
Submitted:
28 August 2023
Posted:
29 August 2023
You are already at the latest version
Abstract
Stellatolides are natural compounds that have shown promising biological activities, including antitumor, antimicrobial, and anti-inflammatory properties, making them potential candidates for drug development. Chemical Reactivity Theory (CRT) is a branch of chemistry that explains and predicts the behavior of chemical reactions based on the electronic structure of molecules. Conceptual Density Functional Theory (CDFT) and Computational Peptidology (CP) are computational approaches used to study the behavior of atoms, molecules, and peptides. In this study, we present the results of our investigation of the chemical reactivity and ADMET properties of Stellatolides A-H using a novel computational approach called Conceptual DFT-based Computational Peptidology (CDFT-CP). Our study uses CDFT and CP to predict the reactivity and stability of molecules and to understand the behavior of peptides at the molecular level. We also predict the ADMET properties of the Stellatolides A-H to provide insight into their effectiveness, potential side effects, and optimal dosage and route of administration. This study sheds light on the potential of Stellatolides A-H as promising candidates for drug development and highlights the potential of CDFT-CP for the study of other natural compounds and peptides.
Keywords:
Drug discovery
; computational chemistry
; conceptual DFT
; marine cyclopeptides
; chemical reactivity properties
; bioavailability scores
1. Introduction
Stellatolides are a group of natural compounds that have been isolated from a variety of marine organisms, including sponges and tunicates. They are characterized by their distinctive chemical structure, which features a complex macrolide ring system with multiple functional groups. Stellatolides have generated significant interest in the pharmaceutical industry due to their potential as a source of novel drugs for the treatment of various diseases [1,2,3,4,5,6].
The chemistry of stellatolides is complex and fascinating. These compounds are composed of a large macrolide ring system, which is formed through a series of complex biosynthetic pathways. The exact mechanisms by which stellatolides are synthesized in nature are not yet fully understood, but it is believed that they are produced by a combination of enzymatic and non-enzymatic processes [1,2,3,4,5,6].
Stellatolides have been shown to have a range of biological activities, including antitumor, antimicrobial, and anti-inflammatory properties. These activities are believed to be mediated by the ability of stellatolides to interact with specific biological targets in the body, such as enzymes or receptors [1,2,3,4,5,6].
One of the most exciting aspects of stellatolides is their potential as pharmacological drugs. Researchers have identified several stellatolide derivatives that show promising activity against a range of diseases, including cancer and infectious diseases. These compounds have been shown to have potent anticancer activity against a range of tumor cell lines, and they may offer a promising alternative to traditional chemotherapy drugs [7,8,9,10].
Stellatolides also have potential as a source of new antibiotics. With the rise of antibiotic-resistant bacteria, there is a growing need for new drugs that can effectively treat infections [11]. Stellatolides have been shown to have potent antimicrobial activity against a range of bacteria, including methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococcus (VRE) [12].
In summary, stellatolides are a fascinating group of natural compounds with a wide range of biological activities and potential as pharmacological drugs. Further research is needed to fully understand their mechanisms of action and to develop effective drug candidates, but they represent a promising area of research in the pharmaceutical industry [1,2,3,4,5,6,7,8,9,10].
In spite of the allegations that peptides have no future as therapeutic drugs based on the violation of the Lipinski’s rules [13] related to their size, solubility, and flexibility, leading to poor bioavailability, it should be mentioned that this assertion does not hold for cyclopeptides [14]. There is a lot of published research validating this phenomena and also explaining the mechanisms for the drug delivery into the body [15,16,17,18,19,20,21]. Moreover, there are ongoins studies on the use of different substances as nanocarriers allowing the correct delivery of the potential therapeutic peptides [22,23,24,25].
Chemical Reactivity Theory (CRT) is a branch of chemistry that seeks to explain and predict the behavior of chemical reactions based on the electronic structure of molecules. The basic idea behind CRT is that the reactivity of a molecule depends on its electronic structure, specifically the distribution and arrangement of electrons in the molecule. This electronic structure determines how the molecule interacts with other molecules or ions, leading to chemical reactions [26]. One of the key concepts in CRT is the concept of frontier molecular orbitals. These are the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of a molecule. The HOMO represents the electron density that is most likely to participate in chemical reactions, while the LUMO represents the electron density that is most easily available to accept electrons [27].
One of the most succesfull approaches to CRT is the so-called Conceptual Density Functional Theory (CDFT) which is a theoretical framework used to study the behavior of atoms and molecules in chemistry and physics. The conceptual aspect of DFT refers to the interpretation of the electronic density in terms of chemical concepts, such as bond formation and reactivity. For example, the electron density can be used to predict chemical reactions, identify reactive sites, and explain the properties of materials. Thus, by interpreting the electronic density in terms of chemical concepts, CDFT provides insight into the behavior of molecules and materials and can be used to predict and understand chemical reactions and material properties [27,28,29,30,31,32,33,34].
Computational Peptidology (CP) is a field of research that uses computational methods to study the properties and behavior of peptides, which are small chains of amino acids that are important in many biological processes. The basic idea behind CP is to use computer simulations and modeling to understand the behavior of peptides at the molecular level. This involves studying the interactions between individual amino acids, as well as the interactions between peptides and other molecules in their environment, such as proteins or membranes. By considering both approaches, CDFT and CP, for the study of peptides, we have developed Conceptual DFT-based Computational Peptidology (CDFT-CP), which is a computational approach that uses DFT, CDFT and CP to study the properties and behavior of peptides at the molecular level. CDFT-CP uses concepts such as electronegativity, hardness, softness, and electrophilicity to predict the reactivity and stability of molecules [35,36,37,38,39,40,41,42].
The objective of this research is to report the results of CDFT-CP study of the chemical reactivity properties of the Stellatolides A-H marine cyclopeptides [7,9]. This will be complemented by the computational prediction of their ADMET properties [43]. ADMET is an acronym that stands for Absorption, Distribution, Metabolism, Excretion, and Toxicity. It refers to a set of processes that a drug undergoes in the body [44,45]. Comprehending the ADMET characteristics of a pharmaceutical compound holds significance in the realm of drug development. This understanding aids in foreseeing the drug’s efficacy, potential adverse reactions, and the most suitable dosage and method of delivery [46].
2. Materials and Methods
2.1. Density Functional Theory (DFT) Calculations
The Kohn-Sham (KS) methodology involves assessing the energy and density of a specific molecular system, along with the orbital energies associated with the frontier orbitals such as the Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO) [47,48,49,50]. This approach is particularly useful for evaluating quantitative characteristics associated with Conceptual DFT descriptors [27,28,29,30,31,32,33,34].
In the present investigation, the identification of the molecular conformers was carried out utilizing MarvinView 17.15, a software provided by ChemAxon (http://www.chemaxon.com). Molecular Mechanics calculations were performed using the MMFF94 force field [51,52,53,54,55]. Subsequently, a geometry optimization and frequency calculation were conducted using the Density Functional Tight Binding (DFTBA) approach [56]. The purpose of this final stage was to ensure the stability of the optimized structures by confirming the absence of imaginary frequencies, which serves as a criterion for determining their status as energy minima in the overall energy landscape.
The electronic properties and chemical reactivity descriptors of the molecules under investigation were analyzed using the MN12SX/Def2TZVP/H2O model chemistry [57,58,59]. The molecular structures were previously optimized as mentioned earlier, and this particular model chemistry was selected because it allows for the validation of the ’Koopmans in DFT’ (KID) procedure [36,37,38,39,40,41,42]. The calculations were performed using Gaussian 16 software 56 and the SMD solvation model [60]. The chosen model chemistry comprises the MN12SX screened-exchange density functional [57] and the Def2TZVP basis set [58,59]. Throughout the analysis, the molecules were treated as neutral, while the radical anion and cation were considered in the doublet spin state.
2.2. In Silico Pharmacokinetics Analysis and ADMET Study
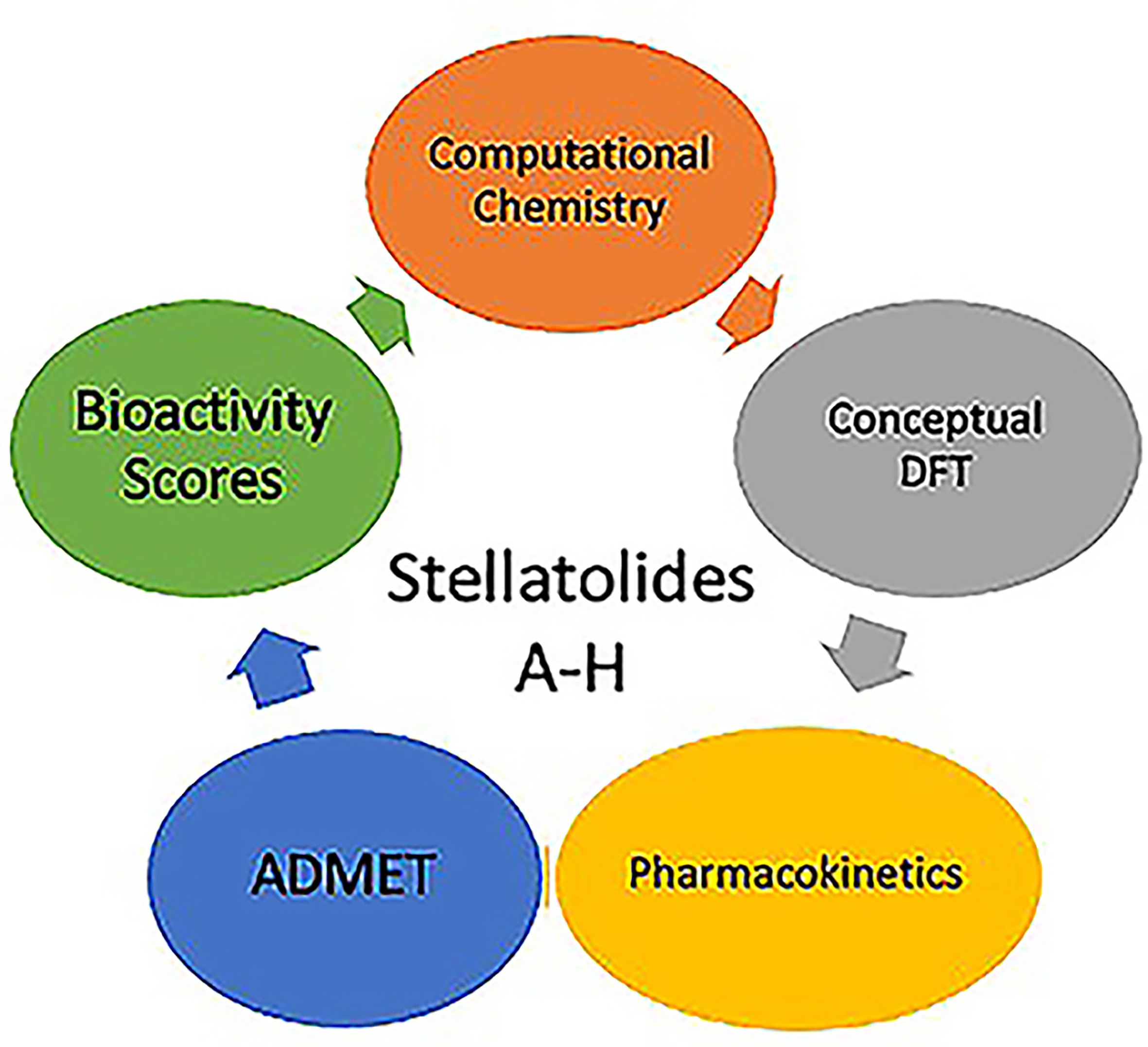
The initial molecular configurations of the investigated antimicrobial marine cyclopeptides slated for examination were acquired from ChemSpider (https://www.chemspider.com). ChemSpider is a no-cost chemical structure repository that offers rapid text and structural search capabilities for more than 100 million structures sourced from numerous data outlets. It also encompasses details about physical, chemical, and biological attributes, interactive spectra, and references to scholarly works. These initial molecular structures are shown in Figure 1.
To gain insight into the potential therapeutic attributes of the peptides under investigation, we utilized the SMILES (Simplified Molecular Input Line Entry Specification) notation of each compound. This notation was obtained by utilizing ChemDoodle 11.3.0 software. Subsequently, we employed the Molinspiration software from Molinspiration Cheminformatics (https://www.molinspiration.com) to compute the molecular properties associated with drug-like characteristics.
The objective of exploring similarity searches within the chemical realm of compounds, comparing them to the compounds under investigation and known pharmacological properties, was successfully achieved by utilizing the online Molinspiration software. This software facilitated the prediction of bioactivity scores for various drug targets, including GPCR ligands, kinase inhibitors, ion channel modulators, enzymes, and nuclear receptors.
In order to assess the potential bioactivity of the antimicrobial marine cyclopeptides examined in this research, we employed a webtool called SwissTargetPrediction [61]. This tool efficiently predicts protein targets for small molecules. By utilizing this associated website, it was possible to estimate the most likely macromolecular targets of a small molecule assumed to be bioactive. The prediction process incorporates a combination of 2D and 3D similarity analysis using a comprehensive library of over 370,000 known active compounds targeting more than 3,000 proteins from three distinct species.
During the process of developing a novel medication, it is crucial to understand the potential path of the therapeutic compound within the body, a concept referred to as pharmacokinetics. This involves assessing various aspects known as Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) parameters, which provide insights into the compound’s effects. Rather than relying solely on experimental methods, computer models can serve as an alternative approach to determine these parameters. In our study, we utilized the SwissADME software, which is accessible online, to estimate a subset of ADME parameters [44,62].
Additional information about the Pharmacokinetics and ADMET properties were obtained by resorting to pkCSM [63], a software for the prediction of small-molecule pharmacokinetic properties using graph-based signatures or SMILES and that can be accessed through it associated webpage (https://biosig.unimelb.edu.au/pkcsm/). Chemicalize, which is a software developed by ChemAxon (http://www.chemaxon.com), was used for name to structure generation and the prediction of several properties related to Chemoinformatics (http://chemicalize.com/) (March, 2023).
3. Results and Discussion
It is often assumed that the quality of a density functional can be assessed by comparing its results to experimental values or to the results of post-Hartree-Fock calculations. However, this is not always possible, as experimental data may not be available for the systems being studied, or the molecules may be too large for accurate calculations to be feasible.
To address this issue, a protocol called KID (Koopmans in DFT) has been developed [35,36,37,38,39]. This protocol aims to validate a density functional by assessing its internal consistency. The KID protocol has been previously shown to be effective for peptides, but it is worth further validating for the molecules considered in this study.
The KID protocol was implemented using the in-house developed CDFT software tool. The results of this analysis are shown in Table 1:
The J and J descriptors consider differences between the HOMO and ionization energy (I), and between the LUMO and electron affinity (A) respectively. According to the Ionization Energy Theorem, A can be predicted by the SOMO, which is equivalent to the anion’s HOMO. To align with Koopmans’ theorem, we introduced the SL descriptor, comparing SOMO and LUMO values. The J descriptor verifies the difference between the H-L gap and the I-A gap calculated from energy differences of neutral, positive, and negative species.
The results in Table 1 show that the KID descriptor values are nearly 0, indicating that the MN12SX/Def2TZVP/H2O model chemistry is effective for verifying Janak and Ionization Energy theorems. Although further calculations involve energy differences, accurate predictions for HOMO, LUMO, and H-L gap provide confidence in our estimations of chemical reactivity properties. These estimations are theoretically grounded, not accidental as often seen with other density functionals.
The optimized molecular structures of the eight members of the Stellatolides A-H family of cylopeptides are displayed in Figure 2. These optimized structures have been obtained by resorting to the procedure described in subSection 2.1 of the Materials and Methods section.
The usual way of analyzing the chemical reactivity of molecular systems through the consideration of Conceptual DFT is by reporting the values of the global and local descriptors that arise from this theory [27,28,29,30,31,32,33,34]. The three basic descriptors are the electronegativity , the global hardnesss , and the global electrophilicity . The first one, is a measure of the tendency of an atom to attract electrons. It is a fundamental property of atoms that affects their chemical bonding and reactivity. In CDFT, electronegativity is defined as the Lagrange multiplier associated with the constraint that the total electronic chemical potential of a system is equal to a constant. This means that electronegativity is a measure of the resistance of an atom to losing electrons, and it can be estimated through the operational formula [27,28,29,30,31,32,33,34].
Global hardness is a crucial concept within the realm of CDFT, which imeasures the resistance of a system to changes in its electron density when subjected to external perturbations. The global hardness can be used to predict the reactivity of a molecule. A molecule with a high global hardness is less reactive than a molecule with a low global hardness. This is because a molecule with a high global hardness is more difficult to polarize, and therefore less likely to react with other molecules. The formula is used for the evaluation of , either considering energy differences or frontier orbitals [27,28,29,30,31,32,33,34].
The third basic descriptor, gobal electrophilicity [64] is particularly useful in predicting reactions and interactions between molecules. It can be expressed as a relation between and as = . A more electrophilic species tends to react with more nucleophilic ones, as electron flow will be from the nucleophile to the electrophile. This concept aids in understanding reaction mechanisms, molecular binding, and site-selectivity in chemical processes [27,28,29,30,31,32,33,34].
In addition to the global reactivity descriptors originating from Conceptual Density Functional Theory (DFT), Domingo and his colleagues [65,66,67,68,69] introduced a Nucleophilicity Index labeled as N. This index is established by evaluating the HOMO energy using the KS approach, wherein the reference point is shifted arbitrarily, centered around the tetracyanoethylene (TCE) molecule.
Newer developed global reactivity descriptors are the electrodonating and electroaccepting powers: = , = , respectively [29], and the net electrophilicity [30] as a comparison of the former two.
By considering the mentioned CDFT software tool as well as the MultiWFN package [70,71] applied to the results of the calculation of the electronic properties of the studied cyclopeptides, the values of the defined global reactivity descriptors could be obtained and they are displayed in Table 2:
From Table 2, the values of the electronegativity range from 3.2610 eV for Stellatolide A to 3.9451 eV for Stellatolide F. A little inferior result is obtained for Stellatolide G, while the other considered peptides have similar intermediate values for this descriptor. If we now turn our attention to global hardness , then it can be appreciated that the lowest value of this property is for the case of Stellatolide F, with 4.6145 eV. The results from both reactivity descriptors agree in that Stellatolide F will be the most reactive cyclopeptide of this family. The other members display larger values of , meaning that they will be less reactive than the H species. Notwithstanding, there are differences between Stellatolides F and G, that will be a bit more reactive than Stellatolides A-E. As mentioned earlier, the global electrophilicity represents a balance between and . The results for this descriptor from Table 2 ranges bteween 1.1759 eV for Stellatolide C to 1.6864 eV for Stellatolide F. Stellatolides A-E together with Stellatolide H display the lowest values of , while Stellatolide G presents a bit greater number. The analysis of the first three basic chemical reactivity descriptors lead us to the conclusion that Stellatolide F will the most reactive of all the cyclopeptides considred in this research.
As mentioned earlier, the nucleophilic index N is another important quantity that serves as a descriptor of chemical reactivity. From Table 2, their values ranges from 2.8066 eV for Stellatolide C to 3.1565 eV for Stellatolide H. As a matter of fact, all the member of this family have similar values for the nucleophilicity index N, with the exception of stellatolide H which is a bit larger. According to the clasification proposed by Domingo et al [65,66,67,68,69], while Stellatolide H may be regarded as a strong nucleophile, the others can be considered as moderated ones.
The last three descriptors in Table 2 are interrelated between them. As it is usual for this kind of molecules, the electrodonating power is large that the corresponding electroaccepting power . This behavior could be attributed to their cyclic structure and the large number of conjugated bonds, as well as the presence of several O and N atoms. Stellatolide F possesses the larges value of , followed by Stellatolide G, while the others display almost the same values for this chemical reactivity descriptor. For the case of the electroaccepting power , again Stellatolide F is the most important among the whole family of cyclopeptides, with significative differences with the other molecules, with the exception of Stellatolide G. As expected, the net electrophilicity , being defines as a way to compare with , shows the same behavior for the cyclopeptides as for the other two reactivity descriptors, allowing the conclusion that Stellatolide F will be the most reactive cyclopeptide of all those considered in this work.
The chemical reactivity descriptors that have been reported are classified as global indicators, as they play a crucial role in predicting the overall properties of molecules. Nevertheless, there are instances where delving into the characteristics of specific regions within molecular systems becomes intriguing. By recognizing that distinct chemical reactivity is confined to various zones within these molecules, we gain insights into their behavior. This prompts the use of what are known as local reactivity descriptors, which stem from the principles of Conceptual DFT (CDFT) [27,28,29,30,31,32,33,34].
The Fukui functions (FF), cited in references such as [27,28,29,30,31,32,33,34], stand out as the most prevalent local descriptors in Conceptual Density Functional Theory (CDFT). These functions can be categorized into nucleophilic, electrophilic, and radical Fukui functions, their classification contingent on the incoming reagent and the specific attack site. This classification enables FF to meticulously characterize individual atoms within a molecule where reactions might take place.
While this approach has proven incredibly valuable and achieved significant success in the context of small molecules, its application to larger molecular systems poses challenges. This is due to the tendency for the resulting values to become exceedingly minute, making it arduous to differentiate among them.
In CDFT, the dual descriptor, often abbreviated as DD, stands as a powerful tool utilized for the examination of molecule reactivity [72,73]. This descriptor furnishes a quantification of a molecule’s nucleophilic and electrophilic traits, thus establishing its worth as a predictive instrument in anticipating chemical reactivity.
The derivation of the dual descriptor DD emanates from the frontier molecular orbitals, namely, the HOMO and LUMO of the molecule. çVia the HOMO and LUMO, one can obtain the corresponding electrophilic and nucleophilic Fukui functions, which in turn allow for the computation of the dual descriptor DD [72,73,74,75].
The dual descriptor DD boasts an array of chemistry applications, encompassing catalyst design, chemical reaction prognosis, and chemical reactivity analysis. This tool serves as a valuable asset for comprehending the electronic structure of molecules, enabling the prediction of their conduct in chemical reactions.
By harnessing the dual descriptor DD, one can foresee a molecule’s reactivity toward nucleophilic or electrophilic assaults. A markedly positive value of this descriptor signifies the molecule’s proficiency as an electrophile, while a notably negative value suggests its competence as a nucleophile [72,73,74,75].
Figure 3 and Figure 4 display a graphical representation of the dual descriptor DD of the Stellatolides A-H in a separate way: the left pictures indicates the zones where DD > 0, while the right ones corresponds to the areas where DD < 0.
Upon reviewing Figure 3 and Figure 4, it becomes evident that it is indeed possible to discern specific regions within the peptides that are prone to susceptibility towards either nucleophilic or electrophilic attacks. This capability proves to be immensely valuable, as it lays the groundwork for the prospective evaluation of these molecular systems as potential therapeutic agents.
The online platform for open bioactivity prediction, such as Molinspiration by Molinspiration Cheminformatics in Slovensky Grob, Slovak Republic, has proven instrumental in assessing molecular properties and bioactivity scores. This is achieved through an analysis of the chemicals in question, utilizing the Simplified Molecular Input Line Entry Specification (SMILES) notation. Molinspiration serves as an adept system for calculating drug-likeness information, while SwissTargetPrediction also contributes to this endeavor [61]. Notably, SwissTargetPrediction aids in predicting various targets, including g-protein coupled receptors (GPCR) ligands, ion channel modulators, kinase inhibitors, nuclear receptor ligands, protease inhibitors, and other enzyme targets (http://www.molinspiration.com/cgi-bin/properties).
In line with the information garnered from a CDFT analysis to gauge the chemical reactivity of the studied marine cyclopeptides, Table 3 furnishes bioactivity scores for these peptides across a spectrum of targets. These encompass GPCR ligands, ion channel modulators, nuclear receptor ligands, kinase inhibitors, protease inhibitors, and enzyme inhibitors. These scores play a pivotal role in predicting the peptides’ corresponding biological targets, an indispensable facet of drug development that offers profound insights into the potential therapeutic applications of these compounds [46].
The bioactivity scores associated with the analyzed cyclopeptides can be comprehended in the following manner: considered active when the bioactivity score exceeds 0, categorized as moderately active if the score falls within the range of -5.0 to 0.0, and deemed inactive when the bioactivity score is below -5.0. This categorization system allows for a clear interpretation of the cyclopeptides’ bioactivity levels.
ADMET properties, standing for Absorption, Distribution, Metabolism, Excretion, and Toxicity, play a pivotal role in the realm of drug discovery. These properties collectively determine how a potential drug candidate interacts within the human body, influencing its efficacy and safety profile [43,44,46].
Computational prediction of ADMET properties has emerged as an indispensable tool in the drug discovery process. Traditional experimental methods for assessing these properties are time-consuming, costly, and often carried out in later stages of drug development. In contrast, computational models utilize data-driven algorithms to swiftly evaluate a compound’s ADMET characteristics, even in the early stages of drug design.
By harnessing computational approaches, researchers can rapidly analyze vast chemical libraries to identify promising candidates with favorable ADMET profiles. This accelerates the drug discovery timeline and reduces the likelihood of investing resources into compounds that might encounter significant hurdles in clinical trials due to unfavorable ADMET properties.
Furthermore, computational prediction assists in identifying potential toxicity issues, enabling researchers to eliminate compounds with a higher risk of adverse effects. This proactive strategy not only minimizes the chances of late-stage failures but also enhances patient safety.
In summary, the computational prediction of ADMET properties revolutionizes the drug discovery landscape by providing timely insights into a compound’s behavior within the body. This predictive power not only expedites the identification of viable drug candidates but also contributes to more efficient, cost-effective, and safer drug development processes.
The ADMET parameters for the eight members of the Stellatolides A-H family estimated by considering the SwissADME [62] and pkCSM [63] software are displayed in Table 4, Table 5, Table 6, Table 7 and Table 8:
From Table 4 it can be seen that the values of the excretion parameters are the same for the eight cyclopeptides with the exception of Caco-2 Permeability.
The Caco-2 cell line plays a pivotal role in the field of ADMET within the realm of drug development. Caco-2 cells are derived from human colon carcinoma and are widely used as an in vitro model to simulate the behavior of the intestinal epithelium, specifically for drug absorption studies [43,44,46]. These cells form a monolayer that mimics the intestinal lining, allowing researchers to assess how well a drug can be absorbed through the gastrointestinal tract. This information is crucial because a drug’s bioavailability, or the extent to which it reaches systemic circulation, heavily depends on its ability to be absorbed by the intestines. In drug development, Caco-2 assays are employed to predict a drug’s potential for oral absorption thus allowing the estimation of the likelihood of a drug being absorbed efficiently or encountering obstacles due to efflux transporters. This information guides medicinal chemists and pharmaceutical scientists in optimizing drug structures to enhance their bioavailability. In this regards, Stellatolides C, D and G excels over the others, while Stellatolide H presents a very different small value.
Distribution is a critical aspect of ADMET. Once a drug is absorbed into the bloodstream, it needs to be effectively distributed to its target tissues and organs to exert its therapeutic effects. Understanding a drug’s distribution properties is essential for optimizing its efficacy and minimizing potential side effects. Distribution can also be influenced by factors like tissue perfusion rates and the presence of drug transporters. Tissues with high blood flow receive more drug, while those with low blood flow receive less [43,44,46].
Volume of Distribution at Steady State (VDss) is a pharmacokinetic parameter used in the field of clinical pharmacology to describe the theoretical volume into which a drug would need to distribute in order to account for its total amount in the body at steady state concentration. VDss provides valuable insights into a drug’s distribution characteristics within the human body. The concept of VDss is particularly useful for understanding how extensively a drug distributes beyond the bloodstream into various tissues and compartments. It helps determine whether a drug tends to stay predominantly within the blood plasma or if it has a tendency to accumulate in specific tissues or organs. VDss is a crucial parameter in drug dosing calculations. It aids in determining the appropriate dosage needed to achieve the desired therapeutic effect based on the drug’s distribution characteristics. Drugs with high VDss may require larger loading doses to rapidly establish therapeutic levels, while drugs with low VDss may need lower loading doses due to their tendency to remain in the bloodstream [44].
Fraction unbound, also known as the free fraction or unbound fraction, is a crucial concept in the field of ADMET within drug development. It refers to the proportion of a drug that exists in its pharmacologically active, unbound form within the bloodstream. In pharmacology, only the unbound fraction of a drug is capable of interacting with its target receptors and exerting therapeutic effects. The fraction unbound is particularly relevant in understanding a drug’s distribution, as only the unbound drug can pass through cell membranes and diffuse into tissues to reach its target site.
Blood-Brain Barrier (BBB) permeability is a fundamental concept in neuroscience and pharmacology, playing a critical role in understanding how substances, including drugs, interact with the central nervous system (CNS). The BBB is a specialized barrier that separates the bloodstream from the brain and spinal cord, tightly regulating the passage of molecules between these two compartments. Understanding BBB permeability is of utmost importance in drug development, especially for drugs targeting neurological disorders. A drug’s ability to cross the BBB is critical for it to exert therapeutic effects on the brain. Some drugs are designed to have high BBB permeability, while others are engineered to stay outside the CNS to prevent unwanted side effects.
Central Nervous System (CNS) permeability refers to the ability of substances, particularly drugs, to cross the blood-brain barrier (BBB) and gain access to the brain and spinal cord. The CNS is a highly protected and vital area of the body, and its permeability properties significantly influence the efficacy of drugs targeting neurological disorders. CNS permeability is a pivotal consideration in drug development for neurological disorders. Understanding the factors influencing CNS permeability and designing drugs that can effectively cross the blood-brain barrier are essential for developing treatments that target the central nervous system with precision and efficacy.
In summary, distribution properties play a vital role in determining a drug’s effectiveness and potential side effects. By studying how a drug is distributed within the body, researchers can optimize drug formulations, dosing regimens, and delivery strategies to ensure that the right concentration reaches the intended target while minimizing the risk of adverse effects in non-target tissues. Thus, the results from Table 5 may be of great when developing therapeutic drugs through the consideration of Stellatolides A-H.
Metabolism properties are a crucial aspect of ADMET studies in the field of drug development [43,44,46]. Metabolism refers to the biochemical processes that transform a drug into different compounds, often resulting in its inactivation and elimination from the body. Understanding a drug’s metabolism properties is essential for optimizing its efficacy, safety, and dosing regimens. Drug metabolism primarily occurs in the liver, where enzymes break down drugs into metabolites that are more water-soluble and can be excreted through urine or bile.
From Table 6, it can be appreciated that all the studied cyclopeptides will display a negative behavior on being either substrate or inhibitor of different cytochrome enzymes.
Excretion is one of the key properties within ADMET that focuses on the elimination of drugs and their metabolites from the body. This process primarily occurs through the kidneys via urine, but can also involve other routes like bile, sweat, and exhaled air. The excretion property of ADMET plays a pivotal role in determining a drug’s dosing regimen and potential toxicity. Efficient excretion helps maintain appropriate drug levels in the body, preventing accumulation that could lead to adverse effects. Impaired excretion, on the other hand, can lead to drug accumulation, prolonged therapeutic effects, or even harmful side effects [43,44,46].
Clearance is a fundamental pharmacokinetic parameter as it directly influences a drug’s dosing regimen. It helps determine how often a drug needs to be administered to maintain therapeutic levels in the body and avoid toxicity due to drug accumulation. Total clearance takes into account two primary processes: renal clearance and hepatic clearance. Renal clearance refers to the removal of a drug via the kidneys, primarily through urine. Hepatic clearance involves the metabolism and subsequent elimination of a drug by the liver. These two processes are often the dominant contributors to a drug’s total clearance.
Renal excretion is especially crucial, as the kidneys filter the bloodstream to eliminate drug molecules. This filtration process is influenced by factors such as the drug’s size, charge, and lipid solubility. Active transporters in the kidneys can also affect excretion, leading to variations in drug clearance among individuals. Renal OCT2 (Organic Cation Transporter 2) substrate refers to a drug or compound that can be transported into and out of cells in the kidneys by the OCT2 transporter protein. OCT2 is a type of transporter that plays a crucial role in the renal excretion of various organic cations, which are positively charged molecules.
It can be seen from Table 7, that all the cyclopeptides present a negative behavior for being a Renal OC2 susbtrate. On the contrary, they display different activity under the Total Clearence test, with Stellatolides F and G showing positive values while they are negative for all the others.
Toxicity is a significant aspect of the ADMET framework that focuses on assessing the potential harmful effects of a drug or compound on living organisms [43,44,46]. It plays a critical role in drug development, as understanding and mitigating potential toxicities is essential for ensuring the safety and efficacy of pharmaceutical products. Toxicity can manifest in various ways, including adverse effects on organs, tissues, cells, and biochemical processes. There are different types of toxicity, such as acute toxicity (rapid and severe effects shortly after exposure) and chronic toxicity (long-term and cumulative effects). The toxic effects can be dose-dependent, where higher doses lead to more severe reactions, or idiosyncratic, occurring unpredictably in certain individuals due to genetic variations. In summary, toxicity assessment within the ADMET framework is crucial for identifying and managing potential harmful effects of drugs and compounds. It guides drug development by ensuring the safety of pharmaceutical products and contributing to better patient outcomes. Thus, we believe that the results from Table 8 will be of great help to know in advance the possible toxicities of the Stellatolides A-H to guide future developments of therapeutic drugs based in the marine cyclopeptides.
4. Conclusions
In conclusion, the investigation presented in this study offers a comprehensive understanding of the chemical reactivity, stability, and ADMET properties of Stellatolides A-H, utilizing the innovative computational approach known as Conceptual DFT-based Computational Peptidology (CDFT-CP). The promising biological activities exhibited by Stellatolides, such as their antitumor, antimicrobial, and anti-inflammatory properties, underscore their potential as candidates for drug development.
The integration of Chemical Reactivity Theory (CRT), Conceptual Density Functional Theory (CDFT), and Computational Peptidology (CP) has provided valuable insights into the electronic structure and behavior of Stellatolides A-H. This approach not only advances our understanding of these natural compounds at the molecular level but also sets a precedent for the study of other natural compounds and peptides.
The predictions of ADMET properties based on the CDFT-CP approach offer significant implications for the potential therapeutic application of Stellatolides A-H. By elucidating their effectiveness, potential side effects, and appropriate dosage and administration routes, this study contributes to informed decision-making in drug development.
The outcomes of this research highlight the potential of Stellatolides A-H as promising drug candidates and emphasize the role of computational methodologies in accelerating the drug discovery process. The synergistic approach of combining various computational techniques enables a more holistic evaluation of these compounds, bridging the gap between theoretical insights and practical applications.
In conclusion, the findings presented in this study offer a glimpse into the future of drug development, where computational methods like CDFT-CP play a pivotal role in unveiling the potential of natural compounds and peptides. As the field of ADMET continues to evolve, studies like this pave the way for safer and more effective therapeutic interventions.
Author Contributions
Conceptualization, NFH and JSSL; methodology, NFH and DGM; software, NFH and DGM; validation, NFH and DGM ; formal analysis, NFH and DGM; investigation, NFH, JSSL, and DGM; resources, NFH and DGM; data curation, NFH and DGM; writing–original draft preparation, NFH and DGM; visualization, NFH and DGM; supervision, NFH and DGM; project administration, NFH; funding acquisition, NFH. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by grant number 25017/23 from CIMAV.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not appplicable.
Data Availability Statement
All generated data will be available from the authors under request.
Acknowledgments
This study received support from the Centro de Investigación en Materiales Avanzados, S.C. (CIMAV) and the Consejo Nacional de Humanidades Ciencia y Tecnología (CONAHCYT). NFH, JSSL, and DGM are researchers affiliated with CIMAV and CONAHCYT.
Conflicts of Interest
The authors declare no conflict of interest
References
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine Natural Products. Natural Product Reports 2016, 33, 382–431. [Google Scholar] [CrossRef] [PubMed]
- Rotter, A.; Barbier, M.; Bertoni, F.; Bones, A.M.; Cancela, M.L.; Carlsson, J.; Carvalho, M.F.; Cegłowska, M.; Chirivella-Martorell, J.; Dalay, M.C.; Cueto, M.; Dailianis, T.; Deniz, I.; Díaz-Marrero, A.R.; Drakulovic, D.; Dubnika, A.; Edwards, C.; Einarsson, H.; Erdogân, A.; Eroldogân, O.T.; Ezra, D.; Fazi, S.; FitzGerald, R.J.; Gargan, L.M.; Gaudêncio, S.P.; Udovič, M.G.; DeNardis, N.I.; Jónsdóttir, R.; Kataržytė, M.; Klun, K.; Kotta, J.; Ktari, L.; Ljubešić, Z.; Bilela, L.L.; Mandalakis, M.; Massa-Gallucci, A.; Matijošytė, I.; Mazur-Marzec, H.; Mehiri, M.; Nielsen, S.L.; Novoveská, L.; Overlingė, D.; Perale, G.; Ramasamy, P.; Rebours, C.; Reinsch, T.; Reyes, F.; Rinkevich, B.; Robbens, J.; Röttinger, E.; Rudovica, V.; Sabotič, J.; Safarik, I.; Talve, S.; Tasdemir, D.; Schneider, X.T.; Thomas, O.P.; Toruńska-Sitarz, A.; Varese, G.C.; Vasquez, M.I. The Essentials of Marine Biotechnology. Frontiers in Marine Science 2021, 8, 1–53. [Google Scholar] [CrossRef]
- Pelay-Gimeno, M.; Albericio, F.; Tulla-Puche, J. Synthesis of Complex Head-to-Side-Chain Cyclodepsipeptides. Nature Protocols 2016, 11, 1924–1947. [Google Scholar] [CrossRef] [PubMed]
- Sarabia, F.; Chammaa, S.; Ruiz, A.; Ortiz, L.; Herrera, F. Chemistry and Biology of Cyclic Depsipeptides of Medicinal and Biological Interest. Current Medicinal Chemistry 2004, 11, 1309–1332. [Google Scholar] [CrossRef]
- Pelay-Gimeno, M.; Tulla-Puche, J.; Albericio, F. Head-to-Side-Chain Cyclodepsipeptides of Marine Origin. Marine Drugs 2013, 11, 1693–1717. [Google Scholar] [CrossRef]
- Vitali, A. Antimicrobial Peptides Derived from Marine Sponges. American Journal of Clinical Microbiology and Antimicrobials 2018, 1, 1–11. [Google Scholar]
- Martín, M.J.; Rodríguez-Acebes, R.; García-Ramos, Y.; Martínez, V.; Murcia, C.; Digón, I.; Marco, I.; Pelay-Gimeno, M.; Fernández, R.; Reyes, F.; Francesch, A.M.; Munt, S.; Tulla-Puche, J.; Albericio, F.; Cuevas, C. Stellatolides, a New Cyclodepsipeptide Family from the Sponge Ecionemia acervus Isolation, Solid-Phase Total Synthesis, and Full Structural Assignment of Stellatolide A. Journal of the American Chemical Society 2014, 136, 6754–6762. [Google Scholar] [CrossRef]
- Nakamukai, S.; Takada, K.; Furihata, K.; Ise, Y.; Okada, S.; Morii, Y.; Yamawaki, N.; Takatani, T.; Arakawa, O.; Gustafson, K.R.; Matsunaga, S. Stellatolide H, A Cytotoxic Peptide Lactone from a Deep-Sea Sponge Discodermia sp. Tetrahedron Letters 2018, 59, 2532–2536. [Google Scholar] [CrossRef]
- Zeng, M.; Tao, J.; Xu, S.; Bai, X.; Zhang, H. Marine Organisms as a Prolific Source of Bioactive Depsipeptides. Marine Drugs 2023, 21, 120. [Google Scholar] [CrossRef]
- Macedo, M.W.F.S.; da Cunha, N.B.; Carneiro, J.A.; da Costa, R.A.; de Alencar, S.A.; Cardoso, M.H.; Franco, O.L.; Dias, S.C. Marine Organisms as a Rich Source of Biologically Active Peptides. Frontiers in Marine Science 2021, 8, 667764. [Google Scholar] [CrossRef]
- León-Buitimea, A.; Garza-Cárdenas, C.R.; Garza-Cervantes, J.A.; Lerma-Escalera, J.A.; Morones-Ramírez, J.R. The Demand for New Antibiotics: Antimicrobial Peptides, Nanoparticles, and Combinatorial Therapies as Future Strategies in Antibacterial Agent Design. Frontiers in Microbiology 2020, 11, 1669. [Google Scholar] [CrossRef] [PubMed]
- Phyo, Y.; Ribeiro, J.; Fernandes, C.; Kijjoa, A.; Pinto, M. Marine Natural Peptides: Determination of Absolute Configuration Using Liquid Chromatography Methods and Evaluation of Bioactivities. Molecules 2018, 23, 306. [Google Scholar] [CrossRef]
- Lipinski, C.; Lombardo, F.; Dominy, B.; Feeney, P. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Advanced Drug Delivery Reviews 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Santos, G.B.; Ganesan, A.; Emery, F.S. Oral Administration of Peptide-Based Drugs: Beyond Lipinski’s Rule. ChemMedChem 2016, 11, 2245–2251. [Google Scholar] [CrossRef] [PubMed]
- Linker, S.M.; Schellhaas, C.; Kamenik, A.S.; Veldhuizen, M.M.; Waibl, F.; Roth, H.J.; Fouché, M.; Rodde, S.; Riniker, S. Lessons for Oral Bioavailability: How Conformationally Flexible Cyclic Peptides Enter and Cross Lipid Membranes. Journal of Medicinal Chemistry 2023, 66, 2773–2788. [Google Scholar] [CrossRef]
- Giri, N.C. Protein and Peptide Drug Delivery. In Smart Drug Delivery; Ahmad, U., Haider, M.F., Akhtar, J., Eds.; IntechOpen: London, UK, 2022. [Google Scholar] [CrossRef]
- Ilangala, A.B.; Lechanteur, A.; Fillet, M.; Piel, G. Therapeutic Peptides for Chemotherapy: Trends and Challenges for Advanced Delivery Systems. European Journal of Pharmaceutics and Biopharmaceutics 2021, 167, 140–158. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic Peptides: Historical Perspectives, Current Development Trends, and Future Directions. Bioorganic & Medicinal Chemistry 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic Peptides: Current Applications and Future Directions. Signal Transduction and Targeted Therapy 2022, 7. [Google Scholar] [CrossRef]
- Lamers, C. Overcoming the Shortcomings of Peptide-based Therapeutics. Future Drug Discovery 2022, 4. [Google Scholar] [CrossRef]
- Zhu, Q.; Chen, Z.; Paul, P.K.; Lu, Y.; Wu, W.; Qi, J. Oral Delivery of Proteins and Peptides: Challenges, Status Quo and Future Perspectives. Acta Pharmaceutica Sinica B 2021, 11, 2416–2448. [Google Scholar] [CrossRef]
- Haddadzadegan, S.; Dorkoosh, F.; Bernkop-Schnürch, A. Oral Delivery of Therapeutic Peptides and Proteins: Technology Landscape of Lipid-based Nanocarriers. Advanced Drug Delivery Reviews 2022, 182, 114097. [Google Scholar] [CrossRef]
- Verma, S.; Goand, U.K.; Husain, A.; Katekar, R.A.; Garg, R.; Gayen, J.R. Challenges of Peptide and Protein Drug Delivery by Oral Route: Current Strategies to Improve the Bioavailability. Drug Development Research 2021, 82, 927–944. [Google Scholar] [CrossRef] [PubMed]
- Naim, J.; Sharmin, N.; Shuma, M.L.; Halder, S. Lipid-Based Nanocarriers for Oral Delivery of Proteins and Peptides: Opportunities, Challenges, and Future Prospects. Dhaka University Journal of Pharmaceutical Sciences 2022, 395–416. [Google Scholar] [CrossRef]
- Räder, A.F.; Reichart, F.; Weinmüller, M.; Kessler, H. Improving Oral Bioavailability of Cyclic Peptides by N-Methylation. Bioorganic & Medicinal Chemistry 2018, 26, 2766–2773. [Google Scholar] [CrossRef]
- Chattaraj, P.K. (Ed.) Chemical Reactivity Theory - A Density Functional View; CRC Press. Taylor & Francis Group: Boca Raton, FL, 2009. [Google Scholar]
- Parr, R.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, 1989. [Google Scholar]
- Geerlings, P.; Proft, F.D.; Langenaeker, W. Conceptual Density Functional Theory. Chemical Reviews 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- Gázquez, J.; Cedillo, A.; Vela, A. Electrodonating and Electroaccepting Powers. Journal of Physical Chemistry A 2007, 111, 1966–1970. [Google Scholar] [CrossRef]
- Chattaraj, P.; Chakraborty, A.; Giri, S. Net Electrophilicity. Journal of Physical Chemistry A 2009, 113, 10068–10074. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, P.; Chamorro, E.; Chattaraj, P.K.; Proft, F.D.; Gázquez, J.L.; Liu, S.; Morell, C.; Toro-Labbé, A.; Vela, A.; Ayers, P. Conceptual Density Functional Theory: Status, Prospects, Issues. Theoretical Chemistry Accounts 2020, 139, 36. [Google Scholar] [CrossRef]
- Liu, S. (Ed.) Conceptual Density Functional Theory: Towards a New Chemical Reactivity Theory; Wiley-VCH Verlag: Weinheim, Germany, 2022. [Google Scholar]
- Glossman-Mitnik, D. (Ed.) Density Functional Theory - Recent Advances, New Perspectives and Applications; IntechOpen: London, UK, 2022. [Google Scholar]
- Kaya, S.; von Szentpaly, L.; Serdaroglu, G.; Guo, L. (Eds.) Chemical Reactivity; Elsevier - Health Sciences Division: Philadelphia, PA, 2023. [Google Scholar]
- Frau, J.; Flores-Holguín, N.; Glossman-Mitnik, D. Chemical Reactivity Properties, pKa Values, AGEs Inhibitor Abilities and Bioactivity Scores of the Mirabamides A–H Peptides of Marine Origin Studied by Means of Conceptual DFT. Marine Drugs 2018, 16, 302–19. [Google Scholar] [CrossRef]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Chemical-Reactivity Properties, Drug Likeness, and Bioactivity Scores of Seragamides A–F Anticancer Marine Peptides: Conceptual Density Functional Theory Viewpoint. Computation 2019, 7, 52. [Google Scholar] [CrossRef]
- Frau, J.; Flores-Holguín, N.; Glossman-Mitnik, D. Chemical Reactivity Theory and Empirical Bioactivity Scores as Computational Peptidology Alternative Tools for the Study of Two Anticancer Peptides of Marine Origin. Molecules 2019, 24, 1115. [Google Scholar] [CrossRef] [PubMed]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Computational Prediction of Bioactivity Scores and Chemical Reactivity Properties of the Parasin I Therapeutic Peptide of Marine Origin Through the Calculation of Global and Local Conceptual DFT Descriptors f. Theoretical Chemistry Accounts 2019, 138. [Google Scholar] [CrossRef]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. A Fast and Simple Evaluation of the Chemical Reactivity Properties of the Pristinamycin Family of Antimicrobial Peptides. Chemical Physics Letters 2020, 739, 137021. [Google Scholar] [CrossRef]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Conceptual DFT-Based Computational Peptidology of Marine Natural Compounds: Discodermins A–H. Molecules 2020, 25, 4158. [Google Scholar] [CrossRef] [PubMed]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Virtual Screening of Marine Natural Compounds by Means of Chemoinformatics and CDFT-Based Computational Peptidology. Marine Drugs 2020, 18, 478. [Google Scholar] [CrossRef]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Conceptual DFT as a Helpful Chemoinformatics Tool for the Study of the Clavanin Family of Antimicrobial Marine Peptides. In Density Functional Theory; De Lazaro, S.R., Da Silveira Lacerda, L.H., Pontes Ribeiro, R.A., Eds.; IntechOpen: London, UK, 2021. [Google Scholar]
- Wang, J.; Urban, L. Predictive ADMET; John Wiley & Sons: Nashville, TN, 2014. [Google Scholar]
- Tsaioun, K.; Kates, S.A. (Eds.) ADMET for Medicinal Chemists; Wiley-Blackwell: Hoboken, NJ, 2011. [Google Scholar]
- Clark, D.E. Chapter 10 Computational Prediction of ADMET Properties: Recent Developments and Future Challenges. In Annual Reports in Computational Chemistry; Elsevier, 2005; pp. 133–151. [Google Scholar] [CrossRef]
- Kallen, A. Computational Pharmacokinetics; CRC Press: London, England, 2019. [Google Scholar]
- Lewars, E. Computational Chemistry - Introduction to the Theory and Applications of Molecular and Quantum Mechanics; Kluwer Academic Publishers: Dordrecht, 2003. [Google Scholar]
- Young, D. Computational Chemistry - A Practical Guide for Applying Techniques to Real-World Problems; John Wiley & Sons: New York, 2001. [Google Scholar]
- Jensen, F. Introduction to Computational Chemistry, 2nd ed.; John Wiley & Sons: Chichester, England, 2007. [Google Scholar]
- Cramer, C. Essentials of Computational Chemistry - Theories and Models, 2nd ed.; John Wiley & Sons: Chichester, England, 2004. [Google Scholar]
- Halgren, T.A. Merck Molecular Force Field. I. Basis, Form, Scope, Parameterization, and Performance of MMFF94. Journal of Computational Chemistry 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck Molecular Force Field. II. MMFF94 van der Waals and Electrostatic Parameters for Intermolecular Interactions. Journal of Computational Chemistry 1996, 17, 520–552. [Google Scholar] [CrossRef]
- Halgren, T.A. MMFF VI. MMFF94s Option for Energy Minimization Studies. Journal of Computational Chemistry 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Halgren, T.A.; Nachbar, R.B. Merck Molecular Force Field. IV. Conformational Energies and Geometries for MMFF94. Journal of Computational Chemistry 1996, 17, 587–615. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck Molecular Force field. V. Extension of MMFF94 Using Experimental Data, Additional Computational Data, and Empirical Rules. Journal of Computational Chemistry 1996, 17, 616–641. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C.01, 2016. Gaussian Inc.: Wallingford CT.
- Peverati, R.; Truhlar, D.G. Screened-Exchange Density Functionals with Broad Accuracy for Chemistry and Solid-State Physics. Physical Chemistry Chemical Physics 2012, 14, 16187–16191. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Physical Chemistry Chemical Physics 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting Basis Sets for H to R. Physical Chemistry Chemical Physics 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.; Cramer, C.; Truhlar, D. Universal Solvation Model Based on Solute Electron Density and a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. Journal of Physical Chemistry B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated Data and New Features for Efficient Prediction of Protein Targets of Small Molecules. Nucleic Acids Research 2019, 47, W357–W364. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Scientific Reports 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. Journal of Medicinal Chemistry 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Parr, R.; Szentpaly, L.; Liu, S. Electrophilicity Index. Journal of the American Chemical Society 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Perez, P. Understanding the Reactivity of Captodative Ethylenes in Polar Cycloaddition Reactions. A Theoretical Study. The Journal of Organic Chemistry 2008, 73, 4615–4624. [Google Scholar] [CrossRef]
- Jaramillo, P.; Domingo, L.R.; Chamorro, E.; Pérez, P. A Further Exploration of a Nucleophilicity Index Based on the Gas-Phase Ionization Potentials. Journal of Molecular Structure: THEOCHEM 2008, 865, 68–72. [Google Scholar] [CrossRef]
- Domingo, L.R.; Sáez, J.A. Understanding the Mechanism of Polar Diels-Alder Reactions. Organic and Biomolecular Chemistry 2009, 7, 3576–3583. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Perez, P. The Nucleophilicity N Index in Organic Chemistry. Organic and Biomolecular Chemistry 2011, 9, 7168–7175. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. Journal of Computational Chemistry 2011, 33, 580–592. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Realization of Conceptual Density Functional Theory and Information-Theoretic Approach in Multiwfn Program. In Conceptual Density Functional Theory: Towards a New Chemical Reactivity Theory; Liu, S., Ed.; Wiley: Weinheim, Germany, 2022. [Google Scholar] [CrossRef]
- Morell, C.; Grand, A.; Toro-Labbé, A. New Dual Descriptor for Chemical Reactivity. The Journal of Physical Chemistry A 2004, 109, 205–212. [Google Scholar] [CrossRef]
- Morell, C.; Grand, A.; Toro-Labbé, A. Theoretical Support for Using the Δf(r) Descriptor. Chemical Physics Letters 2006, 425, 342–346. [Google Scholar] [CrossRef]
- Martínez-Araya, J.I. Explaining Reaction Mechanisms Using the Dual Descriptor: A Complementary Tool to the Molecular Electrostatic Potential. Journal of Molecular Modeling 2012, 19, 2715–2722. [Google Scholar] [CrossRef]
- Martínez-Araya, J.I. Why is the Dual Descriptor a More Accurate Local Reactivity Descriptor than Fukui Functions? Journal of Mathematical Chemistry 2015, 53, 451–465. [Google Scholar] [CrossRef]
Figure 1.
Graphical sketches of the molecular structures of the Stellatolides A-H.
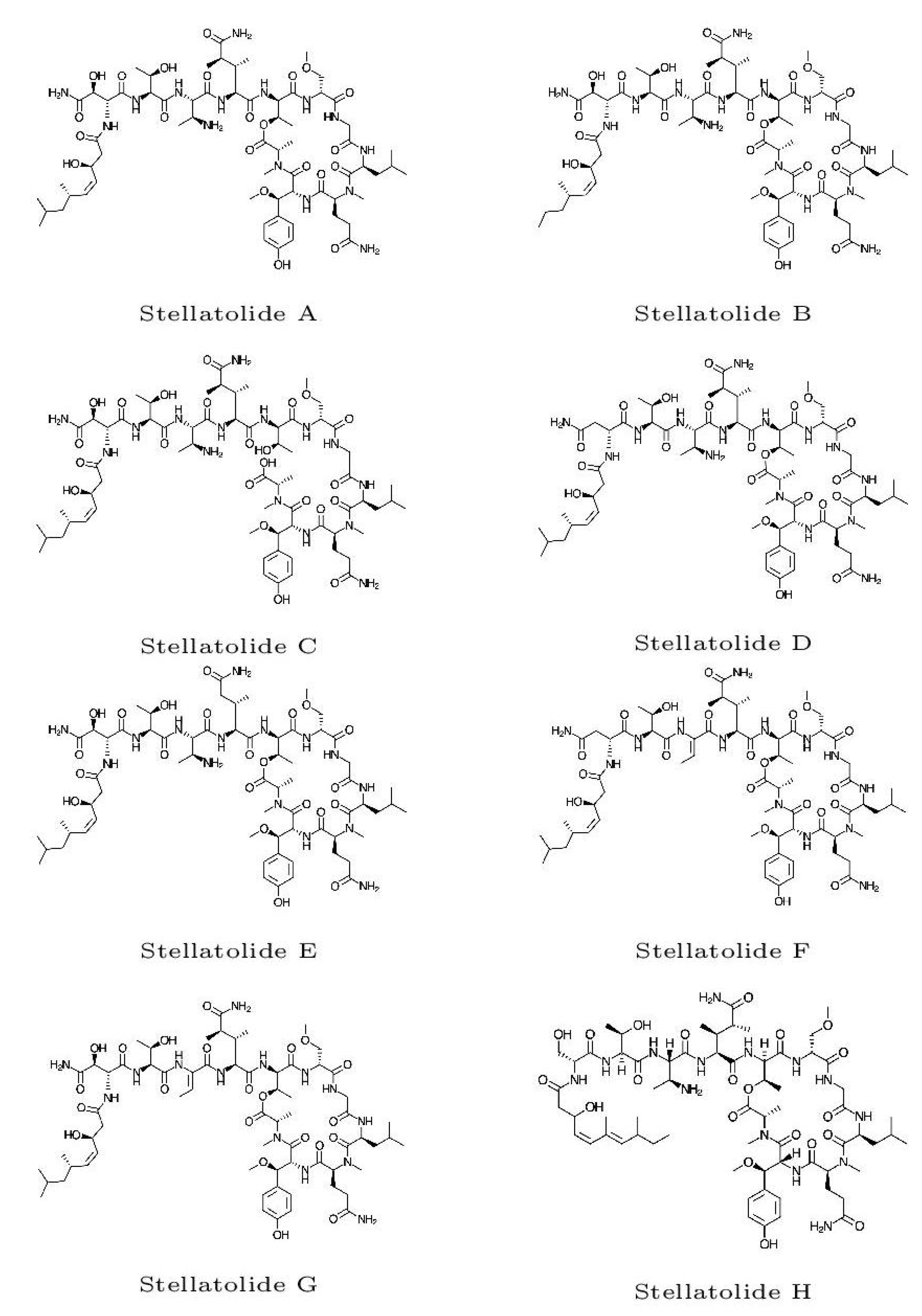
Figure 2.
Optimized molecular structures of the Stellatolides A-H.
Figure 3.
Graphical representation of the dual descriptor DD of the Stellatolides A-D. Left: DD > 0, Right: DD < 0.
Figure 3.
Graphical representation of the dual descriptor DD of the Stellatolides A-D. Left: DD > 0, Right: DD < 0.
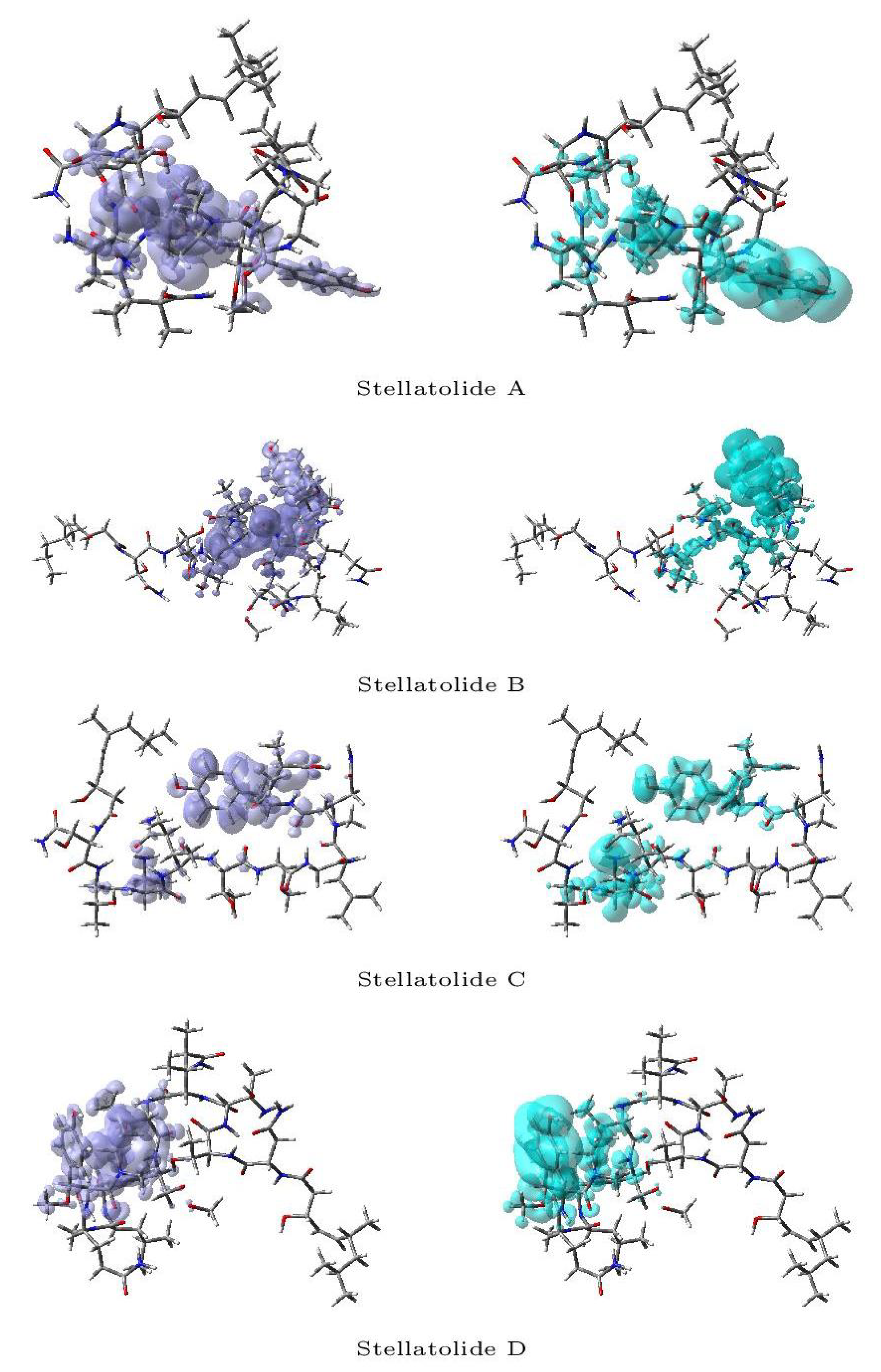
Figure 4.
Graphical representation of the dual descriptor DD of the Stellatolides E-H. Left: DD > 0, Right: DD < 0.
Figure 4.
Graphical representation of the dual descriptor DD of the Stellatolides E-H. Left: DD > 0, Right: DD < 0.
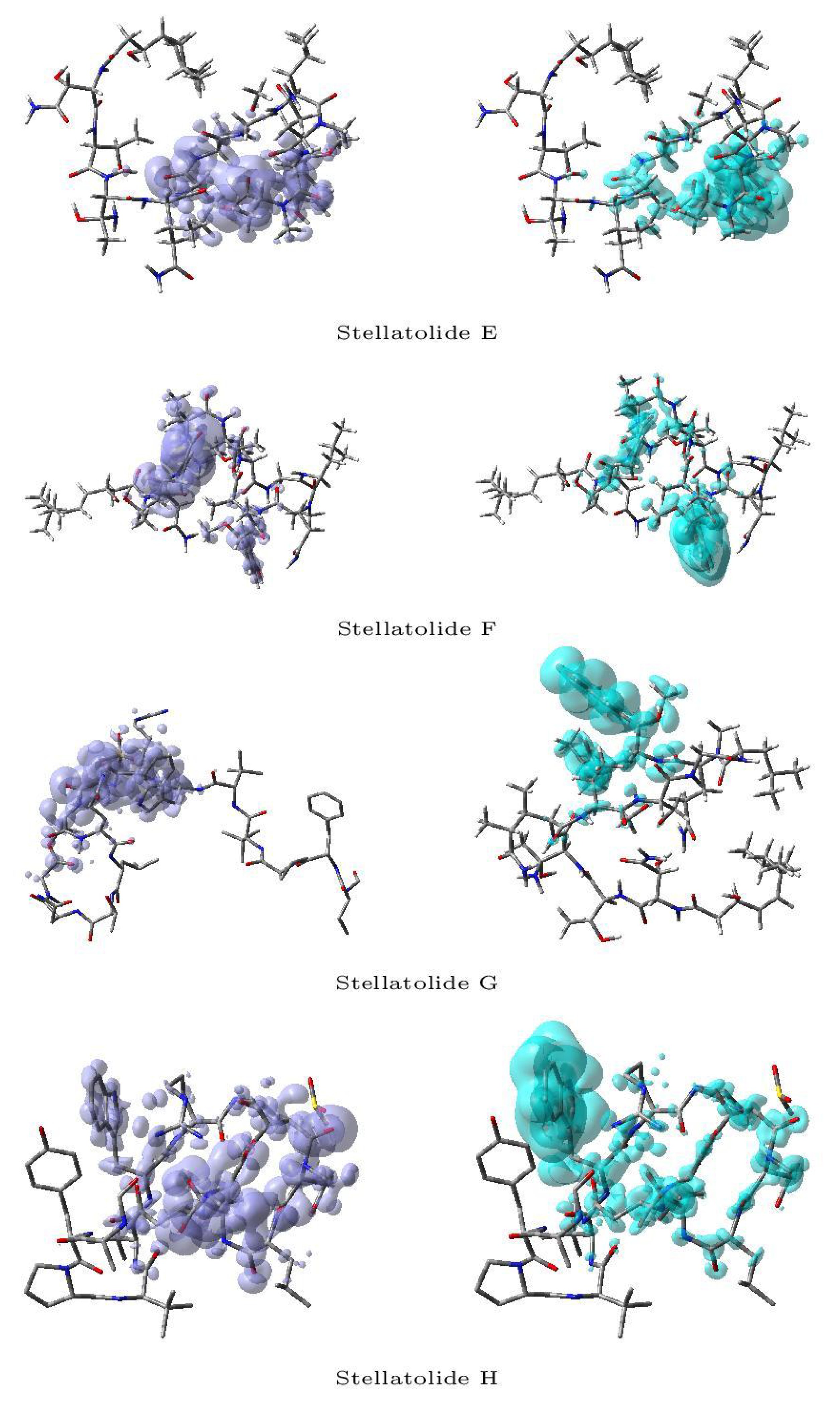

Table 1.
Predicted Frontier Orbital Energies, H-L Gap and the KID Descriptors (all in eV) for the Stellatolides A-H.
Table 1.
Predicted Frontier Orbital Energies, H-L Gap and the KID Descriptors (all in eV) for the Stellatolides A-H.
| Molecule | HOMO | LUMO | SOMO | H-L Gap | J | J | J | SL |
|---|---|---|---|---|---|---|---|---|
| Stellatolide A | -6.2605 | -0.9396 | -0.9192 | 5.3209 | 0.026 | 0.011 | 0.028 | 0.020 |
| Stellatolide B | -6.2418 | -1.0234 | -1.0226 | 5.2183 | 0.035 | 0.001 | 0.035 | 0.001 |
| Stellatolide C | -6.3147 | -0.8531 | -0.9241 | 5.4616 | 0.061 | 0.029 | 0.067 | 0.071 |
| Stellatolide D | -6.2681 | -1.1480 | -1.1061 | 5.1201 | 0.034 | 0.015 | 0.037 | 0.042 |
| Stellatolide E | -6.2118 | -1.0876 | -1.0787 | 5.1242 | 0.032 | 0.006 | 0.032 | 0.009 |
| Stellatolide F | -6.2524 | -1.6379 | -1.6349 | 4.6145 | 0.026 | 0.001 | 0.026 | 0.003 |
| Stellatolide G | -6.2722 | -1.4128 | -1.3690 | 4.8594 | 0.038 | 0.017 | 0.042 | 0.044 |
| Stellatolide H | -5.9647 | -1.1255 | -1.1676 | 4.8393 | 0.027 | 0.019 | 0.033 | 0.042 |
Table 2.
Global Reactivity Descriptors for the Stellatolides A-H: Electronegativity (), Global Hardness (), Electrophilicity (), Nucleophilicity N, Electrodonating Power (), Electroaccepting Power () and Net Electrophilicity () (all in eV)
Table 2.
Global Reactivity Descriptors for the Stellatolides A-H: Electronegativity (), Global Hardness (), Electrophilicity (), Nucleophilicity N, Electrodonating Power (), Electroaccepting Power () and Net Electrophilicity () (all in eV)
| Molecule | N | ||||||
|---|---|---|---|---|---|---|---|
| Stellatolide A | 3.2610 | 5.3209 | 1.2179 | 2.8608 | 4.5684 | 0.9683 | 5.5366 |
| Stellatolide B | 3.6326 | 5.2183 | 1.2644 | 2.8795 | 4.6712 | 1.0386 | 5.7097 |
| Stellatolide C | 3.5839 | 5.4616 | 1.1759 | 2.8066 | 4.4850 | 0.9011 | 5.3861 |
| Stellatolide D | 3.7081 | 5.1201 | 1.3427 | 2.8531 | 4.8595 | 1.1515 | 6.0110 |
| Stellatolide E | 3.6497 | 5.1242 | 1.2998 | 2.9094 | 4.7447 | 1.0949 | 5.8396 |
| Stellatolide F | 3.9451 | 4.6145 | 1.6864 | 2.8688 | 5.6338 | 1.6887 | 7.3224 |
| Stellatolide G | 3.8425 | 4.8594 | 1.5192 | 2.8490 | 5.2634 | 1.4209 | 6.6843 |
| Stellatolide H | 3.5451 | 4.8393 | 1.2985 | 3.1565 | 4.6720 | 1.1269 | 5.7990 |
Table 3.
Bioactivity Scores of the Stellatolides A-H Calculated on the Basis of the GPCR Ligand, Ion Channel Modulator, Nuclear Receptor Ligand, Kinase Inhibitor, Protease Inhibitor, and Enzyme Inhibitor Interactions.
Table 3.
Bioactivity Scores of the Stellatolides A-H Calculated on the Basis of the GPCR Ligand, Ion Channel Modulator, Nuclear Receptor Ligand, Kinase Inhibitor, Protease Inhibitor, and Enzyme Inhibitor Interactions.
| Property | A | B | C | D | E | F | G | H |
|---|---|---|---|---|---|---|---|---|
| GPCR Ligand | -3.95 | -3.95 | -3.96 | -3.95 | -3.94 | -3.95 | -3.95 | -3.94 |
| Ion Channel Modulator | -4.01 | -4.01 | -4.01 | -4.01 | -4.00 | -4.01 | -4.02 | -4.00 |
| Nuclear Receptor Ligand | -4.03 | -4.03 | -4.03 | -4.02 | -4.02 | -4.03 | -4.03 | -4.01 |
| Kinase Inhibitor | -4.02 | -4.02 | -4.02 | -4.01 | -4.02 | -4.02 | -4.03 | -4.00 |
| Protease Inhibitor | -3.90 | -3.90 | -3.90 | -3.89 | -3.89 | -3.89 | -3.90 | -3.89 |
| Enzyme Inhibitor | -3.96 | -3.96 | -3.97 | -3.96 | -3.95 | -3.96 | -3.96 | -3.95 |
Table 4.
Absorption Properties of the Stellatolides A-H
| Property | A | B | C | D | E | F | G | H |
|---|---|---|---|---|---|---|---|---|
| Caco-2 Permeability | -0.760 | -0.760 | -0.893 | -0.804 | -0.648 | -0.792 | -0.810 | -0.254 |
| Intestinal Absorption (Human) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Skin Permeability | -2.735 | -2.735 | -2.735 | -2.735 | -2.735 | -2.735 | -2.735 | -2.735 |
| P-glycoprotein Substrate | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| P-glycoprotein I Inhibitor | No | No | No | No | No | No | No | No |
| P-glycoprotein II Inhibitor | No | No | No | No | No | No | No | No |
Table 5.
Distribution Properties of the Stellatolides A-H
| Property | A | B | C | D | E | F | G | H |
|---|---|---|---|---|---|---|---|---|
| VDss (Human) | -0.413 | -0.413 | -0.605 | -0.338 | -0.429 | -0.191 | -0.315 | -0.361 |
| Fraction Unbound (Human) | 0.312 | 0.312 | 0.340 | 0.301 | 0.317 | 0.302 | 0.305 | 0.334 |
| BBB Permeability | -3.081 | -3.081 | -3.661 | -2.973 | -3.076 | -2.795 | -3.053 | -2.902 |
| CNS Permeability | -7.266 | -7.266 | -7.945 | -6.861 | -7.300 | -6.409 | -6.835 | -6.691 |
Table 6.
Metabolism Properties of the Stellatolides A-H
| Property | A | B | C | D | E | F | G | H |
|---|---|---|---|---|---|---|---|---|
| CYP2D6 Substrate | No | No | No | No | No | No | No | No |
| CYP3A4 Substrate | No | No | No | No | No | No | No | No |
| CYP1A2 Inhibitor | No | No | No | No | No | No | No | No |
| CYP2C19 Inhibitor | No | No | No | No | No | No | No | No |
| CYP2C9 Inhibitor | No | No | No | No | No | No | No | No |
| CYP2D6 Inhibitor | No | No | No | No | No | No | No | No |
| CYP3A4 Inhibitor | No | No | No | No | No | No | No | No |
Table 7.
Excretion Properties of the Stellatolides A-H
| Property | A | B | C | D | E | F | G | H |
|---|---|---|---|---|---|---|---|---|
| Total Clearance | -1.731 | -1.731 | -0.965 | -1.704 | -1.694 | 0.611 | 0.623 | -1.755 |
| Renal OCT2 Substrate | No | No | No | No | No | No | No | No |
Table 8.
Toxicity Properties of the Stellatolides A-H
| Property | A | B | C | D | E | F | G | H |
|---|---|---|---|---|---|---|---|---|
| AMES Toxicity | No | No | No | No | No | No | No | No |
| Maximum Tolerated Dose (Human) | 0.527 | 0.527 | 0.51 | 0.519 | 0.540 | 0.502 | 0.503 | 0.557 |
| hERG I Inhibitor | No | No | No | No | No | No | No | No |
| hERG II Inhibitor | Yes | Yes | No | Yes | Yes | Yes | Yes | Yes |
| Oral Rat Acute Toxicity (LD50) | 2.475 | 2.475 | 2.481 | 2.482 | 2.478 | 2.494 | 2.477 | 2.482 |
| Oral Rat Chronic Toxicity (LOAEL) | 4.837 | 4.837 | 4.473 | 4.354 | 5.070 | 4.616 | 4.673 | 5.887 |
| Hepatotoxicity | Yes | Yes | Yes | Yes | Yes | Ye | Yes | Yes |
| Skin Sensitisation | No | No | No | No | No | No | No | No |
| T. Pyriformis Toxicity | 0.285 | 0.285 | 0.285 | 0.285 | 0.285 | 0.285 | 0.285 | 0.285 |
| Minnow Toxicity | 18.264 | 18.264 | 18.517 | 17.368 | 17.894 | 16.506 | 17.155 | 19.686 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



