice[21,22]. In addition to genetic predisposition, SNHL may also be caused by external noxious events. As yet, more than 150 established ototoxic substances have been identified, the most widely used being aminoglycoside antibiotics (AABs), loop diuretics, and antitumor medications. The administration of these substances to animals recapitulates indeed cochlear degenerative events observed in humans, and are helpful to study degenerative mechanisms and neuroprotective strategies[18]. Additionally, the exposure to loud sounds is an important risk factor for noise-induced hearing loss (NIHL), hence noise trauma can be used to successfully reproduce NIHL in animals as well. Of note, rodents are more susceptible to noise trauma compared to non-human primates, suggesting different degenerative mechanisms with important implications from a translational point of view[23].
2.3. New models created by CRISPR/Cas9 technology
The CRISPR/Cas9 technology has been developed following years of research on the adaptive immunity in prokaryotes[24,25,26]. Engineering of this machinery has allowed to perform gene-editing in terms of base and prime editing, as well as knock-out/ knock-in of genes. Therefore, the CRISPR/Cas9 technology represents a powerful tool for basic molecular studies in hearing research, and a promising strategy for therapeutic approaches of SNHL[27,28,29]. In particular, CRISPR/Cas9 has been successfully applied for creating new in vitro and in vivo models of cochlear diseases[30]. For instance, this technology has allowed to study genes associated with ototoxicity, via the knock-out of Lim-domain only 4 (LMO4), for cisplatin, and HtrA Serine Peptidase 2 (htra2), for aminoglycosides, in in vitro and in vivo models respectively[31,32]. It also allowed the study of inherited hearing loss genes, such as MYO7A, CIB2 and CDH23, for the Usher syndrome[30]. Moreover, zebrafish models to study genes involved in the development of the auditory system, such as POU4F3, have recently been developed based on CRISPR/Cas9 [33] .
3. Omics techniques
3.1. Introduction to omics: principles and advancements
The term omics refers to a rapidly evolving and expanding group of techniques aimed at investigating pools of biological molecules of an organism, including nucleic acids, proteins and metabolites[34]. Hence, the main branches of omics techniques are known as genomics, epigenomics, transcriptomics, proteomics and metabolomics. The most advanced omics techniques include single cell omics and spatial omics, which allows to investigate the molecular events occurring at single cell resolution and to retain the spatial information[35,36]. There are also other advanced and upcoming sequencing-based omics, such as epitranscriptomics, epiproteomics and interactomics (DNA-RNA, RNA-RNA, RNA-protein, protein-protein, protein-metabolite), which give detailed information on the complex interactions and dynamics of regulation in a biological system[34]. The number of omics studies in cochlear research is relatively low compared to other sensory systems, since sampling of the cochlear tissues has only recently advanced, and some techniques are incompatible with the obtained small sample quantity[37]. Nonetheless, the studies performed so far have significantly advanced the knowledge of cochlear physio-pathology.
In this review, we focus on the omics techniques that target nucleic acids, that are genomics, transcriptomics and epigenomics in bulk, single and spatial resolution. We also provide evidence that the availability of these techniques has been transformative in unraveling novel molecular signatures in hearing research, advancing our understanding of the treatment of cochlear degenerative diseases.
3.2. Principles of single cell omics
The term single cell omics refers to the process of profiling the genome, transcriptome, epigenome, proteome, and metabolome in individual cells. As a consequence, single cell techniques were shown to be useful in several biological fields, including cancer[38], developmental biology[39], stem cell research[40], neuroscience[41], and hearing[8].
The first step of all these technologies requires isolation of individual cells and setting up of libraries. Multiple methodologies have been designed to isolate single cells from pooled cell populations/tissues through a variety of techniques[42], that span from the most straightforward - using pipettes and cell isolation by dilution - to the more sophisticated - using advanced microfluidic technologies[43]. The latter include hydrodynamic trapping, droplet-based isolation, valve-based isolation, microwell-based isolation and dielectrophoresis trapping[44], as well as magnetic activated cell sorting (MACS), flow activated cell sorting (FACS)[42], laser capture microdissection (LCM) - which also preserves spatial context, and nanowell-based cell sorting[45]. Details on the isolation methods of abundant or rare cells have been described by Wang and Navin[46].
After isolation of the single cells, the genome, the epigenome and the transcriptome can be profiled[8,47]. Notably, single cell multi-omics approaches have recently been developed to investigate the molecular events that occur in individual cells under physiological or pathological conditions in a wider overview, at once. An example of this cutting-edge methodology is the single-cell triple omics sequencing (scTrio-seq), which simultaneously gathers data from the genome, DNA methylome, and transcriptome of a single cell[48].
3.3. Spatial omics
The study of omics at single cell resolution has been transformative in the identification of novel biomarkers and molecular regulators of tissues, yet it cannot deliver information on the tissue or sub-cellular localization of the isolated cells. For this reason, spatial omics have been developed with the aim of identifying molecular events while maintaining the spatial information. Multiple spatial omics approaches exist, and they vary depending on the biomolecules of interest. In the cochlea, spatial omics are of particular relevance due to the complex anatomical architecture. Indeed, the cochlea exhibits a tonotopic organization from its base (high frequency perception) to the apex (low frequency perception), which requires appropriate cellular structures and expression patterns[49]. Moreover, different cell types are also present from the medial [i.e., greater epithelial ridge (GER), IHCs and their associated SCs] to the lateral (i.e., Deiters cells, pillar cells and OHCs) compartment of the cochlea[49]. Hence, entering the spatial era can deepen our understanding of the cellular organization and interplay in regions of interest.
4. Genomics
4.1. Principles of sequencing
Genomics investigates somatic and germ line inter-individual variations of the genome. The currently most used genomics are based on sequencing, and they have been used to identify several genetic disorders, and to disclose novel alleles in multiple inherited human diseases[34], including hearing loss[50,51]. It is possible to perform sequencing of targeted genes (targeted panel sequencing), whole exome sequencing (WES)[52,53]or whole genome sequencing (WGS)[54,55] (Figure 2). The first sequencing method, known as chain-termination method, was first developed by Sanger in 1977, and was based on capillary electrophoresis of fragmented DNA bound to a single-stranded DNA template. The main drawback of Sanger sequencing relied on the possibility to sequence only a low amount of DNA at one time[56]. To date, more advanced sequencing technologies have been developed and allow massive, faster and more precise sequencing of nucleic acids. These are the next generation sequencing (NGS) - more widely used - and third generation sequencing (TGS). In Table 1, we summarize details of the existing advanced sequencing methods and how they work.
4.2. Single cell and spatial genomics
Single cell DNA sequencing (scDNAseq) allows DNA profiling in individual cells[62]. The whole genome of single cells can be primarily amplified by three methods: the degenerate oligonucleotide primed PCR (DOP-PCR), the multiple displacement amplification (MDA), and the multiple annealing and looping-based amplification cycles (MALBAC)[63]. scDNAseq is generally based on NGS. Recently, a single cell WGS method based on TGS was also developed and is known as single-molecule real-time sequencing of long fragments amplified through transposon insertion (SMOOTH-seq). SMOOTH-seq has greatly improved the identification of structural variants (SVs) and extra-chromosomal DNA compared to NGS, thanks to the possibility to sequence long reads[64]. Also, spatial genomics have recently been developed but they are mostly used in cancer research to dissect the cellular genome heterogeneity of tumoral cells[65].
4.3. Genomic studies have delivered unprecedented knowledge on the genetic background and early diagnosis of inherited hearing loss
Genetic hearing loss that affects any part of the auditory system accounts for ~50% of the deaf population, and can be either non-syndromic (70%)[66,67] or syndromic (30%)[68]. The large heterogeneity of genes engaged in deafness makes it difficult to study and diagnose it[52]. However, thanks to the advancements in genomics, to date several variants have been identified in genes associated with hearing loss. For instance, the combination of WES, qPCR and TGS was able to unravel for the first-time novel SVs of centrosomal protein 78 (CEP78), a key gene responsible for hearing loss associated with cone-rod dystrophy (CRDHL)[55]. Thanks to advanced genomics, new variants have recently been outlined in important hearing loss-related genes, that are: myosin 15 A (MYO15A), otoferlin (OTOF), radixin (RDX)[69], TATA-box binding protein associated factor 1 (TAF1)[70], atonal BHLH transcription factor 1 (ATOH1)[71], and centrosomal protein 78 (CEP78)[55]. The discovery of novel variants represents a fundamental step forward in the understanding of the molecular basis of cochlear diseases, and indeed it has improved the diagnosis of genetic hearing loss, as well as the prediction of its severity and prognosis. So far, a number of studies have benefitted from genome sequencing (via either WES or WGS) for early detection of hearing loss[72,73,74]. For instance, a recent study has shown that the combination of conventional hearing screening and extended genetic sequencing improves the early diagnosis of inherited hearing loss in newborns, with important implications for their clinical management[75]. Yet, genetic conducive and SNHL are a common occurrence among newborns, whose diagnosis was often missed due to the lack of proper genetic screening at birth.
Overall, the use of genomics has been useful in revealing novel gene variants linked to hearing loss, and thus it represents a potent diagnostic tool for the genetic screening of inherited deafness.
5. Transcriptomics
Transcriptomics enable the analysis of gene expression at the RNA level, including messenger RNAs (mRNAs), transfer RNAs (tRNAs), ribosomal RNAs (rRNAs), and other non-coding RNAs (ncRNAs) [e.g., microRNAs (miRNAs), long-non-coding RNAs (lncRNAs), and circular RNAs (circRNAs)][34,76]. As for genomics, the currently most used transcriptomics technologies are based on sequencing (described above in paragraph 4.1); however, transcriptome profiling is more challenging compared to genome sequencing, due to the highly dynamic nature of the transcriptome in biological processes. Sequencing of the entire transcriptome in a tissue or cell population is known as bulk-RNAseq and can be performed either by direct RNA sequencing (dRNA-seq) or by cDNA sequencing[77]. Additionally, the transcriptome can also be investigated at single cell resolution or through spatial transcriptomics, as detailed in the following paragraphs. A schematic overview of the transcriptomics techniques is shown in Figure 3.
5.1. Single cell transcriptomics
Single cell RNA sequencing (scRNA seq) allows RNA profiling of individual cells. Notably, most of the single cell omics developed so far have been focused on the transcriptome[46,47,78]. The currently available high-throughput platforms for scRNAseq require retro-transcription of the RNA into cDNA, which is then amplified for the preparation of sequencing libraries. The amplification methods may be based on PCR or in vitro transcription (IVT) and are followed by sequencing through different platforms[78,79]. Among the most widely used scRNAseq platforms, it is important to mention: SMART/SMARTseq2, CEL-seq/CEL-seq2, 10X Genomics, Drop-seq, inDrop, seq-well[47,79]. To date, the most used commercial platform is the 10X Genomics Chromium, that is a droplet-based scRNAseq technology (10X Genomics, Pleasanton, CA). Thanks to the development of microfluidics-based [e.g., Drop-seq[80] and inDrop[42,81] and of nanowell-based (such as the seq-well) methods, an important improvement has been made in scRNAseq, since it is now possible to sequence dozens of thousands of cells individually[82] Recently, single nuclei RNA sequencing (snRNA-Seq) has also been developed to improve the quality of scRNAseq by mitigating the expression changes that can be induced by enzymatic cellular dissociation methods; snRNA-Seq is also used to study gene expression under particular conditions, such as those where it is difficult to recover intact cells[83,84]. However, it is important to outline that snRNA-Seq does not include cytoplasmatic RNAs, hence it could hide important information needed to fully characterize the cell transcriptome.
5.2. Spatial transcriptomics
The spatial omics for transcriptome studies are broadly divided in imaging-based technologies (where RNA is detected using fluorophores on intact tissues and then detected by microscopy) and sequencing-based technologies (based on RNA capturing from the tissue, followed by NGS)[65,85,86,87].
Imaging-based technologies
One of the major imaging-based methodologies for spatial transcriptomics is fluorescent in situ hybridization (FISH), which includes SeqFISH[88], SeqFISH+ , and MERFISH[89]. The principle of FISH is the hybridization of fluorescent probes to nucleic acids on a tissue section directly, which is then analyzed through microscopy. A recent improvement of FISH is the enhanced electric FISH (EEL-FISH), in which tissue mRNAs are electrophoretically transferred onto glass coverslips before the hybridization step, a step which accelerates data collection due to reduced need of acquired images compared to the other FISH techniques[90]. Notably, today it is also possible to visualize 3D gene expression in a tissue thanks to the development of the expansion-assisted iterative fluorescence in situ hybridization (EASI-FISH) methodology[87,91] Another imaging-based approach for spatial transcriptomics is in situ sequencing (ISS), where nucleic acids are first amplified (preserving spatial localization) and then profiled through microscopy (1-2 nucleotides at a time) by using distinct fluorophores[86,87,92]
Sequencing-based technologies
Sequencing-based technologies allow the sequencing of the RNA from a tissue section through NGS. The spatial information is also retained because of the recording of the specific location where the RNA is captured. Sequencing-based techniques include microdissection-based and array-based methodologies[87]. Microdissection-based methods allow sequencing of a specific portion of a tissue by microdissection. Hence, the main limitation is the low spatial resolution that can be obtained. Microdissection-based technologies include laser capture microdissection combined with NGS (LCM-NGS)[93], Tomo-seq[94], Geo-seq[95], GeoMx DSP[96,97] and STRP-seq[98].
Differently, array-based technologies employ arrays with spatially-barcoded probes, that enable retro-transcription of local RNAs into cDNAs and then sequencing. In this case, the spatial resolution depends on the area of the barcode[87]. To date, Visium by 10X Genomics has achieved a spatial resolution of 2 μm[99,100], while the Stereo-seq , is capable to trigger even a lower resolution, up to 0.5 μm[101]. Other notable techniques for spatial transcriptomic analysis also include slide-seq and slide-seqV2[65,102,103], and the deterministic barcoding in tissue for spatial omics sequencing (DBiT-seq), which employs microfluidic channels used to print the array directly onto the tissue. Here, the spatial resolution depends on the diameter of the microfluidic channel used (no less than 10 μm)[104].
5.3. scRNAseq is a key tool for deciphering the complex cellular heterogeneity of the cochlea
Previous studies on the auditory transcriptome were performed using microarray technologies and bulk RNA-seq[105], providing significant knowledge and information on the differential gene expression in physiological and pathological conditions of the cochlea, with important implications for the development of new therapies[106,107]). Studies on the transcriptome have also given insights into the developmental processes of the inner ear[108], on the transcriptional changes associated with ageing[109] and on cochlear cell damage/degeneration[110]. However, information on the specific cell populations is not possible with bulk RNA-seq, and this is a major limit for studies on the cochlea due to its heterogenic cell types[105] .
In this context, scRNAseq has given unprecedented information on cochlear cell diversity and alternative signaling mechanisms[111]. Profiling the transcriptome at single cell resolution helped to unravel novel populations of cells in the cochlea. For instance, subtypes of SCs (lateral and medial) with a distinct cluster of regenerative-associated markers were discovered in the avian cochlea[112]. Intriguingly, some of these discovered markers were also found to be expressed in specific regions of the mammalian cochlea[113], further supporting the stem cell-like potential of SCs[114]. Moreover, scRNAseq has also allowed the identification of new specific markers of HCs, like sorcin (sri) for OHCs[115], that was then discovered to be implicated in calcium dynamics and somatomotility of OHCs[115]. Another important application of scRNAseq has been the study of exons and genes associated with deafness[115,116]. In this context, new genes associated with apoptosis, calcium regulation, and extra cellular matrix (ECM) were found to be modulated in HCs of inner ear organoids in association to type II transmembrane protease 3 (tmprss3), a key gene for hearing loss[116]. Likewise, differential gene expression patterns among the cells of the lateral wall, of the stria vascularis, of the immune system, and SGNs have been identified is association to acoustic trauma by means of scRNAseq, delineating a cell-specific transcriptomic map of the cochlea upon noise damage[117].
Overall, due to the complexity and heterogeneity of the cochlea, scRNAseq has provided umatched opportunites to further our current understanding of its molecular underpinnings in health and disease.
5.4. Spatial transcriptomics have enabled to understand the cellular and molecular architecture of the cochlea
The cochlea is spatially organized with distinct and localized functions. Thanks to the development of spatial transcriptomics methodologies, it is now possible to study the localization of specific gene expression patterns in relationship to the different anatomical structures of the cochlea[118,119]. For instance, the combination of scRNAseq and FISH has allowed the identification of two subpopulations of SCs (named SC1 and SC2) retaining distinct transcriptomes in specific anatomic locations of the cochlea: medial for SC1 and lateral for SC2[120]. Spatial transcriptomics are also particularly important for studies of the developing cochlea, since the cochlea’s cellular organization during development is regulated by several spatiotemporal dependent key signalling mechanisms. For instance, Munnamalai and co-workers investigated the spatiotemporal cadence of Wnt, NOTCH and BMP signaling in the developing cochlea, and found that they are differentially regulated depending on the cochlear location (from lateral to medial) and on the developmental stage. This study emphasizes the spatiotemporal signaling necessary to modulate the development of the cochlea in its radial axis and further supports the importance of spatial transcriptomics for cochlear research[121]. Another study used LCM-NGS to profile the transcriptome in different regions of the cochlea (e.g., the organ of Corti, spiral ganglion, lateral wall, and spiral limbus) and provided quantitative information of the transcripts of each region with important findings on deafness-associated genes[93].
To our knowledge, more advanced spatial transcriptomics technologies, like the Visium or the Stereo-seq technologies, have not yet been applied to cochlear research. However, it is expected that they could provide unmatched opportunities for future studies in the field.
6. Epigenomics
6.1. Principles of epigenomics
The term epigenomics refers to the techniques used to investigate the epigenome, that is the set of regulatory processes that modify the activity of gene expression without modifications in the DNA sequence. Epigenomics can be classified depending on the target: DNA methylation, histone modifications, chromatin accessibility and chromosome interactions. The methodologies to study bulk epigenomics can be further classified as array-based and sequencing-based techniques[122]. Array-based technologies use hybridization to pre-designed microarrays, while sequencing-based methods use sequencing techniques like NGS. It is important to highlight that for DNA methylation, it is required a first step for the exposure of the methylated DNA through one of the following methods: (i) DNA digestion by methylation-sensitive restriction enzymes (MSREs)[123], (ii) affinity enrichment of DNA by antibodies targeting methylated CpGs[124], (iii) conversion of unmethylated cytosines to uracil by bisulfite treatment[123]. To date, the bisulfite sequencing (BS-seq) method is considered the gold standard technique for studies on DNA methylation, because of its single-base resolution[122]. Important techniques used for monitoring histone modifications include chromatin immunoprecipitation (ChIP), which is based on antibodies targeting histone modifications of interest, and the cleavage under targets & release using nuclease (CUT&RUN)[125] and cleavage under targets and tagmentation (CUT&TAG)[126] methods, which both rely on the same principle of recognizing DNA bound proteins of interest through specific antibodies. Multiple techniques for chromatin accessibility studies have also been developed. Among these, the most recent is the accessible chromatin using sequencing technology (ATAC-seq). It employs tagmentation (inserting adapter sequences by using the hyperactive mutant Tn5 transposase) to open target regions of the chromatin, that are then amplified and sequenced[127]. Other widely used techniques for chromatin accessibility include the DNase I hyper-sensitive sites sequencing (DNAse-seq)[128], the micrococcal nuclease digestion with deep sequencing (MNase seq)[129], and the formaldehyde-assisted identification of regulatory elements followed by sequencing (FAIRE-seq)[130]. The higher order organization of the nucleus is also important for the epigenetic regulation of cellular processes, hence techniques able to analyze chromosomal interactions have also been developed. They include the chromatin conformation capture technique (3C), Hi-C, the chromatin interaction analysis by paired-end tag sequencing (ChIA PET) and the proximity ligation-assisted ChIP-seq (PLAC-seq)[122]. Further details of available epigenomics methodologies have already been extensively reviewed (see[122,131]), and those applied to cochlear research are summarized in Figure 4.
6.2. Single-cell epigenomics
Single-cell epigenomics enable a detailed analysis of the epigenetic regulation at the single cell resolution, and include: single-cell DNA methylation profiling, single-cell chromatin mapping, single-cell Hi-C and single-cell replication dynamics[132]. Single cell DNA methylation profiling can in turn be performed using a variety of methods, the most recent of which are single-cell combinatorial indexing for methylation analysis (sci-MET) and single-cell CGI methylation sequencing (scCGI-seq)[122,133,134]Histone modifications in single cells can also be studied using: sc-ChIP-seq, single-cell droplet–based chromatin immunoprecipitation (drop-ChIP)[47], single-cell chromatin immune-cleavage sequencing technique (scChIC-seq), antibody-guided chromatin tagmentation sequencing (ACT-seq), combinatorial barcoding and targeted chromatin release (COBATCH), and single-cell chromatin integration labeling sequencing (scChIL-seq)[122]. Finally, single-cell chromatin accessibility can be investigated by sc-DNAse-seq and sc-ATAC-seq. The available single cell epigenetic methods have been recently reviewed in detail (see[132,135]).
6.3. Spatial epigenomics
To fully appreciate the influence of epigenetic variations in patho-physiological processes, it is essential to know their spatial context. However, the development of spatial epigenomics techniques has been challenging for a long time due to the limited spatial resolution available[136,137]. The first spatial epigenomic technology was developed in 2021 and is now beginning to open new possibilities in the field of biology and medicine. The high-spatial-resolution chromatin modification state profiling by sequencing (hsrChST-seq) is the first spatial epigenomic technique that has been developed, and is based on the spatial transcriptomic technique DBiT-seq. This method is based on the combination of CUT&TAG and tissue deterministic barcoding with fluorescence microscopy[138]. Another technique developed later to resolve spatially the chromatin accessibility is the spatial-ATAC seq, which is based on the combination of in situ Tn5 transposase chemistry with microfluidic deterministic barcoding[139]. Recently, thanks to the advent of epigenomic MERFISH (which combines CUT&TAG and MERFISH) - a spatial epigenomic technique for the analysis of histone modifications- it has been possible to analyze the active and inactive promoters/enhancers associated with histone modifications in single cells while maintaining spatial information[140]. Furthermore, LCM can be applied to epigenomics in order to analyze spatially modifications in the epigenome[141]. Finally, the most recent epigenomic technique is the spatial chromatin accessibility sequencing (SCA-seq), which provides simultaneous knowledge on the chromatin accessibility, epigenomics marks (e.g., CpG methylation) and higher order genome architecture[142].
6.4. Epigenetic profiling of the cochlea has provided new insights into the mechanisms whereby genes responsible for auditory function are regulated
Hearing loss can be caused by epigenetic alterations or by mutations in the genes encoding for the epigenetic machinery, affecting DNA methylation dynamics[143,144,145], histone modifications[146,147,148], and chromatin remodeling[144,149,150]. Thus, investigating epigenetic mechanisms could eventually pave the way towards new approaches to therapeutics. To date, most of the studies on cochlear epigenome are based on bulk epigenomic profiling and only a few were performed with single cell epigenomics, namely scATAC-seq[151,152]. Instead, spatial epigenomics have not yet been applied in this field, though the epigenomics studies conducted until now have given profound insights into the regulatory mechanisms of development, trans-differentiation and regeneration of the auditory system. Application of ChIP-seq and ChIP-qPCR have led to the identification of fundamental epigenetic modifications in the promoters of two key genes involved in SGNs differentiation (Cdk2 and NeuroD1), which can affect the binding of the regulatory transcription factor neurogenin 1 (neurog1)[153]. Also, ChIP-qPCR allowed to describe the histone modifications associated with the epigenetic regulation of Atonal bHLH transcription factor 1 (Atoh1), that is an evolutionary conserved transcription factor for the development of the auditory system[154]. Yet, histone modifications of Atoh1, that are characteristic of HCs during their development, are suppressed in the same cells after birth, but they persist in perinatal SCs. This is an important finding, which gives new information on the mechanisms underlying the regenerative potential of SCs[154]. Likewise, ATAC-seq provided new findings on specific variations in chromatin accessibility during the reprogramming of SCs into HCs in cochlear organoids[155], and scATAC-seq has unraveled important information on the mechanisms which limit the capacity of SCs trans-differentiation into HCs in the adult mammalian cochlea[151]. Furthermore, the combination of scATAC-seq with scRNA-seq has recently allowed to identify molecular regulators of key transcription factors (such as Sox and Six) involved in HCs regeneration from SCs in the zebrafish inner ear[152].
Overall, the epigenome profiling conducted until now in the cochlea has given new insights into the regulatory mechanisms of cochlear development, regeneration and disease. It is expected that application of the most advanced spatial epigenomics techniques could provide an even better understanding of those processes in the next future.
7. Discussion
The cochlea is a complex sensory organ, whose degeneration may be caused by multiple damaging conditions that lead to irreversible hearing loss. The neuro-sensory epithelium - the organ of Corti - is particularly susceptible to degeneration as a consequence of inherited or environmental conditions. Moreover, SGNs degenerate as a consequence of damage to the organ of Corti, resulting in reduced performance of cochlear implants[156,157,158]. To date, there are no effective treatments to prevent cochlear degeneration or to promote its regeneration. All these issues call for an urgent need of new therapeutic approaches, which ultimately depend on the understanding of the detailed molecular mechanisms that underlie cochlear physiology and pathology. The appreciation of the molecular bases (both genetic and non-genetic) of hearing loss and of developmental processes of the cochlea, and studies on the stem cell-like regenerative potential of SCs in the organ of Corti are among the most active fields of cochlear research. Their significant improvement has been possible due to advanced omics, and Table 2 summarizes the advanced genomics, transcriptomics and epigenomics techniques that have been applied in cochlear research, alongside the improvement that they have provided in the field. For instance, genome sequencing has allowed to identify new variants in genetic hearing loss and represents a great improvement in the diagnosis of genetic deafness in newborns[72,73,74]. Nonetheless, studies on the cochlea are particularly challenging compared to other sensory organs, because of several practical limitations. Indeed, the cochlea is enveloped in the bony labyrinth, and cannot be visualized directly, requiring imaging techniques like MRI and CT[159,160], or histological techniques of post-mortem tissues in humans and animals[161,162]. The high heterogeneity of the tissue[163], as well as the low number of cells especially in the organ of Corti (~3.500 IHCs and ~12.000 OHCs in the human ear)[164], represent additional major limitations for studies in the field, especially with the conventional molecular techniques (like real time PCR or western blotting). On the one hand, due to the low amount of cells, the quantity of nucleic acids or other molecules (proteins and metabolites) is very limited and it is often necessary to pool tissues together to reach a sufficient amount of material for the analysis; on the other hand, since there are several sub-populations with different functional and morphological properties, and since also the same cell types differ in expression and phenotype along the tonotopic axis of the cochlea, interpretation of the molecular results that derive from the whole tissue could be difficult. In fact, bulk studies in the whole cochlea may easily hide some molecular information, eventually diluted in the pool of the whole genome/transcriptome/epigenome. More recently, due to the advancements in single cell omics techniques, these limitations have been successfully overcome[8]. In cochlear research, most of the single cell studies have been performed to investigate the transcriptome through scRNAseq[8,165]. Single cell analysis does not allow to retain the spatial information, and the application of spatial transcriptomics seems of particular relevance in the field because of the peculiar cellular heterogeneity of the cochlea[118,121]. Thanks to spatial transcriptomics, it has been possible to identify differential expression patterns for the development and regeneration of the cochlea in distinct cell populations of specific anatomical locations[166]. For instance, Waldhaus and colleagues profiled SCs in the apex and base of the murine cochlea and found that SCs - especially Pillar cells - express regenerative and proliferative genes potentially relevant for HCs regeneration in mouse apical cochlea. However, the most advanced spatial omics techniques have not yet been applied to cochlear research. Therefore, it is expected that using high throughput resolution spatial omics will enable a further dissection of the cochlear patho-physiology in more detail. Moreover, thanks to the application of scRNAseq in species able to self-regenerate HCs (like zebrafish and birds), researchers have identified key expression patterns in SCs, which could potentially induce their reprogramming into HCs also in the mammalian cochlea[167,168]. However, although some HC-like cells have been successfully regenerated in the murine cochlea by forcing the expression of the identified patterns, fully differentiated and functional HCs in the mammalian cochlea have not yet been developed. Notably, recent studies seem to indicate that this is due to epigenetic patterns. Yet, epigenomic data obtained through scATAC-seq have identified specific epigenetic modifications in the zebrafish SCs important for their reprogramming into HCs[152]. Likewise, scATAC-seq also revealed the epigenetic mechanisms responsible of the inability of SCs to trans-differentiate into HCs in the mammalian cochlea[151]
Despite the huge amount of information provided by high-throughput omics methodologies, a major limitation relies in the storage of data and their interpretation. Indeed, multiple platforms are used for data storage and each retains a different format; thus, a pre-processing of data is necessary before the analysis. Moreover, artifacts may be generated for multiple reasons, such as the low amount of input material (especially for genome sequencing die to the presence of only two DNA copies), and the induction of stress genes due to the dissociation methods used for cell isolation. Therefore, orthogonal validation with targeted approaches is needed, and specialized bio-informaticians are necessary to properly read and interpret the data[46,78]
Overall advanced genomics, epigenomics and transcriptomics techniques represent the state-of-the-art approaches in cochlear research and are providing unprecedented information on the molecular bases of cochlear patho-physiology. From the available literature, it is increasingly evident that multi-omics approaches are necessary to achieve an integrated view of the biological processes of the cochlea in health and disease. As yet, spatial omics have been poorly used in cochlear research, hence it is expected that further studies in this direction, as well as the combination of genomics/transcriptomics/epigenomics with other omics - like proteomics and metabolomics , will give unmatched opportunities to decipher the complex molecular underpinnings of such a complex sensory organ.
Author Contributions
MM: conceptualization; AT and SP: writing-original draft preparation; MM: resources; AT, SP and MM: writing – review and editing; AT and MM: supervision; AT and SP: figures and tables preparation. All the authors contributed to the article and approved the submitted version.
Funding
SP was partly supported by Dompé Farmaceutici S.p.A. (Milan, Italy), under a PhD programme in Experimental Medicine that is co-shared with University of L’Aquila and supervised by MM.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Deafness and Hearing Loss Available online:. Available online: https://www.who.int/news-room/fact-sheets/detail/deafness-and-hearing-loss (accessed on 23 August 2023).
- Ma, Y.; Wise, A.K.; Shepherd, R.K.; Richardson, R.T. New Molecular Therapies for the Treatment of Hearing Loss. Pharmacol Ther 2019, 200, 190–209. [Google Scholar] [CrossRef]
- Liberman, M.C.; Kujawa, S.G. Cochlear Synaptopathy in Acquired Sensorineural Hearing Loss: Manifestations and Mechanisms. Hear Res 2017, 349, 138–147. [Google Scholar] [CrossRef]
- Wise, A.K.; Pujol, R.; Landry, T.G.; Fallon, J.B.; Shepherd, R.K. Structural and Ultrastructural Changes to Type I Spiral Ganglion Neurons and Schwann Cells in the Deafened Guinea Pig Cochlea. JARO: Journal of the Association for Research in Otolaryngology 2017, 18, 751. [Google Scholar] [CrossRef] [PubMed]
- Smith-Cortinez, N.; Tan, A.K.; Stokroos, R.J.; Versnel, H.; Straatman, L. V. Regeneration of Hair Cells from Endogenous Otic Progenitors in the Adult Mammalian Cochlea: Understanding Its Origins and Future Directions. International Journal of Molecular Sciences 2023, Vol. 24, Page 7840 2023, 24, 7840. [Google Scholar] [CrossRef]
- Santos-Cortez, R.L.P.; Yarza, T.K.L.; Bootpetch, T.C.; Tantoco, M.L.C.; Mohlke, K.L.; Cruz, T.L.G.; Perez, M.E.C.; Chan, A.L.; Lee, N.R.; Tobias-Grasso, C.A.M.; et al. Identification of Novel Candidate Genes and Variants for Hearing Loss and Temporal Bone Anomalies. Genes (Basel) 2021, 12, 566. [Google Scholar] [CrossRef] [PubMed]
- Kelley, M.W. Cochlear Development; New Tools and Approaches. Front Cell Dev Biol 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Xia, M.; Li, W.; Li, H. Single-Cell Sequencing Applications in the Inner Ear. Front Cell Dev Biol 2021, 9. [Google Scholar] [CrossRef]
- Bommakanti, K.; Iyer, J.S.; Stankovic, K.M. Cochlear Histopathology in Human Genetic Hearing Loss: State of the Science and Future Prospects. Hear Res 2019, 382, 107785. [Google Scholar] [CrossRef]
- Yamakami, I.; Ito, S.; Higuchi, Y. Retrosigmoid Removal of Small Acoustic Neuroma: Curative Tumor Removal with Preservation of Function: Clinical Article. J Neurosurg 2014, 121, 554–563. [Google Scholar] [CrossRef]
- Nicoli, T.K.; Atula, T.; Sinkkonen, S.T.; Korpi, J.; Vnencak, M.; Tarkkanen, J.; Mäkitie, A.A.; Jero, J. Ear Canal and Middle-Ear Tumors: A Single-Institution Series of 87 Patients. 2022, 142, 132–139. [CrossRef]
- Gao, S.S.; Xia, A.; Applegate, B.E.; Shelton, R.L.; Yuan, T.; Raphael, P.D.; Oghalai, J.S. Quantitative Imaging of Cochlear Soft Tissues in Wild-Type and Hearing-Impaired Transgenic Mice by Spectral Domain Optical Coherence Tomography. Optics Express, Vol. 19, Issue 16, pp. 15415-15428 2011, 19, 15415–15428. [Google Scholar] [CrossRef]
- Rivolta, M.N.; Holley, M.C. Cell Lines in Inner Ear Research. J Neurobiol 2002, 53, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.J.; Moon, S.K. Establishment of Cell Lines from the Human Middle and Inner Ear Epithelial Cells. Adv Exp Med Biol 2011, 720, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Haque, K.D.; Pandey, A.K.; Kelley, M.W.; Puligilla, C. Culture of Embryonic Mouse Cochlear Explants and Gene Transfer by Electroporation. JoVE (Journal of Visualized Experiments) 2015, e52260. [Google Scholar] [CrossRef]
- Mazzarda, F.; D’Elia, A.; Massari, R.; De Ninno, A.; Bertani, F.R.; Businaro, L.; Ziraldo, G.; Zorzi, V.; Nardin, C.; Peres, C.; et al. Organ-on-Chip Model Shows That ATP Release through Connexin Hemichannels Drives Spontaneous Ca2+ Signaling in Non-Sensory Cells of the Greater Epithelial Ridge in the Developing Cochlea. Lab Chip 2020, 20, 3011–3023. [Google Scholar] [CrossRef]
- Roccio, M.; Edge, A.S.B. Inner Ear Organoids: New Tools to Understand Neurosensory Cell Development, Degeneration and Regeneration. Development (Cambridge) 2019, 146. [Google Scholar] [CrossRef]
- Lin, X.; Luo, J.; Tan, J.; Yang, L.; Wang, M.; Li, P. Experimental Animal Models of Drug-Induced Sensorineural Hearing Loss: A Narrative Review. Ann Transl Med 2021, 9, 1393–1393. [Google Scholar] [CrossRef]
- Legan, P.K.; Rau, A.; Keen, J.N.; Richardson, G.P. The Mouse Tectorins. Modular Matrix Proteins of the Inner Ear Homologous to Components of the Sperm-Egg Adhesion System. J Biol Chem 1997, 272, 8791–8801. [Google Scholar] [CrossRef]
- Verhoeven, K.; Van Laer, L.; Kirschhofer, K.; Legan, P.K.; Hughes, D.C.; Schatteman, I.; Verstreken, M.; Van Hauwe, P.; Coucke, P.; Chen, A.; et al. Mutations in the Human Alpha-Tectorin Gene Cause Autosomal Dominant Non-Syndromic Hearing Impairment. Nat Genet 1998, 19, 60–62. [Google Scholar] [CrossRef]
- Vreugde, S.; Erven, A.; Kros, C.J.; Marcotti, W.; Fuchs, H.; Kurima, K.; Wilcox, E.R.; Friedman, T.B.; Griffith, A.J.; Bailing, R.; et al. Beethoven, a Mouse Model for Dominant, Progressive Hearing Loss DFNA36. Nature Genetics 2002 30:3 2002, 30, 257–258. [Google Scholar] [CrossRef]
- Friedman, L.M.; Dror, A.A.; Avraham, K.B. Mouse Models to Study Inner Ear Development and Hereditary Hearing Loss. Int J Dev Biol 2007, 51, 609–631. [Google Scholar] [CrossRef]
- Le Prell, C.G.; Hammill, T.L.; Murphy, W.J. Noise-Induced Hearing Loss: Translating Risk from Animal Models to Real-World Environments. J Acoust Soc Am 2019, 146, 3646–3651. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome Engineering Using the CRISPR-Cas9 System. Nature Protocols 2013 8:11 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yang, Y.; Qi, H.; Cui, W.; Zhang, L.; Fu, X.; He, X.; Liu, M.; Li, P. feng; Yu, T. CRISPR/Cas9 Therapeutics: Progress and Prospects. Signal Transduction and Targeted Therapy 2023 8:1 2023, 8, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Akram, F.; Sahreen, S.; Aamir, F.; Haq, I. ul; Malik, K.; Imtiaz, M.; Naseem, W.; Nasir, N.; Waheed, H.M. An Insight into Modern Targeted Genome-Editing Technologies with a Special Focus on CRISPR/Cas9 and Its Applications. Mol Biotechnol 2023, 65, 227–242. [Google Scholar] [CrossRef]
- Zou, B.; Mittal, R.; Grati, M.; Lu, Z.; Shu, Y.; Tao, Y.; Feng, Y.; Xie, D.; Kong, W.; Yang, S.; et al. The Application of Genome Editing in Studying Hearing Loss. Hear Res 2015, 327, 102–108. [Google Scholar] [CrossRef]
- Gao, X.; Tao, Y.; Lamas, V.; Huang, M.; Yeh, W.H.; Pan, B.; Hu, Y.J.; Hu, J.H.; Thompson, D.B.; Shu, Y.; et al. Treatment of Autosomal Dominant Hearing Loss by in Vivo Delivery of Genome Editing Agents. Nature 2018 553:7687 2017, 553, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Farooq, R.; Hussain, K.; Tariq, M.; Farooq, A.; Mustafa, M. CRISPR/Cas9: Targeted Genome Editing for the Treatment of Hereditary Hearing Loss. Journal of Applied Genetics 2020 61:1 2020, 61, 51–65. [Google Scholar] [CrossRef]
- Wu, J.; Tao, Y.; Deng, D.; Meng, Z.; Zhao, Y. The Applications of CRISPR/Cas-Mediated Genome Editing in Genetic Hearing Loss. Cell & Bioscience 2023 13:1 2023, 13, 1–25. [Google Scholar] [CrossRef]
- Rathinam, R.; Rosati, R.; Jamesdaniel, S. CRISPR/Cas9-Mediated Knockout of Lim-Domain Only Four Retards Organ of Corti Cell Growth. J Cell Biochem 2018, 119, 3545–3553. [Google Scholar] [CrossRef]
- Gu, X.; Wang, D.; Xu, Z.; Wang, J.; Guo, L.; Chai, R.; Li, G.; Shu, Y.; Li, H. Prevention of Acquired Sensorineural Hearing Loss in Mice by in Vivo Htra2 Gene Editing. Genome Biol 2021, 22, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Duan, X.; Li, M.; Liu, D.; Bai, X. CRISPR/Cas9-Mediated Pou4f3 Knockout Induces Defects in the Development of the Zebrafish Inner Ear. J BioX Res 2021, 4, 163–170. [Google Scholar] [CrossRef]
- Dai, X.; Shen, L. Advances and Trends in Omics Technology Development. Front Med (Lausanne) 2022, 9, 911861. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.; Li, R.; Tian, Y.; Zhang, Y.; Lu, Y.; Ou, Q.; Gao, P.; Li, K.; Zhang, Y. Single-Cell Omics: A New Direction for Functional Genetic Research in Human Diseases and Animal Models. Front Genet 2023, 13, 1100016. [Google Scholar] [CrossRef] [PubMed]
- Bingham, G.C.; Lee, F.; Naba, A.; Barker, T.H. Spatial-Omics: Novel Approaches to Probe Cell Heterogeneity and Extracellular Matrix Biology. Matrix Biology 2020, 91–92, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.Y.; Rozanas, C.R.; Thalmann, I.; Chance, M.R.; Alagramam, K.N. Inner Ear Proteomics of Mouse Models for Deafness, a Discovery Strategy. Brain Res 2006, 1091, 113–121. [Google Scholar] [CrossRef]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour Evolution Inferred by Single-Cell Sequencing. Nature 2011, 472, 90–95. [Google Scholar] [CrossRef]
- Pollen, A.A.; Nowakowski, T.J.; Shuga, J.; Wang, X.; Leyrat, A.A.; Lui, J.H.; Li, N.; Szpankowski, L.; Fowler, B.; Chen, P.; et al. Low-Coverage Single-Cell MRNA Sequencing Reveals Cellular Heterogeneity and Activated Signaling Pathways in Developing Cerebral Cortex. Nat Biotechnol 2014, 32, 1053–1058. [Google Scholar] [CrossRef]
- Ealy, M.; Ellwanger, D.C.; Kosaric, N.; Stapper, A.P.; Heller, S. Single-Cell Analysis Delineates a Trajectory toward the Human Early Otic Lineage. Proc Natl Acad Sci U S A 2016, 113, 8508–8513. [Google Scholar] [CrossRef]
- Tasic, B. Single Cell Transcriptomics in Neuroscience: Cell Classification and Beyond. Curr Opin Neurobiol 2018, 50, 242–249. [Google Scholar] [CrossRef]
- Macaulay, I.C.; Ponting, C.P.; Voet, T. Single-Cell Multiomics: Multiple Measurements from Single Cells. Trends in Genetics 2017, 33, 155–168. [Google Scholar] [CrossRef]
- Hwang, B.; Lee, J.H.; Bang, D. Single-Cell RNA Sequencing Technologies and Bioinformatics Pipelines. Experimental & Molecular Medicine 2018 50:8 2018, 50, 1–14. [Google Scholar] [CrossRef]
- Xu, X.; Wang, J.; Wu, L.; Guo, J.; Song, Y.; Tian, T.; Wang, W.; Zhu, Z.; Yang, C. Microfluidic Single-Cell Omics Analysis. Small 2020, 16, 1903905. [Google Scholar] [CrossRef] [PubMed]
- Menze, L.; Duarte, P.A.; Haddon, L.; Chu, M.; Chen, J. Selective Single-Cell Sorting Using a Multisectorial Electroactive Nanowell Platform. ACS Nano 2022, 16. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Navin, N.E. Advances and Applications of Single-Cell Sequencing Technologies. Mol Cell 2015, 58, 598–609. [Google Scholar] [CrossRef]
- Mincarelli, L.; Lister, A.; Lipscombe, J.; Macaulay, I.C. Defining Cell Identity with Single-Cell Omics. Proteomics 2018, 18, 1700312. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Guo, H.; Cao, C.; Li, X.; Hu, B.; Zhu, P.; Wu, X.; Wen, L.; Tang, F.; Huang, Y.; et al. Single-Cell Triple Omics Sequencing Reveals Genetic, Epigenetic, and Transcriptomic Heterogeneity in Hepatocellular Carcinomas. Cell Research 2016 26:3 2016, 26, 304–319. [Google Scholar] [CrossRef]
- Raphael, Y.; Altschuler, R.A. Structure and Innervation of the Cochlea. Brain Res Bull 2003, 60, 397–422. [Google Scholar] [CrossRef] [PubMed]
- Rehman, A.U.; Morell, R.J.; Belyantseva, I.A.; Khan, S.Y.; Boger, E.T.; Shahzad, M.; Ahmed, Z.M.; Riazuddin, S.; Khan, S.N.; Riazuddin, S.; et al. Targeted Capture and Next-Generation Sequencing Identifies C9orf75, Encoding Taperin, as the Mutated Gene in Nonsyndromic Deafness DFNB79. Am J Hum Genet 2010, 86, 378–388. [Google Scholar] [CrossRef]
- Yan, D.; Tekin, M.; Blanton, S.H.; Liu, X.Z. Next-Generation Sequencing in Genetic Hearing Loss. 2013, 17, 581–587. [Google Scholar] [CrossRef]
- Shearer, A.E.; Smith, R.J.H. Massively Parallel Sequencing for Genetic Diagnosis of Hearing Loss. Otolaryngology–Head and Neck Surgery 2015, 153, 175–182. [Google Scholar] [CrossRef]
- Dunn, P.; Albury, C.L.; Maksemous, N.; Benton, M.C.; Sutherland, H.G.; Smith, R.A.; Haupt, L.M.; Griffiths, L.R. Next Generation Sequencing Methods for Diagnosis of Epilepsy Syndromes. Front Genet 2018, 9, 314696. [Google Scholar] [CrossRef]
- Hegde, M.; Santani, A.; Mao, R.; Ferreira-Gonzalez, A.; Weck, K.E.; Voelkerding, K. V. Development and Validation of Clinical Whole-Exome and Whole-Genome Sequencing for Detection of Germline Variants in Inherited Disease. Arch Pathol Lab Med 2017, 141, 798–805. [Google Scholar] [CrossRef]
- Ascari, G.; Rendtorff, N.D.; De Bruyne, M.; De Zaeytijd, J.; Van Lint, M.; Bauwens, M.; Van Heetvelde, M.; Arno, G.; Jacob, J.; Creytens, D.; et al. Long-Read Sequencing to Unravel Complex Structural Variants of CEP78 Leading to Cone-Rod Dystrophy and Hearing Loss. Front Cell Dev Biol 2021, 9, 664317. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA Sequencing with Chain-Terminating Inhibitors. Proc Natl Acad Sci U S A 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed]
- Shendure, J.; Porreca, G.J.; Reppas, N.B.; Lin, X.; McCutcheon, J.P.; Rosenbaum, A.M.; Wang, M.D.; Zhang, K.; Mitra, R.D.; Church, G.M. Accurate Multiplex Polony Sequencing of an Evolved Bacterial Genome. Science 2005, 309, 1728–1732. [Google Scholar] [CrossRef] [PubMed]
- Drmanac, R.; Drmanac, S.; Chui, G.; Diaz, R.; Hou, A.; Jin, H.; Jin, P.; Kwon, S.; Lacy, S.; Moeur, B.; et al. Sequencing by Hybridization (SBH): Advantages, Achievements, and Opportunities. Adv Biochem Eng Biotechnol 2002, 77, 75–101. [Google Scholar] [CrossRef] [PubMed]
- Blazej, R.G.; Kumaresan, P.; Mathies, R.A. Microfabricated Bioprocessor for Integrated Nanoliter-Scale Sanger DNA Sequencing. Proc Natl Acad Sci U S A 2006, 103, 7240–7245. [Google Scholar] [CrossRef]
- Quail, M.A.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A Tale of Three next Generation Sequencing Platforms: Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq Sequencers. BMC Genomics 2012, 13, 1–13. [Google Scholar] [CrossRef]
- Jain, M.; Fiddes, I.T.; Miga, K.H.; Olsen, H.E.; Paten, B.; Akeson, M. Improved Data Analysis for the MinION Nanopore Sequencer. Nature Methods 2015 12:4 2015, 12, 351–356. [Google Scholar] [CrossRef]
- Evrony, G.D.; Hinch, A.G.; Luo, C. Applications of Single-Cell DNA Sequencing. Annu Rev Genomics Hum Genet 2021, 22, 171. [Google Scholar] [CrossRef] [PubMed]
- Gawad, C.; Koh, W.; Quake, S.R. Single-Cell Genome Sequencing: Current State of the Science. Nature Reviews Genetics 2016 17:3 2016, 17, 175–188. [Google Scholar] [CrossRef]
- Fan, X.; Yang, C.; Li, W.; Bai, X.; Zhou, X.; Xie, H.; Wen, L.; Tang, F. SMOOTH-Seq: Single-Cell Genome Sequencing of Human Cells on a Third-Generation Sequencing Platform. Genome Biol 2021, 22, 1–19. [Google Scholar] [CrossRef]
- Tang, L. Spatially Resolved DNA Sequencing. Nature Methods 2022 19:2 2022, 19, 139–139. [Google Scholar] [CrossRef]
- Zhong, L.X.; Kun, S.; Jing, Q.; Jing, C.; Denise, Y. Non-Syndromic Hearing Loss and High-Throughput Strategies to Decipher Its Genetic Heterogeneity. J Otol 2013, 8, 6–24. [Google Scholar] [CrossRef]
- Aldè, M.; Cantarella, G.; Zanetti, D.; Pignataro, L.; Mantia, I. La; Maiolino, L.; Ferlito, S.; Mauro, P. Di; Cocuzza, S.; Lechien, J.R.; et al. Autosomal Dominant Non-Syndromic Hearing Loss (DFNA): A Comprehensive Narrative Review. Biomedicines 2023, Vol. 11, Page 1616 2023, 11, 1616. [Google Scholar] [CrossRef]
- Koffler, T.; Ushakov, K.; Avraham, K.B. Genetics of Hearing Loss – Syndromic. Otolaryngol Clin North Am 2015, 48, 1041. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Nian, S.; Feng, L.; Ruan, Q.; Luo, X.; Wu, M.; Yan, Z. Identification of Novel Variants in MYO15A, OTOF, and RDX with Hearing Loss by next-Generation Sequencing. Mol Genet Genomic Med 2019, 7, e808. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Capponi, S.; Wakeling, E.; Marchi, E.; Li, Q.; Zhao, M.; Weng, C.; Stefan, P.G.; Ahlfors, H.; Kleyner, R.; et al. Missense Variants in TAF1 and Developmental Phenotypes: Challenges of Determining Pathogenicity. Hum Mutat 2020, 41, 449–464. [Google Scholar] [CrossRef]
- Brownstein, Z.; Gulsuner, S.; Walsh, T.; Martins, F.T.A.; Taiber, S.; Isakov, O.; Lee, M.K.; Bordeynik-Cohen, M.; Birkan, M.; Chang, W.; et al. Spectrum of Genes for Inherited Hearing Loss in the Israeli Jewish Population, Including the Novel Human Deafness Gene ATOH1. Clin Genet 2020, 98, 353–364. [Google Scholar] [CrossRef]
- Costales, M.; Diñeiro, M.; Cifuentes, G.A.; Capín, R.; Otero, A.; Viejo-Díaz, M.; Plasencia, A.; Núñez, F.; Gómez, J.R.; Llorente, J.L.; et al. Utilidad Clínica de La Secuenciación de Nueva Generación En El Diagnóstico Etiológico de La Hipoacusia Neurosensorial En Una Unidad de Hipoacusia Infantil. Acta Otorrinolaringol Esp 2020, 71, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Van Heurck, R.; Carminho-rodrigues, M.T.; Ranza, E.; Stafuzza, C.; Quteineh, L.; Gehrig, C.; Hammar, E.; Guipponi, M.; Abramowicz, M.; Senn, P.; et al. Benefits of Exome Sequencing in Children with Suspected Isolated Hearing Loss. Genes (Basel) 2021, 12, 1277. [Google Scholar] [CrossRef]
- Tropitzsch, A.; Schade-Mann, T.; Gamerdinger, P.; Dofek, S.; Schulte, B.; Schulze, M.; Battke, F.; Fehr, S.; Biskup, S.; Heyd, A.; et al. Diagnostic Yield of Targeted Hearing Loss Gene Panel Sequencing in a Large German Cohort With a Balanced Age Distribution from a Single Diagnostic Center: An Eight-Year Study. Ear Hear 2022, 43, 1049–1066. [Google Scholar] [CrossRef]
- Zhu, Y.; Hu, L.; Yang, L.; Wang, L.; Lu, Y.; Dong, X.; Xiao, T.; Xu, Z.; Wu, B.; Zhou, W. Association Between Expanded Genomic Sequencing Combined With Hearing Screening and Detection of Hearing Loss Among Newborns in a Neonatal Intensive Care Unit. JAMA Netw Open 2022, 5, e2220986–e2220986. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.C.; Chen, Y. Transcriptomics: Advances and Approaches. Sci China Life Sci 2013, 56, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Wongsurawat, T.; Jenjaroenpun, P.; Wanchai, V.; Nookaew, I. Native RNA or CDNA Sequencing for Transcriptomic Analysis: A Case Study on Saccharomyces Cerevisiae. Front Bioeng Biotechnol 2022, 10, 842299. [Google Scholar] [CrossRef] [PubMed]
- Jovic, D.; Liang, X.; Zeng, H.; Lin, L.; Xu, F.; Luo, Y.; Correspondence, Y.; Luo, D.; Jovic, L. Single-Cell RNA Sequencing Technologies and Applications: A Brief Overview. Clin Transl Med 2022, 12, e694. [Google Scholar] [CrossRef] [PubMed]
- Olsen, T.K.; Baryawno, N. Introduction to Single-Cell RNA Sequencing. Curr Protoc Mol Biol 2018, 122. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-Wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef]
- Zilionis, R.; Nainys, J.; Veres, A.; Savova, V.; Zemmour, D.; Klein, A.M.; Mazutis, L. Single-Cell Barcoding and Sequencing Using Droplet Microfluidics. Nature Protocols 2016 12:1 2016, 12, 44–73. [Google Scholar] [CrossRef]
- Gierahn, T.M.; Wadsworth, M.H.; Hughes, T.K.; Bryson, B.D.; Butler, A.; Satija, R.; Fortune, S.; Christopher Love, J.; Shalek, A.K. Seq-Well: Portable, Low-Cost RNA Sequencing of Single Cells at High Throughput. Nat Methods 2017, 14, 395–398. [Google Scholar] [CrossRef]
- Grindberg, R. V.; Yee-Greenbaum, J.L.; McConnell, M.J.; Novotny, M.; O’Shaughnessy, A.L.; Lambert, G.M.; Araúzo-Bravo, M.J.; Lee, J.; Fishman, M.; Robbins, G.E.; et al. RNA-Sequencing from Single Nuclei. Proc Natl Acad Sci U S A 2013, 110, 19802–19807. [Google Scholar] [CrossRef]
- Slyper, M.; Porter, C.B.M.; Ashenberg, O.; Waldman, J.; Drokhlyansky, E.; Wakiro, I.; Smillie, C.; Smith-Rosario, G.; Wu, J.; Dionne, D.; et al. A Single-Cell and Single-Nucleus RNA-Seq Toolbox for Fresh and Frozen Human Tumors. Nature Medicine 2020 26:5 2020, 26, 792–802. [Google Scholar] [CrossRef]
- Fangma, Y.; Liu, M.; Liao, J.; Chen, Z.; Zheng, Y. Dissecting the Brain with Spatially Resolved Multi-Omics. J Pharm Anal 2023. [Google Scholar] [CrossRef]
- Payne, A.C.; Chiang, Z.D.; Reginato, P.L.; Mangiameli, S.M.; Murray, E.M.; Yao, C.C.; Markoulaki, S.; Earl, A.S.; Labade, A.S.; Jaenisch, R.; et al. In Situ Genome Sequencing Resolves DNA Sequence and Structure in Intact Biological Samples. Science (1979) 2021, 371. [Google Scholar] [CrossRef]
- Williams, C.G.; Lee, H.J.; Asatsuma, T.; Vento-Tormo, R.; Haque, A. An Introduction to Spatial Transcriptomics for Biomedical Research. Genome Med 2022, 14, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Lubeck, E.; Coskun, A.F.; Zhiyentayev, T.; Ahmad, M.; Cai, L. Single-Cell in Situ RNA Profiling by Sequential Hybridization. Nature Methods 2014 11:4 2014, 11, 360–361. [Google Scholar] [CrossRef]
- Chen, K.H.; Boettiger, A.N.; Moffitt, J.R.; Wang, S.; Zhuang, X. Spatially Resolved, Highly Multiplexed RNA Profiling in Single Cells. Science (1979) 2015, 348. [Google Scholar] [CrossRef]
- Borm, L.E.; Mossi Albiach, A.; Mannens, C.C.A.; Janusauskas, J.; Özgün, C.; Fernández-García, D.; Hodge, R.; Castillo, F.; Hedin, C.R.H.; Villablanca, E.J.; et al. Scalable in Situ Single-Cell Profiling by Electrophoretic Capture of MRNA Using EEL FISH. Nature Biotechnology 2022 41:2 2022, 41, 222–231. [Google Scholar] [CrossRef]
- Wang, Y.; Eddison, M.; Fleishman, G.; Weigert, M.; Xu, S.; Wang, T.; Rokicki, K.; Goina, C.; Henry, F.E.; Lemire, A.L.; et al. EASI-FISH for Thick Tissue Defines Lateral Hypothalamus Spatio-Molecular Organization. Cell 2021, 184, 6361–6377.e24. [Google Scholar] [CrossRef] [PubMed]
- Ke, R.; Mignardi, M.; Pacureanu, A.; Svedlund, J.; Botling, J.; Wählby, C.; Nilsson, M. In Situ Sequencing for RNA Analysis in Preserved Tissue and Cells. Nat Methods 2013, 10, 857–860. [Google Scholar] [CrossRef]
- Nishio, S. ya; Takumi, Y.; Usami, S. ichi Laser-Capture Micro Dissection Combined with next-Generation Sequencing Analysis of Cell Type-Specific Deafness Gene Expression in the Mouse Cochlea. Hear Res 2017, 348, 87–97. [Google Scholar] [CrossRef]
- Kruse, F.; Junker, J.P.; van Oudenaarden, A.; Bakkers, J. Tomo-Seq: A Method to Obtain Genome-Wide Expression Data with Spatial Resolution. Methods Cell Biol 2016, 135, 299–307. [Google Scholar] [CrossRef]
- Chen, J.; Suo, S.; Tam, P.P.; Han, J.D.J.; Peng, G.; Jing, N. Spatial Transcriptomic Analysis of Cryosectioned Tissue Samples with Geo-Seq. Nature Protocols 2017 12:3 2017, 12, 566–580. [Google Scholar] [CrossRef]
- Hernandez, S.; Lazcano, R.; Serrano, A.; Powell, S.; Kostousov, L.; Mehta, J.; Khan, K.; Lu, W.; Solis, L.M. Challenges and Opportunities for Immunoprofiling Using a Spatial High-Plex Technology: The NanoString GeoMx® Digital Spatial Profiler. Front Oncol 2022, 12, 890410. [Google Scholar] [CrossRef]
- Merritt, C.R.; Ong, G.T.; Church, S.E.; Barker, K.; Danaher, P.; Geiss, G.; Hoang, M.; Jung, J.; Liang, Y.; McKay-Fleisch, J.; et al. Multiplex Digital Spatial Profiling of Proteins and RNA in Fixed Tissue. Nature Biotechnology 2020 38:5 2020, 38, 586–599. [Google Scholar] [CrossRef] [PubMed]
- Schede, H.H.; Schneider, C.G.; Stergiadou, J.; Borm, L.E.; Ranjak, A.; Yamawaki, T.M.; David, F.P.A.; Lönnerberg, P.; Tosches, M.A.; Codeluppi, S.; et al. Spatial Tissue Profiling by Imaging-Free Molecular Tomography. Nature Biotechnology 2021 39:8 2021, 39, 968–977. [Google Scholar] [CrossRef]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and Analysis of Gene Expression in Tissue Sections by Spatial Transcriptomics. Science (1979) 2016, 353, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Maynard, K.R.; Collado-Torres, L.; Weber, L.M.; Uytingco, C.; Barry, B.K.; Williams, S.R.; Catallini, J.L.; Tran, M.N.; Besich, Z.; Tippani, M.; et al. Transcriptome-Scale Spatial Gene Expression in the Human Dorsolateral Prefrontal Cortex. Nat Neurosci 2021, 24, 425–436. [Google Scholar] [CrossRef]
- Chen, A.; Liao, S.; Cheng, M.; Ma, K.; Wu, L.; Lai, Y.; Qiu, X.; Yang, J.; Xu, J.; Hao, S.; et al. Spatiotemporal Transcriptomic Atlas of Mouse Organogenesis Using DNA Nanoball-Patterned Arrays. Cell 2022, 185, 1777–1792. [Google Scholar] [CrossRef] [PubMed]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.M.; Chen, F.; Macosko, E.Z. Slide-Seq: A Scalable Technology for Measuring Genome-Wide Expression at High Spatial Resolution. Science 2019, 363, 1463–1467. [Google Scholar] [CrossRef] [PubMed]
- Stickels, R.R.; Murray, E.; Kumar, P.; Li, J.; Marshall, J.L.; Di Bella, D.J.; Arlotta, P.; Macosko, E.Z.; Chen, F. Highly Sensitive Spatial Transcriptomics at Near-Cellular Resolution with Slide-SeqV2. Nature Biotechnology 2020 39:3 2020, 39, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, M.; Deng, Y.; Su, G.; Enninful, A.; Guo, C.C.; Tebaldi, T.; Zhang, D.; Kim, D.; Bai, Z.; et al. High-Spatial-Resolution Multi-Omics Sequencing via Deterministic Barcoding in Tissue. Cell 2020, 183, 1665. [Google Scholar] [CrossRef] [PubMed]
- Schimmang, T.; Maconochie, M. Gene Expression Profiling of the Inner Ear. J Anat 2016, 228, 255–269. [Google Scholar] [CrossRef]
- Ramírez-Gordillo, D.; Powers, T.R.; Van Velkinburgh, J.C.; Trujillo-Provencio, C.; Schilkey, F.; Serrano, E.E. RNA-Seq and Microarray Analysis of the Xenopus Inner Ear Transcriptome Discloses Orthologous OMIM® Genes for Hereditary Disorders of Hearing and Balance. BMC Res Notes 2015, 8, 1–9. [Google Scholar] [CrossRef]
- Schrauwen, I.; Hasin-Brumshtein, Y.; Corneveaux, J.J.; Ohmen, J.; White, C.; Allen, A.N.; Lusis, A.J.; Van Camp, G.; Huentelman, M.J.; Friedman, R.A. A Comprehensive Catalogue of the Coding and Non-Coding Transcripts of the Human Inner Ear. Hear Res 2016, 333, 266–274. [Google Scholar] [CrossRef]
- Cao, R.; Takechi, M.; Wang, X.; Furutera, T.; Nojiri, T.; Koyabu, D.; Li, J. Temporal and Regulatory Dynamics of the Inner Ear Transcriptome during Development in Mice. Scientific Reports 2022 12:1 2022, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Giffen, K.P.; Chen, L.; Henderson, H.J.; Cao, T.A.; Kozeny, G.A.; Beisel, K.W.; Li, Y.; He, D.Z. Molecular and Cytological Profiling of Biological Aging of Mouse Cochlear Inner and Outer Hair Cells. Cell Rep 2022, 39, 110665. [Google Scholar] [CrossRef] [PubMed]
- Cederroth, C.R.; Park, J. sub; Basinou, V.; Weger, B.D.; Tserga, E.; Sarlus, H.; Magnusson, A.K.; Kadri, N.; Gachon, F.; Canlon, B. Circadian Regulation of Cochlear Sensitivity to Noise by Circulating Glucocorticoids. Current Biology 2019, 29, 2477–2487. [Google Scholar] [CrossRef]
- Lush, M.E.; Diaz, D.C.; Koenecke, N.; Baek, S.; Boldt, H.; St Peter, M.K.; Gaitan-Escudero, T.; Romero-Carvajal, A.; Busch-Nentwich, E.M.; Perera, A.G.; et al. Scrna-Seq Reveals Distinct Stem Cell Populations That Drive Hair Cell Regeneration after Loss of Fgf and Notch Signaling. Elife 2019, 8. [Google Scholar] [CrossRef]
- Janesick, A.; Scheibinger, M.; Benkafadar, N.; Kirti, S.; Ellwanger, D.C.; Heller, S. Cell-Type Identity of the Avian Cochlea. Cell Rep 2021, 34, 108900. [Google Scholar] [CrossRef] [PubMed]
- Geng, R.; Noda, T.; Mulvaney, J.F.; Lin, V.Y.W.; Edge, A.S.B.; Dabdoub, A. Comprehensive Expression of Wnt Signaling Pathway Genes during Development and Maturation of the Mouse Cochlea. PLoS One 2016, 11, e0148339. [Google Scholar] [CrossRef]
- Lagarde, M.M.M.; Wan, G.; Zhang, L.L.; Gigliello, A.R.; McInnis, J.J.; Zhang, Y.; Bergles, D.; Zuo, J.; Corfas, G. Spontaneous Regeneration of Cochlear Supporting Cells after Neonatal Ablation Ensures Hearing in the Adult Mouse. Proc Natl Acad Sci U S A 2014, 111, 16919–16924. [Google Scholar] [CrossRef]
- Ranum, P.T.; Goodwin, A.T.; Yoshimura, H.; Kolbe, D.L.; Walls, W.D.; Koh, J.Y.; He, D.Z.Z.; Smith, R.J.H. Insights into the Biology of Hearing and Deafness Revealed by Single-Cell RNA Sequencing. Cell Rep 2019, 26, 3160–3171. [Google Scholar] [CrossRef]
- Tang, P.C.; Alex, A.L.; Nie, J.; Lee, J.; Roth, A.A.; Booth, K.T.; Koehler, K.R.; Hashino, E.; Nelson, R.F. Defective Tmprss3-Associated Hair Cell Degeneration in Inner Ear Organoids. Stem Cell Reports 2019, 13, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Milon, B.; Shulman, E.D.; So, K.S.; Cederroth, C.R.; Lipford, E.L.; Sperber, M.; Sellon, J.B.; Sarlus, H.; Pregernig, G.; Shuster, B.; et al. A Cell-Type-Specific Atlas of the Inner Ear Transcriptional Response to Acoustic Trauma. Cell Rep 2021, 36, 109758. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.M.; Cheah, K.S.E.; Huh, S.H.; Ornitz, D.M. Sox2 and FGF20 Interact to Regulate Organ of Corti Hair Cell and Supporting Cell Development in a Spatially-Graded Manner. PLoS Genet 2019, 15. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Eckrich, S. Quantitative Fluorescent in Situ Hybridization Reveals Differential Transcription Profile Sharpening of Endocytic Proteins in Cochlear Hair Cells Upon Maturation. Front Cell Neurosci 2021, 15, 643517. [Google Scholar] [CrossRef] [PubMed]
- Hoa, M.; Olszewski, R.; Li, X.; Taukulis, I.; Gu, S.; DeTorres, A.; Lopez, I.A.; Linthicum, F.H.; Ishiyama, A.; Martin, D.; et al. Characterizing Adult Cochlear Supporting Cell Transcriptional Diversity Using Single-Cell RNA-Seq: Validation in the Adult Mouse and Translational Implications for the Adult Human Cochlea. Front Mol Neurosci 2020, 13, 491389. [Google Scholar] [CrossRef]
- Munnamalai, V.; Fekete, D.M. Notch-Wnt-Bmp Crosstalk Regulates Radial Patterning in the Mouse Cochlea in a Spatiotemporal Manner. Development (Cambridge) 2016, 143, 4003–4015. [Google Scholar] [CrossRef]
- Mehrmohamadi, M.; Sepehri, M.H.; Nazer, N.; Norouzi, M.R. A Comparative Overview of Epigenomic Profiling Methods. Front Cell Dev Biol 2021, 9, 714687. [Google Scholar] [CrossRef]
- Oakes, C.C.; La Salle, S.; Robaire, B.; Trasler, J.M. Evaluation of a Quantitative DNA Methylation Analysis Technique Using Methylation-Sensitive/Dependent Restriction Enzymes and Real-Time PCR. 2007, 1, 146–152. [CrossRef]
- Weber, M.; Davies, J.J.; Wittig, D.; Oakeley, E.J.; Haase, M.; Lam, W.L.; Schübeler, D. Chromosome-Wide and Promoter-Specific Analyses Identify Sites of Differential DNA Methylation in Normal and Transformed Human Cells. Nature Genetics 2005 37:8 2005, 37, 853–862. [Google Scholar] [CrossRef]
- Skene, P.J.; Henikoff, S. An Efficient Targeted Nuclease Strategy for High-Resolution Mapping of DNA Binding Sites. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Kaya-Okur, H.S.; Wu, S.J.; Codomo, C.A.; Pledger, E.S.; Bryson, T.D.; Henikoff, J.G.; Ahmad, K.; Henikoff, S. CUT&Tag for Efficient Epigenomic Profiling of Small Samples and Single Cells. Nature Communications 2019 10:1 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-Seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 2015, 109, 21–29. [Google Scholar] [CrossRef]
- Song, L.; Crawford, G.E. DNase-Seq: A High-Resolution Technique for Mapping Active Gene Regulatory Elements across the Genome from Mammalian Cells. Cold Spring Harb Protoc 2010, 2010, pdb–prot5384. [Google Scholar] [CrossRef]
- Pajoro, A.; Muiño, J.M.; Angenent, G.C.; Kaufmann, K. Profiling Nucleosome Occupancy by MNase-Seq: Experimental Protocol and Computational Analysis. Methods Mol Biol 2018, 1675, 167–181. [Google Scholar] [CrossRef]
- Davie, K.; Jacobs, J.; Atkins, M.; Potier, D.; Christiaens, V.; Halder, G.; Aerts, S. Discovery of Transcription Factors and Regulatory Regions Driving In Vivo Tumor Development by ATAC-Seq and FAIRE-Seq Open Chromatin Profiling. PLoS Genet 2015, 11, e1004994. [Google Scholar] [CrossRef]
- Li, Y. Modern Epigenetics Methods in Biological Research. Methods 2021, 187, 104–113. [Google Scholar] [CrossRef]
- Schwartzman, O.; Tanay, A. Single-Cell Epigenomics: Techniques and Emerging Applications. Nature Reviews Genetics 2015 16:12 2015, 16, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Wu, H.J.; Zhu, H.; Kim, K.Y.; Marjani, S.L.; Riester, M.; Euskirchen, G.; Zi, X.; Yang, J.; Han, J.; et al. Bisulfite-Independent Analysis of CpG Island Methylation Enables Genome-Scale Stratification of Single Cells. Nucleic Acids Res 2017, 45, e77–e77. [Google Scholar] [CrossRef]
- Mulqueen, R.M.; Pokholok, D.; Norberg, S.J.; Torkenczy, K.A.; Fields, A.J.; Sun, D.; Sinnamon, J.R.; Shendure, J.; Trapnell, C.; O’Roak, B.J.; et al. Highly Scalable Generation of DNA Methylation Profiles in Single Cells. Nat Biotechnol 2018, 36, 428–431. [Google Scholar] [CrossRef]
- Clark, S.J.; Lee, H.J.; Smallwood, S.A.; Kelsey, G.; Reik, W. Single-Cell Epigenomics: Powerful New Methods for Understanding Gene Regulation and Cell Identity. Genome Biology 2016 17:1 2016, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Su, J.H.; Zheng, P.; Kinrot, S.S.; Bintu, B.; Zhuang, X. Genome-Scale Imaging of the 3D Organization and Transcriptional Activity of Chromatin. Cell 2020, 182, 1641–1659. [Google Scholar] [CrossRef]
- Takei, Y.; Yun, J.; Zheng, S.; Ollikainen, N.; Pierson, N.; White, J.; Shah, S.; Thomassie, J.; Suo, S.; Eng, C.H.L.; et al. Integrated Spatial Genomics Reveals Global Architecture of Single Nuclei. Nature 2021 590:7845 2021, 590, 344–350. [Google Scholar] [CrossRef]
- Deng, Y.; Bartosovic, M.; Kukanja, P.; Zhang, D.; Liu, Y.; Su, G.; Enninful, A.; Bai, Z.; Castelo-Branco, G.; Fan, R. Spatial-CUT&Tag: Spatially Resolved Chromatin Modification Profiling at the Cellular Level. Science (1979) 2022, 375, 681–686. [Google Scholar] [CrossRef]
- Deng, Y.; Bartosovic, M.; Ma, S.; Zhang, D.; Kukanja, P.; Xiao, Y.; Su, G.; Liu, Y.; Qin, X.; Rosoklija, G.B.; et al. Spatial Profiling of Chromatin Accessibility in Mouse and Human Tissues. Nature 2022, 609, 375–383. [Google Scholar] [CrossRef]
- Lu, T.; Ang, C.E.; Zhuang, X. Spatially Resolved Epigenomic Profiling of Single Cells in Complex Tissues. Cell 2022, 185, 4448–4464. [Google Scholar] [CrossRef] [PubMed]
- Bhamidipati, T.; Sinha, M.; Sen, C.K.; Singh, K. Laser Capture Microdissection in the Spatial Analysis of Epigenetic Modifications in Skin: A Comprehensive Review. Oxid Med Cell Longev 2022, 2022. [Google Scholar] [CrossRef]
- Yeming, X.; Fengying, R.; Yaning, L.; Meng, L.; Chen, Z.; Zhichao, C.; Zhe, X.; Zhe, W.; Weitian, C.; Wenfang, C.; et al. Spatial Chromatin Accessibility Sequencing Resolves High-Order Spatial Interactions of Epigenomic Markers. Elife 2023, 12. [Google Scholar] [CrossRef]
- Klein, C.J.; Bird, T.; Ertekin-Taner, N.; Lincoln, S.; Hjorth, R.; Wu, Y.; Kwok, J.; Mer, G.; Dyck, P.J.; Nicholson, G.A. DNMT1 Mutation Hot Spot Causes Varied Phenotypes of HSAN1 with Dementia and Hearing Loss. Neurology 2013, 80, 824–828. [Google Scholar] [CrossRef]
- Balendran, V.; Ritter, K.E.; Martin, D.M. Epigenetic Mechanisms of Inner Ear Development. Hear Res 2022, 426, 108440. [Google Scholar] [CrossRef]
- Seyama, R.; Tsuchida, N.; Okada, Y.; Sakata, S.; Hamada, K.; Azuma, Y.; Hamanaka, K.; Fujita, A.; Koshimizu, E.; Miyatake, S.; et al. Two Families with TET3-Related Disorder Showing Neurodevelopmental Delay with Craniofacial Dysmorphisms. J Hum Genet 2022, 67, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Streit, A. Lsd1 Interacts with CMyb to Demethylate Repressive Histone Marks and Maintain Inner Ear Progenitor Identity. Development (Cambridge) 2018, 145. [Google Scholar] [CrossRef]
- Shin, J.O.; Lee, J.J.; Kim, M.; Chung, Y.W.; Min, H.; Kim, J.Y.; Kim, H.P.; Bok, J. CTCF Regulates Otic Neurogenesis via Histone Modification in the Neurog1 Locus. Moleucles and Cells 2018, 41, 695–702. [Google Scholar] [CrossRef]
- Tao, L.; Yu, H. V.; Llamas, J.; Trecek, T.; Wang, X.; Stojanova, Z.; Groves, A.K.; Segil, N. Enhancer Decommissioning Imposes an Epigenetic Barrier to Sensory Hair Cell Regeneration. Dev Cell 2021, 56, 2471–2485. [Google Scholar] [CrossRef]
- Vissers, L.E.L.M.; Van Ravenswaaij, C.M.A.; Admiraal, R.; Hurst, J.A.; De Vries, B.B.A.; Janssen, I.M.; Van Der Vliet, W.A.; Huys, E.H.L.P.G.; De Jong, P.J.; Hamel, B.C.J.; et al. Mutations in a New Member of the Chromodomain Gene Family Cause CHARGE Syndrome. Nature Genetics 2004 36:9 2004, 36, 955–957. [Google Scholar] [CrossRef]
- Dawe, C.E.; Kooistra, M.K.; Fairbridge, N.A.; Pisio, A.C.; McDermid, H.E. Role of Chromatin Remodeling Gene Cecr2 in Neurulation and Inner Ear Development. Developmental Dynamics 2011, 240, 372–383. [Google Scholar] [CrossRef]
- Iyer, A.A.; Hosamani, I.; Nguyen, J.D.; Cai, T.; Singh, S.; McGovern, M.M.; Beyer, L.; Zhang, H.; Jen, H.I.; Yousaf, R.; et al. Cellular Reprogramming with ATOH1, GFI1, and POU4F3 Implicate Epigenetic Changes and Cell-Cell Signaling as Obstacles to Hair Cell Regeneration in Mature Mammals. Elife 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, E.; Slevin, C.C.; Song, W.; Chen, Z.; Frederickson, S.C.; Gildea, D.; Wu, W.; Elkahloun, A.G.; Ovcharenko, I.; Burgess, S.M. A Regulatory Network of Sox and Six Transcription Factors Initiate a Cell Fate Transformation during Hearing Regeneration in Adult Zebrafish. Cell Genomics 2022, 2, 100170. [Google Scholar] [CrossRef]
- Song, Z.; Jadali, A.; Fritzsch, B.; Kwan, K.Y. NEUROG1 Regulates CDK2 to Promote Proliferation in Otic Progenitors. Stem Cell Reports 2017, 9, 1516–1529. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Yuan, S.S.; Zhang, L.J.; Ji, Z.L.; Quan, X.J. Atonal BHLH Transcription Factor 1 Is an Important Factor for Maintaining the Balance of Cell Proliferation and Differentiation in Tumorigenesis (Review). Oncol Lett 2020, 20, 2595–2605. [Google Scholar] [CrossRef]
- Kalra, G.; Lenz, D.; Abdul-Aziz, D.; Hanna, C.; Herb, B.R.; Colantuoni, C.; Milon, B.; Saxena, M.; Shetty, A.C.; Hertzano, R.P.; et al. Cochlear Organoids Reveal Epigenetic and Transcriptional Programs of Postnatal Hair Cell Differentiation from Supporting Cells. bioRxiv 2021, 2021.09.19.460948. [CrossRef]
- Webster, M.; Webster, D.B. Spiral Ganglion Neuron Loss Following Organ of Corti Loss: A Quantitative Study. Brain Res 1981, 212, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, R.K.; Hardie, N.A. Deafness-Induced Changes in the Auditory Pathway: Implications for Cochlear Implants. Audiology and Neurotology 2002, 6, 305–318. [Google Scholar] [CrossRef]
- Versnel, H.; Agterberg, M.J.H.; de Groot, J.C.M.J.; Smoorenburg, G.F.; Klis, S.F.L. Time Course of Cochlear Electrophysiology and Morphology after Combined Administration of Kanamycin and Furosemide. Hear Res 2007, 231, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Maheshwari, S.; Kirtane, M.; Shrivastav, N. Pictorial Review of MRI/CT Scan in Congenital Temporal Bone Anomalies, in Patients for Cochlear Implant. Indian J Radiol Imaging 2009, 19, 99. [Google Scholar] [CrossRef]
- Burwood, G.W.S.; Fridberger, A.; Wang, R.K.; Nuttall, A.L. Revealing the Morphology and Function of the Cochlea and Middle Ear with Optical Coherence Tomography. Quant Imaging Med Surg 2019, 9, 85881–85881. [Google Scholar] [CrossRef] [PubMed]
- Rask-Andersen, H.; Liu, W.; Erixon, E.; Kinnefors, A.; Pfaller, K.; Schrott-Fischer, A.; Glueckert, R. Human Cochlea: Anatomical Characteristics and Their Relevance for Cochlear Implantation. The Anatomical Record: Advances in Integrative Anatomy and Evolutionary Biology 2012, 295, 1791–1811. [Google Scholar] [CrossRef] [PubMed]
- Tisi, A.; Ramekers, D.; Flati, V.; Versnel, H.; Maccarone, R. MTOR Signaling in BDNF-Treated Guinea Pigs after Ototoxic Deafening. Biomedicines 2022, 10, 2935. [Google Scholar] [CrossRef] [PubMed]
- Tisi, A.; Rovers, J.; Vink, H.A.; Ramekers, D.; Maccarone, R.; Versnel, H. No Protective Effects of Hair Cells or Supporting Cells in Ototoxically Deafened Guinea Pigs upon Administration of BDNF. Brain Sci 2021, 12. [Google Scholar] [CrossRef]
- Pujol, R.; Lavigne-rebillard, M.; Uziel, A. Development of the Human Cochlea. 2009, 111, 7–13. 13. [CrossRef]
- Jean, P.; Tai, F.W.J.; Singh-Estivalet, A.; Lelli, A.; Scandola, C.; Megharba, S.; Schmutz, S.; Roux, S.; Mechaussier, S.; Sudres, M.; et al. Single-Cell Transcriptomic Profiling of the Mouse Cochlea: An Atlas for Targeted Therapies. Proceedings of the National Academy of Sciences 2023, 120, e2221744120. [Google Scholar] [CrossRef]
- Waldhaus, J.; Durruthy-Durruthy, R.; Heller, S. Quantitative High-Resolution Cellular Map of the Organ of Corti. Cell Rep 2015, 11, 1385–1399. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.G.; Huang, M.; Obholzer, N.D.; Sun, S.; Li, W.; Petrillo, M.; Dai, P.; Zhou, Y.; Cotanche, D.A.; Megason, S.G.; et al. Myc and Fgf Are Required for Zebrafish Neuromast Hair Cell Regeneration. PLoS One 2016, 11, e0157768. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Jin, R.; Xu, J.; Ji, Y.; Zhang, M.; Zhang, X.; Zhang, X.; Han, Z.; Zeng, S. Hair Cell Regeneration or the Expression of Related Factors That Regulate the Fate Specification of Supporting Cells in the Cochlear Ducts of Embryonic and Posthatch Chickens. Hear Res 2016, 332, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Romero-Carvajal, A.; Haug, J.S.; Seidel, C.W.; Piotrowski, T. Gene-Expression Analysis of Hair Cell Regeneration in the Zebrafish Lateral Line. Proc Natl Acad Sci U S A 2014, 111, E1383–E1392. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, X.; Bi, Z.; Sugino, K.; Wang, G.; Zhu, T.; Liu, Z. Comprehensive Transcriptome Analysis of Cochlear Spiral Ganglion Neurons at Multiple Ages. Elife 2020, 9. [Google Scholar] [CrossRef]
- Chen, J.; Gao, D.; Chen, J.; Hou, S.; He, B.; Li, Y.; Li, S.; Zhang, F.; Sun, X.; Jin, Y.; et al. Pseudo-Temporal Analysis of Single-Cell RNA Sequencing Reveals Trans-Differentiation Potential of Greater Epithelial Ridge Cells Into Hair Cells During Postnatal Development of Cochlea in Rats. Front Mol Neurosci 2022, 15, 832813. [Google Scholar] [CrossRef]
Figure 1.
Schematic illustration of the available experimental models for cochlear research. The available models for cochlear research include cell lines of otocyst, HCs, organ of Corti and stria vascularis. Explants of cochlear tissues may also be used, more recently via microfluidic chambers for organ-on-chip cultures. Cochlear organoids are an additional in vitro possibility and can be derived from induced pluripotent stem cells (iPSC) or from embryonic stem cells (ESC). Animal models can be generated by the exposure to ototoxic drugs or by noise trauma; also age-related and transgenic models of hearing loss have been developed. Finally, all the models may be subjected to CRISPR/Cas9 to achieve targeted gene editing. Abbreviations: IMO; immortomouse; HC; hair cell, SC; supporting cells, iPSC; induced pluripotent stem cells, ESC; embryonic stem cells, Dmm; disproportionate micromelia; sedc, spondyloepiphyseal dysplasia congenital; USH: usher; Cho: chondrodysplasia.
Figure 1.
Schematic illustration of the available experimental models for cochlear research. The available models for cochlear research include cell lines of otocyst, HCs, organ of Corti and stria vascularis. Explants of cochlear tissues may also be used, more recently via microfluidic chambers for organ-on-chip cultures. Cochlear organoids are an additional in vitro possibility and can be derived from induced pluripotent stem cells (iPSC) or from embryonic stem cells (ESC). Animal models can be generated by the exposure to ototoxic drugs or by noise trauma; also age-related and transgenic models of hearing loss have been developed. Finally, all the models may be subjected to CRISPR/Cas9 to achieve targeted gene editing. Abbreviations: IMO; immortomouse; HC; hair cell, SC; supporting cells, iPSC; induced pluripotent stem cells, ESC; embryonic stem cells, Dmm; disproportionate micromelia; sedc, spondyloepiphyseal dysplasia congenital; USH: usher; Cho: chondrodysplasia.
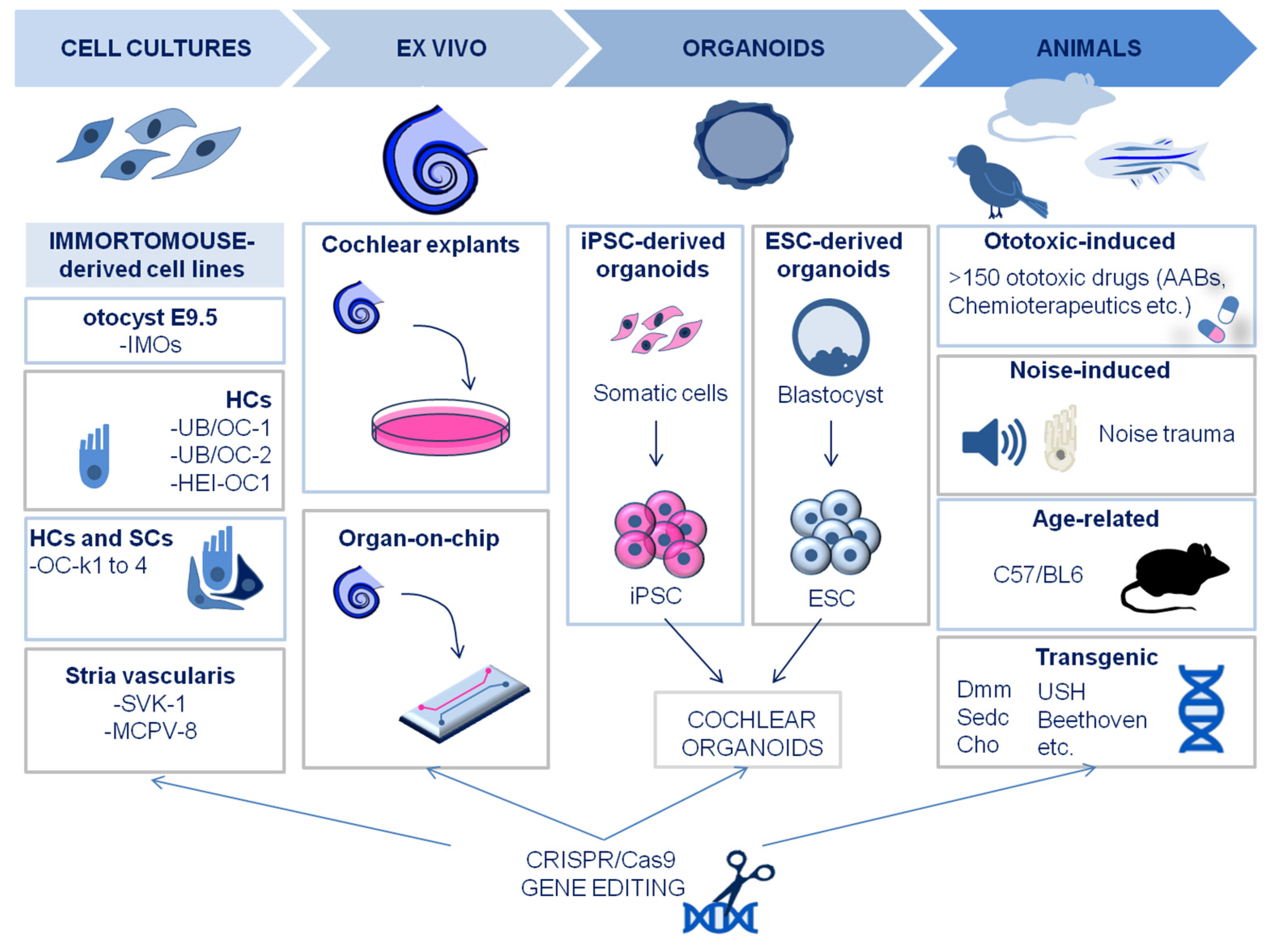
Figure 2.
Schematic illustration of genomics. DNA is isolated from cells or tissues and is fragmented in order to create DNA libraries by using DNA adapters. Sequencing can then be performed on targeted sequences (panels), on the whole exosome (WES), or on the whole genome (WGS). Genomics has provided important advancements in the diagnosis and discovery of genetic hearing loss.
Figure 2.
Schematic illustration of genomics. DNA is isolated from cells or tissues and is fragmented in order to create DNA libraries by using DNA adapters. Sequencing can then be performed on targeted sequences (panels), on the whole exosome (WES), or on the whole genome (WGS). Genomics has provided important advancements in the diagnosis and discovery of genetic hearing loss.
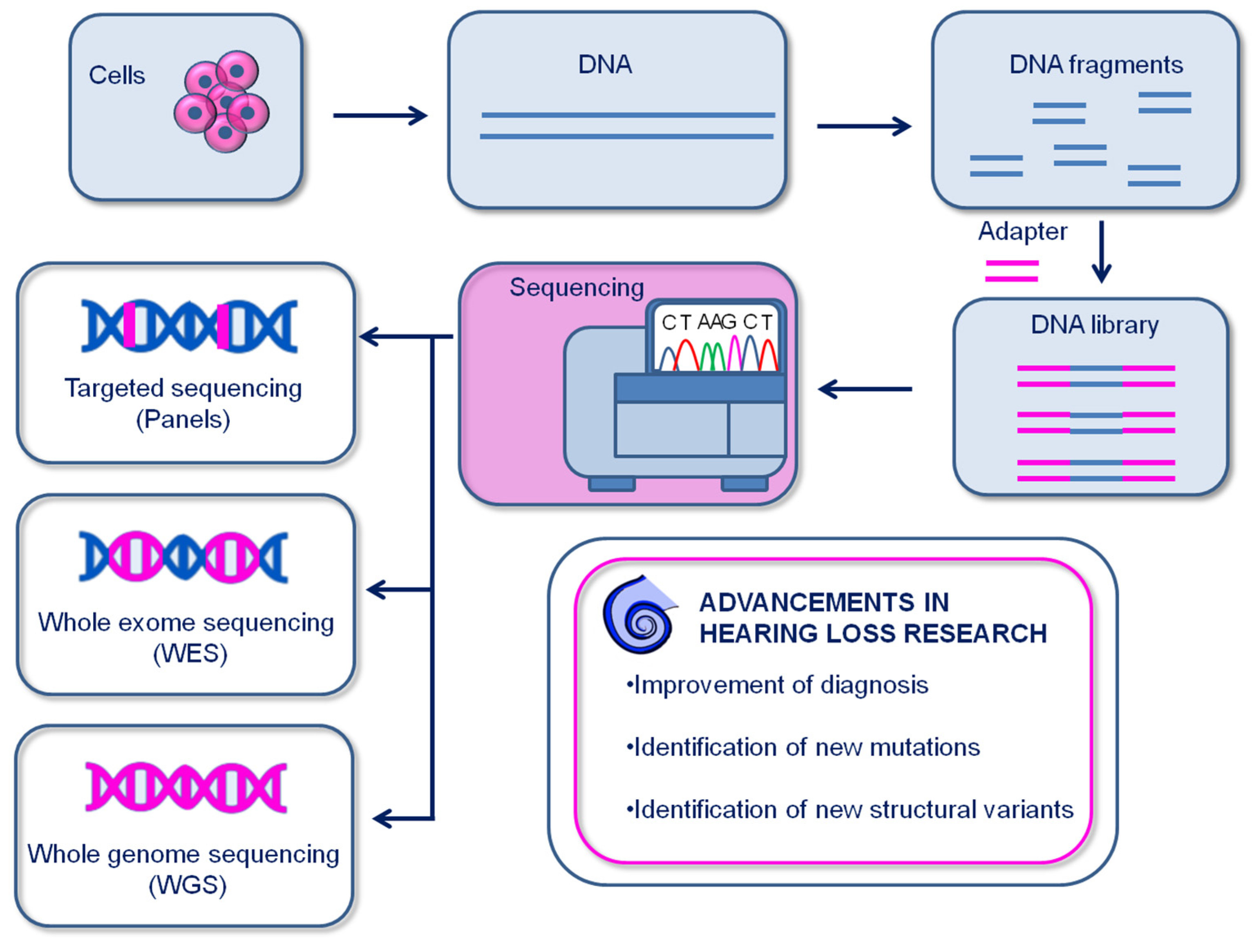
Figure 3.
Schematic illustration of transcriptomics. Transcriptomics can be performed in bulk, single cell or spatial resolution. In bulk RNAseq, total RNA is extracted from the tissue and can be directly sequenced or converted into cDNA, and then sequenced. In scRNAseq, the sequencing of the RNA is limited to single cells that are isolated from the tissue and analyzed individually. In spatial transcriptomics, the transcriptome may be analyzed by imaging-based methods, by using fluorescent labeled probes which bind to the RNA on tissue slides, followed by microscopic analysis; spatial transcriptomics may also be performed through sequencing-based methods by using arrays of barcoded probes or microdissection of target tissue areas, both followed by sequencing. Array-based spatial transcriptomics have not yet been applied in cochlear research. The other transcriptomics techniques have provided important new insights into the gene regulatory networks of the cochlea, under both physiological and pathological conditions. Abbreviations: Bulk RNA seq, Bulk RNA sequencing; scRNA seq, single cell RNA sequencing.
Figure 3.
Schematic illustration of transcriptomics. Transcriptomics can be performed in bulk, single cell or spatial resolution. In bulk RNAseq, total RNA is extracted from the tissue and can be directly sequenced or converted into cDNA, and then sequenced. In scRNAseq, the sequencing of the RNA is limited to single cells that are isolated from the tissue and analyzed individually. In spatial transcriptomics, the transcriptome may be analyzed by imaging-based methods, by using fluorescent labeled probes which bind to the RNA on tissue slides, followed by microscopic analysis; spatial transcriptomics may also be performed through sequencing-based methods by using arrays of barcoded probes or microdissection of target tissue areas, both followed by sequencing. Array-based spatial transcriptomics have not yet been applied in cochlear research. The other transcriptomics techniques have provided important new insights into the gene regulatory networks of the cochlea, under both physiological and pathological conditions. Abbreviations: Bulk RNA seq, Bulk RNA sequencing; scRNA seq, single cell RNA sequencing.
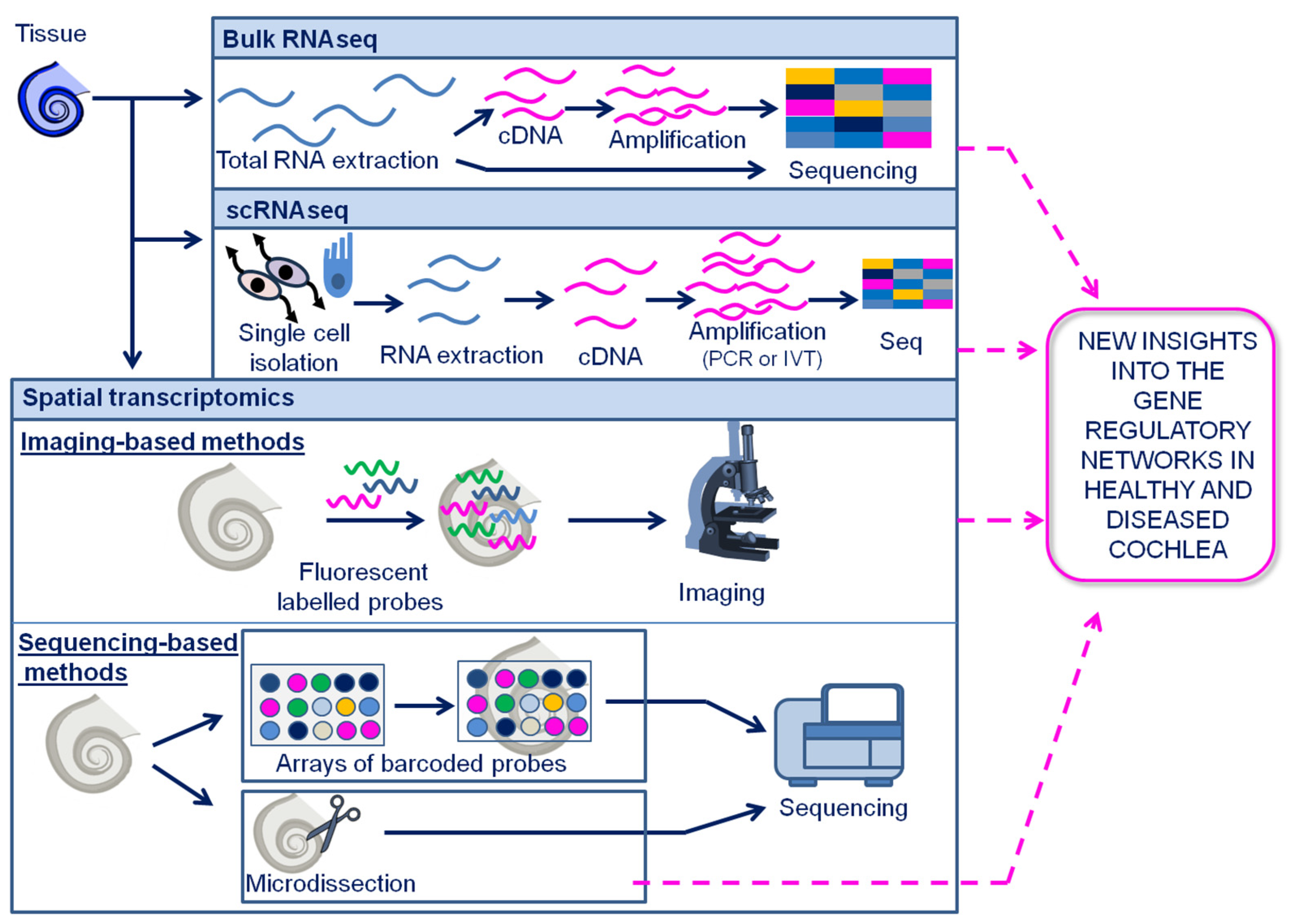
Figure 4.
Schematic illustration of epigenomics. Epigenomics can be performed in bulk, single and spatial resolution. Bulk epigenomics and single cell epigenomics have been applied in the cochlea. The study of epigenomics includes the assessment of DNA methylation dynamics, histone modifications, chromatin accessibility and chromosome conformations. These epigenomic methods can be performed either by arrays or by sequencing. The techniques ChIP and ATAC-seq (in bulk and single-cells) have been applied in the cochlea. ChIP relies on immunoprecipitating DNA-protein complexes via specific antibodies, and ATAC-seq uses Tn5 transposase chemistry and NGS to analyze open or accessible chromatin regions. These techniques have provided novel insights into the molecular mechanisms underlying the developmental and regenerative processes in the cochlea. Abbreviations: BS-seq, bisulfite sequencing; RRBS, reduced representation bisulfite sequencing; MeDIP, methylated DNA immunoprecipitation; ChIP, chromatin immunoprecipitation; CUT&RUN, cleavage under target & release using nuclease; CUT&Tag, ATAC-seq cleavage under targets and tagmentation; DNAse-seq, DNase I hyper-sensitive sites sequencing; FAIRE-seq, formaldehyde-assisted identification of regulatory elements followed by sequencing; 3C, conformation capture technique; PLAC-seq, proximity ligation-assisted ChIP-seq; ChIA PET, chromatin interaction analysis by paired-end tag sequencing; sci-MET, single-cell combinatorial indexing for methylation analysis; scCGI-seq, single-cell CGI methylation sequencing; scChIC-seq, single-cell chromatin immune-cleavage sequencing technique; ACT-seq, antibody-guided chromatin tagmentation sequencing; COBATCH, combinatorial barcoding and targeted chromatin release; scChIL-seq, single-cell chromatin integration labeling sequencing.
Figure 4.
Schematic illustration of epigenomics. Epigenomics can be performed in bulk, single and spatial resolution. Bulk epigenomics and single cell epigenomics have been applied in the cochlea. The study of epigenomics includes the assessment of DNA methylation dynamics, histone modifications, chromatin accessibility and chromosome conformations. These epigenomic methods can be performed either by arrays or by sequencing. The techniques ChIP and ATAC-seq (in bulk and single-cells) have been applied in the cochlea. ChIP relies on immunoprecipitating DNA-protein complexes via specific antibodies, and ATAC-seq uses Tn5 transposase chemistry and NGS to analyze open or accessible chromatin regions. These techniques have provided novel insights into the molecular mechanisms underlying the developmental and regenerative processes in the cochlea. Abbreviations: BS-seq, bisulfite sequencing; RRBS, reduced representation bisulfite sequencing; MeDIP, methylated DNA immunoprecipitation; ChIP, chromatin immunoprecipitation; CUT&RUN, cleavage under target & release using nuclease; CUT&Tag, ATAC-seq cleavage under targets and tagmentation; DNAse-seq, DNase I hyper-sensitive sites sequencing; FAIRE-seq, formaldehyde-assisted identification of regulatory elements followed by sequencing; 3C, conformation capture technique; PLAC-seq, proximity ligation-assisted ChIP-seq; ChIA PET, chromatin interaction analysis by paired-end tag sequencing; sci-MET, single-cell combinatorial indexing for methylation analysis; scCGI-seq, single-cell CGI methylation sequencing; scChIC-seq, single-cell chromatin immune-cleavage sequencing technique; ACT-seq, antibody-guided chromatin tagmentation sequencing; COBATCH, combinatorial barcoding and targeted chromatin release; scChIL-seq, single-cell chromatin integration labeling sequencing.
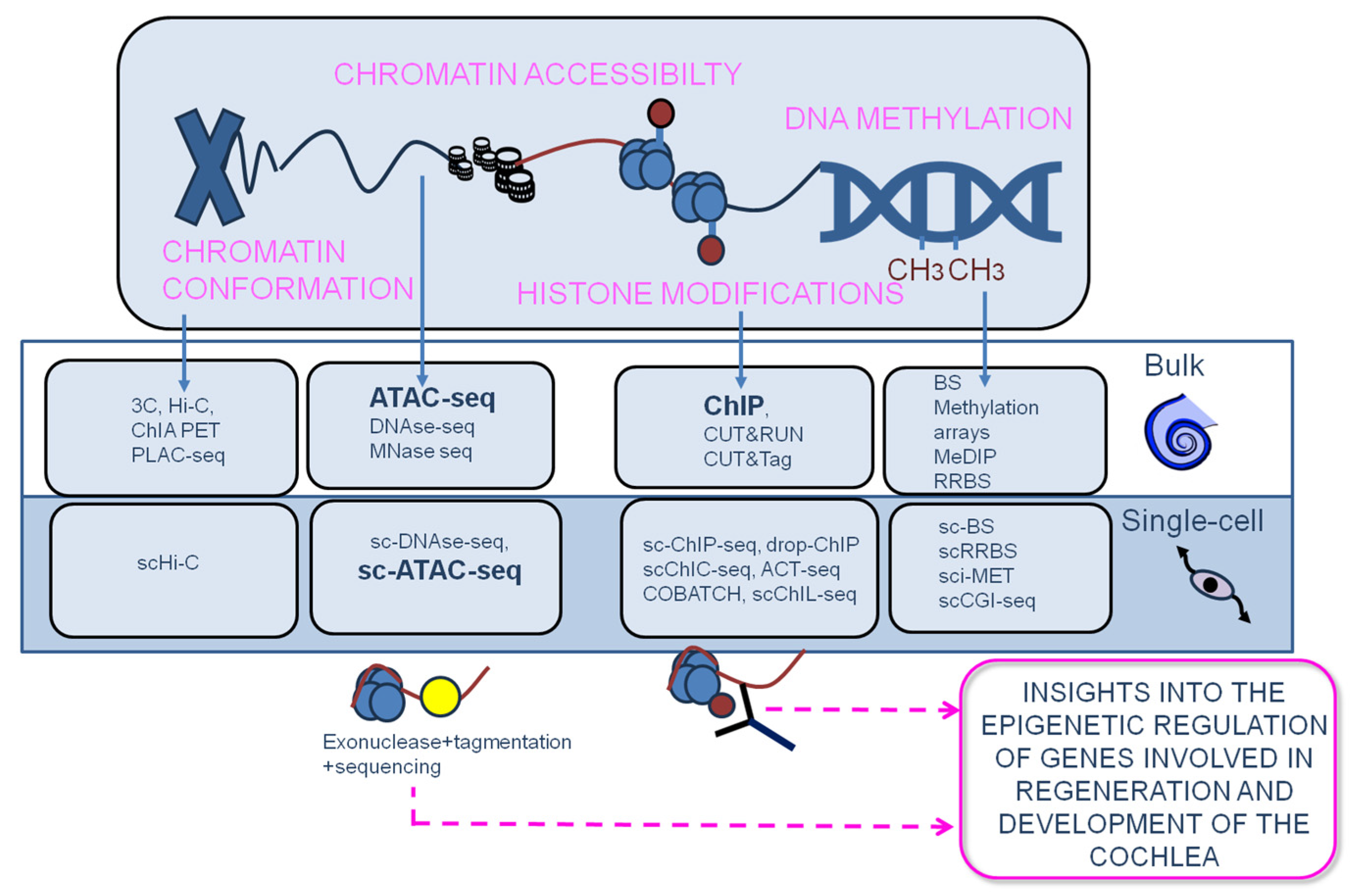

Table 1.
Advanced sequencing methodologies of nucleic acids.
| Sequencing technology | Category | Principle | Reads length | Reference |
|---|---|---|---|---|
| NGS | Cyclic-array sequencing (Illumina and Ion Torrent) | Repeated cycles of enzymatic catalytic reactions | Short | [57] |
| NGS | Hybridization-based sequencing | Multiple oligonucleotides are hybridized with complementary sequences of the target genome/transcriptome. | Short | [58] |
| NGS | microelectrophoretic-based | Lab-on-a-chip-level which combines all the Sanger sequencing steps together for a more efficient sequencing. | Short | [59] |
| TGS | Pacific Biosciences (PacBio) | Laser-induced fluorescence signals that are activated during the incorporation of dNTPs into DNA, alongside recording the color and duration of the signals in real time. | Long | [60] |
| TGS | Oxford Nanopore technology (ONT) | Nanopore-based technology in which sequencing is allowed by determination of current change induced by nucleotides passing through the nanopore. | Long | [61] |
Table 2.
Advanced genomics, epigenomics and transcriptomics techniques that have been applied in cochlear research.
Table 2.
Advanced genomics, epigenomics and transcriptomics techniques that have been applied in cochlear research.
| Omics Categories | Techniques | Applications in hearing research | Models utilized | Reference |
|---|---|---|---|---|
| Genomics | WGS, WES | Identification of novel structural variants and rare mutations in genes associated with deafness | Humans (Affected individuals with the CRDHL) | [55] |
| WES | Early detection of hearing loss for diagnostic purposes | Humans (individuals with diagnosis of hearing loss) | [73] | |
| Target exome panel | Improvement in the clinical diagnostic yield and thereby routine genetic screening | Humans (deaf patients suspected with underlying genetic causes of deafness) | [74] | |
| Humans (patients diagnosed with SNHL) | [72] | |||
| Transcriptomics | TruSeq | Identification of differential and preferential gene expression patterns and characterization of novel molecular pathways of the cochlea | Humans (patients with tumors of the skull base with normal hearing) | [107] |
| Engineered mouse models of genes related to circadian rhythm with noise damage | [110] | |||
| Comprehension of mechanisms involved in hair cell regeneration. | Ototoxic (neomycin) treated zebrafish | [169] | ||
| SMART-Seq v4 | Insights into the transcriptional changes of HCs during the process of ageing and damage | CBA/J mice 1,9, 18, 22 and 26 months-old | [109] | |
| RNA-Seq V2 | Unraveling the genes specific to SGNs and their dynamicity in developmental processes. | Mouse at different stages: E15.5, P1, P8, P14 and P30 | [170] | |
| Single-cell transcriptomics | SMART-Seq2 | Identification of novel subtypes of cochlear cells | Chicken | [112] |
| Identification of new markers of HCs | Mouse (C3HeB/FeJ) | [115] | ||
| 10x Genomics | Identification of gene regulatory networks involved in HCs regeneration. | Zebrafish (transgenic model for HCs ablation) | [152] | |
| Identification of genes associated with Tmprss3-related hearing loss | Mouse (Tmprss3-KO organoids) | [116] | ||
| Delineation of key regulatory mechanisms in HCs regeneration | Rats | [171] | ||
| Spatial transcriptomics | Single molecule FISH (smFISH) | Annotating distinct transcriptome of SCs populations in specific anatomic locations of the cochlea | Mouse | [120] |
| Whole mount ISH | Spatio-temporal cadence of key signalling pathways in the context of developmental processes of the cochlea | Mouse organotypic cultures | [121] | |
| Genetically engineered mouse models of genes related to developmental processes | [118] | |||
| Fluor | Uncovering quantitative differential transcriptional profile in pre-mature and mature HCs, revealing novel role of genes in the differentiation process | Mice (P4 and 3 weeks old) | [119] | |
| LCM-NGS | Discovery of quantitative information of transcripts relevant in deafness in the organ of Corti, spiral ganglion, lateral wall, and spiral limbus | Mice (C57BL/6J) | [93] | |
| Epigenomics | ChIP-seq and ChIP-qPCR | Epigenetic modifications in the promoters of genes involved in SGNs differentiation | In vitro immortalized multipotent otic progenitors (iMOP cells) | [153] |
| ATAC-seq | Identification of dynamics in chromatin accessibility of key transcriptional factors during the reprogramming of SCs into HCs | Mouse (Atoh1-nGFP, Sox2-GFP or Lgr5-GFP) and cochlear organoids | [155] | |
| Single-cell epigenomics | scATAC seq | Regulation of chromatin accessibility during the process of regeneration and identification of genetically conserved regenerative response elements necessary for injury/regenerative responses | Zebrafish (transgenic model for HCs ablation) | [152] |
| Identification of the epigenetic mechanisms responsible for the inability of SCs to trans-differentiation in HCs in the adult mammalian cochlea | Transgenic mouse models expressing transcription factors | [151] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



