
Submitted:
28 August 2023
Posted:
29 August 2023
You are already at the latest version
Abstract
Interactions between proteins and ions are essential for various biological functions like structural stability, metabolism, and signal transport. Given that more than half of all proteins bind to ions, it becomes crucial to identify ion-binding sites. Accurate identification of protein-ion binding sites helps us to understand proteins’ biological functions and plays a significant role in drug discovery. While several computational approaches have been proposed, this remains a challenging problem due to the small size and high versatility of metals and acid radicals. In this study, we propose IonPred, a sequence-based approach that employs ELECTRA (Efficiently Learning an Encoder that Classifies Token Replacements Accurately) to predict ion binding sites using only raw protein sequences. We successfully fine-tuned our pretrained model to predict the binding sites for nine metal ions (Zn2+, Cu2+, Fe2+, Fe3+, Ca2+, Mg2+, Mn2+, Na+, and K+) and four acid radical ion ligands (CO32−, SO42−, PO43−, NO2−). IonPred surpassed six current state-of-the-art tools by over 44.65% and 28.46% respectively in F1 score and MCC when compared on an independent test dataset. Our method is more computationally efficient than existing tools producing prediction results for a hundred sequences for a specific ion in under ten minutes.
Keywords:
Deep Learning
; ELECTRA
; Ion binding site prediction
; Transformer
; Natural Language Processing
; Sequence-based prediction
1. Introduction
Many biological processes are facilitated by the interactions between proteins and ligand ions [1]. These interactions are necessary for the proteins to carry out their functions properly [2,3]. More than fifty percent of proteins, when observed, interact with metal ions (cations) and acid radicals to stabilize their structure, and regulate their biological functions [4,5]. Fe3+ binding to hemoglobin is critical for transporting oxygen through the blood [6]. Ca2+ intracellular signaling triggers T cell activation, development of B cells response to antigen, differentiation, and development [7,8]. Zn2+ maintains the stability of the protein’s tertiary structure and is also essential for over 300 enzyme activities [9] – lack or excess may cause central nervous system diseases [10]. The interaction of proteins with phosphate ions (PO43−) can result in phosphorylation which switches enzymes on and off thereby altering their function and activity [11]. Sulfate ions (SO42−) play a variety of structural roles as well as bind to a variety of cytokines, growth factors, cell surface receptors, adhesion molecules, enzymes, and fibrillary glycoproteins to carry out various essential biological functions [12]. From these examples, we see that ions play significant roles in a wide range of cellular processes. Hence, accurate identification of the protein–ion-binding sites is important for understanding the mechanism of protein function and new drug discovery.
To understand the mechanism of protein-ion interactions, biological experiments such as Nuclear Magnetic Resonance (NMR) spectroscopy [13] and fluorescence [14] methods are carried out to measure structure information of protein-ligand complexes, target ligand-binding proteins, and corresponding binding sites. As this is a very tedious and time-consuming process, computational methods to identify protein-ion binding sites are essential. The various computational methods proposed for predicting protein-ion binding sites can be grouped into sequence-based [15,16] and structure-based methods [17,18]. IonCom [19] proposed a new ligand-specific approach to predict the binding sites of nine metal ions (Zn2+, Cu2+, Fe2+, Fe3+, Ca2+, Mg2+, Mn2+, Na+ and K+ and four acid radical ion ligands (CO2−,PO3−,NO2−)using a sequence-based ab initio model that was first trained on sequence profiles, then extended by a modified AdaBoost algorithm to balance binding and non-binding residue samples. Sobolev and Edelman predicted the binding sites of protein chains and transition metal ions by implementing the ‘CHED’ algorithm; obtaining a specificity of 96% when predicting 349 whole proteins, 95% specificity was obtained [20]. Lu et al. used the fragment transformation method to predict metal ions (Ca2+, Mg2+, Cu2+, Fe2+, Mn2+, Zn2+) ligand binding sites, and obtained an overall accuracy of 94.6% and a sensitivity of 60.5 % [21]. Hu et al. identified four metal ions in the BioLip database [23] by implementing both sequence-based and template-based methods and obtained Matthew’s correlation coefficient (MCC) greater than 0.5 [22]. Cao et al. used the SVM algorithm to identify ten metal ion binding sites based on amino acid sequences, which obtained a good result by 5-fold cross-validation [24]. Greenside et al. used an interpretable confidence-rated boosting algorithm to predict protein-ligand interactions with high accuracy from ligand chemical substructures and protein 1D sequence motifs, which produced decent results [25].
A major drawback of some of the existing computational tools is that they involve complex 3D computations, threading of protein sequences to potential structural templates, and integrating multiple data types from both sequences and structures that are computationally intensive and time-consuming. In addition, several sequence-based tools have limited predictive performance (i.e., low precision) since they do not include tertiary structure information.
In this work, we propose IonPred, a Deep Learning framework based on ELECTRA [26] for predicting ion binding in proteins. The model adopts a sequence-based method for predicting the binding sites of nine metal ions and four acidic radicals. It takes raw protein sequences from all the protein chains with at least one binding site as input for the model. IonPred is based on the Transformer architecture which adopts a two-stage pre-training and fine-tuning process. In the initial pretraining phase, it employs the replacement token detection technique to learn contextual representations within the protein sequences from unlabeled protein sequence fragments. In contrast, in the fine-tuning phase, the model is trained with labeled sequence fragments to perform various binary classification tasks for various types of ion binding sites.
2. Results
To benchmark the performance of our method, we compared its predictive performance with existing tools and selected the Zinc dataset as a case study to understand how its performance is affected by different model configurations.
2.1. Comparison with other tools
We compared IonPred with six state-of-the-art tools. Three of these are sequence-based tools (i.e., TargetS [27], ZinCaps [28], and LMetalSite [29]) while the other three are structure-based (i.e., MIB [30], IonCom and DELIA [31]) predictors. ZinCaps only supports the prediction of Zn2+ while DELIA does not. We also compared the performance of our tool for predicting the binding sites of non-metal ions with IonCom. For the metals, as reported in Table 1, alkali metals (Na+ and K+) are the hardest to differentiate according to their low-performance scores followed by the alkali earth metals (Ca2+, Mg2+). This could probably be due to the wide variability of ion binding in these ion categories even among the homologous proteins and subtle differences in their binding affinities across various amino acid residues.
Except for Zn2+, where LMetalSite surpassed IonPred by 1.8% and 26.67% in the F1 and MCC, respectively, IonPred significantly outperformed all the sequence and structure-based tools. In most of the ion categories. The performance of LMetalSite is comparable to that of IonPred as both tools are based on similar architecture (i.e., pretrained language models) and it shows the sequence representation obtained from pretrained language models is both more insightful and more robust than the evolutionary and structural information incorporated by other tools.
For the non-metal ion category as seen in Table 2, IonPred outperforms IonCom in all metrics for all the acid radical by 50% – 117% for recall, 8.03% - 38.07% for precision, 44.65% – 67.03% in F1 score, and 28.46% - 67.13% in MCC.
We also plotted the ROC curves for the metal ions to further illustrate the superior performance of our method. As seen in Figure 1, except for Zn2+, the ROC curves for IonPred are all located at the upper portion of the plots to show more coverage and a higher AUC score. This indicates that IonPred has a greater capability to distinguish between positive and negative classes.
IonPred is way more computationally efficient than other tools, as it takes about 5 minutes to generate prediction results for 50 - 100 protein sequences of various lengths. It takes about 8 minutes to predict the same number of sequences with ZinCaps. It takes about 3 minutes to obtain the prediction results on just one protein sequence with TargetS, whereas it takes several hours to get one prediction result on just one protein sequence on IonCom and MIB.
2.2. Ablation tests
To understand the efficiency of IonPred with different configurations, we used Zn2+ dataset as a case study. This is because of its abundance in nature and the availability of quality datasets available for this ion. We evaluated the effect of the number of pretraining steps and discriminator size on model performance. We pretrained and fine-tuned several ELECTRA models with various configurations for generator discriminator sizes namely ELECTRA-0.25G-100K (the generator is 25% the size of the discriminator with 100,000 training steps), ELECTRA-0.25G-200K (the generator is 25% the size of the discriminator with 200,000 training steps), IonPred-0.25G-1M (the generator is 25% the size of the discriminator with 1 million pretraining steps), ELECTRA-0.5G-200K (the generator is 50% the size of the discriminator with 100,000 training steps) and ELECTRA-no-pretraining. We report their performance on the validation dataset for Zinc in Table 3.
From the results, we see that of all the three models created with 200K training steps, ELECTRA-0.25G-200K has a higher performance. This indicates that a generator size of 25% gives optimal performance. Then for all the configurations with a generator size of 0.25, we see that IonPred-0.25G-1M provides better and overall superior performance. This indicates that a higher number of training steps gives better performance. While ELECTRA-0.25G-100K has the same generator size as IonPred-0.25G-1M, it reports lower metric scores due to a lack of convergence of the model during pretraining. The model ELECTRA-no-pretraining, which is created without pretraining, reports the lowest performance for both AUC and AUPR.
2.3. Tool
The pretrained ELECTRA model for ion binding site prediction is provided as an open-source command-line tool available at https://github.com/clemEssien/IonPred. It takes a Fasta file containing one or more protein sequences. The instructions for use have been properly documented and the test datasets used are made available in the code repository. The output of the tool is a text file that contains the probability scores for each candidate site of the specified ion. The residues whose probability scores are higher than 0.5 are considered binding sites. IonPred was trained on a GPU, and it needs a GPU to run the prediction. The development environment requirements are Python 3, TensorFlow-GPU 1.15. CUDA 10, NumPy, Pandas, scikit-learn, and SciPy. The default batch size for running predictions is 128.
3. Discussion
In this work, we present IonPred, a pretrained ELECTRA model for predicting some of the most frequently seen ion binding sites that have significant impact on protein structures and functions. Our method uses raw sequence-based prediction because many proteins have no known structures or reliably predicted structures. The model is pretrained on a large corpus of unlabeled protein fragments in an unsupervised method and fine-tuned on a smaller quantity of non-redundant semi-manually curated labeled datasets. The model provides better predictive performance on alkali and alkali earth metal ions, which are typically difficult to predict. This is because the self-attention mechanism is adept at understanding the structural contexts of amino acid residues within protein sequences. This mechanism excels at assimilating conserved protein information by inherently focusing on neighboring residues and utilizes the transformer architecture to discern long-range dependencies.
However, there’s room for improvement for both metal categories. The attention mechanism of IonPred learns from the imbalanced dataset and provides improvement in the recall. We compared different ELECTRA configurations of training steps and generator sizes before we settled on the best configuration. IonPred significantly outperforms existing sequence and structure-based tools in all ion categories except Zinc, where LMetalSite slightly outperforms. Here, we see that directly fine-tuning the pre-trained model on each specific binding site gives a better performance than just using it for feature extraction, as was demonstrated in LMetalSite.
The performance of the fine-tuning stage is mainly dependent on the availability of large high quality labeled datasets. For ion-binding sites that have limited labels, the performance would not be as good. For future work, meta-learning could be explored as this could speed up the adaptation of binding sites with very limited labels. Also, the use of large protein information like ESM [32] or Sequence profile and predicted structures from alpha fold [33] could also be incorporated to improve context-dependent biological properties learned by the Discriminator with the purpose of significantly improving the recall.
4. Materials and Methods
4.1. Data and data processing
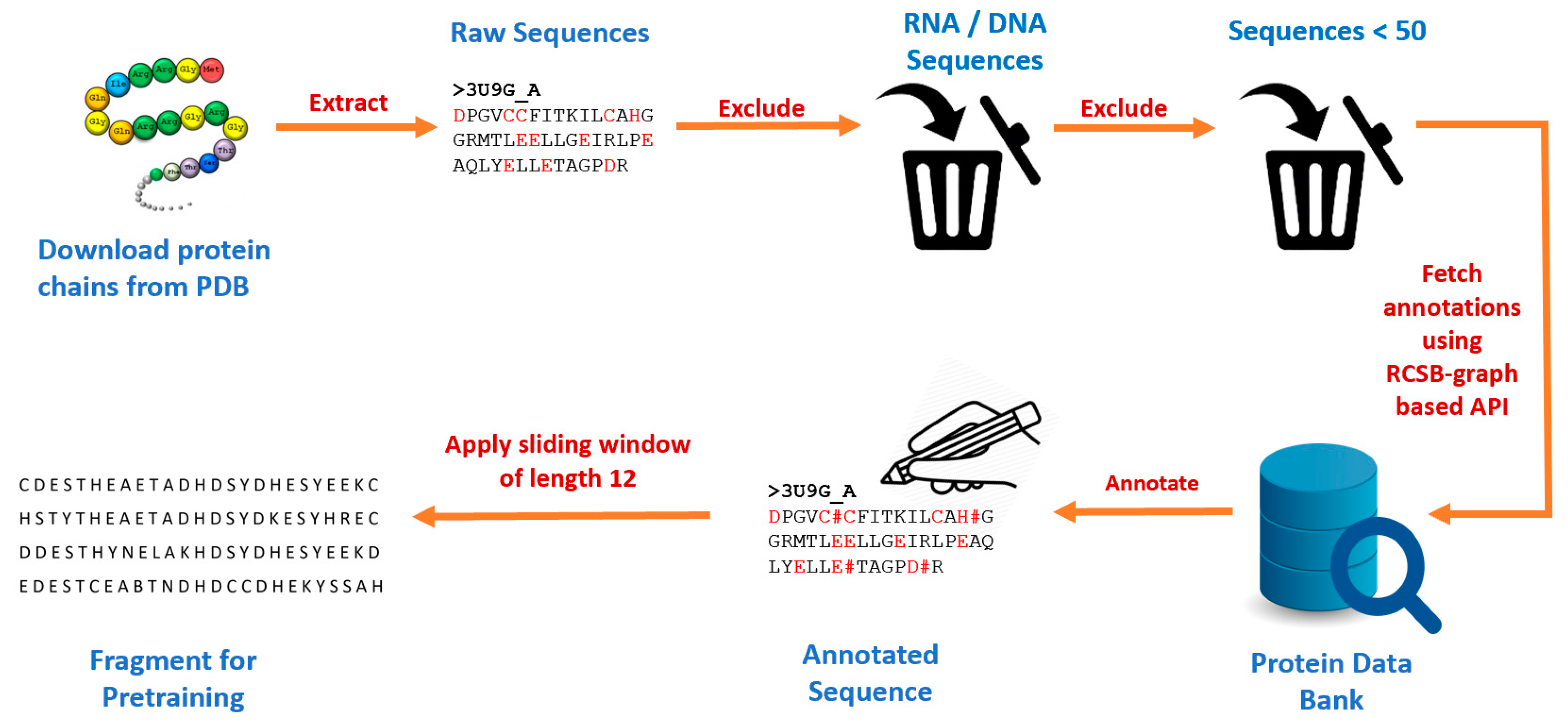
This study developed a new pretraining dataset by first downloading all protein chains from RCSB Protein Data Bank [34] using Biopython [35]. A total of 521,419 chains with their corresponding protein sequences were obtained. We excluded RNA and DNA components and protein chains that had less than 50 amino acid residues. We then made a series of API calls to the RCSB graph-based API [36], passing each protein chain ID as a parameter and then parsing the output to obtain annotations for the ligand binding sites. We obtained a total of 515,957 chains with at least one binding site residue and 27,626 unique ligand binding sites. We identified various categories of binding sites such as anions, cations, organic compounds, etc. Then we used the sliding window technique to extract fragments of length 25 (i.e., 12 amino acid residues on each side of the candidate binding residue).
Figure 2.
Data preprocessing pipeline for generating protein fragments used for pretraining.
4.2. Candidate residue selection
Almost all the amino acid residues are potential binding sites to varying degrees. A few of them participate more frequently in ion-binding than others. Some of these residues are regular candidates for specific ions. To determine the candidate binding residues to inform our decision on extracting fragments for pretraining, we use a binary heatmap to plot the distribution of each amino acid residue with respect to the number of ion binding sites, as shown in Figure 3. The represents the twenty amino acid residues, while the represents the ion ligands.
The plot is a frequency distribution of amino acid residues in relation to the number of ion binding sites. From the Figure, we determined that a residue is a binding site if its frequency is greater than or equal to the mean of the total residues in each row for a particular binding site. For the metal ions in Figure 3a, every amino acid residue is a candidate residue, but we observe the highest representation of candidate residues at Aspartate (D), Glutamate (E), and Histidine (H) followed by Leucine (L) and Cysteine (C). For the acid radical ions, we observe the highest representation of candidate residues at Glycine (G), Histidine (H), Lysine (K), Arginine (R), and Serine (S). Using the sliding window technique, we extract protein fragments of length 25 (i.e., 12 amino acid residues to the left and right of each candidate residue of interest) as implemented in [30] around the following amino acid residues: CYS (C), ASP (D), GLU (E), GLY (G), HIS (H), LYS (K), ARG (R) and SER (S) at the center. If the amino acid residue at the center is an ion-binding site, the whole fragment is considered a positive sample; otherwise, it is regarded as a negative sample. We excluded negative fragments that contained a binding residue.
A total of 2,284,717 fragments were generated of which 83,526 were positive. The complete process for obtaining the protein sequences used, annotation, and input fragment generation for pretraining is summarized in Figure 1. For the second stage, where we needed labeled data for finetuning, we obtained the labeled data of nine metal ions and four acid radicals from IonCom. This consists of a non-redundant set of ion-binding proteins downloaded from the BioLip database, which have a pairwise sequence identity of less than 30%, all having a sequence length of at least 50 amino acid residues and a resolution less than 3Å. This choice was made because not all ligands in the Protein Data Bank have biological significance. CD-HIT [37] tool was used to split the fine-tuning dataset into training, test and validation sets using a 40% similarity threshold. The distribution of proteins used for fine-tuning is displayed in Table 4.
4.3. Problem definition
The ion binding site prediction in this study is formulated as a binary classification problem. For example, given a protein sequence for which the binding sites are unknown, we select a particular ion (i.e., Zn2+, Cu2+, Fe2+, Fe3+, etc.) for which we want to determine binding sites. Then the aim would be to ascertain if the candidate binding residues (from Figure 3.) for the selected ion(s) are binding site(s) or not. This will output probabilities for each candidate residue. A probability of 0.5 and above is considered a positive prediction (i.e., an ion binding site), while a probability less than 0.5 is regarded as a negative prediction (i.e., not a binding site).
4.4. Deep Learning model
The architecture of the proposed IonPred as shown in Figure 4 is based on ELECTRA (i.e., “Efficiently Learning an Encoder that Classifies Token Replacements Accurately”) learning model. This architecture comprises two neural networks, a Generator, and a Discriminator. These networks basically map a sequence of input tokens into a sequence of contextualized vector representations
So, for any given position t, where xt is a masked amino acid residue [MASK], the generator uses a SoftMax layer to produce the probability of generating a particular token xt.
In the equation above, denotes the embeddings for the amino acid residues. The generator is trained using masked language modeling (MLM). For a given input, , MLM selects a random set of positions ranging from 1 to n to mask out. This produces the vector . The residues in these positions are replaced with a [MASK] token, which is represented as = . The generator learns to predict the original amino acid residues. The discriminator predicts whether the amino acid residue is originally from the input data or if it is a replacement from the generator distribution using a sigmoid output layer, as shown in the equation below:
The masked-out residues have been replaced by samples from the generator. This sample is represented as .The discriminator is trained to predict which residues in match the original input. The model inputs are described as shown below:
And the loss functions used for the generator and discriminator are shown in Equations 5 and 6 below:
The minimized combined loss for both the Generator and Discriminator is given as:
4.5. Pretraining
As shown in Figure 5, the pretraining consists of the Generator and Discriminator, which are essentially two transformer models. Here, the Generator corrupts a percentage of the tokens (i.e., amino acid residues) from the input fragments, and the Discriminator is trained to detect the replaced tokens. This enables the model to learn context-dependent biological properties of protein sequence fragments from a large-scale task-independent and unlabeled protein dataset. The patterns learned during this stage are then embedded into a smaller task-specific and labeled dataset in the downstream tasks, i.e., binary classification prediction for various protein-ion binding sites. This significantly reduces the amount of labeled data needed since the pretrained model has already learned the underlying patterns related to classification. We selected the ELECTRA-small model, which comprises 12 layers, 256 hidden layers, and 128-dimension embedding.
This model was chosen due to the relatively small size of our pretraining corpus and the fact that a larger size model would be computationally expensive to train, which may not lead to any significant improvement. The vocabulary size used is 25, which includes all 20 amino acid residues, the ‘-’ character to pad positions at the protein terminus, [MASK] as the masking character, [CLS] to mark the start of a fragment, [SEP] to mark the end of a fragment, and [UNK] for out of vocabulary words i.e., unknown amino acid residues. We mask 15% of each input fragment in the embedding layer which is then encoded into the token embeddings matrix having a dimension of [27,128]. Both the token and position embeddings are summed and presented as input tokens i.e. x = [x1, . . .., x27] into the Generator. The Generator used is 25% the size of the Discriminator [32] with 12 layers and a hidden size of 64. The Generator trains using maximum likelihood to predict the original masked-out amino acid residues based on the contextual information from neighboring amino acid residues in the protein fragment. The model was trained over 1 million training steps using a batch size of 128 and a learning rate of 0.0001.
4.6. Fine-tuning
After pretraining, the Generator is discarded, and the Discriminator is then fine- tuned using labeled data for various specific classification tasks. For this, a fully connected layer was built over the pre-trained ELECTRA model and the entire network was fine-tuned with 12 layers of the discriminator. This was done to ensure the error was backpropagated throughout the whole architecture and that the weights of the discriminator were updated based on the fragments in the fine-tuned dataset. We fine-tuned separate models for each ligand-ion binding site using labeled fragments generated from the protein sequence described in Table 1. The candidate binding residues used for the metals are C, H, E, and D, while the ones used for acidic radicals are G, H, K, R, and S. The training and testing and dev fragments were split by a ratio of 80%, 10%, and 10% respectively. We added a fully connected layer at the end of the pre-trained ELECTRA model and fine-tuned the entire network consisting of 12 layers of the discriminator so that the error was backpropagated across the entire architecture and the discriminator weights were updated using the labeled data as shown in Figure 6. Similar hyperparameters used in pretraining were implemented at this stage except for the learning rate and the number of training steps, which were set at 0.00001 and 200 epochs, respectively. Fine-tuning runs much quicker than pretraining.
4.7. Model Assessment
We evaluated IonPred using the following metrics: Recall, Precision, F1 score, and Matthew’s correlation coefficient (MCC) which are defined below:
where TP represents the number of binding residues correctly predicted as binding residues, TN is the number of non-binding residues that are correctly predicted as non-binding residues, FP is the number of non-binding residues that are incorrectly predicted as binding residues, and FN represents the number of binding residues incorrectly predicted as non-binding residues. We also reported the AUC score and AUPR score. These results are reported in Table 1 and Table 2.
Author Contributions
Conceptualization, D.X., D.W. and C.E.; methodology, C.E. and L.J.; software, C.E.; validation, C.E., L.J. and D.X.; formal analysis, C.E.; data curation, C.E.; writing—original draft preparation, C.E.; writing—review and editing, D.X.; supervision, D.X.; funding acquisition, D.X. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the US National Institutes of Health grant R35-GM126985. In addition, this work used the high-performance computing infrastructure provided by Research Computing Support Services at the University of Missouri, as well as the Pacific Northwest National Laboratory (PNNL). We would like to thank Negin Manshour for technical assistance.
Data Availability Statement
All the source codes and data used for this project are available at https://github.com/clemEssien/IonPred.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular biology of the cell. Scand. J. Rheumatol. 2003, 32, 125–125. [Google Scholar]
- Gao, M.; Skolnick, J. The distribution of ligand-binding pockets around protein-protein interfaces suggests a general mechanism for pocket formation. Proc. Natl. Acad. Sci. USA 2012, 109, 3784–3789. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Skolnick, J. A comprehensive survey of small-molecule binding pockets in proteins. PLoS Comput. Biol. 2013, 9, e1003302. [Google Scholar] [CrossRef] [PubMed]
- Tainer, J.A.; Roberts, V.A.; Getzoff, E.D. Metal-binding sites in proteins. Curr. Opin. Biotechnol. 1991, 2, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.J.; Gray, H.B. Bio-inorganic chemistry. Curr. Opin. Chem. Biol. 1998, 2, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Hsia, C.C.W. Respiratory function of hemoglobin. N. Engl. J. Med. 1998, 338, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Fracchia, K.M.; Pai, C.; Walsh, C.M. Modulation of t cell metabolism and function through calcium signaling. Front. Immunol. 2013, 4, 324. [Google Scholar] [CrossRef]
- Baba, Y.; Kurosaki, T. Role of calcium signaling in b cell activation and biology. In B Cell Receptor Signaling; 2015; pp. 143–174.
- McCall, K.A.; Huang, C.-c.; Fierke, C.A. Function and mechanism of zinc metalloenzymes. J. Nutr. 2000, 130, 1437S–1446S. [Google Scholar] [CrossRef]
- Gower-Winter, S.D.; Levenson, C.W. Zinc in the central nervous system: from molecules to behavior. BioFactors 2012, 38, 186–193. [Google Scholar] [CrossRef]
- Wang, J.P.; Chuang, L.; Loziuk, P.L.; Chen, H.; Lin, Y.C.; Shi, R.; Qu, G.Z.; Muddiman, D.C.; Sederoff, R.R.; Chiang, V.L. Phosphorylation is an on/off switch for 5-hydroxyconiferaldehyde o-methyl-transferase activity in poplar monolignol biosynthesis. Proc. Natl. Acad. Sci. USA 2015, 112, 8481–8486. [Google Scholar] [CrossRef]
- Zhang, B.; Chi, L. Chondroitin sulfate/dermatan sulfate-protein interactions and their biological functions in human diseases: Implications and analytical tools. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Sletten, E. The binding of transition metal ions to dna oligonucleotides studied by nuclear magnetic resonance spectroscopy. In Cytotoxic, Mutagenic and Carcinogenic Potential of Heavy Metals Related to Human Environment; Springer: 1997; pp. 493–509.
- Yonezawa, M.; Doi, N.; Higashinakagawa, T.; Yanagawa, H. Dna display of biologically active proteins for in vitro protein selection. J. Biochem. 2004, 135, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Huang, J.Z.; Gao, X. Ligandrfs: random forest ensemble to identify ligand-binding residues from sequence information alone. BMC Bioinformatics 2014, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Hu, S.; Zhang, J.; Gao, X.; Li, J.; Xia, J.; Wang, B. A sequence-based dynamic ensemble learning system for protein ligand-binding site prediction. IEEE/ACM Trans. Comput. Biol. Bioinf. 2015, 13, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Yang, J.; Zhang, Y. Cofactor: an accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res. 2012, 40, W471–W477. [Google Scholar] [CrossRef]
- Yang, J.; Roy, A.; Zhang, Y. Protein–ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013, 29, 2588–2595. [Google Scholar] [CrossRef]
- Hu, X.; Dong, Q.; Yang, J.; Zhang, Y. Recognizing metal and acid radical ion-binding sites by integrating ab initio modeling with templatebased transferals. Bioinformatics 2016, 32, 3260–3269. [Google Scholar] [CrossRef]
- Sobolev, V.; Edelman, M. Web tools for predicting metal binding sites in proteins. Isr. J. Chem. 2013, 53, 166–172. [Google Scholar] [CrossRef]
- Lu, C.H.; Lin, Y.F.; Lin, J.J.; Yu, C.S. Prediction of metal ion–binding sites in proteins using the fragment transformation method. PLoS ONE 2012, 7, e39252. [Google Scholar] [CrossRef]
- Hu, X.; Wang, K.; Dong, Q. Protein ligand-specific binding residue predictions by an ensemble classifier. BMC Bioinformatics 2016, 17, 1–12. [Google Scholar] [CrossRef]
- Yang, J.; Roy, A.; Zhang, Y. Biolip: a semi-manually curated database for biologically relevant ligand–protein interactions. Nucleic Acids Res. 2012, 41, D1096–D1103. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Hu, X.; Zhang, X.; Gao, S.; Ding, C.; Feng, Y.; Bao, W. Identification of metal ion binding sites based on amino acid sequences. PLOS ONE 2017, 12, e0183756. [Google Scholar] [CrossRef] [PubMed]
- Greenside, P.; Hillenmeyer, M.; Kundaje, A. Prediction of protein-ligand interactions from paired protein sequence motifs and ligand sub- structures. In PACIFIC SYMPOSIUM ON BIOCOMPUTING 2018: Proceedings of the Pacific Symposium; World Scientific: 2018; pp. 20–31.
- Clark, K.; Luong, M.T.; Le, Q.V.; Manning, C.D. ELECTRA: Pre-training Text Encoders as Discriminators Rather Than Generators. arXiv 2020, arXiv:2003.10555. [Google Scholar]
- Yu, D.J.; Hu, J.; Yang, J.; Shen, H.B.; Tang, J.; Yang, J.Y. Designing template-free predictor for targeting protein-ligand binding sites with classifier ensemble and spatial clustering. IEEE/ACM Trans. Comput. Biol. Bioinf. 2013, 10, 994–1008. [Google Scholar] [CrossRef] [PubMed]
- Essien, C.; Wang, D.; Xu, D. Capsule network for predicting zinc binding sites in metalloproteins. In 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM); IEEE: 2019; pp. 2337–2341.
- Yuan, Q.; Chen, S.; Wang, W. Prediction of ligand binding residues in protein sequences using machine learning. In 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM); IEEE: 2019; pp. 2298–2304.
- Lin, Y.-F.; Cheng, C.-W.; Shih, C.-S.; Hwang, J.-K.; Yu, C.-S.; Lu, C.-H. Mib: metal ion-binding site prediction and docking server. J. Chem. Inf. Model. 2016, 56, 2287–2291. [Google Scholar] [CrossRef]
- Xia, C.-Q.; Pan, X.; Shen, H.-B. Protein–ligand binding residue prediction enhancement through hybrid deep heterogeneous learning of sequence and structure data. Bioinformatics 2020, 36, 3018–3027. [Google Scholar] [CrossRef]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Costa, A.d.S.; Fazel-Zarandi, M.; Sercu, T.; Candido, S.; et al. Language models of protein sequences at the scale of evolution enable accurate structure prediction. bioRxiv 2022.
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Z´ıdek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with alphafold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: freely available python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Segura, J.; Rose, Y.; Westbrook, J.; Burley, S.K.; Duarte, J.M. Rcsb protein data bank 1d tools and services. Bioinformatics 2020, 36, 5526–5527. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Jaroszewski, L.; Godzik, A. Clustering of highly homologous sequences to reduce the size of large protein databases. Bioinformatics 2001, 17, 282–283. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Comparison of ROC curves of IonPred with existing tools for the metal ion predictions.
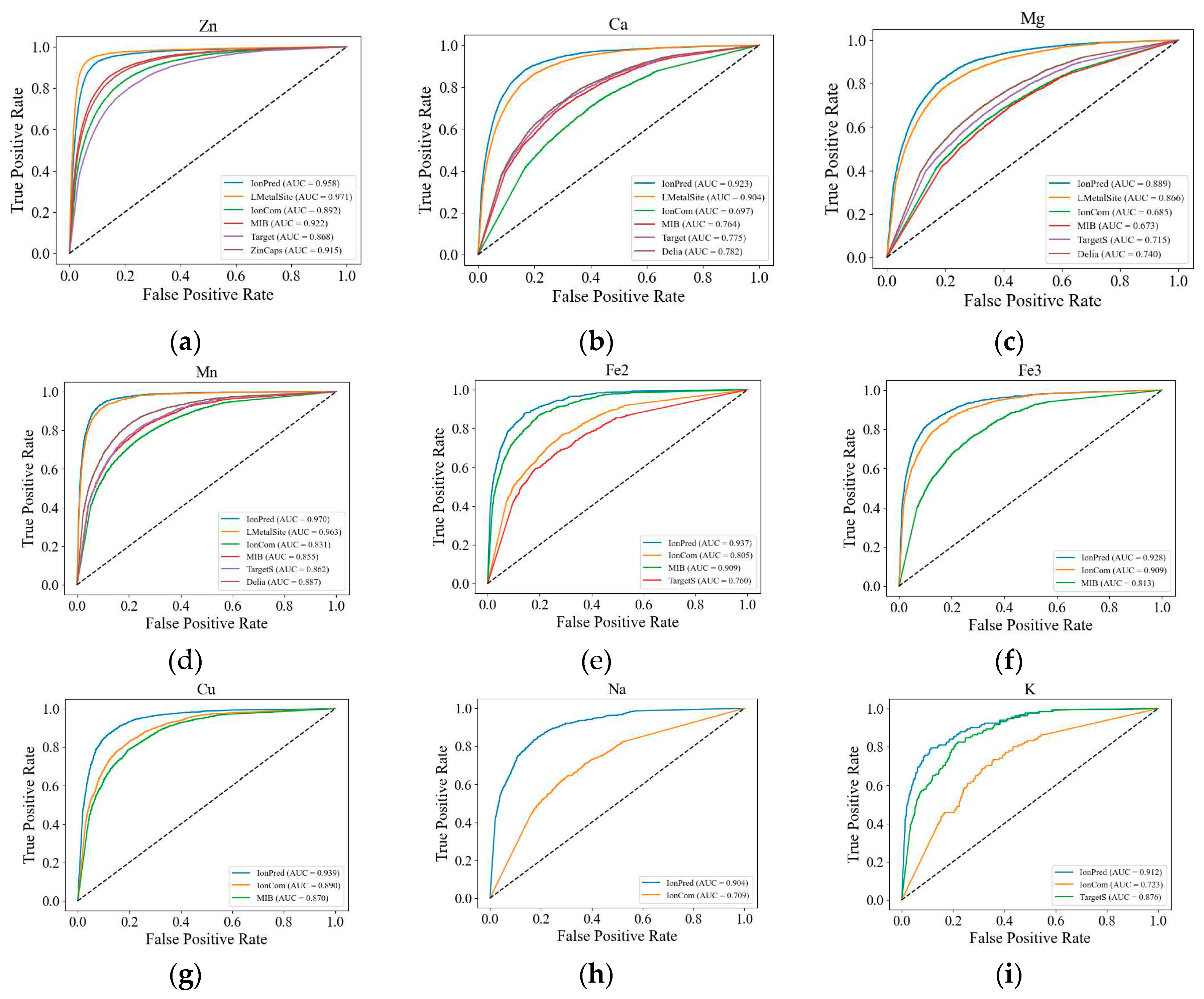
Figure 3.
Frequency distribution of amino acid residues with respect to ion ligands.
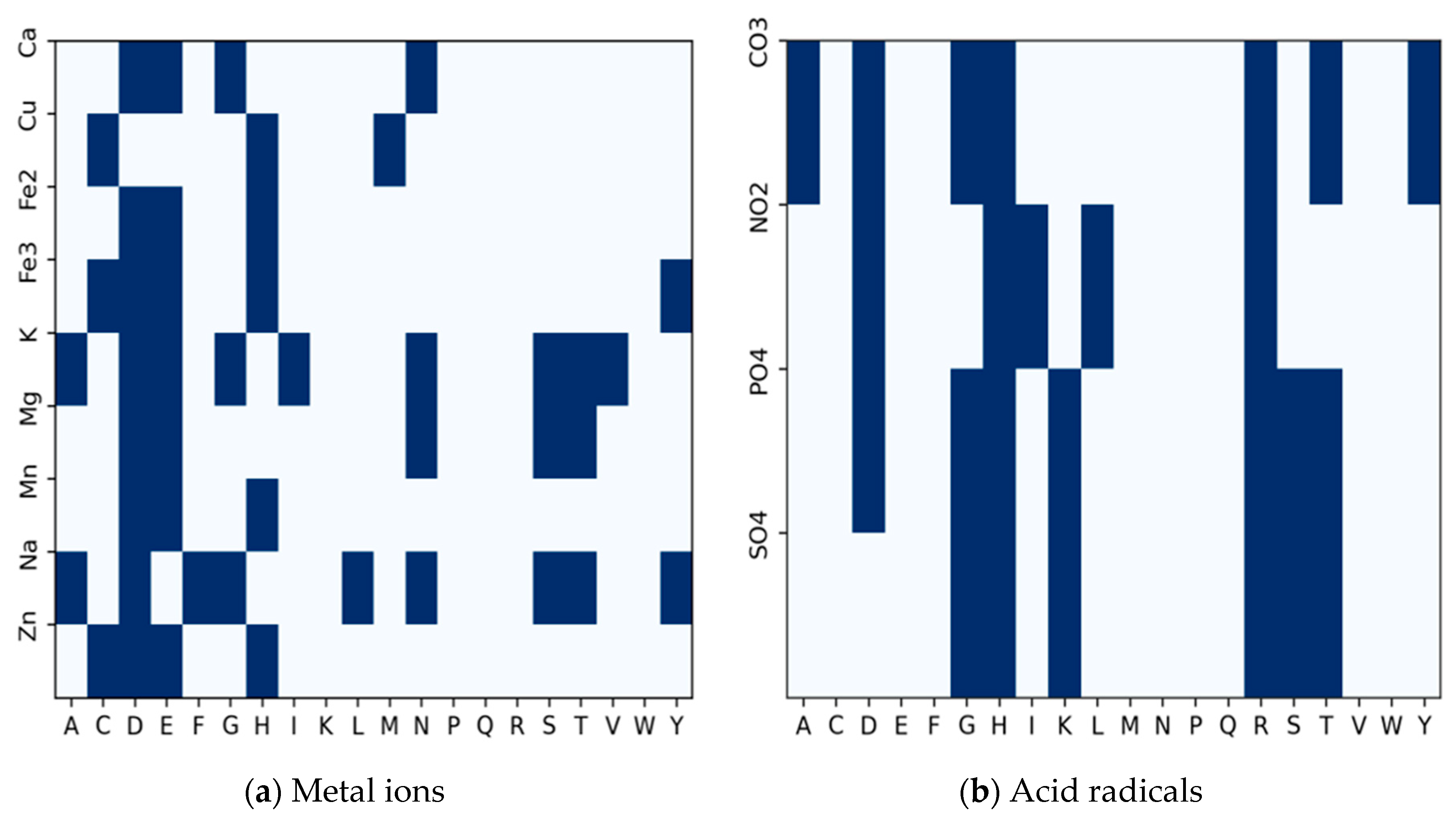
Figure 4.
Electra architecture which illustrates token corruption and replacement by Generator and Discriminator.
Figure 4.
Electra architecture which illustrates token corruption and replacement by Generator and Discriminator.
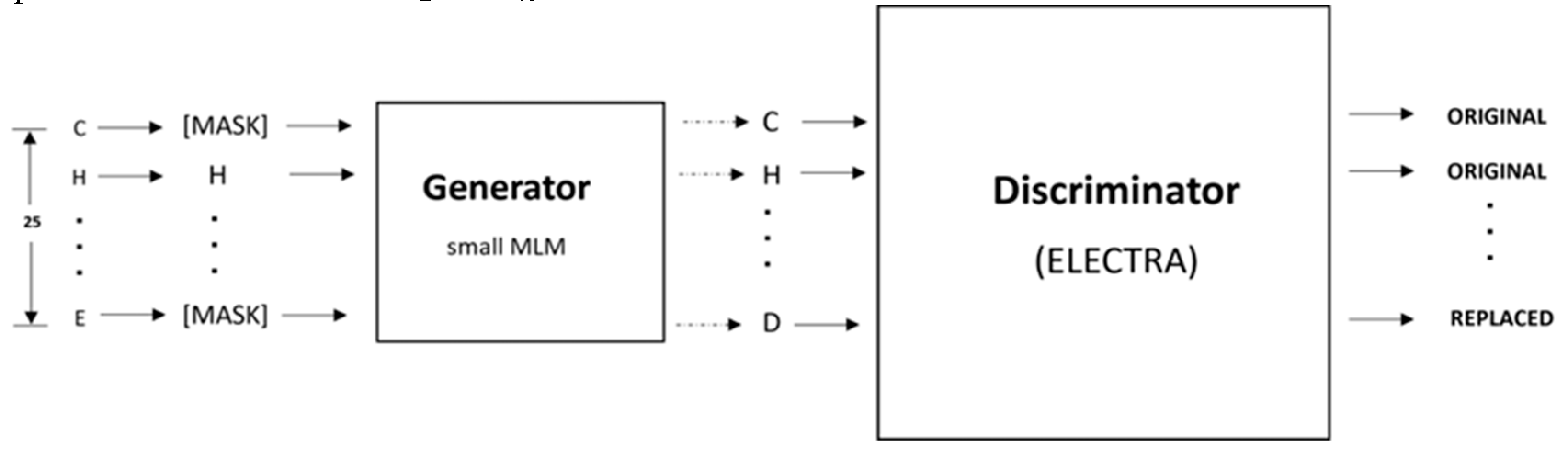
Figure 5.
Pretraining process taking in raw protein fragments as input.
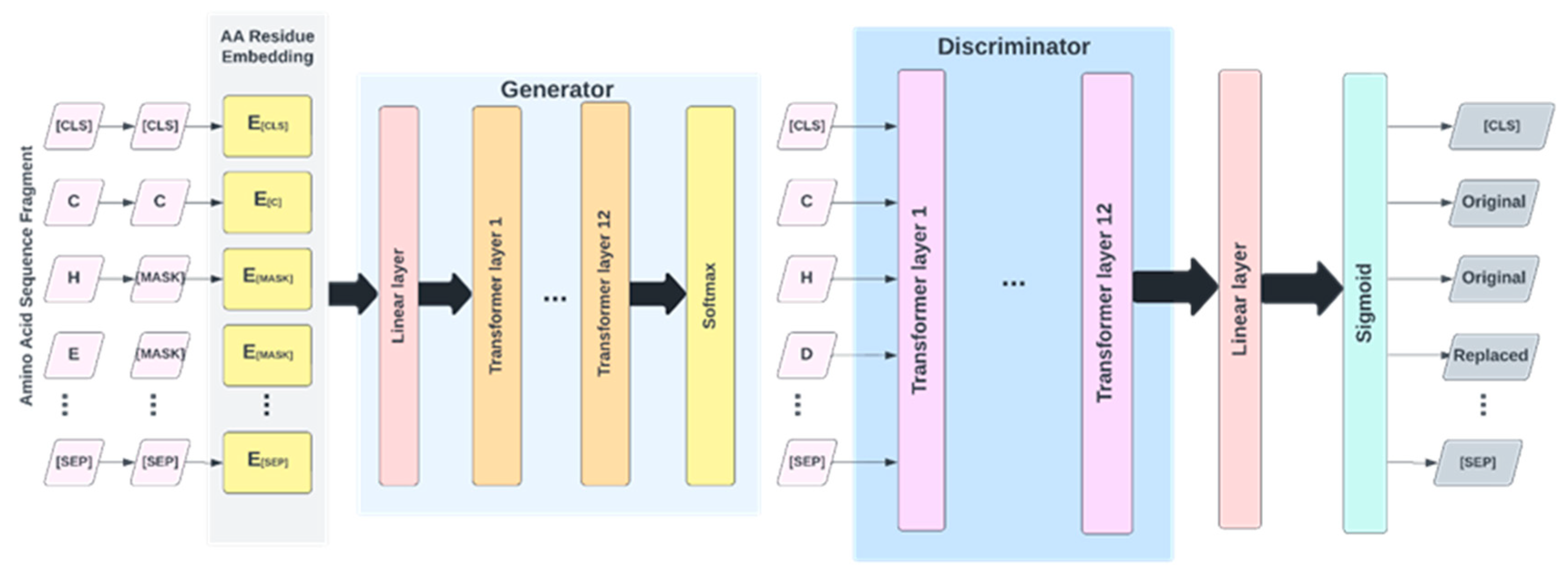
Figure 6.
Fine-tuning with labeled dataset to generate probabilities for binary classification.
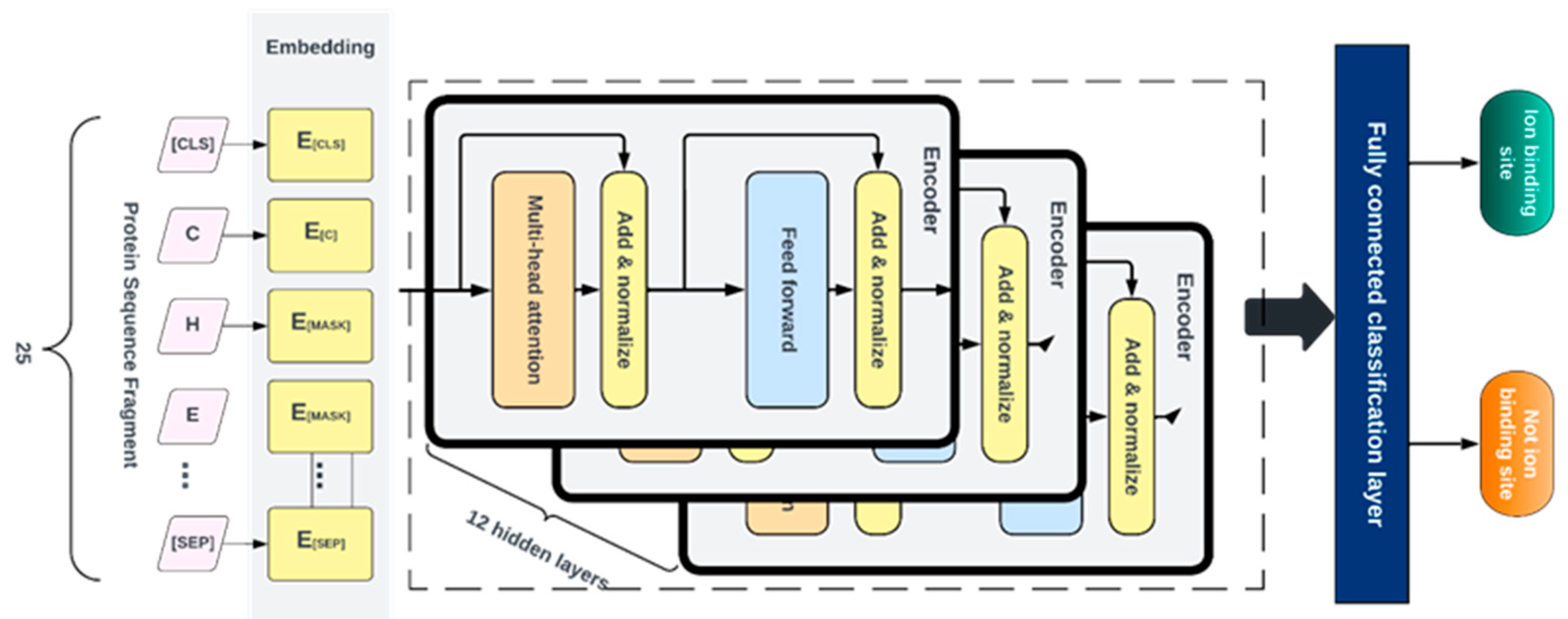

Table 1.
Performance comparison of IonPred with other tools on metal ions test sets.
| Ion | Method | Rec | Pre | F1 | MCC | AUC | AUPR |
|---|---|---|---|---|---|---|---|
| MIB | 0.739 | 0.220 | 0.339 | 0.389 | 0.922 | 0.388 | |
| TargetS | 0.450 | 0.750 | 0.563 | 0.578 | 0.868 | 0.594 | |
| Zn2+ | ZinCaps | 0.753 | 0.780 | 0.766 | 0.601 | 0.915 | 0.768 |
| IonCom | 0.779 | 0.137 | 0.233 | 0.317 | 0.892 | 0.671 | |
| LMetalSite | 0.722 | 0.859 | 0.785 | 0.760 | 0.971 | 0.801 | |
| IonPred | 0.790 | 0.840 | 0.814 | 0.600 | 0.958 | 0.780 | |
| MIB | 0.341 | 0.082 | 0.132 | 0.139 | 0.764 | 0.105 | |
| TargetS | 0.119 | 0.487 | 0.191 | 0.244 | 0.775 | 0.165 | |
| Ca2+ | DELIA | 0.172 | 0.630 | 0.270 | 0.330 | 0.782 | 0.251 |
| IonCom | 0.297 | 0.247 | 0.270 | 0.258 | 0.697 | 0.166 | |
| LMetalSite | 0.413 | 0.720 | 0.525 | 0.540 | 0.904 | 0.490 | |
| IonPred | 0.467 | 0.759 | 0.578 | 0.615 | 0.923 | 0.520 | |
| MIB | 0.246 | 0.043 | 0.073 | 0.082 | 0.673 | 0.053 | |
| TargetS | 0.118 | 0.491 | 0.190 | 0.237 | 0.715 | 0.148 | |
| Mg2+ | IonCom | 0.240 | 0.250 | 0.245 | 0.237 | 0.685 | 0.184 |
| DELIA | 0.129 | 0.065 | 0.086 | 0.287 | 0.740 | 0.198 | |
| LMetalSite | 0.245 | 0.728 | 0.367 | 0.419 | 0.866 | 0.316 | |
| IonPred | 0.400 | 0.780 | 0.529 | 0.470 | 0.889 | 0.450 | |
| MIB | 0.462 | 0.096 | 0.159 | 0.193 | 0.855 | 0.168 | |
| TargetS | 0.271 | 0.496 | 0.350 | 0.362 | 0.862 | 0.322 | |
| Mn2+ | DELIA | 0.502 | 0.665 | 0.572 | 0.574 | 0.887 | 0.489 |
| IonCom | 0.511 | 0.245 | 0.331 | 0.344 | 0.831 | 0.304 | |
| LMetalSite | 0.613 | 0.719 | 0.662 | 0.661 | 0.963 | 0.625 | |
| IonPred | 0.620 | 0.700 | 0.658 | 0.670 | 0.970 | 0.670 | |
| IonCom | 0.635 | 0.379 | 0.475 | 0.449 | 0.805 | 0.411 | |
| MIB | 0.586 | 0.620 | 0.603 | 0.573 | 0.909 | 0.354 | |
| Fe2+ | TargetS | 0.345 | 0.254 | 0.293 | 0.245 | 0.760 | 0.299 |
| IonPred | 0.749 | 0.728 | 0.738 | 0.723 | 0.937 | 0.771 | |
| IonCom | 0.610 | 0.498 | 0.548 | 0.579 | 0.909 | 0.567 | |
| Fe3+ | MIB | 0.474 | 0.399 | 0.433 | 0.383 | 0.813 | 0.438 |
| IonPred | 0.743 | 0.612 | 0.671 | 0.652 | 0.928 | 0.724 | |
| IonCom | 0.596 | 0.398 | 0.477 | 0.592 | 0.890 | 0.399 | |
| Cu2+ | MIB | 0.466 | 0.280 | 0.350 | 0.358 | 0.870 | 0.419 |
| IonPred | 0.789 | 0.634 | 0.703 | 0.620 | 0.939 | 0.677 | |
| IonCom | 0.210 | 0.178 | 0.193 | 0.160 | 0.723 | 0.156 | |
| K+ | TargetS | 0.389 | 0.411 | 0.400 | 0.341 | 0.876 | 0.336 |
| IonPred | 0.498 | 0.672 | 0.572 | 0.524 | 0.912 | 0.478 | |
| Na+ | IonCom | 0.451 | 0.292 | 0.355 | 0.218 | 0.709 | 0.233 |
| IonPred | 0.523 | 0.731 | 0.610 | 0.595 | 0.904 | 0.487 |
Table 2.
Performance comparison of IonPred with other tools on non-metal ions test sets.
| Radicals | Method | Rec | Pre | F1 | MCC | AUC | AUPR |
|---|---|---|---|---|---|---|---|
| CO32- | IonCom | 0.610 | 0.498 | 0.548 | 0.579 | 0.909 | 0.567 |
| IonPred | 0.743 | 0.612 | 0.671 | 0.652 | 0.928 | 0.724 | |
| NO2- | IonCom | 0.596 | 0.398 | 0.477 | 0.592 | 0.890 | 0.399 |
| IonPred | 0.789 | 0.634 | 0.703 | 0.620 | 0.939 | 0.677 | |
| SO43- | IonCom | 0.210 | 0.178 | 0.193 | 0.160 | 0.723 | 0.156 |
| IonPred | 0.389 | 0.411 | 0.400 | 0.341 | 0.876 | 0.336 | |
| PO43- | IonCom | 0.451 | 0.292 | 0.355 | 0.218 | 0.709 | 0.233 |
| IonPred | 0.523 | 0.731 | 0.610 | 0.595 | 0.904 | 0.487 |
Table 3.
Performance evaluation of several ELECTRA model configurations on Zinc dataset.
| Configuration | AUC | AUPR |
|---|---|---|
| ELECTRA -0.25G-100K | 0.916 | 0.698 |
| ELECTRA -0.25G-200K | 0.951 | 0.756 |
| IonPred-0.25G-1M | 0.958 | 0.780 |
| ELECTRA -0.5G-200K | 0.926 | 0.739 |
| ELECTRA-1.0G-200K | 0.904 | 0.676 |
| ELECTRA-no-pretraining | 0.857 | 0.519 |
Table 4.
Statistics of the residue distribution of each ion dataset used for fine-tuning.
| Category | Ion | Nprot | Rpos | Rneg |
|---|---|---|---|---|
| Ca2+ | 179 | 1,360 | 119,192 | |
| Cu2+ | 110 | 535 | 38,488 | |
| Fe2+ | 227 | 1,115 | 73,813 | |
| Fe3+ | 103 | 439 | 34,113 | |
| Metal ions | K+ | 53 | 536 | 18,776 |
| Mg2+ | 103 | 391 | 76,382 | |
| Mn2+ | 379 | 1,778 | 148,618 | |
| Na+ | 78 | 489 | 27,408 | |
| Zn2+ | 142 | 697 | 93,952 | |
| CO32− | 62 | 316 | 22,766 | |
| Acid radicals | NO2− | 22 | 98 | 8,144 |
| PO43− | 303 | 2,125 | 99,729 | |
| SO42− | 339 | 2,168 | 112,279 |
* Nprot represents the number of protein chains while Rpos and Rneg represent the number of binding residues and the number of non-binding residues respectively.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



