
Submitted:
25 August 2023
Posted:
29 August 2023
You are already at the latest version
Abstract
Background: Down Syndrome is the most well-studied aneuploidy condition in humans. It is associated with multiple disease phenotypes including cardiovascular, neurological, haemato-logical, and immunological disease processes. In this review paper we aim to discuss the research into gene expression studies performed at the fetal stage of development. Methods: A descriptive review was performed including all papers published on the PubMed database between Septem-ber 1960- September 2022. Results: In the amniotic fluid, genes such as COL6A1 and DSCR1 are affected, causing the phenotypical craniofacial changes. Other genes affected in amniotic fluid in-cluding: GSTT1, CLIC6, ITGB2, C21orf67, C21orf86 and RUNX1. In the placenta, MEST, SNF1LK and LOX were dysregulated, affecting nervous system development. In the brain DYRK1A, DNMT3L, DNMT3B, TBX1, olig2 and AQP4 were found to be dysregulated, affecting the nervous system and intellectual disability. In the cardiac tissues GART, EST2 and ERG found to be dysregulated causing secondary heart field abnormalities. XIST, RUNX1, SON, ERG and STAT1 dysregulated causing myeloproliferative disorders. Conclusions: Differential expression of genes provides clues to the genetic consequences of DS. A better understanding of these processes could eventually to lead to the development of genetic and pharmacological therapies.
Keywords:
down syndrome
; trisomy 21
; gene expression
; disease phenotypes
; T21
; DS
1. Introduction
Down Syndrome (DS), caused by complete or partial trisomy of chromosome 21, is one of the most well-known and studied aneuploid conditions in humans. The incidence is approximately 1 in 700 births and increases with higher maternal age [1]. It is associated with heart and gastrointestinal malformations, endocrine and immunological deficiencies, typical facies, an increased risk of leukaemia and early onset Alzheimer’s disease [2]. It has been long established that the DS phenotype is due to the additional chromosome T21. As such, DS is thought to be caused by multiple different genes, with a generalized disruption of early developmental pathways [3]. However, the precise genetic determinants of individual clinical manifestations of DS are not yet fully understood. It is unknown how many of the ~300 genes on chromosome 21 have any phenotypic effect when present in 3 copies [4].
Research aimed at developing therapeutics for DS has been stalled by the lack of clarity of which genes cause the phenotypes associated with the condition. One confirmed genetic change is that of amyloid precursor protein (APP), a key element in the pathogenesis of Alzheimer’s disease and the source of amyloid plaque deposition. This has been found to be significantly over-expressed in trisomy 21 trophoblasts [5].
There are many viable ways of investigating genetic or whole genome gene expression changes in DS mouse or foetal cell models. Microarray analysis of DS cells, with validation via real-time PCR, DNA methylation changes, RNA-sequencing technology, and chromosome silencing are all reasonable approaches, but each have strengths and weaknesses in identifying key gene targets. Although a large number of differentially expressed genes have been detected in various studies, no changes in genes, sets of genes or changes in pathways have been consistently identified to suggest a causative role. One issue with these studies is that most have been performed in adults or children. The developmental changes leading to DS, however, occur early in foetal development. Neural differentiation occurs from 10 weeks and continues in the hippocampus and cerebellum after birth [6]. Understanding the gene expression changes at different stages of foetal development would help to identify potential targets and the optimal timing for future gene therapies.
The aim of this review is to assess the studies conducted into gene expression in DS and identify candidate targets for gene therapy. It will focus on foetal tissue to study how the changes in gene expression will affect growth of the foetus, development of organs, and expression of the DS phenotype. Papers including mouse models are also reviewed to identify how gene expression pattern changes lead to differences in cellular function and structural changes. We first highlight the different techniques used to analyse gene expression in DS samples and tissues and their relative strengths and weaknesses. We then discuss the gene expression changes in different stages of foetal development, and then move the focus to specific changes seen in different DS tissues and organs. Finally, we discuss how the data from these studies can be used to develop future gene therapies.
2. Methods
The studies for this review were identified by searching PubMed between September 1960 and September 2022. The reference lists of these articles were searched to identify any further relevant publications. The free text search terms were: ((Down syndrome) OR (DS) OR (Trisomy 21) OR (T21) AND ((Microarray) OR (Gene Expression) AND (foetal). The inclusion criteria included papers in the English language, or that could be translated, with the relevant search terms and relevant abstracts. The exclusion criteria included: inability to access the full article text; papers which did not report clear outcomes; papers which could not be translated into the English language.
3. Assessment of gene expression analysis techniques
There are various methods used in the following studies to analyse gene differences in DS mouse models or foetal cells. A summary of these different methods can be seen in Table 1. The discrepancies in the study methods make it difficult to directly compare and validate the genes that have been identified using different techniques, due to studies varying in power and validity.
Microarray analysis of DS foetal cells and tissues has extensively been investigated. However, as an isolated tool, there are potential limitations in accuracy due to potential difficulties interpreting copy number variations of unknown significance, incomplete penetrance, or variable expressivity in absence of a clear phenotype. Additionally, obtaining sufficient RNA for good quality microarray results from prenatal samples can be very difficult [7]. These results can be validated using real-time PCR, which increases the accuracy. Shi et al., found some genes highlighted in the Affymetrix assay were not validated by real-time PCR highlighting the problems with microarray analysis alone [8]. MicroRNAs play an important role in regulating embryonic development, cell differentiation, proliferation, and apoptosis. Assessing their expression is useful to understand the genetic changes in DS [8]. Microarrays analysis is now commonly used to assess MicroRNAs content in DS.
Single cell RNA sequencing enables investigation of cellular transcriptomics and profiling gene expression [9]. This analysis is commonly used to study molecular pathophysiology underlying DS and other aneuploidies. In trisomic cells, single cell RNA sequencing analysis has evidenced the additional allele is independently transcribed and “the specific transcriptional profile for each gene contributes to the phenotypic variability of trisomies” [10].
During development DNA methylation in the genome follows a dynamic process in-volving both de novo DNA methylation and demethylation. It plays an important role in genomic imprinting, X-chromosome inactivation, and transposition when DNA is dysregulated. DS is known to have a profound impact on DNA methylation, especially in haematopoietic cells in early life and is the most studied form of epigenetic regulation in DS [11]. The technique of bisulfite sequencing is considered ‘gold standard’ in DNA methylation studies and uses techniques such as methylation-specific PCR, PCR and sequencing and bead array. The use of bead arrays is an approach that is cost effective and allows for specific regions of interest to be highlighted [11]. The manufacturers state that assays can detect DNA methylation levels as low as 0.5% using PCR [12]. It is therefore very accurate in quantification and can be used to identify tissue-specific biomarkers [12].
Quantitative transcriptome map analysis integrates gene expression profile data from different tissues enabling an overview of changes in whole organs. Changes in gene expression seen can be validated by RT-PCR to increase reliability and allow an overview of multiple organs and tissues. Antonaros et al., used the TRAM softward to create a T21 blood cell transcriptome map and use the Samtools software to indentify and read the maps on the HR-DSCR [13]. A transcriptome mapper (TRAM) software can be used, allowing for comparison between transcript expression levels and profiles between DS and normal brain, lymphoblastoid cell lines, blood cells, fibroblasts, thymus and induced pluripotent stem cells, respectively.
All gene analysis techniques have their own relative advantages and disadvantages. Using multiple methods can help validate and increase the accuracy of identifying gene expression changes in DS foetal tissues.
4. Gene expression changes in amniocytes and amniotic fluid
The screening process for DS includes a non-invasive prenatal test (NIPT) which isolates cell free fetal DNA in the maternal blood. It aims to determine the likelihood of aneuploidy by assessing the aberrant copy number for whole or segments of chromosomes specific to the test [14]. An invasive test such as amniocentesis in which amniotic fluid (AF) is acquired, is still necessary to confirm the diagnosis for those identified to be at high risk to develop aneuploidy following initial NIPT screening [15]. Amniotic fluid can be split into two fractions: supernatant (cell-free components, placenta-derived microparticles, protein, cell-free foetal DNA and cell-free foetal RNA from foetus) and amniocytes [14]. Amniocytes are cells which can be derived from several foetal tissues. These cells can be cultured and subsequently used for a variety of purposes.
Chung et al., screened cultured amniocytes for expression changes, using a custom array containing 102 genes on chromosome 21, only 1 of which was differentially over-expressed [16]. GSTT1 showed increased expression and is thought to play a role in carcinogenesis [17]. In the amniocytes in DS cases two genes out of 24 were down-regulated, COL6A1 and PRSS7. COL6A1 from the collagen superfamily, plays a role in integrity of tissues [18]. This may explain some of the physical characteristics seen in DS. COL6A1 has been shown in previous studies to be downregulated in the brain but expressed in the atrioventricular (AV) canal. This change of expression has been thought to be linked to abnormalities in the brain of DS a contributed to AV-related cardiac defects [19,20,21]. Studies such as this highlight the use of the AF transcriptome, which might reflect foetal and placental development and can therefore aid in monitoring of abnormal and normal development [22].
Altug-Teber et al., cultured amniocytes and chorionic villus cells focusing on chromosome 21 and found 33 and 16 over-expressed genes, respectively [18]. None showed under-expression. Cultured amniocytes and chorionic villus sampling (CVS) are used due to needing a high yield of high-quality mRNA for the array, however this itself may differ from the environment within the womb. One gene over-expressed included DSCR1, which is in the DS critical region. The DS critical region (DSCR) is a segment on chromosome 21 that contains genes responsible for many features of DS, including craniofacial dysmorphology [23]. DSCR1 gene on chromosome 21 is a developmental regulator and involved in neurogenesis. Its overexpression may contribute to brain abnormalities seen in DS [24].
Amniotic fluid supernatant has been used to detect genome wide expression changes using Affymetrix microarrays [24]. 414 probes showed significant changes in expression, 5 of which were present on chromosome 21 genes. The genes present were CLIC6, ITGB2, and 2 ORFs (C21orf67 and C21orf86) which were up regulated and RUNX1, which was downregulated. CLIC6 is a member of the chloride intracellular channel family of proteins. It is involved in the activation of cAMP-Dependent PKA pathway this usually regulates pathogenicity, hyphal growth, and stress tolerance [25,26]. ITGB2 encodes an integrin beta chain which play an important role in immune response, therefore if upregulated expressing a difference in immune response in people with DS. These results were not validated via PCR, confirming the limitations of the Affymetrix microarray probes, as seen the study by Rozovski et al., which highlights different expression profiles once validated [5]. The samples were however matched for sex and gestational age, improving accuracy, as these factors have previously been shown to impact results [26].
Huang et al., analysed metabolites present in DS using amniotic fluid from foetuses [27]. The amniotic fluid (AF) was processed, and metabolomic fingerprinting was conducted using UPLC-MS. Alterations in porphyrin metabolism, bile acid metabolism, hormone metabolism and amino acid metabolism were validated for the 2 experimental sets. There were significant changes in metabolites of coproporphyrin III, glycocholic acid, taurochenodeoxycholate, taurocholate, hydrocortisone, pregnenolone sulfate, L-histidine, L-arginine, L-glutamate and L-glutamine. Analysis of these metabolic alterations was linked to aberrant gene expression at chromosome 21 of PDE9A, GART and FTCD genes. Specifically, the decrease is coproporhyrin III in the DS fetus may be linked to abnormal erythropoiesis and the unbalanced glutamine-glutamate was found to be closely associated with abnormal brain development in the DS foetus. It is important to note that there was a small sample size of 10-15 controls and cases, reducing the statistical power of the study.
Studies of AF cannot only diagnose DS but can highlight the specific organ systems or tissues that might be affected after birth. As there is a very heterogeneous expression of the DS phenotype in people with the condition, further work could explore if the magnitude of the above-described changes in gene expression associated with neural development, immune competency, or collagen stability, correlate with the severity of the changes seen. This would allow the development of individualised patient therapies in the future if the risk of the AF sampling could be justified.
5. Gene expression changes in the placenta
The placenta, specifically the chorionic villus, is also commonly used for prenatal testing. Placental tissue can also be obtained in the event of pregnancy termination following a diagnosis of DS.
Gross et al., used seven 2nd trimester placentas of foetuses with T21 and seven matched and seven non-matched cDNA samples from normal karyotype placentas as controls [28]. These were used to evaluate the feasibility of differences in gene expression using microarray technology. Their custom array contained approximately 9,000 cDNA clones. 643 cDNAs were overexpressed in T21 vs controls and 3 cDNAs found to be under expressed. When compared with age-matched controls, only 13 differentially expressed cDNAs were detected. The use of microarrays prenatally from placental samples has been slow, due to potential difficulties interpreting copy number variations of unknown significance, and incomplete penetrance or variable expressivity in absence of a clear phenotype [7]. This is highlighted in this study where different genes were expressed in the age-matched controls vs non-age matched, highlighting the dynamic nature of gene expression during gestation and the importance of studying different time points and using age-matched controls.
A more recent study by Lee et al., focused on finding novel epigenetic markers on chromosome 21 that shows a hypermethylated pattern in foetal placenta vs blood using PCR [7]. They performed a high-resolution tiling array analysis of chromosome 21 using methylated-CpG binding domain protein-based method. 93 epigenetic regions were identified as showing placenta-specific differential methylation patterns, three regions showed foetal placenta-specific methylation patterns in T21 placenta samples. These three regions were detectable with high diagnostic accuracy as early as the first trimester, when further statistical analysis was conducted. Therefore, showing clear genetic changes in the placenta, that can be targeted to aid with diagnosis and help increase our understanding of aetiology.
Rozovski et al., measured detectable expression of 5,334 genes out of over 10,000 on an oligonucleotide microarray, from cultured trophoblasts derived from placental samples, obtained by prenatal testing in the first trimester [5]. The sample consisted of 4 normal male foetuses and 4 T21 males. 65 genes were found to be significantly altered in the DS cases, after correction for multiple comparisons, 51 were over-expressed and 14 were under-expressed. The three genes with the highest significant fold change were MEST, SNF1LK and LOX. MEST is thought to play a role in development and is usually expressed in mesoderm-derivatives [29]. Cultured trophoblasts were needed for ethical reasons and to ensure there is sufficient and good quality DNA, which can be difficult from prenatal samples [7]. The results in this study were validated using qRT-PCR, improving the accuracy of the results. This highlighted some differences, for example MAT2A was found to be under-expressed in microarray and over-expressed in qRT-PCR. This is due to non-specific hybridization of MAT2A transcripts to Affymetrix microarray and the probe on the Affymetrix array shares complete identity to a sequence in early endosome antigen 1 gene on chromosome 12, leading to skewed expression and highlighting a limitation of the Affymetrix microarray. Despite this, this study highlights specific genes found to be over-expressed in DS vs controls and can be compared against other studies to increase their power and studied further to see if viable for diagnosis or therapeutic targets.
Studies of gene changes in the placenta highlight the dynamic nature of gene expression during gestation and the importance of studying different time points within development. There are of course ethical issues with studying early tissue, however the gene changes it highlights are invaluable in ongoing research for potential biomarkers.
6. Gene expression changes affecting brain development.
Development of the brain is drastically affected in DS and is the cause of the most prominent hallmarks of the disease. There are known changes in gross brain structure and microdysgenetic changes. These manifest as hypotonia at birth, with gait and ligamentous laxity, seizures, intellectual disability, and the neuropathological changes of Alzheimer’s disease often by 40 years-old [30].
El Hajj et al., investigated changes in gene expression in the developing DS foetal cortex, using 16 DS and 27 foetal controls’ frontal cortexes, 8 DS and 8 control foetal temporal cortexes and 2 DS and 9 control adult frontal cortexes [31]. The used Illumina 450K arrays which showed that 1.85% of all analysed CpG sites were significantly hypermethylated and 0.31% hypomethylated in foetal DS cortex. Methylation values were quantified using Pyro Q-CpG software and methylation PCR. Specifically, there were reduced NRSF/REST expression due to upregulation of DYRK1A (21qq22.13). REST is a transcriptional repressor, involved in the repression of neural genes. DYRK1A is a kinase which functions to help the development of the nervous system. DYRK1A gene expression changes are linked to intellectual disability, speech development and autism, therefore possibly explaining some of the phenotypical features of DS [32]. Methylation of REST binding sites during early development may contribute to genome-wide excess of hypermethylated sites. There was upregulation of DNMT3L, which is hypothesised to lead to de novo methylation of neuroprogenitors, which persists in the foetal DS brain, whereas DNMT3A and DNMT3B are downregulated in DS. DNMT3A and DNMT3B are responsible for establishing DNA methylation patterns during embryogenesis, therefore this alteration will contribute to phenotypic characteristics in DS [33]. Another finding was that a large amount of differentially methylated promoters on chromosomes other than 21. The PCDHG cluster on chromosome 5 was hypermethylated and downregulated in DS, this is involved in the neural circuit formation in the developing brain, this downregulation causes a reduction in dendrite arborization and growth in cortical neurons. This study used gestational age matched controls, to reduce error in result interpretation caused by differentiation in expression at different stages of development.
Olmos-Serrano et al., conducted a multi-region transcriptome analysis of DS and euploid control brains, looking from mid-foetal development (14 weeks post-conception) until adulthood (42 years old) [34]. This was to focus on the complexity of brain development and changes over many decades, therefore describing how genes involved in brain development may modulate overtime. Dysregulated genes are found throughout the genome and not solely on chromosome 21. The transcriptome profiling performed using total RNA was extracted from 11 regions, including multiple regions of cerebral neocortex, hippocampus, and cerebellar cortex. There was a genome-wide alteration in expression of large number of genes, which exhibited temporal and spatial specificity, associated with distinct co-expression networks, with distinct biological categories, providing novel insight into multiple biological processes affected in developing and adult DS brain. M43 relating to regulation of action potential and axon ensheathment was downregulated in DS neocortex and hippocampus over development, associated with myelination. Myelination is one the of the most prolonged neurodevelopmental processes continuing until third decade of life [35,36]. If this is impacted, it implies that the neurodevelopmental process in DS is affected throughout the first few decades of life. The protocol used for analysis was standardised from high quality post-mortem human brains, therefore increasing reliability of the results. Co-dysregulation of genes associated with oligodendrocyte differentiation and myelination was validated via cross-species comparison in Ts65Dn trisomy mice.
Shi et al., extracted total RNA from foetal hippocampal tissues to analyse miRNA and mRNA expression via Affymetrix miRNA 4.0 and PrimeView Human Gene Expression Array, which were validated by real-time PCR [8]. They found a specific repertoire of miRNAs involved in the hippocampus in trisomy 21. These included hsa-miR-138, hsa-miR-409 and hsa-miR-138 -5, eluding that there altered activity in the hippocampus was a factor in the alteration of intellectual disability in DS. Further studies have been conducted by Deng et al., and Lim et al., into miRNAs [37,38]. Deng et al., found that miR-125b-2 on chromosome 21 is overexpressed in DS patients with cognitive impairment [37]. miR-125b-2 is known to promote specific types of neuronal differentiation, however its full function in the developing embryo is unknown. The study looked at overexpression of miR-125b-2 and found that it inhibited the differentiation of mouse embryonic stem cells (mESCs) into endoderm and ectoderm and impaired all-trans-retinoic acid-induced neuron development in embryoid bodies. Therefore, important in affecting the rate at which the brain and neurons develop. Lim et al., looked at the possibility of using miRNAs as potential non-invasive biomarkers for detection of foetal trisomy 21 [38]. They used a microarray-based based genome-wide expression profiling comparing expression levels of miRNAs in whole blood samples from non-pregnant women, pregnant women with euploid T21 foetuses and placenta samples from euploid or T21 foetuses. They found that 150 RNAs were up-regulated in placenta in T21 and 149 down-regulated. miRNAs mir-1973 and mir-3196 were expressed at higher levels in T21 placenta compared to euploid placenta. These miRNA studies are useful at increasing our understanding of the changes in a developing DS brain and how these changes cause the well-known phenotype.
A study by Shimizu et al., found that DS mouse model had defective early neuron production in prenatal life and hippocampal hypoplasia [39]. A transcriptomic analysis was conducted on Ts1Cje mice, they were compared against a transcriptomic profile of prenatal forebrain at embryonic day 14.5. There was a decreased expression of the TBX1 mRNA in both prenatal forebrain and adult hippocampus Ts1Cje mice. This was confirmed in other DS mouse models, including the Dp (16)1Yey/+ (longer trisomic regions) and Ts1Rhr (shorter trisomic regions) mouse, validating the results. The TBX1 region HSA22q11 heterozygous deletion is present in individuals with DiGeorge syndrome. In DiGeorge syndrome it is involved in the conotruncal outflow tract cardiac defects seen in over 70% of this population. Despite both conditions having cardiac defects relating to decreased expression of TBX1, they are distinct in DS from DiGeorge, as are endocardiac cushion AVSD and AV canal defects. It is thought to be involved in delayed foetal brain development and postnatal psychiatric phenotypes in DS. Other findings included dysregulation of the interferon-related molecular networks in the hippocampus of Ts1Cje, causing overexpression of Ifnar1 and Ifnar2 genes, which may be a factor in immunodeficiency in DS.
Olig2 is a basic helix-loop-helix transcription factor which is crucial for mammalian CNS development and in the critical region of trisomy 21 [40]. In the previous study by Shimizu et al., Olig1 and Olig2 genes were increased in Ts1Cje mice in comparison to wild type [39]. It is not clear what phenotypical features these changes would lead to. Liu et al., focused on this specifically by developing an Olig2-overexpressing transgenic mice line with a Cre/loxP system, this caused the phenotype of microcephaly, cortical dyslamination, hippocampus malformation and profound motor deficits [40]. They found massive neuronal cell death, downregulation of neuronal specification factors (Ngn1, Ngn2 and Pax6) in the developing cortex of Olig2 misexpressing mice. Olig2 was significantly upregulated in DS frontal cortices at 14 week and 18-week gestation ages [40]. A chromatin-immunoprecipitation and sequencing analysis confirmed that Olig2 directly targets the promoter and/or enhancer regions of Nfatc4, Dscr1/Rcan1 and Dyrk1a, the critical neurogenic genes that contribute to Down syndrome phenotypes and inhibits their expression. This study highlights the importance of Olig2 and how it affects other genes, causing the phenotype.
The DSCR genes have been found to be differentially expressed in other studies. Esposito et al., found gene expression changes in neural progenitors derived from frontal cerebral cortex. Expression of DSCR genes was increased in DS cases [41]. The study analysed 608 probes differentially expressed, representing 334 genes and 46 functional networks. Further analysis found that upregulation of S100B and APP in this critical region activate the stress response kinase pathways and are linked to upregulation of aquaporin 4 (AQP4). Changes in AQP4 have been linked to epilepsy, oedema, Alzheimer’s disease, and other CNS disorders, which may explain the phenotypical changes in DS [42,43].
These studies all increase our understanding of the phenotypical manifestations of DS, including intellectual ability and the neuropathological changes in Alzheimer’s disease. Some of the data dispartitie come from the fact that different studies identified different genes that are up or downregulated and different methods used. Hence the purpose of this review to analyse the advantages and limitations of the different techniques used. Many of the studies are of small scale, due to ethical reasons, which must be considered when interpreting results.
7. Gene expression changes affecting cardiac tissues
Congenital heart defects (CHDs) occur in ~50% of individuals with DS, the most common being atrioventricular septal defect (AVSD) [44]. The mechanism by which these occur is currently unknown. In 2006, Li et al., screened approximately 10,000 genes in heart tissue and skin fibroblast cultures [45]. In the heart, 110 genes were differentially expressed in DS cases. The number of chromosome 21 genes was 17 and 7, respectively, all of which showed increased expression. GART had the largest over-expression on chromosome 21. GART encode a trifunctional enzyme that catalyses the ‘de novo’ inosine monophosphate biosynthetic pathway. This is involved in de novo purine synthesis, which may lead to the phenotypical features such as intellectual disability, hypotonia and increased sensorineural deafness.
Bosman et al., conducted a study using a human embryonic stem cell model of DS [46]. From their study they proposed two candidate genes on chromosome 21, EST2 and ERG, whose overexpression during the early stages of cardiogenesis, most likely account for the disruption of the secondary heart field development and therefore the CHD AVSDs. These could be a target for gene therapy in the future. The sibling hESC model was used for recapitulating early cardiogenesis in DS. This allows for small differences in differentiation and development of the disease itself, rather than small differences in genetic or epigenetic backgrounds. Despite this, they also found an electrophysiological abnormality in the function of T21 cardiomyocytes. Supported and validated by mRNA expression data acquired using RNA-Seq. This could be investigated further to see if reliably found again.
A study by Liu et al., studied mouse mutants carrying different genomic rearrangements in human chromosome 21 (Hsa21) syntenic regions [47]. They found a triplication of the Tiam1-Kcnj6 region on mouse chromosome 16 (Mmu16) resulted in DS-related cardiovascular abnormalities. The mouse chromosome 16 is syntenic with both human chromosomes 21 and 22, thus some of the genes from 22q11 region, may be the same in this mouse models as those from chromosome 21, hence a useful DS model. They then developed two tandem duplications spanning this region, to see if they would develop DS-associated heart defects. They used recombinase-mediated genome engineering and found that Dp (16)4Yey spanning 3.7 Mb Ifnar1-Kcnj6 led to heart defects, but not Dp(16)3Yey triplication at 2.1 Mb Tiam1-Il10rb. They stated that the 3.7 Mb genomic region was the smallest critical genomic region for DS-associated heart defects.
MiRNAs are thought to play an important role in regulating cardiac development. Five chromosome 21 miRNAs have been studied previous miR-99a-5p, miR-125b-2-5p, let-7c-5p, miR-155-5p, and miR-802-5p, but their expression in trisomy tissues has not been explored. Izzo et al., showed that miR-99a-5p, miR-155-5p and let-7c-5p were downregulated in trisomic hearts from DS foetuses and let-7c-5p and miR-155-5p are involved in mitochondrial function [48]. Mitochondrial dysfunction is a hallmark of down syndrome; therefore, these miRNAs may affect mitochondrial dysfunction and cardiogenesis [49].
In all the above-mentioned studies methods vary, and different DS models are used. Differing targets have been identified that effect different organs. Larger scale studies would need to be conducted to assess whether these changes in gene expression can be targeted singularly or need to be targeted as a group to stop the cardiac changes seen in DS.
8. Gene expression changes that lead to haematopoietic cells/myeloproliferative disease
Several studies have shown that individuals with DS have an increased risk of developing certain cancers. In children there is 10-20 times increase in relative risk when compared to the general population of developing acute leukaemia [49,50,51]. This strongly suggests a link with DS and neoplastic formation of haematopoietic cells, in particular the megakaryocyte lineage cells. Interestingly, acute leukemic cells have been seen and presented in people without DS, but with trisomy 21, highlighting the trisomy 21 in the aetiology of the neoplastic transformation of haematopoietic cells [52].
Chiang et al., assessed whether XIST-induced trisomy silencing can largely normalise DS-related haematopoietic phenotypes or not [4]. XIST is an X-linked gene that naturally controls X-chromosome inactivation in human female cells. A doxycycline inducible full-length XIST cDNA was inserted into one of three chromosomes 21s in induced pluripotent stem cells (iPSCs), derived from a male DS patient. They found that XIST induction in 4 independent transgenic clones reproducibly corrected over-production of megakaryocytes and erythrocytes, which are key to producing myeloproliferative disorder and leukaemia.
Further studies found specific genes that were over-expressed in DS, that are linked to myeloproliferative disease. Kubota et al., conducted an integrated genetic/epigenetic analysis and found hypermethylation of RUNX1 on chromosome 21 was found in DS-acute lymphoblastic leukaemia (ALL), not in ALL without DS [53]. Muskens et al., conducted an epigenome-wide associated study on neonatal bloodspots and found that the top two differentially methylated regions overlap RUNX1 and FLI1 [54]. These studies were both using DNA methylation analysis carried out using a chip-based analysis. The use of bloodspots allowed for a large sample size when compared to other studies, with 196 DS patients compared to 439 without DS. Analysis of the cohort was also adjusted for heterogeneity, increasing the power of the study. RUNX1 is essential for differentiation of blood cells, especially B cells. Therefore, hypermethylation of RUNX1 may be associated with a higher incidence of B-cell precursor ALL in DS patients.
A study conducted by Belmonte et al., focused on the gene SON on chromosome 21 [55]. They looked at reduced expression in a zebrafish homolog of SON, this led to lowers amounts of red bloods cells being produced, brain and spinal malformations, reduced thrombocytes and myeloid cells and a significant decrease in T cells. This may highlight a cause for immunodeficiency and myeloproliferative disorders that arise in DS. Ishihara et al., conducted a transcriptomic and flow cytometry analysis of E14.5 Ts1Cje mouse embryo brain, to allow a study of multiple genes and their impacts on the myeloproliferative system [56]. They found that neutrophil and monocyte ratios in CD45-positive haematopoietic cells increased, and macrophages decreased. They used multiple methods of analysis, including microarray, informatics analysis, validated with quantitative RT-PCR, western blotting, flow cytometry, immunohistochemistry, and image analyses, with in vivo BrdU labelling. The DNA microarray analysis of E14.5 Ts1Cje embryo brain revealed elevated expression of S100a8, S100a9, MPO and Ly6c1 mRNAs, which are abundant in neutrophils and/or monocytes. They also found that the triplication of Erg plays a role in self-renewal of haematopoietic stem cells and haematopoiesis in liver during embryogenesis [57,58]. In DS Erg triplication contributes to dysregulation of homeostatic proportion of populations of immune cells in embryonic brain and decreased prenatal cortical neurogenesis in the prenatal brain. This study only examined male mice; therefore these changes may not be consistent in female DS mice. Previous studies have highlighted there is sex-specific abnormalities, suggesting a possibility of a sex-specific phenotypic features of DS, therefore female mice should also be used [26].
Other studies have also focused on the mechanism in DS that causes immunodeficiency. A recent study by Kong et al., studied 45 DS patients and found high levels of IFN-αR1, IFN-αR2, and IFN-γR2 expression on the surface of monocytes and EBV-transformed-B cells [59]. Total and phosphorylated STAT1 (STAT1 and pSTAT1) levels were constitutively high in unstimulated and IFN-α- and IFN-γ-stimulated monocytes from DS patients but lower than those in patients with GOF STAT1 mutations. GOF STAT1 mutations lead to enhanced cellular response to IFN. Normal levels Th17 and high proportion of terminally differentiated CD8+ T cells and low levels of STAT1 expression were found in DS participants. These results may further our understanding of the mechanisms which cause immunodeficiency in DS patients. Table 2 highlights key genes in different organ systems that maybe targeted for gene therapy in DS.
The higher incidence of certain cancers, including acute leukaemia, in DS highlights a clear underlying genetic difference. These studies highlight that the changes in XIST and RUNX1 may play an important role in this. These studies also increased our understanding in how these gene changes can affect the immune system in DS. Table 2 displays the genes identified in different organ systems in DS that are thought to contribute to the changes seen in development. Position of the gene on a particular chromosome and whether it is down or up regulated is displayed.
9. Gene therapy for future implications
In the event that an individual gene or group of genes is conclusively determined to play a role in DS, the question arises of what forms of intervention could be used. Prevention is a more logical course of action; however, this would require treatment early in foetal development. Although individual genes can be inhibited by several methods, such as small interfering RNAs, the situation becomes more complicated once multiple genes, large chromosomal regions, or entire chromosomes are involved. One possibility is to inactivate the extra chromosome 21. This type of approach occurs naturally with the inactivation of the extra X chromosome in females. Normally, RNA produced by the XIST gene interacts with and inactivates one copy of the X chromosome in females. In the mouse, however, ectopic XIST RNA has shown the ability to inactivate autosomes as well [60]. If this process could be targeted, it is possible that DS could be prevented in trisomic individuals by inactivation of the extra chromosome in utero [61].
A previous study by Jiang et al., found that XIST non-coding RNA coats chromosome 21 and triggers stable heterochromatin modifications, causing wide-transcriptional silencing and DNA methylation to form a ‘chromosome 21 Barr body’ [54]. In doing this they found that the deficits in proliferation and neural rosette formation were rapidly reversed upon silencing one chromosome 21. Trisomy silencing in vitro is a major first step towards a potential development of chromosome therapy for DS, this process however is seen as controversial and unethical by many [62].
Czerminski et al., focused on XIST RNA induced in differentiated neural cells and found that they can trigger chromosome-wide silencing of chromosome 21 in DS patient-derived cells [63]. They found a deficiency in differentiations of DS neural stem cells to neurons, which was corrected by inducing XIST at different stages of neurogenesis. These results have not always been replicated and previous studies have been unable to silence the gene [64]. Therefore, larger scale studies into specifics of how the gene would be silenced and at what point need to be researched. Overall, this increases our understanding of the mechanism of DS and development of the phenotype and increases the evidence for the development of potentially transformative therapeutic interventions.
There have been many studies over the past years focusing on the potential of gene silencing in DS. To prevent DS completely genome editing would need to be done early in the womb. Li et al., introduced a TKNEO fusion transgene via a modified adenovirus at 21q21.3 of the gene APP [65]. This resulted in successful targeted removed of the third chromosome. As above XIST silencing has also resulted in disomic cells. A further study by Amano et al., used a biologic made gene ZSCAN4 via a synthetic messenger RNA and Sendai virus vector, after a few weeks causing up to 40% of normal karyotypes in iPSCs [66]. These studies need to be replicated in mouse and then human models. The practicalities of utilising gene silencing is currently difficult due to having to confirm T21 very early on and then successfully delivering gene therapy before phenotypical features begin to develop. Rondal highlights that to normalize embryonic development ideally gene therapy would be implicated day 3 post insemination, as the cells are multipotent [67]. As mentioned about there is also a lot of ethical debate surrounding this topic, in 2014 Inglis et al., produced a questionnaire to allow for parents to have their opinions on whether they would want gene therapy [62]. 41% responded that they would treat their child, if possible, with 27% saying they would not. Despite, the majority saying they would treat their child, this study was on a small scale with only 101 parents. It remains to be seen if gene therapy after birth in DS could modulate the likelihood of developing one of the many comorbid disease processes that people with DS unfortunately develop often early in life. Being able to modulate disease risk as opposed to stopping the development of DS in utero may also be more accepted gene therapy strategy for those people living with DS. Figure 1 highlights some potential gene therapy targets and in which organs they may have their effect.
10. Conclusions
In this review we have highlighted how the study of gene expression at the very earliest stages of the development of DS can inform which genes are likely causative in the development of certain disease states. We have also highlighted how these genes could be targeted in the future to modulate or stop disease in people living with the condition, providing eventual therapeutic avenues for treatment of this most common genetic cause of intellectual disability. Multiple barriers still exist when studying gene expression changes in DS. As highlighted in the different sections of this review, the expression of likely casual genes changes through the course of development, extending into early adulthood. Studies can be limited by sample size, for obvious ethical reasons, and developing treatments for DS has its own ethical considerations. Genetic therapies could potentially fundamentally change what it means to be a person living with DS. Future work would benefit from trying to understand what causes the heterogeneity in expression of the condition, and how to silence key genes to prevent the individual disease features in DS. As gene therapy work develops it will also be important to include people living with DS in the discussion. As the complex ethical, medical and scientific questions that will arise from this line of scientific enquiry will affect the people living with DS and their families the most.
Author Contributions
Conceptualization, AM, DZ.; methodology, LC and DZ.; investigation, LC, SMB and AM.; data curation, LC and SMB.; writing—original draft preparation, LC, SMB and DZ.; writing—review and editing, LC, DZ, SMB, AM.; supervision, AM and SMB.; project administration, LC, and SMB.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Ethical review and approval were waived for this study due to this article being a review article the study being a literature review of studies which have already been granted ethical approval.
Informed Consent Statement
Patient consent was not needed for this paper this study being a literature review, patient consent would have be gained in the individual published studies.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Carothers, A.D.; A Hecht, C.; Hook, E.B. International variation in reported livebirth prevalence rates of Down syndrome, adjusted for maternal age. J. Med Genet. 1999, 36, 386–393. [Google Scholar] [PubMed]
- Korenberg, J.R.; Chen, X.N.; Schipper, R.; Sun, Z.; Gonsky, R.; Gerwehr, S.; et al. Down syndrome phenotypes: the consequences of chromosomal imbalance. Proc Natl Acad Sci USA 1994, 91, 4997. [Google Scholar] [CrossRef] [PubMed]
- Sidman, R.L.; Rakic, P. Neuronal migration, with special reference to developing human brain: a review. Brain Res. 1973, 62, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.-C.; Jiang, J.; Newburger, P.E.; Lawrence, J.B. Trisomy silencing by XIST normalizes Down syndrome cell pathogenesis demonstrated for hematopoietic defects in vitro. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rozovski, U.; Jonish-Grossman, A.; Bar-Shira, A.; Ochshorn, Y.; Goldstein, M.; Yaron, Y. Genome-wide expression analysis of cultured trophoblast with trisomy 21 karyotype. Hum. Reprod. 2007, 22, 2538–2545. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Cho, H.Y.; Cha, D.H. The Amniotic Fluid Cell-Free Transcriptome Provides Novel Information about Fetal Development and Placental Cellular Dynamics. Int. J. Mol. Sci. 2021, 22, 2612. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.E.; Lim, J.H.; Kim, M.H.; Park, S.Y.; Ryu, H.M. Novel Epigenetic Markers on Chromosome 21 for Noninvasive Prenatal Testing of Fetal Trisomy 21. J. Mol. Diagn. 2016, 18, 378–387. [Google Scholar] [CrossRef]
- Shi, W.-L.; Liu, Z.-Z.; Wang, H.-D.; Wu, D.; Zhang, H.; Xiao, H.; Chu, Y.; Hou, Q.-F.; Liao, S.-X. Integrated miRNA and mRNA expression profiling in fetal hippocampus with Down syndrome. J. Biomed. Sci. 2016, 23, 1–12. [Google Scholar] [CrossRef]
- Haque, A.; Engel, J.; Teichmann, S.A.; Lönnberg, T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017, 9, 1–12. [Google Scholar] [CrossRef]
- Stamoulis, G.; Garieri, M.; Makrythanasis, P.; Letourneau, A.; Guipponi, M.; Panousis, N.; Sloan-Béna, F.; Falconnet, E.; Ribaux, P.; Borel, C.; et al. Single cell transcriptome in aneuploidies reveals mechanisms of gene dosage imbalance. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Kurdyukov, S.; Bullock, M. DNA Methylation Analysis: Choosing the Right Method. Biology 2016, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Letourneau, A.; Santoni, F.A.; Bonilla, X.; Sailani, M.R.; Gonzalez, D.; Kind, J.; Chevalier, C.; Thurman, R.; Sandstrom, R.S.; Hibaoui, Y.; et al. Domains of genome-wide gene expression dysregulation in Down’s syndrome. Nature 2014, 508, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Antonaros, F.; Zenatelli, R.; Guerri, G.; Bertelli, M.; Locatelli, C.; Vione, B.; Catapano, F.; Gori, A.; Vitale, L.; Pelleri, M.C.; et al. The transcriptome profile of human trisomy 21 blood cells. Hum. Genom. 2021, 15, 25. [Google Scholar] [CrossRef] [PubMed]
- Allyse, M.; Minear, M.; Rote, M.; Hung, A.; Chandrasekharan, S.; Berson, E.; Sridhar, S. Non-invasive prenatal testing: a review of international implementation and challenges. Int. J. Women's Heal. 2015, 7, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Practice Bulletin, No. 162: Prenatal Diagnostic Testing for Genetic Disorders. The American College of Obstet Gynecol 2016, 127, e108–e122. [Google Scholar]
- Chung, I.-H.; Lee, S.-H.; Lee, K.-W.; Park, S.-H.; Cha, K.-Y.; Kim, N.-S.; Yoo, H.-S.; Kim, Y.S.; Lee, S. Gene Expression Analysis of Cultured Amniotic Fluid Cell with Down Syndrome by DNA Microarray. J. Korean Med Sci. 2005, 20, 82–87. [Google Scholar] [CrossRef]
- COL6A1 Gene - GeneCards | CO6A1 Protein | CO6A1 Antibody. https://www.genecards.org/cgi-bin/carddisp.pl?gene=COL6A1. (accessed on 6 September 2022).
- Altug-Teber, Ö.; Bonin, M.; Walter, M.; Mau-Holzmann, U.; Dufke, A.; Stappert, H.; Tekesin, I.; Heilbronner, H.; Nieselt, K.; Riess, O. Specific transcriptional changes in human fetuses with autosomal trisomies. Cytogenet. Genome Res. 2007, 119, 171–184. [Google Scholar] [CrossRef]
- Davies, G.E.; Howard, C.M.; Farrer, M.J.; Coleman, M.M.; Bennett, L.B.; Cullen, L.M.; et al. Genetic variation in the COL6A1 region is associated with congenital heart defects in trisomy 21 (Down's syndrome). Ann Hum Genet 1995, 59, 253–269. [Google Scholar] [CrossRef]
- Anlar, B.; Atilla, P.; Çakar, A.N.; Kose, M.F.; Beksaç, M.S.; Dagdeviren, A.; Akçören, Z. Expression of adhesion and extracellular matrix molecules in the developing human brain. J. Child Neurol. 2002, 17, 707–713. [Google Scholar] [CrossRef]
- Loftis, M.J.; Sexton, D.; Carver, W. Effects of collagen density on cardiac fibroblast behavior and gene expression. J. Cell. Physiol. 2003, 196, 504–511. [Google Scholar] [CrossRef]
- Fuentes, J.J.; Genescà, L.; Kingsbury, T.J.; Cunningham, K.W.; Pérez-Riba, M.; Estivill, X.; de la Luna, S. DSCR1, overexpressed in Down syndrome, is an inhibitor of calcineurin-mediated signaling pathways. Hum. Mol. Genet. 2000, 9, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Olson, L.E.; Richtsmeier, J.T.; Leszl, J.; Reeves, R.H. A Chromosome 21 Critical Region Does Not Cause Specific Down Syndrome Phenotypes. Science 2004, 306, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Slonim, D.K.; Koide, K.; Johnson, K.L.; Tantravahi, U.; Cowan, J.M.; Jarrah, Z.; et al. Functional genomic analysis of amniotic fluid cell-free mRNA suggests that oxidative stress is significant in Down syndrome fetuses. Proc Natl Acad Sci USA 2009, 106, 9425–9429. [Google Scholar] [CrossRef]
- Zhu, W.; Zhou, M.; Xiong, Z.; Peng, F.; Wei, W. The cAMP-PKA signaling pathway regulates pathogenicity, hyphal growth, appressorial formation, conidiation, and stress tolerance in Colletotrichum higginsianum. Front Microbiol 2017, 8, 1416. [Google Scholar] [CrossRef] [PubMed]
- Block, A.; Ahmed, M.; Dhanasekaran, A.R.; Tong, S.; Gardiner, K.J. Sex differences in protein expression in the mouse brain and their perturbations in a model of Down syndrome. Biol. Sex Differ. 2015, 6, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Mo, J.; Zhao, G.; Lin, Q.; Wei, G.; Deng, W.; Chen, D.; Yu, B. Application of the amniotic fluid metabolome to the study of fetal malformations, using Down syndrome as a specific model. Mol. Med. Rep. 2017, 16, 7405–7415. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.J.; Ferreira, J.C.; Morrow, B.; Dar, P.; Funke, B.; Khabele, D.; Merkatz, I. Gene expression profile of trisomy 21 placentas: A potential approach for designing noninvasive techniques of prenatal diagnosis. Am. J. Obstet. Gynecol. 2002, 187, 457–462. [Google Scholar] [CrossRef]
- Nishita, Y.; Yoshida, I.; Sado, T.; Takagi, N. Genomic imprinting and chromosomal localization of the human MEST gene. Genomics 1996, 36, 539–542. [Google Scholar] [CrossRef]
- Lott, I.T. Neurological phenotypes for Down syndrome across the life span. 197. [CrossRef]
- El Hajj, N.; Dittrich, M.; Böck, J.; Kraus, T.F.J.; Nanda, I.; Müller, T.; Seidmann, L.; Tralau, T.; Galetzka, D.; Schneider, E.; et al. Epigenetic dysregulation in the developing Down syndrome cortex. Epigenetics 2016, 11, 563–578. [Google Scholar] [CrossRef]
- Qiao, F.; Shao, B.; Wang, C.; Wang, Y.; Zhou, R.; Liu, G.; Meng, L.; Hu, P.; Xu, Z. A De Novo Mutation in DYRK1A Causes Syndromic Intellectual Disability: A Chinese Case Report. Front. Genet. 2019, 10. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, J. DNA methyltransferases and their roles in tumorigenesis. Biomark. Res. 2017, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Olmos-Serrano, J.L.; Kang, H.J.; Tyler, W.A.; Silbereis, J.C.; Cheng, F.; Zhu, Y.; Pletikos, M.; Jankovic-Rapan, L.; Cramer, N.P.; Galdzicki, Z.; et al. Down Syndrome Developmental Brain Transcriptome Reveals Defective Oligodendrocyte Differentiation and Myelination. Neuron 2016, 89, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.J.; Duka, T.; Stimpson, C.D.; Schapiro, S.J.; Baze, W.B.; McArthur, M.J.; Fobbs, A.J.; Sousa, A.M.M.; Šestan, N.; Wildman, D.E.; et al. Prolonged myelination in human neocortical evolution. Proc. Natl. Acad. Sci. 2012, 109, 16480–16485. [Google Scholar] [CrossRef] [PubMed]
- Benes, F.M.; Turtle, M.; Khan, Y.; Farol, P. Myelination of a Key Relay Zone in the Hippocampal Formation Occurs in the Human Brain During Childhood, Adolescence, and Adulthood. Arch. Gen. Psychiatry 1994, 51, 477–484. [Google Scholar] [CrossRef]
- Deng, S.; Zhang, Y.; Xu, C.; Ma, D. MicroRNA-125b-2 overexpression represses ectodermal differentiation of mouse embryonic stem cells. Int. J. Mol. Med. 2015, 36, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Lee, D.E.; Kim, S.Y.; Kim, H.J.; Kim, K.S.; Han, Y.J.; Kim, M.H.; Choi, J.S.; Kim, M.Y.; Ryu, H.M.; et al. MicroRNAs as potential biomarkers for noninvasive detection of fetal trisomy 21. J. Assist. Reprod. Genet. 2015, 32, 827–837. [Google Scholar] [CrossRef]
- Shimizu, R.; Ishihara, K.; Kawashita, E.; Sago, H.; Yamakawa, K.; Mizutani, K.-I.; Akiba, S. Decrease in the T-box1 gene expression in embryonic brain and adult hippocampus of down syndrome mouse models. Biochem. Biophys. Res. Commun. 2020, 535, 87–92. [Google Scholar] [CrossRef]
- Lu, J.; Lian, G.; Zhou, H.; Esposito, G.; Steardo, L.; Delli-Bovi, L.C.; Hecht, J.L.; Lu, Q.R.; Sheen, V. OLIG2 over-expression impairs proliferation of human Down syndrome neural progenitors. Hum. Mol. Genet. 2012, 21, 2330–2340. [Google Scholar] [CrossRef]
- Esposito, G.; Imitola, J.; Lu, J.; De Filippis, D.; Scuderi, C.; Ganesh, V.S.; Folkerth, R.; Hecht, J.; Shin, S.; Iuvone, T.; et al. Genomic and functional profiling of human Down syndrome neural progenitors implicates S100B and aquaporin 4 in cell injury. Hum. Mol. Genet. 2007, 17, 440–457. [Google Scholar] [CrossRef]
- Moftakhar, P.; Lynch, M.D.; Pomakian, J.L.; Vinters, H.V. Aquaporin Expression in the Brains of Patients With or Without Cerebral Amyloid Angiopathy. J. Neuropathol. Exp. Neurol. 2010, 69, 1201–1209. [Google Scholar] [CrossRef]
- Vandebroek, A.; Yasui, M. Regulation of AQP4 in the Central Nervous System. Int. J. Mol. Sci. 2020, 21, 1603. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, S.E.; Skotko, B.G.; Rafii, M.S.; Strydom, A.; Pape, S.E.; Bianchi, D.W.; et al. Down syndrome. Nat Rev Dis Primers 2020, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Li, C.M.; Guo, M.; Salas, M.; Schupf, N.; Silverman, W.; Zigman, W.B.; et al. Cell type-specific over-expression of chromosome 21 genes in fibroblasts and fetal hearts with trisomy 21. BMC Med Genet 2006, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Bosman, A.; Letourneau, A.; Sartiani, L.; Del Lungo, M.; Ronzoni, F.; Kuziakiv, R.; Tohonen, V.; Zucchelli, M.; Santoni, F.; Guipponi, M.; et al. Perturbations of Heart Development and Function in Cardiomyocytes from Human Embryonic Stem Cells with Trisomy 21. STEM CELLS 2015, 33, 1434–1446. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Morishima, M.; Jiang, X.; Yu, T.; Meng, K.; Ray, D.; Pao, A.; Ye, P.; Parmacek, M.S.; Yu, Y.E. Engineered chromosome-based genetic mapping establishes a 3.7 Mb critical genomic region for Down syndrome-associated heart defects in mice. Hum. Genet. 2013, 133, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Izzo, A.; Manco, R.; de Cristofaro, T.; Bonfiglio, F.; Cicatiello, R.; Mollo, N.; De Martino, M.; Genesio, R.; Zannini, M.; Conti, A.; et al. Overexpression of Chromosome 21 miRNAs May Affect Mitochondrial Function in the Hearts of Down Syndrome Fetuses. Int. J. Genom. 2017, 2017, 8737649. [Google Scholar] [CrossRef]
- Valenti, D.; Braidy, N.; De Rasmo, D.; Signorile, A.; Rossi, L.; Atanasov, A.; Volpicella, M.; Henrion-Caude, A.; Nabavi, S.; Vacca, R. Mitochondria as pharmacological targets in Down syndrome. Free. Radic. Biol. Med. 2018, 114, 69–83. [Google Scholar] [CrossRef]
- Hitzler, J.K.; Zipursky, A. Origins of leukaemia in children with Down syndrome. Nat. Rev. Cancer 2005, 5, 11–20. [Google Scholar] [CrossRef]
- Massey, G.V. Transient leukemia in newborns with Down syndrome. Pediatr. Blood Cancer 2004, 44, 29–32. [Google Scholar] [CrossRef]
- Hasle, H.; Haunstrup Clemmensen, I.; Mikkelsen, M. Risks of leukaemia and solid tumours in individuals with Down’s syndrome. Lancet 2000, 355, 165–169. [Google Scholar] [CrossRef]
- Kubota, Y.; Uryu, K.; Ito, T.; Seki, M.; Kawai, T.; Isobe, T.; Kumagai, T.; Toki, T.; Yoshida, K.; Suzuki, H.; et al. Integrated genetic and epigenetic analysis revealed heterogeneity of acute lymphoblastic leukemia in Down syndrome. Cancer Sci. 2019, 110, 3358–3367. [Google Scholar] [CrossRef] [PubMed]
- Muskens, I.S.; Li, S.; Jackson, T.; Elliot, N.; Hansen, H.M.; Myint, S.S.; Pandey, P.; Schraw, J.M.; Roy, R.; Anguiano, J.; et al. The genome-wide impact of trisomy 21 on DNA methylation and its implications for hematopoiesis. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Belmonte, R.L.; Engbretson, I.L.; Kim, J.-H.; Cajias, I.; Ahn, E.-Y.E.; Stachura, D.L. son is necessary for proper vertebrate blood development. PLOS ONE 2021, 16, e0247489. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, K.; Shimizu, R.; Takata, K.; Kawashita, E.; Amano, K.; Shimohata, A.; Low, D.; Nabe, T.; Sago, H.; Alexander, W.S.; et al. Perturbation of the immune cells and prenatal neurogenesis by the triplication of the Erg gene in mouse models of Down syndrome. Brain Pathol. 2019, 30, 75–91. [Google Scholar] [CrossRef]
- Taoudi, S.; Bee, T.; Hilton, A.; Knezevic, K.; Scott, J.; Willson, T.A.; Collin, C.; Thomas, T.; Voss, A.K.; Kile, B.T.; et al. ERG dependence distinguishes developmental control of hematopoietic stem cell maintenance from hematopoietic specification. Minerva Anestesiol. 2011, 25, 251–262. [Google Scholar] [CrossRef]
- Xie, Y.; Koch, M.L.; Zhang, X.; Hamblen, M.J.; Godinho, F.J.; Fujiwara, Y.; Xie, H.; Klusmann, J.-H.; Orkin, S.H.; Li, Z. Reduced Erg Dosage Impairs Survival of Hematopoietic Stem and Progenitor Cells. STEM CELLS 2017, 35, 1773–1785. [Google Scholar] [CrossRef]
- Kong, X.-F.; Worley, L.; Rinchai, D.; Bondet, V.; Jithesh, P.V.; Goulet, M.; Nonnotte, E.; Rebillat, A.S.; Conte, M.; Mircher, C.; et al. Three Copies of Four Interferon Receptor Genes Underlie a Mild Type I Interferonopathy in Down Syndrome. J. Clin. Immunol. 2020, 40, 807–819. [Google Scholar] [CrossRef]
- Lee, J.T.; Jaenisch, R. Long-range cis effects of ectopic X-inactivation centres on a mouse autosome. Nature 1997, 386, 275–279. [Google Scholar] [CrossRef]
- Jiang, J.; Jing, Y.; Cost, G.J.; Chiang, J.-C.; Kolpa, H.J.; Cotton, A.M.; Carone, D.M.; Carone, B.R.; Shivak, D.A.; Guschin, D.Y.; et al. Translating dosage compensation to trisomy 21. Nature 2013, 500, 296–300. [Google Scholar] [CrossRef]
- Inglis, A.; Lohn, Z.; Austin, J.C.; Hippman, C. A “cure” for Down syndrome: What do parents want? Clin Genet 2014, 86, 310. [Google Scholar] [CrossRef]
- Czermiński, J.T.; Lawrence, J.B. Silencing Trisomy 21 with XIST in Neural Stem Cells Promotes Neuronal Differentiation. Dev. Cell 2020, 52, 294–308. [Google Scholar] [CrossRef]
- Wutz, A.; Jaenisch, R. A Shift from Reversible to Irreversible X Inactivation Is Triggered during ES Cell Differentiation. Mol. Cell 2000, 5, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Li, L.B.; Chang, K.-H.; Wang, P.-R.; Hirata, R.K.; Papayannopoulou, T.; Russell, D.W. Trisomy Correction in Down Syndrome Induced Pluripotent Stem Cells. Cell Stem Cell 2012, 11, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Amano, T.; Jeffries, E.; Amano, M.; Ko, A.C.; Yu, H.; Ko, M.S.H. Correction of Down syndrome and Edwards syndrome aneuploidies in human cell cultures. DNA Res. 2015, 22, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Rondal, J.A. From the lab to the people: major challenges in the biological treatment of Down syndrome. AIMS Neurosci. 2021, 8, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Evans, T.G. Considerations for the use of transcriptomics in identifying the ‘genes that matter’ for environmental adaptation. J. Exp. Biol. 2015, 218, 1925–1935. [Google Scholar] [CrossRef] [PubMed]
- Lacoma, T. Electrophoresis Process. Sciencing. Available online: https://sciencing.com/electrophoresis-process-5481819.html (accessed on 18 January 2023).
- Wu, B. Immunohistochemistry stains. DermNet. 2015. Available online: https://dermnetnz.org/topics/immunohistochemistry-stains (accessed on 18 January 2023).
- Marmiroli N and Maestri E. Polymerase Chain Reaction (PCR). Science Direct. Food Toxicants Analysis: Techniques, Strategies and Developments, 2007. Available online: https://www.sciencedirect.com/topics/agricultural-and-biological-sciences/real-time-polymerase-chain-reaction#:~:text=Significant%20advantages%20of%20real%2Dtime,expensive%20than%20traditional%20PCR%20machines (accessed on 18 January 2023).
- Cole, M. What are the advantages & disadvantages of flow cytometry? Sciencing. 2018. Available online: https://sciencing.com/calculate-cell-concentration-2788.html (accessed on 18 January 2023).
- Luo, Q.; Zhang, H. Emergence of Bias During the Synthesis and Amplification of cDNA for scRNA-seq. Adv Exp Med Biol 2018, 1068, 149–158. [Google Scholar]
- Knox, A.J.; Graham, C.; Bleskan, J.; Brodsky, G.; Patterson, D. Mutations in the Chinese hamster ovary cell GART gene of de novo purine synthesis. Gene 2009, 429, 23–30. [Google Scholar] [CrossRef]
- Cho, H.-J.; Lee, J.-G.; Kim, J.-H.; Kim, S.-Y.; Huh, Y.H.; Kim, H.-J.; Lee, K.-S.; Yu, K.; Lee, J.-S. Vascular defects of DYRK1A knockouts are ameliorated by modulating calcium signaling in zebrafish. Dis. Model. Mech. 2019, 12. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Hazen, S.L. Myeloperoxidase and Cardiovascular Disease. Arter. Thromb. Vasc. Biol. 2005, 25, 1102–1111. [Google Scholar] [CrossRef]
- Ly6c1 lymphocyte antigen 6 complex, locus C1 [Mus musculus (house mouse)] - Gene – NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/17067 (accessed on 18 January 2023).
- Danopoulos, S.; Bhattacharya, S.; Deutsch, G.; Nih, L.R.; Slaunwhite, C.; Mariani, T.J.; Al Alam, D. Prenatal histological, cellular, and molecular anomalies in trisomy 21 lung. J. Pathol. 2021, 255, 41–51. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Diagram highlighting some of the genes that are upregulated (upwards arrow) and downregulated (downwards arrow). The genes are by the organ in which they were found in testing, or their dysregulation affects. The red highlights the genes that have been studied for silencing T21. A study by Li et al., worked on silencing APP to remove the third chromosome of trisomy 21 [58]. APP upregulation causes an increase in Aquaporin 4 channel (AQP4). Changes in AQP4 have been linked to epilepsy, oedema, Alzheimer's disease and other CNS disorders [35,36]. A further study by Czerminski et al., focused on XIST RNA induced in differentiated neural cells to trigger chromosome-wide silencing of chromosome 21 in DS patient-derived cells [56]. ALL = acute lymphoblastic leukaemia. “Created with BioRender.com.”.
Figure 1.
Diagram highlighting some of the genes that are upregulated (upwards arrow) and downregulated (downwards arrow). The genes are by the organ in which they were found in testing, or their dysregulation affects. The red highlights the genes that have been studied for silencing T21. A study by Li et al., worked on silencing APP to remove the third chromosome of trisomy 21 [58]. APP upregulation causes an increase in Aquaporin 4 channel (AQP4). Changes in AQP4 have been linked to epilepsy, oedema, Alzheimer's disease and other CNS disorders [35,36]. A further study by Czerminski et al., focused on XIST RNA induced in differentiated neural cells to trigger chromosome-wide silencing of chromosome 21 in DS patient-derived cells [56]. ALL = acute lymphoblastic leukaemia. “Created with BioRender.com.”.
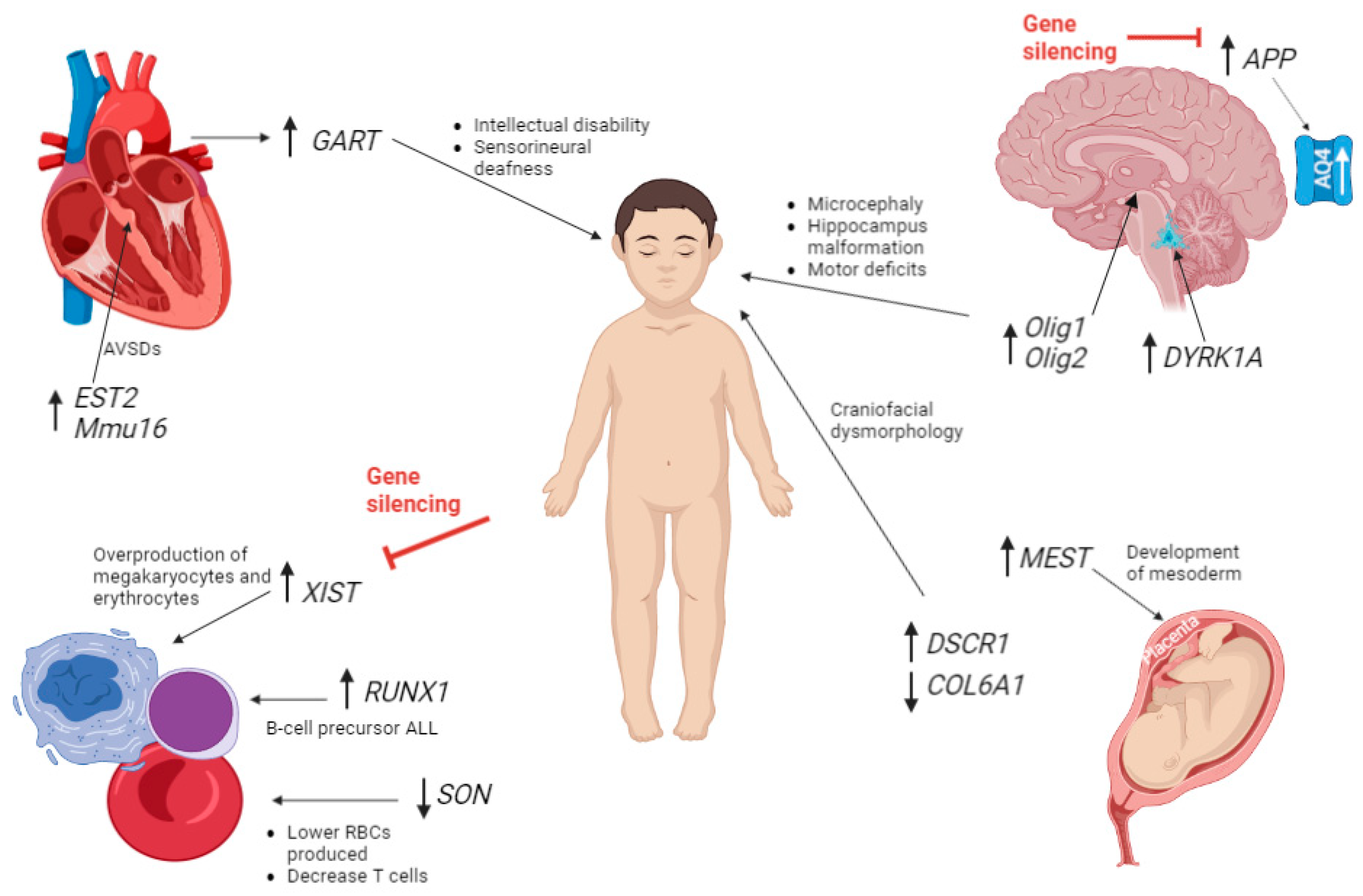

Table 1.
A summary of the advantages and disadvantages of different techniques used for analysis.
| Technique | Advantages | Disadvantages |
|---|---|---|
| Microarray analysis | • Results can be validated using real-time PCR High throughput method allowing expression levels of thousands of genes at once |
• Poor accuracy due to difficultly interpreting copy number variants of unknown significance7 Limited to genomic sequences Problems with probes cross-hybridization or sub-standard hybridization |
| DNA methylation analysis |
• Highly sensitive - can detect DNA methylation levels as low as 0.5% Very accurate in quantification Increase understanding of gene regulation and identify potential biomarkers33 |
• Multiple different ways of analysing DNA methylation with some disadvantages to each |
| Quantitative transcriptome map |
• Allows overview of changes in a whole organ Can be further validated by RT-PCR |
• Inappropriate for identifying genes with large impacts on adaptive responses to the environement68 mRNA abundance is an unreliable indicator of protein activity68 Standard practice in analysis is limited by prioritising highly differentially expressed genes over those who have moderate fold-changes and can’t be annotated68 |
| Western blot | • Sensitivity, able to detect 0.1 nanograms of protein, can be used in early diagnosis69 Specificity due to gel electrophoresis and the specificity of the antibody-antigen interaction69 |
• Time-consuming process Skilled analysts and laboratory equipment, minor error in the process can cause incorrect results, false negatives if proteins are not given enough incubation time69 It is non-quantitative Primary antibodies needed can be expensive. Antibodies can sometimes bind off-target. False-positive results due to antibodies reacting with a non-intended protein69 |
| Immunohistochemistry | • Relatively low cost70 Quick Can be done on fresh/frozen tissue samples70 Allows in-situ verification of various antibodies at the same time in organs, tissues and cells Can be done on fresh/frozen tissue samples70 |
• Not standardised worldwide70 The process is cheap, but the initial equipment to run it is expensive70 It is non-quantitative70 High chance of human error and relies on antibody staining optimization70 |
| Real-time PCR (RT-qPCR) | • Measure RNA concentrations over a large range Sensitive Process multiple samples simultaneously71 Provides immediate information71 |
• Requires optimization of good primers and correct reaction conditions |
| Flow cytometry analysis | • Fast single cell multiparametric analysis Very accurate and can be used on very small populations of cells72 Good at highlighting non-uniformity72 Produces very detailed data72 |
• Very slow analysis72 More expensive than alternate assays.72 It is non-quantitative; it provides average densities but not specific amounts72 • Relies on antibody staining optimization and requires very specialized instrumentation for the analysis |
| Single cell RNA sequencing | • Assess quantification and sequence of RNA using Next Generation Sequencing (NGS)73 Uses short reads of mRNA and reveal which genes are turned on73 Allows detection of novel transcripts and is quantifiable73 |
• Isolation of sufficient high quality RNA, low throughput73 RNA degrades rapidly. Subjected to amplification bias73 |
Table 2.
Table summarising the different organ systems and detailing the gene expression changes seen in each tissue. DSCR: down syndrome critical region of chromosome 21, RT-PCR: reverse transcriptase PCR and qRT-PCR: quantitative reverse transcriptase PCR.1.3
Table 2.
Table summarising the different organ systems and detailing the gene expression changes seen in each tissue. DSCR: down syndrome critical region of chromosome 21, RT-PCR: reverse transcriptase PCR and qRT-PCR: quantitative reverse transcriptase PCR.1.3
| Gene/miRNA | Chromosome position | Gene expression change | How this affects development | |
|---|---|---|---|---|
| Nervous system | NRSF/REST23 | 4q12 | Downregulated | Transcriptional repressor represses neuronal genes in non-neuronal tissues.40 |
| Ngn135 | 14 | Downregulated | Neuronal cell death | |
| Ngn235 | 4 | Downregulated | ||
| Pax635 | 11 | Downregulated | ||
| DNMT3A23 | 2q23 | Downregulated | DNA methylation in late stage of embryonic development | |
| DNMT3B23 | 20q11.2 | Downregulated | DNA methylation in broader range of genes in early embryonic development.8 | |
| PCDHG23 | 5q31 | Downregulated | Reduction in dendrite arborization and growth in cortical neurons | |
| M4338 | Downregulated | Regulation of action potential and axon ensheathment, neocortex and hippocampus over development | ||
| TBX135 | HSA22q11 | Downregulated | Fetal brain development and postnatal psychiatric phenotypes in DS | |
| Hsa-miR-138 39 | 16q13 | Upregulated | Hippocampus development | |
| hsa-miR-40939 | 14 | Upregulated | ||
| hsa-miR-138 -5p39 | 3 and 13 | Upregulated | Intellectual disability | |
| miR-125b-2 42 | 21 | Upregulated | Cognitive impairment, promotes neuronal differentiation | |
| mir-197349 | 21 | Upregulated | Regulating CNS and nervous systems | |
| mir-319649 | 20 | Upregulated | ||
| Olig135 | Critical region 21 | Upregulated | Microcephaly, cortical dyslamination, hippocampus malformation, profound motor deficits. Promotes enhancer regions of Nfact4, Dscr1/Rcan1 and Dyrk1a > DS phenotype. |
|
| Olig235 | Critical region 21 | Upregulated | ||
| S100B45 | DSCR | Upregulated | Activate the stress response kinase pathways and upregulated aquaporin 4. | |
| APP45 | DSCR | Upregulated | ||
| DYRK1A23 | 21qq22.13 | Upregulated | Reduces NRSF/REST | |
| DNMT3L23 | 21q22.4 | Upregulated | De novo methylation in neuroprogenitors, persist in foetal DS brain | |
| Cardiac | miR-99a-5p 49 | 21q21.1 | Downregulated | Congenital heart defects |
| miR-155-5p49 | 21 | Downregulated | Mitochondrial dysfunction | |
| Let-7c-5p 49 | 21q21.1 | Downregulated | ||
| GART 45 | 21 | Upregulated | De novo purine synthesis > intellectual disability, hypotonia, increased sensorineural deafness.74 | |
| EST2 47 | 21q22 | Upregulated | Most likely cause 2nd heart field development, AVSDs. | |
| Mmu16 48 | Tiam1-Kcnj6 region of 16 | Triplication | AVSDs | |
| Blood | SON 56 | 21 | Downregulated | Lower RBCs produced, brain and spinal malformations, reduced thrombocytes and myeloid cells, significant decrease in T cells. |
| STAT1 75 | 2q32.2 | Downregulated | Low = reduced Enhanced cellular response to IFN | |
| XIST 4 | Xq | Upregulated | X-chromosome inactivation in females, Induction corrected over-production of megakaryocytes and erythrocytes |
|
| RUNX1 54,55 | 21 | Hypermethylation | Differentiation of blood cells, B cells. | |
| S100a857 | 1q21 | Upregulated | Abundant in neutrophils/monocytes | |
| S100a957 | 1q21 | Upregulated | ||
| MPO57 | 17q12-24 | Upregulated | Creates reactive oxidant species, part of innate immune response and contributes to tissue damage during inflammation.70 | |
| Ly6c157 | 15 | Upregulated | Part of inflammatory response in atherosclerosis, regulates endothelial adhesion of CD8 T cells.71 | |
| IFN-αR1 40,,75 | 21 | Upregulated | Expressed on surface of monocytes, EBV-transformed B-cells. Immunodeficiency. | |
| IFN-αR240,,75 | 21 | Upregulated | ||
| IFN-γR2 75 | 12 | Upregulated | ||
| ERG57 | 21 | Triplication | Self-renewal of haematopoietic stem cells and haematopoiesis in liver during embryogenesis Dysregulation of homeostatic proportion of population of immune cells in embryonic brain and decreased prenatal cortical neurogenesis |
|
| SOX278 | 3q26.33 | Downregulated | Reduction in airway smooth muscle discontinuous in proximal airway | |
| Lung | DYRK1A78 | DSCR | Upregulated | Reduced incidence of solid tumours (neuroblastoma) and defects in angiogenesis of central arteries developing in hindbrain |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



