
Submitted:
28 August 2023
Posted:
29 August 2023
You are already at the latest version
Abstract
Ebola virus disease (EVD) represents a global health threat. The etiological agents of EVD are 6 species of Orthoebolaviruses, with Orthoebolavirus zairense (EBOV) having the greatest public health and medical significance. EVD disease pathogenesis occurs as a result of broad cellular tropism of the virus, robust viral replication and a potent and dysregulated production of cytokines. In vivo, tissue macrophages are some of the earliest cells infected and contribute significantly to virus load and cytokine production. While EBOV is known to infect macrophages and to generate high titer virus in the liver, EBOV infection of liver macrophages, Kupffer cells, has not previously been examined in tissue culture or experimentally manipulated in vivo. Here, we employed primary murine Kupffer cells (KC) and an immortalized murine Kupffer cell line (ImKC) to assess EBOV-eGFP replication in liver macrophages. KCs and ImKCs were highly permissive for EBOV infection and IFN-γ polarization of these cells suppressed their permissiveness to infection. The kinetics of IFN-γ-elicited antiviral responses were examined using a biologically-contained model of EBOV infection termed EBOV ΔVP30. The antiviral activity of IFN-γ was transient, but a modest ~3-fold reduction of infection persisted for as long as 6 days post treatment. To assess the interferon stimulated gene products (ISGs) responsible for protection, the efficacy of secreted ISGs induced by IFN-γ were evaluated. Secreted ISGs blocked recombinant VSV expressing EBOV GP (rVSV/EBOV GP) infection, but failed to block EBOV ΔVP30. Our studies define new cellular tools for the study of EBOV infection that can potentially aid the development of new antiviral therapies. Furthermore, our data underscore the importance of macrophages in EVD pathogenesis and those IFN-γ-elicited ISGs that help to control EBOV infection.
Keywords:
Macrophage
; Kupffer cell
; interferon gamma
; Ebola virus
; filovirus
1. Introduction
Filoviruses are important viral pathogens that represent a serious global health concern. The family Filoviridae belong to the order Mononegavirales and the Orthoebolavirus comprised one genus composed of six viral species: Orthoebolavirus zairense (EBOV), Orthoebolavirus Sudanense, Orthoebolavirus Bundibugyoense, Orthoebolavirus Taiense, Orthoebolavirus Restonense and Orthoebolavirus Bombaliense (1–3). Of these, EBOV has the greatest public health and medical significance (4). Orthoebolaviruses are enveloped, pleomorphic viruses that contain a negative-sense single-stranded RNA genome of ~19 kb. Infection with EBOV induces a wide range of clinical manifestations encompassing fever, rash, gastrointestinal distress, malaise and myalgia. Patients who subsequently develop fatal disease can manifest haemorrhagic fever, hypovolemic shock and/or organ failure with a mortality rate up to 90% (4, 5). In 2019, the FDA approved the first vaccine for the prevention of Ebola virus disease (EVD) which consists of recombinant vesicular stomatitis virus (rVSV) that expresses the EBOV glycoprotein (GP). This vaccine confers >95% against EBOV (6); however, this vaccine provides little to no cross-protection against other ebolaviruses in animal models (7–9). A pan-filovirus vaccine is needed and such vaccines are currently under development (10, 11).
Tissue mononuclear phagocytes, e.g., macrophages and dendritic cells (DCs), are thought to be the first cells in the body infected (12–14). These cells both respond to and elicit innate immune responses that, depending on the situation, ameliorate or exacerbate associated disease (15–17). Peritoneal macrophages polarized with interferon gamma (IFN-γ) (M1 polarization) stimulates the production of a large group of interferon stimulated genes (ISGs), suppressing viral replication in this cell population and protecting mice from EBOV disease (16). However, M1 polarization of tissue macrophages can be a double-edged sword as the production of proinflammatory soluble factors at late times of EBOV infection is associated with worse outcomes (18, 19). In contrast, IL-4/IL-13 treatment of peritoneal macrophages that induces M2a polarization enhances virus infection of the cells early on and sustains them as viral targets via upregulation of C-type lectins on the cell surface (15). Hence, the microenvironment of tissue mononuclear phagocytes affects both the ability of these cells to support EBOV infection and the cytokines produced. While macrophage infection affects both the control of EBOV replication and the immunopathogenesis associated with infection, details of the role of these cells during infection remains incompletely understood. In part, this is due to the limited availability of cell lines that are easy to work with and accurately recapitulate various aspects of tissue macrophages.
Tissue phagocytes also serve as vehicles for EBOV spread. Infected phagocytes (i.e., DCs) travel to the regional lymph nodes where viral replication occurs followed by viremia and viral dissemination to a variety of organs and tissues (14). The liver is one such organ that becomes infected early during EBOV infection where the tissue resident macrophages, Kupffer cells (KCs), support infection as well as drive inflammatory responses, leading to liver damage (12, 13, 20). However, the interaction of EBOV with KCs has been poorly explored to date.
Here, we phenotypically characterize murine KCs and an adherent, easily manipulatable macrophage model line, immortalized mouse Kupffer cells (ImKCs), and found that this line expresses macrophage-specific and, more specifically, Kupffer cell-specific genes. Further, cytokine-induced polarization-specific markers were comparable between the two cell populations, demonstrating that ImKCs serve as an easily manipulatable proxy for Kupffer cells. Under non-polarized conditions, KCs and ImKCs were highly permissive for EBOV-eGFP and the use of ImKCs allowed us to study EBOV infection kinetics and quality of the associated macrophage immune response using both authentic EBOV and EBOV model systems. As we previously observed in murine peritoneal macrophages (16), infection was robustly inhibited by IFN-γ pre-treatment of the cells. The duration of IFN-γ-elicited antiviral activity was examined and we found that the profound inhibitory effect of IFN-γ on EBOV infection of ImKCs was transient, much of the inhibition conferred by IFN-γ waned within a 24-hour period. However, a more modest ~three-fold inhibition of virus infection persisted for as long as 6 days following IFN-γ treatment. We also assessed if secreted interferon stimulated genes (ISGs) contributed to the IFN-γ-induced protection and found that the secretome was not effective at blocking EBOV infection. These data provide insights into the ISGs and the duration of the antiviral effect of IFN-γ and underscore the importance of macrophages in EVD pathogenesis.
2. Materials and Methods
2.1. Ethics statement
The study was conducted in strict accordance with the Animal Welfare Act and the recommendations in the Guide for the Care and Use of Laboratory animals of the national institutes of Health (University of Iowa (UI) Institutional Assurance Number: #A3021-01). All animal procedures were approved by the UI Institutional Animal Care and Use Committee (IACUC) which oversees the administration of the IACUC protocols, and the study was performed in accordance with the IACUC guidelines (Protocol #1031280).
2.2. Primary Kupffer cell isolation
Wild-type C57BL/6 mice were a kind gift from Dr. John Harty (University of Iowa). Mice were maintained in agreement to IACUC guidelines at the UI. Primary Kupffer cells were isolated from wild type C57B/6 mice as previously reported (21). Briefly, livers were excised, finely chopped, and digested in 10 mL RPMI media containing 1 mg/mL of type IV collagenase (ThermoFisher Scientific, #17104019) for 30 min at 37°C. Digested tissue was mashes through 100 μM cell strainers. Hepatocytes were separated from liver suspensions by low brake low speed centrifugation (500 rpm, 3 min, room temperature). Hepatocytes-free suspensions were spun for 7 min, 1500 rpm, room temperature in a 20% Percoll (Millipore Sigma, #P4937-100ML)/80% HBSS (Gibco, #14025-092) gradient. Supernatants were removed and cell pellets were subject to RBC lysis with lab made lysis buffer. For macrophage polarization cells were plated and treated with polarizing cytokines as described below. Cells were plated and send to the BSL4 for macrophage polarization and infection with EBOV-eGFP. For KCs phenotyping cells were surface staining for macrophage and KCs associated markers as described below.
2.3. Cell lines
ImKCs are commercially available (https://www.sigmaaldrich.com/deepweb/assets/sigmaaldrich/product/documents/371/711/scc119ds.pdf) and were maintained in RPMI supplemented with 10% FBS, penicillin (100U/mL) and streptomycin (100μg/mL). Vero cells were grown in DMEM supplemented with 10% FBS, penicillin (100U/mL) and streptomycin (100μg/mL). EBOV VP30-expressing Vero cells have been described (22) and were maintained and cultured as regular Vero cells. EBOV VP30 expressing ImKCs were generated by transduction of a VP30-encoding pLV-EF1α-VP30-IRES-hygro_v2.1-hygromycin into ImKCs. Cells were selected with hygromycin (300μg/mL). VP30 expression of the bulk population was confirmed by Western blot using a rabbit anti EBOV VP30 polyclonal antisera (#0301-048, IBT Bioservices). VP30 expression in ImKCs was further validated by their ability to support EBOV ΔVP30 dependent infection. EBOV VP30 expressing ImKCs were maintained as parental ImKCs. All cell lines used in our experiments were tested negative for mycoplasma spp. using a commercially available PCR assay (Bulldog Bio; 25233).
2.4. Primary KC and ImKCs phenotyping
Primary KCs and ImKCs were staining for surface markers using the following antibodies: Fixable Viability Dye eFluor 780 (1:1000, #65-0865), CD45.2 PE (1;75, clone 104, #109808), CD45.2 BV421 (1;75, clone 104, #109831), F4/80 APC (1:75, clone BM8, #123116), CLEC4F AF647 (1;75, clone 3E3F9, #156803), TIM-4 PerCP eFlour 710 (1:75, clone RMT4-54, #129906), CLEC2 PE (1:75, clone 17D9/CLEC-2, #146103), CD14 PE (1:75, clone Sa-14-2, #123309), TLR4 APC (1:75, clone SA15-21, #145405), CD11b BV421 (1:75, clone M1/70, #404-0112-80). Antibodies utilized in our studies were purchased from BioLegend and/or Invitrogen. Unstained cells as well as fluorescent minus one (FMO) control samples were used as controls on every staining. All stainings were performed in the presence of the Fc receptors blocker monoclonal antibody (BioxCell, 20ug/mL, clone 2.4G2, #BE0307). Samples were measure on a CytoFLEX cytometer (Beckman Coulter). Data were analyzed using the FlowJo software (BD Biosciences).
2.5. Viruses
The All experiments with the replication-competent EBOV were performed in the National Emerging Infectious Diseases Laboratories (NEIDL) Biosafety Level 4 (BSL4) laboratory (Boston, MA). The recombinant EBOV variant Mayinga expressing enhanced GFP (EBOV-eGFP) was generated and characterized as previously described (23). EBOV ΔVP30 was kindly provided by Peter Halfmann (University of Wisconsin) and propagated and characterized as previously reported (22). Briefly, virus was propagated by infecting EBOV VP30-expressing Vero cells at low MOI (~0.005) and collecting supernatants at 5dpi. The resulting supernatants were filtered through a 45-micron filter and purified by ultra-centrifugation (28,000 g, 4°C, 2 hr) through a 20% sucrose cushion. Recombinant vesicular stomatitis virus encoding and expressing the glycoprotein from EBOV (Mayinga) (rVSV/EBOV GP) was generated as previously described (24). Virus was propagated by infecting Vero cells at low MOI (~0.05) and collecting supernatants at 48hpi. The resulting supernatants were filtered through a 45-micron filter and purified by ultra-centrifugation (28,000 g, 4°C, 2 hr) through a 20% sucrose cushion. The resulting stocks were resuspended in a small volume of PBS and those used for in vivo studies were further purified by treatment with an endotoxin removal kit (Detoxi-Gel Endotoxin Removing Gel, ThermoFisher Scientific 20339) before being aliquoted and stored at -80°C until use. rVSV/EBOV GP viral titers were determined by TCID50 assay on Vero cells. Recombinant vesicular stomatitis virus expressing wild-type G glycoprotein (rVSV/G) was generated and titered in the same manner as rVSV/EBOV GP. All four viruses used in these studies encoded a reporter gene, GFP, that was used to assess virus infection.
2.6. Macrophage polarization
Polarization of primary KCs and ImKCs was achieved by culturing cells for 24 hours in media containing 20 ng/mL IFN-γ (Cell Sciences, #CRI001B) or 20ng/mL IL-4 (BioLegend, #574302) + 20ng/mL of IL-13 (BioLegend, #575902). Following polarization, media was removed and replaced with culturing media without cytokines and harvested for RNA or infected with virus. Macrophage polarization was validated by qRT-PCR.
2.7. In vitro infections
For maximum biocontainment laboratory studies, 96-well plates containing KCs (105 cells) and/or ImKCs (105 cells) were mock-treated or treated with IFN-γ (20 ng/μL) 48 h prior to infection with 104 or 105 particles of EBOV- eGFP for 48 h. For EBOV ΔVP30 experiments under BSL2+ conditions, 24-well plates containing EBOV VP30-expressing ImKCs (2.5x105 cells) were treated with 20 ng/ml of IFN-γ for 24 hours followed by infection with a MOI of 1 or 10 of EBOV ΔVP30 for 48 to 60 hours as noted in figure legends. For RNA analysis, cells were harvested on TRIzol and stored at 4 °C. To assess infection, infected populations were lifted, wash 1x and resuspended in FACS buffer. GFP expression was measured by a Calibur (BD) or CytoFLEX cytometer (Beckman Coulter). Data were analyzed using the FlowJo software (BD Biosciences).
2.8. IFN-γ protection over time
For assessing the protection provided by IFN-γ over time, 24 well-plates containing EBOV VP30 expressing ImKCs were treated with IFN-γ for 24 hours and infected with an MOI of 10 of EBOV ΔVP30 for 60h starting at 0 h, 24 h, 48 h, 96 h, and 144 h after removing IFN-γ. Analysis of RNA and GFP expression was carried out as described above. To understand the impact of IFN-γ on cell viability throughout the duration of the experiment, a luciferase-based ATPlite™ assay was used (PerkinElmer). Briefly, 96 well-plates containing EBOV VP30 expressing ImKCs (1x104 cells) were treated with IFN-γ for 24 hours and lysed at 0 h, 24 h, 48 h, 96 h, and 144 h after removing IFN-γ. Luciferase containing-substrate provided by the manufacturer was added directly to cells in the plate, transferred to white bottom plates and luminescence was measured by plate reader (Tecan) according to the manufacturers protocol.
2.9. Focus-forming assay
EBOV VP30-expressing Vero cells were seeded at a density of 2.5 × 104 cells per well in flat-bottom 96-well tissue culture plates. The following day, medium was removed and replaced with 100 μL of 10-fold serial dilutions of ΔVP30-EBOV. Two hours later, 135 μL of methylcellulose overlay was added. Plates were incubated for 3 days and then fixed with 4% paraformaldehyde in phosphate-buffered saline for 10 min, followed by permeabilization with saponin-containing buffer. Plates were incubated overnight at 4 °C in 100 μL of permeabilization buffer containing a monoclonal anti-EBOV glycoprotein (clone 15H10, BEI resources) at 1:3200 dilution followed by washing and a two-hour room-temperature incubation with secondary anti-mouse-HRP (Jax 115-035-062) diluted 1:1000. Foci were scanned and quantitated on a Biospot plate reader (C.T.L).
2.10. RNA isolation and qRT-PCR
RNA was isolated using TRIzol reagent from Invitrogen following the manufacturer’s instructions. RNA was subsequently converted to cDNA with the High-Capacity cDNA Reverse Transcription kit (#4368814). A total of 1 μg of RNA was used as input for each reaction. Quantitative PCR was performed using the PowerUp™ SYBR™ Green Master Mix (#A25742) from Applied Biosystems according to the manufacturer’s specifications and utilizing a QuantStudio™ 3 Real-time PCR machine from Applied Biosystems. 20 ng of cDNA were amplified. Duplicate qRT-PCR analyses were performed for each sample, and the obtained threshold cycle (CT) values were averaged. Gene expression was normalized to the expression of the housekeeping gene (Cyclophilin A, CypA) resulting in the ΔCT value. The relative mRNA or viral RNA was calculated by 2-ΔCT. The primers utilized in this study are as follows, 5’ to 3’ in format: CypAfor: GCT GGA CCA AAC ACA AAC GG, CypArev: ATG CTT GCC ATC CAG CCA TT, EBOV NP for: CAG TGC GCC ACT CAC GGA CA, EBOV NP rev: TGG TGT CAG CAT GCG AGG GC, Clec4f for: ACA ACT CTG GAC ACG ACA ATC A, Clec4f rev: ATC TGT ACC TCC TTG TGA CAG C, Timd4 for: GGG GAA GGT CCA GTT TGG TG, Timd4 rev: TCC AAG CGC ACA TTC TTC TTG, Clec2a for: GCG GAA CCT GCC TCT TCT TG, Clec2a rev: GAT ACT TTT GCT GTG TGA CCG ACA T, Irf1 for: GCC ATT CAC ACA GGC CGA TAC, Irf1 rev: GCC CTT GTT CCT ACT CTG ATC C, Gbp5 for: CCC AGG AAG AGG CTG ATA G, Gbp5 rev: TCT ACG GTG GTG GTT CAT TT, Gbp2a for: CTG GCT CTG AGA AAA GGA ACT GA, Gbp2a rev: GAA AGT TGC TTC CTG TCT CCA, Arg1 for: CAA ATT GTG AAG AAC CCA CGG, Arg1 rev: CTT CCA ACT GCC AGA CTG TG, Ym1 for: AGC TTT TGA GGA AGA ATC TGT GG, Ym1 rev: CCT GAA TAT AGT CAA GAG ACT GAG A, Clec10a for: CCA AGA GCC TGG TAA AGC AGC, Clec10a rev: ATC CAA TCA CGG AGA CGA CC
2.11. Generation of and studies using IFN-γ conditioned media
ImKC-VP30 cells were plated at 50,000 cells/well in a 48-well format in RPMI with 5% FCS and pen/strep. The following day, some wells were treated with 20 ng/ml of IFN-γ for 24 hours. IFN-γ containing media was removed after 24 hours, cells were washed once with media and maintained for another 24-hour period in media. This media, called conditioned media, was filtered through a 0.45 μ filter and either used directly or frozen at -80°C until use. Prior to infection, ImKC-VP30 cells in a 48 well format were held in media or treated for 24 hours with 20 ng/ml of IFN-γ. Prior to infection, IFN-γ was removed and media refreshed. At the time of infection, additional wells of cells were treated with 20 ng/ml of IFN-γ or conditioned media. These cells were infected with rVSV/EBOV GP, rVSV/G or EBOV ΔVP30 at the MOIs noted in the figures. All three viruses express GFP and infectivity was assessed by determining GFP positive cells in the culture by flow cytometry. The rVSV/EBOV GP and rVSV/G infections were assessed at 20-24 hours, whereas the EBOV ΔVP30 cells were assessed at 48 hours following infection.
2.12. Statistical analysis
Data analysis was performed using GraphPad Prism 9.4.1 (GraphPad, USA). Unless indicated otherwise, data are show as media +- SD. Unpaired one-tailed Student t-test was used to determine statistical significance of single experiments. One-way ANOVA with Dunnett post hoc was used to perform multiple comparisons against reference controls. Tukey post hoc was used to perform multiple comparisons against every condition. In all tests, values of *P<0.05, ** P <0.01, *** P <0.001, **** P<0.0001 were considered significant.
3. Results
3.1. Characterization of primary and immortalized Kupffer cells
To assess the expression of macrophage and KS-specific associated markers, we purified myeloid cells from other liver cells of a C57BL/6 mouse by a Percoll gradient. We phenotypically characterized these cells by flow cytometric analysis following the gating strategy on Figure S1. Approximately 50% of live cells were positive for CD45 (lymphocyte common antigen). Further analysis of the CD45+ cells indicated that ~30% of the cells were KCs, characterized by the expression of the KC-specific markers, CLEC4F (c-type lectin domain family 4 member F) and TIM-4 (T cell immunoglobulin and mucin domain containing 4) (Figures 1A and S1) (25–28). Analysis of the CLEC4+ or TIM-4+cells demonstrated expression of macrophage markers F4/80, TLR4 and CD14 as well as the KC-associated marker CLEC2 (c-type lectin domain family 2, coded by Clec1b) (Figure 1A).
Expression of these markers was also assessed in a murine Kupffer cell line (ImKC) that was established from transgenic mice expressing the thermolabile mutant tsA58 of simian virus 40 large T antigen (gating strategy on Figure S2). As others have previously shown (29), ImKCs were found to express F4/80, CD11B, TLR4 and CD14 (Figure 1B). Additionally, ImKCs expressed CLEC4F and a portion of the population expressed TIM-4 and CLEC2 (Figure 1B).
3.2. Primary KCs and ImKCs respond to polarizing cytokine treatments
Exposure of macrophages to certain cytokines drives macrophage polarization. In our earlier work, we demonstrated that interferon-γ (IFN-γ) generates a proinflammatory M1 phenotype in resident peritoneal macrophages as assessed by elevated production of interferon stimulated genes (ISGs) such as interferon regulatory factor 1 (IRF-1) and other proinflammatory proteins (16). In contrast, interleukin-4/interleukin-13 (IL-4/IL-13) treatment generates a M2a phenotype that is notable for arginase-1 (ARG-1) expression (15). We evaluated the effect of these cytokines on primary KC polarization. Primary KCs were enriched from the bulk population obtained after the Percoll gradient by adherence on tissue culture plates for 2 hours and mock-treated or cultured in the presence of IFN-γ or IL4/IL-13. Treatment of these cells with IFN-γ for 24 h stimulated IRF-1 and guanylate binding protein 5 (GBP5) transcript levels as anticipated (Figure 2A). The KCs that received a 24-hour treatment of IL-4/IL-13 expressed Arg-1 and chitinase-like protein 3 (YM1) (Figure 2A). These data indicate that these cells were appropriately responsive to the immunomodulatory cytokines.
Similarly, following 24h IFN-γ and/or IL-4/IL-13 treatment, ImKCs polarized towards M1-like or M2-like phenotype, respectively (Figure 2B). Treatment with IFN-γ significantly elevated levels of IRF-1 and GBP5 when compared to non-treated ImKCs. Moreover, significantly elevated levels of Arg-1 transcripts were found in M2a polarized ImKCs. Macrophage galactose-type lectin (CLEC10A), another known M2a associated marker (30), was significantly upregulated in M2a ImKCs when compared to M0 and M1 polarized ImKCs. Of note, basal (M0) levels of these activation markers were notably higher in the primary KCs compared to the immortalized cells. This may be due to a generalized activation of primary KCs that occurs during the isolation procedure. In total, these findings show that, similarly to primary macrophages, ImKCs can be polarized towards M1- and M2a-like cells.
Figure 1.
Phenotypic characterization of primary and immortal Kupffer cells. (A) Myeloid cell populations were isolated from livers of C57BL/6 mice and analyzed by flow cytometry following the gating strategy on Figure S1. Live CD45+ CLEC4F+ or Live CD45+ TIM-4+ primary KC cells were analyzed for the expression of CLEC4F, TIM-4, F4/80, TLR4, CD14 and CLEC2 by flow cytometry (n=3; two-livers pooled per group). (B) ImKCs were phenotypically characterized by flow cytometry for the expression of macrophage and Kupffer cell-specific markers F4/80, CD11b, TLR4, CD14, CLEC4F, TIM-4 and CLEC2 following the gating strategy depicted in Figure S2. Shown are representative flow cytometry plots from three independent biological experiments. Fluorescent minus one (FMO) controls were used to delineate gates and served as negative controls for their respective marker expression comparison.
Figure 1.
Phenotypic characterization of primary and immortal Kupffer cells. (A) Myeloid cell populations were isolated from livers of C57BL/6 mice and analyzed by flow cytometry following the gating strategy on Figure S1. Live CD45+ CLEC4F+ or Live CD45+ TIM-4+ primary KC cells were analyzed for the expression of CLEC4F, TIM-4, F4/80, TLR4, CD14 and CLEC2 by flow cytometry (n=3; two-livers pooled per group). (B) ImKCs were phenotypically characterized by flow cytometry for the expression of macrophage and Kupffer cell-specific markers F4/80, CD11b, TLR4, CD14, CLEC4F, TIM-4 and CLEC2 following the gating strategy depicted in Figure S2. Shown are representative flow cytometry plots from three independent biological experiments. Fluorescent minus one (FMO) controls were used to delineate gates and served as negative controls for their respective marker expression comparison.
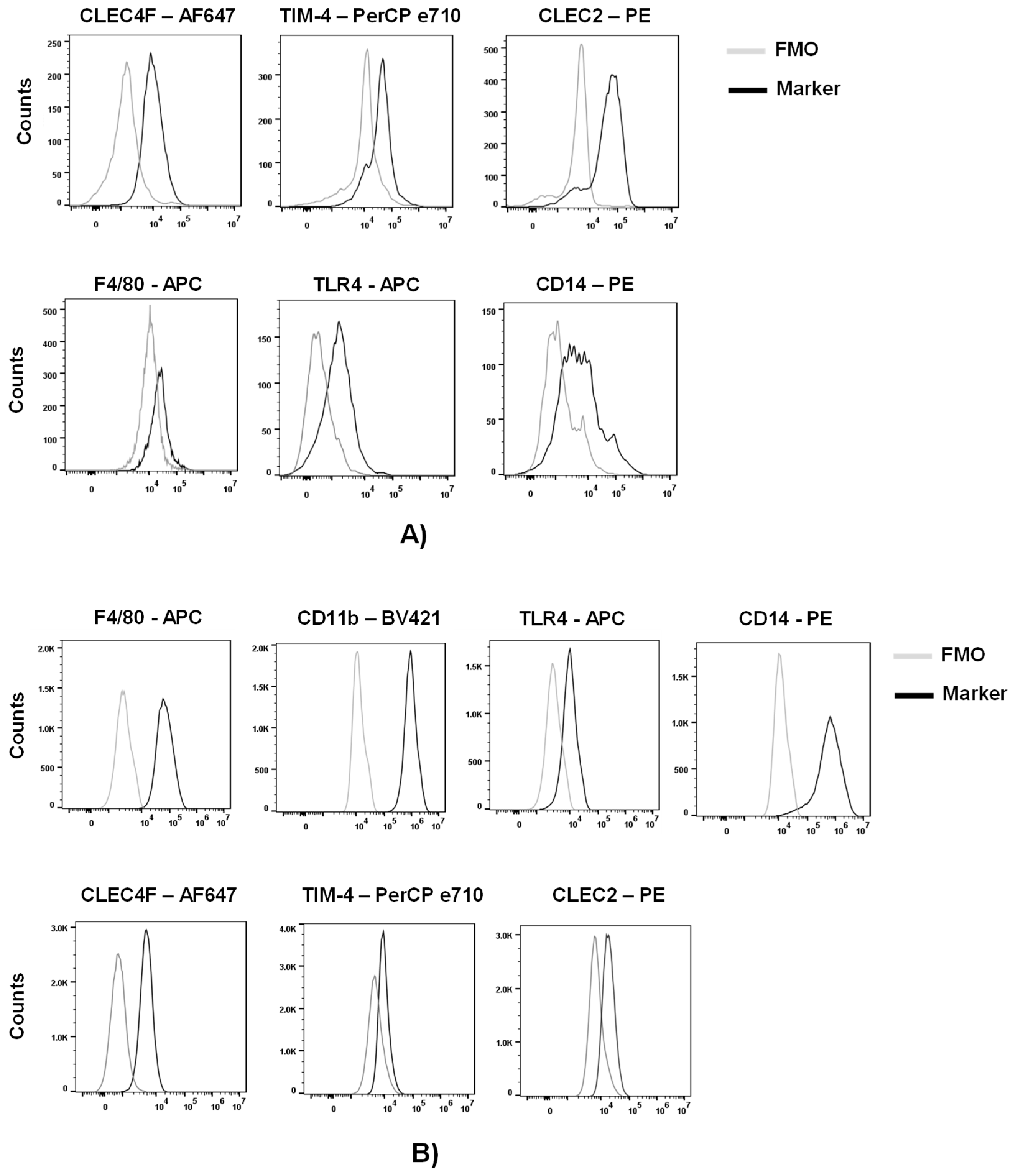
Figure 2.
Primary and immortal Kupffer cells polarize towards M1- and M2a- like cells following treatment with appropriate cytokines. Gene expression changes in (A) primary KCs or (B) ImKCs following 20 ng/ml IFN-γ (M1) or 20 ng/ml of IL-4/IL-13 (M2a) treatment of KCs. Gene expression levels of Irf1, Gbp5, Arg1, Ym1 and Clec10a were determined by qRT-PCR. Cyclophilin A was used as a reference gene. P values were determined by unpaired one-tailed t-test comparing individual treatments with unstimulated (M0) controls. (*P<0.05; **P< 0.01; ***P<0.001; ****P<0.0001).
Figure 2.
Primary and immortal Kupffer cells polarize towards M1- and M2a- like cells following treatment with appropriate cytokines. Gene expression changes in (A) primary KCs or (B) ImKCs following 20 ng/ml IFN-γ (M1) or 20 ng/ml of IL-4/IL-13 (M2a) treatment of KCs. Gene expression levels of Irf1, Gbp5, Arg1, Ym1 and Clec10a were determined by qRT-PCR. Cyclophilin A was used as a reference gene. P values were determined by unpaired one-tailed t-test comparing individual treatments with unstimulated (M0) controls. (*P<0.05; **P< 0.01; ***P<0.001; ****P<0.0001).
3.3. Primary and immortalized murine Kupffer cells support EBOV infection
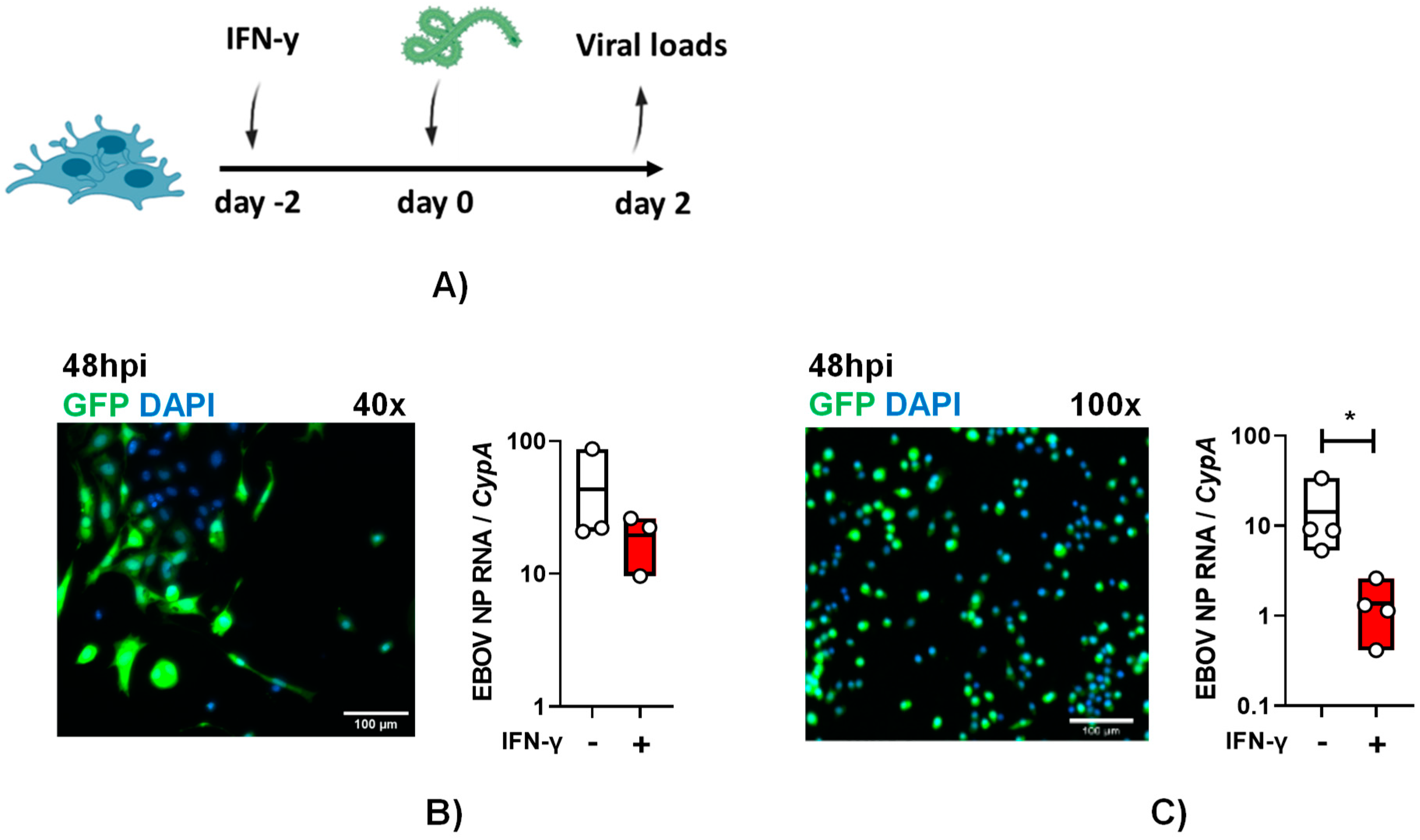
To examine if these cells are susceptible to authentic EBOV infection and the impact of IFN-γ on infection, primary KCs were pre-treated with IFN-γ. Cytokine treated and untreated cells were infected with a multiplicity of infection (MOI) of 0.1 of EBOV-eGFP particles under maximum biocontainment for 48-hours and GFP expression was observed in infected cells and virus load was readily detected (Figure 3A and B). These findings demonstrate for the first time that cultured KCs support EBOV infection. Following IFN-γ treatment, 48 hpi EBOV virus loads in primary KCs trended lower, but the drop in viral load did not achieve statistical significance.
Similar studies were performed with ImKCs. As evidenced in the micrographs, these cells were appreciably smaller than the primary KCs, but they also readily supported EBOV-eGFP infection (Figure 3B and C). In these cells, IFN-γ significantly diminished EBOV-GFP virus load by more than 10-fold (Figure 3C). Altogether, our data show that immortal and primary KCs are permissive for EBOV infection and that ImKCs permissiveness to EBOV is significantly suppressed by prior IFN-γ treatment.
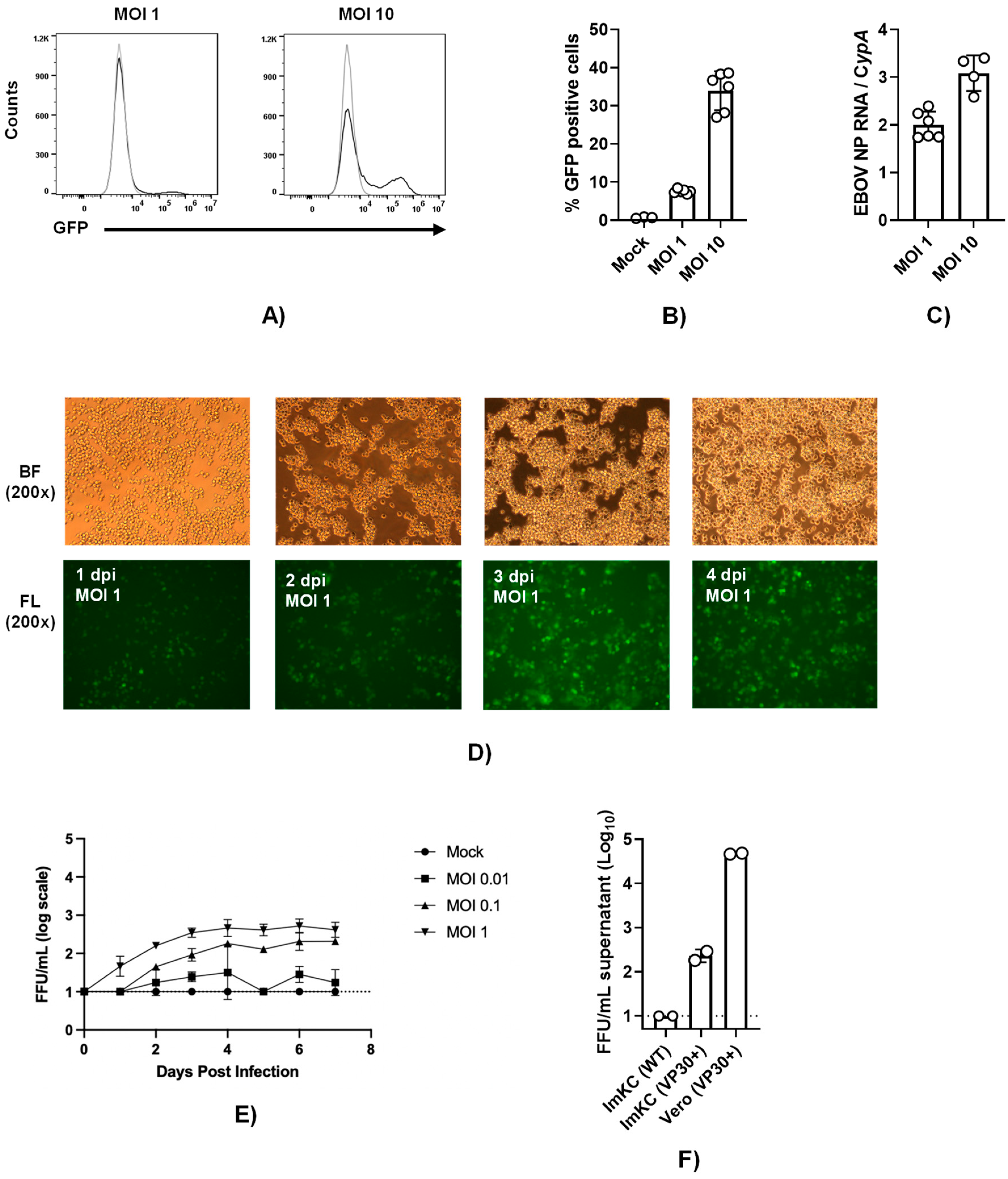
3.5. EBOV VP30-expressing ImKCs support EBOV ΔVP30 infection
To establish a system to study EBOV infection using infectious virus without requiring access to a maximum biocontainment laboratory, we utilized the biological contained, previously developed model of EBOV infection referred to as EBOV ΔVP30 (22). The biologically contained virions express a GFP reporter instead of the VP30 gene and the requisite VP30 gene is supplied in trans in the target cell. EBOV VP30-expressing ImKCs that were generated and biologically cloned were termed ImKCs-VP30. EBOV ΔVP30 stocks that were generated and titered in previously characterized Vero-VP30 cells (22) were evaluated in ImKC-VP30 cells. While a multiplicity of infection MOI of 1 resulted in modest levels of GFP-positive cells at 60 hours, a higher MOI of 10 resulted in ~40% of the cells infected as assessed by flow cytometry. (Figure 4A and B). Viral loads trended similarly as assessed by qRT-PCR (Figure 4C). To examine the ability of EBOV ΔVP30 to spread in ImKC-VP30 cells, virus was added at several lower MOIs and monitored over time. Spread within the culture was observed and by day 4 most of the cells were infected (Figure 4D). Production of new EBOV ΔVP30 was also assessed in these cells. Virus production was dependent on the quantity of input virus and by 3-4 days of infection the quantity of new virus in the supernatant plateaued at a modest level of ~103 iu/ml (Figure 4E). Comparative studies of the infectious virus produced in supernatants on day 5 of infection demonstrated the importance of VP30 expression for the generation of EBOV ΔVP30 and the difference in production of virus in the ImKC-VP30 line versus the previously described Vero-VP30 line (22) (Figure 4F). Thus, while ImKC-VP30 cells support EBOV ΔVP30 infection that spreads through the culture, low levels of virion input resulted in modest generation of new infectious virions in supernatants.
Figure 3.
Primary and immortal Kupffer cells support EBOV infection. (A and B) Primary KCs and (A and C) ImKCs were left untreated or treated with IFN-γ (20ng/mL) for 48h and then infected with 1x104 EBOV-eGFP particles for 48h (n=3, three independent biological experiments conducted at maximum containment laboratory). (B and C) GFP expression after 48hpi. Gene expression levels of EBOV NP were determined by qRT-PCR of cell lysates. Cyclophilin A was used as a reference gene. P values were obtained by unpaired one-tailed t test. (*P<0.05).
Figure 3.
Primary and immortal Kupffer cells support EBOV infection. (A and B) Primary KCs and (A and C) ImKCs were left untreated or treated with IFN-γ (20ng/mL) for 48h and then infected with 1x104 EBOV-eGFP particles for 48h (n=3, three independent biological experiments conducted at maximum containment laboratory). (B and C) GFP expression after 48hpi. Gene expression levels of EBOV NP were determined by qRT-PCR of cell lysates. Cyclophilin A was used as a reference gene. P values were obtained by unpaired one-tailed t test. (*P<0.05).
3.5. IFN-γ inhibits EBOV ΔVP30 infection of ImKCs
In a manner similar to authentic EBOV in the parental ImKCs, 24-hour pre-treatment of ImKCs-VP30 with IFN-γ significantly downregulated viral loads of EBOV ΔVP30 as well as virus-driven GFP expression (Figure 5A-C, and Figure S3). The duration of the antiviral effect of IFN-γ was evaluated in this infection system using confluent wells of ImKC-VP30 cells incubated with low serum-containing media to reduce overgrowth of the culture. ImKCs-VP30 cells were treated for 24 hours with IFN-γ (20 ng/mL). The cytokine was removed and fresh media was added. At 0-144 hours following the completion of the IFN-γ treatment, cells were infected with a MOI of 10 of EBOV ΔVP30 and infection was assessed by virus load or GFP expression at 60 hours. When virus infection was initiated immediately after IFN-γ treatment, IFN-γ elicited a ~30-fold reduction in EBOV ΔVP30 virus load on ImKC-VP30 cells (Figure 5A). With time, the inhibitory effect of IFN-γ on EBOV ΔVP30 infection was reduced, with a ~3-fold reduction in virus load observed by 96 hours after treatment. This ~3-fold inhibition persisted for at least 144 hours (~6 days) following IFN-γ treatment. Similar, but more modest, trends were observed if virus infection was measured by the number of GFP+ cells in the infected cultures (Figure 5C). Cell viability was assessed over the time and was not impacted by the length of the experiment or treatment with IFN-γ (Figure S4). These results indicate that much of the anti-EBOV activity elicited by IFN-γ is lost within 24 hours; however, a more modest antiviral.
Figure 4.
ImKCs-VP30 are susceptible and permissive to EBOV ΔVP30 infection. (A and B) ImKCs-VP30 cells were infected with a MOI of 1 or 10 with EBOV ΔVP30 for 60h (n=3, three independent biological experiments). (A-B) GFP expression was determined by flow cytometry. (C) Gene expression levels of EBOV NP were determined by qRT-PCR. Cyclophilin A was used as a reference gene. Data represents the mean ± SD. (D) GFP expression over the course of a 4 day infection. Shown are 200X micrographs of white (top panel) and fluorescent (bottom panel) light images. (E) Infectious virus present in supernatants collected over time beginning with a MOI of 0.01 to 1. Supernatants were titered on Vero-VP30 cells. (F) A comparison of infectious virus produced in supernatants on day 5 from ImKCs, ImKC-VP30s and Vero-VP30 cells infected with an MOI of 0.1 of EBOV ΔVP30.
Figure 4.
ImKCs-VP30 are susceptible and permissive to EBOV ΔVP30 infection. (A and B) ImKCs-VP30 cells were infected with a MOI of 1 or 10 with EBOV ΔVP30 for 60h (n=3, three independent biological experiments). (A-B) GFP expression was determined by flow cytometry. (C) Gene expression levels of EBOV NP were determined by qRT-PCR. Cyclophilin A was used as a reference gene. Data represents the mean ± SD. (D) GFP expression over the course of a 4 day infection. Shown are 200X micrographs of white (top panel) and fluorescent (bottom panel) light images. (E) Infectious virus present in supernatants collected over time beginning with a MOI of 0.01 to 1. Supernatants were titered on Vero-VP30 cells. (F) A comparison of infectious virus produced in supernatants on day 5 from ImKCs, ImKC-VP30s and Vero-VP30 cells infected with an MOI of 0.1 of EBOV ΔVP30.
effect persists for as long as 5 days. A likely scenario to explain this is that some IFN-γ-elicited interferon stimulated genes (ISGs) are only transiently expressed, whereas others continue to be expressed for a longer time
To investigate if some common IFN-γ-elicited ISGs were quite transiently expressed or had prolonged expression, we measured Irf1, Isg15, Gbp5 (guanylate binding protein 5) and Gbp2a (guanylate binding protein 2a) transcripts levels over time (Figure 5D-G). We previously demonstrated that IFN-γ stimulates the production of these transcripts in IFN-γ-treated murine peritoneal macrophages and overexpression of IRF1 and GPB5 inhibit EBOV infection (16). In ImKC-VP30 cells, expression of all four ISGs was dramatically elevated by 24-hour IFN-γ treatment when compared to untreated cells. Unexpectedly, the elevated levels of ISG15 transcripts elicited by IFN-γ treatment did not change over the course of 6-day experiment, indicating prolonged expression of these transcripts. Levels of the transcription factor Irf1 only modestly decreased (3-fold decrease) and was only statistically significant in infected cells at days 4 and 6 following IFN-γ treatment. With evidence of persistence of Irf1 expression over the 6 day period and as Irf1 is a transcription factor that drives expression of many IFN-γ-elicited ISGs (31), this suggests that the ISGs important for robust EBOV inhibition may be Irf1-independent. Levels of Gbp2a did decrease, with a ~9-fold drop by day 4, but transcript levels still remained orders of magnitude higher than levels found in the untreated cells. Expression of Gbp5 also trended downward, but the decrease was not statistically significant.
3.6. Secreted ISGs do not contribute to protection against EBOV ΔVP30 conferred by IFN-γ
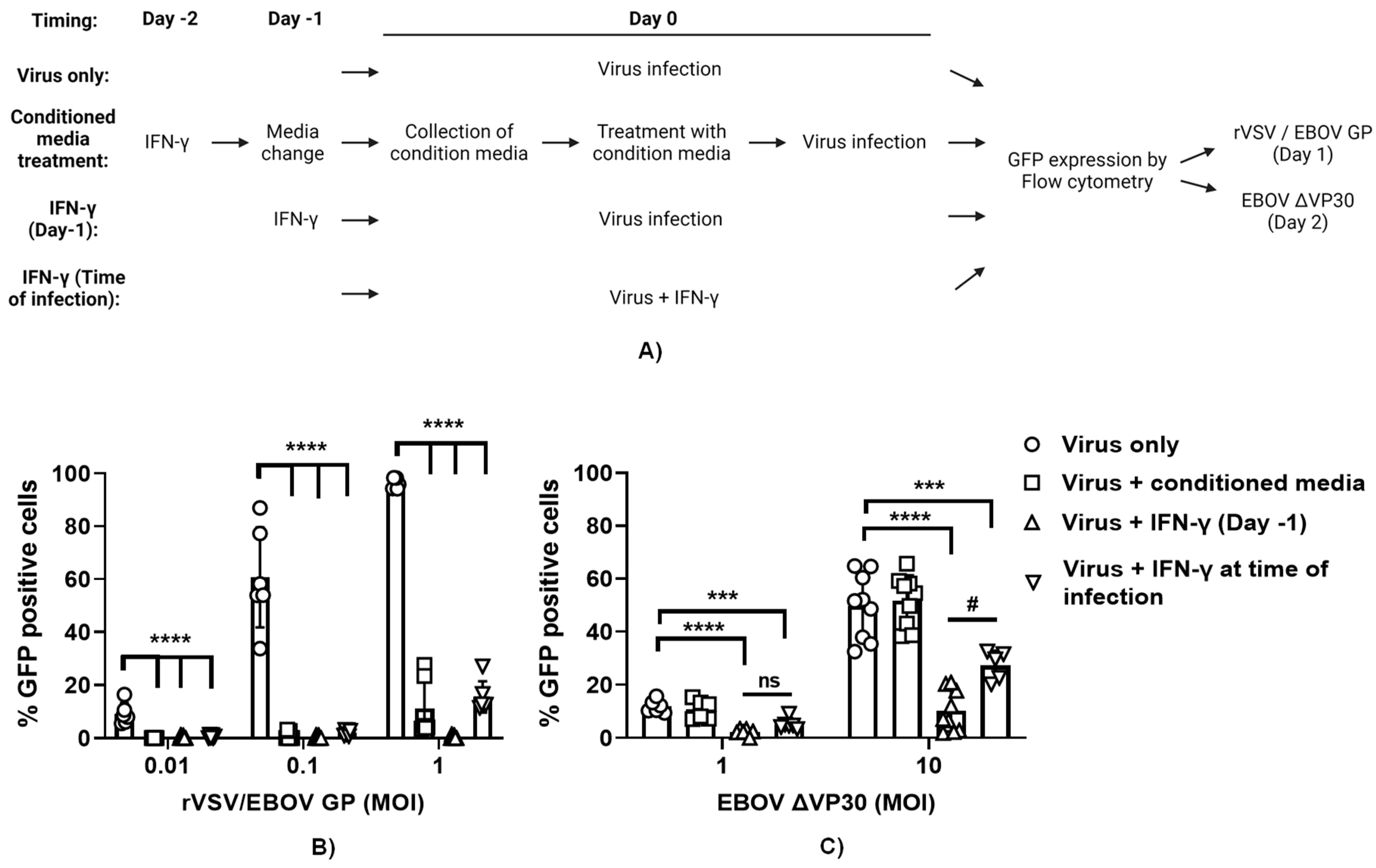
It is appreciated that expression of hundreds of ISGs are elicited upon IFN-γ treatment of macrophages (16). Many of the proteins made from the ISGs remain cell-associated and are cytosolic, nuclear or membrane associated. In contrast, some ISGs are secreted. A number of secreted ISG proteins are chemokines that do not have direct antiviral activity, but instead recruit adaptive immune cells to sites of infection. Other secreted proteins have direct antiviral activity which can be measured in tissue culture. To determine the role of secreted ISG proteins in the direct antiviral effect of IFN-γ, ImKC-VP30 cells were treated with IFN-γ for 24 hours. Cytokine-containing media was removed, cells were washed and fresh cytokine-free media added back for 24 hours. This conditioned media was collected and, in parallel with IFN-γ treatment, was evaluated for its antiviral efficacy in ImKC-VP30 against three different viruses: wild-type vesicular stomatitis virus (rVSV/G), recombinant VSV expressing EBOV GP in place of G glycoprotein (rVSV/EBOV GP) and EBOV ΔVP30 (Figure 6A). As anticipated, infection by all three viruses was inhibited by a 24-hour pretreatment with IFN-γ. In contrast, while conditioned media demonstrated strong antiviral activity against rVSV/G and rVSV/EBOV GP, it had no effect against EBOV ΔVP30 (Figure 6B and C, and Figure S5). These findings indicate that secreted ISGs from ImKCs have direct antiviral activity against VSVs, but do not contribute to the antiviral effect conferred by type II IFN against EBOV ΔVP30.
In these studies, we also assessed the efficacy of a 24-hour IFN-γ treatment when the cytokine was added to cultures 24 hours (IFN-γ on day-1) prior to infection compared to addition at the time of infection (IFN-γ at time of infection). We found that pretreatment with IFN-γ was more effective at inhibiting virus replication than the addition of IFN-γ at the time of infection, providing evidence that the ISGs elicited by prior IFN-γ treatment were highly effective at controlling EBOV ΔVP30 and VSV-based infections (Figure 6B and C, and Figure S5).
To further examine if the antiviral activity present in the conditioned media that inhibited VSV-based viruses, the inhibitory effect of the conditioned media on rVSV/EBOV GP and VSV infection of ImKC-VP30 cells was examined in the presence of 20 μg/ml of interferon α/β receptor (IFNAR) monoclonal antibody (mAb), MAR1-21 or an isotype control IgG. MAR1-21 had no effect on the antiviral activity, indicating that type I IFNs were not contributing to the antiviral activity observed. These findings will guide us in the future identification of secreted ISGs critical for controlling VSV infections. The studies also provide insights into ImKCs cell-associated ISGs elicited by IFN-γ control of EBOV infection, demonstrating that the importance of cell-associated rather than secreted ISGs.
Figure 5.
EBOV ΔVP30 infection in ImKC-VP30 cells is inhibited by IFN-γ treatment and the effect is gradually reduced over the time. EBOV VP30 expressing ImKCs were polarized with IFN-γ (20 ng/mL) for 24 h, media replaced and infection and expression of 4 ISGs were analyzed at the different times noted. (A) Schematic of the experiment. (B) Gene expression of EBOV NP following infection with EBOV ΔVP30 (MOI=10) for 60h at a MOI of 10 (n=3, three independent biological experiments). Cyclophilin A was used as a reference gene. (C) EBOV ΔVP30 infection as measured by eGFP expression which was assessed by flow cytometry. Data shown represent the mean ± SD. (D-G) Gene expression levels of Isg15, Irf1, Gbp2a and Gbp5 were determined by qRT-PCR in EBOV ΔVP30 infected and uninfected cells following IFN-γ treatment. Cyclophilin A was used as a reference gene. (B-C) P values were obtained by unpaired one-tailed t test. (D-G) P values were obtained by one-way ANOVA, Dunnett post hoc test. (*P<0.05; **P<0.01; ***P<0.001; ***P<0.0001; ns=no significance). BL: basal levels of expression on mock-treated cells.
Figure 5.
EBOV ΔVP30 infection in ImKC-VP30 cells is inhibited by IFN-γ treatment and the effect is gradually reduced over the time. EBOV VP30 expressing ImKCs were polarized with IFN-γ (20 ng/mL) for 24 h, media replaced and infection and expression of 4 ISGs were analyzed at the different times noted. (A) Schematic of the experiment. (B) Gene expression of EBOV NP following infection with EBOV ΔVP30 (MOI=10) for 60h at a MOI of 10 (n=3, three independent biological experiments). Cyclophilin A was used as a reference gene. (C) EBOV ΔVP30 infection as measured by eGFP expression which was assessed by flow cytometry. Data shown represent the mean ± SD. (D-G) Gene expression levels of Isg15, Irf1, Gbp2a and Gbp5 were determined by qRT-PCR in EBOV ΔVP30 infected and uninfected cells following IFN-γ treatment. Cyclophilin A was used as a reference gene. (B-C) P values were obtained by unpaired one-tailed t test. (D-G) P values were obtained by one-way ANOVA, Dunnett post hoc test. (*P<0.05; **P<0.01; ***P<0.001; ***P<0.0001; ns=no significance). BL: basal levels of expression on mock-treated cells.
Figure 6.
IFN-γ-induced ISG secretome does not contribute to protection conferred by IFN-γ against EBOVΔVP30. (A) Schematic of the experiment. (B-C) IFN-γ treatment and conditioned media from ImKCs treated with IFN-γ block rVSV/EBOV GP infection (MOI of 0.01, 0.1 and 1) (B), but the conditioned media is not effective at blocking EBOV ΔVP30 infection (MOI of 1 and 10) (C). All studies used a concentration of 20 ng/ml of IFN-γ. Data represents the mean ± SD. P values were obtained by one-way ANOVA (Dunnett post hoc test, ***P<0.001; ***P<0.0001;) (Tukey post hoc test, #P<0.05; ns=no significance).
Figure 6.
IFN-γ-induced ISG secretome does not contribute to protection conferred by IFN-γ against EBOVΔVP30. (A) Schematic of the experiment. (B-C) IFN-γ treatment and conditioned media from ImKCs treated with IFN-γ block rVSV/EBOV GP infection (MOI of 0.01, 0.1 and 1) (B), but the conditioned media is not effective at blocking EBOV ΔVP30 infection (MOI of 1 and 10) (C). All studies used a concentration of 20 ng/ml of IFN-γ. Data represents the mean ± SD. P values were obtained by one-way ANOVA (Dunnett post hoc test, ***P<0.001; ***P<0.0001;) (Tukey post hoc test, #P<0.05; ns=no significance).
4. Discussion
Here, we established an immortalized murine macrophage model to study innate immune responses during EBOV infection outside BSL4 facilities. We demonstrate that ImKCs express macrophage markers and treatment with IFN-γ or IL-4/IL-13 polarizing cytokines increased the expression of respective M1 and M2 markers on these cells, indicating that this cell line serves as an excellent macrophage model for studying cytokine microenvironment. We further demonstrate that ImKCs and EBOV VP30-expressing ImKCs are permissive to EBOV-eGFP and EBOV ΔVP30, respectively, and show that IFN-γ treatment of these cells reduced viral loads and GFP viral gene expression.
We have previously shown that IFN-γ treatment of primary mouse peritoneal macrophages robustly inhibits EBOV infection (16). Here, in our ImKCs models, IFN-γ treatment reduced EBOV-eGFP and EBOV ΔVP30 in similar fashion. We examine the duration of IFN-γ antiviral activity in our IFN-γ-treated ImKCs. Our data supports the contention that there are at least two subsets of ISGs driving anti-EBOV activity. The first subset profoundly inhibited virus infection and is transient following IFN-γ treatment; within 24 to 48 hours, this activity wanes. A second subset of ISGs had more prolonged, but less effective, inhibitory activity that persisted for the duration of our experiments. Analysis of four well-established IFN-γ-stimulated ISGs indicated that expression of these ISGs remained elevated over the 6-day experiment, with modest decreases of two of the transcripts, gbp5 and gbp2a. However, the transcription factor, Irf1, that stimulates expression of many known IFN-γ-dependent ISGs (31) remained elevated throughout the experiment, suggesting that the first wave of strong antiviral activity may be driven by ISGs that are not regulated by Irf1.
In general, type I and II IFN responses are thought to be quite transient, yet these four IFN-γ-elicited ISGs we examined were increased over basal levels in the ImKC-vp30 cells for as long as 5 days following treatment. Others have also reported prolonged IFN responses in other cell lines following either type I IFN treatment or virus infection (32–34). In Daudi cells, type I IFNs were demonstrated to elicit long term (7 day) expression of ISGs than that observed in several other cell types (33). Studies in HUVECs have also demonstrated that expression of the ISGs, MxA, Irf3 and Irf7, are robustly expressed for as long as 7 days during Hantaan virus infection despite quite transient expression of both IFN-α and IFN-β (34). Future studies to examine the duration of the antiviral activity in primary macrophages and identify the ISGs responsible for antiviral activity against EBOV are warranted.
Our studies also demonstrate that secreted ISGs do not participate in the direct control of EBOV ΔVP30 infection as conditioned media from IFN-γ-treated ImKC-VP30 cells conferred no protection against this virus. This stands in contrast to our findings with VSV-based viruses, wild-type VSV and rVSV/EBOV GP, where strong protection with IFN-γ-conditioned media was observed. These studies provide insights into which ISGs are important for controlling viruses from these two different Mononegavirales families. Our own studies and in others (16, 17, 35, 36) have used rVSV/EBOV GP in type I IFN σβ receptor KO mice as a low containment model for EBOV infection. Our data shown here highlights the care with which such studies must be interpreted as the apparent ISGs contributing to control of the respective viruses differs.
Efforts in improve our understanding of mechanisms driving disease pathogenesis following EBOV infection have been hampered by the necessity of high biocontainment conditions (BSL4). The used of recombinant vesicular stomatitis virus (VSV) expressing the EBOV glycoprotein (rVSV/EBOV GP) has been useful for the study of glycoprotein-mediated processes such as viral entry and fusion, as well adaptive immune responses towards EBOV GP (17, 37, 38). However, as we demonstrate in our conditioned media studies, the data obtained by using this infectious BSL2 model may not always recapitulate infection-mediated responses following authentic EBOV infection. Importantly, the generation of biologically contained EBOV lacking the VP30 gene (EBOV ΔVP30) that recapitulates EBOV morphology and growth properties permits EBOV studies in tissue culture under lower containment conditions (22). However, a small animal model (e.g., mouse) suitable for work with EBOVΔVP30 that could be employed outside of the BSL4 is still needed.
In summary, we show that KCs, ImKCs and EBOV ΔVP30 expressing ImKCs support infection with EBOV and EBOV ΔVP30 respectively. Furthermore, our in vitro studies demonstrated that IFN-γ inhibits EBOV and EBOV ΔVP30 infection in ImKCs and provide insights into the type of ISGs that are responsible for their antiviral activity. Overall, these studies provide new tools for the study of EBOV infection that can potentially aid the development of anti-filovirus therapeutics.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Supplementary Figure 1: Gating strategy for phenotypic characterization of primary KCs; Supplementary Figure 2: Gating strategy for phenotypic characterization of ImKCs; Supplementary Figure 3: EBOV ΔVP30 infection (MOI=1) of ImKC-VP30 cells is inhibited by IFN-γ treatment; Supplementary Figure 4: ImKC-VP30 cell viability following IFN-γ treatment; Supplementary Figure 5: IFN-γ and conditioned media from IFN-γ treated cells blocks rVSV/G infection.
Author Contributions
J.A.A.B.-Conceptualization, investigation, data curation-formal analysis, writing-original draft, writing-review, editing and revision. J.M.E.- investigation, data curation-formal analysis, writing-review, editing and revision. J.J.P.- investigation, writing-review, editing, and revision. L.R.W.- investigation, writing-review, editing, and revision. R.A.D.- writing-review, editing, and revision. A.L.B.- writing-review, editing, and revision. W.J.M.- conceptualization, data curation-formal analysis, writing-review, editing, and revision. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the following US National Institutes of Health grants: R01 AI134733 (WJM), UH2 AI169710 (WJM), R21 144215 (WJM) and DP5 OD029608 (ALB).
Institutional Review Board Statement
The study was conducted in strict accordance with the Animal Welfare Act and the recommendations in the Guide for the Care and Use of Laboratory animals of the national institutes of Health (University of Iowa (UI) Institutional Assurance Number: #A3021-01). All animal procedures were approved by the UI Institutional Animal Care and Use Committee (IACUC) which oversees the administration of the IACUC protocols, and the study was performed in accordance with the IACUC guidelines (Protocol #1031280).
Acknowledgments
We would like to thank Dr. Dina Amin Abdelkhalek for her technical assistance provided in our studies.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Emanuel J, Marzi A, Feldmann H. 2018. Filoviruses: Ecology, Molecular Biology, and Evolution, p. 189–221. In Advances in Virus Research. [CrossRef]
- Goldstein T, Anthony SJ, Gbakima A, Bird BH, Bangura J, Tremeau-Bravard A, Belaganahalli MN, Wells HL, Dhanota JK, Liang E, Grodus M, Jangra RK, DeJesus VA, Lasso G, Smith BR, Jambai A, Kamara BO, Kamara S, Bangura W, Monagin C, Shapira S, Johnson CK, Saylors K, Rubin EM, Chandran K, Lipkin WI, Mazet JAK. 2018. The discovery of Bombali virus adds further support for bats as hosts of ebolaviruses. Nat Microbiol 3:1084–1089. [CrossRef]
- Biedenkopf N, Bukreyev A, Chandran K, Di Paola N, Formenty PBH, Griffiths A, Hume AJ, Mühlberger E, Netesov S V., Palacios G, Pawęska JT, Smither S, Takada A, Wahl V, Kuhn JH. 2023. Renaming of genera Ebolavirus and Marburgvirus to Orthoebolavirus and Orthomarburgvirus, respectively, and introduction of binomial species names within family Filoviridae. Arch Virol 168:220. [CrossRef]
- Baseler L, Chertow DS, Johnson KM, Feldmann H, Morens DM. 2016. The Pathogenesis of Ebola Virus Disease. Annu Rev Pathol Mech Dis 2017 12:387–418. [CrossRef]
- Feldmann H, Geisbert TW. 2011. Ebola haemorrhagic fever. Lancet. Lancet Publishing Group. [CrossRef]
- Henao-Restrepo AM, Camacho A, Longini IM, Watson CH, Edmunds WJ, Egger M, Carroll MW, Dean NE, Diatta I, Doumbia M, Draguez B, Duraffour S, Enwere G, Grais R, Gunther S, Gsell PS, Hossmann S, Watle SV, Kondé MK, Kéïta S, Kone S, Kuisma E, Levine MM, Mandal S, Mauget T, Norheim G, Riveros X, Soumah A, Trelle S, Vicari AS, Røttingen JA, Kieny MP. 2017. Efficacy and effectiveness of an rVSV-vectored vaccine in preventing Ebola virus disease: final results from the Guinea ring vaccination, open-label, cluster-randomised trial (Ebola Ça Suffit!). Lancet 389:505–518. [CrossRef]
- Suder E, Furuyama W, Feldmann H, Marzi A, de Wit E. 2018. The vesicular stomatitis virus-based Ebola virus vaccine: From concept to clinical trials. Hum Vaccines Immunother. Taylor and Francis Inc. [CrossRef]
- Falzarano D, Feldmann F, Grolla A, Leung A, Ebihara H, Strong JE, Marzi A, Takada A, Jones S, Gren J, Geisbert J, Jones SM, Geisbert TW, Feldmann H. 2011. Single immunization with a monovalent vesicular stomatitis virus-based vaccine protects nonhuman primates against heterologous challenge with bundibugyo ebolavirus. J Infect Dis. [CrossRef]
- Marzi A, Ebihara H, Callison J, Groseth A, Williams KJ, Geisbert TW, Feldmann H. 2011. Vesicular stomatitis virus-based Ebola vaccines with improved cross-protective efficacy, p. S1066. In Journal of Infectious Diseases. Oxford University Press. [CrossRef]
- Liu G, He S, Chan M, Zhang Z, Schulz H, Cao W, Rahim MN, Audet J, Garnett L, Wec A, Chandran K, Qiu X, Banadyga L. 2023. A Pan-ebolavirus Monoclonal Antibody Cocktail Provides Protection against Ebola and Sudan Viruses. J Infect Dis https://doi.org/10.1093/infdis/jiad205. [CrossRef]
- Woolsey C, Borisevich V, Agans KN, O’Toole R, Fenton KA, Harrison MB, Prasad AN, Deer DJ, Gerardi C, Morrison N, Cross RW, Eldridge JH, Matassov D, Geisbert TW. 2023. A highly attenuated pan-filovirus VesiculoVax vaccine rapidly protects nonhuman primates against Marburg virus and three species of Ebolavirus. J Infect Dis https://doi.org/10.1093/infdis/jiad157. [CrossRef]
- Gibb TR, Bray M, Geisbert TW, Steele KE, Kell WM, Davis KJ, Jaax NK. 2001. Pathogenesis of experimental Ebola Zaire virus infection in BALB/c mice. J Comp Pathol 125:233–242. [CrossRef]
- Geisbert TW, Hensley LE, Larsen T, Young HA, Reed DS, Geisbert JB, Scott DP, Kagan E, Jahrling PB, Davis KJ. 2003. Pathogenesis of Ebola Hemorrhagic Fever in Cynomolgus Macaques: Evidence that Dendritic Cells are Early and Sustained Targets of Infection. Am J Pathol 163:2347–2370. [CrossRef]
- Feldmann H, Sprecher A, Geisbert TW. 2020. Ebola. N Engl J Med 382:1832–1842.
- Rogers KJ, Brunton B, Mallinger L, Bohan D, Sevcik KM, Chen J, Ruggio N, Maury W. 2019. IL-4/IL-13 polarization of macrophages enhances Ebola virus glycoprotein-dependent infection. PLoS Negl Trop Dis 13:e0007819. [CrossRef]
- Rhein BA, Powers LS, Rogers K, Anantpadma M, Singh BK, Sakurai Y, Bair T, Miller-Hunt C, Sinn P, Davey RA, Monick MM, Maury W. 2015. Interferon-γ Inhibits Ebola Virus Infection. PLoS Pathog 11. [CrossRef]
- Rogers KJ, Shtanko O, Vijay R, Mallinger LN, Joyner CJ, Galinski MR, Butler NS, Maury W. 2020. Acute Plasmodium Infection Promotes Interferon-Gamma-Dependent Resistance to Ebola Virus Infection. Cell Rep 30:4041-4051.e4. [CrossRef]
- Geisbert TW, Young HA, Jahrling PB, Davis KJ, Larsen T, Kagan E, Hensley LE. 2003. Pathogenesis of Ebola Hemorrhagic Fever in Primate Models: Evidence that Hemorrhage Is Not a Direct Effect of Virus-Induced Cytolysis of Endothelial Cells. Am J Pathol 163:2371–2382. [CrossRef]
- Geisbert TW, Young HA, Jahrling PB, Davis KJ, Kagan E, Hensley LE. 2003. Mechanisms underlying coagulation abnormalities in ebola hemorrhagic fever: overexpression of tissue factor in primate monocytes/macrophages is a key event. J Infect Dis 188:1618–1629. [CrossRef]
- Martines RB, Ng DL, Greer PW, Rollin PE, Zaki SR. 2015. Tissue and cellular tropism, pathology and pathogenesis of Ebola and Marburg viruses. J Pathol 235:153–174. [CrossRef]
- Li P zhi, Li J zheng, Li M, Gong J ping, He K. 2014. An efficient method to isolate and culture mouse Kupffer cells. Immunol Lett 158:52–56. [CrossRef]
- Halfmann P, Jin HK, Ebihara H, Noda T, Neumann G, Feldmann H, Kawaoka Y. 2008. Generation of biologically contained Ebola viruses. Proc Natl Acad Sci U S A 105:1129–1133. [CrossRef]
- Ebihara H, Theriault S, Neumann G, Alimonti JB, Geisbert JB, Hensley LE, Groseth A, Jones SM, Geisbert TW, Kawaoka Y, Feldmann H. 2007. In vitro and in vivo characterization of recombinant Ebola viruses expressing enhanced green fluorescent protein, p. S313–S322. In Journal of Infectious Diseases. Oxford Academic. [CrossRef]
- Garbutt M, Liebscher R, Wahl-Jensen V, Jones S, Möller P, Wagner R, Volchkov V, Klenk H-D, Feldmann H, Ströher U. 2004. Properties of Replication-Competent Vesicular Stomatitis Virus Vectors Expressing Glycoproteins of Filoviruses and Arenaviruses. J Virol 78:5458–5465. [CrossRef]
- Guilliams M, Scott CL. 2022. Liver macrophages in health and disease. Immunity 55:1515–1529. [CrossRef]
- Blériot C, Barreby E, Dunsmore G, Ballaire R, Chakarov S, Ficht X, De Simone G, Andreata F, Fumagalli V, Guo W, Wan G, Gessain G, Khalilnezhad A, Zhang XM, Ang N, Chen P, Morgantini C, Azzimato V, Kong WT, Liu Z, Pai R, Lum J, Shihui F, Low I, Xu C, Malleret B, Kairi MFM, Balachander A, Cexus O, Larbi A, Lee B, Newell EW, Ng LG, Phoo WW, Sobota RM, Sharma A, Howland SW, Chen J, Bajenoff M, Yvan-Charvet L, Venteclef N, Iannacone M, Aouadi M, Ginhoux F. 2021. A subset of Kupffer cells regulates metabolism through the expression of CD36. Immunity 54:2101-2116.e6. [CrossRef]
- Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, Lippens S, Abels C, Schoonooghe S, Raes G, Devoogdt N, Lambrecht BN, Beschin A, Guilliams M. 2016. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat Commun 7. [CrossRef]
- Tran S, Baba I, Poupel L, Dussaud S, Moreau M, Gélineau A, Marcelin G, Magréau-Davy E, Ouhachi M, Lesnik P, Boissonnas A, Le Goff W, Clausen BE, Yvan-Charvet L, Sennlaub F, Huby T, Gautier EL. 2020. Impaired Kupffer Cell Self-Renewal Alters the Liver Response to Lipid Overload during Non-alcoholic Steatohepatitis. Immunity 53:627-640.e5. [CrossRef]
- Wang ZY, Burlak C, Klaunig JE, Kamendulis LM. 2014. Development of a cytokine-producing immortalized murine Kupffer cell line. Cytokine 70:165–172. [CrossRef]
- Roszer T. 2015. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflamm. Hindawi Limited. [CrossRef]
- Maloney NS, Thackray LB, Goel G, Hwang S, Duan E, Vachharajani P, Xavier R, Virgin HW. 2012. Essential Cell-Autonomous Role for Interferon (IFN) Regulatory Factor 1 in IFN-γ-Mediated Inhibition of Norovirus Replication in Macrophages. J Virol 86:12655–12664. [CrossRef]
- Speranza E, Caballero I, Honko AN, Johnson JC, Bohannon JK, Dewald LE, Gerhardt DM, Sword J, Hensley LE, Bennett RS, Connor JH. 2020. Previremic identification of ebola or marburg virus infection using integrated host-transcriptome and viral genome detection. MBio 11:1–14. [CrossRef]
- Tiwari RK, Kusari J, Sen GC. 1987. Functional equivalents of interferon-mediated signals needed for induction of an mRNA can be generated by double-stranded RNA and growth factors. EMBO J 6:3373–3378. [CrossRef]
- Kim IW, Hwang JY, Kim SK, Kim JK, Park HS. 2007. Interferon-stimulated genes response in endothelial cells following Hantaan virus infection. J Korean Med Sci 22:987–992. [CrossRef]
- Rogers KJ, Shtanko O, Stunz LL, Mallinger LN, Arkee T, Schmidt ME, Bohan D, Brunton B, White JM, Varga SM, Butler NS, Bishop GA, Maury W. 2020. Frontline Science: CD40 signaling restricts RNA virus replication in Mϕs, leading to rapid innate immune control of acute virus infection. J Leukoc Biol JLB.4HI0420-285RR. [CrossRef]
- Menicucci AR, Jankeel A, Feldmann H, Marzi A, Messaoudi I. 2019. Antiviral Innate Responses Induced by VSV-EBOV Vaccination Contribute to Rapid Protection. MbioAsmOrg 10:e00597-19. [CrossRef]
- Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, Cin PD, Dye JM, Whelan SP, Chandran K, Brummelkamp TR. 2011. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 477:340–343. [CrossRef]
- Brunton B, Rogers K, Phillips EK, Brouillette RB, Bouls R, Butler NS, Maury W. 2019. TIM-1 serves as a receptor for ebola virus in vivo, enhancing viremia and pathogenesis. PLoS Negl Trop Dis 13:e0006983. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



