
Submitted:
28 August 2023
Posted:
30 August 2023
You are already at the latest version
Abstract
Increased insulin levels may support the development of neural circuits involved in cognition, while chronic mild inflammation may also result in cognitive impairment. This study aimed to gain more insight into whether cognition is impacted already during adolescence in a genetic rat model for obesity and type 2 diabetes. Cognitive functioning throughout adolescence and early adulthood was investigated in Zucker Diabetic Fatty (ZDF), ZDF lean and healthy outbred Long Evans rats using operant touchscreens. Blood glucose, insulin and lipids were longitudinally analyzed. Histological analyses were performed in the liver, white adipose tissues and the prefrontal cortex. Adolescent ZDF obese rats outperformed lean rats on visual discrimination performance. During the longitudinal cognitive testing period, insulin levels sharply increased over weeks in ZDF obese rats and were significantly enhanced from 6 weeks of age onwards. Early signs of liver steatosis and enlarged adipocytes in white adipose tissue were observed in ZDF early adult obese rats. Histological analyses at early adulthood showed no group differences in the number of prefrontal cortex neurons and microglia, nor PSD95 and SIRT1 mRNA expression levels. Together, our data show that adolescent obese ZDF rats even display enhanced cognition despite their early diabetic profile.
Keywords:
diabetes
; adolescence
; cognition
; insulin
; inflammation
1. Introduction
Obesity is a chronic health condition that has been ranked as the fifth most common cause of mortality worldwide [1]. Genetic heritability and the obesogenic environment play important roles as causal pathways for obesity [2]. Inflammatory reactions in various critical organs, including the brain, have been associated with the obesogenic condition [3,4,5,6]. Moreover, neuroinflammation may result in reduced neuroproliferation in several brain areas such as the prefrontal cortex and hippocampus [7,8,9,10,11,12]. Experimental studies with rodent models of insulin resistance, type 2 diabetes or dietary obesity models have demonstrated deficits in a wide range of cognitive tasks [13,14,15]. However, these experiments were mainly performed in adult rodents, upon long exposure to either the genetic or dietary obesity/diabetic model.
Cognitive decline is putatively due to neuronal signaling impairment, decreased synaptic plasticity and reduced insulin signaling [16]. Interestingly, insulin itself plays a profound role in the formation of neural circuits and synaptic connections in rodents, non-human primates and humans [17,18]. Several studies have convincingly shown that insulin administration enhances behavioral performances [19], protect against neuroinflammation [20] and inhibits pro-inflammatory factors in obese non-diabetic human subjects [21]. Consequently, decreased sensitivity to insulin caused by insulin resistance due to an obesogenic or diabetic condition, and the resulting interruption of its signal transduction can challenge the positive effect insulin seems to have on brain functioning [13,17].
This study aimed to gain insight into the interplay between cognition and the development of obesity/diabetes during the adolescent period. We were interested whether the enhanced levels of insulin during development had effects on cognition at an age where the consequences of low-grade systemic inflammation due to the production of inflammatory mediators by dysfunctional adipose tissue are slowly starting to emerge. Therefore, the obese Zucker diabetic fatty (ZDF) rat was selected since the homozygotes ZDF rats (Leprfa/Leprfa) spontaneously develop obesity, hyperlipidemia, insulin resistance, and hyperglycemia by 6 to 8 weeks of age [22,23]. Prior to the experiments with the obese ZDF research model, we performed all experimental assays in two groups of outbred Long Evans rats to investigate under which feeding circumstances (ad libitum food and sucrose rewards versus food restriction and grain rewards) the rats could acquire the full operant higher-order cognitive task within the adolescent period. We used a touchscreen-based task, which resemble those used to assess cognition in humans and thereby has high translational impact [24,25]. We chose the visual discrimination task since we expected the rats were able to acquire this long-term memory test within the full adolescent period. We hypothesized that when obese ZDF animals age and the physiological obesogenic phenotype emerged, their cognitive performances would decline over test sessions and that overall a lower cognition would be observed.
2. Materials and Methods
2.1. Animals and Dietary Regime
A total of 36 (18 males and 18 females) ZDF rats from Charles River (France) and 31 (15 males and 16 females) Long Evans (LE) rats from Janvier Labs (France) were used. Upon arrival at 3.5 weeks of age, animals were first socially housed in groups of four in standard Macrolon®, 55x33x20cm cages for 5 days and thereafter were housed individually in Macrolon®, 42x26x20cm cages. Animals were housed in a controlled environment (reversed 12/12 light-dark-cycle where white lights went off at 7.00 a.m., 22⁰C) with water provided ad libitum. All experiments were performed in accordance with the European Parliament and Council Directive (2010/63/EU) and approved by the Centrale Commissie Dierproeven in The Hague, the Netherlands. ZDF rats were fed with a purified diet (AIN-93G, SSNIFF diet, Bio Services BV Uden) and LE rats were fed with standard laboratory chow (V1534-703, SSNIFF diet, Germany). To motivate animals for repeated complex cognitive testing, two main feeding regime strategies were employed in LE rats: 1. ad libitum feeding and sucrose pellets [5TUT] as reward, i.e. the LE-sucrose group, and 2. food restriction and regular grain pellets (Tablet [5TUM] 45mg/pellet, TestDiet, USA) as reward, i.e. the LE-grain group. Based on the results in LE rats, the food restriction combined with grain pellets reward, was chosen as the feeding regime for the ZDF rat model. The LE-sucrose group received ad libitum feeding. The other three groups, i.e. LE-grain group and the lean and obese ZDF groups were food restricted up to 90% of free feeding body weight during weekdays. On Friday afternoon, rats received an excess amount for the test-free weekend. On Monday mornings, the remaining food was weighed and used to determine how much food each individual rat consumed under ad libitum conditions. This amount was used to calculate the 90% dietary food restriction regime for the upcoming week. The animals were daily fed after cognitive testing.
2.2. Behavioural Assessment
General approach
In short, the rats were individually placed in LaborasTM cages for 48h sessions at week 4, 6, 8, 10 and 12 of age to measure locomotor activity, eating and drinking behaviour. The operant behaviour was assessed in touchscreen operant chambers (Bussey Rat Touchscreen Chamber, Campden Instruments, UK) whereby the pretraining and visual discrimination (VD) tests were performed during the adolescent period of the rat (~week 4 to 10 of age) and the motivational tests were performed during either adolescence or early adulthood, depending on the acquisition and performance speed during daily testing for each individual rat. The rats were tested twice a day for 30 minutes sessions, with a minimum interval of 3 hours between sessions for all tests, except for the 60 minutes progressive ratio task, which was performed once per day. In the VD task, the rat is required to discriminate two images and to touch the correct image in order to receive a reward pellet [26,27]. This task measures learning by stimulus discrimination and long-term memory while assessing the performances over sessions across several days [26].
2.2.1. Phenotypic Behavioural Assessment
The Laboratory Animal Behavior Observation, Registration & Analysis System (LABORAS, Metris) possesses transducers that can measure vibrations due to the movement of the rat on the platform. The software can subsequently convert the signals into behavioural parameters, such as locomotion, grooming, eating and drinking. Additionally, the position of the animal can be assessed, and thereby the travelled distance, the maximum speed and other motion parameters were measured [28]. The locomotor activity, eating and drinking behaviours from 5 consecutive measurements at week 4, 6, 8, 10 and 12 were averaged.
2.2.2. Touchscreen Operant Chambers
Touchscreen chambers equipped with a touchscreen, infrared beams, white house light, pellet dispenser and a feeder magazine with magazine light were placed within a sound-attenuating chamber, fitted with a camera positioned above the operant chamber to monitor the rats (Bussey Rat Touchscreen Chamber, Campden Instruments, UK). To register responses, the operant chambers were equipped with infrared beams inside the feeder magazine and on the surface of the touchscreen. A black mask with two apertures was placed in front of the touchscreen to minimize unintended responses on the touchscreen and to guide the responses on the rat. Presentation of the stimuli, reward delivery and data collection were conducted using ABET II Touch Software (Campden Instruments, UK) and a multimedia research control Whisker system [29].
2.2.3. Training Stages
One week after arrival, between 3-4 weeks of age, rats started their cognitive training in the operant chambers. Prior to response training, a handful of reward pellets was provided to the rats in their homecage to reduce potential food neophobia. In the first training step, called habituation, the rats were introduced to the chamber and were provided with 25 reward pellets randomly delivered into the feeder magazine over the course of 30 minutes. The second training step consisted of autoshaping (AT), during which a conditioned stimulus (white circle, diameter 8 cm) on the right side of the touchscreen was introduced and associated with reward delivery. In this step, the rat received the reward either upon touching the image, or when the 30 seconds stimulus presentation passed independently from whether the rat touched the screen or not. Each 30 minutes session consisted of 50 trials and to continue to the next training stage, certain criteria had to be met (Table 1). After a correct response, one reward pellet was provided, the stimulus disappeared and an inter-trial interval of 5 seconds followed. An incorrect response or no response (omission) resulted in no reward delivery. In the third step, defined as must touch (MT), rats were required to press the circular stimulus on the screen to obtain the reward and a new trial would start only after the pellet was collected from the feeder magazine. In the next step called punish incorrect (PI), a negative reinforcement was introduced. When the rat pressed the screen on the wrong location or in the absence of any stimulus presented, the house light turned on (illumination of the house-light for 10 sec) and no pellet was delivered. The last step of the training stages consisted of moving punish incorrect (MPI), during which the stimulus changed position within the left and right aperture of the mask in random order.
2.2.4. Visual Discrimination Task
After the acquisition training during the pre-training stages, the rats directly moved on to the visual discrimination (VD) task. In this task, the rat is presented for the first time with two images, one in each aperture of the mask. The images were horizontal white lines versus vertical white lines (4.5 cm x 4.5 cm) on a black background (10 x 10 cm). Each rat was allocated either the horizontal or vertical image as the correct image. The images randomly changed position on the touchscreen. The rewarded image (horizontal or vertical stripes) was counterbalanced between the rats. A correct response resulted in the direct removal of the stimulus, the delivery of one pellet and subsequently an inter-trial interval. An incorrect response was followed by the illumination of the house light (punishment), no pellet delivery, and a correction trial started after 15s. In a correction trial, the rewarded stimulus was presented at the same location as in the previous trial and this continued until the rat made a correct response. When the rat did not make a response within 30s (omission) a new trial started after 10s. The rats were tested for at least 10 sessions on the visual discrimination task, twice a day, with a minimum interval of 3 hours between the two daily sessions. Data acquisition within the visual discrimination task was completed upon the achievement of an average at least 70% correct responses (correct responses/correct responses + incorrect responses) for three subsequent sessions with less than 15% variation of the percentage of correct responses among those selected three sessions.
2.2.5. Motivation Task
When rats successfully learned the VD, they were subjected to fixed ratio (FR1, FR2 and FR5) and subsequently a progressive ratio (PR) schedule of reinforcement to assess the motivation to work for a reward. In these tasks, a white circle was shown on the right side of the screen that had to be touched 1, 2, 5 or a progressive number of times to receive the reward. In FR1, only a single response on the white circle was needed to receive a reward. The rats were between 9-13 weeks of age during the FR sessions. FR1 was followed by FR2 and FR5, where the rat needed to press the white circle two and five times, respectively, to receive the reward. After receiving the reward, the stimulus was removed from the screen and a new trial began 5s after reward collection. Between the removal of the stimulus on the screen and the reward collection, and between the reward collection and the new stimulus presentation, inter-trial interval responses were detected to assess perseverance in responding. The stimulus remained present until the rat touched it the correct number of times. For FR1, FR2 and FR5 the passing criteria was to obtain 50 rewards. Thus, the rats had to touch the stimulus in total either 50, 100 and 250 times, respectively. When rats were successful in the FR tasks, they advanced to the PR schedule. A PR schedule (between week 10-15 of age) of reinforcement measures reward strength, and has become the golden standard task to assess motivation in touchscreen-based tasks in behavioural neuroscience [30]. Within this task, the rat needs to increase the number of responses on a linear basis of 4 (i.e. 1, 5, 9, 13 etc.) to earn a reward. All rats were required to complete 10 PR sessions for ZDF rats and 20 PR sessions for LE rats. In 3 PR sessions the rats received different reward pellets, to assess whether there was a difference in motivation between grain and sucrose pellets as rewards. The LE-sucrose group received sucrose pellets during all cognitive testing except for 3 PR sessions (15, 17, 19) during which they received grain pellets. The LE-grain group received grain pellets during all cognitive testing except for 3 PR sessions (15, 17, 19) during which they received sucrose pellets. The ZDF rats were rewarded with grain pellets except for 3 PR sessions (6, 8, 10) during which ZDF rats were rewarded with sucrose pellets. Average of complete 10 (ZDF rats) or 20 sessions (LE rats) on PR were analyzed.
2.3. Physiological Assessment
General approach
In short, blood glucose and plasma insulin measurements were performed in blood that was collected via a tail puncture or tail cut in EDTA-coated tubes every two weeks for five times in total starting from week 4 of age. Lipid and Lipoprotein Analysis of total plasma cholesterol and triglycerides were measured in week-12 of age. The rats were sacrificed at early adulthood, around week 14 and week 15 for ZDF rats and between week 14 and 19 for LE rats. After transcardial perfusion, the left hemisphere was post-fixed in 2% paraformaldehyde and 48 hours thereafter transferred into a 30% sucrose solution for a total of 7 days at 4⁰C and finally stored in -80⁰C. The right medial prefrontal cortex and hippocampus was dissected and collected in Eppendorf tubes and stored in -80⁰C. A part of the liver, mesenteric and perigonadal white adipose tissue were post-fixed and embedded in paraffin. Adipocyte morphometry [30] and pathological hallmarks in the liver were scored as detailed previously [31,32]. The number of neurons and microglia, and the soma cell size area of microglia was visualized using immunohistochemistry. Gene expression analyses were performed for primers Rattus norvegicus Ywhaz, β-actin and Gapdh, SIRT1, PSD-95, and were synthesized by Sigma (for primer pair sequences, see Table 2). Hippocampal insulin levels were measured via standard protein concentrations of the homogenized tissue and subsequent insulin ELISA.
2.3.1. Blood Glucose and Plasma Insulin Measurements
The blood collection was performed on Mondays, after providing ad libitum food during the weekends. Five hours prior to the blood collection, food was removed. Blood glucose measurements were directly performed with the glucometer Freestyle Lite (Abbott Diabetes Care, Germany). Blood was centrifuged at 4,000 rpm and 4⁰C for 10 minutes, and plasma was collected and stored at -80 ⁰C until further use. Plasma insulin levels were quantified using an Enzyme-Linked Immunosorbent Assay (ELISA) (Ultra-Sensitive Rat insulin ELISA, Crystal Chem Inc, USA), using 5μl plasma in duplicate for each sample and executed according to manufacturer’s instructions. The absorbance values were obtained with a 450 nm excitation filter and corrected with the absorbance value obtained from both a 630 nm excitation filter and the sample diluent. The corrected and averaged concentrations of 5 measurements (from week 4 to week 12) are presented as nanogram per milliliter.
2.3.2. Lipid and Lipoprotein Analysis
Total plasma cholesterol (very low density lipoprotein; VLDL-C, low density lipoprotein; LDL-C and high density lipoprotein; HDL-C) and plasma triglycerides were measured after 5-hour fasting in week-12 of age using commercially available enzymatic assays (CHOD-PAP and GPO-PAP respectively; Roche Diagnostics, Almere, the Netherlands). Plasma HDL-cholesterol was measured after precipitation of the apoB-containing lipoproteins (VLDL and LDL) from 20 μL EDTA plasma by adding 10μL 0.2 M MnCl2 and 10μL heparin (LEO Pharma, The Netherlands; 500 U/mL). Mixtures were incubated during 20 minutes at room temperature and centrifuged for 15 minutes at 13,000 rpm at 4°C to precipitate VLDL and LDL particles. The remaining cholesterol in the supernatant (HDL-C) was quantified with the aforementioned assay (CHOD-PAP). Furthermore, for ZDF rats, lipoprotein profiles were obtained by using the AKTA-fast protein liquid chromatography system (Pharmacia, Roosendaal, the Netherlands) as described previously [31]. The plasma from ZDF or lean rats for both sexes were pooled and analyzed and the averaged profile of 24 fractions resulting from 2 analysis are presented.
2.3.3. Sacrifice and Tissue Collection
Rats were overdosed with approximately 2 ml pentobarbital (60 mg/ml) via an intraperitoneal injection. After sedation, blood was collected through cardiac puncture. After transcardial perfusion with 0.1M phosphate-buffered saline (pH 7.2), rats were decapitated. The left hemisphere was post-fixed in 2% paraformaldehyde and 48 hours thereafter transferred into a 30% sucrose solution for a total of 7 days at 4⁰C. The sucrose solution was refreshed after 4 days. Thereafter, brains were stored in -80⁰C until use for histological experiments. The medial prefrontal cortex was dissected and collected in Eppendorf tubes and stored in -80⁰C until further use. The liver and mesenteric and perigonadal white adipose tissues were weighted. A part of the liver, mesenteric and perigonadal white adipose tissue were post-fixed in 4% paraformaldehyde for 48 hours. These tissues were subsequently transferred to 70% ethanol in demi water for a total of 7 days at 4⁰C. The ethanol solution was refreshed after 4 days. The liver, mesenteric and perigonadal white adipose tissue parts were embedded in paraffin for later histological experiments.
2.3.4. White Adipose Tissue and Liver Histology
Paraffin-embedded cross sections of adipose tissues (5 µm) and liver (3 µm) were stained with haematoxylin and eosin. Adipose tissue cross-sections were digitized with a slide scanner (Aperio AT2, Leica Biosystems, Amsterdam, the Netherlands) and adipocyte morphometry (average adipocyte size and adipocyte size distribution) was analyzed using an automated image analysis as described previously [32]. Liver cross sections were scored blindly by a board-certified pathologist using a grading method for human non-alcoholic steatohepatitis adapted for rodent models [33] and pathological hallmarks were scored as detailed previously [34]. In brief, macrovesicular steatosis, microvesicular steatosis and hepatocellular hypertrophy were determined as a percentage of the total liver section affected. Hepatic inflammation was quantified by counting the number of inflammatory aggregates in 5 non-overlapping fields per rat (at 100x magnification, field of view 4.15mm2), and expressed as the number of aggregates per field.
2.3.5. Immunohistochemistry
Slices of the medial prefrontal cortex of 16 μM were obtained with a cryostat (Leica CM3050 S). Slices were captured in milliQ water and the same day mounted on coated slides (Thermo Scientific, Menzel-Gläser SuperfrostÒPlus). The slides were air dried overnight and frozen in -20⁰C until further use. Before staining, the slides were thawed for 30 minutes, rehydrated in 1x PBS for 10 minutes, thereafter, washed 3 times for 10 minutes in 1x PBS on the 3D orbital incubation shaker (Stuart gyro rocker SSL3) at 30 rpm. During every incubation step, slides were placed horizontally in a wet chamber (surface of slide box covered with milliQ soaked tissues). After every upcoming incubation step, slides were washed 3 times for 10 minutes in 1x PBS on the 3D orbital incubation shaker at 30 rpm. Slides were blocked with 500μl blocking solution (20% BSA (blockerÔ BSA (10x) in PBS, Thermo Fischer Scientific, 37525), 0.2% Triton X-100 (Sigma-Aldrich, T8787) in 1xPBS) for 2.5 hours. Subsequently, slides were incubated with 500μl primary antibodies: Anti Iba1, Rabbit, (Wako, 019-19741 (1:500)) and Anti NeuN (Chicken, Millipore, ABN91 (1:0000)) diluted in blocking buffer overnight at room temperature. The slides were incubated with 500μl secondary antibodies: Alexa Fluor®Donkey anti-rabbit 647, (Thermo Fischer Scientific) and Alexa Fluor®Goat anti-chicken 555 (Thermo Fischer Scientific) both with 1:200 dilution in 1x PBS for 3 hours at room temperature. Thereafter, slides were incubated with 500μl DAPI (1:1000 diluted in 1% PBS) for 7 minutes at room temperature after which slides were individually dipped in milliQ for approximately 5 seconds to diminish remaining PBS. After air drying, slides were mounted with 2 drops fluorsave (Calbiochem, Millipore), covered with a cover glass and placed horizontally in a dry chamber overnight at room temperature to dry. Finally, slides were stored vertically at room temperature until microscope analysis. One image (2x2 tilescan, 40x magnification) of the medial prefrontal cortex was obtained on an automated high-content microscope (Leica DMI6000B) and analyzed using ImageJ software. The soma cell size area of IBA-1 positive immunoreactive cells were quantified using the average of 5 cells from each rat.
2.3.6. Gene Expression Analyses
RNA Isolation
RNA isolation was done using the RNeasy® Mini Kit (Qiagen, cat. nos. 74104 and 74106) and performed according to manufactures instructions. Samples were checked with NanoDrop 2000/2000c Spectrophotometer (Thermo Fisher Scientific, NanoDrop 2000/2000c software) for concentration and pureness and stored at -20 °C.
cDNA Synthesis
The cDNA synthesis was performed according to manufacturer’s instructions using the SensiFAST™cDNA Synthesis Kit (Bioline, cat. nos. BIO-65053). The amount of nanograms mRNA added to the SensiFAST™cDNA Synthesis Kit was equivalent in terms of quantity. The master mix was prepared on ice and mixed by pipetting. Each sample contained; total RNA (up to 1 μg), 2 μl; 5x TransAmp Buffer, 4 μl; Reverse Transcriptase, 1 μl and DNase/RNase free water, up to 20 μl in total. In the thermal cycler the following program was used: 25 °C for 10 minutes (primer annealing), 42 °C for 15 minutes (reverse transcription) and 85 °C for 5 minutes (inactivation) followed by a 4 °C hold. The cDNA reaction product was stored at -20 °C until further use.
qPCR
The qPCR was performed according to manufacturer’s instructions using the SensiFAST™ SYBR®No-ROX Kit (Bioline, cat. nos. BIO-98005). The master mix was prepared kept away from direct light and mixed by pipetting. Each sample contained; 1x SensiFAST SYBR® No-ROX Mix, 400 nM forward and reverse primer, 2 μl template and DNase/RNase free-water, up to 10 μl in total. Master mixes and templates were pipetted in a Rotor-Disc®100 (cat. No. 981311, QIAGEN) by QIAgility pipetting robot (QIAGEN, build 1 (1.6.61) software version 4.14.2) and afterward sealed using the Rotor-Disc Heat Sealer (QIAGEN). qPCR was performed using the Rotor-Gene Q (serial no: 051226, model: 5-Plex HRM, software version: 2.3.1, QIAGEN), with a cycle of 2 minutes 95 °C for polymerase activation, 40 times 5 sec 95 °C for denaturation, 10 sec 63 °C for annealing and 10 sec 72 °C for extension. Machine kept samples at 4 °C until placed in the 4 °C fridge for storage. Primers Rattus norvegicus Ywhaz, β-actin and Gapdh, SIRT1, PSD-95 were synthesized by Sigma (for primer pair sequences, see Table 2). For the statistical analyses, three reference genes were chosen in first instance (Ywhaz, β-actin and Gapdh). However, since Gapdh expression may be influenced by diet, it was added as an exploratory gene. Therefore, gene expression was normalized to the Ywhaz/β-actin ratio and results were subjected to two-way ANOVA for normally distributed variables with equal variances.
2.3.7. Hippocampal Insulin Levels
Isolated Hippocampi from ZDF rats, stored at -80 °C were weighted before extracting total protein. Tissues were first homogenized by sonication in 10× volume of Tissue Lysis Buffer (N-PER-Halt) supplemented with protease inhibitor cocktails (ThermoFisher). Tissue lysis was centrifuged at 12,000 × g for 10 minutes at 4 °C, and the supernatant was transferred to a new Eppendorf tube. Total protein concentration was determined by BCA method (Thermo Scientific™ Pierce™ 96-Well Plates, USA). Standard curve of BSA using a range of concentrations between 250 and 2000 µg/mL were used in order to determine the tissue protein concentrations. For ELISA, 50 µL of tissue supernatants were used in 96-well antibody coated microplate. Two sets of positive control samples were included in each ELISA: Rat insulin standard (supplied in the Ultra Sensitive Kit, CrystalChem, High Performace Assay, USA). Low range assay (0.1-6.4 ng/mL) of insulin was used. Results were presented as µg/mL wet tissue.
2.4. Statistical Analysis
All analyses were executed using GraphPad Prism version 9 (Graphpad Software, San Diego California, USA). The main effect of sex, genotype (for ZDF) or feeding regime (for LE rats) on subsequent or single measurements were analyzed using either repeated measures ANOVA using a Mixed-effects model or were subjected to two-way ANOVA for normally distributed variables with equal variances. A p-value of ≤0.05 was considered statistically significant. Greenhouse-Geisser correction was used where appropriate for violation of sphericity. Significant interaction effects were followed by Tukey’s post-hoc analysis.
3. Results
3.1. Effects of the Two Dietary Regimes in Healthy Long Evans (LE) Rats on Behaviour and Physiological Outcomes
A detailed statistical description and graphic illustration of the comparison between two groups of LE rats (LE sucrose versus LE grain) are presented in full in the supplementary materials. In sum, the ad libitum feeding and sucrose exposure during operant training resulted in a higher body weight compared to male LE grain rats, whereas no difference was observed between female LE groups (Figure S1A-B). Performance during the operant training stages prior to the VD task showed that LE-sucrose rats needed more training sessions in all stages (Figure S2A-B). During the VD task itself, the LE-sucrose group showed reduced performance during the first 10 sessions (Figure S3A-B), but a comparable level of performance was seen once rats reached the VD criteria of >70% correct (Figure S3C). The number of sessions to reach criterion did not differ between groups (Figure S3D). The number of sessions required during FR stages was enhanced in LE-sucrose rats (Figure S3E), while the motivation during PR was not different between groups (Figure S3F). Blood analyses revealed that LE-sucrose rats had significantly increased glucose levels (Figure S4A-B) and increased total cholesterol and triglycerides levels (Figure S4E-F), but insulin levels were not altered (Figure S4C-D). No effects of the dietary regime were observed on the relative weight of perigonadal and mesenteric white adipose tissue and the relative liver weight (Figure S5A-C). However, the number of immune cell aggregates within the liver was significantly increased in female LE-sucrose rats (Figure S5D). Interestingly, in LE-sucrose rats, we observed a significant decrease in the number of both NeuN and IBA-1 positive cells in the PFC in compared to LE-grain rats (Figure S6A-C), but no differences in the IBA-1 immunoreactive cells area (Figure S6D). No significant group differences were observed in SIRT1, PSD-95 and GAPDH mRNA expression levels in the PFC (Figure S7).
3.2. Body Weight Development, Locomotor Activity and Ad Libitum Food Intake in ZDF Rats
Figure 1A presents the enhanced body weight gain in obese versus lean ZDF rats from week 4 to 14 of age (Fgenotype (1,32) =132.6, p<0.0001). A significant interaction effect between genotype and sex was found (Fgenotype x sex (1, 32)=9.184, p=0.0048) which indicated that the difference in body weight development between the female groups was larger (46.09% at week 14) compared to the difference between the male groups (21.40% at week 14) (Figure 1B). Average locomotor activity was significantly lower in obese compared to lean ZDF rats, both when assessed during 48h sessions in LABORAS (Fgenotype(1, 30)=100.2, p<0.0001) (Figure 1C) and during the 10 first sessions of the VD task (Fgenotype(1, 27)=16.41, p=0.0004) (Figure 1D). Moreover, the difference in locomotor activity between obese and lean ZDF rats in females was larger compared to males in LABORAS (Fsex(1, 30)=26.34, p<0.0001, Fgenotypexsex(1, 30)=22.08, p<0.0001). Food intake (corrected for body weight) decreased over development (Fweeks(1, 51)=82.32, p<0.0001) and showed a significant interaction effect for genotype over weeks (Fgenotype(1,318)=14.91 p=0.0001; Fweeksxgenotype(9,318)=7.65, p=0.001) in which obese ZDF rats consumed more food during ad libitum access in weekends on week 5, 6, and 8 of age (Figure S8A). Overall, no significant effect of genotype on the duration spent on eating and drinking during the 48h sessions in LABORAS was observed (Figure S8B-C).
3.3. Cognitive Performance and Level of Motivation in ZDF Rats
Obese rats needed less training sessions compared to lean ZDF rats during the training stages of autoshaping (Fgenotype(1,31)=10.57 p<0.01), punish incorrect (Fgenotype(1,32)=9.17, p<0.01) and moving punish incorrect (Fgenotype(1,32)=15.54, p<0.001) (Figure S9A-B). Only during must touch training, obese ZDF rats required more training sessions (Fgenotype(1,32)=4.35, p<0.05). The analyses across the first 10 VD sessions revealed that obese rats showed an enhanced learning capacity compared to lean ZDF rats (Fgenotype(1,32)=35.12, p<0.0001; F(9, 285) genotype x session=8.263, p<0.0001) (Figure 4A-B). No significant effect of sex was observed (Fsex (1, 32)=2.448, p = 0.1275). Moreover, once the VD criterium was reached by each individual rat, obese rats still showed a significantly higher percentage of correct responses compared to lean ZDF rats (Fgenotype(1,27)=12.97, p<0.01) (Figure 2C). Obese rats required less sessions to reach the VD criterium (Figure 2D). When the motivation to respond was subsequently assessed, no genotype differences during FR stages (Figure 2E) and the PR schedule of reinforcement were observed (Figure 2F). When altering the reward pellet during PR sessions, we observed no effect of genotype or reward pellet on the average number of presses for the new sucrose reward during PR sessions 6, 8 and 10 versus the average number of presses for the known grain reward on session 5, 7, 9 (Figure 2G).
3.4. Blood Glucose, Insulin and Lipids in ZDF Rats
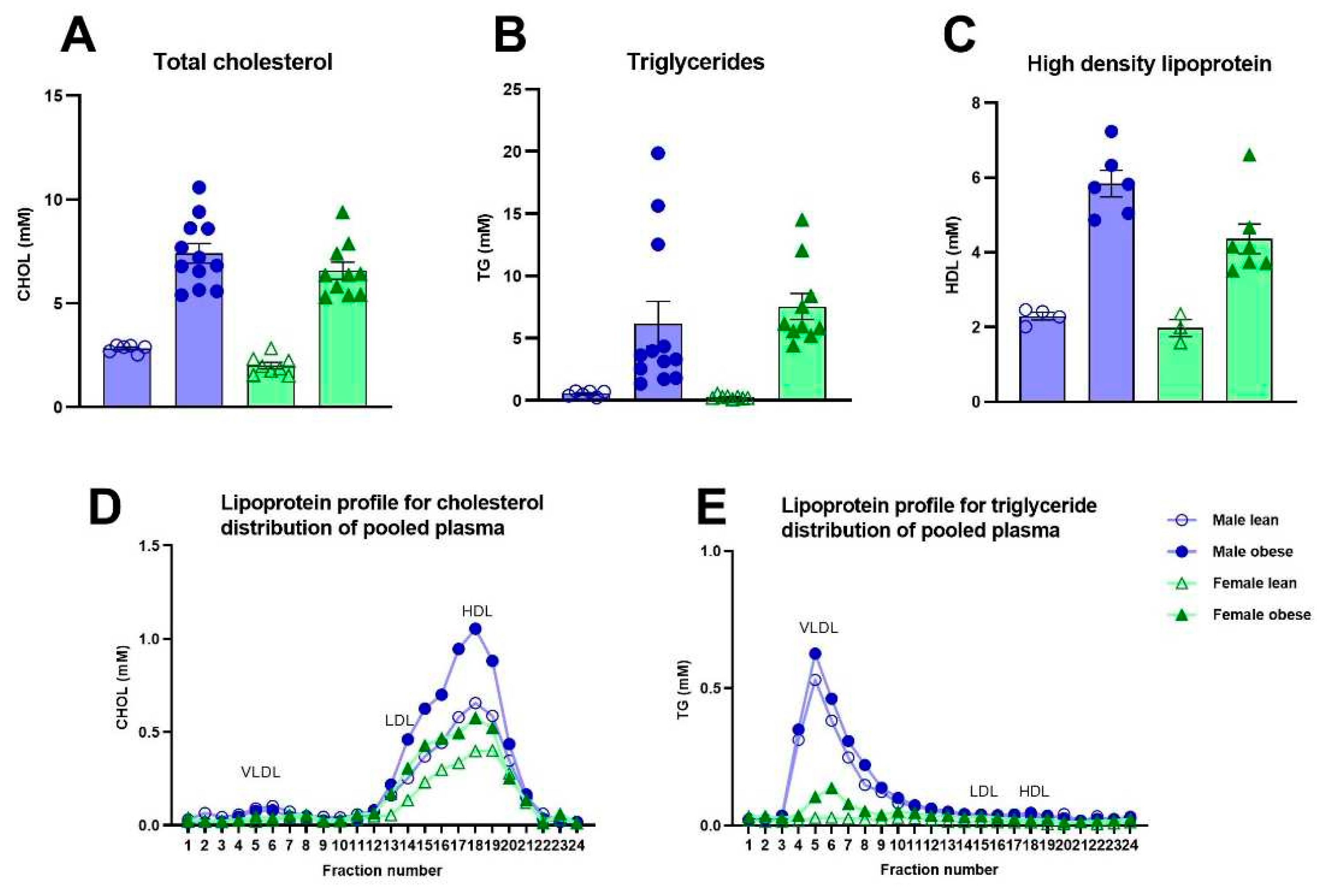
Fasted glucose levels varied over weeks of age (Fweeks(3,84)=5.96, p<0.005) and were significantly higher in obese rats compared to lean ZDF rats (Fgenotype(1,32)=13.03, p=0.001), without a difference in sex (Fsex(1,32)=1.80, p=n.s.) (Figure 3A). Fasted insulin levels increased over weeks of age in the male and female obese group while the insulin levels stayed stable in lean ZDF rats (Fgenotypexweeks(4,123)=13.49, p<0.0001; (Fgenotype(1,32)=45.47, p<0.0001) (Figure 3C). Interestingly, the increment in insulin levels over weeks was higher in obese male rats compared to obese female rats (Fgenotype x sex x weeks(4,123)=4.74, p<0.05; (Fgenotype x sex(1,32)=7.29, p<0.05). The average glucose and insulin values of the five timepoints (week 4-12 of age) are shown in Figure 3B-3D. ZDF obese rats had higher total cholesterol (Fgenotype(1,32)=122.6, p<0.0001), triglycerides (Fgenotype(1,32)=21.48, p<0.0001) as well as high-density lipoprotein levels (Fgenotype(1,16)=53.70, p<0.0001) compared to lean rats (Figure 4A-C). Pooled plasma from ZDF obese and lean rats was investigated for lipoprotein fractionation and showed that obese rats mainly had enhanced high-density lipoprotein fractions in cholesterol and very low-density lipoprotein fractions in triglycerides, compared to lean rats (Figure 4D-E).
Figure 3.
Blood glucose and plasma insulin levels in obese and lean ZDF rats. (A, C) Fasted glucose and insulin levels over time at weeks 4, 6, 8, 10 and 12 of age. (B, D) Average of fasted glucose and insulin levels of the measured 5 timepoints. Values represent the group mean (±SEM).
Figure 3.
Blood glucose and plasma insulin levels in obese and lean ZDF rats. (A, C) Fasted glucose and insulin levels over time at weeks 4, 6, 8, 10 and 12 of age. (B, D) Average of fasted glucose and insulin levels of the measured 5 timepoints. Values represent the group mean (±SEM).
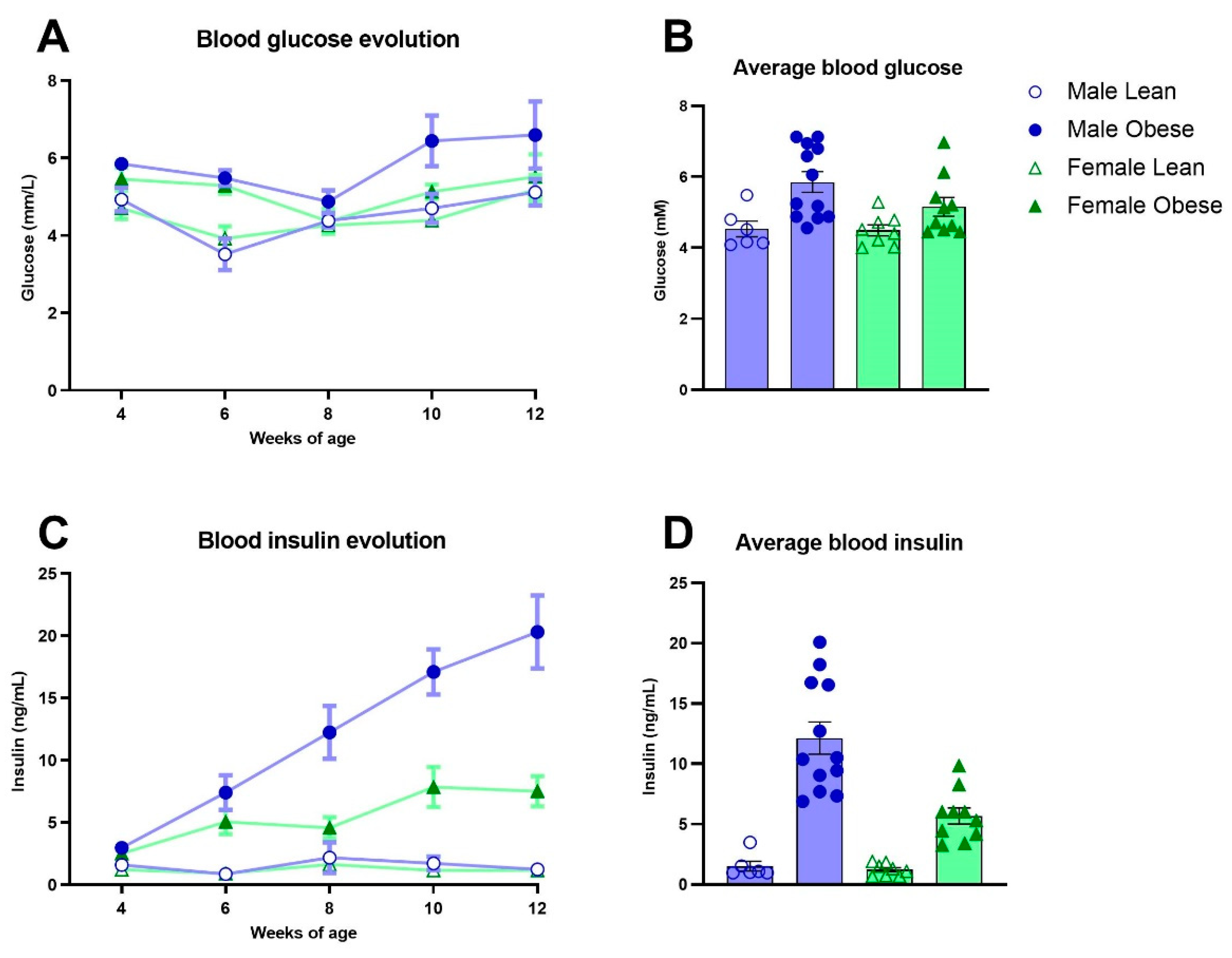
Figure 4.
Blood total cholesterol, triglycerides and high-density lipoprotein levels in obese and lean ZDF rats week 12. (A, B, C) Average total cholesterol, triglycerides and high-density lipoprotein at week 12. (D, E) Lipoprotein profiles of pooled plasma at week 12 with physiological distribution of cholesterol and triglycerides among lipoproteins. Twenty-four fractions were collected and further assessed for cholesterol, and triglyceride concentrations. Fractions 4 to 7 were determined as very low-density lipoprotein, 11 to 16 as low-density lipoprotein, and 17 to 25 as high-density lipoprotein. Values represent the group mean (±SEM). VLDL = very low-density lipoprotein; LDL = low-density lipoprotein; HDL = high-density lipoprotein.
Figure 4.
Blood total cholesterol, triglycerides and high-density lipoprotein levels in obese and lean ZDF rats week 12. (A, B, C) Average total cholesterol, triglycerides and high-density lipoprotein at week 12. (D, E) Lipoprotein profiles of pooled plasma at week 12 with physiological distribution of cholesterol and triglycerides among lipoproteins. Twenty-four fractions were collected and further assessed for cholesterol, and triglyceride concentrations. Fractions 4 to 7 were determined as very low-density lipoprotein, 11 to 16 as low-density lipoprotein, and 17 to 25 as high-density lipoprotein. Values represent the group mean (±SEM). VLDL = very low-density lipoprotein; LDL = low-density lipoprotein; HDL = high-density lipoprotein.
3.5. Histological Analyses of White Adipose Tissues and Liver in ZDF Rats
The relative adipose tissue weights and adipocyte sizes were higher in obese compared to lean ZDF rats. This was the case for both the relative weight of perigonadal (Fgenotype(1,32)=219.4, p<0.0001) and mesenteric white adipose tissue (Fgenotype(1,32)=218.5, p<0.0001) (Figure 5A-B), and the adipocyte sizes (Fgenotype(1,15)=72.49, p<0.0001; Fgenotype(1,16)=73.70, p<0.0001), respectively (Figure 5C-D). Representative images of perigonadal and mesenteric white adipose tissue are presented in Figure 5E-F. The distributions in the percentage of total cells per cell size are presented in Figure 5G-H. The pathological investigation on the liver (Figure 6A) revealed starting hepatic microvascular steatosis (Fgenotype(1,31)=62.23, p<0.0001) and hypertrophy (Fgenotype(1,31)=39.88, p<0.0001) in obese rats compared to lean ZDF rats (Figure 6D). Furthermore, significant higher relative liver weights were found in obese rats (Fgenotype(1,32)=70.05, p<0.0001) (Figure 6B) and the number of immune cell aggregates was significantly increased in obese compared to lean ZDF rats (Fgenotype(1,31)=20.12, p<0.0001) (Figure 6C).
3.6. mPFC NeuN and IBA-1 Positive Cells, mPFC SIRT1 and PSD-95 mRNA Expression, and Hippocampal Insulin Levels in ZDF Rats
No significant differences between ZDF obese and lean rats, or differences between males and females were found for the number of NeuN (Fgenotype(1,16)=0.35, p=n.s.; Fsex(1,16)=0.01, p=n.s.) and IBA-1 positive cells (Fgenotype(1,17)= 0.44, p=n.s.; Fsex(1,17)= 0.77, p=n.s.) (Figure 7A-C). Furthermore, no significant effects of genotype and sex were found for IBA-1 immunoreactive soma cell size (Fgenotype(1,15)=0.224, p=n.s.; Fsex(1,15)=0.7203, p=n.s.) (Figure 7D). The mRNA expression of SIRT1, PSD-95 and GAPDH was not different between groups either (SIRT1: Fgenotype(1,30)=0.2086, p=n.s., Fsex(1,30)=0.5694, p=n.s.; PSD-95: Fgenotype(1,30)=0.2167, p=n.s., Fsex(1,30)= 1.351, p=n.s.; GAPDH: Fgenotype(1,31)= 3.489, p=n.s., Fsex(1,30)=2.412, p=n.s) (Figure 8A-C). The level of brain insulin was measured in the hippocampus and was not different between ZDF obese and lean rats or between sexes (Fgenotype(1,30)=0.045, p=n.s., Fsex(1,30)=0.047, p=n.s).
4. Discussion
This study investigated whether cognitive performance is impacted already during adolescence in a genetic rat model of obesity and diabetes. Interestingly, we observed that adolescent ZDF obese rats cognitively outperformed both ZDF lean rats and healthy LE rats implemented with the same food regime. This enhanced cognition lasted during the entire visual discrimination period. Clear features of type 2 diabetes were observed in ZDF obese rats from week 6 of age onwards. Furthermore, early adult ZDF obese rats presented mild signs of hepatic and white adipose tissue inflammation. Interestingly, no alterations in the number and morphology of neuron and microglia cells, nor differences in PSD-95, SIRT-1 and GAPDH expression were observed between young adult ZDF obese and lean rats. The comparison between the LE-sucrose and LE-grain group showed that the LE-sucrose group had a slower acquisition rate during pre-training stages and a lower cognitive performance during the first ten VD sessions. However, sucrose exposure did not impact the final cognitive performance and neither impacted motivation levels. Sucrose exposure did result in enhanced blood glucose, total cholesterol and triglycerides levels and lower levels of NeuN and IBA-1 positive cells in the PFC, compared to LE-grain rats.
The cognitive superiority of the ZDF obese rats was unexpected, because previous studies reported either no changes in learning [35] or spatial memory deficits [14,15,36]. Since the enhanced performance in both the visual discrimination task and most of the pre-training acquisition phases, it is hard to interpret whether the ZDF obese rats have an enhanced cognition or a different learning capacity. A time response study using a diet-induced obesity model revealed that insulin resistance appeared from the 7th week of fructose feeding, whereas the cognitive dysfunction (memory function in Morris Water Maze) appears only after the 20th week of fructose exposure [15]. Comparable results were found using Zucker rats, in that cognitive alterations in the passive avoidance test (lower capacity to learn and to keep away from unpleasant electric shock) were observed in 20-week-old obese Zucker rats, with no differences in 12 and 16 week-old, compared to lean rats [37]. Moreover, rat strains with a genetic predisposition to obesity did not differ in their ability to experience reward (in a condition place preference task) [38]. It would have been worthwhile to investigate in the current used touchscreen higher-order cognitive task whether older adult ZDF obese rats show cognitive dysfunction. However, this might challenge ethical decisions since we clearly observed that when obese ZDF rats age, the diabetic disease complications started to emerge. In line with literature, at some animals we observed progressive nephropathy, increasing proteinuria resulting in chronic renal insufficiency and impaired wound healing in early adulthood [39,40].
As expected, ZDF obese rats developed obesity as measured by an increased body weight, liver weight and perigonadal- and mesenteric white adipose tissue weights. Given the hyperphagic nature of leptin resistance in genetic animal models, we assessed whether the enhanced cognitive performance could be explained by an increased motivation to obtain reward. We observed no group differences between ZDF obese and lean rats at early adulthood in PR sessions, nor toward a new type of reward pellets. Zucker rats of a comparable age, also showed no differences with lean rats during PR sessions where a relatively high effort was required [38], but when lower effort was required during PR and FR lever press training, a higher motivated behaviour was observed [38,41]. However, a higher acquisition rate, as our ZDF obese rats showed during pre-training sessions for VD and during FR training, is not a true measure of motivation. Therefore, arguing that motivation for receiving reward pellets is the foundation for enhanced cognitive performances of the ZDF obese rats is just speculative. What we did clearly observed in our locomotor activity data, but also based on video recording during operant testing, is that ZDF lean rats, mainly females, were hyperactive. Comparable findings of enhanced locomotor activity have been observed previously in Obese Resistant (OR) rats [38]. This hyperactive behavior of ZDF lean rats was not the case for LE rats. Since ZDF obese rats outperformed both lean and LE rats in the visual discrimination task - and ZDF lean rats did not differ from LE rats in this task - it is not likely that the observed hyperactivity in ZDF lean rats was responsible for the significant difference in cognitive performances between obese and lean rats.
It could also be that the hyper-insulinemic condition of ZDF obese rats influenced cognitive performance. In support, we found a negative correlation between the number of sessions in the visual discrimination task and insulin concentration in the blood (r=-0.4801, p<0.0035), indicating a faster learning acquisition when insulin levels are high. Since insulin can cross the blood-brain-barrier, it is possible that hyperinsulinemia could stimulate cognitive performance via neurogenesis pathways and/or play an adaptative protective role in adolescent ZDF obese rats by directly suppressing pro-inflammatory cytokines and preventing neuronal impairment [18,42]. However, we did not investigate the causal relationship between insulin and cognitive performance. This requires future investigations employing local brain infusions of insulin or a systemic injection with insulin to assess the direct brain effects. We did however measure the brain insulin levels in the hippocampus but have not observed a difference between ZDF obese versus lean rats, when measured at 14-16 of age. Growing evidence supports the role for SIRT1 in the regulation of insulin sensitivity [43] and neuroinflammatory responses [44]. Additionally, decrement of SIRT1 in the brain is associated with cognitive impairment [45]. Remarkably, SIRT1 induced cognitive enhancement even in healthy non-transgenic mice and increased neuronal plasticity proteins such as PSD-95 [46]. Our results do not depict significant differences in the expression of SIRT1 and PDS-95 in the brain of ZDF obese rats compared to lean rats. Furthermore, no significant differences were observed in the number of neurons and microglia or the morphology of the microglia soma sizes, which is in line with a previous study which reported no alteration in the morphology of astrocytes and microglia in ZDF obese versus ZDF lean rats [47]. Therefore, it is unlikely that SIRT1, PDS-95 or the number of neurons and microglia can explain the enhancement of cognitive performances in ZDF obese rats. Previous time response studies showed indeed that a decrease of neurons, morphological changes of microglia with an increase of the area of soma, started to appear in 20-week-old obese Zucker rats (and not at 12 and 16 week old) [37] and 30-week-old ZDF rats (and not at 12 and 20 week old) [48]. A reduction in brain cholinergic (VAChT and α7nAChR) and synaptic markers (synaptophysin and synaptic vesicle glycoprotein 2B) have been observed in the frontal cortex and hippocampus in obese Zucker rats, but these reductions only started to emerge at 20 weeks of age, and were not present at 12 and 16 weeks of age [49]. Since we performed our brain histology and mRNA expression experiments at rats which were 14-16 weeks of age, this supports our idea that negative effects due to the obesity phenotype have not yet occurred within the prefrontal cortex.
Although the genetic ZDF model is a very different disease model compared to diet-induced obesity, the fact that exactly the same experiments were performed in both the LE and ZDF groups allows the valuable possibility to gain insight into the consequences of both models on the same higher-order cognitive performance during development. We observed that in LE rats, food restriction in association with grain pellet reward significantly speeds up the acquisition of a complex cognitive task, relative to ad libitum feeding and sucrose pellet as reward. It has previously been suggested that ad libitum feeding by itself leads to metabolic morbidity, with negative effects on various bodily functions, including cognition [50]. Furthermore, it has been demonstrated that adolescent sucrose exposure in animals reduces cognitive functioning [51]. Blood glucose cholesterol and triglyceride levels were enhanced in LE-sucrose rats compared to LE-grain rats, as was the infiltration of inflammatory cells in the liver. The number of microglia and neurons in the prefrontal cortex were decreased in LE-sucrose rats, which typically has been associated with reduced cognitive performance [52].
5. Conclusions
Taken all data together, ZDF obese rats showed an increased cognitive performance at adolescence despite several physiological symptoms of obesity and type 2 diabetes. At early adulthood, an onset of inflammation in the liver was present while no brain alterations were detected. We propose the possibility that prefrontal function during the induction of obesity and diabetes in a genetic ZDF rodent model was maintained by hyperinsulinism and even resulted in improved cognitive performance. Future research could explore this potential role of insulin further by investigating, for example, the effects of central insulin administration on cognitive performance or the assessment of insulin signaling in adolescent ZDF obese rats.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Fig S1. Body weight development and locomotor activity of male and female LE-sucrose and LE-grain rats. Fig S2. Number of sessions required for male and female LE-sucrose and LE-grain rats to reach criterion during the pretraining stages of the visual discrimination test. Fig S3. Results of the visual discrimination task, fixed ratio and progressive ratio tasks in male and female LE-sucrose and LE-grain rats. Fig S4. Blood glucose, insulin and lipid levels in male and female LE-sucrose and LE-grain rats. Fig S5. Average relative weights of perigonadal and mesenteric white adipose tissues and liver histopathology in male and female LE-sucrose and LE-grain rats. Fig S6. Number of NeuN, IBA-1 positive cells and IBA-1 immunoreactive cells area in the medial prefrontal cortex (PFC) of male and female LE-sucrose and LE-grain rats. Fig S7. mRNA expression levels of SIRT1 (A), PSD-95 (B) and GADPH (C) in the medial prefrontal cortex of male and female LE-sucrose and LE-grain rats. Fig S8. Food intake in male and female ZDF obese and lean rats. Fig S9. Number of sessions required for male and female obese and lean ZDF rats to reach criterion during the pretraining stages.
Author Contributions
MS: GG, RK, JAD and JRH contributed to the study design. MS, YB, VvK, LdG, MB, NK and EK, performed the data collection. MS, YB, and MCM performed the data analyses. MS, YB, MCM, GG, RK, JAD and JRH contributed to the interpretation of the data. MS, YB and JRH drafted the manuscript. All authors were involved in critical revisions of the manuscript. All authors read and approved the final manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the European Regional Development Fund (ERDF), project BriteN: Early nutritional Interventions for healthy brain development, 2016-2020.
Institutional Review Board Statement
All experiments were performed in accordance with the European Parliament and Council Directive (2010/63/EU) and approved by the Centrale Commissie Dierproeven in The Hague, the Netherlands.
Informed Consent Statement
Not applicable.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
We would like to thank Anthonieke Middelman for her technical contributions as well as the help of the biotechnicians for their support in blood draws.
Conflicts of Interest
Authors of Radboudumc and TNO have nothing to disclose. J.A. van Diepen and G. Gross are employees of Reckitt|Mead Johnson Nutrition Institute. R. Bulthuis is an employees of Metris B.V. All authors declare that they have no competing interests.
References
- World Health Organization. Obesity and overweight. Updated , 2021. Available online: https://www.who.int/newsroom/fact-sheets/detail/obesity-and-overweight (accessed on 25 September 2019).
- Blüher, M. Obesity: global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Cildir, G.; Akıncılar, S.C.; Tergaonkar, V. Chronic adipose tissue inflammation: all immune cells on the stage. Trends Mol. Med. 2013, 19, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; Kahraman, A.; Gerken, G.; Canbay, A. Obesity Affects the Liver – The Link between Adipocytes and Hepatocytes. Digestion 2010, 83, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Chooi YC, Ding C, Magkos F. The epidemiology of obesity. Metabolism. 2019;92:6-10. Metabolism 2019, 92, 6–10.
- Cildir, G.; Akıncılar, S.C.; Tergaonkar, V. Chronic adipose tissue inflammation: all immune cells on the stage. Trends Mol. Med. 2013, 19, 487–500. [Google Scholar] [CrossRef]
- Lee, J.W.; Lee, Y.K.; Yuk, D.Y.; Choi, D.Y.; Ban, S.B.; Oh, K.W.; Hong, J.T. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J. Neuroinflammation 2008, 5, 37–37. [Google Scholar] [CrossRef]
- Reichelt, A.C.; Morris, M.J.; Westbrook, R.F. Daily access to sucrose impairs aspects of spatial memory tasks reliant on pattern separation and neural proliferation in rats. Learn. Mem. 2016, 23, 386–390. [Google Scholar] [CrossRef]
- Tozuka, Y, Wada E, Wada K. Diet-induced obesity in female mice leads to peroxidized lipid accumulations and impairment of hippocampal neurogenesis during the early life of their offspring. FASEB J. 2009, 23, 1920–1934. [CrossRef]
- Barber, T.M.; Kyrou, I.; Randeva, H.S.; Weickert, M.O. Mechanisms of Insulin Resistance at the Crossroad of Obesity with Associated Metabolic Abnormalities and Cognitive Dysfunction. Int. J. Mol. Sci. 2021, 22, 546. [Google Scholar] [CrossRef]
- Kothari, V.; Luo, Y.; Tornabene, T.; O’Neill, A.M.; Greene, M.W.; Geetha, T.; Babu, J.R. High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2017, 1863, 499–508. [Google Scholar] [CrossRef]
- Luo, A.; Xie, Z.; Wang, Y.; Wang, X.; Li, S.; Yan, J.; Zhan, G.; Zhou, Z.; Zhao, Y.; Li, S. Type 2 diabetes mellitus-associated cognitive dysfunction: Advances in potential mechanisms and therapies. Neurosci. Biobehav. Rev. 2022, 137, 104642. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Reagan, L.P. Hippocampal insulin resistance and cognitive dysfunction. Nat. Rev. Neurosci. 2015, 16, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Bi, T.; Feng, R.; Zhan, L.; Ren, W.; Lu, X. ZiBuPiYin Recipe Prevented and Treated Cognitive Decline in ZDF Rats With Diabetes-Associated Cognitive Decline via Microbiota–Gut–Brain Axis Dialogue. Front. Cell Dev. Biol. 2021, 9, 651517. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, A.K.; Dharavath, R.N.; Chopra, K. Time-response studies on development of cognitive deficits in an experimental model of insulin resistance. Clin. Nutr. 2018, 38, 1447–1456. [Google Scholar] [CrossRef] [PubMed]
- Grillo, C.A.; Woodruff, J.L.; Macht, V.A.; Reagan, L.P. Insulin resistance and hippocampal dysfunction: Disentangling peripheral and brain causes from consequences. Exp. Neurol. 2019, 318, 71–77. [Google Scholar] [CrossRef]
- Sun, Q.; Li, J.; Gao, F. New insights into insulin: The anti-inflammatory effect and its clinical relevance. World J. Diabetes 2014, 5, 89–96. [Google Scholar] [CrossRef]
- Scherer, T.; Sakamoto, K.; Buettner, C. Brain insulin signalling in metabolic homeostasis and disease. Nat. Rev. Endocrinol. 2021, 17, 468–483. [Google Scholar] [CrossRef]
- Hallschmid, M. Intranasal Insulin for Alzheimer’s Disease. CNS Drugs 2021, 35, 21–37. [Google Scholar] [CrossRef]
- Canteiro, P.B.; Antero, D.C.; Tramontin, N.d.S.; Simon, K.U.; Mendes, C.; Correa, M.E.A.B.; Silveira, P.C.L.; Muller, A.P. Insulin treatment protects the brain against neuroinflammation by reducing cerebral cytokines and modulating mitochondrial function. Brain Res. Bull. 2019, 149, 120–128. [Google Scholar] [CrossRef]
- Aljada A, Ghanim H, Mohanty P, Kapur N, Dandona P. Insulin inhibits the pro-inflammatory transcription factor early growth response gene-1 (egr)-1 expression in mononuclear cells (mnc) and reduces plasma tissue factor (tf) and plasminogen activator inhibitor-1 (pai-1) concentrations. J Clin Endocrinol Metab. 2002, 87, 1419–1422.
- Coimbra, T.M.; Janssen, U.; Gröne, H.J.; Ostendorf, T.; Kunter, U.; Schmidt, H.; Brabant, G.; Floege, J. Early events leading to renal injury in obese Zucker (fatty) rats with type II diabetes. Kidney Int. 2000, 57, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Hoshi S, Shu Y, Yoshida F, Inagaki T, Sonoda J, Watanabe T, Nomoto K, Nagata M: Podocyte injury promotes progressive nephropathy in Zucker diabetic fatty rats. Lab Invest. 2002, 82, 25–35. [CrossRef] [PubMed]
- Bussey, T.J.; Padain, T.L.; Skillings, E.A.; Winters, B.D.; Morton, A.J.; Saksida, L.M. The touchscreen cognitive testing method for rodents: How to get the best out of your rat. Learn. Mem. 2008, 15, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Talpos, J.; Steckler, T. Touching on translation. Cell Tissue Res. 2013, 354, 297–308. [Google Scholar] [CrossRef]
- Horner AE, Heath CJ, Hvoslef-Eide M, Kent BA, Kim CH, Nilsson SR, Alsio J, Oomen CA, Holmes A, Saksida LM et al. The touchscreen operant platform for testing learning and memory in rats and mice. Nat Protoc. 2013, 8, 1961–1984.
- Turner, K.M.; Simpson, C.G.; Burne, T.H.J. BALB/c Mice Can Learn Touchscreen Visual Discrimination and Reversal Tasks Faster than C57BL/6 Mice. Front. Behav. Neurosci. 2017, 11, 16. [Google Scholar] [CrossRef]
- Van de Weerd HA, Bulthuis RJA, Bergman AF, Schlingmann F, Tolboom J, Van Loo PLP, Remie R, Baumans V, Van Zutphen LFM. Validation of a new system for the automatic registration of behaviour in mice and rats. Behavioural Processes 2011, 53, 11–20.
- Cardinal RN and Aitken MRF. Whisker A client—server high-performance multimedia research control system. Behavior Research Methods 2010, 42, 1059–1071. [CrossRef]
- Markou, A.; Salamone, J.D.; Bussey, T.J.; Mar, A.C.; Brunner, D.; Gilmour, G.; Balsam, P. Measuring reinforcement learning and motivation constructs in experimental animals: Relevance to the negative symptoms of schizophrenia. Neurosci. Biobehav. Rev. 2013, 37, 2149–2165. [Google Scholar] [CrossRef]
- Morrison, M.C.; Mulder, P.; Stavro, P.M.; Suárez, M.; Arola-Arnal, A.; van Duyvenvoorde, W.; Kooistra, T.; Wielinga, P.Y.; Kleemann, R. Replacement of Dietary Saturated Fat by PUFA-Rich Pumpkin Seed Oil Attenuates Non-Alcoholic Fatty Liver Disease and Atherosclerosis Development, with Additional Health Effects of Virgin over Refined Oil. PLOS ONE 2015, 10, e0139196. [Google Scholar] [CrossRef]
- Mueller, A.M.; Kleemann, R.; Gart, E.; van Duyvenvoorde, W.; Verschuren, L.; Caspers, M.; Menke, A.; Krömmelbein, N.; Salic, K.; Burmeister, Y.; et al. Cholesterol Accumulation as a Driver of Hepatic Inflammation Under Translational Dietary Conditions Can Be Attenuated by a Multicomponent Medicine. Front. Endocrinol. 2021, 12, 601160. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Menke, A.L.; Driessen, A.; Koek, G.H.; Lindeman, J.H.; Stoop, R.; Havekes, L.M.; Kleemann, R.; van den Hoek, A.M. Establishment of a General NAFLD Scoring System for Rodent Models and Comparison to Human Liver Pathology. PLOS ONE 2014, 9, e115922. [Google Scholar] [CrossRef] [PubMed]
- Tengeler AC, Gart E, Wiesmann M, Arnoldussen IAC, van Duyvenvoorde W, Hoogstad M, Dederen PJ et al. Propionic acid and not caproic acid, attenuates nonalcoholic steatohepatitis and improves (cerebro) vascular functions in obese Ldlr−/−.Leiden mice. FASEB J. 2020, 34, 9575–9593. [CrossRef] [PubMed]
- Bélanger, A.; Lavoie, N.; Trudeau, F.; Massicotte, G.; Gagnon, S. Preserved LTP and water maze learning in hyperglycaemic–hyperinsulinemic ZDF rats. Physiol. Behav. 2004, 83, 483–494. [Google Scholar] [CrossRef]
- Jolivalt, C.G.; Aghanoori, M.R.; Navarro-Diaz, M.C.; Han, M.M.; Sanchez, G.; Guernsey, L.; Quach, D.; Johe, K.; Fernyhough, P.; Calcutt, N.A. Enhancement of Mitochondrial Function by the Neurogenic Molecule NSI-189 Accompanies Reversal of Peripheral Neuropathy and Memory Impairment in a Rat Model of Type 2 Diabetes. J. Diabetes Res. 2022, 2022, 8566970. [Google Scholar] [CrossRef]
- Tomassoni D, Martinelli I, Moruzzi M, Micioni Di Bonaventura MV, Cifani C, Amenta F, Tayebati SK. Obesity and Age-Related Changes in the Brain of the Zucker Lepr fa/fa Rats. Nutrients 2020, 12, 1356. [CrossRef]
- Vogel, H.; Kraemer, M.; Rabasa, C.; Askevik, K.; Adan, R.A.; Dickson, S.L. Genetic predisposition to obesity affects behavioural traits including food reward and anxiety-like behaviour in rats. Behav. Brain Res. 2017, 328, 95–104. [Google Scholar] [CrossRef]
- Chander PN, Gealekman O, Brodsky SV, Elitok S, Tojo A, Crabtree M, Gross SS, Goligorsky MS. Nephropathy in Zucker diabetic fat rat is associated with oxidative and nitrosative stress: prevention by chronic therapy with a peroxynitrite scavenger ebselen. 2004; 15(9):2391-403.
- Slavkovsky, R.; Kohlerova, R.; Tkacova, V.; Jiroutova, A.; Tahmazoglu, B.; Velebny, V.; Rezačová, M.; Sobotka, L.; Kanta, J. Zucker diabetic fatty rat: A new model of impaired cutaneous wound repair with type II diabetes mellitus and obesity. Wound Repair Regen. 2011, 19, 515–525. [Google Scholar] [CrossRef]
- Rasmussen, E.B.; Huskinson, S.L. Effects of rimonabant on behavior maintained by progressive ratio schedules of sucrose reinforcement in obese Zucker (fa/fa) rats. Behav. Pharmacol. 2008, 19, 735–742. [Google Scholar] [CrossRef]
- Kullmann, S.; Kleinridders, A.; Small, D.M.; Fritsche, A.; Häring, H.-U.; Preissl, H.; Heni, M. Central nervous pathways of insulin action in the control of metabolism and food intake. Lancet Diabetes Endocrinol. 2020, 8, 524–534. [Google Scholar] [CrossRef]
- Yoshizaki, T.; Schenk, S.; Imamura, T.; Babendure, J.L.; Sonoda, N.; Bae, E.J.; Oh, D.Y.; Lu, M.; Milne, J.C.; Westphal, C.; et al. SIRT1 inhibits inflammatory pathways in macrophages and modulates insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E419–E428. [Google Scholar] [CrossRef] [PubMed]
- Jiao, F.; Gong, Z. The Beneficial Roles of SIRT1 in Neuroinflammation-Related Diseases. Oxidative Med. Cell. Longev. 2020, 2020, 6782872. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Luo, A.; Sun, R.; Tang, X.; Zhao, Y.; Zhang, J.; Zhou, B.; Zheng, H.; Yu, H.; Li, S. Resveratrol Mitigates Hippocampal Tau Acetylation and Cognitive Deficit by Activation SIRT1 in Aged Rats following Anesthesia and Surgery. Oxidative Med. Cell. Longev. 2020, 2020, 4635163. [Google Scholar] [CrossRef] [PubMed]
- Corpas, R.; Revilla, S.; Ursulet, S.; Castro-Freire, M.; Kaliman, P.; Petegnief, V.; Giménez-Llort, L.; Sarkis, C.; Pallàs, M.; Sanfeliu, C. SIRT1 Overexpression in Mouse Hippocampus Induces Cognitive Enhancement Through Proteostatic and Neurotrophic Mechanisms. Mol. Neurobiol. 2017, 54, 5604–5619. [Google Scholar] [CrossRef]
- Nam, S.M.; Na Kim, Y.; Yoo, D.Y.; Yi, S.S.; Choi, J.H.; Hwang, I.K.; Seong, J.K.; Yoon, Y.S. Hypothyroidism affects astrocyte and microglial morphology in type 2 diabetes. Neural Regen. Res. 2013, 8, 2458–2467. [Google Scholar] [CrossRef]
- Hwang, I.K.; Choi, J.H.; Nam, S.M.; Park, O.K.; Yoo, D.Y.; Kim, W.; Yi, S.S.; Won, M.-H.; Seong, J.K.; Yoon, Y.S. Activation of microglia and induction of pro-inflammatory cytokines in the hippocampus of type 2 diabetic rats. Neurol. Res. 2014, 36, 824–832. [Google Scholar] [CrossRef]
- Martinelli, I.; Tomassoni, D.; Roy, P.; Amenta, F.; Tayebati, S.K. Altered Brain Cholinergic and Synaptic Markers in Obese Zucker Rats. Cells 2021, 10, 2528. [Google Scholar] [CrossRef]
- Martin B, Ji S, Maudsley S, and Mattson MP. “Control” laboratory rodents are metabolically morbid: Why it matters. PNAS 2010, 107, 6127–6133. [CrossRef]
- Noble, E.E.; Olson, C.A.; Davis, E.; Tsan, L.; Chen, Y.-W.; Schade, R.; Liu, C.; Suarez, A.; Jones, R.B.; de La Serre, C.; et al. Gut microbial taxa elevated by dietary sugar disrupt memory function. Transl. Psychiatry 2021, 11, 1–16. [Google Scholar] [CrossRef]
- Friedman, N.P.; Robbins, T.W. The role of prefrontal cortex in cognitive control and executive function. Neuropsychopharmacology 2022, 47, 72–89. [Google Scholar] [CrossRef]
Figure 1.
Body weight and locomotor activity in obese and lean ZDF rats. (A) Body weight development during adolescence and early adulthood between weeks 4 and 14 of age. Significant body weight differences emerged from week 6 (p<0.05) for females and week 7 for males (p<0.05). (B) Body weight comparison at week-14. Values of A and B represent the mean (±SEM). (C) Locomotor activity in Laboras homecages (seconds). Values in C represent the mean of 5 sessions of 48h each in the Laboras homecage ± SEM. (D) Average locomotor activity during the first 10 sessions of the visual discrimination stage of operant testing. Values represent the average number of infrared beam crosses during a session ± SEM.
Figure 1.
Body weight and locomotor activity in obese and lean ZDF rats. (A) Body weight development during adolescence and early adulthood between weeks 4 and 14 of age. Significant body weight differences emerged from week 6 (p<0.05) for females and week 7 for males (p<0.05). (B) Body weight comparison at week-14. Values of A and B represent the mean (±SEM). (C) Locomotor activity in Laboras homecages (seconds). Values in C represent the mean of 5 sessions of 48h each in the Laboras homecage ± SEM. (D) Average locomotor activity during the first 10 sessions of the visual discrimination stage of operant testing. Values represent the average number of infrared beam crosses during a session ± SEM.
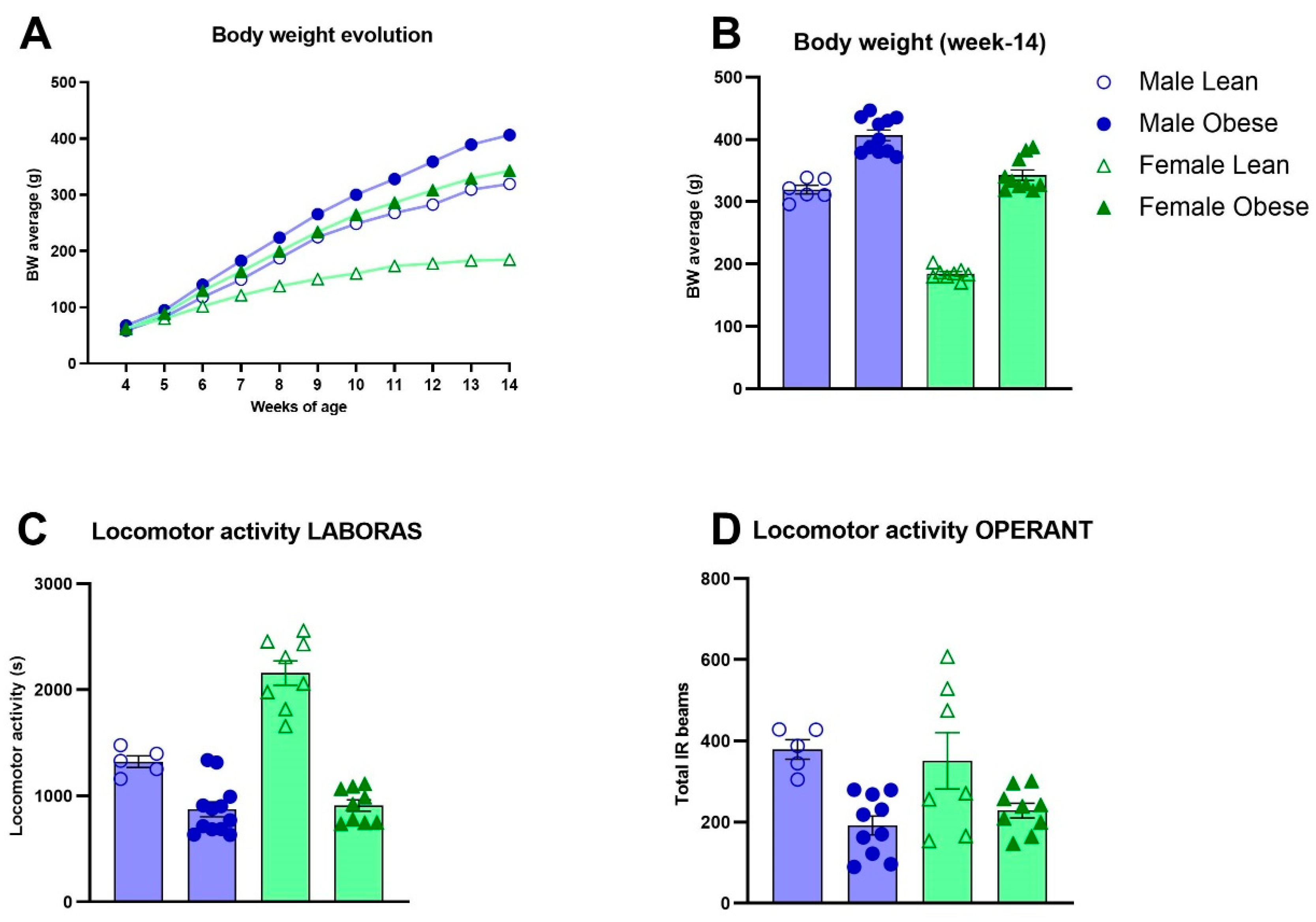
Figure 2.
Cognitive performance and level of motivation in obese and lean ZDF rats. (A) The evolution of percentage of correct trials during the first 10 sessions of the visual discrimination (VD) task. (B) Average values of the percentages of the correct trials during the first 10 VD sessions. (C) Average percentage of correct trials of the 3 consecutive trials when task criterion was reached. (D) Number of sessions required to reach criterion during the VD task. Values during the VD task represent the group mean (±SEM). (E) Number of sessions required to reach criterion in different fixed ratio phases in male and female lean vs obese rats. (F) Average group level of active responses to obtain a reward during the 10 sessions of the progressive ratio in male and female lean vs obese rats. (G) Effects of the reward type (sucrose vs grain pellets) on the motivation to perform active responses to obtain the reward. Values during the FR and PR sessions represent the group mean (±SEM).
Figure 2.
Cognitive performance and level of motivation in obese and lean ZDF rats. (A) The evolution of percentage of correct trials during the first 10 sessions of the visual discrimination (VD) task. (B) Average values of the percentages of the correct trials during the first 10 VD sessions. (C) Average percentage of correct trials of the 3 consecutive trials when task criterion was reached. (D) Number of sessions required to reach criterion during the VD task. Values during the VD task represent the group mean (±SEM). (E) Number of sessions required to reach criterion in different fixed ratio phases in male and female lean vs obese rats. (F) Average group level of active responses to obtain a reward during the 10 sessions of the progressive ratio in male and female lean vs obese rats. (G) Effects of the reward type (sucrose vs grain pellets) on the motivation to perform active responses to obtain the reward. Values during the FR and PR sessions represent the group mean (±SEM).
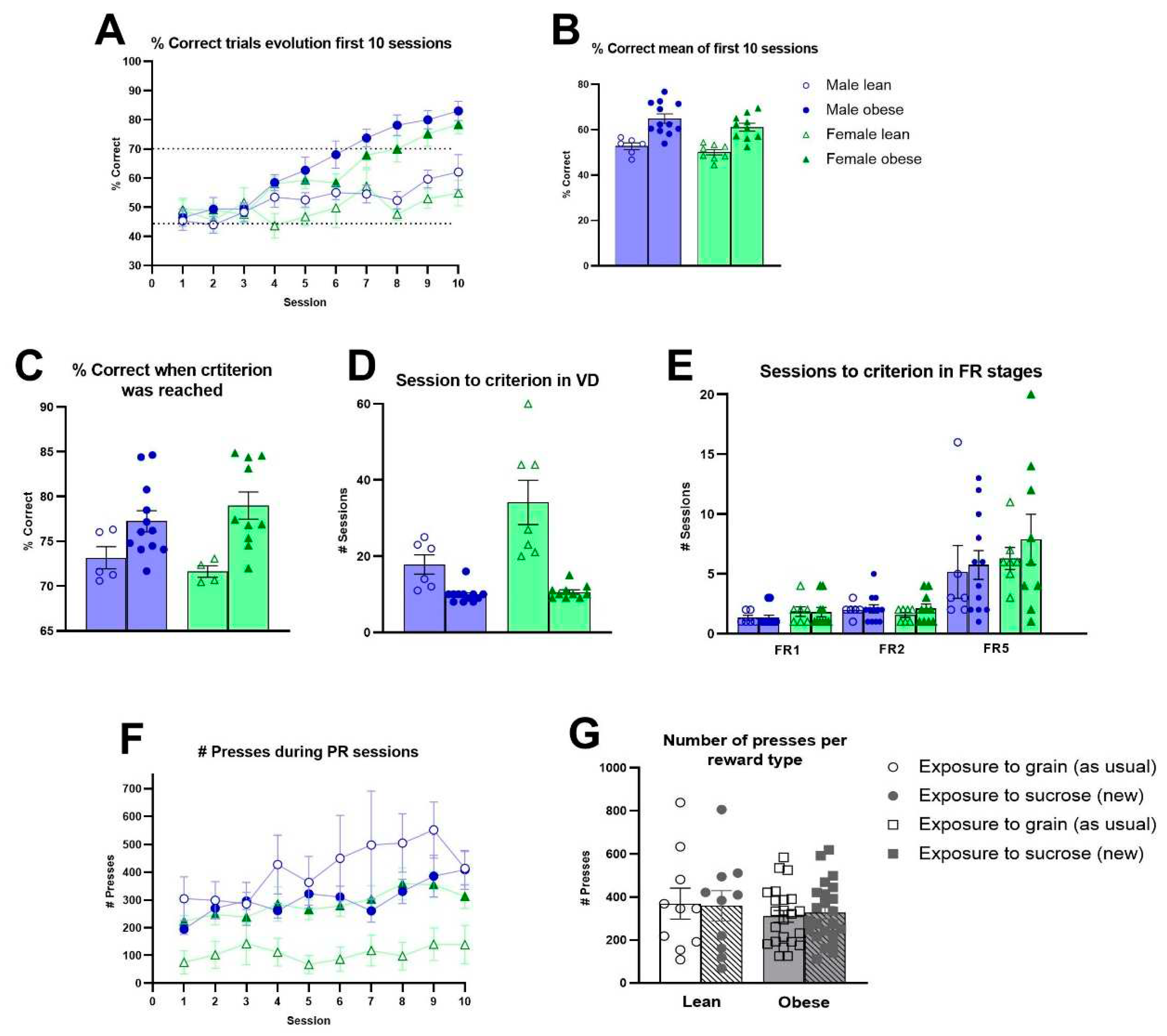
Figure 5.
Histological profiles of white adipose tissue in obese and lean ZDF rats. (A, B) Average relative weights of perigonadal (pgWAT) and mesenteric (mWAT) white adipose tissues. (C-D) pgWAT and mWAT adipocyte size and their percentage distribution per cell size (G-H) in both lean and obese males and females. (E, F) Representative images of pgWAT and mWAT tissues. Values represent the mean group (±SEM).
Figure 5.
Histological profiles of white adipose tissue in obese and lean ZDF rats. (A, B) Average relative weights of perigonadal (pgWAT) and mesenteric (mWAT) white adipose tissues. (C-D) pgWAT and mWAT adipocyte size and their percentage distribution per cell size (G-H) in both lean and obese males and females. (E, F) Representative images of pgWAT and mWAT tissues. Values represent the mean group (±SEM).
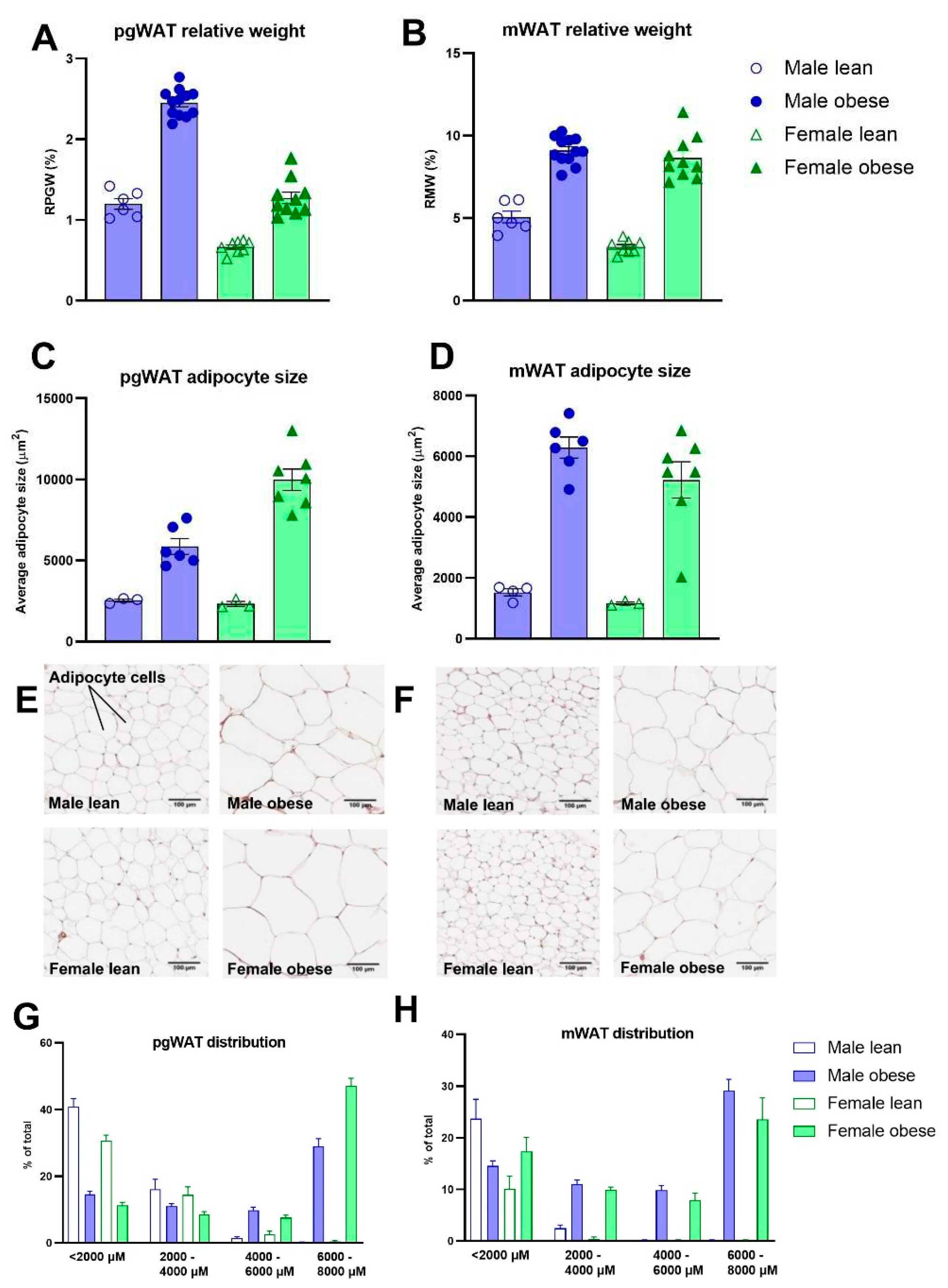
Figure 6.
Liver histopathology in obese and lean ZDF rats. (A) Representative images of the liver H&E stainings showing the histopathological affections in ZDF obese rats. (B) Relative liver weights. (C) Hepatic inflammation expressed as the number of observed aggregates per field. (D) Average percentage of the liver tissue steatosis (macro- and micro vesicular steatosis) and hypertrophy. Values represent the group mean (±SEM).
Figure 6.
Liver histopathology in obese and lean ZDF rats. (A) Representative images of the liver H&E stainings showing the histopathological affections in ZDF obese rats. (B) Relative liver weights. (C) Hepatic inflammation expressed as the number of observed aggregates per field. (D) Average percentage of the liver tissue steatosis (macro- and micro vesicular steatosis) and hypertrophy. Values represent the group mean (±SEM).
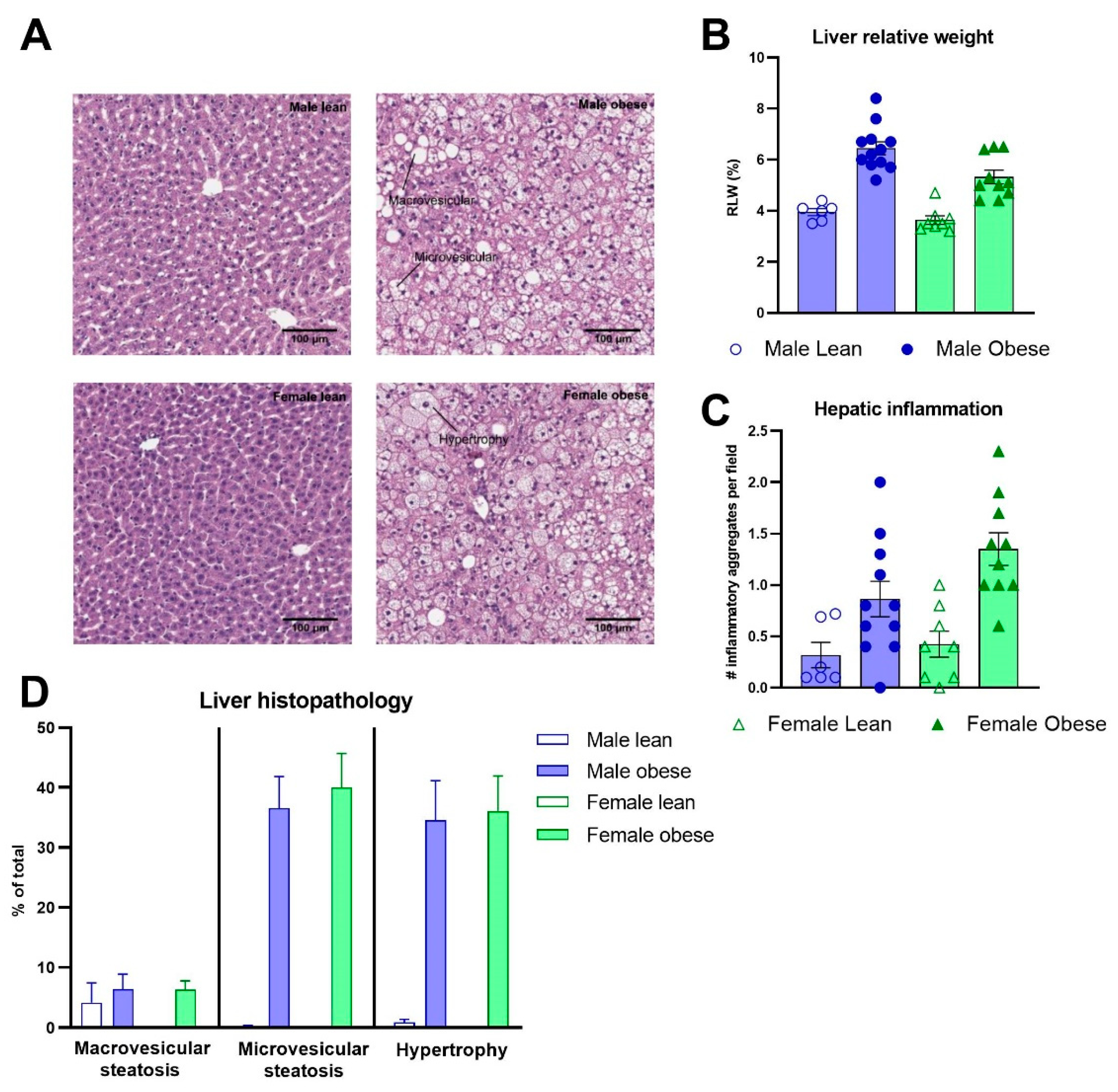
Figure 7.
Evaluation of NeuN, IBA-1 positive cells and IBA-1 immunoreactive soma cell size area by immunohistochemistry in the medial prefrontal cortex in obese and lean ZDF rats. (A) Representative images of NeuN and IBA-1 immunoreactive cells. (B) Number of NeuN positive cells. (C) Number of IBA-1 positive cells. (D) IBA-1 immunoreactive soma cell size. Values represent the group mean (±SEM).
Figure 7.
Evaluation of NeuN, IBA-1 positive cells and IBA-1 immunoreactive soma cell size area by immunohistochemistry in the medial prefrontal cortex in obese and lean ZDF rats. (A) Representative images of NeuN and IBA-1 immunoreactive cells. (B) Number of NeuN positive cells. (C) Number of IBA-1 positive cells. (D) IBA-1 immunoreactive soma cell size. Values represent the group mean (±SEM).
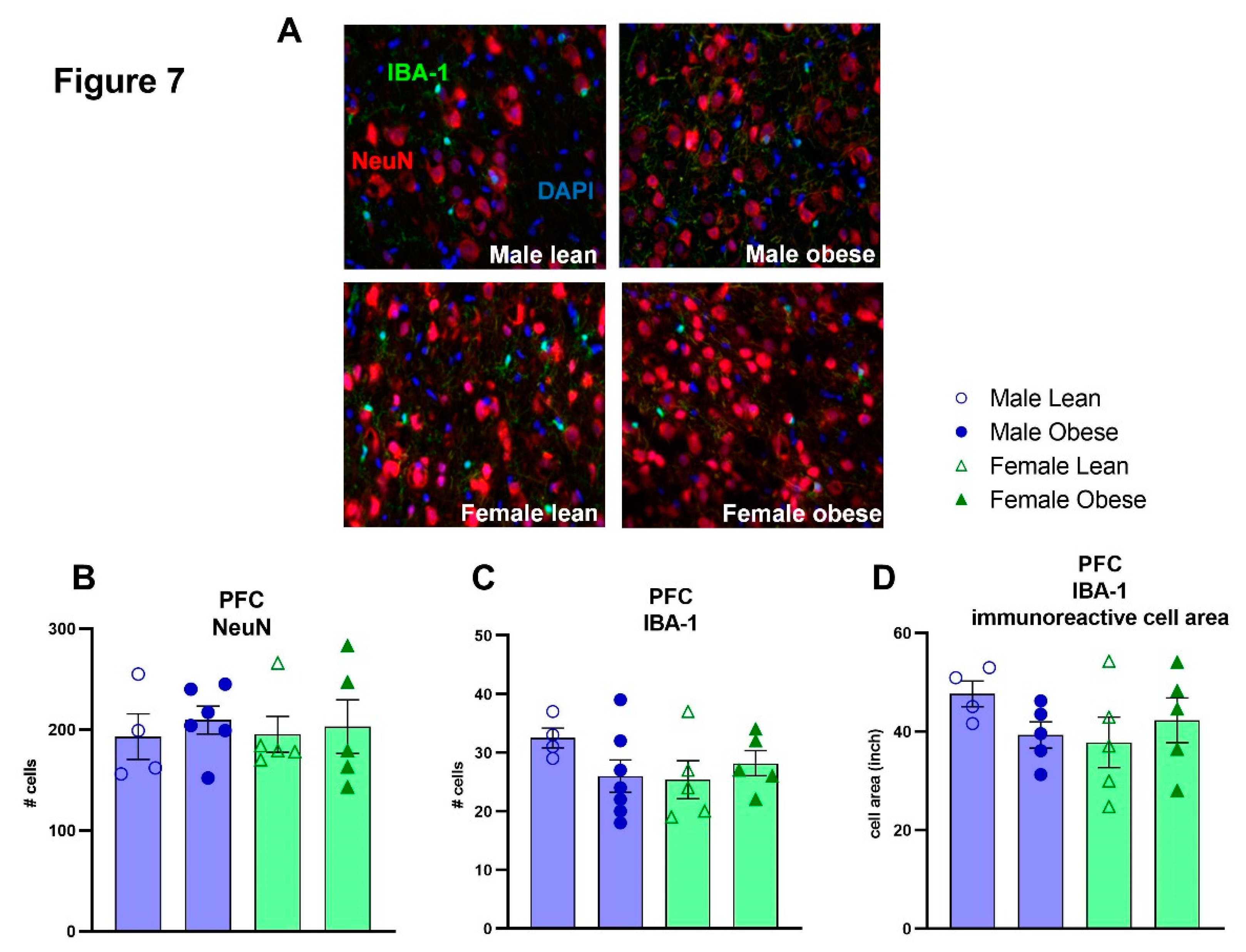
Figure 8.
mRNA exression levels in the medial prefrontal cortex and hippocampal insulin levels in obese and lean ZDF rats. mRNA levels of SIRT1 (A), PSD-95 (B) and GAPDH (C). Hippocampal insulin levels (D). Values represent the group mean (±SEM).
Figure 8.
mRNA exression levels in the medial prefrontal cortex and hippocampal insulin levels in obese and lean ZDF rats. mRNA levels of SIRT1 (A), PSD-95 (B) and GAPDH (C). Hippocampal insulin levels (D). Values represent the group mean (±SEM).
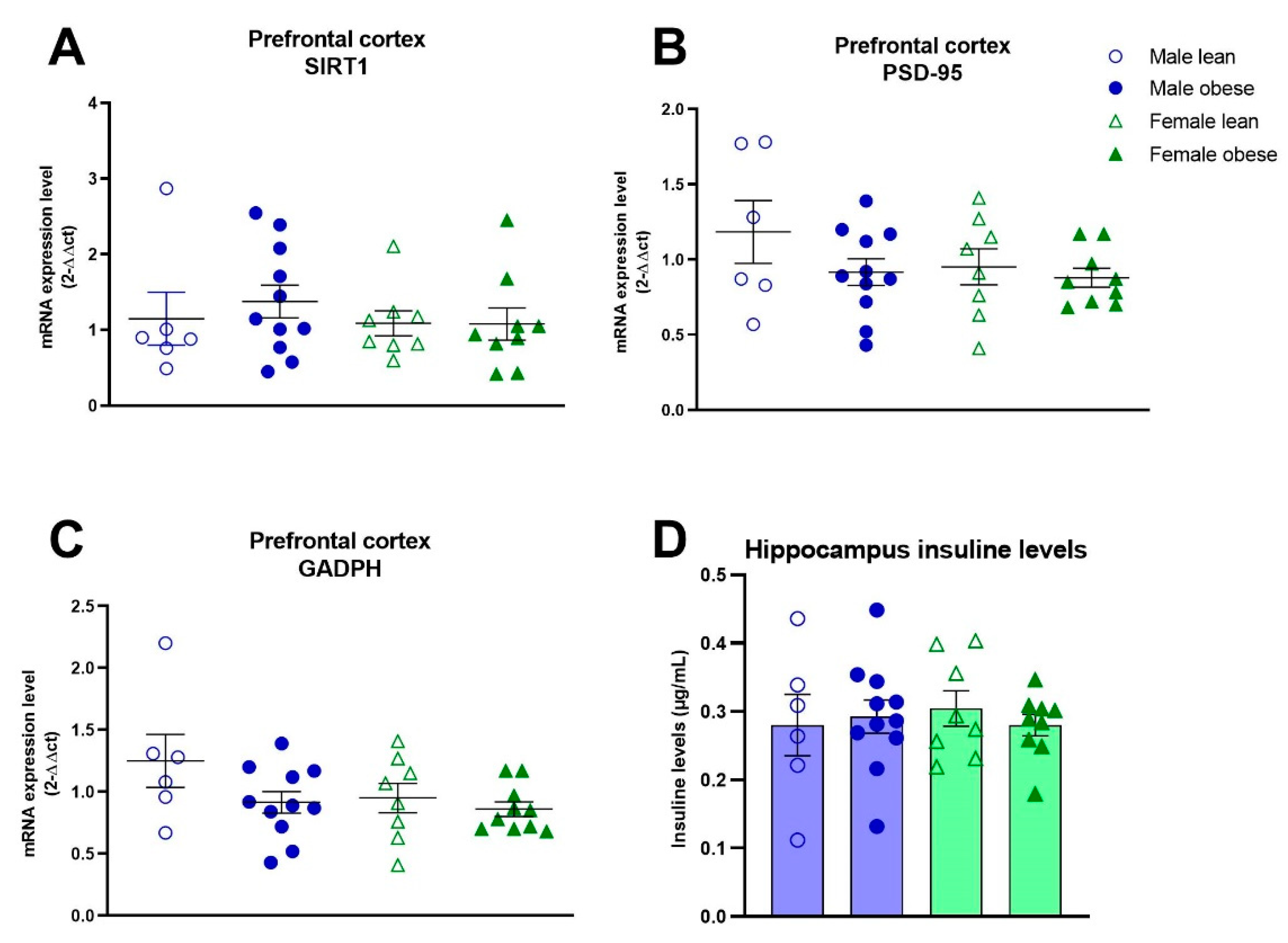

Table 1.
Different criteria during cognitive testing.
| Learning stage | Criteria | Description |
|---|---|---|
| Habituation | Complete one session | The rat was provided with 25 pellets at variable time intervals. |
| Autoshaping | All 25 pellets eaten | In order to learn the association between the stimulus and the reward; the rat received a reward regardless of whether it touched the stimulus and would receive the reward immediately when it touched the image. |
| Must touch | Two sessions 25 correct responses and all 25 pellets eaten | The rat had to touch the stimulus to receive a reward, otherwise no reward was given. |
| Punish incorrect | At least 70% correct responses for two sessions (Nr correct/Nr correct+Nr incorrect)*100 | The rat receives negative feedback (house light on) when it touches the screen in the wrong location. Then, no pellet is given. |
| Moving punish incorrect | At least 70% correct responses for two sessions (Nr correct/Nr correct+Nr incorrect)*100 | Similar to the punish incorrect, but the stimulus changes position from left to right in a pseudorandom order. |
| Visual discrimination | At least 10 sessions & average 70% correct responses across three consecutive sessions (Nr correct/Nrcorrect+Nr incorrect)*100 | Similar to moving punish incorrect, however the circular stimulus is replaced by two striped images. The designated correct stimulus (counterbalanced between rats) changes position in a pseudorandom order. |
| Fixed Ratio 1 | 50 correct responses | The rat had to press once to receive a reward. |
| Fixed Ratio 2 | 100 correct responses | The rat had to press twice to receive a reward. |
| Fixed Ratio 5 | 250 correct responses | The rat had to press five times to receive the reward. |
| Progressive Ratio | Complete 10 (ZDF rats) or 20 sessions (LE rats) | On each subsequent trial the reward response requirement increased on a linear basis (+4). |
Table 2.
Primers used in the qPCR analysis.
| Name | Sequece 5’ -> 3’ | Tm | GC% |
|---|---|---|---|
| YWHAZ | fw: GACAAGAAAGGAATTGTGGACCAGT rv: GGGCCAGACCCAGTCTGATG |
61.44 62.26 |
44.00 65.00 |
| GAPDH | fw: CCACCAACTGCTTAGCCCCC rv: TGGTCATGAGCCCTTCCACG |
63.12 62.18 |
65.00 60.00 |
| SIRT1 | fw: AGATACCTTGGAGCAGGTTGCAG rv: AGATGCTGTTGCAAAGGAACCATGA |
62.51 63.48 |
52.17 44.00 |
| β-actin | fw: CGTGAAAAGATGACCCAGATCA rv: AGAGGCATACAGGGACAACAC |
58.40 59.72 |
45.45 52.38 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



