Submitted:
28 August 2023
Posted:
29 August 2023
You are already at the latest version
Abstract
Glucose is an important carbon sources for microbial growth, and its content in infertile soils is essential for the growth of the bacterium. Since the mechanism of oligotrophic bacterium adaptation in barren soils is unclear, the research employed RNA Seq technology to examine the impact of glucose concentration on the low nutrient bacteria BS-G1 in soil affected by desertification. RNA-Seq revealed that differentially expressed genes(DEG)histidine metabolism, glutamate synthesis, HIF-1 signalling pathway, sporulation, and TCA cycle pathway of BS-G1 were significantly enriched in 0.015 g/L glucose concentration, compared with 10 g/L glucose concentration. DEG amino acids system, two-component system, Metal Ion Transport, and nitrogen metabolism system of B. subtilis BS-G1 were significantly enriched in 5 g/L glucose concentration, compared with 10 g/L glucose concentration. In addition, The present study identified its regulation pattern and key genes under the low glucose environment (7 mRNAs, 16 sRNAs). This study primarily investigates the variances in the regulatory pathways of oligotrophic bacteria BS-G1, which holds substantial importance in comprehending the mechanism underlying the limited sugar tolerance of oligotrophic bacteria.
Keywords:
Oligotrophic bacteria
; Low glucose tolerance
; sRNA
; transcriptome
1. Introduction
The limitation of nutrients in ecosystems is a significant constraint on the vitality of microbial life [1]. Numerous microorganisms that thrive in nutrient-poor environments, including freshwater lakes, deserts, plateaus, and oceans, have been identified[2,3,4,5]. Among these microorganisms, nutrient-poor bacteria can be classified as specialized or parthenogenetic. obligately oligotrophic bacteria can only proliferate in media containing carbon concentrations ranging from 1 to 15 mg/L and are not well-suited to nutrient-rich conditions [6,7]. Conversely, facultatively oligotrophic bacterium is capable of growth and adaptation in both high and low concentrations of organic carbon [8]. Due to their predominant distribution in highly nutrient-depleted environmental settings, this particular assemblage of bacteria exhibits sluggish growth rates, protracted growth cycles, diminutive size, and the ability to traverse 0.45 μm filter membranes. Consequently, researchers have designated them as Filterable bacteria and ultramicrobacteria [9,10]. Given their exacting requirements for survival conditions, the oligotrophic bacterium is difficult to culture in the laboratory [11]. For example,The discovery of Planctomycetes can be attributed to the Hungarian biologist Nador Gimesi in 1924, who initially found it at the lake of Langymanyos. However, due to the unavailability of a pure culture strain, researchers worldwide were limited to studying it solely through morphological description. It was not until 1973 that Staley successfully isolated the strain, marking a significant milestone in the understanding of Planctomycetes [12]. In 1976, Bauld and Staley provided a formal description of this strain as the inaugural species within the phylum Planctomycetes, relying on phenotypic and genetic characterization. Subsequently, from 1987 to 2006, Carl Woese and Strous et al. further substantiated the relationship between Planctomycetes and Chlamydomonas through phenotypic observations and comparisons of protein sequences [13]. It took a total of 76 years (1924-2020) for the precise identification of the strain, with 49 of those years (1924-1973) attributed to the inability to acquire a pure culture of the strain, which hindered comprehensive study by microbiologists [14].
Bacteria are required to swiftly acclimate to ongoing fluctuations in their surroundings, encompassing scarcities in nutrients, variations in oxygen availability, and exposure to abiotic stresses (e.g., alterations in temperature)[7,15,16]. This adaptability is especially crucial for bacteria that exist independently. Glucose is the optimum carbon source for bacteria, as it supplies both energy and material resources necessary for strain growth. Therefore, a diminished concentration of glucose has an adverse effect on strain growth[9,17,18]. Previous studies have demonstrated that propionic acid, a metabolite known for its growth inhibitory properties, accumulates significantly in the culture medium under controlled glucose concentrations of 1 g/L and 0.2 g/L, consequently impacting the growth conditions of B. subtilis [10]. Additionally, it has been proposed that the presence of β-glucosidase (UnBgl1A) enables B. subtilis to withstand a glucose concentration of 0.9 M [9,19]. However, previous research investigating the adaptation of Bacillus subtilis to low sugar conditions has employed glucose concentration as a manipulated variable to examine its impact on strain growth rate, production of fermentation products (such as microbial fibrillases), feedback inhibition of β-glucosidase, and glucose transport [9,10,19,20]. The phosphoenolpyruvate-dependent phosphotransferase system (PTS) is of utmost importance in the bacterial glucose transportation process, facilitating the movement of specific sugars across the bacterial inner membrane through sugar phosphorylation [21,22,23,24,25]. It has been observed that bacteria commonly possess PTS systems for glucose uptake, and it has been discovered that non-glucose PTS systems can transport glucose as well (e.g., the cellobiose PTS system can facilitate intracellular glucose transport) [22]. The study has revealed that the phosphotransferase system (PTS) is capable of detecting variations in nutrient levels within the surrounding environment, thereby facilitating appropriate cellular responses to these fluctuations [26].
Bacterial small regulatory RNAs (sRNAs), which typically range from 50 to 500 nucleotides in length, serve as significant post-transcriptional regulators of gene expression in response to environmental stimuli stressors [27,28,29,30,31,32]. sRNAs, which are transcribed from intergenic regions and do not undergo translation, exhibit a high degree of conservation among homologs [33]. These sRNAs exert their influence on mRNA stability and translation by forming base pairs with the 5'-untranslated region (5'-UTR) of the target mRNA or with bases in the ribosome-binding site (RBS) [28,34,35,36]. sRNA can positively or negatively regulate target genes. For example,sRNAs can negatively regulate their targets by inhibiting translation or stimulating degradation via ribonuclease RNase E[37,38,39,40]. The interaction of sRNAs with target mRNAs generally requires the RNA chaperone Hfq, which binds to sRNAs, promotes sRNA-mRNA base pairing, and directly binds and regulates the translation of certain mRNAs[39,41,42,43]. Furthermore, one sRNA usually regulates multiple mRNAs, and one mRNA can be handled by various sRNAs, thus forming a regulatory network to respond to changing environments[44]. sRNAs have been identified in many bacteria, including Escherichia coli, Staphylococcus aureus, and Vibrio cholerae, where they play key roles in bacterial adaptation to environmental stress and virulence[45,41,42]. sRNAs also affect the pathogenesis of Gram-positive Enterococcus faecalis by regulating their growth and survival under various environmental stresses, including iron stress [43]. Studies have shown that approximately 10-30% of bacterial genes are regulated by sRNAs [42].
Many researchers have comprehensively studied the response of oligotrophic bacteria to low nutrition. For example, Prochlorococcus adapts to the evolutionary strategy of oligotrophy by minimizing the necessary resources by reducing the bacteria's size and the coding genes [43]. ABC transport-binding proteins are critical for the transport and metabolism of nutrients in oligotrophic bacteria in the ocean [46,47]. In addition, eutrophic bacteria mainly transport external nutrients through the ABC transport system, while oligotrophic bacteria prefer to use the PTS system to absorb external nutrients [47,48,49,50]. Oligotrophic bacteria grow slowly in media and pick low molecular weight (LMW) organic molecules for growth compared to high concentrations of organic matter (HMW biopolymers)[51,52,53]. The many researchers have explored the hunger stress response of the oligotrophic bacteria in the water environment.However,There are few reports on the low-sugar tolerance strategies of the oligotrophic bacteria of poor soil, and the mechanism of their adaptation to low sugar is still unclear. Due to the impact of climate change, land use, and human activities, the ecosystem may become unstable, necessitating the implementation of a strategy to detect microbial responses to environmental changes. This approach can serve as an early warning system, enabling the timely detection of environmental changes and facilitating the implementation of appropriate remedial measures. Research has demonstrated that the assessment of cyanobacteria density in aquatic environments can effectively predict alterations in water quality, thereby assisting managers in mitigating water quality issues[54]. In addition, researchers have used qRT-PCR to quantify Microcystis spp. toxin genes in lakes to determine the dynamics of cyanobacterial blooms [55,56]. Ningxia is one of the provinces with the most severe desertification in China, with a desertification area of 2.898 million hm2, accounting for 55.8% of the total land area of Ningxia. Land degradation has seriously affected the local ecological environment and people's lives[19,57]. Hence, the accurate anticipation of alterations in soil nutrient levels holds significant relevance in the context of managing sandy soils.This study employed global transcriptome analysis (RNA-Seq) to elucidate the adaptation patterns of B. subtilis BS-G1 under varying glucose concentrations. In addition, the study aims to identify the crucial mRNAs and sRNAs involved in low-glucose adaptation and establish a theoretical foundation for the development of biogenetic early warning indicators for soil impoverishment.
2. Materials and Methods
2.1. Strain
The oligotrophic bacterium of B. subtilis BS-G1 was isolated from microbial crusts that developed early in the desertification area of Ningxia; B. subtilis W1, B. subtilis MX31, and B. subtilis L1 were derived from desert soil in Ningxia; B. subtilis ACCC11025 was obtained from the biological Provided by the Key Laboratory of Technology and Engineering Ministry of Education; B. subtilis 168 was purchased from China Research Institute of Food and Fermentation Industry Co., Ltd. All six strains of Bacillus subtilis were stored at -80°C in 40% glycerol bacteria.
2.2. Growth curve and determination of residual sugar content
Pick activated bacterial strains and inoculate them in 100 ml glucose concentration of 5 g/L oligotrophic culture medium at 37°C, 150 r/min for 24 hours. Centrifuge the bacterial solution at 5000 r/min for 5 min, discard the supernatant, wash the bacterial cells with physiological saline, suspend the bacterial cells in the oligotrophic medium with the glucose concentration of 0.015 g/L, adjust the OD600 of the bacterial liquid to about 1.2, Reserve as seed solution. With the inoculum amount of 2%, the seed solution was inoculated in an oligotrophic medium with 12 glucose concentrations and cultured at 37°C and 150 r/min. The absorbance of the bacterial liquid at 600nm and the residual sugar content in the medium were measured every 2 hours using an automatic growth curve analyzer and a biosensor analyzer SBA-40D.
2.3. Construction of transcriptome and processing of biological information
2.3.1. Material Handling
The activated B. subtilis BS-G1 seed solution was picked and inoculated with 2% inoculum in the oligotrophic culture medium, culture at 37°C, 150 r/min for 8h. Bacteria cultured for 8 hours were collected separately, and RNA was extracted for later use.
2.3.2. RNA extraction
The Trizol reagent method was employed to extract total RNA from the collected cells, while DNase I (Takara) was used to remove genomic DNA. Subsequently, the quality of the RNA was assessed using an Agilent 2100 Bioanalyzer, and its quantity was determined using an ND-2000 (NanoDrop Technologies). For the subsequent library construction, RNA samples of high quality were utilized, meeting the following criteria: OD260/280 ratio ranging from 1.8 to 2.0, OD260/230 ratio equal to or greater than 2.0, RIN value of at least 6.5, 23S:16S ratio exceeding 1.0, concentration of 100 ng/μl or higher, and a total amount of at least 2μg.
2.3.3. Library Construction and Sequencing
RNA library construction was performed using Illumina (San Diego, CA) TruSeqTM RNA sample preparation Kit. The Ribo-Zero Magnetic kit (epicenter) was employed to eliminate rRNA, followed by the random fragmentation of mRNA into approximately 200 bp fragments. The resulting mRNA was utilized as a template, and the reverse transcription process was carried out using random primers (Illumina) and the SuperScript double-stranded cDNA synthesis kit (Invitrogen, CA) to synthesize double-stranded cDNA. When synthesizing the second strand of cDNA, use dUTP instead of dTTP for synthesis, add the synthesized double-stranded cDNA to End Repair Mix to make it blunt, phosphorylate the 5' end, add an A base to the 3' end, and connect the Y-shaped sequence connector. The second strand of cDNA containing dUTP is then eliminated with the UNG enzyme so that only the first strand of cDNA is contained in the library. The library was amplified by PCR with Phusion DNA polymerase (NEB) for 15 cycles. Following the quantification of TBS380 (Picogreen), RNA-seq paired-end sequencing was performed using Illumina HiSeq X Ten (2×150bp).
2.3.4. Bioinformatics Analysis
The bioinformatics analysis was conducted utilizing data generated by the Illumina platform. The analyses were conducted utilizing the cloud platform (cloud.majorbio.com) provided by Shanghai Majorbio Bio-pharm Technology Co., Ltd.
3. Results
3.1. Growth Curves and Glucose Utilization
By measuring the growth curves of B. subtilis BS-G1 and five control strains at a glucose concentration of 0.015 g/L-10 g/L, it was found that compared with the five control strains, at a sugar concentration of 0.015 g/L, only B. subtilis BS-G1 can grow normally, while 5 control strains are dying, and B. subtilis BS-G1 can grow under the sugar concentration of 0.15g/L-10g/L, and the increase of glucose concentration in the medium is beneficial to the growth of the strain growth (Figure 1). It indicated that B. subtilis BS-G1 is a facultative oligotrophic bacterium.
By determining the sugar uptake curves of B. subtils BS-G1 and five control strains at 0.015 g/L glucose concentration, it was found that the fastest rate of sugar uptake was found in B. subtils BS-G1 (Figure 2A).The rate of sugar uptake of B. subtils BS-G1 in the range of 0.015g/L-10g/L glucose sugar concentration was found to be increased with the increase of glucose concentration. and the strain sugar uptake curve reached a stabilization period at 16 h of incubation (Figure 2B).
3.2. Illumina sequencing, quality filtering, and sequence alignment
In this study, we constructed 15 cDNA libraries, including bacteria from BS-G1 strain with three glucose concentrations, namely; 0.015g/L (Low glucose,L), 5g/L (Medium glucose ,M), and 10g/L (High low glucose ,H) Samples, for processing and controls. These libraries were then sequenced on the Illumina Novaseq 6000 platform. After removing sequencing adapters and low-quality data, 592,812,250 filtered reads were obtained with a Q30 base percentage higher than 96.11% for each sample, and then the filtered reads from each sample were aligned to the reference genome, and the filtered reads for each sample were compared with the reference genome. The mapping efficiency is between 98.91% and 99.37%. Higher mapping percentages indicated good sequencing quality of the samples (Table S1).
3.3. Functional annotation and expression analysis of transcripts
The RSEM software was used to analyze gene expression levels quantitatively, and the quantitative index was TPM, which refers to the analysis of the transcript data of the sample with the number of transcripts as the calculation unit. Transcriptome data analysis showed that 4020 genes (mRNA) were expressed in B. subtilis BS-G1 under different glucose concentrations. The above genes were compared with GO, COG, and KEGG databases. Among them, the COG annotation rate was 77.63%, and the GO annotation rate was 77.63%. The rate is 73.25%, and the KEGG annotation rate is 54.60% from the perspective of the KEGG pathway. Among the top 20 genes expressed in the three sugar concentrations, the 0.015g/L sugar concentration (L) is mainly concentrated in the three KEGG pathways (ribosome, phosphotransferase system, quorum sensing); the 5g/L sugar concentration (M) is primarily enriched in three KEGG pathways (ribosome, flagella assembly, glycolysis/gluconeogenesis system); 10g/L sugar concentration (H) is primarily enriched in five KEGG pathways (ribosome, butyrate metabolism, population induction, two-component systems, and glycolysis/gluconeogenesis systems) (Tables S2-S5).
3.3. Identification of DEGs
We used DESeq2 software to identify differentially expressed genes (p-adjust<0.05, |Log2FC|≥1) for the transcripts of BS-G1 under different glucose concentrations. The results showed 1821 differentially expressed mRNAs (DEGs) in LvsH, of which 1099 DEGs were significantly upregulated, and 722 DEGs were significantly downregulated. At the same time, 1571 DEGs were detected in MvsH, 845 DEGs were significantly upregulated, and 726 DEGs were down-regulated considerably (Figure S1). The DEG fold change distributions of LvsH and MvsH showed that the genes with the most significant fold change of DEG were distributed between 1≤|Log2FC|<2 (Figure S2).
3.4. Functional annotation analysis of differentially expressed genes
DEGs were annotated and classified according to the COG database to gain a deeper understanding of the transcriptional mechanism of BS-G1 strain adaptation to different sugar concentrations. As shown in Figure 3A and Figure 3B, The DEG annotation results of LvsH and MvsH were classified into 20 categories(Figures 3A and 3B,). In six functional categories such as: "Cell cycle control, cell division, chromosome allocation", "Intracellular trafficking, secretion, and vesicular trafficking", "Transcription", "Amino acid transport and metabolism", "Nucleotide transport and Metabolism", "Lipid Transport and Metabolism", the number of DEG induced by 0.015g/L and 5g/L was higher than the number of DEG inhibited. In addition, it was shown in Table S6 that a large number of DEGs in the LvsH and MvsH groups were assigned to "amino acid transport and metabolism", "carbohydrate transport and metabolism", and "cell wall/membrane/envelope biogenesis". It was found that in the "cell movement" functional category, there were no significantly upregulated DEGs and only nine significantly down-regulated DEGs in the LvsH group, while there were 18 upregulated considerably DEGs and four significantly down-regulated DEGs in the MvsH group.
The annotation results of DEGs in KEGG pathways showed that the 4 KEGG functions with the most upregulated DEGs in the LvsH group were "amino acid transport and metabolism" (73 DEGs), "carbohydrate transport and metabolism" (64 DEGs), "membrane transport" (39 DEGs), "lipid metabolism" (26 DEGs); among the down-regulated DEGs in the LvsH group, 65 DEGs were annotated as "carbohydrate metabolism", 60 DEGs were annotated as "cofactor and vitamin metabolism", 50 DEGs were annotated as "membrane transport", and 42 DEGs were annotated as "energy metabolism". In addition, we found that the number of DEGs related to the function of "cofactor and vitamin metabolism" was the most down-regulated in LvsH, with a specific number of 60, while 27 upregulated DEGs and 23 down-regulated DEGs were in the MvsH group (Table S7, Figure 4). The KEGG annotation results are consistent with previous analyses based on COG categories. It also showed that different sugar levels mainly affected the "amino acid metabolism", "carbohydrate metabolism", "membrane transport", "cofactor and vitamin metabolism" and "energy metabolism process" of BS-G1.
3.5. GO functional enrichment and KEGG pathway enrichment
To systematically determine DEGs' possible functions and participating pathways, we performed GO and KEGG pathway enrichment analysis on DEGs using Goatools software and R script, respectively. GO enrichment analysis showed that the GO annotations of DEGs were divided into three parts: biological process (BP), cellular composition (CC), and molecular function (MF). After the screening (p-value ≤ 0.05), it was found that the top twenty GO terms enriched by DEG of LvsH were mainly divided into three categories, namely: transporter activity, sporulation, histidine, and catabolism of acidic organic compounds (Table S8). The top twenty GO terms significantly enriched in DEG in MvsH were mainly enriched in five aspects: nitrogenous inorganic salt metabolism, nucleotide synthesis and metabolism, mobilization, oxidoreductase activity, and histidine synthesis (Table S9). The corresponding numbers of DEGs in the significantly enriched pathways in LvsH and MvsH are shown in Figure 5.
The KEGG enrichment pathway results of DEGs were arranged in ascending order of P-value, and further screening was performed when the P-value≤0.05. KEGG enrichment analysis showed that the DEGs of LvsH were mainly concentrated in porphyrin and chlorophyll metabolism, phosphotransferase system (PTS), histidine metabolism, glycolysis/gluconeogenesis, butyrate metabolism, HIF-1 signaling pathway ( Table S10). The DEGs of MvsH were mainly concentrated in KEGG pathways such as arginine biosynthesis, flagellar assembly, two-component system, and nitrogen metabolism (Table S11).The number of DEGs in pathways enriched by LvsH and MvsH is shown in Figure 6.
3.6. Identification of sRNA
A total of 595 sRNAs were identified in the BS-G1 strain, and these transcripts ranged from 1 nt to 500 nt in length, with an average length of 95 nt (Figure 6B). The sRNA can be divided into two types, one is the sRNA that acts on the intergenic region of the target mRNA, and the other is the antisense sRNA that works on the SD sequence or part of the coding region of the target mRNA. Among the sRNAs identified in this study, there are 471 intergenic sRNAs and 121 antisense sRNAs, of which 51 have been annotated (Figure 7A). Of the 527 unannotated genes, 411 were classified as intergenic region sRNAs, and 116 were classified as antisense sRNAs. Comparing the sRNAs with known functions with the Rfam database, 51 sRNAs were annotated, including 28 rRNAs, 2 tRNAs, 20 CisRNAs, and one annotated as bacterial large signal recognition particle RNA (sRNA0014).
3.7. Predict target genes of differentially expressed sRNAs
In this study, the sRNA changes of BS-G1 in response to different glucose concentrations were measured, and the numbers of significantly differentially expressed sRNAs were summarized in Figure 8, respectively. Analysis of sRNA transcript levels at different sugar levels provides a window into the regulation of mRNA transcription, where glucose-triggered changes increase with lower sugar levels. In this study, a total of 196 differentially expressed sRNAs were identified. Compared with the H group, the L group had the most differentially expressed sRNAs, including 101 upregulated sRNAs, 24 down-regulated sRNAs, and 24 upregulated sRNAs in the M group. Down-regulated sRNAs were 30.
Significantly differentially expressed sRNA target genes were compared with the COG database to obtain clues about the physiological role of the target genes in the M and L groups compared with the H group. The COG classification results showed that "carbohydrate transport and metabolism", "amino acid transport and metabolism", "transcription", and "cell wall/membrane/envelope biogenesis" were the most annotated DEGs (Figures 9A and 9B). This is consistent with the COG annotation results for DEGs. In addition, the target genes in the LvsH group were also involved in "inorganic ion transport and metabolism", "replication, recombination and repair", and "energy production and conversion" (Figure 9A). Target genes were also assigned to "Signal Transduction Mechanisms", "Coenzyme Transport and Metabolism" and "Ribosomal Transport and Metabolism" in the MvsH group (Figure 9B).
3.8. GO/KEGG enrichment analysis of sRNA target genes.
To further evaluate the function of the significantly differentially expressed sRNA target genes, we performed GO and KEGG enrichment analysis on these target genes and set the screening condition as P-value≤0.05. The sRNA target genes significantly differentially expressed in LvsH were greatly enriched. The top twenty GO terms in the set were divided into six categories: ATPase activity, hydrolase activity, substance transport, transporter activity, localization, and nucleoside triphosphatase activity (Table S12). The sRNA target gene enrichment pathways of MvsH are classified into nine categories, namely: nucleoside sugar synthesis, plasma membrane, others, nucleotide synthesis and metabolism, nitrogen metabolism, metal ion transport, ATPase activity, anhydrase, and amino acid synthesis (Tables 1 and S13). In addition, we set the screening condition to be P-value≤0.05, performed KEGG enrichment analysis on significantly differentially expressed sRNA target genes, and found that LvsH significantly differentially expressed sRNA target genes enriched two genes, including photosynthesis and ABC transporter. KEGG pathway. MvsH significantly differentially expressed sRNA target genes and enriched five KEGG pathways, namely: "RNA polymerase, amino sugar and nucleotide sugar metabolism, purine metabolism, ABC transporter, a pentose sugar and glucuronic acid interconversion." (Tables S14-S15 and Figure 10)
3.9. Adaptation patterns and key genes in a low-glucose environment
Based on the enrichment results of BS-G1 strain DEG and significantly differentially expressed sRNA target genes GO/KEGG, we constructed the adaptation model of the BS-G1 strain under different glucose concentrations (Figure 11). As the concentration of glucose in the medium decreased, the activity of the glucose transporter increased, the aerobic oxidation pathway of glucose weakened, the amount of ATP produced decreased, and it gradually transformed into a dormant body. In addition, BS-G1 in group H and group L sensed changes in external nutrient content through the HIF-1 signalling pathway, while BS-G1 in group M received external environmental signals through a two-component system and quickly transmitted them to the bacteria to make more nutritional changes. Positive and effective adaptive responses. Utilization of nitrogen-containing inorganic salts (NH4+, NO3-) and metal ions (Mg2+, K+, Ca2+) in the culture medium by strains in group M. The production and metabolism of amino acids played a positive regulatory role in the strain's adaptation to different glucose environments, and the output of histidine and arginine in the M group increased. The L group of histidine is converted into glutamic acid, and glutamic acid enters the TCA cycle through further metabolism to generate α-ketoglutarate. Among them, the imidazole group of histidine can form coordination compounds with metal ions in the medium, which is beneficial to maintaining cell homeostasis and controlling the transportation of nutrients. The highly helical flagella of strains in the M group increased motility, driving the strains to more beneficial nutritional small molecules or ions and avoiding substances that were not conducive to their growth. At different glucose concentrations, sRNA targets regulate transporter activity, ion transport, amino acid production, and ATP generation pathways.
3.10. Critical genes for low-sugar adaptation.
To explore the adaptation mechanism of B. subtilis BS-G1 under the low concentration of glucose, we determined the vital pathway for the strain to adapt to the environment of 0.015g/L glucose through the GO/KEGG enrichment analysis of the DEG and sRNA target genes of LvsH, namely: the transport pathway, all genes in the transport pathway were screened (screening condition |Log2FC|≥5), and finally crucial mRNAs (7) and sRNAs (15) that were adapted to the 0.015 g/L glucose environment were obtained (Tables S16-S26). The seven key mRNAs were divided into three categories, amino acid ABC transporters, PTS system component proteins, and glutaminase ABC transporters (Table 1 and Table 2).
Taking seven key mRNAs as the central genes and analyzing the interaction with their corresponding sRNAs, the two key mRNAs (gene0435 and gene3876) with the highest degree of network connection were determined. mtlA (gene0435), celB (gene3876) are mannitol and mannose PTS transporter subunits, respectively. Among them, three key sRNAs (sRNA0504, sRNA0362, and sRNA0200) were connected to two key mRNAs (Figure 12).
3.11. qRT-PCR Validation of DEGs
To further verify the reliability of the transcriptome results, we selected five DEGs (gene3877, gene0435, gene0646, gene1536, gene0436) related to carbohydrate transport for qRT-PCR(Figure 13). Although the relative expression values determined by qRT-PCR did not exactly match those obtained from transcriptome sequencing, they were similar to the gene expression trends (up- or down-regulation) obtained from transcriptomes.
4. Discussion
It has been shown in previous reports that the transport system is closely related to stress [58,59]. In this study, we explored possible mechanisms of sugar stress through transcriptome analysis. This study showed that the four pathways and PTS systems related to transporter activity were significantly enriched in the LvsH group by GO functional enrichment and KEGG enrichment (Tables S8 and S10). It was reported that the transporter activity genes of Lactiplantibacillus plantarum ZDY2013 were significantly differentially expressed under acid stress [58]. This is consistent with the results of this study. In this study, the DEGs associated with LvsH transporter activity were mainly divided into ABC transporters, MFS transporters, and PTS transporters. Studies have shown that under acid-stress conditions, the related genes of ABC and PTS transporters in L. plantarum ZDY2013 changed significantly [58]. The MFS transporter is the most important secondary active transporter on the cell membrane, which can selectively transport monosaccharides, oligosaccharides, nucleotides, and other substrates. Studies have shown that under pH stress, the expression of the MFS transporter of Enterococcus faecalis is upregulated, thereby enhancing the efflux capacity of hydrogen ions and enhancing the strain's tolerance to acid [59]. Glucose is mainly transported across membranes through the PTS system. Studies have shown that under butanol stress, the transcription of PTS genes transporting mannitol and cellobiose in L. plantarum WCFS1 is down-regulated [60]. It has been reported that under oxidative stress, the expression of multiple PTS genes in Listeria monocytogenes YjbH is down-regulated, and the down-regulation fold is as high as hundreds of times [61]. In addition, transport proteins are important osmoregulatory responsible for the uptake and excretion of vital substances such as inorganic ions, sugars, and amino acids. This plays a critical role in regulating the osmotic pressure of the BS-G1 strain in a low-concentration glucose environment.
Metal ions play an essential role in maintaining cellular homeostasis and controlling transport. Among them, K+ and Na+ play a vital role in cell homeostasis, membrane transport, and regulation of osmotic pressure [62,63,64]. Ktr system potassium transporter A (gene3042) was significantly upregulated in both LvsH and MvsH, and the up-regulation fold of LvsH was more prominent (Tables S28 and S29). There are two forms of Na+ transporter, symport and anti-transport. Na+ symport may be related to the absorption of amino acids, sugars, organic cations, or anions. The sodium: proton antiporter (gene0984) was significantly upregulated in both LvsH and MvsH. Studies have shown that Dietzia sp. DQ12-45-1b after Na+/H+ antiporter mutation and wild-type Dietzia sp. DQ12-45-1b was cultured in 0.25M-1M NaCl simultaneously and the cells of the wild-type strain. The degree of enrichment was significantly higher than that of the mutant strain [65]. K+ and Na+ are closely related to the spore germination process, and spore germination will stimulate the release of K+ and Na+[66]. In addition, studies have shown that the accumulation of K+ also induces the germination of spores [67]. In this study, "endospore-forming prespores" and "intracellular immature spores" were highly enriched among GO functional enrichments in DEGs of the LvsH group. Under harsh conditions, Bacillus subtilis can be transformed into endospores to survive under the regulation of SigB[68]. When favorable growing conditions return, the spores exit dormancy and germinate. Mg2+ is an essential divalent cation for every cell and an important cofactor in DNA replication, transcription, and translation [69,70,71]. Mg2+ must pass through the biomembrane through Mg2+ transporters. So far, there are four Mg2+ transporters: CorA, MgtA/B, MgtA, and NramP[72]. But in prokaryotes, Mg2+ is mainly transported by CorA and MgtA [69,70,71]. In this study, corA (gene0850) was significantly upregulated in both LvsH and MvsH, and the upregulation degree of LvsH was higher than that of MvsH. Jayanti Saha et al. analyzed the genome and comparative genome of Pseudomonas aeruginosa and believed that corA might be necessary for strains to resist heavy metal toxicity [73]. It indicated that in group L, BS-G1 tried to maintain the homeostasis of cells by strengthening the transport of Mg2+ in the medium. Iron ions are essential cofactors for cellular processes, playing important roles in respiration, nitrogen fixation, DNA synthesis, the TCA cycle, and oxygen transport [74,75,76]. As a regulator of iron uptake, Fur can bind to Fe2+ to control the vehicle of Fe2+[75,77]. In this study, both LvsH and MvsH iron transporters (gene0417, gene0416, gene3287) were significantly down-regulated, but the degree of down-regulation of LvsH was lower than that of MvsH (Tables S28 and S29). Transcriptome analysis of the metal ion uptake system of Clostridium beijerinckii NRRL B-598 by Maryna Vasylkivska's team found that the expression of genes related to the iron uptake system increased during the sporulation stage [62]. Iron has been reported to help facilitate the germination process of spores [78]. When Samuel Plante et al. studied spore germination and the siderophore transporter Str1, they believed siderophores might be secreted when the bacteria transformed from a swollen round reproductive body to a vase-shaped dormant body[78]. As a second messenger, Ca2+ can participate in many physiological activities of organisms, including maintaining cell membrane homeostasis, regulating the growth and development of organisms, and regulating enzyme activities[78,79]. Studies have shown that Ca2+ helps to reduce the accumulation of cadmium in Phanerochaete chrysosporium, thereby reducing the toxic effect of cadmium on the strain [79]. This study showed that the calcium/proton exchanger chaA (gene0837) was significantly upregulated in LvsH but not significantly differentially expressed in MvsH. Studies by Yingkun Wan et al. showed that after chaA-knockout E.coil and wild-type E.coil were treated with gentamicin for six days, the number of cells in the knockout strain was notably reduced compared to the wild-type strain[80].
Chemotaxis in Bacillus subtilis and other bacteria is a widely studied adaptive mechanism by which bacteria detect chemical compounds and exhibit movements towards or away from specific compounds [81,82,83]. This mechanism plays an important role in cell growth, biofilm formation, virulence, and infectivity. The bacterial flagellum is a macromolecular complex consisting of approximately 20,000-30,000 protein subunits, including about 30 different proteins [82]. Our results show that there is a significant up-regulation of flagellar synthesis genes in group M. The study demonstrated that P. extremaustralis exhibited upregulation of flagellar genes (flgB, flgN, flgM) in response to an oxygen stress environment. This upregulation led to an increased motility of the strain, enabling it to evade toxic compounds such as H2O2 [84].
sRNAs are regulators involved in gene expression in organisms. Under adverse circumstances, organisms will regulate gene transcription levels through sRNA or produce new proteins to cope with the stress [85]. In this study, LvsH and MvsH identified 156 and 161 differentially expressed sRNA target genes related to carbohydrate metabolism and transport, respectively (Tables S32 and S33). The researchers placed Salmonella enterica under non-starvation and carbon source starvation treatments and found that the differentially expressed sRNA target genes contained multiple genes related to carbohydrate transport and metabolism[86]. In Caulobacter crescentus with oligotrophic characteristics, an sRNA closely related to carbon starvation was identified, that is, CrfA, which can target and regulate the mRNA of various membrane transporters [87]. In this study, LvsH and MvsH significantly differentially expressed sRNA target genes GO, and KEGG functional enrichment showed that ABC transporters, membrane functions, and fatty acid biosynthesis were enriched considerably, indicating that under different carbon source conditions, sRNA may be transported. Protein diversification regulates the strain's carbon source uptake capacity. In addition, we found that sacA (gene3825, gene3444) in LvsH and sacA (gene3825), sacB (gene4085), and sacC (gene4086) of MvsH strains were all significantly upregulated (Tables S30 and S31). Sac proteins belong to the Bgl-Sac anti-termination protein family, which is essential in responding to carbon source starvation. Sac anti-termination protein consists of 1 RNA-binding domain and two regulatory domains (PRD1 and PRD2), reversible phosphorylation binding sites in response to cognate carbon sources. In the absence of carbon sources, Under this condition, the protein phosphorylates PRD1 through the EII transporter of the PTS system, making it inactive [77]. In the presence of a carbon source, the EII transporter dephosphorylates PRD1, while the HPr protein (a non-sugar-specific component of the PTS system) phosphorylates PRD2. When PRD1 is not phosphorylated and PRD2 is phosphorylated, the anti-terminator protein will form a homodimer, which can combine with the target gene to form an anti-terminator structure, thereby ensuring the regular operation of the PTS system[88].
Histidine is the α-amino β-imidazolyl propionic acid of the eight-gene operon hisGDC[NB]HAF[IE]) [89]. The HisF gene encodes a cyclase that forms a heterodimer holoenzyme imidazole glycerol phosphate synthase (IGPS) with HisH (a transglutaminase), and this enzyme connects three different biological pathways, namely; nitrogen metabolism, histidine biosynthesis, and de novo synthesis of purines [90,91]. At a glucose concentration of 0.015g/L, histidine is converted into glutamic acid under the action of iminomethylglutamic acid, and glutamic acid is converted to glutamic acid by glutamic acid dehydrogenase (GDH) or alanine or aspartic acid Transaminases (TAs) convert TCA cycle intermediate α-ketoglutarate (α-KG). After α-KG enters the tricarboxylic acid cycle, it is converted into oxaloacetate, which is converted into phosphoenolpyruvate by phosphoenolpyruvate carboxylase and enters the gluconeogenesis pathway to generate glucose, thus providing the medium with carbon source. Coxiella burnetiid IC bacteria can use glutamate as the sole carbon source and metabolize glutamate to produce ketoglutarate and enter the TCA cycle and gluconeogenesis pathway[92]. In addition, the 0.015g/L glucose environment strengthens purine metabolism, and purine, as the most abundant metabolic substrate in all organisms, provides essential components for synthesizing DNA and RNA. In addition to being the building blocks of DNA and RNA, purines provide crucial energy and cofactors for cell survival and proliferation. Under the environment of 5g/L glucose, the glutamine produced by nitrogen metabolism of BS-G1 can enter the histidine synthesis pathway through HisF[90]. The imidazole group of histidine forms a coordination compound with metal ions (Ca2+, Mg2+, K+), which is beneficial for the strain to maintain cell homeostasis, membrane transport, and regulation of osmotic pressure. Therefore, the synthesis and metabolism of histidine played a crucial regulatory role in adapting BS-G1 to different grape concentrations.
This study identified seven critical mRNAs adapted to the glucose concentration of 0.015 g/L, among which mtlA (gene0435) and celB (gene3876, gene3860) are used for the specific transport of mannitol and cellobiose, respectively [93,94]. Studies have shown that when the mannitol-specific PTS transporter (mtlA) was overexpressed, the strain enhanced the uptake rate of PTS sugars, including N-acetylglucosamine, methyl α-glucoside and 2-deoxyglucose [95]. The researchers propose that overexpression of PTS transporter genes specifically enhances the uptake rate of PTS substrates, which may be caused by stimulatory protein-protein interactions between MtlA and target PTS transporters, but the specific mechanism is unclear [95]. In the environment of 0.015g/L glucose, the strain can transport ammonium ions in the culture medium into the cell through the glutamine ABC transporter (gene2655, gene2653) across the membrane [96]. Previous research has demonstrated that the utilization of ABC transporter proteins, which are connected to the transportation of heavy metal ions, can serve as biomarkers for the identification of heavy metal pollution in soil through the examination of their expression [50]. Glucose serves as a primary carbon source for microorganisms, and prior empirical findings have demonstrated that the expression of seven crucial mRNAs related to carbohydrate transport is notably elevated under conditions of low glucose levels. Notably, the genes mtlA (gene0435) and celB (gene3876, gene3860) are prevalent across various strains, including Vibrio cholerae, Escherichia coli, Lactococcus lactis, Clavibacter michiganensis, and Listeria monocytogenes[97,98,99,100]. The subsequent research direction of our team involves utilizing mtlA (gene0435) and celB (gene3876, gene3860) as bio-alert indicators to assess the nutrient status of the soil based on their expression.
5. Conclusions
Bacterial adaptation to changing environments is often accompanied by transcriptome remodelling. This study determined the mechanism of B. subtilis BS-G1 adapting to the 0.015g/L glucose environment from the transcriptome perspective. B. subtilis BS-G1 promoted the high expression of PTS transporters (mtlA and celB) through sRNA targeting in the 0.015g/L glucose environment, thereby promoting the glucose uptake rate by B. subtilis BS-G1. In addition, after the histidine produced in the strain is transformed into glutamic acid, the final metabolite α-ketoglutarate produced through further metabolism enters the TCA cycle and the gluconeogenesis pathway successively, providing carbon for the growth of the strain again source. In addition, this study established the adaptation model of BS-G1 to three different sugar concentrations. However, this experiment lacks a verification experiment for seven key mRNAs, and I will conduct further experimental verification by inhibiting the expression of critical genes (mRNA and sRNA). This study offers a theoretical foundation for a comprehensive investigation into the mechanism of oligotrophic microbial adaptation in low-sugar environments. Additionally, it identifies potential avenues for future research aimed at further understanding the mechanisms of oligotrophic microbial adaptation in nutrient-depleted soil environments.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: LvsH and MvsH differentially expressed gene mRNA (DEG)title; Figure S2: Fold change distribution of DEGs present in samples with different treatments;Table S1. Statistical table of raw data of BS-G1 transcriptome induced by different glucose concentrations; Table S2. Gene annotation information of BS-G1 under different glucose concentrations; Table S3. Annotation information of the top 20 genes expressed in BS-G1 in 0.015g/L glucose concentration (L group); Table S4. Annotation information of the top 20 genes expressed in BS-G1 at 5g/L glucose concentration (M group); Table S5. Annotation information of the top 20 genes expressed in BS-G1 at 10g/L glucose concentration (Group H); Table S6. DEG COG functional classification of LvsH and MvsH; Table S7. KEGG functional classification of DEGs of LvsH and MvsH; Table S8. LvsH differentially expressed mRNA GO enrichment (top twenty); Table S9. MvsH differentially expressed mRNA GO enrichment (top twenty); Table S10. LvsH differentially expressed mRNA KEGG enrichment (top twenty); Table S11. MvsH differentially expressed mRNA KEGG enrichment (top twenty); Table S12. GO enrichment of LvsH differentially expressed sRNA target genes (top twenty); Table S13. MvsH differentially expressed sRNA target genes GO enrichment (top twenty); Table S14. LvsH differentially expressed sRNA target gene KEGG enrichment (top 20); Table S15. MvsH differentially expressed sRNA target gene KEGG enrichment (top twenty); Table S16. LvsH differentially expressed mRNA GO enrichment (top twenty) Pvalue ≤0.05 (transmembrane transporter activity GO:0022857); Table S17. LvsH differentially expressed mRNA GO enrichment (top twenty) Pvalue ≤0.05 (transporter activity GO:GO:0005215); Table S18. LvsH differentially expressed mRNA GO enrichment (top 20) Pvalue ≤0.05 (carbohydrate transmembrane transporter activity GO:0015144, carbohydrate transporter activity GO:1901476); Table S19. LvsH differentially expressed mRNA GO enrichment (top 20) Pvalue ≤0.05 (active transmembrane transporter activity GO:0022804); Table S20. LvsH differentially expressed mRNA KEGG enrichment (top twenty) Pvalue ≤0.05 (phosphotransferase system (PTS) map02060); Table S21. GO enrichment of LvsH differentially expressed sRNA target genes (top twenty) Pvalue ≤0.05 (transmembrane transporter activity driven by P-P-bond hydrolysis GO:0015405 primary active transmembrane transporter activity GO:0015399); Table S22. LvsH differentially expressed sRNA target gene GO enrichment (top twenty) Pvalue ≤0.05 (transmembrane transport GO:0055085); Table S23. LvsH differentially expressed sRNA target gene GO enrichment (top twenty) Pvalue ≤0.05 (single organism transport GO:0044765 and single organism localization GO:1902578); Table S24. MvsH differentially expressed sRNA target gene GO enrichment (top 20) Pvalue ≤0.05 (monocarboxylic acid transmembrane transporter activity GO:0008028); Table S25. LvsH differentially expressed sRNA target gene KEGG enrichment (top twenty) Pvalue ≤0.05 (ABC transporter map02010); Table S26. MvsH differentially expressed sRNA target gene KEGG enrichment (top 20) Pvalue ≤0.05 (ABC transporter map02010); Table S27. Genes and primers for validation of DEG expression using qRT-PCR; Table S28. DEG statistics of LvsH-encoded metal ion and vitamin transporters; Table S29. DEG statistics of MvsH-encoded metal ion and vitamin transporters; Table S30. LvsH differentially expressed gene list (mRNA and sRNA); Table S31. MvsH differentially expressed gene list;.
Author Contributions
J.S.; conceptualization, funding acquisition, supervision, writing—review and editing. L.C.; sample collection and processing, writing-original draft,visualization, writing—review and editing.C.W.; investigation, validation. All authors have read and agreed to the published version of the manu-script.
Funding
This research was funded by the Ningxia Natural Science Foundation (Ning Xia Key R&D Project), grant number 2017BY081. The funder is Science and Technology Department of Ningxia.
Data Availability Statement
Data are available upon request from the corresponding author.
Acknowledgments
We thank all members of the Su laboratory for helpful suggestions. This work was supported by research grants from the Ningxia Natural Science Foundation (Ning Xia Key R&D Project, grant number 2017BY081).
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Appendix A
This experiment takes B. subtilis BS-G1 as the research object and explores its transcriptome at glucose concentrations of 0.015g/L, 5g/L, and 10g/L. For easier writing and drawing, I defined 0.015g/L glucose concentration, 5g/L glucose concentration, and 10g/L glucose concentration as low glucose (L group), medium glucose (M group), and high glucose (H group), respectively
References
- Props, R.; Monsieurs, P.; Vandamme, P.; Leys, N.; Denef, V.J.; Boon, N. Gene Expansion and Positive Selection as Bacterial Adaptations to Oligotrophic Conditions. mSphere 2019, 4, e00011-19. [CrossRef]
- Chiriac, M.C.; Haber, M.; Salcher, M.M. Adaptive Genetic Traits in Pelagic Freshwater Microbes. Environ Microbiol 2023, 25, 606–641. [CrossRef]
- Chen, Y.; Neilson, J.W.; Kushwaha, P.; Maier, R.M.; Barberán, A. Life-History Strategies of Soil Microbial Communities in an Arid Ecosystem. ISME J 2021, 15, 649–657. [CrossRef]
- Cao, J.; Jiao, Y.; Che, R.; Holden, N.M.; Zhang, X.; Biswas, A.; Feng, Q. The Effects of Grazer Exclosure Duration on Soil Microbial Communities on the Qinghai-Tibetan Plateau. Sci Total Environ 2022, 839, 156238. [CrossRef]
- Dong, C.; Wei, L.; Wang, J.; Lai, Q.; Huang, Z.; Shao, Z. Genome-Based Taxonomic Rearrangement of Oceanobacter-Related Bacteria Including the Description of Thalassolituus Hydrocarbonoclasticus Sp. Nov. and Thalassolituus Pacificus Sp. Nov. and Emended Description of the Genus Thalassolituus. Front Microbiol 2022, 13, 1051202. [CrossRef]
- Ishida, Y.; Kadota, H. Growth Patterns and Substrate Requirements of Naturally Occurring Obligate Oligotrophs. Microb Ecol 1981, 7, 123–130. [CrossRef]
- de Araújo, H.L.; Martins, B.P.; Vicente, A.M.; Lorenzetti, A.P.R.; Koide, T.; Marques, M.V. Cold Regulation of Genes Encoding Ion Transport Systems in the Oligotrophic Bacterium Caulobacter Crescentus. Microbiol Spectr 2021, 9. [CrossRef]
- Ishida, Y.; Imai, I.; Miyagaki, T.; Kadota, H. Growth and Uptake Kinetics of a Facultatively Oligotrophic Bacterium at Low Nutrient Concentrations. Microb Ecol 1982, 8, 23–32. [CrossRef]
- Ogura, M.; Sato, T.; Abe, K. Bacillus Subtilis YlxR, Which Is Involved in Glucose-Responsive Metabolic Changes, Regulates Expression of TsaD for Protein Quality Control of Pyruvate Dehydrogenase. Frontiers in Microbiology 2019, 10, 1–15. [CrossRef]
- Park, Y.S.; Kai, K.; Iijima, S.; Kobayashi, T. Enhanced Beta-Galactosidase Production by High Cell-Density Culture of Recombinant Bacillus Subtilis with Glucose Concentration Control. Biotechnology & Bioengineering 1992, 40, 686. [CrossRef]
- Dedysh, S.N. Cultivating Uncultured Bacteria from Northern Wetlands: Knowledge Gained and Remaining Gaps. Front Microbiol 2011, 2, 184. [CrossRef]
- Staley, J.T. Budding Bacteria of the Pasteuria-Blastobacter Group. Can J Microbiol 1973, 19, 609–614. [CrossRef]
- Strous, M.; Pelletier, E.; Mangenot, S.; Rattei, T.; Lehner, A.; Taylor, M.W.; Horn, M.; Daims, H.; Bartol-Mavel, D.; Wincker, P.; et al. Deciphering the Evolution and Metabolism of an Anammox Bacterium from a Community Genome. Nature 2006, 440, 790–794. [CrossRef]
- Kaboré, O.D.; Godreuil, S.; Drancourt, M. Planctomycetes as Host-Associated Bacteria: A Perspective That Holds Promise for Their Future Isolations, by Mimicking Their Native Environmental Niches in Clinical Microbiology Laboratories. Front Cell Infect Microbiol 2020, 10, 519301. [CrossRef]
- Zhuang, H.; Wu, Z.; Xu, L.; Leu, S.-Y.; Lee, P.-H. Energy-Efficient Single-Stage Nitrite Shunt Denitrification with Saline Sewage through Concise Dissolved Oxygen (DO) Supply: Process Performance and Microbial Communities. Microorganisms 2020, 8, 919. [CrossRef]
- Koza, N.A.; Adedayo, A.A.; Babalola, O.O.; Kappo, A.P. Microorganisms in Plant Growth and Development: Roles in Abiotic Stress Tolerance and Secondary Metabolites Secretion. Microorganisms 2022, 10, 1528. [CrossRef]
- Carreón-Rodríguez, O.E.; Gosset, G.; Escalante, A.; Bolívar, F. Glucose Transport in Escherichia Coli: From Basics to Transport Engineering. Microorganisms 2023, 11, 1588. [CrossRef]
- Wen, J.; Zhao, X.; Si, F.; Qi, G. Surfactin, a Quorum Sensing Signal Molecule, Globally Affects the Carbon Metabolism in Bacillus Amyloliquefaciens. Metab Eng Commun 2021, 12, e00174. [CrossRef]
- Chamoli, S.; Kumar, P.; Navani, N.K.; Verma, A.K. Secretory Expression, Characterization and Docking Study of Glucose-Tolerant β-Glucosidase from B. Subtilis. International Journal of Biological Macromolecules 2016, 85, 425–433. [CrossRef]
- Pan, S.; Chen, G.; Wu, R.; Cao, X.; Liang, Z. Non-Sterile Submerged Fermentation of Fibrinolytic Enzyme by Marine Bacillus Subtilis Harboring Antibacterial Activity With Starvation Strategy. Frontiers in Microbiology 2019, 10, 1025. [CrossRef]
- Norris, N.; Levine, N.M.; Fernandez, V.I.; Stocker, R. Mechanistic Model of Nutrient Uptake Explains Dichotomy between Marine Oligotrophic and Copiotrophic Bacteria. PLoS Comput Biol 2021, 17, e1009023. [CrossRef]
- Wang, Y.; Hammes, F.; Boon, N.; Egli, T. Quantification of the Filterability of Freshwater Bacteria through 0.45, 0.22, and 0.1 Microm Pore Size Filters and Shape-Dependent Enrichment of Filterable Bacterial Communities. Environ Sci Technol 2007, 41, 7080–7086. [CrossRef]
- Sánchez-Cañizares, C.; Prell, J.; Pini, F.; Rutten, P.; Kraxner, K.; Wynands, B.; Karunakaran, R.; Poole, P.S. Global Control of Bacterial Nitrogen and Carbon Metabolism by a PTSNtr-Regulated Switch. Proc Natl Acad Sci U S A 2020, 117, 10234–10245. [CrossRef]
- Jeckelmann, J.M.; Erni, B. Carbohydrate Transport by Group Translocation: The Bacterial Phosphoenolpyruvate: Sugar Phosphotransferase System. Subcell Biochem 2019, 92, 223–274. [CrossRef]
- Fragoso-Jiménez, J.C.; Baert, J.; Nguyen, T.M.; Liu, W.; Sassi, H.; Goormaghtigh, F.; Van Melderen, L.; Gaytán, P.; Hernández-Chávez, G.; Martinez, A.; et al. Growth-Dependent Recombinant Product Formation Kinetics Can Be Reproduced through Engineering of Glucose Transport and Is Prone to Phenotypic Heterogeneity. Microb Cell Fact 2019, 18, 26. [CrossRef]
- Morabbi Heravi, K.; Altenbuchner, J. Cross Talk among Transporters of the Phosphoenolpyruvate-Dependent Phosphotransferase System in Bacillus Subtilis. J Bacteriol 2018, 200, e00213-18. [CrossRef]
- Storz, G.; Vogel, J.; Wassarman, K.M. Regulation by Small RNAs in Bacteria: Expanding Frontiers. Mol Cell 2011, 43, 880–891. [CrossRef]
- Wagner, E.G.H.; Romby, P. Small RNAs in Bacteria and Archaea: Who They Are, What They Do, and How They Do It. Adv Genet 2015, 90, 133–208. [CrossRef]
- Papenfort, K.; Melamed, S. Small RNAs, Large Networks: Posttranscriptional Regulons in Gram-Negative Bacteria. Annu Rev Microbiol 2023, 77, 36944261. [CrossRef]
- Taneja, S.; Dutta, T. On a Stake-out: Mycobacterial Small RNA Identification and Regulation. Noncoding RNA Res 2019, 4, 86–95. [CrossRef]
- Charbonnier, M.; González-Espinoza, G.; Kehl-Fie, T.E.; Lalaouna, D. Battle for Metals: Regulatory RNAs at the Front Line. Front Cell Infect Microbiol 2022, 12, 952948. [CrossRef]
- Parise, M.T.D.; Parise, D.; Aburjaile, F.F.; Pinto Gomide, A.C.; Kato, R.B.; Raden, M.; Backofen, R.; Azevedo, V.A. de C.; Baumbach, J. An Integrated Database of Small RNAs and Their Interplay With Transcriptional Gene Regulatory Networks in Corynebacteria. Front Microbiol 2021, 12, 656435. [CrossRef]
- Gottesman, S.; Storz, G. Bacterial Small RNA Regulators: Versatile Roles and Rapidly Evolving Variations. Cold Spring Harb Perspect Biol 2011, 3, a003798. [CrossRef]
- Fröhlich, K.S.; Papenfort, K. Regulation Outside the Box: New Mechanisms for Small RNAs. Mol Microbiol 2020, 114, 363–366. [CrossRef]
- Romilly, C.; Deindl, S.; Wagner, E.G.H. The Ribosomal Protein S1-Dependent Standby Site in TisB MRNA Consists of a Single-Stranded Region and a 5′ Structure Element. Proc Natl Acad Sci U S A 2019, 116, 15901–15906. [CrossRef]
- Melson, E.M.; Kendall, M.M. The SRNA DicF Integrates Oxygen Sensing to Enhance Enterohemorrhagic Escherichia Coli Virulence via Distinctive RNA Control Mechanisms. Proc Natl Acad Sci U S A 2019, 116, 14210–14215. [CrossRef]
- Balasubramanian, D.; Vanderpool, C.K. New Developments in Post-Transcriptional Regulation of Operons by Small RNAs. RNA Biol 2013, 10, 337–341. [CrossRef]
- Sheehan, L.M.; Budnick, J.A.; Fyffe-Blair, J.; King, K.A.; Settlage, R.E.; Caswell, C.C. The Endoribonuclease RNase E Coordinates Expression of MRNAs and Small Regulatory RNAs and Is Critical for the Virulence of Brucella Abortus. J Bacteriol 2020, 202, e00240-20. [CrossRef]
- Quendera, A.P.; Seixas, A.F.; dos Santos, R.F.; Santos, I.; Silva, J.P.N.; Arraiano, C.M.; Andrade, J.M. RNA-Binding Proteins Driving the Regulatory Activity of Small Non-Coding RNAs in Bacteria. Front Mol Biosci 2020, 7, 78. [CrossRef]
- McQuail, J.; Carpousis, A.J.; Wigneshweraraj, S. The Association between Hfq and RNase E in Long-Term Nitrogen-Starved Escherichia Coli. Mol Microbiol 2022, 117, 54–66. [CrossRef]
- De Lay, N.; Schu, D.J.; Gottesman, S. Bacterial Small RNA-Based Negative Regulation: Hfq and Its Accomplices. J Biol Chem 2013, 288, 7996–8003. [CrossRef]
- Murina, V.N.; Nikulin, A.D. Bacterial Small Regulatory RNAs and Hfq Protein. Biochemistry (Mosc) 2015, 80, 1647–1654. [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved Seed Pairing, Often Flanked by Adenosines, Indicates That Thousands of Human Genes Are MicroRNA Targets. Cell 2005, 120, 15–20. [CrossRef]
- Melamed, S.; Peer, A.; Faigenbaum-Romm, R.; Gatt, Y.E.; Reiss, N.; Bar, A.; Altuvia, Y.; Argaman, L.; Margalit, H. Global Mapping of Small RNA-Target Interactions in Bacteria. Mol Cell 2016, 63, 884–897. [CrossRef]
- Bobrovskyy, M.; Vanderpool, C.K. Regulation of Bacterial Metabolism by Small RNAs Using Diverse Mechanisms. Annu Rev Genet 2013, 47, 209–232. [CrossRef]
- Ford, B.A.; Sullivan, G.J.; Moore, L.; Varkey, D.; Zhu, H.; Ostrowski, M.; Mabbutt, B.C.; Paulsen, I.T.; Shah, B.S. Functional Characterisation of Substrate-Binding Proteins to Address Nutrient Uptake in Marine Picocyanobacteria. Biochem Soc Trans 2021, 49, 2465–2481. [CrossRef]
- Lauro, F.M.; McDougald, D.; Thomas, T.; Williams, T.J.; Egan, S.; Rice, S.; DeMaere, M.Z.; Ting, L.; Ertan, H.; Johnson, J.; et al. The Genomic Basis of Trophic Strategy in Marine Bacteria. Proc Natl Acad Sci U S A 2009, 106, 15527–15533. [CrossRef]
- Schwalbach, M.S.; Tripp, H.J.; Steindler, L.; Smith, D.P.; Giovannoni, S.J. The Presence of the Glycolysis Operon in SAR11 Genomes Is Positively Correlated with Ocean Productivity. Environ Microbiol 2010, 12, 490–500. [CrossRef]
- Tam, R.; Saier, M.H. Structural, Functional, and Evolutionary Relationships among Extracellular Solute-Binding Receptors of Bacteria. Microbiol Rev 1993, 57, 320–346. [CrossRef]
- Jaskulak, M.; Grobelak, A.; Vandenbulcke, F. Effects of Sewage Sludge Supplementation on Heavy Metal Accumulation and the Expression of ABC Transporters in Sinapis Alba L. during Assisted Phytoremediation of Contaminated Sites. Ecotoxicol Environ Saf 2020, 197, 110606. [CrossRef]
- Giovannoni, S.J.; Tripp, H.J.; Givan, S.; Podar, M.; Vergin, K.L.; Baptista, D.; Bibbs, L.; Eads, J.; Richardson, T.H.; Noordewier, M.; et al. Genome Streamlining in a Cosmopolitan Oceanic Bacterium. Science 2005, 309, 1242–1245. [CrossRef]
- Williams, T.J.; Ertan, H.; Ting, L.; Cavicchioli, R. Carbon and Nitrogen Substrate Utilization in the Marine Bacterium Sphingopyxis Alaskensis Strain RB2256. ISME J 2009, 3, 1036–1052. [CrossRef]
- Orsi, W.D.; Smith, J.M.; Liu, S.; Liu, Z.; Sakamoto, C.M.; Wilken, S.; Poirier, C.; Richards, T.A.; Keeling, P.J.; Worden, A.Z.; et al. Diverse, Uncultivated Bacteria and Archaea Underlying the Cycling of Dissolved Protein in the Ocean. ISME J 2016, 10, 2158–2173. [CrossRef]
- Lofton, M.E.; Brentrup, J.A.; Beck, W.S.; Zwart, J.A.; Bhattacharya, R.; Brighenti, L.S.; Burnet, S.H.; McCullough, I.M.; Steele, B.G.; Carey, C.C.; et al. Using Near-Term Forecasts and Uncertainty Partitioning to Inform Prediction of Oligotrophic Lake Cyanobacterial Density. Ecol Appl 2022, 32, e2590. [CrossRef]
- Chen, K.; Allen, J.; Lu, J. Community Structures of Phytoplankton with Emphasis on Toxic Cyanobacteria in an Ohio Inland Lake during Bloom Season. J Water Resour Prot 2017, 9, 1–29. [CrossRef]
- Rinta-Kanto, J.M.; Konopko, E.A.; Debruyn, J.M.; Bourbonniere, R.A.; Boyer, G.L.; Wilhelm, S.W. Lake Erie Microcystis: Relationship between Microcystin Production, Dynamics of Genotypes and Environmental Parameters in a Large Lake. Harmful Algae 2009, 8, 665–673. [CrossRef]
- Guo, S.; Wang, Y.; Huang, J.; Dong, J.; Zhang, J. Decoupling and Decomposition Analysis of Land Natural Capital Utilization and Economic Growth: A Case Study in Ningxia Hui Autonomous Region, China. Int J Environ Res Public Health 2021, 18, 646. [CrossRef]
- Peng, L.; Zhao, K.; Chen, S.; Ren, Z.; Wei, H.; Wan, C. Whole Genome and Acid Stress Comparative Transcriptome Analysis of Lactiplantibacillus Plantarum ZDY2013. Arch Microbiol 2021, 203, 2795–2807. [CrossRef]
- Zhang, L.; Song, D.; Wu, Z. Transcriptome Analysis of Cyclocarya Paliurus Flavonoids Regulation of Differently Expressed Genes in Enterococcus Faecalis under Low PH Stress. Arch Microbiol 2021, 203, 2147–2155. [CrossRef]
- Petrov, K.; Arsov, A.; Petrova, P. Butanol Tolerance of Lactiplantibacillus Plantarum: A Transcriptome Study. Genes (Basel) 2021, 12, 181. [CrossRef]
- Cheng, C.; Han, X.; Xu, J.; Sun, J.; Li, K.; Han, Y.; Chen, M.; Song, H. YjbH Mediates the Oxidative Stress Response and Infection by Regulating SpxA1 and the Phosphoenolpyruvate-Carbohydrate Phosphotransferase System (PTS) in Listeria Monocytogenes. Gut Microbes 2021, 13, 1–19. [CrossRef]
- Vasylkivska, M.; Jureckova, K.; Branska, B.; Sedlar, K.; Kolek, J.; Provaznik, I.; Patakova, P. Transcriptional Analysis of Amino Acid, Metal Ion, Vitamin and Carbohydrate Uptake in Butanol-Producing Clostridium Beijerinckii NRRL B-598. PLoS One 2019, 14, e0224560. [CrossRef]
- Stautz, J.; Hellmich, Y.; Fuss, M.F.; Silberberg, J.M.; Devlin, J.R.; Stockbridge, R.B.; Hänelt, I. Molecular Mechanisms for Bacterial Potassium Homeostasis. J Mol Biol 2021, 433, 166968. [CrossRef]
- Stülke, J.; Krüger, L. Cyclic Di-AMP Signaling in Bacteria. Annu Rev Microbiol 2020, 74, 159–179. [CrossRef]
- Fang, H.; Qin, X.Y.; Zhang, K.D.; Nie, Y.; Wu, X.L. Role of the Group 2 Mrp Sodium/Proton Antiporter in Rapid Response to High Alkaline Shock in the Alkaline- and Salt-Tolerant Dietzia Sp. DQ12-45-1b. Appl Microbiol Biotechnol 2018, 102, 3765–3777. [CrossRef]
- Dürre, P. Physiology and Sporulation in Clostridium. Microbiol Spectr 2014, 2, TBS-0010-2012. [CrossRef]
- Eisenstadt, E. Potassium Content during Growth and Sporulation in Bacillus Subtilis. J Bacteriol 1972, 112, 264–267. [CrossRef]
- Vohradsky, J.; Schwarz, M.; Ramaniuk, O.; Ruiz-Larrabeiti, O.; Vaňková Hausnerová, V.; Šanderová, H.; Krásný, L. Kinetic Modeling and Meta-Analysis of the Bacillus Subtilis SigB Regulon during Spore Germination and Outgrowth. Microorganisms 2021, 9, 112. [CrossRef]
- Stetsenko, A.; Guskov, A. Cation Permeability in CorA Family of Proteins. Sci Rep 2020, 10, 840. [CrossRef]
- Payandeh, J.; Pfoh, R.; Pai, E.F. The Structure and Regulation of Magnesium Selective Ion Channels. Biochim Biophys Acta 2013, 1828, 2778–2792. [CrossRef]
- Pohland, A.C.; Schneider, D. Mg2+ Homeostasis and Transport in Cyanobacteria - at the Crossroads of Bacterial and Chloroplast Mg2+ Import. Biol Chem 2019, 400, 1289–1301. [CrossRef]
- Trachsel, E.; Redder, P.; Linder, P.; Armitano, J. Genetic Screens Reveal Novel Major and Minor Players in Magnesium Homeostasis of Staphylococcus Aureus. PLoS Genet 2019, 15, e1008336. [CrossRef]
- Saha, J.; Dey, S.; Pal, A. Whole Genome Sequencing and Comparative Genomic Analyses of Pseudomonas Aeruginosa Strain Isolated from Arable Soil Reveal Novel Insights into Heavy Metal Resistance and Codon Biology. Curr Genet 2022, 68, 481–503. [CrossRef]
- Cheng, D.; He, Q. PfsR Is a Key Regulator of Iron Homeostasis in Synechocystis PCC 6803. PLoS One 2014, 9, e101743. [CrossRef]
- Cheng, Y.; Zhang, T.; Cao, Y.; Wang, L.; Chen, W. New Insights into the Function of the Proteins IsiC and IsiD from Synechocystis Sp. PCC 6803 under Iron Limitation. Appl Microbiol Biotechnol 2021, 105, 4693–4707. [CrossRef]
- Seo, S.W.; Kim, D.; Latif, H.; O’Brien, E.J.; Szubin, R.; Palsson, B.O. Deciphering Fur Transcriptional Regulatory Network Highlights Its Complex Role beyond Iron Metabolism in Escherichia Coli. Nat Commun 2014, 5, 4910. [CrossRef]
- Plante, S.; Labbé, S. Spore Germination Requires Ferrichrome Biosynthesis and the Siderophore Transporter Str1 in Schizosaccharomyces Pombe. Genetics 2019, 211, 893–911. [CrossRef]
- Pivato, M.; Ballottari, M. Chlamydomonas Reinhardtii Cellular Compartments and Their Contribution to Intracellular Calcium Signalling. J Exp Bot 2021, 72, 5312–5335. [CrossRef]
- Cao, R.; Qin, P.; Li, W.; Shang, C.; Chai, Y.; Jin, D.; Chen, A. Hydrogen Sulfide and Calcium Effects on Cadmium Removal and Resistance in the White-Rot Fungus Phanerochaete Chrysosporium. Appl Microbiol Biotechnol 2021, 105, 6451–6462. [CrossRef]
- Wan, Y.; Wang, M.; Chan, E.W.C.; Chen, S. Membrane Transporters of the Major Facilitator Superfamily Are Essential for Long-Term Maintenance of Phenotypic Tolerance to Multiple Antibiotics in E. Coli. Microbiol Spectr 2021, 9, e0184621. [CrossRef]
- Wani, A.K.; Akhtar, N.; Sher, F.; Navarrete, A.A.; Américo-Pinheiro, J.H.P. Microbial Adaptation to Different Environmental Conditions: Molecular Perspective of Evolved Genetic and Cellular Systems. Arch Microbiol 2022, 204, 144. [CrossRef]
- Coloma-Rivero, R.F.; Flores-Concha, M.; Molina, R.E.; Soto-Shara, R.; Cartes, Á.; Oñate, Á.A. Brucella and Its Hidden Flagellar System. Microorganisms 2021, 10, 83. [CrossRef]
- Zhou, M.; Liu, Z.; Wang, J.; Zhao, Y.; Hu, B. Sphingomonas Relies on Chemotaxis to Degrade Polycyclic Aromatic Hydrocarbons and Maintain Dominance in Coking Sites. Microorganisms 2022, 10, 1109. [CrossRef]
- Solar Venero, E.C.; Ricardi, M.M.; Gomez-Lozano, M.; Molin, S.; Tribelli, P.M.; López, N.I. Oxidative Stress under Low Oxygen Conditions Triggers Hyperflagellation and Motility in the Antarctic Bacterium Pseudomonas Extremaustralis. Extremophiles 2019, 23, 587–597. [CrossRef]
- Sridhar, J.; Gayathri, M. Transcriptome Based Identification of Silver Stress Responsive SRNAs from Bacillus Cereus ATCC14579. Bioinformation 2019, 15, 474–479. [CrossRef]
- Amin, S.V.; Roberts, J.T.; Patterson, D.G.; Coley, A.B.; Allred, J.A.; Denner, J.M.; Johnson, J.P.; Mullen, G.E.; O’Neal, T.K.; Smith, J.T.; et al. Novel Small RNA (SRNA) Landscape of the Starvation-Stress Response Transcriptome of Salmonella Enterica Serovar Typhimurium. RNA Biol 2016, 13, 331–342. [CrossRef]
- Landt, S.G.; Lesley, J.A.; Britos, L.; Shapiro, L. CrfA, a Small Noncoding RNA Regulator of Adaptation to Carbon Starvation in Caulobacter Crescentus. J Bacteriol 2010, 192, 4763–4775. [CrossRef]
- Babitzke, P.; Lai, Y.-J.; Renda, A.J.; Romeo, T. Posttranscription Initiation Control of Gene Expression Mediated by Bacterial RNA-Binding Proteins. Annu Rev Microbiol 2019, 73, 43–67. [CrossRef]
- Chioccioli, S.; Del Duca, S.; Vassallo, A.; Castronovo, L.M.; Fani, R. Exploring the Role of the Histidine Biosynthetic HisF Gene in Cellular Metabolism and in the Evolution of (Ancestral) Genes: From LUCA to the Extant (Micro)Organisms. Microbiol Res 2020, 240, 126555. [CrossRef]
- Fani, R.; Brilli, M.; Fondi, M.; Lió, P. The Role of Gene Fusions in the Evolution of Metabolic Pathways: The Histidine Biosynthesis Case. BMC Evol Biol 2007, 7 Suppl 2, S4. [CrossRef]
- Martínez-Guitián, M.; Vázquez-Ucha, J.C.; Álvarez-Fraga, L.; Conde-Pérez, K.; Lasarte-Monterrubio, C.; Vallejo, J.A.; Bou, G.; Poza, M.; Beceiro, A. Involvement of HisF in the Persistence of Acinetobacter Baumannii During a Pneumonia Infection. Front Cell Infect Microbiol 2019, 9, 310. [CrossRef]
- Kuba, M.; Neha, N.; De Souza, D.P.; Dayalan, S.; Newson, J.P.M.; Tull, D.; McConville, M.J.; Sansom, F.M.; Newton, H.J. Coxiella Burnetii Utilizes Both Glutamate and Glucose during Infection with Glucose Uptake Mediated by Multiple Transporters. Biochem J 2019, 476, 2851–2867. [CrossRef]
- Byer, T.; Wang, J.; Zhang, M.G.; Vather, N.; Blachman, A.; Visser, B.; Liu, J.M. MtlR Negatively Regulates Mannitol Utilization by Vibrio Cholerae. Microbiology (Reading) 2017, 163, 1902–1911. [CrossRef]
- Zheng, Z.; Jiang, T.; Zou, L.; Ouyang, S.; Zhou, J.; Lin, X.; He, Q.; Wang, L.; Yu, B.; Xu, H.; et al. Simultaneous Consumption of Cellobiose and Xylose by Bacillus Coagulans to Circumvent Glucose Repression and Identification of Its Cellobiose-Assimilating Operons. Biotechnol Biofuels 2018, 11, 320. [CrossRef]
- Aboulwafa, M.; Zhang, Z.; Saier, M.H. Protein-Protein Interactions in the Cytoplasmic Membrane of Escherichia Coli: Influence of the Overexpression of Diverse Transporter-Encoding Genes on the Activities of PTS Sugar Uptake Systems. Microb Physiol 2020, 30, 36–49. [CrossRef]
- Chen, Q.; Li, F.; Zuo, X.; Chen, J.; Qin, P.; Wang, C.; Xu, J.; Yang, D.; Xing, B.; Liu, Y.; et al. Reversible Domain Closure Modulates GlnBP Ligand Binding Affinity. PLoS One 2022, 17, e0263102. [CrossRef]
- Zhang, M.G.; Liu, J.M. Transcription of Cis Antisense Small RNA MtlS in Vibrio Cholerae Is Regulated by Transcription of Its Target Gene, MtlA. J Bacteriol 2019, 201, e00178-19. [CrossRef]
- Poorinmohammad, N.; Hamedi, J.; Masoudi-Nejad, A. Genome-Scale Exploration of Transcriptional Regulation in the Nisin Z Producer Lactococcus Lactis Subsp. Lactis IO-1. Sci Rep 2020, 10, 3787. [CrossRef]
- Jang, H.; Kim, S.T.; Sang, M.K. Suppressive Effect of Bioactive Extracts of Bacillus Sp. H8-1 and Bacillus Sp. K203 on Tomato Wilt Caused by Clavibacter Michiganensis Subsp. Michiganensis. Microorganisms 2022, 10, 403. [CrossRef]
- Cao, T.N.; Joyet, P.; Aké, F.M.D.; Milohanic, E.; Deutscher, J. Studies of the Listeria Monocytogenes Cellobiose Transport Components and Their Impact on Virulence Gene Repression. J Mol Microbiol Biotechnol 2019, 29, 10–26. [CrossRef]
Figure 1.
Growth curve (A) Growth curves of B. subtilis BS-G1 and 5 control strains at 0.015g/L glucose concentration; (B) Growth curves of B. subtilis BS-G1 at different glucose concentrations.
Figure 1.
Growth curve (A) Growth curves of B. subtilis BS-G1 and 5 control strains at 0.015g/L glucose concentration; (B) Growth curves of B. subtilis BS-G1 at different glucose concentrations.
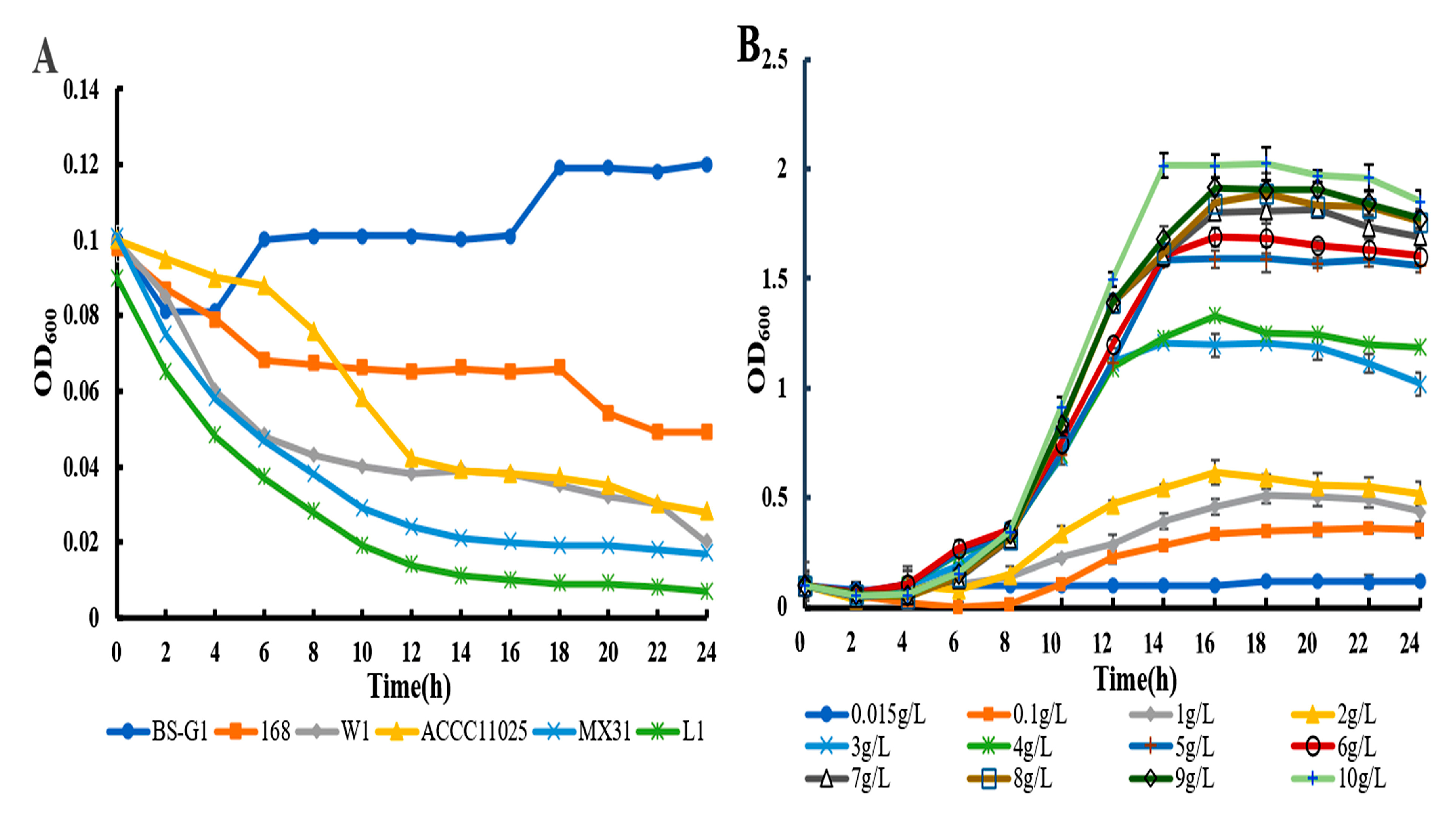
Figure 2.
Glucose uptake curve (A) Glucose uptake curves of B. subtilis BS-G1 and 5 control strains at 0.015g/L-10g/L glucose concentration uptake curve; (B) B. subtilis BS-G1 at 0.015g/L-10g/L glucose concentration glucose uptake curve.
Figure 2.
Glucose uptake curve (A) Glucose uptake curves of B. subtilis BS-G1 and 5 control strains at 0.015g/L-10g/L glucose concentration uptake curve; (B) B. subtilis BS-G1 at 0.015g/L-10g/L glucose concentration glucose uptake curve.
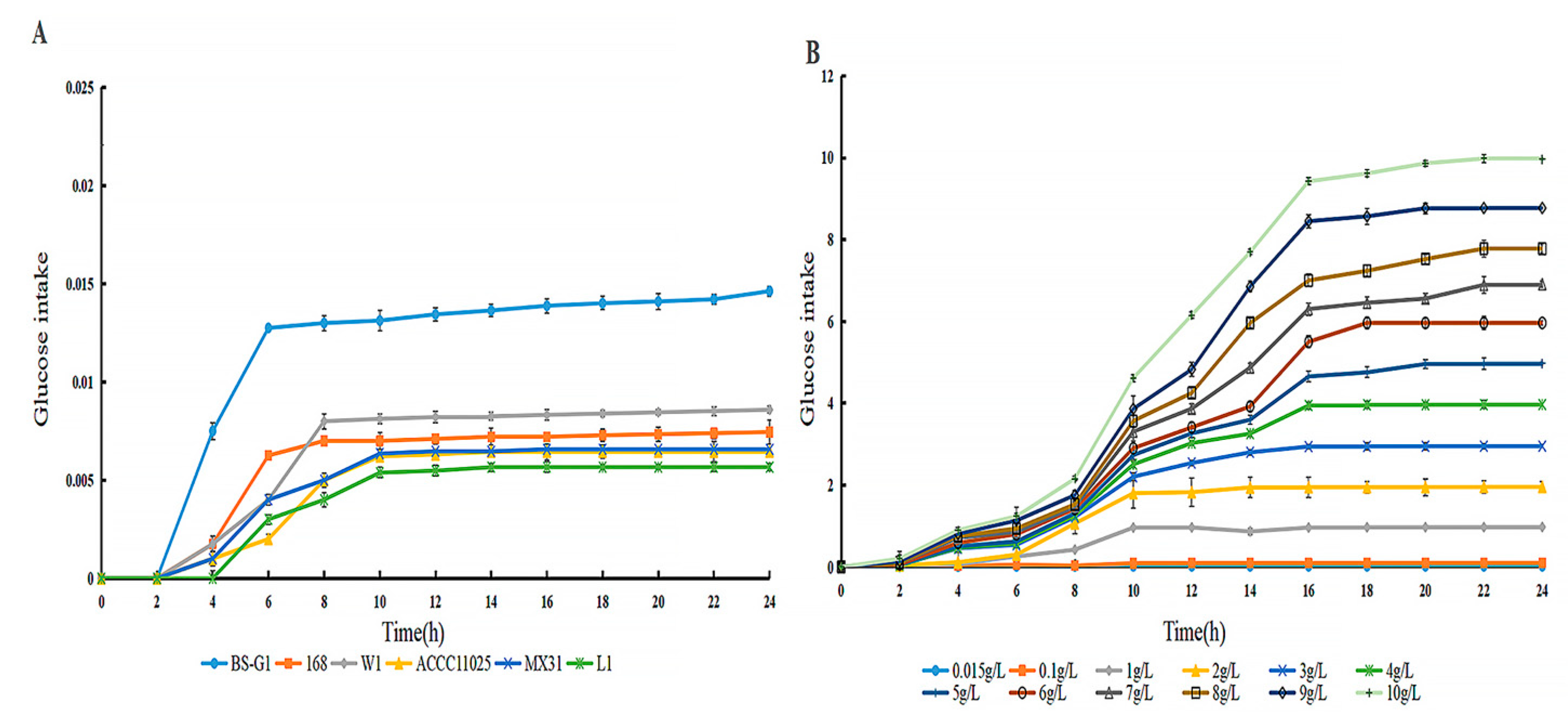
Figure 3.
COG functional classification of DEGs (A) COG functional classification of LvsH differentially expressed genes; (B) COG functional classification of MvsH differentially expressed genes.
Figure 3.
COG functional classification of DEGs (A) COG functional classification of LvsH differentially expressed genes; (B) COG functional classification of MvsH differentially expressed genes.
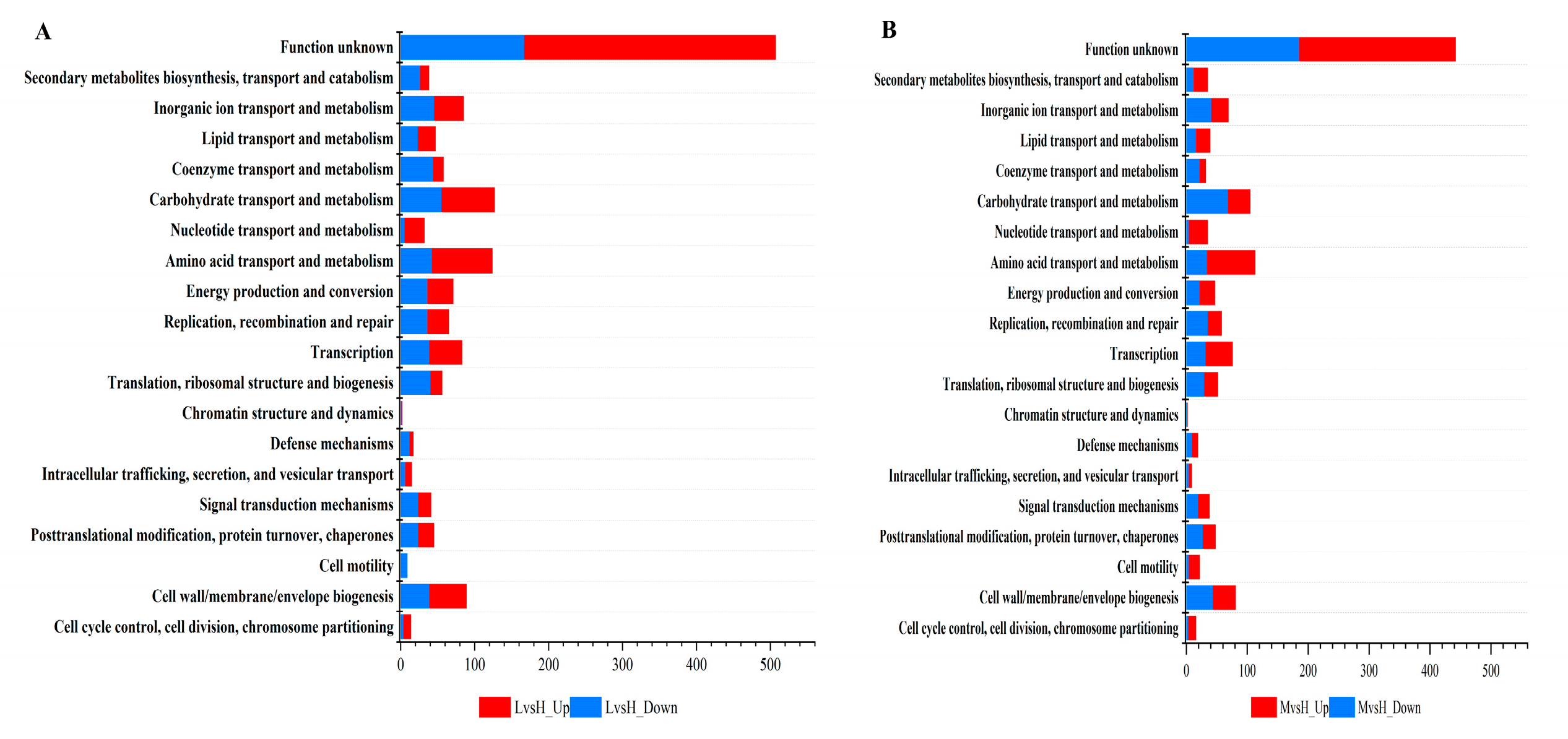
Figure 4.
KEGG functional classification of DEGs (A)KEGG functional classification of LvsH differentially expressed genes (B) KEGG functional classification of MvsH differentially expressed genes.
Figure 4.
KEGG functional classification of DEGs (A)KEGG functional classification of LvsH differentially expressed genes (B) KEGG functional classification of MvsH differentially expressed genes.
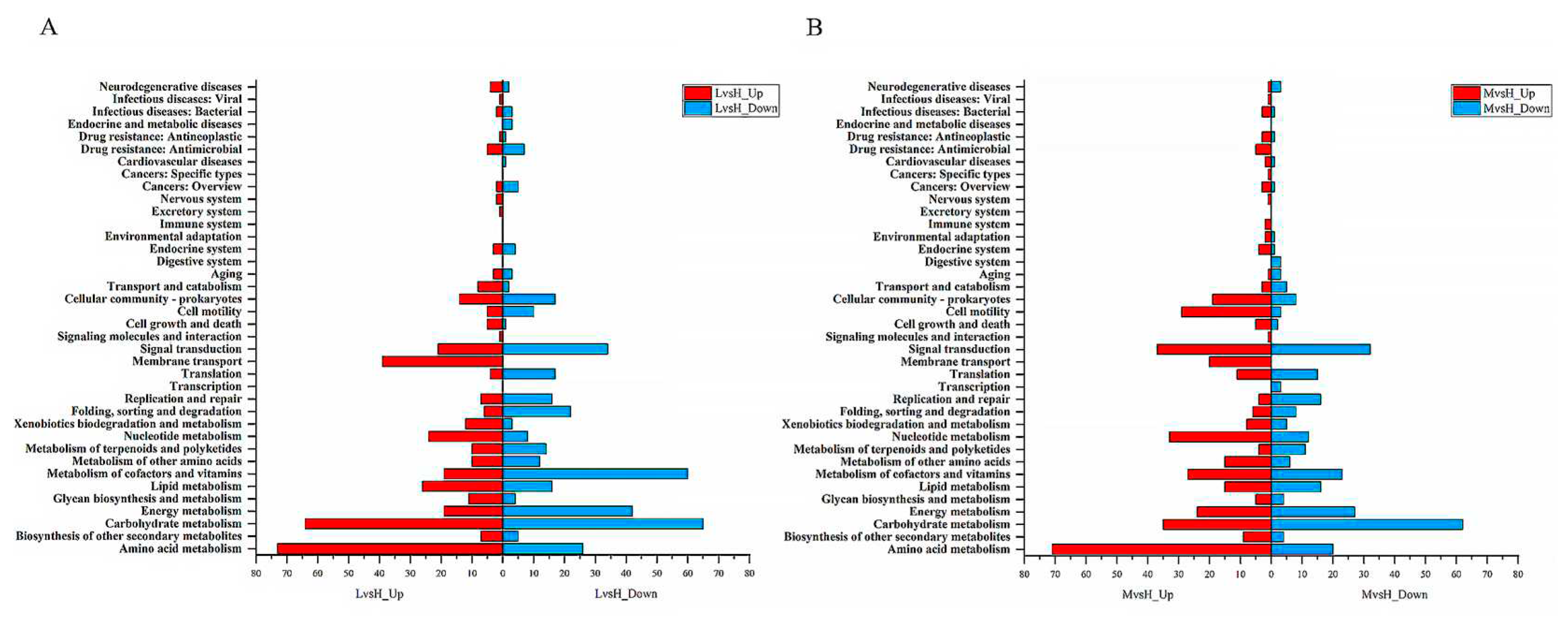
Figure 5.
GO enrichment of LvsH and MvsH differentially expressed mRNA(A) GO enrichment of LvsH differentially expressed mRNAs (B) GO enrichment of MvsH differentially expressed mRNAs. (The diagram of GO enrichment: from outside to inside, the outermost circle is the ID number of GO enrichment, the second circle is the background gene, the third circle is the number of DEGs, and the fourth circle is the enrichment factor, that is: the number of DEGs divided by as the number of background genes).
Figure 5.
GO enrichment of LvsH and MvsH differentially expressed mRNA(A) GO enrichment of LvsH differentially expressed mRNAs (B) GO enrichment of MvsH differentially expressed mRNAs. (The diagram of GO enrichment: from outside to inside, the outermost circle is the ID number of GO enrichment, the second circle is the background gene, the third circle is the number of DEGs, and the fourth circle is the enrichment factor, that is: the number of DEGs divided by as the number of background genes).
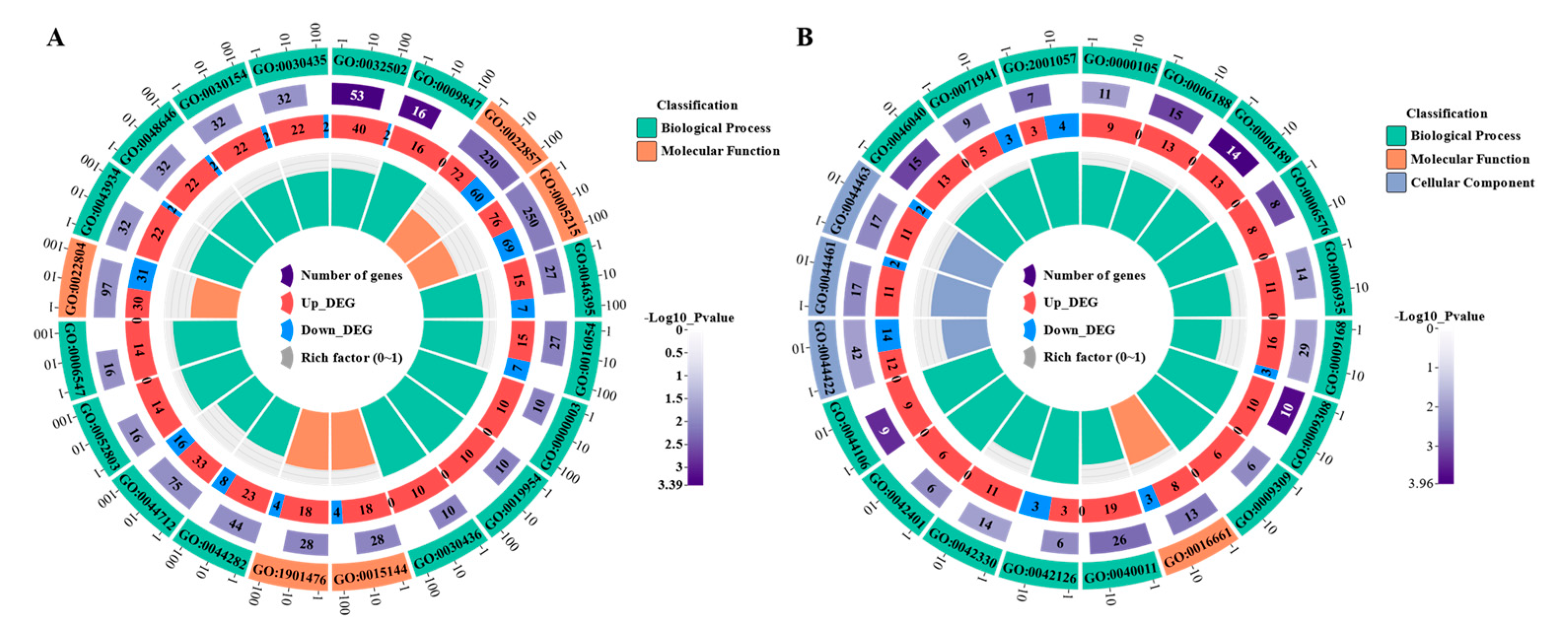
Figure 6.
KEGG enrichment of LvsH and MvsH differentially expressed mRNA (A) KEGG enrichment of LvsH differentially expressed mRNA (B) KEGG enrichment of MvsH differentially expressed mRNA.
Figure 6.
KEGG enrichment of LvsH and MvsH differentially expressed mRNA (A) KEGG enrichment of LvsH differentially expressed mRNA (B) KEGG enrichment of MvsH differentially expressed mRNA.
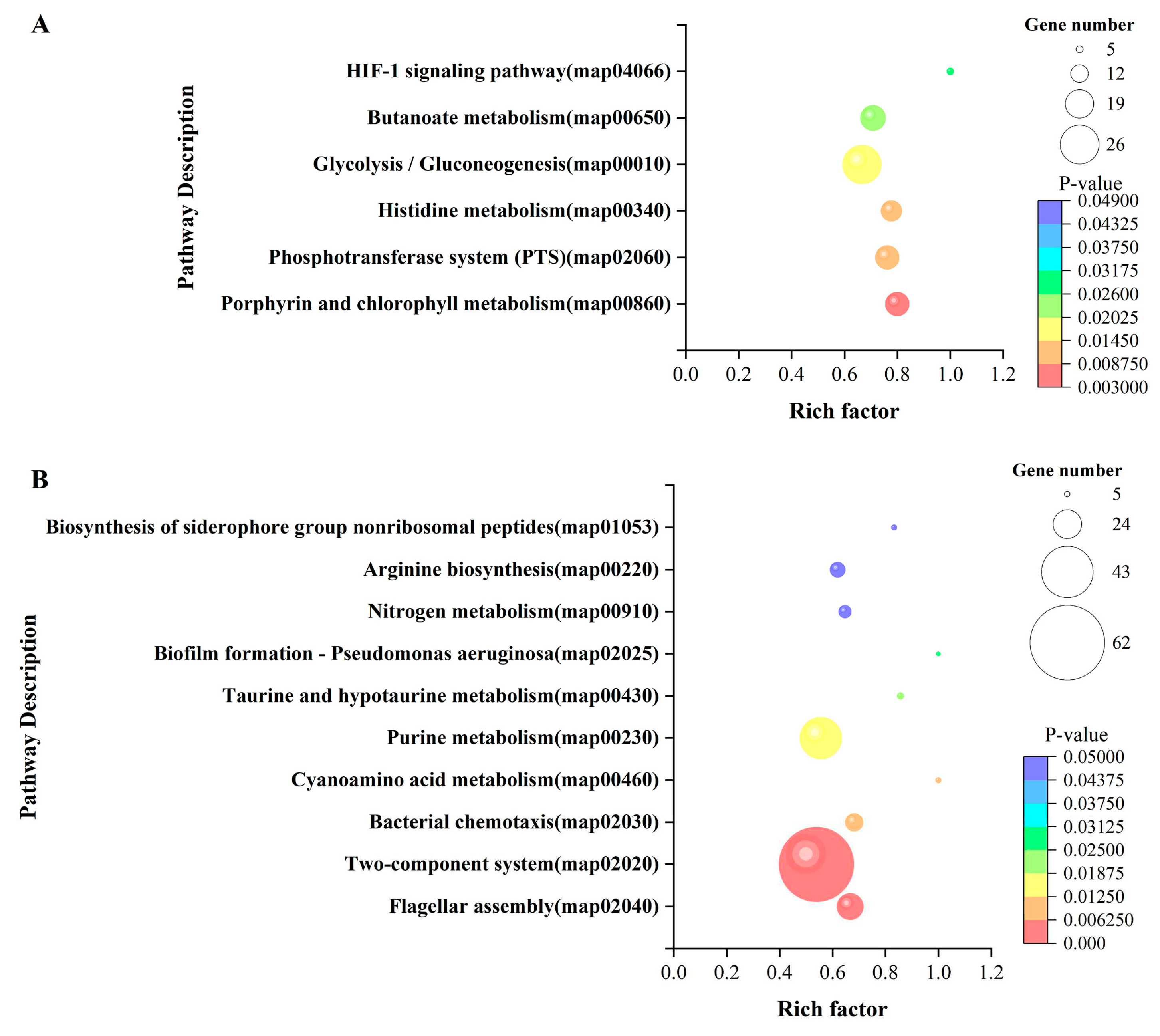
Figure 7.
Characteristics of BS-G1sRNA (A) Statistics of cis-acting sRNA and trans-acting sRNA (B) sRNA length distribution; (C) Rfam annotation statistics of sRNA).
Figure 7.
Characteristics of BS-G1sRNA (A) Statistics of cis-acting sRNA and trans-acting sRNA (B) sRNA length distribution; (C) Rfam annotation statistics of sRNA).
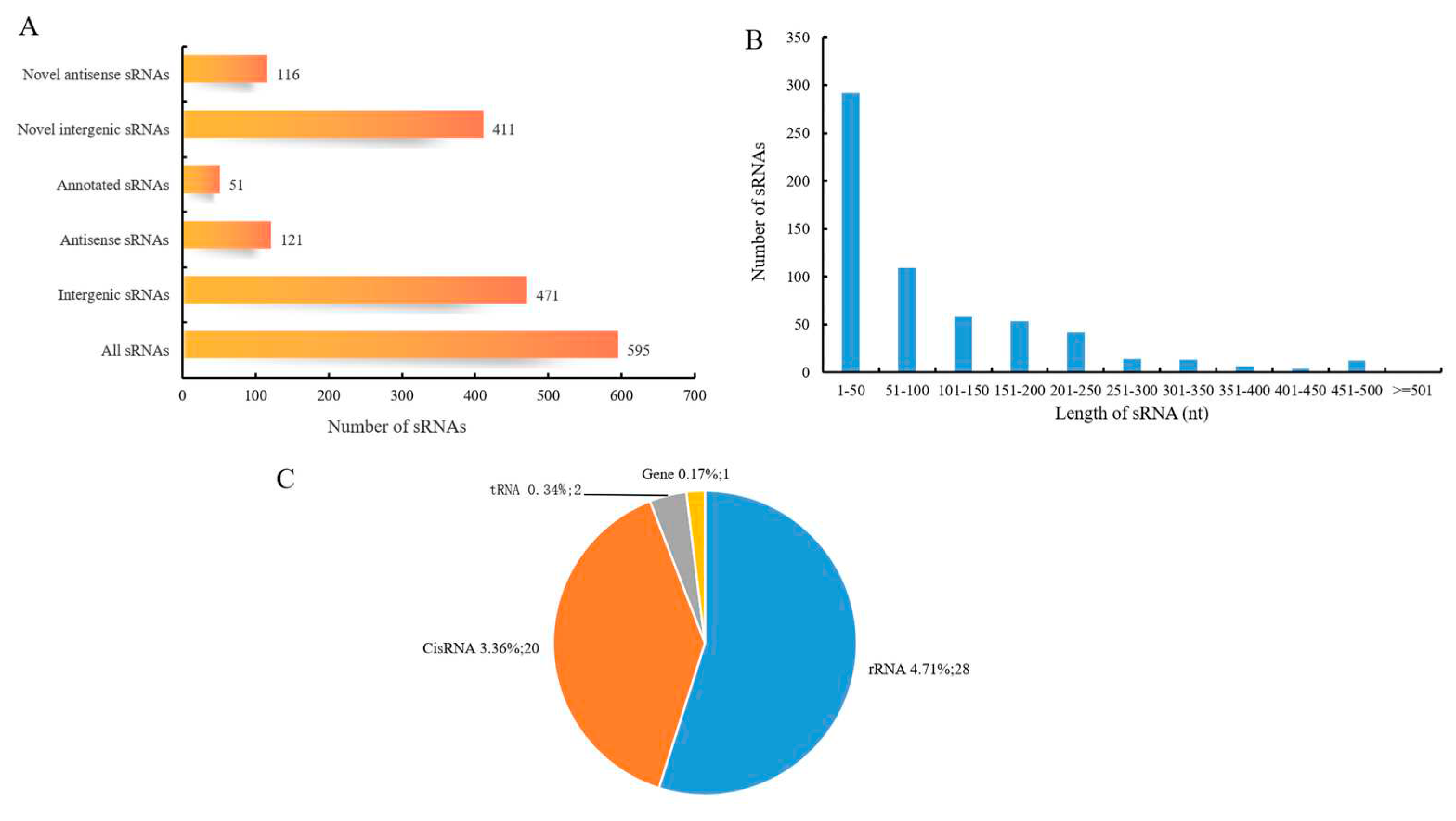
Figure 8.
Significantly differentially expressed sRNA.
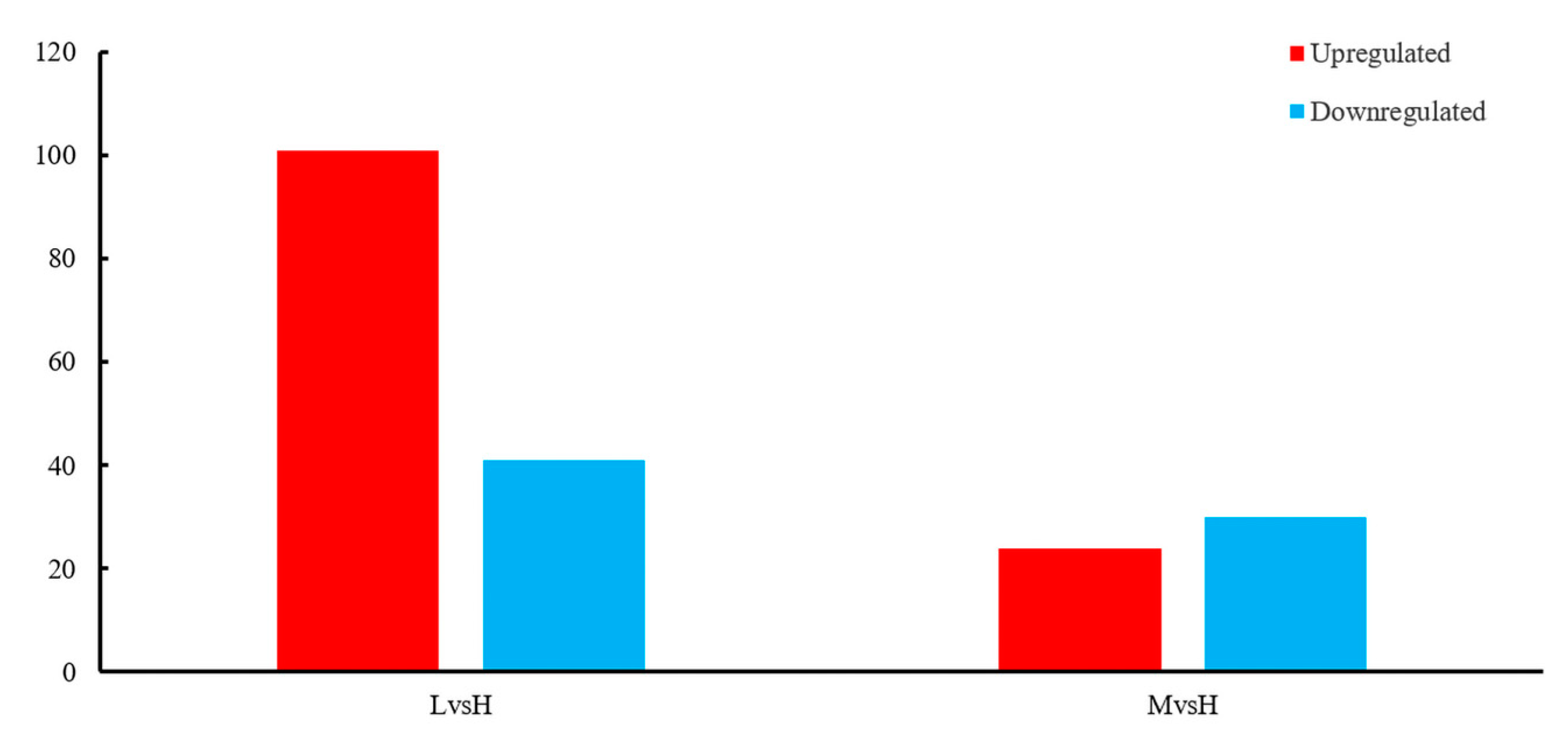
Figure 9.
COG annotation classification of target genes with significant differential expression of sRNA (A) COG annotation classification of LvsH differentially expressed sRNA target genes (B) COG annotation classification of MvsH differentially expressed sRNA target genes.
Figure 9.
COG annotation classification of target genes with significant differential expression of sRNA (A) COG annotation classification of LvsH differentially expressed sRNA target genes (B) COG annotation classification of MvsH differentially expressed sRNA target genes.
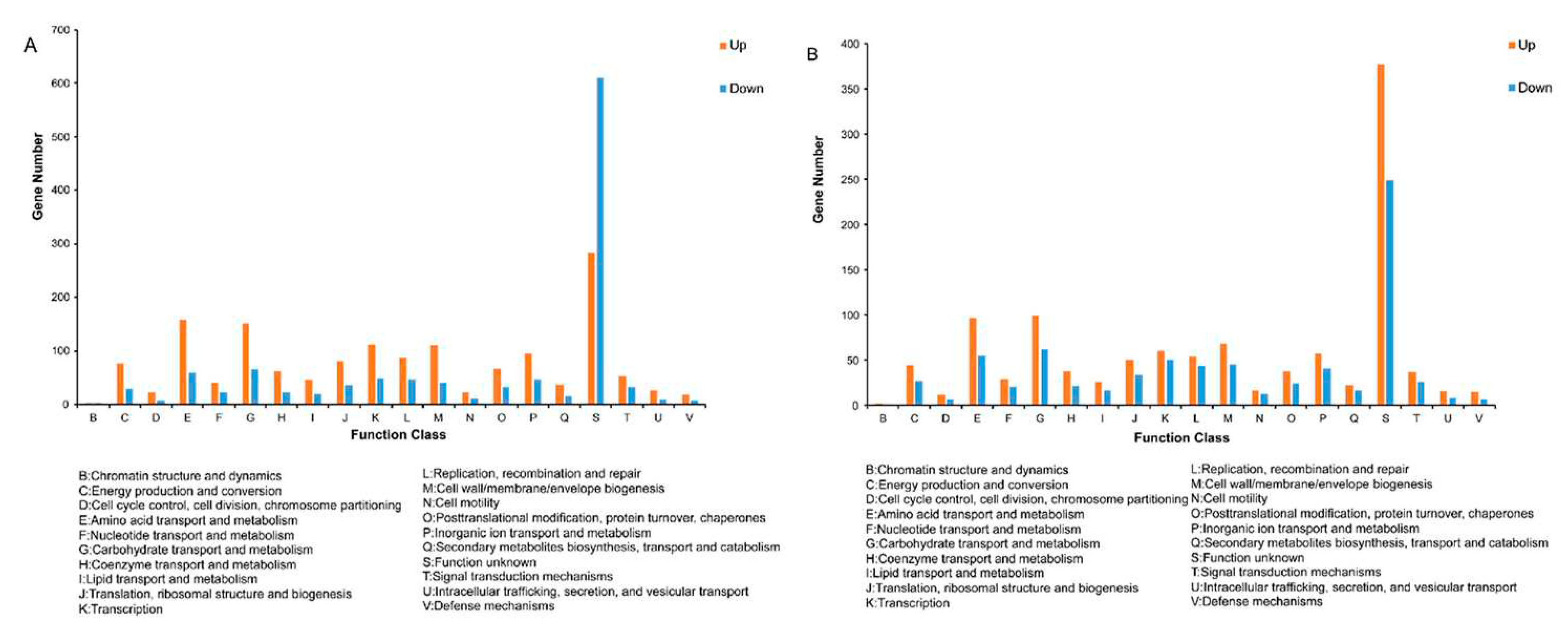
Figure 10.
KEGG enrichment of significantly differentially expressed sRNA target genes.(A)KEGG enrichment of target genes of LvsH differentially expressed sRNA (B) KEGG enrichment of target genes of MvsH expressed sRNA .
Figure 10.
KEGG enrichment of significantly differentially expressed sRNA target genes.(A)KEGG enrichment of target genes of LvsH differentially expressed sRNA (B) KEGG enrichment of target genes of MvsH expressed sRNA .
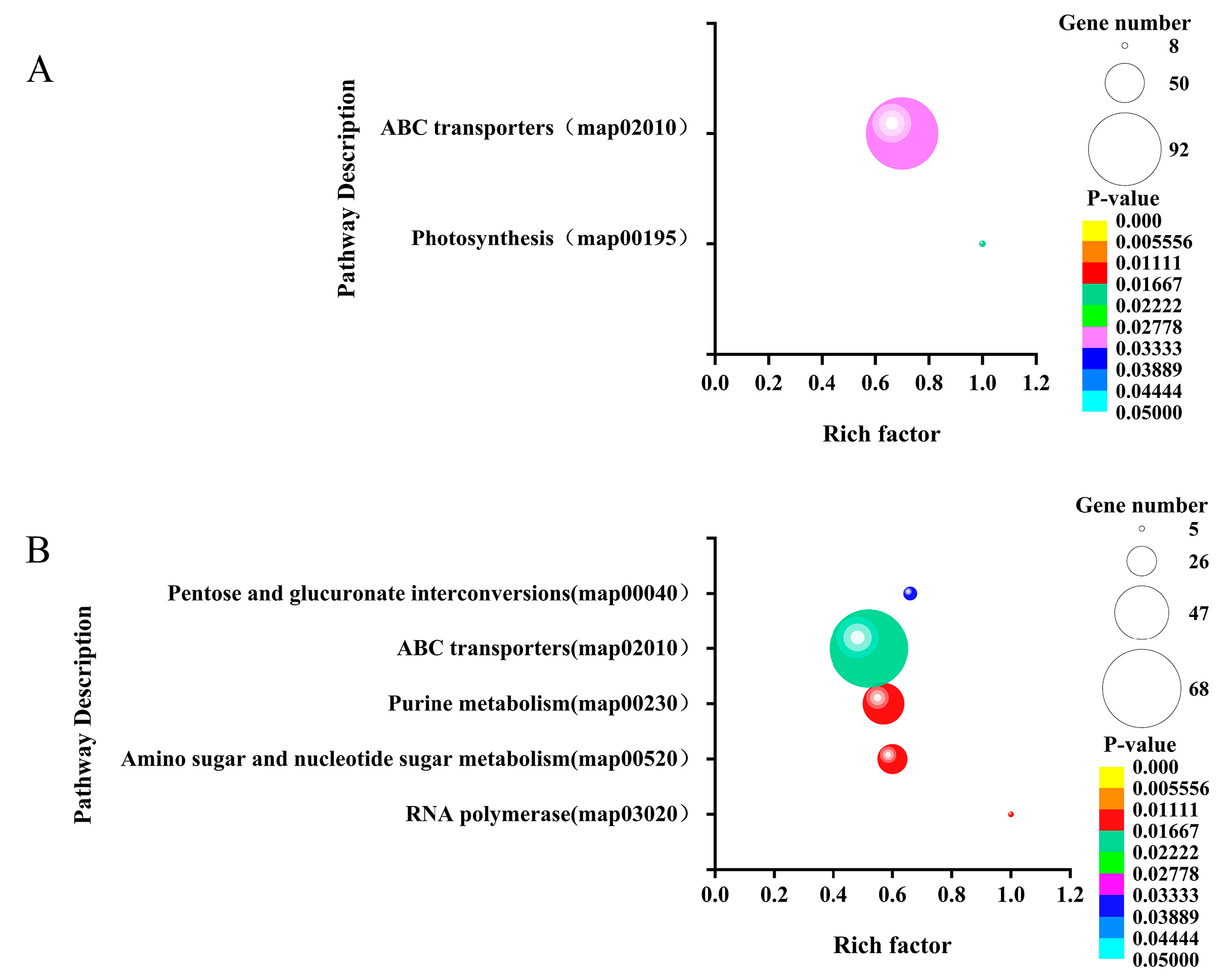
Figure 11.
Adaptation mode of B. subtilis BS-G1 under different glucose concentrations.
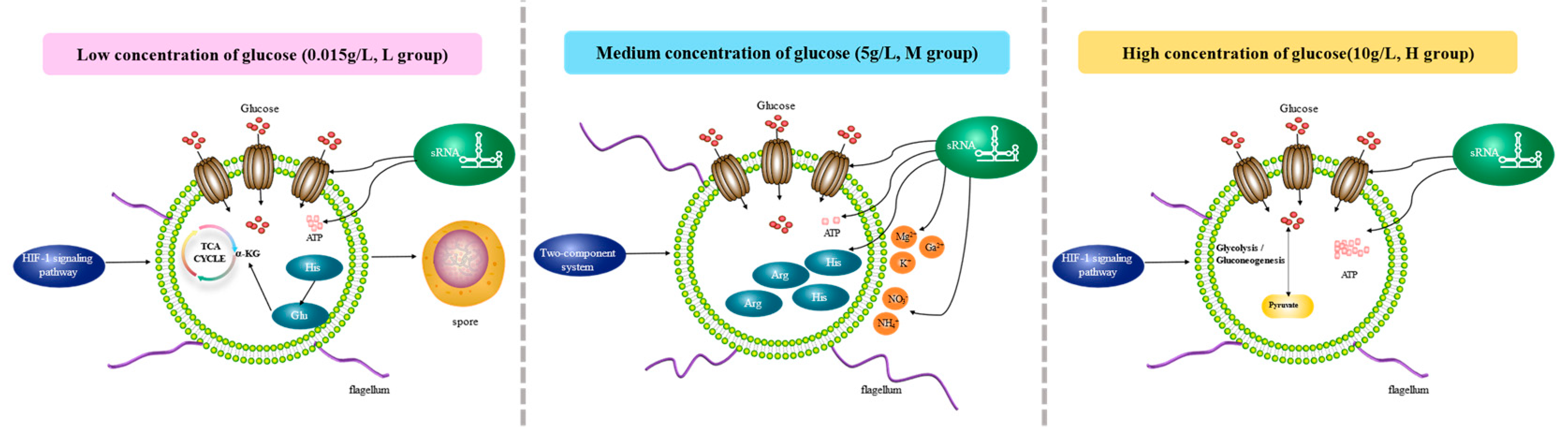
Figure 12.
Crucial mRNA and sRNA interaction network diagram.
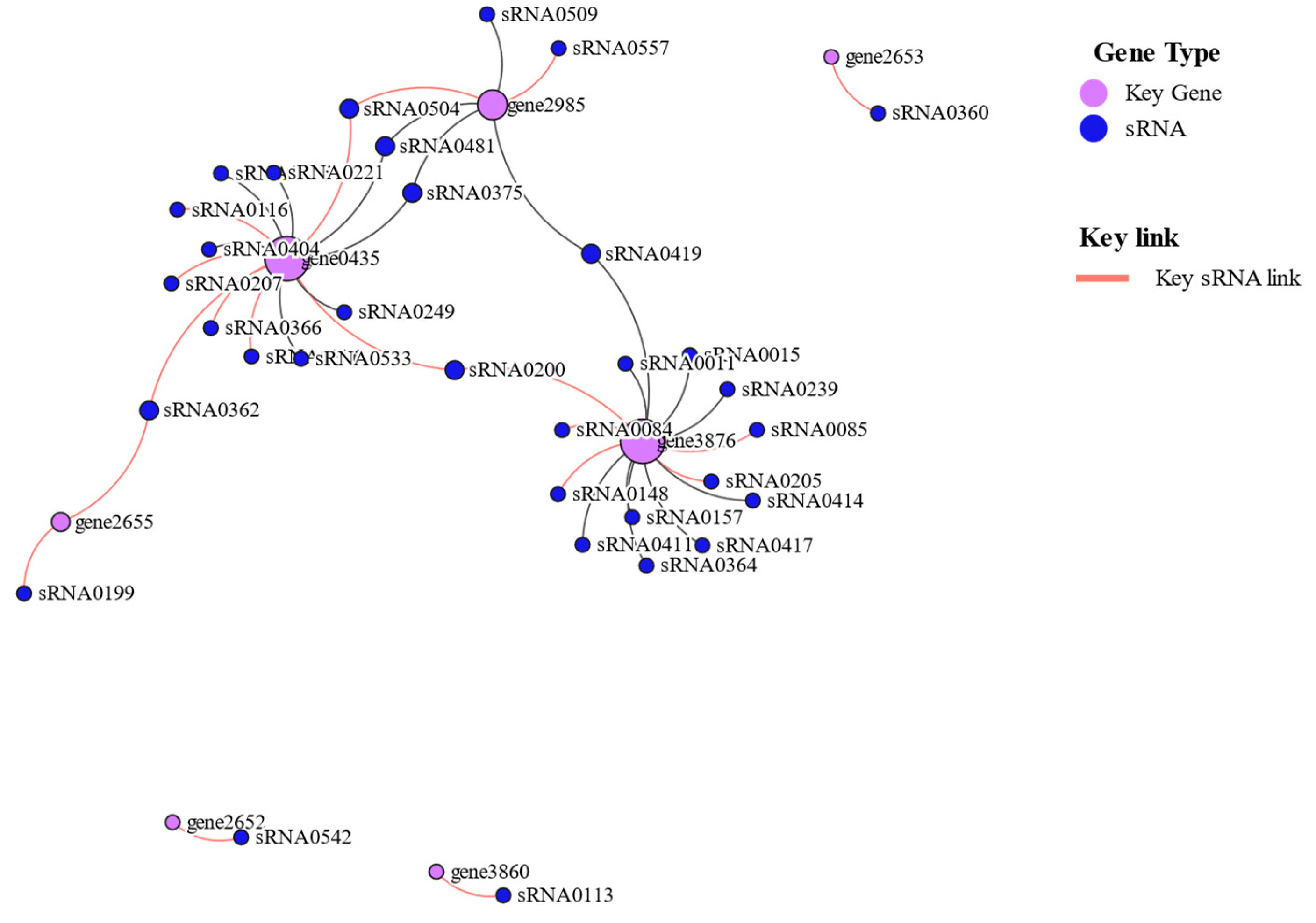
Figure 13.
The relative mRNA expression of key genes determined by qRT-PCR.
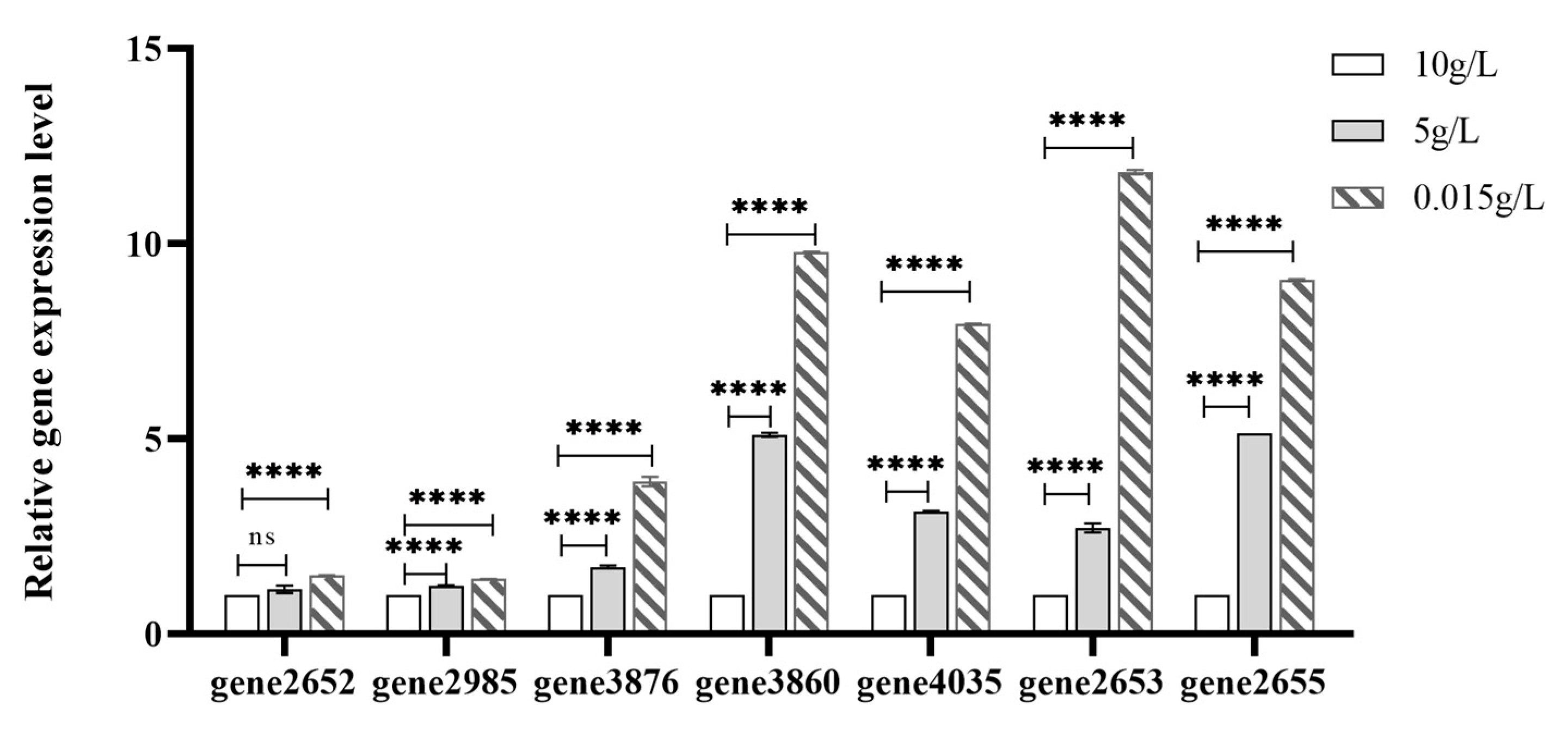

Table 1.
Critical genes (mRNA) adapted to 0.015 g/L glucose environment.
| Key Gene ID | Description | Gene Name | Log2FC(L/H) |
| gene2652 | amino acid ABC transporter ATPase | / | 7.448 |
| gene2985 | ABC transporter permease | / | 10.238 |
| gene3860 | oligo-beta-mannoside permease IIC protein | celB | 9.383 |
| gene0435 | PTS mannitol transporter subunit IIB | mtlA | 7.98 |
| gene3876 | oligo-beta-mannoside permease IIC protein | celB | 7.362 |
| gene2653 | glutamine ABC transporter substrate-binding protein | / | 7.496 |
| gene2655 | glutamine ABC transporter permease | / | 5.051 |
Table 2.
Critical mRNAs and sRNAs adapted to 0.015 g/L glucose environment.
| sRNA_id | Log2FC(L/H) | Target Gene ID |
| sRNA0200 | 11.71890433 | gene0435;gene3876 |
| sRNA0113 | 11.533039 | gene3860 |
| sRNA0084 | 11.40169888 | gene3876 |
| sRNA0085 | 11.40169888 | gene3876 |
| sRNA0148 | 11.40169888 | gene3876 |
| sRNA0207 | 10.90597229 | gene0435 |
| sRNA0205 | 9.776851885 | gene3876 |
| sRNA0366 | 8.533373277 | gene0435 |
| sRNA0336 | 6.681651624 | gene0435 |
| sRNA0362 | 6.434679786 | gene0435;gene2655 |
| sRNA0116 | 6.373911204 | gene0435 |
| sRNA0360 | 6.357940855 | gene2653 |
| sRNA0542 | 6.212579528 | gene2652 |
| sRNA0557 | 6.026353075 | gene2985 |
| sRNA0199 | 5.159987109 | gene2655 |
| sRNA0504 | 5.048301125 | gene2985;gene0435 |
| sRNA0015 | 4.964673587 | gene3876 |
| sRNA0533 | 4.896095911 | gene0435 |
| sRNA0419 | 4.801798083 | gene2985;gene3876 |
| sRNA0239 | 4.618899947 | gene3876 |
| sRNA0249 | 4.578804747 | gene0435 |
| sRNA0495 | 3.975578933 | gene0435 |
| sRNA0411 | 3.854669068 | gene3876 |
| sRNA0509 | 3.764795398 | gene2985 |
| sRNA0221 | 3.520639327 | gene0435 |
| sRNA0414 | 3.022561069 | gene3876 |
| sRNA0481 | 2.967066947 | gene0435、gene2985 |
| sRNA0417 | 1.934699587 | gene3876 |
| sRNA0364 | 1.373518955 | gene3876 |
| sRNA0011 | 1.103524326 | gene3876 |
| sRNA0157 | -1.52566018 | gene3876 |
| sRNA0404 | -1.81557978 | gene0435 |
| sRNA0375 | -2.395421117 | gene2985;gene0435 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



