
Submitted:
29 August 2023
Posted:
30 August 2023
You are already at the latest version
Abstract
Alternative splicing can produce transcripts that affect cancer development and shows potential for cancer diagnosis and treatment. However, intron retention (IR), a type of alternative splicing, has been less systematically studied in cancer biology research. Here, we generated a pan-cancer IR landscape for more than 10,000 samples across 33 cancer types from The Cancer Genome Atlas (TCGA). We characterized differentially retained introns between tumor and normal samples and identified retained introns associated with survival. We discovered 988 differentially retained introns in 14 cancers, some of which demonstrated diagnostic potential in multiple cancer types. We also inferred a large number of prognosis-related introns in 33 cancer types, and the associated genes included well-known cancer hallmarks such as angiogenesis, metastasis, and DNA mutations. Notably, we discovered a novel intron retention event inside 5′UTR of STN1 that is associated with the survival of lung cancer patients. The retained intron reduces translation efficiency by producing upstream open reading frames (uORFs) and thereby inhibits colony formation and cell migration of lung cancer cells. Besides, the IR-based prognostic model achieved good stratification on certain cancers, as illustrated in acute myeloid leukemia. Taken together, we performed a comprehensive IR survey at a pan-cancer level, and the results implied that IR has the potential to be diagnostic and prognostic cancer biomarkers, as well as new drug targets.
Keywords:
intron retention
; RNA-seq
; cancer
; diagnosis
; prognosis
1. Introduction
Alternative splicing (AS) is a way for eukaryotes to produce multiple transcripts from a single gene by changing the composition of exons during RNA processing[1]. Intron retention (IR) is a type of AS and affects about 80% of human genes[2]. IR transcripts tend to be degraded by nonsense-mediated decay (NMD) or exosome pathway. Therefore, IR coupled with RNA surveillance can regulate gene expression and affect various biological processes [3,4]. IR transcripts can be detained in the nucleus and wait for splicing signals, such as in the T cell activation process responding to external stimulus[5]. IR can also produce novel proteins with different functions[6,7,8] or subcellular locations[9], and play essential roles in many key biological conditions[3,10,11]. IR is under sophisticated regulation that can be affected by sequence variations, splicing factors, epigenetic and transcriptional regulatory mechanisms, etc[12].
Tumors generally have 30% more aberrant alternative splicing events than normal tissues [13] and can give rise to many tumor-specific transcripts associated with oncogenic functions and drug resistance[14,15,16,17]. There are numerous relevant studies on exon skipping in cancer, while only a handful of them have identified features and functions of IR. Dvinge and Bradley reported that compared to adjacent normal tissues, more introns were retained in tumor tissues for 15 cancer types, and many of them could be detected in the cytoplasm [18]. Evidence showed that IR can promote cancer development. Somatic mutations in tumors can trigger IR and these mutations are enriched in tumor suppressor genes (TSGs)[19]. Inactivation of histone H3K36 methyltransferase SETD2 reduced DVL2 IR, and as a result activated Wnt signaling to promote colon cancer predisposition[20]. Similarly, ZRSR2 loss increased retention of a minor intron of LZTR1 and resulted in enhanced RAS signaling, potentially driving leukemia[21]. IR produces epitopes presented on MHC I, making it a potential source of tumor-specific antigens (neoantigens)[22]. However, these researches have mainly focused on individual or patient-specific IR events, a systematic survey of recurrent IR alterations at a pan-cancer level, especially from the prognostic perspective, is still lacking.
In the present study, we profiled the IR landscape of 33 cancer types in the Cancer Genome Atlas (TCGA), identified IR events that were differentially regulated between normal and tumor samples, and discovered IR events associated with survival. Some informative introns not only show potential in accurate diagnosis and prognosis with machine learning methods but also are involved in cancer pathology validated in our experiments (Figure 1). Many of these introns were recurrently retained in multiple patients across cancers, serving as a resource of potential diagnostic and prognostic biomarkers, even as promising targets for new therapy.
2. Materials and methods
2.1. RNA-seq bam download and IR quantification
We downloaded RNA-seq bam files using the GDC data transfer tool, including 33 cancer types and over 10,000 tumor and adjacent normal samples in the TCGA project.
Stringtie (version 2.1.3)[23] was used to quantify gene expression. IRFinder (version 1.3.1)[2] was used to identify and quantify IR. And before applying IRFinder, samtools (version 1.9) was used to sort bam files by read pairs. The genome version was hg38 and the gene annotation version was gencode v35.
We performed quality control on both intron and sample levels. Filtering out introns that overlap with known exons (flagged by IRFinder as “known-exon”) resulted in 243,151 introns covering 21,520 genes genome-wide. Before quantifying IR, at least 4 junction reads (split reads) spanning flanking exons were required to make sure that the isoform was expressed. IRratio was used to measure the IR level, and it was calculated by dividing the median read depth of an intron by itself plus the number of reads spanning flanking exons. If the coverage of an intron was less than 20%, it was likely to be completely spliced and its IRratio was assigned to 0. If the coverage of an intron was above 70% and the median read depth was above 3, the intron was likely to be retained and its IRratio was kept, which must exceed 0. Otherwise, the retention state could not be accurately decided for an intron and its IRratio was set as missing. IRFinder automatically decided if an RNA-seq sample was suitable for IR detection based on the ratio of the number of reads that map to intergenic regions to the number of reads that map to coding regions. When the ratio exceeded 10%, it gave a warning message, and such samples were not used for further analysis. Over 90% of TCGA RNA-seq samples passed this QC.
The majority of TCGA RNA-seq was unstranded, so the read orientation could not be determined, which may lead to false positive IR detection[2,24]. Nevertheless, we made use of a small number of strand-specific RNA-seq samples in TCGA which passed sample quality control (n = 34) to generate a “whitelist” of retained introns, meaning they were more likely to be genuine and reliable. Specifically, we obtained a maximum IRratio of each intron in these samples, and introns with a maximum IRratio above 0.08 comprised the “whitelist” (n = 47,026). That is, these introns were retained in at least one stranded RNA-seq sample, which made them more reliable. Thus, differentially retained introns and survival-associated introns were restricted to only “whitelist” introns for higher confidence.
2.2. Differential IR and differential gene expression
Because IR levels did not follow a normal distribution across individuals, we used paired Wilcoxon rank-sum test to detect differential IR events (DIRs) in paired tumor and normal samples (n > 15) for 14 cancer types. A differentially retained intron should have a P value less than 0.05 and the difference of median IRratios between tumor and normal samples should be larger than 0.1.
In terms of differentially expressed genes (DEG) detection, DESeq2[25] was used, and we selected DEGs with Padjusted < 0.05 and |log2FC| > 1.
2.3. Dimensionality reduction and visualization
If the quality control mentioned above was not passed, the IRratio was set to a missing value. We generated IRratio matrices, kept IR events with missing rates less than 30%, and imputed missing values with a mean value from the remaining samples. Principle component analysis was then performed. We extracted the first 100 principal components and further analyzed them with package Rtsne[26] to draw tSNE plots, with perplexity set to 30.
2.4. Functional enrichment
2.5. Sequence features analysis
We classified introns into four types to compare their sequence features. Constitutive introns (n = 48,344) were introns with median IRratio of 0 in tumor and normal samples across all cancers; not regulated IR were introns retained in tumor or normal samples in any cancer but showed no significant difference between tumor and normal samples (n = 10,887); down DIRs were introns with reduced retention level in tumor samples in any cancer (n = 401); up DIRs were introns with increased retention level in tumor samples in any cancer (n = 669).
Conservation analysis: hg38 version of phastCons30way.bw was downloaded from UCSC, and bwtool[30] was used to calculate a mean phastCons[31] score around boundaries of the above 4 types of introns separately.
Splice score of 5′ and 3′ sites were calculated using the maximum entropy modeling method[32].
Intron GC content, length and relative gene position were all analyzed based on hg38 and gencode v35. To analyze the distribution of introns in genes, the relative gene position of an intron was calculated by dividing the rank of the intron (5′ to 3′ orientation) by the total intron count of this transcript.
We adjusted a nonsense-mediated decay (NMD) prediction rule from a previous study[33]. Specifically, when an intron was retained in a protein coding gene, it was very possible to introduce a premature stop codon (PTC) and elicits NMD unless under following conditions. If the intron resides in a 5′ or 3′ untranslated region (UTR), or the intron was close to start codon (< 200 nt), or it was the last intron, NMD would not be elicited. If no PTC was produced, or the PTC was located within 55 nt upstream of the last exon-exon junction, NMD would not be elicited either. Otherwise, the IR transcript was prone to be targeted by NMD.
2.6. Random Forests model
We merged DIRs from different cancer types, and filtered out the ones with pan-cancer missing rates over 30% and the ones that were inconsistently up or down-regulated in different cancers. This resulted in 273 DIRs to be used in Random Forests models for pan-cancer modeling. R package randomForest[34] was applied to the train model, with mtree = 500, mtry = 3 and proximity = TRUE. A hundred times four-fold cross validation was used to calculate a pooled area under the curve (AUC) for the training set (paired tumor and normal samples from 14 cancers), and the receiver operating curve (ROC) in randomly selected one run was drawn using a R package pROC[35]. We used the Rfcv function to predict model performances with a sequentially reduced number of DIRs (ranked by importance), with cv.fold = 5 and step = 0.9.
2.7. Survival analysis
For introns that had valid IRratios in at least 50% of patients and at least 5% of valid IRratios were above 0.1, we restricted our analysis to patients with IRratios over 0 and classified them into IR-high or low groups based on median IRratio. Then we performed Univariate Cox regression and selected intron related to overall survival (OS) or disease-free survival (DFS), and an unadjusted p value less than 0.05 was considered significant. Similarly, gene expression associated with survival was identified by selecting genes with a median expression level over 1 TPM, dividing patients into high and low expression groups based on the median cutoff, and performing univariate Cox regression.
2.8. LASSO regression to build a prognostic model
To build an IR-based prognostic model for each cancer type, we selected introns with missing rates less than 20% (missing values were later filled with mean) and perform LASSO regression with R package glmnet[36,37]. Candidate introns and corresponding coefficients were derived with the λ parameter associated with the minimum mean error or with one standard error. Intron retention risk (IRR) score was calculated as the sum of IRratios multiplied by corresponding coefficients of candidate introns. And then patients were divided into IRR-low and high groups based on median value.
2.9. Cell culture and lentiviral transfection
HEK293 (human embryonic kidney 293 cells), H1299 cells, A549 cells were purchased from National Collection of Authenticated Cell Cultures and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Invitrogen, 11960044) supplemented with 10% FBS (Gibco), streptomycin (100 μg/ml), and penicillin (100 U/ml) at 37℃/5% CO2.
We applied lentivirus transfection-mediated gene-silencing strategy to stably knockdown target gene STN1. HEK293T cells were transfected as described[38]. Specifically, shRNAs (shRNA sequences were listed in Supplementary Table S7) were obtained from Sigma-Aldrich, then annealed and cloned into pLKO.1 vector. HEK293 cells grown in 6-well plates were transfected with 1 μg constructed vectors or empty control vectors pLKO.1 with VSVG and gag/pol encoding plasmids by using Lipofectamine 2000 (Invitrogen). Then, the virus supernatant was harvested in 24 hours to infect A549 cells in six-well plates seeded one day ahead. After incubation for one more day, A549 cells were screened by 2.5 μg/ml puromycin for 24 hours. The surviving cells were cultured for two more days and then harvested for following RNA extraction and cell proliferation assays.
2.10. RNA preparation, RT-PCR and qRT-PCR
Total RNA was extracted from cells with TRIzol (Invitrogen) according to the manufacturer’s instructions (Invitrogen) and reversely transcribed into cDNA with oligo-(dT) primer (Supplementary Table S7) using FastQuant RT Kit (Tiangen).
For RT-PCR, 100 ng cDNA was used for each PCR reaction, and PCR products were detected by agarose gel electrophoresis. Gene expression at the RNA level was quantified by qRT-PCR using a 2× SYBR mix (Vazyme). GAPDH served as an internal control. Then, the reaction was run on the Bio-Rad CFX manager machine. The IR and spliced transcripts of STN1 were quantified by qRT-PCR using primers specifically targeting the retained intron and exon junction, respectively. All primer sequences were listed in Supplementary Table S7.
2.11. RNA stability assay, and isolation of nuclear and cytoplasmic fractions
A549s cells were treated with 10µg/ml actinomycin-D (Act D; Sigma-Aldrich, Inc., St. Louis, MO, USA, A4262) for 0, 2, 4, and 6 hours, respectively. The total RNAs were extracted, and RNA levels were quantified by qRT-PCR as described above.
For nuclear and cytoplasmic fractionation, A549 cells were cultured in a T25 flask until 90% confluence. Then, cells were trypsinized, washed twice with cold PBS, and then cells were fractioned by Nuclear/Cytosol Fractionation Kit (Phygene, PH1466). Following RNA extraction and qRT-PCR were carried out as described above.
2.12. Psi-CHECK2 constructs and dual luciferase assay
Luciferase reporter gene expression vectors were bought from Promega and were prepared according to the manufacturer’s protocol. Briefly, the 590bp intron retained 5′ UTR and 146 spliced 5′ UTR was PCR amplified using STN1_5′ UTR_Forward (Nhe I) 5’-TACGACTCACTATAGgctagcggggtcgtcgccgccag -3’ and STN1_5′ UTR_Reverse (Nhe I) 5’-TTGGAAGCCATGGTGgctagccaggctgcatcaagaggca-3’. PCR fragments were cloned into the NheI-restricted site of the psi-CHECK2 vector. The STN1 5′ UTR was inserted directly upstream of the Renilla gene. The psi-CHECK2 construct containing the mutated 5′ UTR IR fragment was synthesized by Tsingke company, where the three start codons within the intron (181ATG, 283ATG, and 392ATG) were all mutated to AGC. DH5α cells were transformed with the three distinct constructs and cultured on ampicillin-containing media.
One day before transfection, 1 × 105 HEK293T cells were seeded into each well of a 24-well culture plate in 500μl DMEM supplemented with 10% FBS. Cells at 70% confluency were transfected with three psi-CHECK2 constructs with Lipofectamine 2000 reagent. 24 hours after transfection, growth media were removed and cells were washed gently with PBS. Passive lysis buffer (Promega) (100ul/well) was added and gently shaken for 15 min at room temperature, then cell lysates were harvested for dual luciferase assay. The activities of firefly and Renilla luciferase were measured using the Dual-Luciferase® Reporter 1000 Assay System (Promega) according to the manufacturer’s instructions. A total of 25μl cell lysate was transferred into a white opaque 96-well plate. The luminescence obtained for the mutated and wild-type constructs was normalized with the internal control firefly luciferase signal. Each experiment was performed in triplicate, and three independent experiments were performed. Quantitation of the reporter gene assay was calculated as mean ± SEM. Student’s t test was used to determine significant differences between each mutated construct compared with the wild-type construct.
2.13. Colony formation assay, transwell migration assay and cell proliferation assay
For the colony formation assay, cells were cultured in the six-well plate at a density of 800 cells per well. Cells were cultured under normal culture conditions for 15 days. For fixation of the cell, after the medium supernatant was removed, the cells were treated with 4% paraformaldehyde and were stained with 1% crystal violet (SigmaAldrich) for 15 min. Then, the plates were washed with phosphate-buffered saline (PBS) and photographed.
For the transwell migration assay, we used a 24-well transwell chamber (Corning). Cells were suspended in non-serum DMEM and then seeded in the top chamber of the transwell with a density of 1 × 104 per chamber, and 300 μl fresh complete DMEM (10% FBS) was added to the bottom chamber. After incubating for 48 hours, use PBS to wash the cells in the top chamber twice, and then fix the cells with 4% paraformaldehyde for 15 min, followed by staining with 1% crystal violet (Sigma-Aldrich) for 30 min. After washing and wiping off the cells in the inner side of the top chamber, the migratory cells adhering to the bottom surface of the membrane were observed, photographed and then counted by ImageJ software.
Cell proliferation assay was performed on cultured cells at four time points (24, 48, 72, and 96 h, respectively). A total of 100 μL cells were seeded in a 96-well plate with at least 1000 cells per well and four replicates for each time point. Cell Counting Kit-8 (CCK-8) reagent (Dojindo) was added according to the manufacturer’s protocol. Then, cells were incubated at 37°C for 2 hours and the absorbance at 450 nm was measured with a microplate reader (TECAN).
3. Results
3.1. Landscape of intron retention in 33 cancer types
RNA-seq data (in bam format) of the primary tumor and adjacent normal tissues from 33 TCGA cancer types, were downloaded and processed with IRFinder[2]. A total of 10,189 samples passed quality control and were used in this study (Figure 2A, Supplementary Table S1). IRratio was used to measure the retention level of each intron. Median IRratios of each intron among tumor samples were used to represent the IR level in each cancer type, and the correlation coefficients between different cancers were generally higher than 0.6. While acute myeloid leukemia (LAML), esophageal carcinoma (ESCA), and stomach adenocarcinoma (STAD) had lower similarity IR patterns with the other cancer types (Figure 2B).
Next, we computed the average number of retained introns (IRratio > 0.1) for each sample (Figure 2C, Supplementary Table S1). Notably, LAML tumor samples had an average of 27,148 retained introns, which was an-order-of-magnitude more than many other cancers. The sequencing depth and read length of LAML were comparable to that of other cancers (Supplementary Table S1), and no significant degradation of RNA was observed (Supplementary Figure S1A and B). Manual inspection showed that often multiple introns were retained within a gene and these signals did not appear to come from genomic contamination (Supplementary Figure S1C-E). In line with this, Dvinge and Bradley have also observed a much stronger IR increase in LAML than in other cancers [18]. In total, 13 cancer types exhibited significantly increased numbers of retained introns comparing to normal samples, while breast invasive carcinoma (BRCA) showed the opposite trend. Our results validate the findings of Dvinge and Bradley, and again suggest that inefficient intron removal is a common phenomenon in many cancers except breast cancer.
3.2. Differentially retained introns between tumor and normal tissues
To accurately compare IR between normal and tumor samples, we analyzed 14 cancer types each has at least 15 paired normal and tumor samples (Supplementary Figure S2A). Principle component analysis (PCA) showed varying degrees of difference in global IR patterns for normal and tumor samples across cancer types (Supplementary Figure S2B). We used paired Wilcoxon rank-sum test to detect introns significantly up- or downregulated in tumor tissues compared to matched adjacent normal tissues for each cancer type (P < 0.05) and required a difference of median values between normal and tumor samples greater than 10%.
A total of 988 differential introns were detected across 14 cancers, most of which were retained in both normal and tumor samples but with altered retention levels (Supplementary Figure S3A), while a handful of them were mainly retained in tumor samples and spliced in normal samples (cancer associated IR), or the other way around (i.e. normal associated IR) (Figure 3A). Colon Adenocarcinoma (COAD) and BRCA were detected with the most differential IR events (DIRs), but most DIRs were upregulated in COAD and downregulated in BRCA. DIRs were often specific to one type of cancer (Supplementary Figure S3B), and DIR genes usually had only one intron that was differentially retained (Supplementary Figure S3C). IR alteration between tumor and normal samples demonstrated a weak negative correlation with mRNA expression change (R = -0.17, P = 2.7e-11, Spearman correlation), and on average less than 15% of DIR genes were differentially expressed according to IR change (Figure 3B). Their mild correlation reflects the known function for IR to finetune gene expression through RNA degradation[3,5,10,11,39,40,41].
Mutation-induced IR has been widely reported to inactivate tumor suppressor genes (TSGs)[19], but such mutations were found in only a minority of patients. However, DIRs affected multiple patients. Although not enriched in COSMIC TSGs or oncogenes[42], DIRs were found in some cancer-related genes. For example, retention of intron 15 of LZTR1 was upregulated in COAD, STAD, and uterine corpus endometrial carcinoma (UCEC) tumor samples (Supplementary Figure S4A). LZTR1 is a TSG that negatively regulates RAS signaling through ubiquitination[43,44,45], and its minor intron retention is associated with tumorigenesis in leukemias[21]. Since the expression of LZTR1 did not significantly change between tumor and normal samples, elevated IR, implying a decreased level of normally spliced functional products. Another TSG that was affected by DIR was ERCC4 (Supplementary Figure S4B), which functions in nucleotide excision repair[46,47]. DIR was also found in oncogenes. Tumor samples of BRCA exhibited lower retention of intron 3 in CSF3R (Supplementary Figure S5), which is a highly mutated oncogene in chronic myeloid leukemia[48,49]. DIRs in different cancers could affect various biological pathways, such as DNA damage and cell cycle checkpoint in lung squamous cell carcinoma (LUSC) (Supplementary Figure S3C). These results indicate that differentially retained introns detected in multiple patients were found in genes with various biological functions including those relevant to carcinogenesis, though their functional consequences and detailed mechanisms require further study.
3.3. Sequence features of differentially retained introns
Many sequence features that are known to predispose introns to retention were also discovered in our retained introns, including higher GC content, shorter intron lengths, and biased distribution at the 3′ end of the gene, and lower likelihood of inducing NMD (Figure 3C,D; Supplementary Figure S6A). Notably, introns differentially retained in cancers were shorter compared to not regulated ones (the median intron lengths of the down-regulated, up-regulated, and unregulated IR were 456, 452, and 934 bp, respectively) (Figure 3C). On the other hand, no significant difference was observed in splice signal strength between different groups of introns (Figure 3D). This may be puzzling because many previous studies have found that retained introns usually have weaker splice sites[3,18,50]. However, Zhang and colleagues have also reported that introns differentially retained between prostate cancer and normal tissues have splice signals comparable with control introns[51]. Retained and constitutive introns demonstrated almost the same conservation within the intronic boundaries sequence, again supporting their comparable splice signals strength. However, constitutive introns had the most conserved flanking exonic boundaries, followed by down and up DIRs, indicating their higher selective pressure than unregulated retained introns (Supplementary Figure S6B).
For some cancers, up- and down-DIRs had different sequence features which may underlie different behaviors of these introns (increased or decreased retention levels) (Supplementary Figure S6). For instance, COAD and liver hepatocellular carcinoma (LIHC) had weaker 3′ splice signals in up-DIRs than down-DIRs. Differences in GC content or intron length were also observed in several cancers, such as lung adenocarcinoma (LUAD) and thyroid carcinoma (THCA).
3.4. Differential IR events have diagnostic potential
We used tSNE[26] to visualize IR patterns in over 10,000 samples from 33 TCGA cancer types. Genome-wide introns (n = 37,845) had limited tissue origin specificity and tumor vs. normal specificity (Figure 3E,F), whereas DIRs (n = 375) exhibited stronger specificity (Figure 3G,H). Similar results were seen when we restricted to 14 cancer types where DIRs were detected (Supplementary Figure S8). This suggests that DIRs may be able to distinguish tumor and normal samples, and then Random Forests[34] were applied to test IR’s potential as cancer biomarkers. After removing DIRs that were inconsistently up and downregulated in different cancer types and DIRs that had a missing rate exceeding 30% in all samples, 273 DIRs were left for model training. All TCGA samples were divided into 3 groups: 1) a training set including 1,210 paired tumor and normal samples from 14 cancers, which were the same samples used for the DIRs detection; 2) the first validation set including 5,738 unpaired tumor and normal samples from the same 14 cancers; 3) the second validation set including 3,190 samples from the rest 19 cancer types (Figure 4A). A hundred times 4-fold cross validation with the training set yielded a pooled area under curve (AUC) of 0.958, and the result of one randomly selected run was shown in Figure 4B. When the whole training set was used to train the model, the AUCs for the first and second validation sets were 0.936 and 0.838 respectively (Figure 4C,D). In addition, comparable performance can be achieved with the top 30 DIRs (Figure 4E,F). Two representative DIRs were displayed here, which were down-regulated (ZDHHC8) and up-regulated (ZNF800) in 5 cancer types respectively (Figure 4G,H). We also test the ability of DIRs as biomarkers for each cancer type, and the AUC of Random Forests models was all above 0.9 (Supplementary Figure S9). Taken together, DIRs demonstrated powerful diagnosis potential in a pan-cancer level and for a specific cancer type.
3.5. Identify prognostic IR events across cancers
We expanded our analysis to all primary tumor samples from 33 cancers in TCGA, to explore the prognosis potential of introns with varying degrees of retention in patients. Specifically, we investigated introns that had a valid IRratio for over 50% of patients within a cancer type, and the IRratio should be larger than 0.1 in at least 5% of these patients. And then univariate Cox regression was applied to patients with IRratio > 0. The results showed that 29 cancers had over 100 IR events associated with overall survival or disease-free survival (P < 0.05), of which LAML has the highest number of prognostic IR events (Figure 5A). Interestingly, prognostic introns of some cancers were predominantly associated with favorable or adverse outcomes, such as LAML and prostate adenocarcinoma (PRAD). A total of 11,522 prognostic IR events were detected among 33 cancers, of which 67.4% were specific to one cancer type, and 32.6% were shared by different cancers (Figure 5B, Supplementary Table S4).
Noteworthy, the number of prognostic IR events was not proportional to the number of genes whose expression was associated with survival (Supplementary Table S3). Moreover, only ~19% of prognostic IR genes had expression levels also associated with prognosis (Supplementary Figure S9). The discordance suggests that IR has unique prognostic features independent of gene expression. On average 42.8% of prognostic introns were negatively correlated with RNA expression across all cancer types, and 6.5% strong (r ≤ -0.5) negative association (Supplementary Figure S9).
3.6. Prognostic introns affect genes involved in tumorigenesis
Gene Ontology (GO) terms enriched for prognostic IR genes from 33 cancer types include chromatin organization, histone modification, cell cycle arrest, and GTPase activity regulation (Figure 5C). From the perspective of individual cancer types, there were 25 cancers with prognostic IR genes enriched for GO terms associated with cancer hallmarks (Figure 5D, Supplementary Table S5). Sustained angiogenesis, tissue invasion and metastasis, and genome instability and mutation were the three most frequently altered hallmarks. Prognostic IR genes in many cancer types were also enriched in KEGG pathways that are cancer related, such as the MAPK signaling pathway and PD-1 checkpoint pathway (Supplementary Figure S11). Compared with non-prognostic IR genes, prognostic IR genes in lymphoid neoplasm diffuse large B-cell lymphoma (DLBC), kidney renal papillary cell carcinoma (KIRP) and LAML were significantly enriched for COSMIC cancer genes (Supplementary Figure S12). The increased last intron retention of MYC, a famous oncogene, was associated with a favorable prognosis in BLCA and negatively correlated with expression (Supplementary Figure S13). In a less studied gene, STN1, while the mRNA level did not associate with survival in LUAD (Supplementary Figure S14A), intron retention in its 5′ UTR demonstrated a significant connection with improved survival (Figure 6A). Moreover, this intron was retained at a lower level in tumor samples (Supplementary Figure S14C). To investigate the function of this 5′ UTR intron retention, we carried out a series of experiments as below.
A high IR level in the STN1 5′ UTR was also observed in the A549 lung adenocarcinoma cell line, with a spliced transcript to IR transcript ratio of approximately 2:1 (Figure 6B,C). The IR transcripts had a similar degradation rate as the spliced transcripts, which was consistent with our observation in TCGA LUAD samples that IR levels did not correlate with mRNA levels (Figure 6D and Supplementary Figure S14B). Although IR transcripts were more frequently distributed in the nucleus than spliced transcripts, more than 50% of IR transcripts were also localized in the cytoplasm (Figure 6E). By closely examining the intronic sequence of STN1 5′ UTR, we found three potential upstream open reading frames (uORFs) as predicted by ORFinder of NCBI (Supplementary Figure S15). Because uORFs are known to reduce protein expression[52], we speculated that this IR might diminish the translation efficiency of STN1, thereby reducing its protein levels. This hypothesis was confirmed by dual luciferase assay, which demonstrated that the IR in STN1 5′ UTR significantly impeded the translation of the main ORF, and mutating all three start codons within the intron to AGC nearly restored the translation efficiency (Figure 6F and G).
Given that STN1 is a telomere replication-related gene and that cells rely on telomerase for maintaining telomere levels[53], we postulated that the cell viability may be affected by STN1 levels, which is readily influenced by its 5′ UTR IR. To validate this, we knocked down STN1 to simulate a high level of 5′ UTR IR. STN1 knockdown significantly reduced the level of proliferation markers, such as CDK1 and MKI67 in A549 cells (Figure 6H), ultimately leading to impaired colony formation, deficient cell migration, and cell proliferation (Figure 6I and J). These results provide evidence for the regulatory role of intron retention of the STN1 5′ UTR in lung adenocarcinoma cells. Through the production of uORFs, as well as more detailed transcripts in the nucleus, the 5′ UTR IR reduced translated protein levels which may ultimately affect tumor growth and progression.
3.7. IR enables accurate risk stratification in multiple cancers
The current standard of care assessment of many cancers relies on clinical biomarkers and the detection of specific genetic mutations or chromosomal structural variation. Take acute myeloid leukemia, for example, cytogenetic screening and target gene sequencing-based stratification is effective for half of LAML patients. The rest of the patients are difficult to assign to a defined risk group since they are cytogenetically normal and do not carry mutations in known risk genes[54]. As a result, revising prognostic models using additional molecular features to improve stratification is an ongoing effort for LAML as well as other cancers.
Because LAML has the highest number of prognostic introns, we first tried to build an IR prognostic model with the least absolute shrinkage and selection operator (LASSO) regression[36] for LAML. The resultant model was made up of 4 IR signatures and was designated as IR risk (IRR) (Figure 7A, Supplementary Figure S16). Based on the IRR score, we can divide LAML patients from TCGA into two risk groups with significantly diverged overall survival outcomes (HR = 5.24, P = 1.36e-10, Figure 7B). We used univariate Cox regression to compare the contribution of IRR and other reported clinical and molecular predictors[54,55], and IRR turned out to be the most powerful predictor (Figure 7C, Supplementary Table S6). Although age over 60 at diagnosis is a known risk for LAML, IRR still enabled effective stratification in patients under or over 60 years old (Figure 7D). Similarly, we were able to construct a prognostic model based on 4~30 IR events with LASSO regression for 32 cancer types, except for testicular germ cell tumors (TGCT), achieving good risk stratification (Figure 8). This demonstrates the potential of IR as powerful prognostic markers for many cancers.
4. Discussion
Aberrant AS is one of the hallmarks of cancer, and tumor tissues commonly have about 30% more AS events than normal tissues[13]. By producing tumor-specific proteins or by altering the production of normal proteins, aberrant AS can lead to the activation of proto-oncogenes or inactivation of TSGs, ultimately affecting cell growth and differentiation, angiogenesis, tissue invasion, and metastasis[56]. Therefore, the study of aberrant splicing not only helps to understand the mechanisms of cancer initiation and development but also has potential clinical implications[57]. There is an intriguing imbalanced IR pattern in multiple cancers where tumors exhibit a significant increase in IR compared to normal samples, which is not observed in other AS types[18]. IR may contribute to cancer development by inactivating TSGs in cancer patients[19]. Moreover, IR can encode novel proteins and may be an important source of tumor-specific antigens[22]. In this study, we systematically quantified and profiled IRs in 33 cancers from TCGA and tentatively explored their clinical relevance. So far as we know, this is a comprehensive analysis of IR regarding the largest number of cancer types.
We identified differential intron retention events (DIRs) by comparing paired tumor and normal tissues in multiple cancer types. We found that the splicing signals of differentially retained introns were almost as strong as constitutive introns, which was consistent with Zhang et al.’s finding[51]. One possible explanation is that DIRs were recurrent in tumor and/or normal samples, and they may have similar biological importance as constitutive introns. Interestingly, DIRs were shorter than both constitutive introns and unregulated retained introns. Zhang et al. have reported that shorter exons are more likely to be excluded in cancers, and are possibly regulated by elevated transcription and dysregulation of some RBPs in cancer cells[58]. In addition, most genes are spliced co-transcriptionally[59], and increased RNA Pol II accumulation in retained introns has also been reported[3,60]. Whether shorter introns are more sensitive to transcription and other splicing alterations in cancer deserves further study.
Compared with adjacent normal tissues, dozens to hundreds of introns per cancer showed up or down-regulation in tumor tissues, resulting in a total of 988 DIRs across 14 cancer types. Some of them were cancer-type specific (such as CSF3R), while some others (such as LZTR1 and ERCC4) showed consistent alterations across multiple cancers. We further identified 30 DIRs that stratified tumor samples and normal samples, demonstrating their diagnostic potential. A recent study exerted intron splicing events generated by SF3B1 mutations that are specific to tumor patients, to design synthetic introns and achieve targeted clearance of tumor cells [61]. Similarly, DIRs in our study may also serve as promising candidates for therapeutic synthetic introns, since they were widespread in multiple patients and even in multiple cancers (e.g., IR of ZDHHC8 in Figure 4G). On the other hand, because retained introns can also encode peptides located on cancer cell surfaces[22], commonly retained introns may be potential targets for “off-the-shelf” immunotherapy.
We discovered several introns with the potential to be survival indicators. For example, we experimentally validated a functional prognostic intron in LUAD. Specifically, the 5′ UTR intron regulates the translation efficiency of STN1, which is essential for cancer cell proliferation through maintaining telomere replication and genome stability (Supplementary Figure S17). These insightful results also indicate potential novel therapeutic strategies to combat LUAD.
There have been ongoing efforts in exploring new biomarkers for risk stratification in cancers, such as gene expression[55,62,63,64,65]. However, splicing has been reported to outperform gene expression analysis in predicting survival in multiple tumor types[66,67]. The potential of alternative splicing (AS) in prognosis has also been demonstrated in various cancers, including ovarian cancer[68], colorectal cancer[69], lung cancer[70], esophageal cancer[71], liver cancer[72] and adrenocortical carcinoma[73]. We explored the prognostic power of IR in 33 cancer types, and most of the cancers (n=32) can be stratified with less than 30 introns. The performance of the IR-based prognostic model in TCGA LAML cohorts outperformed clinical and other molecular predictors. IR has been reported to indicate prostate cancer aggressiveness[51] and pancreatic cancer clinical outcomes[66,74]. Our results further suggest that IR can serve as an accurate and powerful biomarker in multiple cancers. Moreover, IR (including other AS types) is usually quantified in a relative rather than an absolute method, which may be less influenced by different processing procedures.
DIRs and prognostic IR events were two types of informative IR characterized in this study. Differentially retained introns across many cancer types were heterogenous and influenced genes of various functional categories. Prognostic introns affected genes involved in cancer-related pathways, including DNA damage and cell cycle regulation, angiogenesis, cancer cell invasion and metastasis. In DLBC, KIRP and LAML, prognostic IR genes were significantly enriched for COSMIC cancer genes. IR has been recognized as a widespread mechanism of TSG inactivation[19], and we found that IR may also regulate the activity of oncogenes, such as MYC (Supplementary Figure S13). Of note, one of the limitations of our study is that we still lack experimental assessments of the IR functions, since IR transcripts have diverse fates[12]. The second limitation is that only a few patients in TCGA dataset have matched normal tissues. More normal samples as background or negative controls will add to finding more informative IR events in future analysis.
6. Conclusions
Overall, we systematically characterized IR at a pan-cancer level, explored its clinical relevance and provided experimental evidence of IR in cancer pathology. Our results revealed a rich resource for IR that had potential clinical applications including cancer biomarkers and even therapeutic targets.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Acknowledgments
This work is financially supported by the National Key R&D Program of China (2021YFA0909300), the National Natural Science Foundation of China (91949107), and the Natural Science Foundation Project of Shanghai (21ZR1407000).
Conflicts of Interest
The authors declare no conflict of interest.
Data accessibility
This study used TCGA RNA-seq dataset (https://portal.gdc.cancer.gov/repository).
References
- Johnson JM, Castle J, Garrett-Engele P, Kan Z, Loerch PM, Armour CD, Santos R, Schadt EE, Stoughton R, Shoemaker DD: Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science 2003, 302:2141-2144. [CrossRef]
- Middleton R, Gao D, Thomas A, Singh B, Au A, Wong JJ, Bomane A, Cosson B, Eyras E, Rasko JE, Ritchie W: IRFinder: assessing the impact of intron retention on mammalian gene expression. Genome Biol 2017, 18:51. [CrossRef]
- Braunschweig U, Barbosa-Morais NL, Pan Q, Nachman EN, Alipanahi B, Gonatopoulos-Pournatzis T, Frey B, Irimia M, Blencowe BJ: Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res 2014, 24:1774-1786.. [CrossRef]
- Jacob AG, Smith CWJ: Intron retention as a component of regulated gene expression programs. Hum Genet 2017, 136:1043-1057. [CrossRef]
- Ni T, Yang W, Han M, Zhang Y, Shen T, Nie H, Zhou Z, Dai Y, Yang Y, Liu P, et al: Global intron retention mediated gene regulation during CD4+ T cell activation. Nucleic Acids Res 2016, 44:6817-6829.. [CrossRef]
- Gontijo AM, Miguela V, Whiting MF, Woodruff RC, Dominguez M: Intron retention in the Drosophila melanogaster Rieske Iron Sulphur Protein gene generated a new protein. Nat Commun 2011, 2:323. [CrossRef]
- Bell TJ, Miyashiro KY, Sul JY, Buckley PT, Lee MT, McCullough R, Jochems J, Kim J, Cantor CR, Parsons TD, Eberwine JH: Intron retention facilitates splice variant diversity in calcium-activated big potassium channel populations. Proc Natl Acad Sci U S A 2010, 107:21152-21157.
- Bell TJ, Miyashiro KY, Sul JY, McCullough R, Buckley PT, Jochems J, Meaney DF, Haydon P, Cantor C, Parsons TD, Eberwine J: Cytoplasmic BKCa channel intron-containing mRNAs contribute to the intrinsic excitability of hippocampal neurons. Proc Natl Acad Sci U S A 2008, 105:1901-1906.. [CrossRef]
- Buckley PT, Lee MT, Sul JY, Miyashiro KY, Bell TJ, Fisher SA, Kim J, Eberwine J: Cytoplasmic intron sequence-retaining transcripts can be dendritically targeted via ID element retrotransposons. Neuron 2011, 69:877-884. [CrossRef]
- Wong JJL, Ritchie W, Ebner OA, Selbach M, Wong JWH, Huang YZ, Gao DD, Pinello N, Gonzalez M, Baidya K, et al: Orchestrated intron retention regulates normal granulocyte differentiation. Cell 2013, 154:583-595. [CrossRef]
- Yap K, Lim ZQ, Khandelia P, Friedman B, Makeyev EV: Coordinated regulation of neuronal mRNA steady-state levels through developmentally controlled intron retention. Genes Dev 2012, 26:1209-1223.
- Monteuuis G, Wong JJL, Bailey CG, Schmitz U, Rasko JEJ: The changing paradigm of intron retention: regulation, ramifications and recipes. Nucleic Acids Res 2019, 47:11497-11513.. [CrossRef]
- Kahles A, Lehmann KV, Toussaint NC, Huser M, Stark SG, Sachsenberg T, Stegle O, Kohlbacher O, Sander C, Cancer Genome Atlas Research N, Ratsch G: Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer Cell 2018, 34:211-224 e216.. [CrossRef]
- Oltean S, Bates DO: Hallmarks of alternative splicing in cancer. Oncogene 2014, 33:5311-5318.
- Okumura N, Yoshida H, Kitagishi Y, Nishimura Y, Matsuda S: Alternative splicings on p53, BRCA1 and PTEN genes involved in breast cancer. Biochem Biophys Res Commun 2011, 413:395-399. [CrossRef]
- Sebestyen E, Zawisza M, Eyras E: Detection of recurrent alternative splicing switches in tumor samples reveals novel signatures of cancer. Nucleic Acids Res 2015, 43:1345-1356. [CrossRef]
- Rossi A, Kontarakis Z: Beyond Mendelian Inheritance: Genetic Buffering and Phenotype Variability. Phenomics 2022, 2:79-87. [CrossRef]
- Dvinge H, Bradley RK: Widespread intron retention diversifies most cancer transcriptomes. Genome Med 2015, 7:45. [CrossRef]
- Jung H, Lee D, Lee J, Park D, Kim YJ, Park WY, Hong D, Park PJ, Lee E: Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nat Genet 2015, 47:1242-1248.
- Yuan H, Li N, Fu D, Ren J, Hui J, Peng J, Liu Y, Qiu T, Jiang M, Pan Q, et al: Histone methyltransferase SETD2 modulates alternative splicing to inhibit intestinal tumorigenesis. J Clin Invest 2017, 127:3375-3391. [CrossRef]
- Inoue D, Polaski JT, Taylor J, Castel P, Chen S, Kobayashi S, Hogg SJ, Hayashi Y, Pineda JMB, El Marabti E, et al: Minor intron retention drives clonal hematopoietic disorders and diverse cancer predisposition. Nat Genet 2021, 53:707-718.. [CrossRef]
- Smart AC, Margolis CA, Pimentel H, He MX, Miao D, Adeegbe D, Fugmann T, Wong KK, Van Allen EM: Intron retention is a source of neoepitopes in cancer. Nat Biotechnol 2018, 36:1056-1058. [CrossRef]
- Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL: StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 2015, 33:290-295.. [CrossRef]
- Broseus L, Ritchie W: Challenges in detecting and quantifying intron retention from next generation sequencing data. Comput Struct Biotechnol J 2020, 18:501-508. [CrossRef]
- Love MI, Huber W, Anders S: Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014, 15:550. [CrossRef]
- van der Maaten L, Hinton G: Visualizing data using t-SNE. Journal of Machine Learning Research 2008, 9:2579-2605.
- Yu GC, Wang LG, Han YY, He QY: clusterProfiler: an R package for comparing biological themes among gene clusters. Omics-a Journal of Integrative Biology 2012, 16:284-287. [CrossRef]
- Plaisier CL, Pan M, Baliga NS: A miRNA-regulatory network explains how dysregulated miRNAs perturb oncogenic processes across diverse cancers. Genome Res 2012, 22:2302-2314. [CrossRef]
- Li YS, Sahni N, Pancsa R, McGrail DJ, Xu J, Hua X, Coulombe-Huntington J, Ryan M, Tychhon B, Sudhakar D, et al: Revealing the determinants of widespread alternative splicing perturbation in cancer. Cell Rep 2017, 21:798-812.. [CrossRef]
- Pohl A, Beato M: bwtool: a tool for bigWig files. Bioinformatics 2014, 30:1618-1619.
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou MM, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al: Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 2005, 15:1034-1050. [CrossRef]
- Yeo G, Burge CB: Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 2004, 11:377-394. [CrossRef]
- Lindeboom RG, Supek F, Lehner B: The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat Genet 2016, 48:1112-1118. [CrossRef]
- Breiman L: Random forests. Machine Learning 2001, 45:5-32.
- Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, Muller M: pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 2011, 12:77.. [CrossRef]
- Friedman J, Hastie T, Tibshirani R: Regularization paths for generalized linear models via coordinate descent. J Stat Softw 2010, 33:1-22.
- Simon N, Friedman J, Hastie T, Tibshirani R: Regularization paths for Cox’s proportional hazards model via coordinate descent. Journal of Statistical Software 2011, 39:1-13. [CrossRef]
- Yao J, Ding D, Li X, Shen T, Fu H, Zhong H, Wei G, Ni T: Prevalent intron retention fine-tunes gene expression and contributes to cellular senescence. Aging Cell 2020, 19.. [CrossRef]
- Jaillon O, Bouhouche K, Gout JF, Aury JM, Noel B, Saudemont B, Nowacki M, Serrano V, Porcel BM, Segurens B, et al: Translational control of intron splicing in eukaryotes. Nature 2008, 451:359-362.
- Gudipati RK, Xu Z, Lebreton A, Seraphin B, Steinmetz LM, Jacquier A, Libri D: Extensive degradation of RNA precursors by the exosome in wild-type cells. Mol Cell 2012, 48:409-421. [CrossRef]
- Pimentel H, Parra M, Gee SL, Mohandas N, Pachter L, Conboy JG: A dynamic intron retention program enriched in RNA processing genes regulates gene expression during terminal erythropoiesis. Nucleic Acids Res 2016, 44:838-851. [CrossRef]
- Sondka Z, Bamford S, Cole CG, Ward SA, Dunham I, Forbes SA: The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nature Reviews Cancer 2018, 18:696-705. [CrossRef]
- Piotrowski A, Xie J, Liu YF, Poplawski AB, Gomes AR, Madanecki P, Fu C, Crowley MR, Crossman DK, Armstrong L, et al: Germline loss-of-function mutations in LZTR1 predispose to an inherited disorder of multiple schwannomas. Nat Genet 2014, 46:182-+. [CrossRef]
- Paganini I, Chang VY, Capone GL, Vitte J, Benelli M, Barbetti L, Sestini R, Trevisson E, Hulsebos TJ, Giovannini M, et al: Expanding the mutational spectrum of LZTR1 in schwannomatosis. Eur J Hum Genet 2015, 23:963-968.
- Bigenzahn JW, Collie GM, Kartnig F, Pieraks M, Vladimer GI, Heinez LX, Sedlyarov V, Schischlik F, Fauster A, Rebsamen M, et al: LZTR1 is a regulator of RAS ubiquitination and signaling. Science 2018, 362:1171-+. [CrossRef]
- Koberle B, Ditz C, Kausch I, Wollenberg B, Ferris RL, Albers AE: Metastases of squamous cell carcinoma of the head and neck show increased levels of nucleotide excision repair protein XPF in vivo that correlate with increased chemoresistance ex vivo. Int J Oncol 2010, 36:1277-1284. [CrossRef]
- Manandhar M, Boulware KS, Wood RD: The ERCC1 and ERCC4 (XPF) genes and gene products. Gene 2015, 569:153-161.
- Maxson JE, Gotlib J, Pollyea DA, Fleischman AG, Agarwal A, Eide CA, Bottomly D, Wilmot B, McWeeney SK, Tognon CE, et al: Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med 2013, 368:1781-1790. [CrossRef]
- Maxson JE, Luty SB, MacManiman JD, Paik JC, Gotlib J, Greenberg P, Bahamadi S, Savage SL, Abel ML, Eide CA, et al: The colony-stimulating factor 3 receptor T64ON mutation is oncogenic, sensitive to JAK inhibition, and mimics T618I. Clin Cancer Res 2016, 22:757-764. [CrossRef]
- Sakabe NJ, de Souza SJ: Sequence features responsible for intron retention in human. BMC Genomics 2007, 8:59. [CrossRef]
- Zhang D, Hu Q, Liu X, Ji Y, Chao HP, Liu Y, Tracz A, Kirk J, Buonamici S, Zhu P, et al: Intron retention is a hallmark and spliceosome represents a therapeutic vulnerability in aggressive prostate cancer. Nat Commun 2020, 11:2089. [CrossRef]
- Calvo SE, Pagliarini DJ, Mootha VK: Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc Natl Acad Sci U S A 2009, 106:7507-7512. [CrossRef]
- Lu H, Lei Z, Lu Z, Lu Q, Lu C, Chen W, Wang C, Tang Q, Kong Q: Silencing tankyrase and telomerase promotes A549 human lung adenocarcinoma cell apoptosis and inhibits proliferation. Oncol Rep 2013, 30:1745-1752.
- Dohner H, Weisdorf DJ, Bloomfield CD: Acute myeloid leukemia. N Engl J Med 2015, 373:1136-1152.
- Docking TR, Parker JDK, Jadersten M, Duns G, Chang L, Jiang J, Pilsworth JA, Swanson LA, Chan SK, Chiu R, et al: A clinical transcriptome approach to patient stratification and therapy selection in acute myeloid leukemia. Nature Communications 2021, 12. [CrossRef]
- Sveen A, Kilpinen S, Ruusulehto A, Lothe RA, Skotheim RI: Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene 2016, 35:2413-2427.. [CrossRef]
- Singh B, Eyras E: The role of alternative splicing in cancer. Transcription 2017, 8:91-98.
- Zhang S, Mao M, Lv Y, Yang Y, He W, Song Y, Wang Y, Yang Y, Al Abo M, Freedman JA, et al: A widespread length-dependent splicing dysregulation in cancer. Sci Adv 2022, 8:eabn9232. [CrossRef]
- Bentley DL: Coupling mRNA processing with transcription in time and space. Nat Rev Genet 2014, 15:163-175. [CrossRef]
- Wong JJ, Gao D, Nguyen TV, Kwok CT, van Geldermalsen M, Middleton R, Pinello N, Thoeng A, Nagarajah R, Holst J, et al: Intron retention is regulated by altered MeCP2-mediated splicing factor recruitment. Nat Commun 2017, 8:15134.
- North K, Benbarche S, Liu B, Pangallo J, Chen S, Stahl M, Bewersdorf JP, Stanley RF, Erickson C, Cho H, et al: Synthetic introns enable splicing factor mutation-dependent targeting of cancer cells. Nat Biotechnol 2022, 40:1103-1113.
- Gentles AJ, Plevritis SK, Majeti R, Alizadeh AA: Association of a leukemic stem cell gene expression signature with clinical outcomes in acute myeloid leukemia. JAMA 2010, 304:2706-2715. [CrossRef]
- He L, Chen J, Xu F, Li J, Li J: Prognostic Implication of a Metabolism-Associated Gene Signature in Lung Adenocarcinoma. Mol Ther Oncolytics 2020, 19:265-277. [CrossRef]
- Lou S, Meng F, Yin X, Zhang Y, Han B, Xue Y: Comprehensive Characterization of RNA Processing Factors in Gastric Cancer Identifies a Prognostic Signature for Predicting Clinical Outcomes and Therapeutic Responses. Front Immunol 2021, 12:719628. [CrossRef]
- Shi R, Bao X, Unger K, Sun J, Lu S, Manapov F, Wang X, Belka C, Li M: Identification and validation of hypoxia-derived gene signatures to predict clinical outcomes and therapeutic responses in stage I lung adenocarcinoma patients. Theranostics 2021, 11:5061-5076.
- Tan DJ, Mitra M, Chiu AM, Coller HA: Intron retention is a robust marker of intertumoral heterogeneity in pancreatic ductal adenocarcinoma. Npj Genomic Medicine 2020, 5.. [CrossRef]
- Shen S, Wang Y, Wang C, Wu YN, Xing Y: SURVIV for survival analysis of mRNA isoform variation. Nat Commun 2016, 7:11548. [CrossRef]
- Zhu J, Chen Z, Yong L: Systematic profiling of alternative splicing signature reveals prognostic predictor for ovarian cancer. Gynecol Oncol 2018, 148:368-374. [CrossRef]
- Xiong YF, Deng Y, Wang K, Zhou H, Zheng XR, Si LY, Fu ZX: Profiles of alternative splicing in colorectal cancer and their clinical significance: A study based on large-scale sequencing data. Ebiomedicine 2018, 36:183-195.
- Li Y, Sun N, Lu ZL, Sun SG, Huang JB, Chen ZL, He J: Prognostic alternative mRNA splicing signature in non-small cell lung cancer. Cancer Lett 2017, 393:40-51. [CrossRef]
- Mao S, Li Y, Lu Z, Che Y, Sun S, Huang J, Lei Y, Wang X, Liu C, Zheng S, et al: Survival-associated alternative splicing signatures in esophageal carcinoma. Carcinogenesis 2019, 40:121-130. [CrossRef]
- Zhu GQ, Zhou YJ, Qiu LX, Wang B, Yang Y, Liao WT, Luo YH, Shi YH, Zhou J, Fan J, Dai Z: Prognostic alternative mRNA splicing signature in hepatocellular carcinoma: a study based on large-scale sequencing data. Carcinogenesis 2019, 40:1077-1085.
- Xu W, Anwaier A, Liu W, Tian X, Zhu WK, Wang J, Qu Y, Zhang H, Ye D: Systematic Genome-Wide Profiles Reveal Alternative Splicing Landscape and Implications of Splicing Regulator DExD-Box Helicase 21 in Aggressive Progression of Adrenocortical Carcinoma. Phenomics 2021, 1:243-256. [CrossRef]
- Dong C, Reiter JL, Dong E, Wang Y, Lee KP, Lu X, Liu Y: Intron-retention neoantigen load predicts favorable prognosis in pancreatic cancer. JCO Clin Cancer Inform 2022, 6:e2100124. [CrossRef]
Figure 1.
Schematic overview of data set and analysis. We downloaded over 10,000 RNA-seq samples originating from 33 cancer types from the TCGA data portal and detected genome-wide intron retention events for each sample. Differential analysis between adjacent normal tissues and cancerous tissues was conducted to find differential IR events, and survival analysis was conducted to find prognostic IR events. We demonstrated that some informative introns were potential diagnostic or prognostic biomarkers with machine learning methods. Importantly, our molecular and functional experiments validated that IR plays a role in cancer pathology.
Figure 1.
Schematic overview of data set and analysis. We downloaded over 10,000 RNA-seq samples originating from 33 cancer types from the TCGA data portal and detected genome-wide intron retention events for each sample. Differential analysis between adjacent normal tissues and cancerous tissues was conducted to find differential IR events, and survival analysis was conducted to find prognostic IR events. We demonstrated that some informative introns were potential diagnostic or prognostic biomarkers with machine learning methods. Importantly, our molecular and functional experiments validated that IR plays a role in cancer pathology.
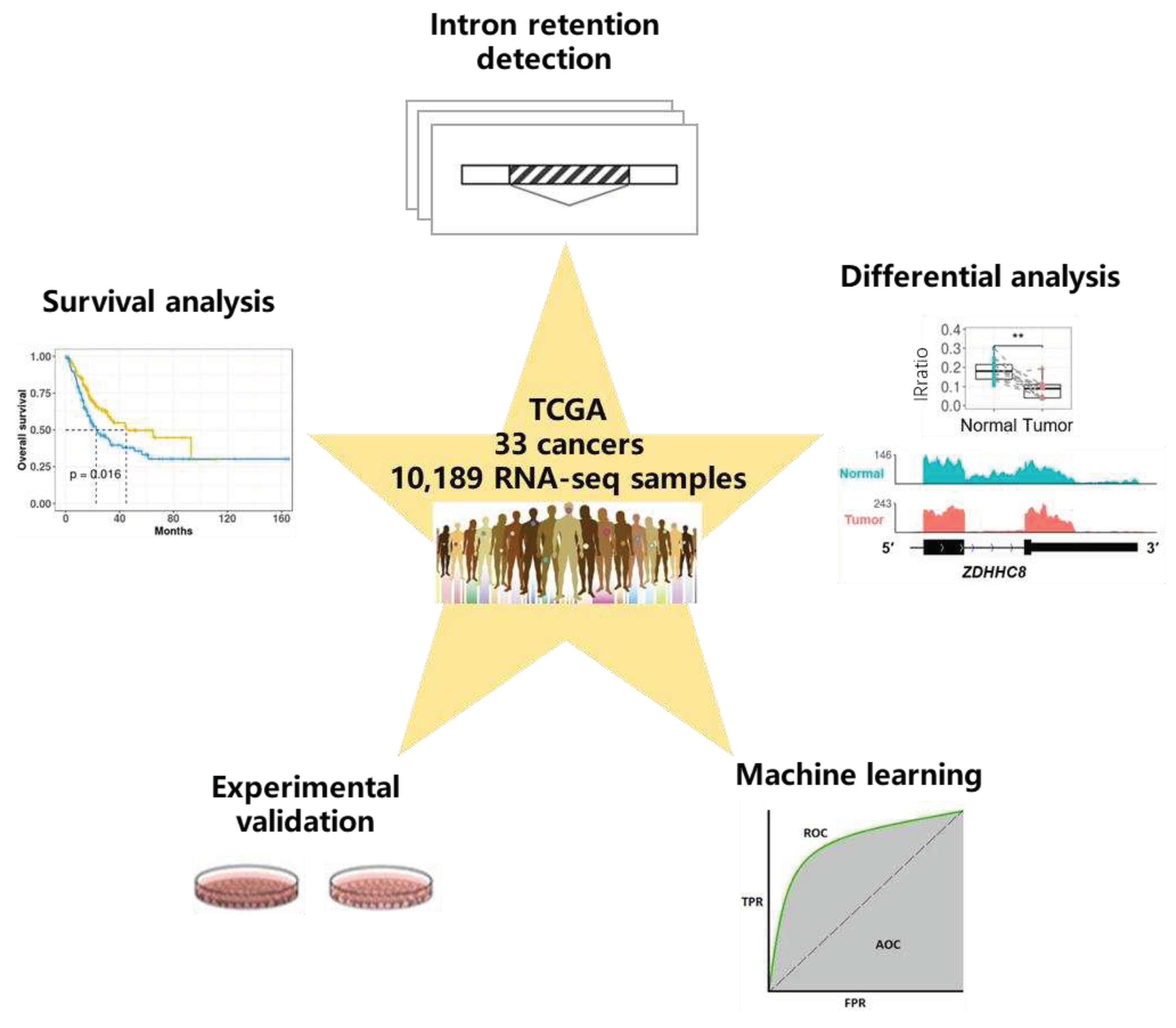
Figure 2.
Overview of intron retention across 33 cancer types. (A) Sample statistics for a total of 10,189 samples from 33 cancer types. Only samples that passed IRFinder QC were analyzed. (B) Hierarchical clustering of median IRratio across cancer types based on pair-wise Spearman correlation. (C) The average number of retained introns (IRratio > 0.1) in tumor and normal samples for each cancer type. Some cancers lack normal samples, and as a result, only tumor samples were shown. Error bars indicate standard deviation. *, P < 0.05; **, P < 0.01; ***, P < 0.001; two-tailed student’s t test.
Figure 2.
Overview of intron retention across 33 cancer types. (A) Sample statistics for a total of 10,189 samples from 33 cancer types. Only samples that passed IRFinder QC were analyzed. (B) Hierarchical clustering of median IRratio across cancer types based on pair-wise Spearman correlation. (C) The average number of retained introns (IRratio > 0.1) in tumor and normal samples for each cancer type. Some cancers lack normal samples, and as a result, only tumor samples were shown. Error bars indicate standard deviation. *, P < 0.05; **, P < 0.01; ***, P < 0.001; two-tailed student’s t test.
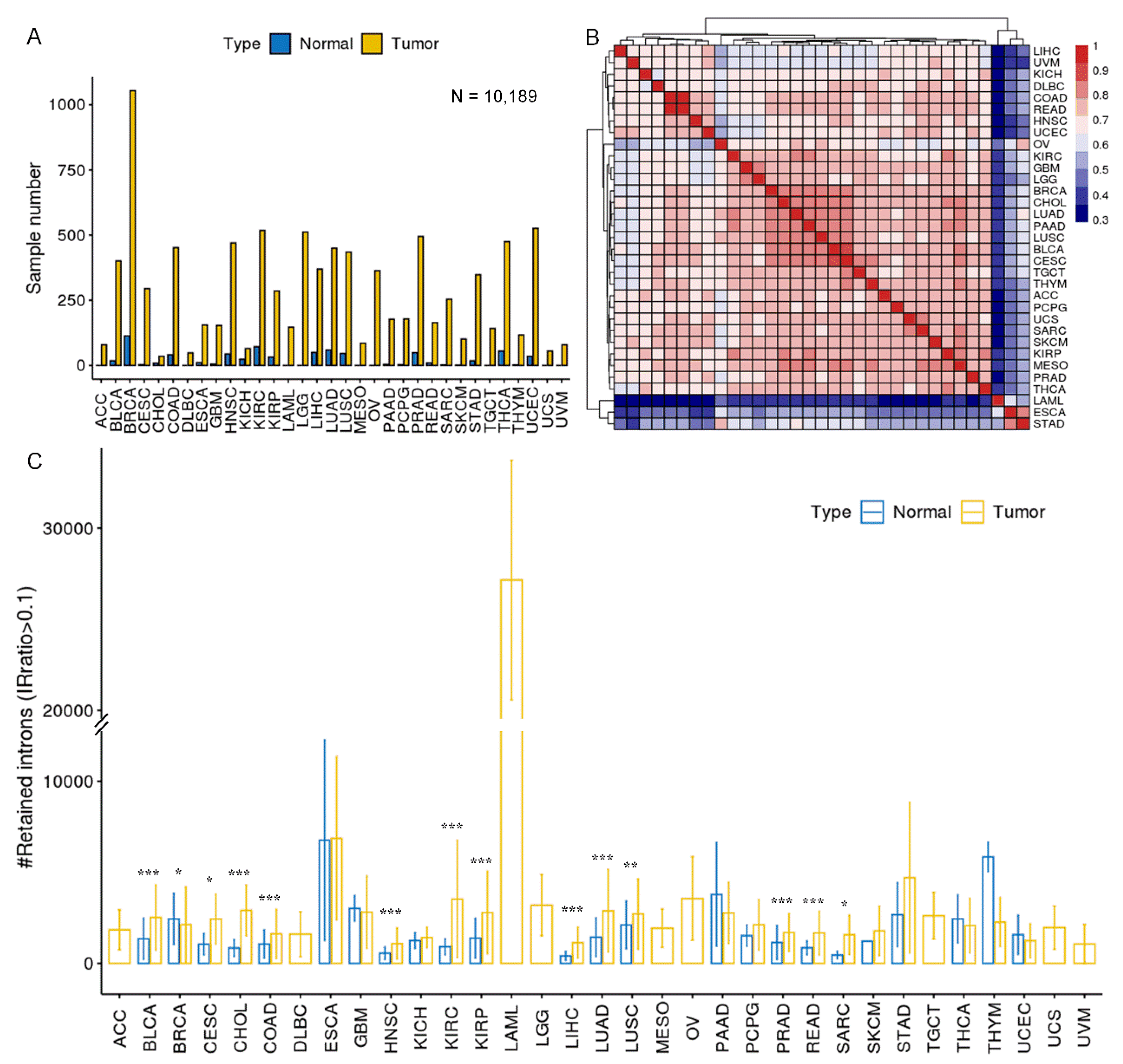
Figure 3.
Differential IR events between tumor and adjacent normal samples.(A) Statistics of up and down-regulated differential IR events (DIRs) in 14 cancer types. Cancer associated IR referred to introns that tend to be spliced in normal samples (median IRratio = 0) and retained in tumor samples (median IRratio > 0.1). The opposite standed for normal associated IR. (B) The relationship between DIRs and gene expression changes. The left panel showed a comparison of median IRratio change and corresponding mRNA fold change between tumor and normal samples. Grey area masked genes whose expression fold change were between 1/2 and 2. The right panel shows the proportion of differentially expressed genes that were anti-concordant with IR change (i.e. expression increases and IR decreases, and vice versa). (C) Length distribution of four types of introns. ***, P < 0.001, two-tailed Mann–Whitney U test. (D) Comparison of GC content, relative position in genes (intron number divided by the total number of introns) and splice signals across four types of introns. (E, F) tSNE plot of genome-wide introns (n = 37,845 after missing rate filter) colored by cancer types (E) or by tumor or normal sample (F). (G, H) tSNE plot of DIRs (n = 375 after missing rate filter) colored by cancer types (G) or by tumor or normal sample (H).
Figure 3.
Differential IR events between tumor and adjacent normal samples.(A) Statistics of up and down-regulated differential IR events (DIRs) in 14 cancer types. Cancer associated IR referred to introns that tend to be spliced in normal samples (median IRratio = 0) and retained in tumor samples (median IRratio > 0.1). The opposite standed for normal associated IR. (B) The relationship between DIRs and gene expression changes. The left panel showed a comparison of median IRratio change and corresponding mRNA fold change between tumor and normal samples. Grey area masked genes whose expression fold change were between 1/2 and 2. The right panel shows the proportion of differentially expressed genes that were anti-concordant with IR change (i.e. expression increases and IR decreases, and vice versa). (C) Length distribution of four types of introns. ***, P < 0.001, two-tailed Mann–Whitney U test. (D) Comparison of GC content, relative position in genes (intron number divided by the total number of introns) and splice signals across four types of introns. (E, F) tSNE plot of genome-wide introns (n = 37,845 after missing rate filter) colored by cancer types (E) or by tumor or normal sample (F). (G, H) tSNE plot of DIRs (n = 375 after missing rate filter) colored by cancer types (G) or by tumor or normal sample (H).
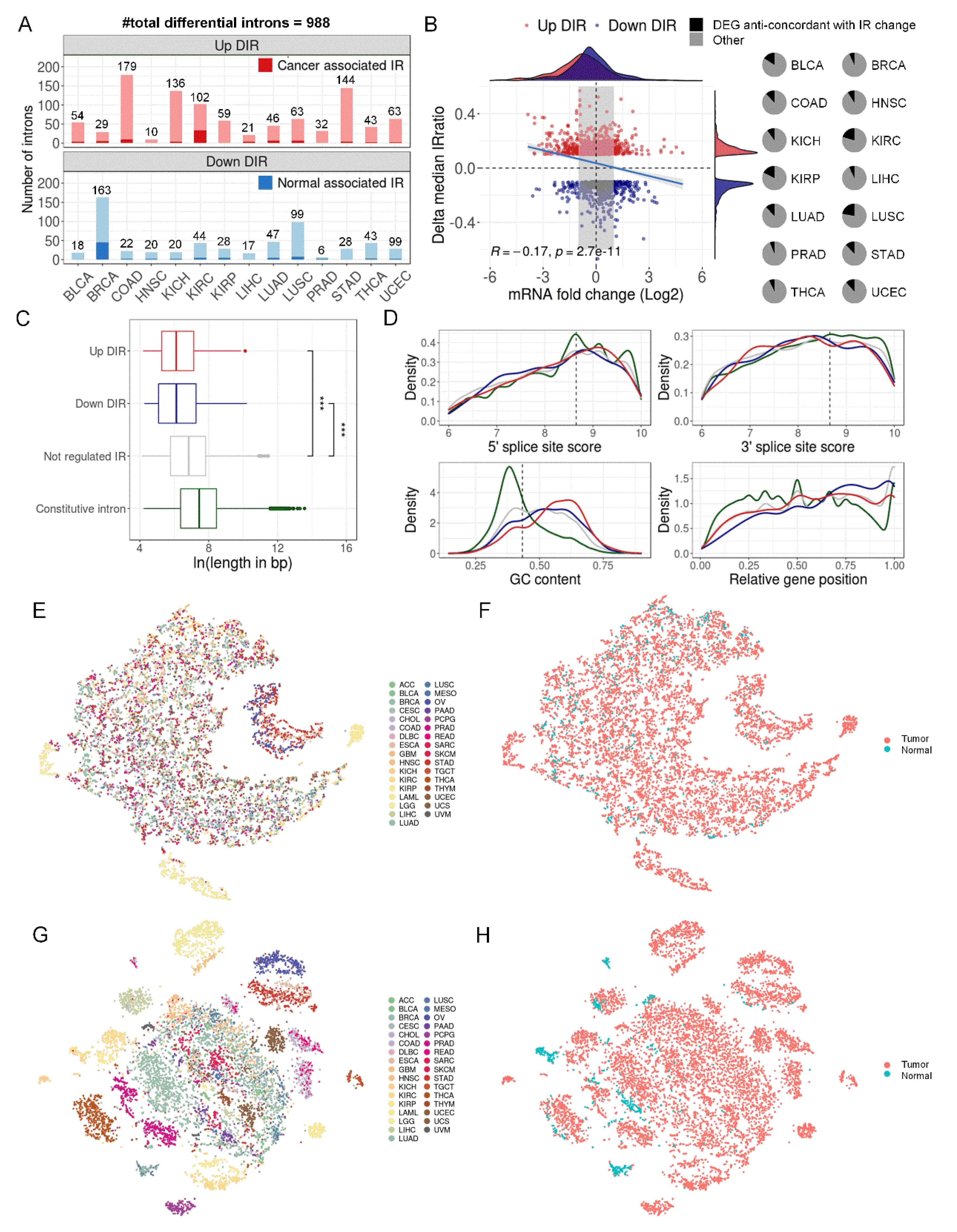
Figure 4.
Differential introns can distinguish tumors from normal samples. (A) TCGA samples were divided into three groups: paired tumor and normal samples in 14 cancer types that were previously used to detect DIRs (training set for Random Forests model, n = 1,210), unpaired samples in the same 14 cancer types (validation set 1, n = 5,789), and all samples from 19 other cancer types (validation set 2, n = 3,190). (B) Random Forests model performance in training set in a 4-fold cross validation run. The upper right panel showed the sample distribution. The left panel showed the ROC of the model and the bottom right panel shows the confusion matrix. (C, D) Random Forests model performance in validation set 1 (C) and set 2 (D) when 1,210 paired samples in 14 cancers were all used to train the model. (E) Five-fold cross-validated prediction performance of models in training set with a sequentially reduced number of DIR (ranked by importance). (F) Top 30 DIRs that contributed most to model accuracy and their alteration in 14 cancer types. Blue and red standed for down and up-regulated introns, respectively. (G, H) ZDHHC8 (G) and ZNF800 (H) last introns were differentially retained in paired tumor (red) and normal (blue) samples in 5 cancer types. The left panels were IRratio boxplots and the right panels were RNA-seq read coverage from example patients. *, P < 0.05; ***, P < 0.001; paired Wilcoxon rank-sum test.
Figure 4.
Differential introns can distinguish tumors from normal samples. (A) TCGA samples were divided into three groups: paired tumor and normal samples in 14 cancer types that were previously used to detect DIRs (training set for Random Forests model, n = 1,210), unpaired samples in the same 14 cancer types (validation set 1, n = 5,789), and all samples from 19 other cancer types (validation set 2, n = 3,190). (B) Random Forests model performance in training set in a 4-fold cross validation run. The upper right panel showed the sample distribution. The left panel showed the ROC of the model and the bottom right panel shows the confusion matrix. (C, D) Random Forests model performance in validation set 1 (C) and set 2 (D) when 1,210 paired samples in 14 cancers were all used to train the model. (E) Five-fold cross-validated prediction performance of models in training set with a sequentially reduced number of DIR (ranked by importance). (F) Top 30 DIRs that contributed most to model accuracy and their alteration in 14 cancer types. Blue and red standed for down and up-regulated introns, respectively. (G, H) ZDHHC8 (G) and ZNF800 (H) last introns were differentially retained in paired tumor (red) and normal (blue) samples in 5 cancer types. The left panels were IRratio boxplots and the right panels were RNA-seq read coverage from example patients. *, P < 0.05; ***, P < 0.001; paired Wilcoxon rank-sum test.
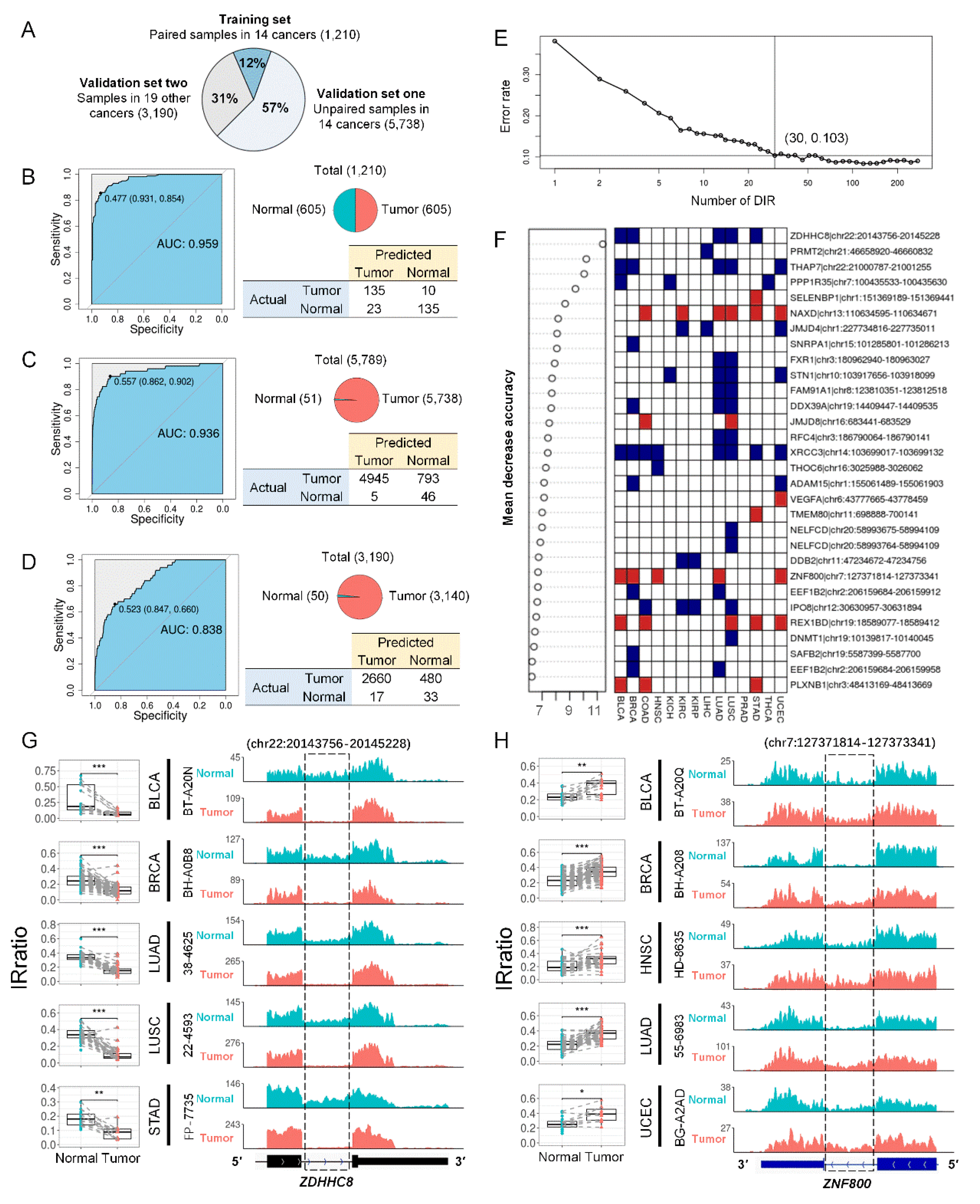
Figure 5.
Statistics and functional enrichment of prognostic introns. (A) Statistics of prognostic introns. The upper panel showed the number of prognostic introns for each cancer type. The bottom panel showed the percentage of prognostic introns associated with longer (favorable prognosis, blue) or shorter (adverse prognosis, red) survival for each cancer type. (B) Proportion of two types of prognostic introns. (C) GO term enrichment of total prognostic IR genes. (D) Enrichment of prognostic IR genes in each cancer for GO terms associated with cancer hallmarks. GO terms (rows) were sorted according to cancer hallmarks on the left. Cell color indicated P values (hypergeometric test; white cells indicate P values larger than 0.1).
Figure 5.
Statistics and functional enrichment of prognostic introns. (A) Statistics of prognostic introns. The upper panel showed the number of prognostic introns for each cancer type. The bottom panel showed the percentage of prognostic introns associated with longer (favorable prognosis, blue) or shorter (adverse prognosis, red) survival for each cancer type. (B) Proportion of two types of prognostic introns. (C) GO term enrichment of total prognostic IR genes. (D) Enrichment of prognostic IR genes in each cancer for GO terms associated with cancer hallmarks. GO terms (rows) were sorted according to cancer hallmarks on the left. Cell color indicated P values (hypergeometric test; white cells indicate P values larger than 0.1).
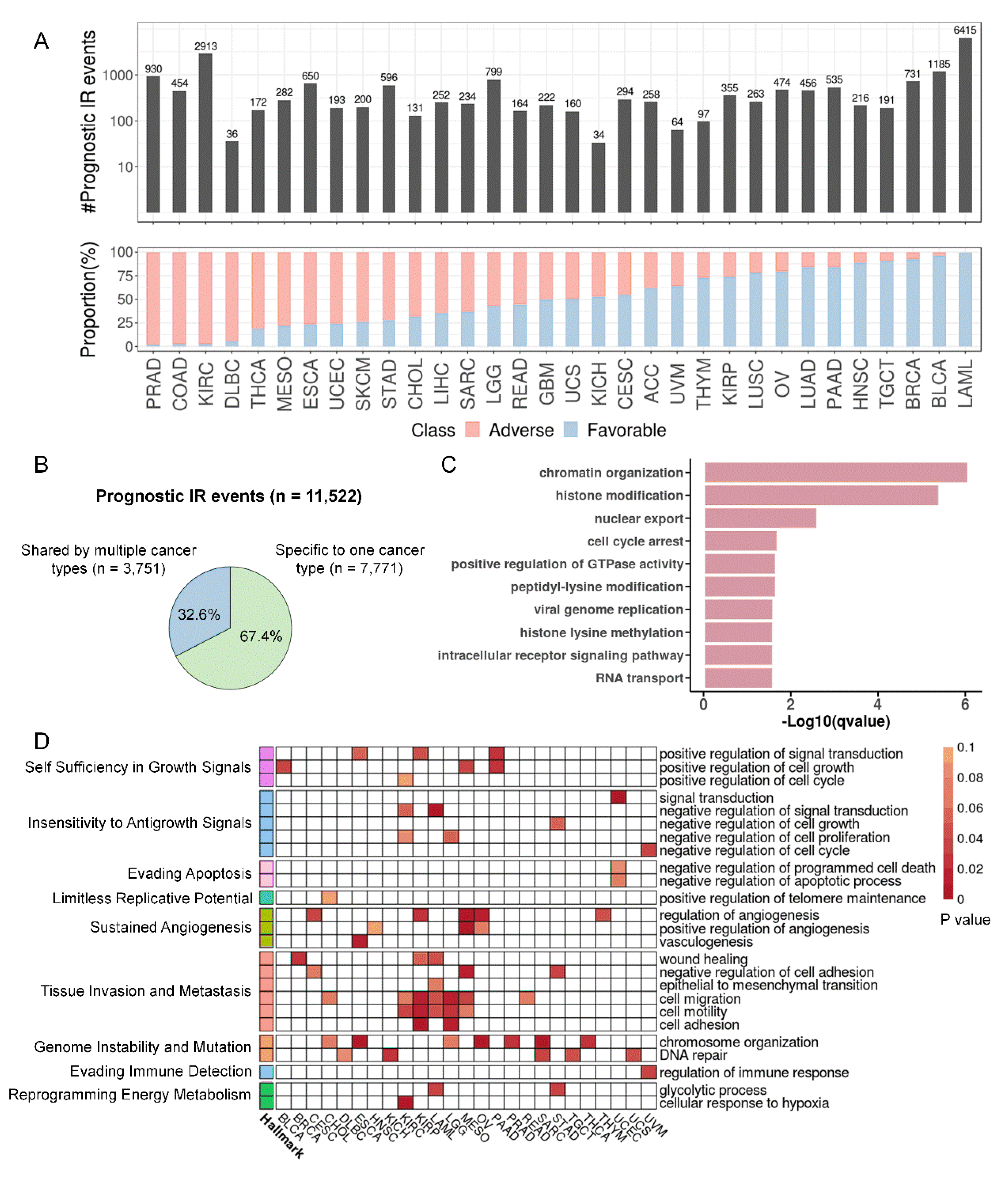
Figure 6.
A 5′UTR IR reduces the translation efficiency of STN1 and may suppress tumor growth. (A) Increased IR in 5′ UTR of STN1 was associated with longer survival in lung adenocarcinoma patients. (B) RT-PCR using primers amplifying exons 1-2 (‘e1-e2′) as well as specific to IR isoform in H1299 and A549 cells. (C) Relative intron retention ratio of STN1 in A549 cells detected by qRT-PCR. (D) Relative expression of IR and spliced transcripts of STN1 detected by qRT-PCR after inhibition of transcription in actinomycin D (10 µg/ ml)-treated A549 cells. (E) qRT-PCR for STN1 spliced and IR transcripts, following nuclear and cytoplasmic fractionation of A549 cell lysates. NEAT1 and ACTB serve as markers (or the internal controls) for the nucleus and cytoplasm, respectively. (F) Schematic diagram illustrating the design of the dual-luciferase reporter vector, where 3 different 5′ UTRs of STN1 (spliced 5′ UTR, IR 5′ UTR, and mutated IR 5′ UTR (ATG to AGC)) were attached to the psi-CHECK2 vector, located in front of Renilla. (G) The translation efficiency of three 5′ UTRs was detected by the relative fluorescence intensity of Renilla to Firefly. (H) Expression levels of STN1, CDK1, MKI67, were detected by qPT-PCR upon STN1 knockdown. (I) Effect of STN1 knockdown on cell viability in the clonogenic assay (upper panel) and cell migration assay (lower panel). (J) The cell proliferation rate of A549 cells evaluated by CCK-8 assay upon STN1 knockdown. *, P < 0.05; **, P < 0.01; ***, P < 0.001; two-tailed student’s t test.
Figure 6.
A 5′UTR IR reduces the translation efficiency of STN1 and may suppress tumor growth. (A) Increased IR in 5′ UTR of STN1 was associated with longer survival in lung adenocarcinoma patients. (B) RT-PCR using primers amplifying exons 1-2 (‘e1-e2′) as well as specific to IR isoform in H1299 and A549 cells. (C) Relative intron retention ratio of STN1 in A549 cells detected by qRT-PCR. (D) Relative expression of IR and spliced transcripts of STN1 detected by qRT-PCR after inhibition of transcription in actinomycin D (10 µg/ ml)-treated A549 cells. (E) qRT-PCR for STN1 spliced and IR transcripts, following nuclear and cytoplasmic fractionation of A549 cell lysates. NEAT1 and ACTB serve as markers (or the internal controls) for the nucleus and cytoplasm, respectively. (F) Schematic diagram illustrating the design of the dual-luciferase reporter vector, where 3 different 5′ UTRs of STN1 (spliced 5′ UTR, IR 5′ UTR, and mutated IR 5′ UTR (ATG to AGC)) were attached to the psi-CHECK2 vector, located in front of Renilla. (G) The translation efficiency of three 5′ UTRs was detected by the relative fluorescence intensity of Renilla to Firefly. (H) Expression levels of STN1, CDK1, MKI67, were detected by qPT-PCR upon STN1 knockdown. (I) Effect of STN1 knockdown on cell viability in the clonogenic assay (upper panel) and cell migration assay (lower panel). (J) The cell proliferation rate of A549 cells evaluated by CCK-8 assay upon STN1 knockdown. *, P < 0.05; **, P < 0.01; ***, P < 0.001; two-tailed student’s t test.
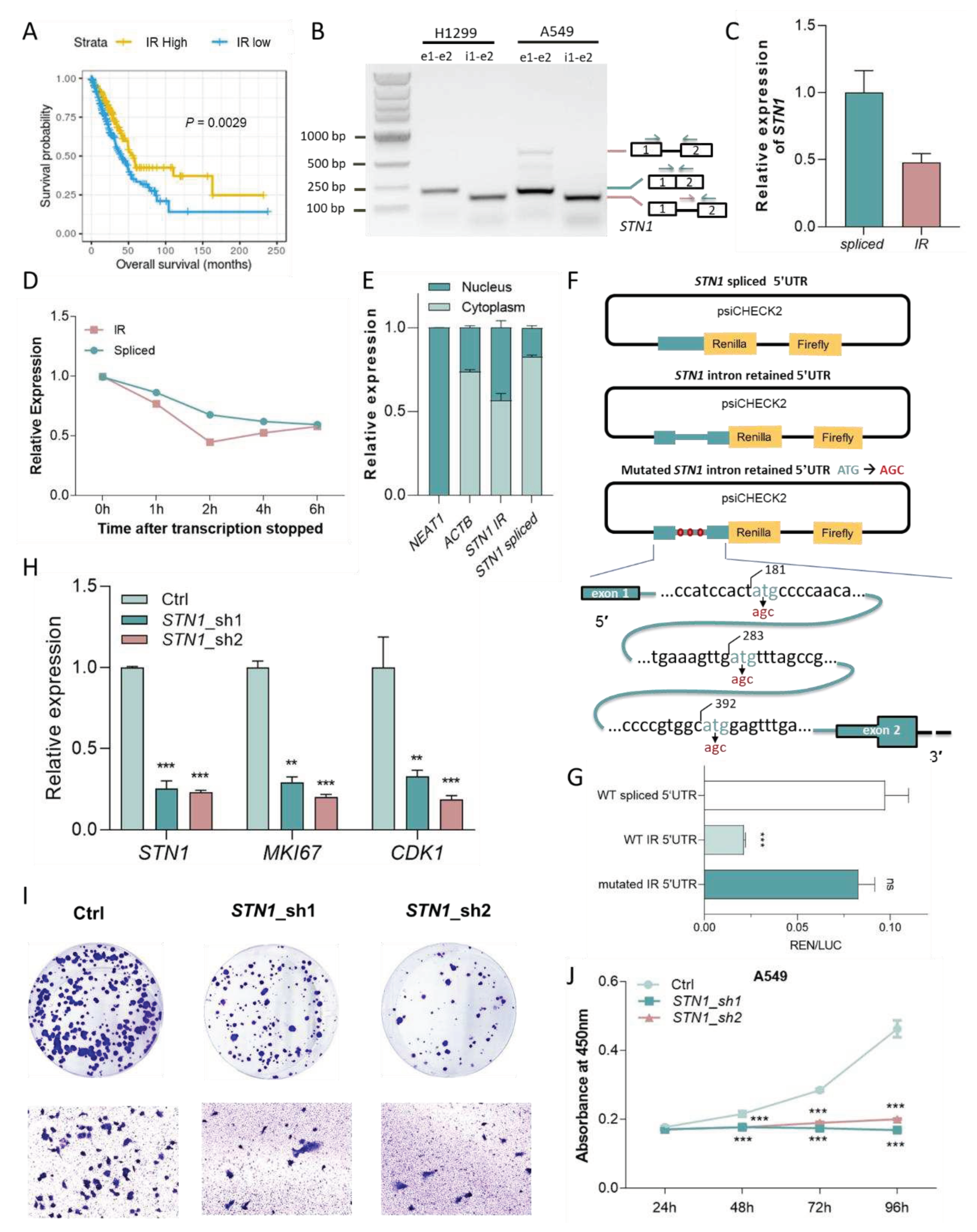
Figure 7.
IR prognostic model stratifies patients in LAML. (A) IRR model coefficients. Y-axis indicates introns and corresponding genes that were used to build this model, and the x-axis indicates their LASSO Cox coefficients. (B) Kaplan-Meier curves of high and low risk groups divided based on median IRR. P value was calculated with a log rank test. (C) IR (orange), clinical (green), mutational (yellow), cytogenetic (blue), and expression (purple) features that were significantly associated with prognosis in univariate COX regression. X-axis and y-axis indicate hazard ratios and the corresponding P values, respectively (both were log-transformed). Point size reflected the percentage of patients affected by the investigated feature.
Figure 7.
IR prognostic model stratifies patients in LAML. (A) IRR model coefficients. Y-axis indicates introns and corresponding genes that were used to build this model, and the x-axis indicates their LASSO Cox coefficients. (B) Kaplan-Meier curves of high and low risk groups divided based on median IRR. P value was calculated with a log rank test. (C) IR (orange), clinical (green), mutational (yellow), cytogenetic (blue), and expression (purple) features that were significantly associated with prognosis in univariate COX regression. X-axis and y-axis indicate hazard ratios and the corresponding P values, respectively (both were log-transformed). Point size reflected the percentage of patients affected by the investigated feature.
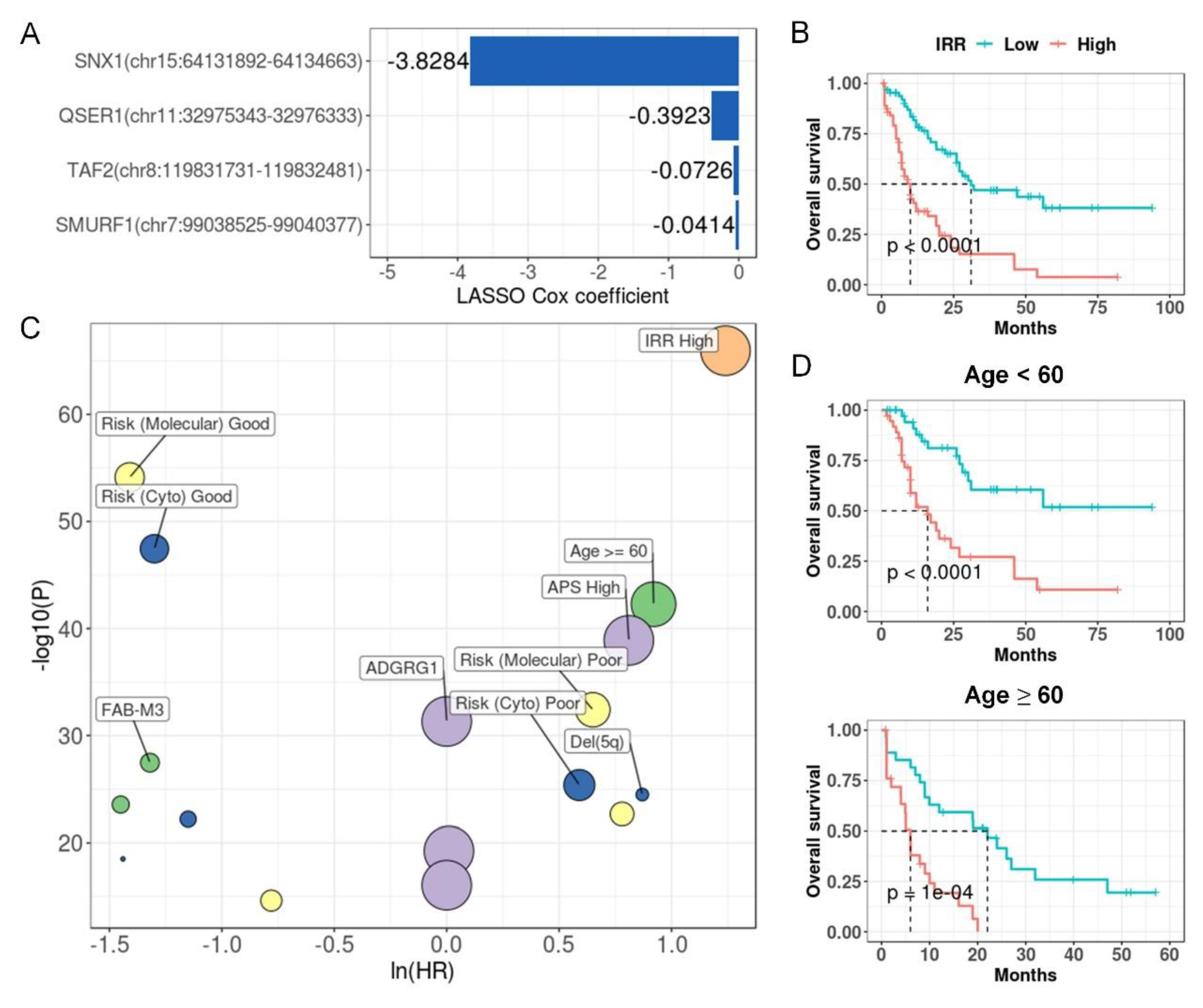
Figure 8.
Kaplan-Meier curves of IR-based prognostic models in 32 cancer types. For each cancer, LASSO regression was performed to build an IR-based prognostic model which we called IRR. Patients in each cancer type were classified into high and low risk groups based on median IRR, and Kaplan-Meier curves were drawn. P values were calculated with a log rank test.
Figure 8.
Kaplan-Meier curves of IR-based prognostic models in 32 cancer types. For each cancer, LASSO regression was performed to build an IR-based prognostic model which we called IRR. Patients in each cancer type were classified into high and low risk groups based on median IRR, and Kaplan-Meier curves were drawn. P values were calculated with a log rank test.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



