
Submitted:
28 August 2023
Posted:
29 August 2023
You are already at the latest version
Abstract
Non-systemic oral drugs, which do not get absorbed through the gastrointestinal membrane, are an important group of compounds for targeting diseases residing within the gastrointestinal lumen. However, they require a set of design principles not sufficiently covered by traditional druglikeness metrics, such as Lipinski’s Rule of 5. These druglikeness metrics fail to accurately identify true negative outcomes; such is the requirement for novel cheminformatic based approaches for the discovery of non-systemic small molecules. One such example of these newer metrics is the fraction of SP3-hybridised carbon atoms (fSP3) which has shown promising application as an identifier of poor oral-bioavailability. Herein, we discuss the application of fSP3 in the drug discovery process in a number of case studies, including our own work discovering non-systemic fructose scavengers for the treatment of fructose malabsorption.
Keywords:
fSP3
; drug design
; bioavailability
; non-systemic drugs
; druglikeness
; cheminformatics
1. Introduction
In the field of medicinal chemistry lead optimisation, there has been an increased focus upon improving the high attrition rates of candidate drugs, where about 90 percent of clinical trials result in failure [1]. Much of this attention has centred upon the concept of optimising Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) profiles of molecules. Particular attention has centred on the absorption aspect, thus designing drugs that possess a high degree of oral bioavailability has become a priority. This has led to the development of simple filters to identify compounds that satisfy this particular requirement. Starting with Lipinski’s seminal ‘Rule of 5’ criteria [2], a number of filters have been designed to identify compounds with desirable ADMET profiles. These filters include the Ghose, Veber, Mugge, and Egan filters [3,4,5,6]. These ‘rule of thumb’ approaches are facile to understand, and apply, and therefore enable medicinal chemists to readily incorporate them into in their drug design workflows. The art of medicinal chemistry is mutiparametric in nature and lead optimisation is a complicated business. This is because teams need to not only take account of potency, but factors such as drug stability, toxicity, patentability, level of binding to plasma proteins, and pharmacodynamics need to be assessed. Consequently, methods aimed at predicting drug properties like pharmacokinetic profiles are attractive to drug hunters.
Also, learning about the profile of compounds as early as possible can also help to save on research expenditure (because of the traditional high attrition rates seen in drug development). Estimates of pre-launch research and development costs range from several hundred to multibillion US dollar levels.
Lipinski postulated that in most cases, orally active compounds tend to violate no more than one of the following criteria: a molecular weight of less than or equal to 500 gmol-1, a lipophilicity (logP) of less than or equal to 5, less than or equal to 5 hydrogen-bond donators, and less than or equal to 10 hydrogen-bond acceptors [2]. It is interesting to note that these rules were originally framed as a way of selecting compounds that are more likely to be orally bioavailable, rather to solve any wider medicinal chemistry problems. This seminal research inspired further development of these criteria. These interations included the aforementioned Ghose filter, that constrained the molecular weight to being in-between 160 and 480 gmol-1, and included a criterion of molar refractivity between 40 and 130 [3]. Veber’s rules included a criterion for a topological polar surface area (TPSA) no greater than 140 Å2, and no more than 10 rotatable bonds [5]. Egan’s ‘boiled egg’ filter further constrained the logP and TPSA criteria to no greater than 5.88 and 131.6 Å2 respectively for GI absorption, as well as including an additional set of criteria for drugs that are required to permeate through the blood-brain barrier [6]. Mugge’s filter further restricted the criteria to a molecular weight of 200 gmol-1, a logP between 2 and 5, a TSPA no greater than 150 Å2, less than or equal to 15 rotatable bonds, and no greater than 4 rings [4].
These filters are a highly valuable tool in the toolkit of a medicinal chemist; however, many researchers working in drug discovery have come to over rely on such filters, almost treating them as set in stone. This has resulted in the reduced exploration of molecular space, as chemists are confined to the rules set forth by druglikeness filters. Although a number of chemists have begun to explore beyond rule of five chemical space [7], our understanding of this space is still relatively limited. It is also important to understand how overreliance of druglikeness filters may adversely affect the progress of drug development projects. The concept of molecular obesity has been proposed to illustrate a recent trend in the drug design process that artificially inflates the lipophilicity of compounds (in attempts to increase potency) [8]. Although this may have the desired effect of increasing drug potency, the increasing rate of drug attrition in clinical trials has partially been attributed to molecular obesity. In other words, the inappropriate use of lipophilicity may lead to the progress of potential drug candidates becoming stunted.
However, these approaches are only suited for compounds that need to be absorbed through the gastrointestinal (GI) tract. It is important to flip modern ways of thinking around the properties needed for orally bioavailable molecules when considering agents suitable for non-systemic circulation. Also, there are a number of pathological targets that reside within the intestinal lumen, and thus require drugs exhibiting a low likelihood of passing through the GI tract. These include bacteria such as Vibrio cholerae [9] which produce absorption and secretion altering toxins, as well as Shingella and E.coli [10,11] which result in the destruction of the colonic epithelium. Several different methodologies have been developed for the design of ‘non-systemic’ drugs; however, this class of compounds are typically characterised by high molecular weights, resulting in difficult, and expensive synthetic methodologies [12]. The ability to design small (molecular weight <500 Daltons) molecules with ‘non-systemic characteristics’ could facilitate the development of new drug classes suitable for non-systemic exposure. However, although standard druglikeness filters are effective at predicting druglikeness, they are not able to predict non-druglikeness with the same accuracy [13]. So, there is a need to development new filters that can evaluate non-systemic drugs, and the identification of novel chemical descriptors may enable this to become possible. The concept of developing drugs with high gut residence is a vital tool in the treatment of diseases located in the small intestine, such as Clostridium Difficile (C. Diff) infections [14,15,16,17], and in our research into the discovery of non-systemic, fructose-selective aryl boronic acid scavengers for the treatment of fructose malabsorption. Drugs can pass through the small intestine through different ways [18]. Most often, this is achieved via passive diffusion as described by Fick’s law. In the context of drug absorption through the small intestine, lipid diffusion is the most important type of diffusion in ascertaining a drug’s permeability, hence the importance of logP in determining a drug’s oral bioavailability. Alternatively, drugs can be absorbed via the use of carrier-mediated membrane transporters that utilise both active transport and facilitated diffusion. Active transport has the benefit of being able to transport drugs from areas of low concentration to areas of higher concentration, unlike passive diffusion. However, this process requires energy, unlike passive diffusion. Actively transported drugs are required to form a complex with the carrier molecule. One example of facilitated diffusion is shown by the behaviour of the antidiabetic drug Metformin, which forms a complex with organic cation transporter 1 (OCT1) before passage through the small intestine.
Lovering’s seminal paper introduced the chemical descriptor fraction of sp3 carbon atoms (fSP3), as shown by the formula in Figure 1. The concept of ‘flatness’ [19] has encouraged the design of more complex compounds that emulate natural products more than their counterparts that adhere solely towards traditional druglikeness filters, such as Lipinski’s Rule of Five [2]. A select number of compounds are present in the literature with low fSP3, resulting in poor oral bioavailability despite a low molecular weight. These compounds are ideal for the treatment of non-systemic targets. Lovering discovered that the average fSP3 for marketed drugs is 0.47, and 0.36 for hit compounds. The introduction of fSP3 was intended to design drugs with increased oral bioavailability, by taking into account drug design trends found in natural products. One of these trends is the incorporation of spirocycles. These compounds feature two molecular rings that share one atom between them, resulting in an increase in fSP3 relative to compounds bearing two rings and sharing two atoms between them. Spirocycles have the added benefits of potential chirality [20], resulting in increased affinity for protein binding and specificity [21]. A number of recent compound libraries have been developed that incorporate spirocyclic groups in order to increase fSP3 [22,23].
In this paper, we present the following new hypothesis; that fSP3 can be applied as an indicator for drugs with poor bioavailability. This may lead to the development of new chemical motifs for the design of non-systemic small molecules that are localised in the gut. The introduction of new metrics that are able to predict non-systemic exposure may lead to the development of new ADMET filters that can accurately identify non-systemic drugs, enabling medicinal chemists to develop suitable compound libraries. We look forward to the drug discovery community helping us to further validate this hypothesis.
Flaws in Lipinski’s Rule of Five and Quantifying Druglikeness
Lipinski’s seminal Rule of Five (Ro5) has provided medicinal chemists with a methodology to develop compounds with predicted high oral bioavailability [2]. However, this rule is not perfect, and despite Lipinski’s indications that his filter should be only used as a ‘rule of thumb’ approach, it is treated by many medicinal chemists as absolute. This results in an over restriction in chemical space by medicinal chemists in drug design when compared to the larger structural variety found in natural products, which may lead to potential therapeutic strategies being omitted in drug discovery [24]. No filter can be perfect at identifying orally bioavailable compounds, and Benet showed that despite only 2.8 percent of compounds with high solubility and permeability violating Ro5, 13.6 percent of compounds with low solubility and permeability violate Ro5 [13]. This indicates that although Ro5 is an effective indicator for compounds with high oral bioavailability, it performs less well as an indicator for non-systemic compounds, which suggests a need for new metrics for the discovery of non-systemic compounds. It is important to also recognise that as drug discovery incorporates more computationally-lead methodologies, such as de novo design [25] and the application of Machine Learning (ML) models, the need to move on from binary (pass or fail) filters to more quantitative methods for evaluating drugs-likeness are needed, such as Bickerton’s Quantitative Estimate of Drug-likeness (QED) [26]. Quantitative methods enable the comparison of druglikeness between different compounds and put them into greater context. This methodology is effective at quantifying oral bioavailability; however, the methodology is not as effective at identifying non-systemic compounds and is not as readily applicable as Ro5, and other more simple druglikeness filters. This is because the metrics used to identify orally bioavailable compounds are not as effective at identifying non-systemic compounds. Benet determined that 14.3 percent of highly permeable oral drugs violate Ro5, and 11.9 percent of highly soluble drug violate the filter, indicating there is scope to evolve the filter [13]. Fraction of sp3 carbon atoms has be used to develop new metrics, such as MCE-18 [27], shown below in Figure 2, which aims to identify novelty and lead potential in molecules. MCE-18 stands for Medicinal Chemistry Evolution, 2018,
and represents a useful tool.
MCE-18 aims to iterate upon the initial formula for fSP3 by considering a major weakness in fSP3, which is that fSP3 fails to identify out of plane carbon atoms. This results in the 3-dimensional shape of a molecule not always being described by fSP3 [24].
Design of non-systemic compounds
A number of approaches have been discovered for the design of non-systemic drugs, resulting from Chamot’s proposal for five classes of non-systemic drugs: I) sequestering agents; II) ligands of soluble intestinal enzymes; III) enzymes; IV) minimally absorbed and rapidly metabolised drugs; and V) ligands of apical targets [12]. Each of these classes present a different approach to the design of non-systemic drugs, by preventing absorption through the GI tract. Examples of classes I-II are shown in Figure 3.
Class I compounds, also referred to as sequestering agents, bind to small molecules within the lumen of the small intestine, resulting in the formation of an insoluble complex that passes through the GI tract, and eventually eliminated through faeces. These compounds are typically synthesised as crosslinked polymeric materials of varying morphologies. Contact with intestinal fluids results in the resin swelling, and transfer of endogenous ion from the GI lumen with counter ions from the resin. Compounds in this class include bile acid sequestrants, such as Cholestramine, Colestipol, and Colesevelam [28,29,30].
Class II compounds bind to host proteins in the intestinal lumen, resulting in the inhibiting of digestive enzymes due to blocking digestion of nutrients to easily absorbable molecules, reducing nutrient absorption. Example compounds in this class include the appetite suppressants Orlistat [31] and Sibutramine [32], both of which are lipase inhibitors, and the disaccharidase inhibitors Acarbose, and Miglitol [33,34]; these are also shown in Figure 3. Both Acarbose and Miglitol are used to regulate levels of glucose in the blood of patients affected by diabetes.
Class III compounds compensate for enzyme deficiencies in the host GI tract or metabolise bacterial and metabolic toxins. This class of compounds has been used in the administration of porcine pancreatic enzymes for the treatment of Cystic Fibrosis [35]. Although offering a useful approach for the treatment of some conditions, this class of compounds falls more into the realm of biologics, and thus approaches related to the design of small molecules are not as applicable for this class of compounds.
Class IV compounds target the inner wall of the GI tract. The parent drugs of this type are broken down quickly by first pass metabolism in the enterocytes of the liver, resulting in low concentration in the blood stream. These compounds have also been referred to as ‘soft-drugs’. Buchwald published a thorough review of this class of rapidly metabolised compounds [36], such as the β-blockers Esmolol and Landiolol [37,38]. It is important to note that these compounds may have a high degree of GI tract permeability; it is their rapid metabolism that leads to a low concentration of the drug found in blood.
Class V compounds target membrane proteins on the gut epithelia’s surface. These have a wide range of applications, including transporter inhibition, G-protein coupled-receptor binding, and tight junction permeability modulation. These classifications aim to provide detail for the various types of targets that exist prior to intestinal absorption. However, the classification system does not indicate how the compounds achieve non-druglikeness. There is a need for the creation of classes of small molecular scaffolds with poor druglikeness in order to investigate non-systemic targets and distribution.
A potential application of non-systemic compounds is the treatment of bacterial infections residing primarily within the lumen of the small intestine. Compounds in this category must be taken orally, however, excessively high oral-bioavailability, and subsequent absorption into the bloodstream, will hinder the therapeutic effect of these compounds. One such example of this kind of infection is caused by the bacteria C. Diff [14], often occurring because of the changes in intestinal bowel microbiota caused by antibiotic use [15]. Current treatment of C .Diff is carried out using very large, polar molecules such as Vancomycin (molecular weight = 1447.30 gmol-1; Topological Polar Surface Area (TPSA) = 530.490 Å), and Fidaxomicin (molecular weight = 1056.425 gmol-1; TPSA = 266.660 Å) [17,42]. However, recent research into a new, smaller compound, Ridinilazole, [16] has shown promise in initial clinical trials for the treatment of C. Diff. Paradoxically, this drug (shown in Figure 4) is not absorbed through the small intestine despite passing all traditional druglikeness filters. Summit Therapeutics developed this compound and demonstrated superior performance and clinical response rates versus Vancomycin, the standard method of care for this type of infection [16]. Although drug absorption is complex and cannot be explained simply using a singular metric [43], it is worth considering alternative physiochemical properties to identify drugs with the potential for poor oral bioavailability. Ridinilazole has a fCSP3 of 0, and it is possible that its high degree of ‘flatness’ may be a key factor in its surprisingly low absorption. This may lower the surface area of the molecule relative to molecules of a similar molecular weight and atom count. This may result in a reduction in solubility, due to the formation of a weaker solvation shell.
An additional application for non-systemic compounds is represented by Cholecystokinin 1 receptor (CCK1R) agonists. CCK is a hormone secreted into the gastrointestinal lumen by duodenal endocrine I-cells after the consumption of food, resulting in a number of physiological responses related to the feeling of satiation [44,45]. It has therefore been determined that CCK1R modulation could be used in the treatment of obesity [46,47]. Since CCK1R is expressed on the epithelium of the GI tract, it is necessary to develop agonists with poor GI tract permeation. GI181771X (MW 606, cLogP = 5.1, cLogDpH6.5 = 4.1, TPSA = 139 Å2, fSP3 = 0.15) was found to act as a CCK1R agonist, while also possessing low oral bioavailability (0.4%) in rats [48]. Further developments identified CE-326597 (MW = 595, cLogP = 6.3, LogDpH7.4 = 4.9, fSP3 = 0.19) [49] which was also found to have poor oral bioavailability in rats (F = 1%). It is worth noting that the very low fSP3 values in these compounds may play a significant role in their poor oral bioavailability, despite their other physiological properties meeting, or coming close to meeting, the values prescribed by traditional druglikeness metrics.
The treatment of irritable bowel syndrome often requires drugs with a high degree of gut residence. One such example is Otilonium bromide (cation: MW 484, cLogP = 4.0, TPSA= 65 Å2, rotatable bonds = 17, fSP3 = 0.52) [50], an antispasmodic agent which inhibits muscarinic receptors, resulting in the smoothing in the rate of GI muscle relaxation. The poor permeation of Otilonium bromide (F < 3 %) has been attributed the presence of a charged nitrogen atom as part of a quaternary ammonium group. The therapeutic space around the design of drugs to alleviate GI complications is of great interest to us.
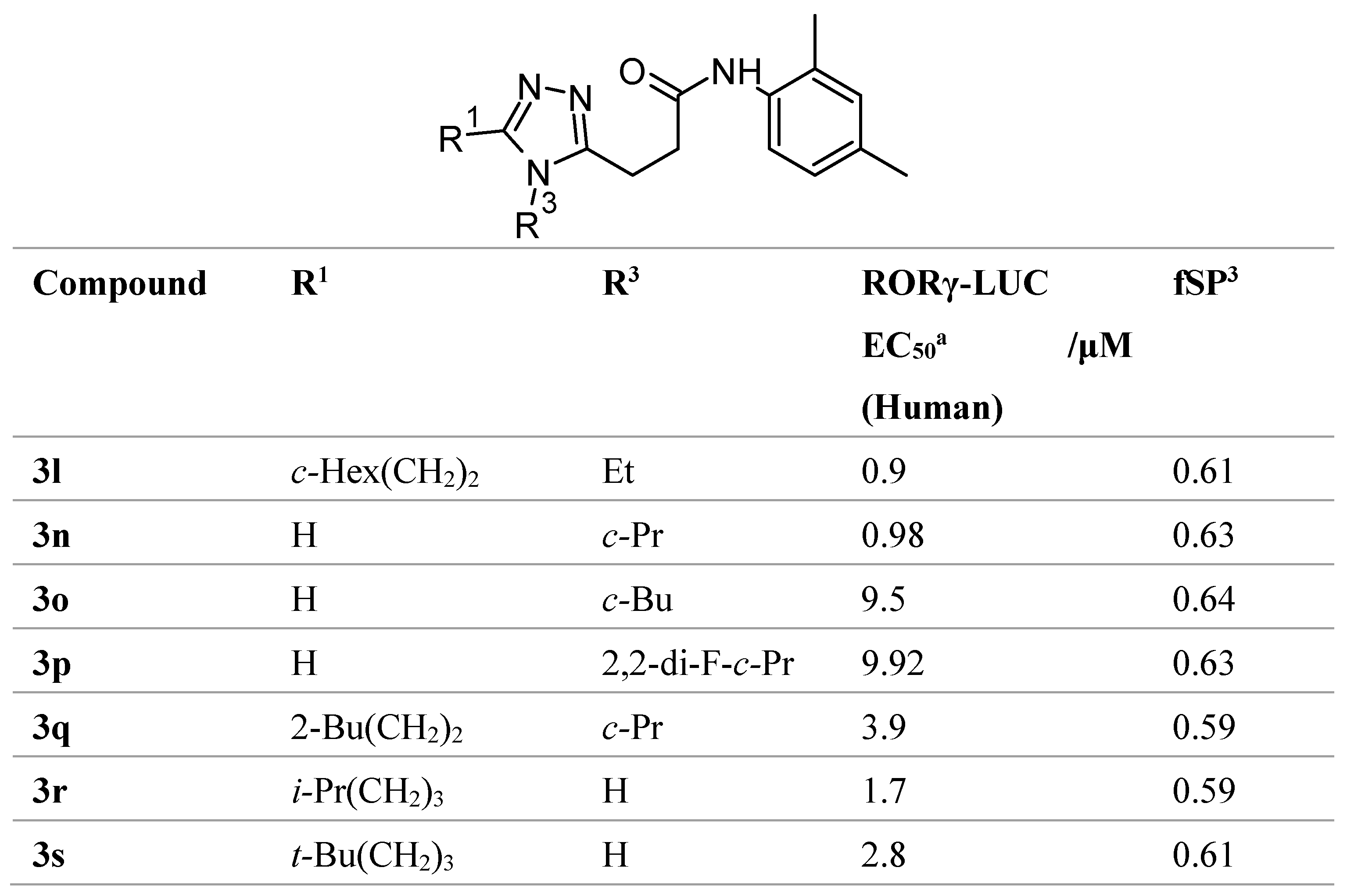
The compounds listed herein show that fSP3 parameter, among other physiological properties, presents a highly useful indicator for poor oral bioavailability that goes beyond the descriptors used by traditional druglikeness filters. There is a need for further study to ascertain their ability to describe druglikeness in the development of non-systemic drugs, thereby aiding the design such compounds. There is limited understanding of the role of fSP3 regarding medicinal chemistry, especially in the context of oral bioavailability. However, one such study was conducted by Hirata et al. [51] in the development of orally bioavailable RORγ inhibitors, finding that, as shown in Table 1, substituents with high values of fSP3 values correlated with increased potency and higher liver permeance, which despite being less relevant in regards to our work, are noteworthy observations. Iusupov et al. also incorporated fSP3 in their drug design philosophy, introducing spirocyclic substituents to increase fSP3 [52]. This resulted in more sterically rigid molecules, that are more able to undergo stereochemically precise interactions with their targets, due to a reduction in the degrees of rotational freedom. These studies show the potential of considering fSP3 in developing more potent lead compounds; however, there is still much more room for further development of the concept, and there is no literature regarding whether low values of fSP3 can result in a reduction of oral bioavailability.
Case study – Phenylboronic acid Fructose Scavengers
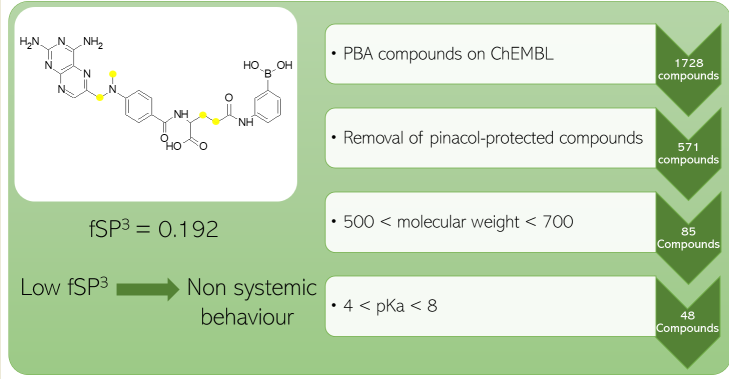
Phenylboronic acids are known to be able to readily bind to 1,2-cis diols, such as those found in monosaccharides [53]. This property presents the opportunity to utilise phenylboronic acids as sugar sequestering agents in the treatment of gastrointestinal agents, such as fructose malabsorption [54]. In general, boronic acids have been widely studied for medicinal chemistry purposes [55]. Since a build up of fructose is located within the gastrointestinal lumen itself, it is necessary that the sequestering agent has limited absorption through the small intestine and remains in the lumen to bind to fructose. With this requirement in mind, we aimed to explore pre-existing chemical databases to identify reported and novel phenylboronic acids with potential for poor oral bioavailability, using fCSP3 as an indicator. A substructure search of phenylboronic acid on the ChEMBL database [56,57,58,59,60] resulted in 1728 compounds being found. The removal of pinacol-protected compounds and benzoboroxoles, which are beyond the scope of our study, left 571 phenylboronic acid compounds to be analysed. Since we were exploring beyond Ro5 chemical space to further promote non-systemic behaviour [61], we filtered the compounds to select those with a molecular weight between 500 and 700, leaving 85 compounds.
O’Donovan postulated that a pKa less than 4 is desired for permeation of compounds with a molecular weight in our chosen range [62]; however, lower pKa values are needed to promote good Lewis acidity for sugar binding at physiological pH [63]. Setting a final filter for compounds with a pKa range between 4 and 8, using the pKa prediction software OPERA [64] resulted in a final 48 compounds for semi-empirical sugar binding analysis to identify the most suitable candidates for non-systemic, fructose-selective scavenging. Our sugar recognition work will be reported on in due course, and we plan to check other compound databases to identify potential chemical starting points.
The aforementioned 48 compounds are listed in Table S1 (see supporting information). A range of heterocyclic rings, carbocyclic rings, and functional groups are represented in the listed molecules. In addition, a couple of these compounds bear two boronic acid motifs.
Conclusion
The application of novel physiochemical metrics to describe molecules, such as fSP3, brings new insight and opportunities to design compounds that may not have been considered previously. The traditional drug discovery process is fraught with problems that limit scope in exploring molecular space. As the drug design process continues to incorporate artificial intelligence and machine learning into its methodologies, there is a growing need to identify and evaluate quantifiable values associated with molecules, in order to predict their medicinal properties. More research is needed around the evaluation of the impact of fSP3 both medicinally, and in terms of GI absorption, especially in relation to its potential for poor GI absorption in small molecules. We intend to incorporate fSP3 into our workflows to identify novel fructose scavengers for the treatment of fructose malabsorption and other gastrointestinal complaints. Our ongoing compound design also features an integrated bias for fructose binding (over other monosaccharides) in order to maximise fructose scavenging, and thereby reducing the passage of fructose into the large intestine. We aim to report on further studies concerning the correlation between gut residence and fSP3.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: Results from non-systemic phenylboronic acid filtering.
Funding
This research received no external funding.
Author Contributions:
Conceptualisation, M.B. and S.P.W.; methodology, M.B.; data curation, M.B.; writing—original draft preparation, M.B and S.P.W.; writing—review and editing, M.B., A. Le G., A. E., and S.P.W.; supervision, A. Le G., A. E., and S.P.W.; project administration, S.P.W. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Readers are encouraged to contact the authors to discuss/see our data.
Acknowledgments
We would like to thank the Wren research group for useful discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hingorani AD, Kuan V, Finan C, Kruger FA, Gaulton A, Chopade S, et al. Improving the odds of drug development success through human genomics: modelling study. Sci Rep 2019;9:18911. [CrossRef]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 1997;23:3–25. [CrossRef]
- Ghose AK, Viswanadhan VN, Wendoloski JJ. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J Comb Chem 1999;1:55–68. [CrossRef]
- Muegge I, Heald SL, Brittelli D. Simple Selection Criteria for Drug-like Chemical Matter. J Med Chem 2001;44:1841–6. [CrossRef]
- Veber DF, Johnson SR, Cheng H-Y, Smith BR, Ward KW, Kopple KD. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J Med Chem 2002;45:2615–23. [CrossRef]
- Egan WJ, Merz Kenneth M., Baldwin JJ. Prediction of Drug Absorption Using Multivariate Statistics. J Med Chem 2000;43:3867–77. [CrossRef]
- DeGoey DA, Chen H-J, Cox PB, Wendt MD. Beyond the Rule of 5: Lessons Learned from AbbVie’s Drugs and Compound Collection. J Med Chem 2018;61:2636–51. [CrossRef]
- Hann MM. Molecular obesity, potency, and other addictions in drug discovery. Medchemcomm 2011;2:349–55. [CrossRef]
- Wong FYK, Desmarchelier PM. VIBRIO | Vibrio Cholerae. In: Robinson RK, editor. Encyclopedia of Food Microbiology, Oxford: Elsevier; 1999, p. 2242–8. [CrossRef]
- Percival SL, Williams DW. Chapter Eleven - Shigella. In: Percival SL, Yates M V, Williams DW, Chalmers RM, Gray NF, editors. Microbiology of Waterborne Diseases (Second Edition), London: Academic Press; 2014, p. 223–36. [CrossRef]
- Poolman JT. Escherichia coli. In: Quah SR, editor. International Encyclopedia of Public Health (Second Edition), Oxford: Academic Press; 2017, p. 585–93. [CrossRef]
- Charmot D. Non-systemic drugs: a critical review. Curr Pharm Des 2012;18:1434–45. [CrossRef]
- Benet LZ, Hosey CM, Ursu O, Oprea TI. BDDCS, the Rule of 5 and Drugability. Adv Drug Deliv Rev 2016;101:89. [CrossRef]
- Gale AR, Wilson M. Clostridioides difficile Infection. Gastrointestinal Emergencies: Evidence-Based Answers to Key Clinical Questions 2023:353–6. [CrossRef]
- Mullish BH, Williams HRT. Clostridium difficile infection and antibiotic-associated diarrhoea. Clinical Medicine 2018;18:237. [CrossRef]
- Cho JC, Crotty MP, Pardo J. Ridinilazole: a novel antimicrobial for Clostridium difficile infection. Ann Gastroenterol 2019;32:134–40. also see Summit Therapeutics press release. Available online: https://www.globenewswire.com/news-release/2015/11/23/789529/10157064/en/Summit-Therapeutics-Announces-Novel-Antibiotic-Ridinilazole-SMT19969-Achieves-Statistical-Superiority-Over-Vancomycin-in-CoDIFy-Phase-2-Clinical-Trial-for-C-difficile-Infection.html (accessed on 24 August 2023). [CrossRef]
- Zhanel GG, Walkty AJ, Karlowsky JA. Fidaxomicin: A novel agent for the treatment of Clostridium difficile infection. The Canadian Journal of Infectious Diseases & Medical Microbiology 2015;26:305. [CrossRef]
- Alagga AA, Gupta V. Drug Absorption. 2023.
- Lovering F, Bikker J, Humblet C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J Med Chem 2009;52:6752–6. [CrossRef]
- Feher M, Schmidt JM. Property Distributions: Differences between Drugs, Natural Products, and Molecules from Combinatorial Chemistry. J Chem Inf Comput Sci 2003;43:218–27. [CrossRef]
- Clemons PA, Wilson JA, Dančík V, Muller S, Carrinski HA, Wagner BK, et al. Quantifying structure, and performance diversity for sets of small molecules comprising small-molecule screening collections. Proceedings of the National Academy of Sciences 2011;108:6817–22. [CrossRef]
- Sepsey Für C, Bölcskei H. New Spiro[cycloalkane-pyridazinone] Derivatives with Favorable Fsp3 Character. Chemistry (Easton) 2020;2:837–48. [CrossRef]
- Stotani S, Lorenz C, Winkler M, Medda F, Picazo E, Ortega Martinez R, et al. Design and Synthesis of Fsp3-Rich, Bis-Spirocyclic-Based Compound Libraries for Biological Screening. ACS Comb Sci 2016;18:330–6. [CrossRef]
- Wei W, Cherukupalli S, Jing L, Liu X, Zhan P. Fsp3: A new parameter for drug-likeness. Drug Discov Today 2020;25:1839–45. [CrossRef]
- Xie W, Wang F, Li Y, Lai L, Pei J. Advances and Challenges in De Novo Drug Design Using Three-Dimensional Deep Generative Models. J Chem Inf Model 2022;62:2269–79. [CrossRef]
- Bickerton GR, Paolini G V., Besnard J, Muresan S, Hopkins AL. Quantifying the chemical beauty of drugs. Nat Chem 2012;4:90. [CrossRef]
- Ivanenkov YA, Zagribelnyy BA, Aladinskiy VA. Are We Opening the Door to a New Era of Medicinal Chemistry or Being Collapsed to a Chemical Singularity? J Med Chem 2019;62:10026–43. [CrossRef]
- Riaz S, John S. Cholestyramine Resin. Handbook of Nuclear, Biological, and Chemical Agent Exposures 2023:60–1.
- Hameed MH, Farzam K. Colestipol. StatPearls 2022.
- Patel PH, Can AS. Colesevelam. StatPearls 2023.
- Jain SS, Ramanand SJ, Ramanand JB, Akat PB, Patwardhan MH, Joshi SR. Evaluation of efficacy and safety of orlistat in obese patients. Indian J Endocrinol Metab 2011;15:99–104. [CrossRef]
- R. Araujo J, Martel F. Sibutramine Effects on Central Mechanisms Regulating Energy Homeostasis. Curr Neuropharmacol 2012;10:49. [CrossRef]
- McIver LA, Preuss C V., Tripp J. Acarbose. XPharm: The Comprehensive Pharmacology Reference 2022:1–3. [CrossRef]
- Miglitol. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury 2021.
- Somaraju UR, Solis-Moya A. Pancreatic enzyme replacement therapy for people with cystic fibrosis. Cochrane Database Syst Rev 2016;2016. [CrossRef]
- Buchwald P. Soft drugs: design principles, success stories, and future perspectives. Expert Opin Drug Metab Toxicol 2020;16:645–50. [CrossRef]
- Pevtsov A, Kerndt CC, Ahmed I, Fredlund KL. Esmolol. XPharm: The Comprehensive Pharmacology Reference 2023:1–5. [CrossRef]
- Domanovits H, Wolzt M, Stix G. Landiolol: pharmacology and its use for rate control in atrial fibrillation in an emergency setting. Eur Heart J Suppl 2018;20:A1. [CrossRef]
- Levy ES, Samy KE, Lamson NG, Whitehead KA, Kroetz DL, Desai TA. Reversible inhibition of efflux transporters by hydrogel microdevices. European Journal of Pharmaceutics and Biopharmaceutics 2019;145:76–84. [CrossRef]
- Makagiansar IT, Avery M, Hu Y, Audus KL, Siahaan TJ. Improving the Selectivity of HAV-Peptides in Modulating E-Cadherin-E-Cadherin Interactions in the Intercellular Junction of MDCK Cell Monolayers. Pharm Res 2001;18:446–53. [CrossRef]
- Song K-H, Kim S-B, Shim C-K, Chung S-J, Kim D-D, Rhee S-K, et al. Paracellular permeation-enhancing effect of AT1002 C-terminal amidation in nasal delivery. Drug Des Devel Ther 2015:1815. [CrossRef]
- Álvarez R, López Cortés LE, Molina J, Cisneros JM, Pachón J. Optimizing the Clinical Use of Vancomycin. Antimicrob Agents Chemother 2016;60:2601–9. [CrossRef]
- Wang NN, Dong J, Deng YH, Zhu MF, Wen M, Yao ZJ, et al. ADME Properties Evaluation in Drug Discovery: Prediction of Caco-2 Cell Permeability Using a Combination of NSGA-II and Boosting. J Chem Inf Model 2016;56:763–73. [CrossRef]
- Liddle RA, Goldfine ID, Rosen MS, Taplitz RA, Williams JA. Cholecystokinin bioactivity in human plasma. Molecular forms, responses to feeding, and relationship to gallbladder contraction. Journal of Clinical Investigation 1985;75:1144–52. [CrossRef]
- Little TJ, Horowitz M, Feinle-Bisset C. Role of cholecystokinin in appetite control and body weight regulation. Obesity Reviews 2005;6:297–306. [CrossRef]
- Herranz R. Cholecystokinin antagonists: Pharmacological and therapeutic potential. Med Res Rev 2003;23:559–605. [CrossRef]
- Szewczyk J, Laudeman C. CCK1R Agonists: A Promising Target for the Pharmacological Treatment of Obesity. Curr Top Med Chem 2003;3:837–54. [CrossRef]
- Sugg EE, Birkemo L, Gan L-SL, Tippin TK. Orally Active Nonpeptide CCK-A Agonists. Integration of Pharmaceutical Discovery and Development, Boston: Kluwer Academic Publishers; n.d., p. 507–24. [CrossRef]
- Elliott RL, Cameron KO, Chin JE, Bartlett JA, Beretta EE, Chen Y, et al. Discovery of N-benzyl-2-[(4S)-4-(1H-indol-3-ylmethyl)-5-oxo-1-phenyl-4,5-dihydro-6H-[1,2,4]triazolo[4,3-a][1,5]benzodiazepin-6-yl]-N-isopropylacetamide, an orally active, gut-selective CCK1 receptor agonist for the potential treatment of obesity. Bioorg Med Chem Lett 2010;20:6797–801. [CrossRef]
- Evangelista S. Quaternary Ammonium Derivatives as Spasmolytics for Irritable Bowel Syndrome. Curr Pharm Des 2004;10:3561–8. [CrossRef]
- Hirata K, Kotoku M, Seki N, Maeba T, Maeda K, Hirashima S, et al. SAR Exploration Guided by LE and Fsp(3): Discovery of a Selective and Orally Efficacious RORγ Inhibitor. ACS Med Chem Lett 2015;7:23–7. [CrossRef]
- Iusupov IR, Lukyanenko ER, Altieri A, Kurkin A V. Design and Synthesis of Fsp3-Enriched Spirocyclic-Based Biological Screening Compound Arrays via DOS Strategies and Their NNMT Inhibition Profiling. ChemMedChem 2022;17:e202200394. [CrossRef]
- Dowlut M, Hall DG. An Improved Class of Sugar-Binding Boronic Acids, Soluble and Capable of Complexing Glycosides in Neutral Water. J Am Chem Soc 2006;128:4226–7. [CrossRef]
- Benardout M, Le Gresley A, ElShaer A, Wren SP. Fructose malabsorption: causes, diagnosis, and treatment. British Journal of Nutrition 2022;127:481–9. [CrossRef]
- Silva MP, Saraiva L, Pinto M, Sousa ME. Boronic acids and their derivatives in medicinal chemistry: synthesis and biological applications. Molecules 2020, 25(18), 4323. [CrossRef]
- Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res 2012;40:D1100-7. [CrossRef]
- Bento AP, Gaulton A, Hersey A, Bellis LJ, Chambers J, Davies M, et al. The ChEMBL bioactivity database: an update. Nucleic Acids Res 2014;42:D1083-90. [CrossRef]
- Gaulton A, Hersey A, Nowotka M, Bento AP, Chambers J, Mendez D, et al. The ChEMBL database in 2017. Nucleic Acids Res 2017;45:D945–54. [CrossRef]
- Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG, et al. A comprehensive map of molecular drug targets. Nat Rev Drug Discov 2017;16:19–34. [CrossRef]
- Mendez D, Gaulton A, Bento AP, Chambers J, De Veij M, Félix E, et al. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res 2019;47:D930–40. [CrossRef]
- DeGoey DA, Chen H-J, Cox PB, Wendt MD. Beyond the Rule of 5: Lessons Learned from AbbVie’s Drugs and Compound Collection. J Med Chem 2018;61:2636–51. [CrossRef]
- O’ Donovan DH, De Fusco C, Kuhnke L, Reichel A. Trends in Molecular Properties, Bioavailability, and Permeability across the Bayer Compound Collection. J Med Chem 2023;66:2347–60. [CrossRef]
- Yan J, Springsteen G, Deeter S, Wang B. The relationship among pKa, pH, and binding constants in the interactions between boronic acids and diols—it is not as simple as it appears. Tetrahedron 2004;60:11205–9. [CrossRef]
- Mansouri K, Grulke CM, Judson RS, Williams AJ. OPERA models for predicting physicochemical properties and environmental fate endpoints. J Cheminform 2018;10:10. [CrossRef]
Figure 1.
General formula for fSP3 calculation.
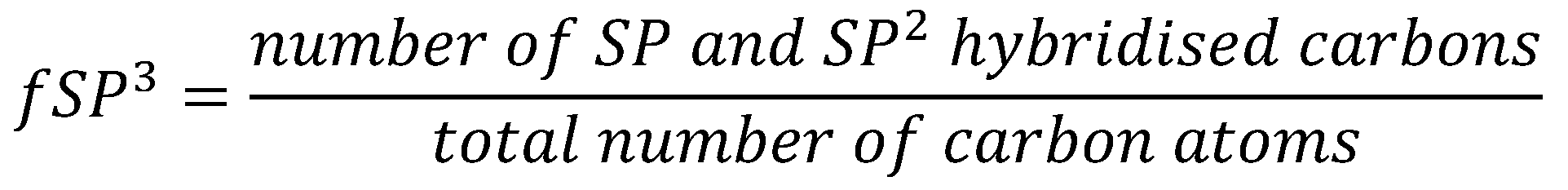
Figure 2.
Formula for MCE-18, where AR is equal to 1 if an aromatic ring is present in the molecule, or 0 if absent; NAR is equal to 1 if an aromatic or heterocyclic ring is present, or 0 if absent; CHIRAL is equal to 1 if a spiro atom is present, or 0 if absent. Cyc is the fraction of cyclic carbons that are SP3 hybridised; Acyc is the fraction of acyclic carbons that are SP3 hybridised. Q1 is the normalised quadratic index.
Figure 2.
Formula for MCE-18, where AR is equal to 1 if an aromatic ring is present in the molecule, or 0 if absent; NAR is equal to 1 if an aromatic or heterocyclic ring is present, or 0 if absent; CHIRAL is equal to 1 if a spiro atom is present, or 0 if absent. Cyc is the fraction of cyclic carbons that are SP3 hybridised; Acyc is the fraction of acyclic carbons that are SP3 hybridised. Q1 is the normalised quadratic index.
Figure 3.
Examples of Class I and II compounds as described by Charmot [10].
Figure 3.
Examples of Class I and II compounds as described by Charmot [10].
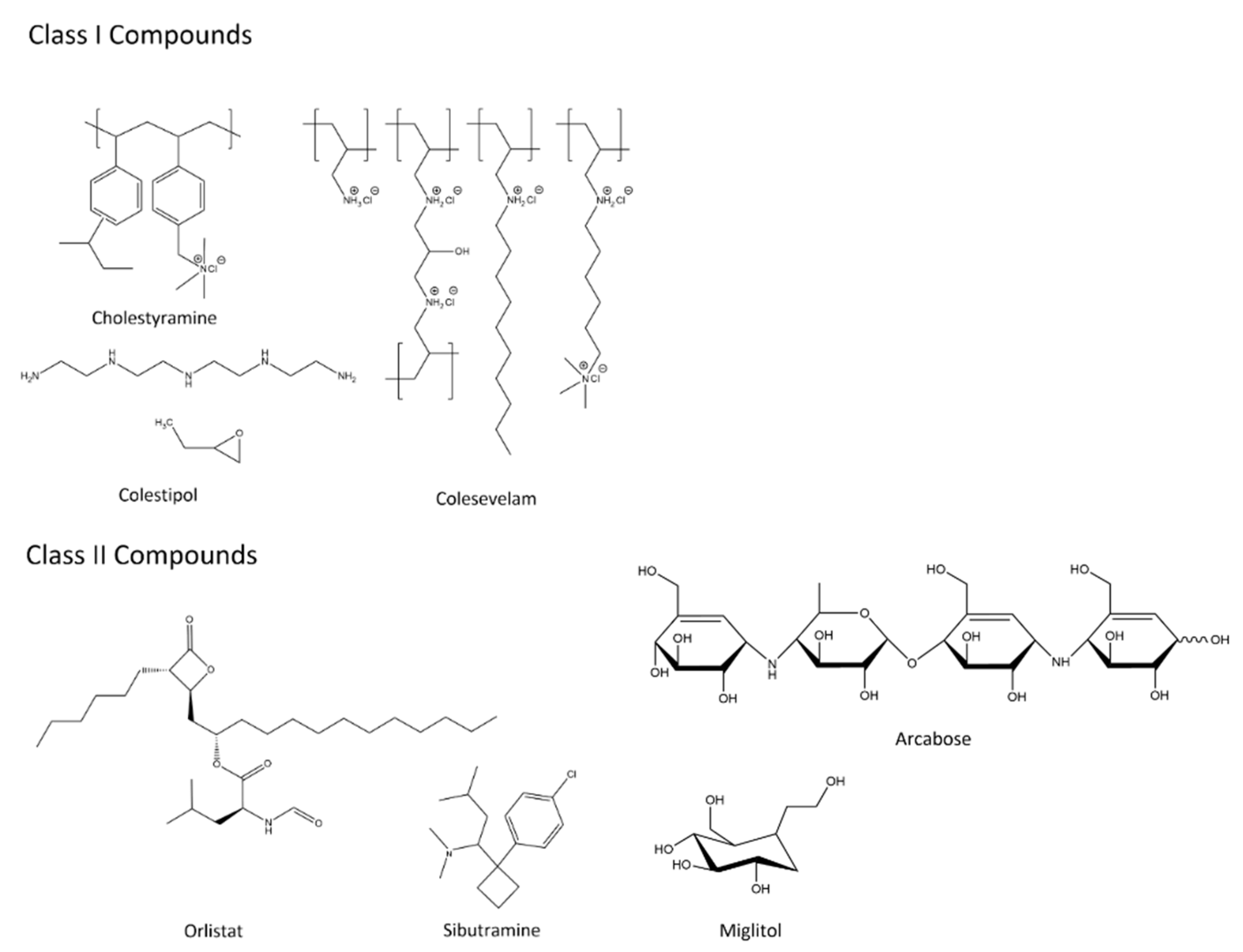
Figure 4.
Structure of Ridinilazole.
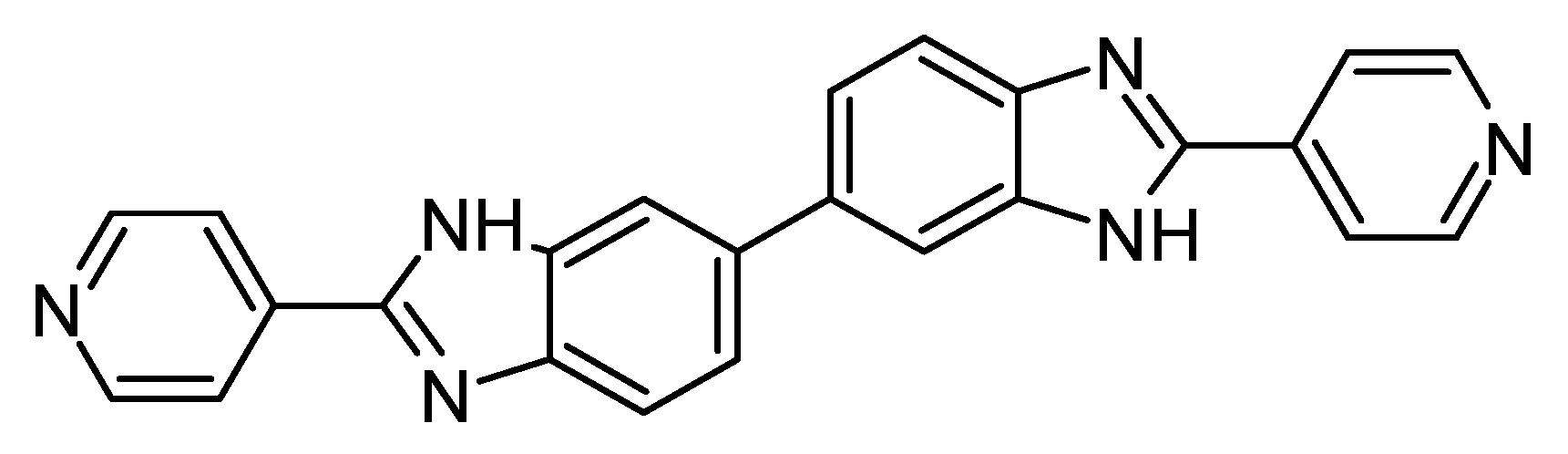

Table 1.
Structure activity relationship study in RORγ inhibitor development. Adapted from Hirata [51].
Table 1.
Structure activity relationship study in RORγ inhibitor development. Adapted from Hirata [51].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



