
Submitted:
29 August 2023
Posted:
31 August 2023
You are already at the latest version
Abstract
Kidney diseases are worldwide public health problems affecting millions of people. However, there are still limited therapeutic options against kidney diseases. Semaphorin 3A (SEMA3A) is the secreted and membrane-associated proteins, which regulate diverse functions, including immune regulation, cell survival, migration and angiogenesis, thus involving in the several pathogeneses of diseases, including eyes and neurons, as well as kidneys. SEMA3A is expressed in podocytes and tubular cells in the normal adult kidney, and recent evidence revealed that excess SEMA3A expression and subsequent signaling pathway aggravates kidney injury in a variety of kidney diseases, including nephrotic syndrome, diabetic nephropathy, acute kidney injury, and chronic kidney disease. In addition, several reports demonstrated that inhibition of SEMA3A ameliorated kidney injury via reduction of cell apoptosis, fibrosis and inflammation, thus SE-MA3A may be a potential therapeutic target for kidney diseases. In this review article, we summarized current knowledges regarding the role of SEMA3A signaling in kidney pathophysiology and their potential use in kidney diseases.
Keywords:
semaphorin 3A
; neuropilin-1
; podocyte
; diabetic nephropathy
; acute kidney injury
; chronic kidney injury
; lupus nephritis
; fibrosis
; apoptosis
; inflammation
1. Introduction
Semaphorins are guidance proteins regulating cellular morphology and functions, thus have important roles in developments and diseases, including cancers and metabolic diseases [1]. Semaphorin belongs to the super semaphorin family, consisting of 30 glycoproteins and divided into 8 classes [2]. Class 1 and class 2 semaphorins were identified invertebrates, class 3-7 semaphorins were in vertebrates. In vertebrates, class 3 semaphorin (SEMA3A-3G) are the only member of secreted proteins [3]. In 1990, Raper and colleagues isolated a molecule from embryonic chick brain that induced collapse of neuronal growth cones in culture, and the molecule was originally named Collapsin [4,5], and it was renamed as SEMA3A. Since then, SEMA3A has been well studied not only in axon guidance, but also in pleiotropic functions, including in angiogenesis, immune cell regulation and cell migration [6]. SEMA3A is expressed in kidneys, as well as nervous system, heart, lung, eyes, bone and immune cells [7,8,9,10,11,12], and regulates tissue development and maintenance of homeostasis, for example through the regulation of cell proliferation, migration and immune system [13,14,15]. Several reports identified increase or decrease in SEMA3A expression under several disease conditions [16,17,18,19], thus SEMA3A was suggested as a possible biomarker, as well as a therapeutic target. For example, it is reported that retinal SEMA3A expression was increased in a retinal vein occlusion mouse model, where anti-SEMA3A neutralizing antibody BI-X mediated protective role in intraretinal edema and retinal blood flow [12]. SEMA3A inhibitors was also reported to protect retinal ganglion cells in animal models of optic nerve injury, retinal ischemia and glaucoma [13]. In a neuro system, inhibition of SEMA3A enhanced functional recovery during subacute stroke recovery period [20]. SEMA3A not only regulated organization of brain structures affected by autism spectrum disorder (ASD), but also was related to neuron inflammatory processes in ASD [21]. Moreover, lipopolysaccharide (LPS)-induced vascular endothelial cells activation, vascular inflammation, and vascular oxidative stress were substantially improved by inhibition of SEMA3A via siRNA [22]. Serum SEMA3A level was proposed as biomarkers for diabetic retinopathy in patients with type 2 diabetes [23]. Importantly it also reflected the severity of diabetic retinopathy, suggesting a value for evaluating patients’ prognosis. The quantitative real-time PCR analysis using glioma tissues verified that SEMA3(A-G) together with other six genes may be useful as biomarkers in prognosis of glioma patient’s outcome [24]. Serum SEMA3A was decreased in patients with systemic lupus erythematosus (SLE) and was increased in patients with rheumatoid arthritis and sjögren’s syndrome, suggesting the important role of SEMA3A in immune-related diseases [25]. SEMA3A expression in cancer tissues was shown as an independent prognostic factor of overall survival of various types of cancer, such as oral cancer, gastric cancer, breast cancer, prostate cancer, glioblastoma and ovarian cancer. Of note, SEMA3A was significantly correlated with the stage and grade of the disease, depth of invasion, presence of metastases or survival [26,27]. In addition, recent accumulated evidence also revealed important roles of SEMA3A in kidney diseases.
Kidney diseases, characterized as kidney dysfunction, are growing public health burden with a huge economic cost on healthcare systems worldwide [28]. There are several important causes for kidney diseases, including diabetes mellitus (DM), hypertension, glomerulonephritis, obesity and aging. In 2017, a survey showed that 697.5 million cases of all-stage chronic kidney disease (CKD) were recorded, for a global prevalence of 9.1% [29]. Therefore, elucidating the complicated molecular pathology of kidney disease is an urgent task for developing new therapeutic strategies to manage patients with kidney diseases. Over the past few decades, many efforts were devoted to investigating and evaluating the progression of CKD. One of the important advents for CKD therapy is a use of sodium-glucose co-transporter-2 (SGLT-2) inhibitor [30,31], which slowers CKD progression. In addition, glucagon-like peptide (GLP) 1 receptor agonists are reported to reduce the incidence of kidney events in the patients with DM [32]. Several promising therapies for CKD, including apoptosis signal regulating kinase 1 (ASK1) inhibitor, endothelin receptor antagonist, phosphodiesterase inhibitor, Janus kinases (JAK)1/2 inhibitor and nuclear factor erythroid 2-related factor 2 (Nrf2) activator were undergoing clinical trials [33,34,35,36,37]. Nevertheless, its etiology and pathogenesis are still far from being elucidated. Accumulated evidence suggested importance roles of SEMA3A signaling in kidney development and several kidney diseases, including proteinuric diseases, acute kidney injury (AKI) and CKD. Several reports indicated potential use of inhibitory drugs of SEMA3A signaling against kidney diseases, and also identified SEMA3A expression as the potential biomarkers for early detection and prognosis for AKI, and relapse of nephrotic syndromes. In this review article, we summarize a current overview of evidence about SEMA3A in the kidney development and diseases.
2. SEMA3A and its receptors, and expression in kidneys
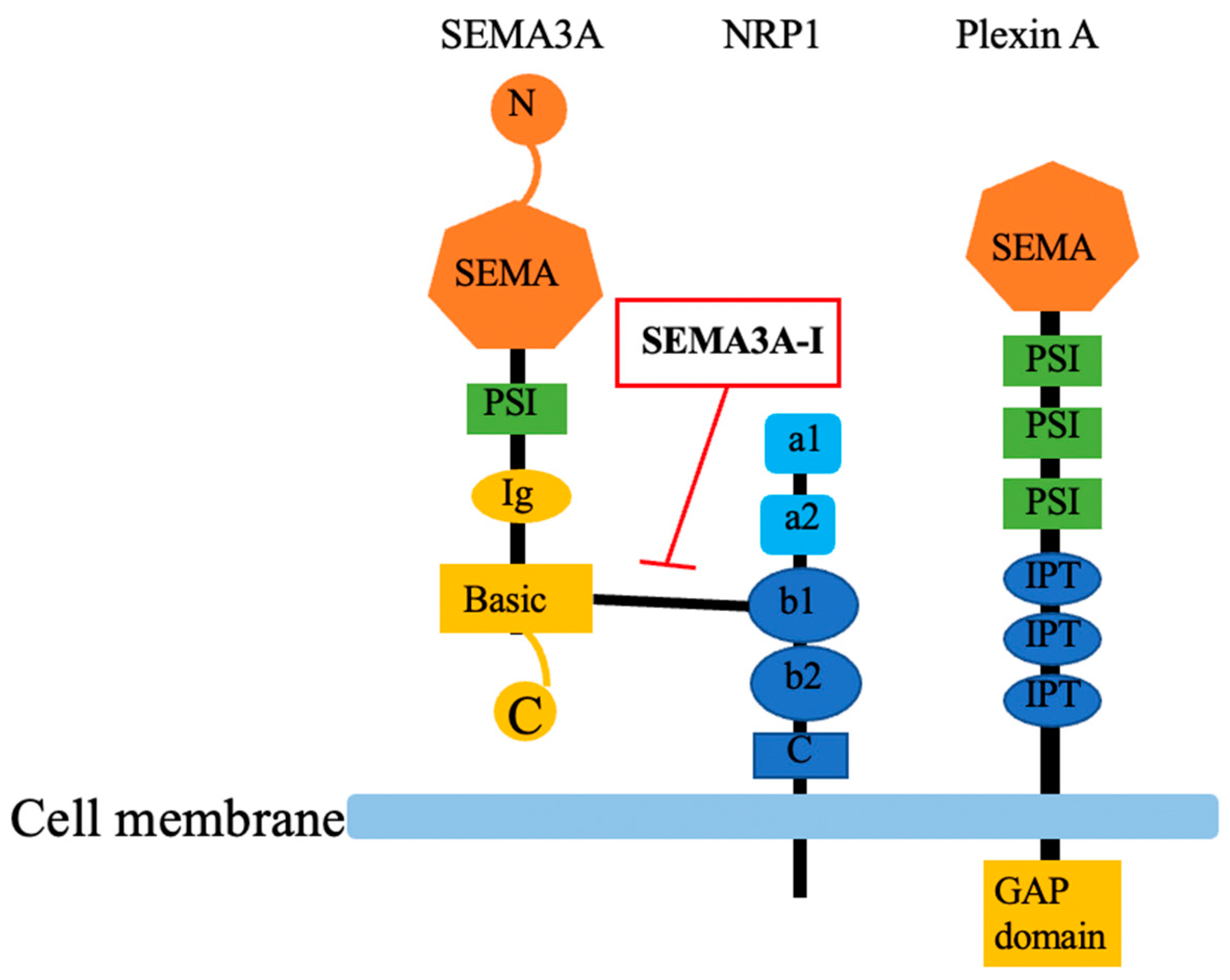
SEMA3A, as other semaphorin family members, has a conserved N-terminal SEMA domain which contained 500 amino acids and diverges C -terminus. The central feature of SEMA domain is a disulfide-rich seven blade β-propeller fold. A small cysteine-rich plexin-semaphorin-integrin (PSI) domain is between SEMA-domain and immunoglobulin (Ig)-like domain. Basic domain is located at the C -terminus of SEMA3A. The C-terminus determines affinity to Neuropilin-1 (NRP1) (Figure 1) [3,38]. SEMA3A has two receptors; NRP1 is necessary for ligand binding while plexin A is important to subsequent signal transduction. NRP1 is a transmembrane protein with molecular weight of 120-130kDa. The extracellular part contains 3 kinds of domains which are two complement binding domains a1/a2, two coagulation factor V/VIII domains, and c-domain, then connect with NRP1 cytoplasmic part (Figure 1) [3]. The signaling receptor PlexinA is a transmembrane glycoprotein with SEMA3A similar extracellular part which implying their common evolutionary origin, it is followed by three PSI domains and three Ig-like, plexin and transcription factor (IPT domains). Importantly, plexin is the only receptor possessing highly conserved GAP domain (GTPase activating protein), regulating diverse function in cell activity. Plexin A downstream signaling is complex, involving GTPases: R-Ras, M-Ras and Rap (from the Ras family), RhoA, Ras-related C3 botulinum toxin substrate 1 (Rac1), and Rnd (from the Rho family) as well as Fyn phosphorylation [39]. During signal transduction, SEMA3A-NRP1-Plexin complex formed a holoreceptor [40]. Three-dimensional structure of the SEMA3A-NRP1-Plexin complex was recently reported [41]. Together, their characteristics adding more crosstalk to the pathways involved in SEMA3A signaling. It is reported that pleiotropic signaling pathways were involved in SEMA3A/NRP1/PlexinA signaling, including c-Jun N-terminal kinase (JNK) and Akt signaling pathway.
It is reported that excess SEMA3A expression may accelerate kidney injury in variety of kidney diseases via JNK or Akt signaling pathway [42,43,44,45,46], suggesting that targeting SEMA3A signaling may be the potential therapy against kidney injury. In the mammalian adult kidneys, SEMA3A is expressed in podocytes and tubular cells [47], while SEMA3A receptors, NRP1 and PlexinA, are expressed throughout life in podocytes, endothelial cells and tubular cells [48,49]. In addition, a recent analysis using single-cell RNA sequence revealed increased NRP1 expression in activated fibroblasts, suggesting a possible involvement of SEMA3A signaling during a progression of kidney fibrosis [50]. NRP1 is also known as co-receptors for the vascular endothelial growth factor A isoform 165 (VEGF165); thus, SEMA3A plays a role in suppressing angiogenesis by competitive inhibition of VEGF signaling through inhibition of binding to NRP1 [51]. It is reported that a SEMA3A inhibitor (SM-345431) inhibited connections between SEMA3A and NRP1, thereby blocking SEMA3A signaling [46,52] (Figure 1). Indeed, SM-345431 was shown to antagonize SEMA3A-induced axonal growth cone collapse in embryonic neurons [53]. In addition, SM-345431 was shown to preserve corneal nerve and epithelial integrity in rodent dry eye model [54]. To specify an interaction between a peptide inhibitor and SEMA3A-NRP1 system, a peptide inhibitor was modified with the photoactivatable amino acids-4-benzoyl-l-lphenylalaine or photo-l-leucine by solid-phase peptide synthesis. SEMA3A-peptide interaction were found in a defined area of the SEMA domain, which was also involved in NRP1 [55].
3. SEMA3A in kidney development
SEMA3A has roles in axon pathfinding, cardiovascular, lung and kidney patterning during organogenesis [56,57]. During kidney development, SEMA3A plays an important role in patterning the ureteric bud branching. Recombinant SEMA3A decreased the number of developing glomeruli in vitro and inhibited ureteric bud branching via downregulation of glial-cell-line-derived neurotrophic factor (GDNF) signaling, competition with VEGF165 and decreased activity of Akt survival pathways. Conversely, deletion of SEMA3A in mice enhanced ureteric bud branching [58]. Thus, SEMA3A function as a negative regulator of ureteric bud branching during normal kidney development. Reidy et al. established loss- and gain- of function mouse models, which revealed that SEMA3A-/- mice showed increased endothelial cells and defects in renal vascular patterning, whereas SEMA3A+/+ mice had normal wide-open capillary loops observed by light microscopy. Morphometric analysis of transmission electron microscopy (TEM) revealed that SEMA3A-/- mice had effaced podocyte foot processes, which were associated with albuminuria. On the other hand, overexpression of podocyte SEMA3A resulted in glomerular hypoplasia, undifferentiated podocytes and congenital proteinuria [48]. Taken together, balanced SEMA3A expression may be essential for normal glomerular development, glomerular filtration barrier function and ureteric bud branching. In addition to kidney development, the role of SEMA3A in matured kidneys has also been explored. Recombinant SEMA3A injection in adult mice induced nephrotic range proteinuria [44]. In addition, it is also reported that excess SEMA3A increased starvation-induced apoptosis in cultured podocytes and in the developing kidney in vivo [48,49]. Taken together, SEMA3A not only regulates kidney development, but also may be involved in kidney diseases.

Table 1.
Expression of SEMA3A in kidney diseases.
| Disease | Etiology | Species | Sample | SEMA3A expression |
Ref. |
|---|---|---|---|---|---|
| Proteinuric diseases | MCNS | Human | Urine | Increase | [59] |
| PAN | Rats | Kidney | Increase | [57] | |
| DN | - | Human | Urine | Increase | [60] |
| db/db | Mice | Kidney | Increase | [60] | |
| db/db | Mice | Kidney | Increase | [57] | |
| Streptozotocin | Mice | Kidney | Increase | [61] | |
| AKI | IRI | Mice | Kidney | Increase | [62] |
| LPS | Mice | Kidney | Increase | [45] | |
| contrast | Human | Urine | Increase | [63] | |
| Cardiac operation |
Human | Serum/ Urine | Increase | [43] | |
| CKD | - | Human | Urine | Increase | [64] |
| LN | - | Human | Kidney | Increase | [65] |
| - | Human | Urine | Decrease | [66] |
AKI: Acute kidney injury; MCNS: Minimal change nephrotic syndrome; DN: Diabetic nephropathy; LN: lupus nephritis; PAN: puromycin; IRI: ischemia-reperfusion injury; LPS: lipopolysaccharide.
4. SEMA3A and kidney diseases
4.1. Podocytopathy and diabetic nephropathy
Podocytes, which have crucial roles in the kidney filtration barrier, line out of glomerular basement membrane (GBM) preventing urinary protein loss [67]. Podocyte foot processes linked by slit diaphragms which regulate cell shape and work as filtration barrier [68]. Hence, podocyte injury is associated with proteinuria. As indicated above, recombinant SEMA3A injection in adult mice induced nephrotic range proteinuria [44], and the increase was within 4 hrs and was resolved by 24 hrs. TEM analysis revealed extensive fusion and effacement of podocyte foot processes in kidneys examined 4 hrs after SEMA3A injection, which were recovered at 48 hrs, demonstrating that excess circulating SEMA3A may cause podocyte ultrastructural abnormalities, and the permeability of the glomerular filtration barrier are transient and reversible, providing proof of principle of excess SEMA3A and glomerular disease. In addition, it is also reported that SEMA3A induced downregulation of podocin in a dose-dependent manner, decreases the interactions between nephrin, podocin, and CD2-assocaited protein (CD2AP) in cultured podocytes [49]. SEMA3A induced a 10-fold increase in podocyte apoptosis by decreasing Akt survival pathway [49]. Excess SEMA3A was shown to induce endothelial cell swelling, thickening and lamination of GBM, and podocyte foot process effacement, all of which were transient and reversible upon withdrawal of transgene induction. SEMA3A disrupted podocyte shape in an autocrine fashion, based on podocyte contraction and F-actin collapse [69]. It is reported that the mechanism under the GBM phenotype change was through increased matrix metalloproteinase 9 expression or collagen and laminin chain composition [47]. Excess SEMA3A caused reversible nephrin downregulation, while podocin expression and WT1+ nuclei counts were not altered, suggesting that SEMA3A caused decreased nephrin expression without podocyte loss. In addition, GST binding assays revealed a direct interaction between plexin A and nephrin [69], indicating that extracellular SEMA3A signaling may directly link to slit-diaphragm signaling complex. Increased SEMA3A mRNA and protein expression were found in experimental models of puromycin (PAN)-induced podocyte injury [57]. In our previous study, we investigated the pathological roles of SEMA3A signaling on podocyte injury using a doxorubicin (Dox)-induced podocytopathy mouse model and examined the therapeutic effect of a SEMA3A inhibitor, SM-345431 [70]. We indicted that Dox-induced massive albuminuria and podocyte apoptosis via JNK signaling, as well as an increase of SEMA3A expression in podocytes, all of these were improved by treatments with SM-345431 [46]. We also examined serum and urinary SEMA3A levels in 72 patients who underwent kidney biopsies and shown that urinary SEMA3A levels in minimal change nephrotic syndrome (MCNS) patients were higher compared to other patients [59]. Furthermore, we evaluated urinary SEMA3A and MCNS activity, and found that levels of urinary SEMA3A at onset were significantly higher than those at remission in patients with MCNS. These results suggested the urinary SEMA3A might be useful as a biomarker for MCNS.
Diabetic nephropathy (DN) is one of important complications of DM [71], and is a leading cause of CKD and end-stage renal disease (ESRD) worldwide [72,73]. DN recognizes with podocyte injury and generally starts with microalbuminuria, which progresses to GBM thickness, mesangial expansion, macroalbuminuria, finally decreasing glomerular filtration rate [74,75]. Multifactorial interaction of factors are involved in DN, such as advanced glycation end products (AGEs) formation and the renin-angiotensin system (RAAS), further stimulating protein kinase C, generation of reactive oxygen species (ROS) [76,77] and microRNAs. It is reported that urinary SEMA3A excretion was increased early after induction of diabetes in diabetic mouse models and in diabetic patients with albuminuria, particularly in those with macroalbuminuria [60]. In diabetic mice, podocyte-specific SEMA3A overexpression (SEMA3A+) caused Kimmelstiel-Wilson-like nodular glomerulosclerosis, massive proteinuria and kidney insufficiency [42]. Increased SEMA3A expression was found in db/db kidneys [57], as well as streptozotocin-induced diabetes mouse kidneys [61]. Genetic ablation and inhibition of SEMA3A signaling ameliorated diabetes induced kidney dysfunction [60]. Importantly, a SEMA3A inhibitor, xanthofulvin treatment or deletion of podocyte plexinA1 abrogated diabetic nodular glomerulosclerosis induced by SEMA3A+ gain of function [42]. Recent study revealed that microRNAs play important roles in DN pathogenesis. miR-15b-5p restored cell proliferation in high glucose (HG)-triggered podocytes by downregulating proapoptotic protein markers, Bax and cleavaged caspase-3, and upregulating antiapoptotic protein, Bcl-2 [78]. miR-15b-5p remarkably decreased HG-induced inflammatory response via downregulation of cytokines, IL-1β, TNF-α and IL-6. In addition, it is reported that SEMA3A is a direct target of miR-15b-5p, and beneficial effects of miR-15b-5p were impeded by excess SEMA3A [78]. SEMA3A is also targeted by miR-23b-3p [79] and miR-16-5p [80]. KCNQ1 opposite strand/antisense transcript 1(KCNQ1OT1), a long non-coding RNA (IncRNA) was recognized as a miR-23b-3p sponge, and KCNQ1OT1 inhibition ameliorated DN by absorbing miR-23b-3p and regulating SEMA3A [79]. It is also reported that serum LncRNA T-cell factor7 (TCF7) were elevated in patients with DN and TCF7 silencing ameliorated HG-induced podocyte injury by downregulating SEMA3A via miR-16-5p [80]. Collectively, excess SEMA3A is involved in the progression of DN and SEMA3A targeting may be the potential therapy against DN.
4.2. Acute kidney injury
AKI is recognized as a major public health problem affecting millions of patients worldwide and leading to higher mortality [81], CKD progression, and sometimes to new onset of CKD, called AKI-CKD transition. Kidney ischemia-reperfusion injury (IRI), nephrotoxic agents such as LPS or cisplatin, infection leading to sepsis, and contrast-related are major causes of AKI [82]. Currently, AKI is diagnosed according to serum creatinine levels and urine volume [83]. Early prediction before increase in serum creatinine levels and early treatment are important for patients at risk of AKI. Urinary SEMA3A is shown to be increased within 6 hrs after IRI, whereas serum creatinine is increased at 24 hrs in animals [43]. Urinary SEMA3A was also shown to be increased and peaked at 2 hrs after liver transplantation-induced AKI [84]. Serum SEMA3A in cisplatin-induced AKI was upregulated at 24 hrs and 48 hrs. In pediatric patients, AKI was detected on 48 hrs after cardiopulmonary bypass (CPB) by serum creatinine [43], while urine SEMA3A was elevated 2 hrs after CPB and peaked at 6 hrs. Moreover, early increase in urinary SEMA3A levels were associated with clinical outcomes, such as severity of AKI and length of hospital stay [43]. Urinary SEMA3A was compared with other urinary biomarkers of IL-18 [85], L-type fatty acid-binding protein(L-FABP) [86], gelatinase-associated lipocalin (NGAL) [87] and N-acetyl-β-d-glucosamininidase (NAG) in intensive care unit (ICU) admission. These biomarkers showed similar performance in detecting established AKI, later-onset AKI and AKI progression, while urinary SEMA3A was not increased in non-progressive established AKI. Finally, urinary SEMA3A was not increased in sepsis-induced AKI, while levels of other urinary biomarkers were increased [88,89]. Increase in urinary SEMA3A was also reported in contrast-induced acute kidney injury [63]. Among 168 patients underwent percutaneous coronary intervention (PCI), 20 patients developed AKI. Both urinary SEMA3A and NGAL levels were significantly elevated at 2 hrs and 6 hrs post-PCI procedure, and peak at 2 hrs post-PCI in the AKI patients, which were much earlier than the rise in serum creatinine levels at 48-72 hrs post-PCI. Further receiver operating characteristic (ROC) analyses of SEMA3A at 2 hrs after PCI showed better predictive sensitivity and specificity compared to NGAL. These results indicate that urinary SEMA3A may be useful as an early and predictive biomarker for AKI.
In addition to biomarkers, SEMA3A signaling is also expected as a therapeutic target. SEMA3A expression was increased after LPS-induced AKI in mice tubular epithelial cells, as well as in LPS-treated rat kidney proximal tubular epithelial cell line in vitro via Rac1/ nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB) p65 and JNK pathways [45]. In addition, inhibition of SEMA3A by (-)-epigallocatechin-3-gallate (EGCG) could significantly ameliorate LPS-induced kidney inflammation and apoptosis [45]. It is also reported that genetic silencing and pharmacological inhibition of SEMA3A ameliorated kidney injury from IRI by inhibiting inflammation and epithelial cell apoptosis [62]. These observations indicate an underling signaling pathway of SEMA3A and the potential utility of SEMA3A inhibitor as a therapeutic agent for regulating inflammation and apoptosis in AKI. G-protein-coupled receptors (GPCRs) is known to participate in plenty of physiologic functions and some GPCRs have critical roles in the regulation of kidney function. Among them, Gpr97 is a newly identified adhesion GPCR. Gpr97 was upregulated in IRI-induced AKI kidneys from mice [90]. Both in vivo and vitro study revealed that Gpr97 deficiency attenuated AKI-induced kidney injury by regulating SEMA3A signaling. It is also reported that curcumin, well known for its antioxidant and anti-inflammatory properties, and 12/15 lipoxygenase inhibitor-LOXblock-1 ameliorated IRI-induced AKI by reducing inflammatory processes, oxidative stress and apoptosis, and the effects were through suppression of SEMA3A signaling pathway [91]. Another study highlighted protective effects of human-bone-marrow-derived mesenchymal stem cells exosomes in kidney IRI by delivering miR-199a-3p to kidney cells [92]. The mechanism involved downregulating Sema3A expression and activating Akt and extracellular signal-regulated kinase (ERK) signaling pathways, ultimately leading to reduced apoptosis and improved kidney function. These reports indicate a potential avenue for developing new therapeutic strategies as targeting SEMA3A signaling for AKI.
4.3. Chronic kidney disease
CKD is characterized by progressive damage and loss of kidney function, where parenchymal cell loss, chronic inflammation, fibrosis and reduced regenerative capacity of the kidney are involved in the progression [93]. It is reported that urinary SEMA3A levels were positively correlated with urine albumin-to-creatinine ratio and serum creatinine levels in hypertensive patients [94]. In the study, patients with CKD showed higher urinary SEMA3A levels as compared to those without CKD. Kidney fibrosis is the common pathological pathway of kidney diseases. In our previous study, we evaluated SEMA3A signaling by using a kidney fibrosis model, unilateral ureteral obstruction (UUO) mouse model [64]. After UUO surgery, SEMA3A expression in proximal tubular area and NRP1 expression in fibroblast and tubular cells were increased. The expression of a myofibroblast marker, tenascin-C and kidney fibrosis were increased in UUO kidneys, all of which were ameliorated by SEMA3A inhibitor through regulation of JNK signaling [64]. One of important mechanisms in kidney tubulointerstitial fibrosis is kidney tubular epithelial-mesenchymal transition (EMT) process, where kidney tubular epithelial cells lose their cell-to-cell membrane connection and their structural polarity to become spindle-shaped mesenchymal-like phenotype [95]. Our study indicated that injection of a SEMA3A inhibitor, SM-345431 could attenuate UUO-induced EMT in vivo [64]. We also demonstrated that recombinant SEMA3A caused tubular cell EMT and SM-345431 treatment was able to ameliorate TGF-β1-induced EMT in vitro. We also indicated a positive correlation between urinary SEMA3A and tubular injury marker, urinary NAG, in patients who underwent kidney biopsy. Collectively, SEMA3A signaling may be involved in progression of kidney fibrosis under CKD, and inhibition of SEMA3A signaling might be a therapeutic option for protecting from kidney fibrosis [64].
4.4. Systemic lupus erythematosus
SEMA3A also regulates immune systems, especially enhancing T-cell and B-cell regulatory properties [3]. Hence, it is reported that SEMA3A is involved in pathogenesis of autoimmune diseases, including SLE [66], rheumatoid arthritis [96] and sjögren’s syndrome [97]. SLE is a multi-system autoimmune disease characterized by aberrant activity of the immune system and presents with a wide range of clinical manifestation including skin, synovia, brain and kidney [98]. Of note, an analysis of SEMA3A immunostaining in kidneys from patients with lupus nephritis (LN) revealed an increase in SEMA3A expression in patients with LN while SEMA3A expression was negatively associated with clinic-pathological parameters, including proteinuria and kidney function [65]. In contrast, another study indicated that serum SEMA3A levels in SLE patients were lower than normal individuals [66]. Aiming to establish a regulatory/protective role for SEMA3A in SLE, serum SEMA3A was assessed in patients with SLE and compared this level with SLE disease activity [66], where serum SEMA3A levels were lower in SLE patients compared to that in normal controls. In addition, altered serum SEMA3A levels were found to be inversely correlated with SLE disease activity, mainly with kidney damage and the presence of anti-cardiolipin antibodies. These findings suggest an important role of SEMA3A in SLE.
It is reported that SEMA3A downregulated autoimmune responses by suppressing over-activity of both B and T cells [99,100]. SEMA3A in B-regulatory cells of patients with SLE was smaller compared to those of normal individuals. Toll-like receptor-9 (TLR-9) expression could possibly be modulated in memory B cells of SLE patients, which is associated with production of inflammatory cytokines, such as IL-6 and anti-dsDNA [101,102]. SEMA3A co-cultured with purified B cells from SLE patients significantly reduced TLR-9 expression, supporting the idea that SEMA3A may regulate B cell autoimmunity in SLE [66]. Serum SEMA3A levels were decreased in SLE while increase in RA and SS compared to healthy controls. How about urinary SEMA3A levels? A study analyzed urinary SEMA3A levels in 38 patients with SLE [103]. Among them, 13 patients had kidney involvements. Urinary SEMA3A levels were lower in SLE patients compared to healthy volunteers, especially lower in SLE patients with LN than in patients without nephritis, indicating that urinary SEMA3A is inversely correlated with proteinuria and SLE disease activity. Aberrant expression of SEMA3A level in urine and serum in SLE may suggest important roles of SEMA3A in SLE disease activity. Indeed, it is reported that SEMA3A injection in the NZB/W mice, a model of LN, delayed an appearance of proteinuria and reduced kidney damage, as well as a decrease in deposition of immune complex deposition in the glomeruli, indicating the protective effect of SEMA3A in LN [104].
Table 2.
Kidney outcome in reports targeting SEMA3A signaling.
| Etiology | Model | SEMA3A- targeting |
Targeting Method |
Species | Outcome | Target | Function | Ref |
|---|---|---|---|---|---|---|---|---|
| Podocyte injury | Dox | Down | SEMA3A inhibitor |
Mice | Proteinuria↓ | JNK | Anti-apoptosis | [70] |
| - | - | Up | Recombinant SEMA3A | Mice | Nephrotic proteinuria |
- | Podocytopathy | [44] |
| - | - | Up | Podocyte SEMA3A+ |
Mice | Proteinuria↑ | dysregulation of nephrin, MMP9, αvβ3 integrin | Podocytopathy | [69] |
| DN | STZ | Down | SEMA3A- | Mice | Proteinuria↓ | - | - | [60] |
| STZ | Down | SEMA3A inhibitor |
Mice | Proteinuria↓ Kidney fibrosis↓ Kidney dysfunction↓ |
- | - | [60] | |
| High- glucose |
Down | miR-15b-5p | Podocyte | - | - | Anti-apoptosis Anti-inflammation |
[78] | |
| High- glucose |
Up | KCNQ1OT1 | Podocyte | - | miR-23b-3p | Inflammation Apoptosis |
[79] | |
| High- glucose |
Down | TCF7 silence / SEMA3A- siRNA |
Podocyte | Cytotoxicity↓ | miR-16-5p | - | [80] | |
| Podocyte SEMA3A+ & STZ |
Down | SEMA3A inhibitor (xanthofulvin) |
Mice | Proteinuria↓ Kidney dysfunction↓ |
MICAL1 | - | [42] | |
| AKI | IRI | Down | SEMA3A- / SEMA3A -inhibitor | Mice | Kidney dysfunction↓ Neutrophil infiltration↓ |
- | Anti-apoptosis, Anti-inflammation |
[62] |
| IRI / cisplatin |
Down | Gpr97- | Mice | Kidney dysfunction↓ Neutrophil infiltration↓ |
HuR | Aiti-apoptosis anti-inflammation |
[90] | |
| IRI | Down | LOXblock-I / Curcumin |
Rats | - | - | Anti-apoptosis Anti-inflammation |
[91] | |
| IRI | Down | miR-199a-3p | Mice | Kidney dysfunction↓ | AKT / ERK pathway |
Aiti-apoptosis | [92] | |
| LPS | Down | EGCG | Mice | Kidney dysfunction↓ Neutrophil infiltration↓ |
Rac1/NF-κB p65 / JNK pathway | Anti-apoptosis, Anti-inflammation |
[45] | |
| CKD | UUO | Down | SEMA3A Inhibitor (SM-345431) |
Mice | Kidney fibrosis↓ | JNK | Anti-apoptosis, Anti-EMT |
[64] |
| LN | NZB | Up | Recombinant SEMA3A |
Mice | Proteinuria↓ Immune complex- deposition↓ |
- | - | [104] |
DN: Diabetic nephropathy; AKI: Acute kidney injury; CKD: chronic kidney disease; LN: lupus nephritis; SEMA3A: semaphorin3A; Dox: doxorubicin; STZ: Streptozotocin; IRI: ischemia-reperfusion injury; LPS: lipopolysaccharide; UUO: unilateral ureteral obstruction; NZB: New Zealand black mouse; JNK: c-Jun N-terminal kinase; MMP9: matrix metalloproteinase 9; MICAL1: Molecules interaction with CasL1; HuR: RNA-binding protein human antigen R; AKT:activated the protein kinase B;ERK :extracellular signal regulated kinase pathways; Rac1: Ras-related C3 botulinum toxin substrate 1; NF-κB: nuclear factor kappa-light-chain enhancer of activated B cells; EMT: epithelial-mesenchymal transition; KCNQ1OT1: KCNQ1 opposite strand/antisense transcript 1; TCF7: long non-coding RNA T-cell factor 7; EGCG: (-)-epigallocatechin-3-gallate.
5. Conclusion
On the basis of these studies, SEMA3A plays an important role in kidney morphogenesis and kidney diseases. SEMA3A loss- and gain- of function studies indicate that SEMA3A is required for maintenance of structure and function in glomerular filtration barrier. Excess SEMA3A causes progression of a variety of kidney diseases, including DN, AKI and CKD, through the increase in albuminuria, kidney fibrosis, apoptosis and inflammation. SEMA3A mutant mice or pharmacological-based inhibition of SEMA3A protected from these kidney diseases. Therefore, SEMA3A targeting therapy may be a novel therapeutic option for treating against kidney diseases in clinic use. On the other hand, SEMA3A deficiency may lead to the progression of LN through upregulating autoimmune responses by activation of T cells and B cells, suggesting the concern of side effect of inflammatory response in kidneys under SEMA3A inhibitory therapy. In addition, detail mechanisms of SEMA3A underlying kidney diseases are not fully understood, there are several possible signaling pathways, involving SEMA3A signaling, such as Rac1/NF-κB p65, JNK and TLR4 signaling, which may regulate inflammation and cell apoptosis. Of note, the Rho family of GTPases acts downstream of plexin A, regulating adhesion, proliferation, migration and survival in different cell types and interact with diverse signaling pathways, suggesting that Rho family may be involved as a downstream effector of SEMA3A. Further studies are still needed for the deep understanding of SEMA3A signaling.
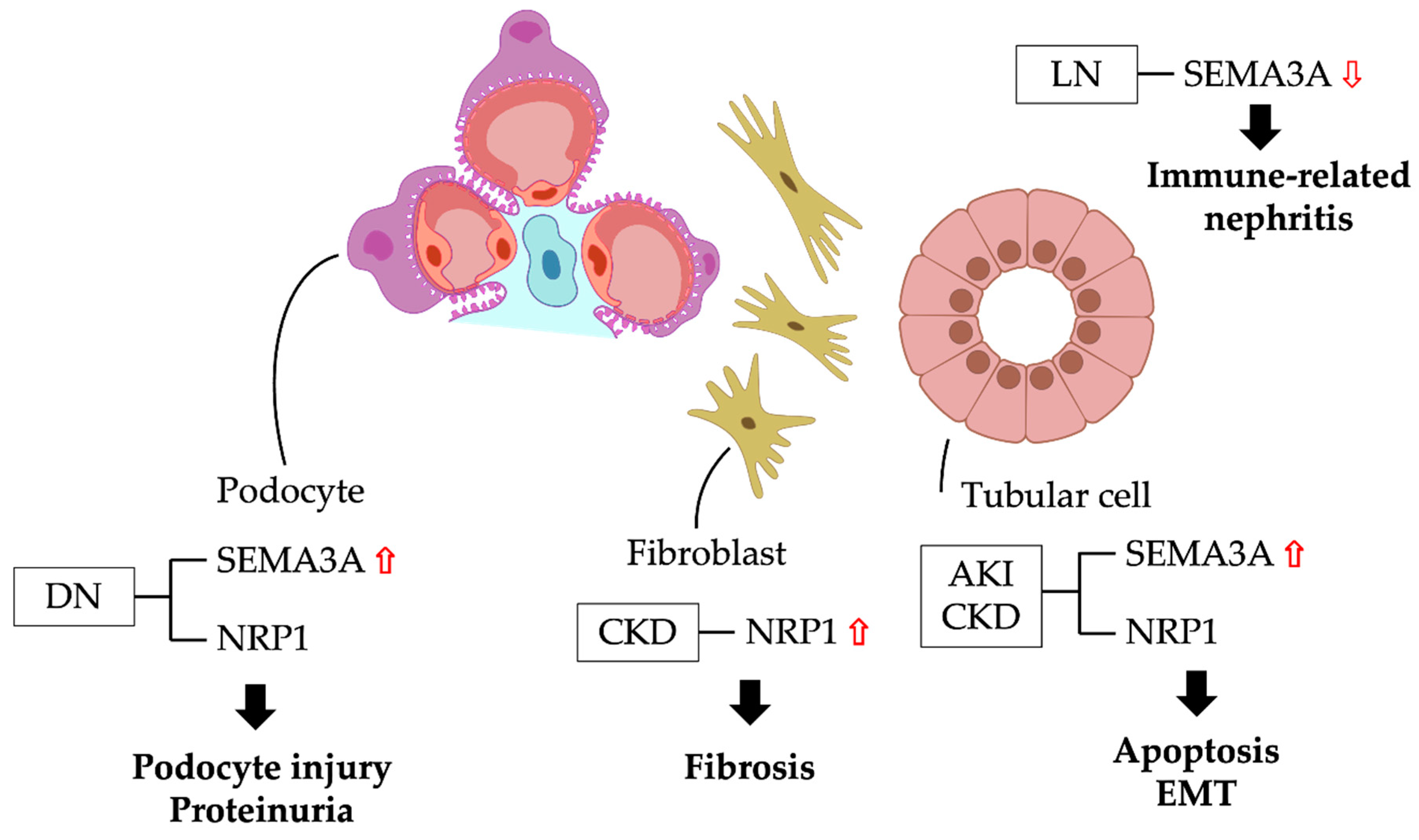
Figure 2.
Semaphroin3A signaling and pathophysiology in kidney diseases. DN: diabetic nephropathy; CKD: chronic kidney disease; AKI: acute kidney injury; LN: lupus nephritis; EMT: epithelial-mesenchymal transition; SEMA3A: semaphorin3A; NRP1: neuropillin-1.
Figure 2.
Semaphroin3A signaling and pathophysiology in kidney diseases. DN: diabetic nephropathy; CKD: chronic kidney disease; AKI: acute kidney injury; LN: lupus nephritis; EMT: epithelial-mesenchymal transition; SEMA3A: semaphorin3A; NRP1: neuropillin-1.
Author Contributions
Conceptualization, K.T.; formal analysis, S.Y., K.T., H.N. and K.F.; investigation, K.Ts., H.N. and K.F.; writing—original draft preparation, S.Y. and K.T.; writing—review and editing, K.T., H.N., K.F. S.K. and J.W.; supervision, J.W.; All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Japanese Society for the Promotion of Science (JSPS)/Grants-in-Aid for Young Scientists 18K15978 and 20K17283 (to K.T.) and Shanghai Pujiang Young Rheumatologists Training Program (SPROG2201, to Y.S.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
There are no datasets generated during and/or analyzed during the current study.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lu, Q.; Zhu, L. , The Role of Semaphorins in Metabolic Disorders. Int J Mol Sci 2020, 21, (16). [Google Scholar] [CrossRef] [PubMed]
- Alto, L. T.; Terman, J. R. , Semaphorins and their Signaling Mechanisms. Methods Mol Biol 2017, 1493, 1–25. [Google Scholar] [PubMed]
- Kiseleva, E. P.; Rutto, K. V. , Semaphorin 3A in the Immune System: Twenty Years of Study. Biochemistry (Mosc) 2022, 87, 640–657. [Google Scholar] [CrossRef]
- Raper, J. A.; Kapfhammer, J. P. , The enrichment of a neuronal growth cone collapsing activity from embryonic chick brain. Neuron 1990, 4, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Raible, D.; Raper, J. A. , Collapsin: a protein in brain that induces the collapse and paralysis of neuronal growth cones. Cell 1993, 75, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Roth, L.; Koncina, E.; Satkauskas, S.; Cremel, G.; Aunis, D.; Bagnard, D. , The many faces of semaphorins: from development to pathology. Cell Mol Life Sci 2009, 66, 649–666. [Google Scholar] [CrossRef]
- Kim, O.; Tran, P. T.; Gal, M.; Lee, S. J.; Na, S. H.; Hwangbo, C.; Lee, J. H. , RAS-stimulated release of exosomal miR-494-3p promotes the osteolytic bone metastasis of breast cancer cells. Int J Mol Med 2023, 52, (3). [Google Scholar] [CrossRef]
- Kurokawa, S.; Kashimoto, M.; Hagikura, K.; Shimodai-Yamada, S.; Otsuka, N.; Wakamatsu, Y.; Nagashima, K.; Matsumoto, T.; Hao, H.; Okumura, Y. , Intravenous Semaphorin 3A Administration Maintains Cardiac Contractility and Improves Electrical Remodeling in a Mouse Model of Isoproterenol-Induced Heart Failure. Int Heart J 2023, 64, 453–461. [Google Scholar] [CrossRef]
- Qiu, Q.; Yu, X.; Chen, Q.; He, X. , Sema3A inactivates the ERK/JNK signalling pathways to alleviate inflammation and oxidative stress in lipopolysaccharide-stimulated rat endothelial cells and lung tissues. Autoimmunity 2023, 56, 2200908. [Google Scholar] [CrossRef]
- Tian, T.; Chen, L.; Wang, Z.; Zhu, M.; Xu, W.; Wu, B. , Sema3A Drives Alternative Macrophage Activation in the Resolution of Periodontitis via PI3K/AKT/mTOR Signaling. Inflammation 2023, 46, 876–891. [Google Scholar] [CrossRef]
- Ieguchi, K.; Funakoshi, M.; Mishima, T.; Takizawa, K.; Omori, T.; Nakamura, F.; Watanabe, M.; Tsuji, M.; Kiuchi, Y.; Kobayashi, S.; Tsunoda, T.; Maru, Y.; Wada, S. , The Sympathetic Nervous System Contributes to the Establishment of Pre-Metastatic Pulmonary Microenvironments. Int J Mol Sci 2022, 23, (18). [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Nishinaka, A.; Hidaka, Y.; Shimazawa, M.; Thomas, L.; Bakker, R. A.; Hara, H. , Efficacy of an Anti-Semaphorin 3A Neutralizing Antibody in a Male Experimental Retinal Vein Occlusion Mouse Model. Invest Ophthalmol Vis Sci 2022, 63, 14. [Google Scholar] [CrossRef] [PubMed]
- Nitzan, A.; Corredor-Sanchez, M.; Galron, R.; Nahary, L.; Safrin, M.; Bruzel, M.; Moure, A.; Bonet, R.; Perez, Y.; Bujons, J.; Vallejo-Yague, E.; Sacks, H.; Burnet, M.; Alfonso, I.; Messeguer, A.; Benhar, I.; Barzilai, A.; Solomon, A. S. , Inhibition of Sema-3A Promotes Cell Migration, Axonal Growth, and Retinal Ganglion Cell Survival. Transl Vis Sci Technol 2021, 10, 16. [Google Scholar] [CrossRef]
- Kanth, S. M.; Gairhe, S.; Torabi-Parizi, P. , The Role of Semaphorins and Their Receptors in Innate Immune Responses and Clinical Diseases of Acute Inflammation. Front Immunol 2021, 12, 672441. [Google Scholar] [CrossRef] [PubMed]
- Higgins, D. M. O.; Caliva, M.; Schroeder, M.; Carlson, B.; Upadhyayula, P. S.; Milligan, B. D.; Cheshier, S. H.; Weissman, I. L.; Sarkaria, J. N.; Meyer, F. B.; Henley, J. R. , Semaphorin 3A mediated brain tumor stem cell proliferation and invasion in EGFRviii mutant gliomas. BMC Cancer 2020, 20, 1213. [Google Scholar] [CrossRef]
- Karpuz, T.; Araz, M.; Korkmaz, L.; Kilinc, I.; Findik, S.; Karaagac, M.; Eryilmaz, M. K.; Artac, M. , The Prognostic Value of Serum Semaphorin3A and VEGF Levels in Patients with Metastatic Colorectal Cancer. J Gastrointest Cancer 2020, 51, 491–497. [Google Scholar] [CrossRef]
- Sabag, A. D.; Dias-Polak, D.; Bejar, J.; Sheffer, H.; Bergman, R.; Vadasz, Z. , Altered expression of regulatory molecules in the skin of psoriasis. Immunol Res 2018, 66, 649–654. [Google Scholar] [CrossRef]
- Gao, H.; Ma, X. X.; Guo, Q.; Xie, L. F.; Zhong, Y. C.; Zhang, X. W. , Expression of circulating Semaphorin3A and its association with inflammation and bone destruction in rheumatoid arthritis. Clin Rheumatol 2018, 37, 2073–2080. [Google Scholar] [CrossRef]
- Jacob, S.; Al-Kandari, A.; Alroughani, R.; Al-Temaimi, R. , Assessment of plasma biomarkers for their association with Multiple Sclerosis progression. J Neuroimmunol 2017, 305, 5–8. [Google Scholar] [CrossRef]
- Hira, K.; Ueno, Y.; Tanaka, R.; Miyamoto, N.; Yamashiro, K.; Inaba, T.; Urabe, T.; Okano, H.; Hattori, N. , Astrocyte-Derived Exosomes Treated With a Semaphorin 3A Inhibitor Enhance Stroke Recovery via Prostaglandin D(2) Synthase. Stroke 2018, 49, 2483–2494. [Google Scholar] [CrossRef]
- Matrone, C.; Ferretti, G. , Semaphorin 3A influences neuronal processes that are altered in patients with autism spectrum disorder: Potential diagnostic and therapeutic implications. Neurosci Biobehav Rev 2023, 153, 105338. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Liu, J. W.; Wu, J.; Wu, Z. X.; Li, J.; Ji, H. F.; Liang, N. P.; Zhang, H. J.; Lai, Z. Q.; Dong, Y. F. , Inhibition of semaphorin-3a alleviates lipopolysaccharide-induced vascular injury. Microvasc Res 2022, 142, 104346. [Google Scholar] [CrossRef] [PubMed]
- Mokhtar, E. R.; Mahmoud, D. A.; Ebrahim, G. E.; Al Anany, M. G.; Seliem, N.; Hassan, M. M. , Serum metabolomic profiles and semaphorin-3A as biomarkers of diabetic retinopathy progression. Egypt J Immunol 2023, 30, 83–98. [Google Scholar] [CrossRef]
- Valiulyte, I.; Steponaitis, G.; Kardonaite, D.; Tamasauskas, A.; Kazlauskas, A. , A SEMA3 Signaling Pathway-Based Multi-Biomarker for Prediction of Glioma Patient Survival. Int J Mol Sci 2020, 21, (19). [Google Scholar] [CrossRef]
- Wang, P.; Mao, Y. M.; Liu, L. N.; Zhao, C. N.; Li, X. M.; Pan, H. F. , Decreased Expression of Semaphorin 3A and Semaphorin 7A Levels and Its Association with Systemic Lupus Erythematosus. Immunol Invest 2020, 49, (1–2), 69. [Google Scholar] [CrossRef] [PubMed]
- Izycka, N.; Sterzynska, K.; Januchowski, R.; Nowak-Markwitz, E. , Semaphorin 3A (SEMA3A), protocadherin 9 (PCdh9), and S100 calcium binding protein A3 (S100A3) as potential biomarkers of carcinogenesis and chemoresistance of different neoplasms, including ovarian cancer - review of literature. Ginekol Pol 2019, 90, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Qi, L.; Wang, F.; Sun, Z.; Huang, Z.; Xi, Q. , Decreased semaphorin 3A expression is associated with a poor prognosis in patients with epithelial ovarian carcinoma. Int J Mol Med 2015, 35, 1374–1380. [Google Scholar] [CrossRef]
- Luyckx, V. A.; Tonelli, M.; Stanifer, J. W. , The global burden of kidney disease and the sustainable development goals. Bull World Health Organ 2018, 96, 414–422D. [Google Scholar] [CrossRef]
- Collaboration, G. B. D. C. K. D. , Global, regional, and national burden of chronic kidney disease, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar]
- The, E.-K. C. G.; Herrington, W. G.; Staplin, N.; Wanner, C.; Green, J. B.; Hauske, S. J.; Emberson, J. R.; Preiss, D.; Judge, P.; Mayne, K. J.; Ng, S. Y. A.; Sammons, E.; Zhu, D.; Hill, M.; Stevens, W.; Wallendszus, K.; Brenner, S.; Cheung, A. K.; Liu, Z. H.; Li, J.; Hooi, L. S.; Liu, W.; Kadowaki, T.; Nangaku, M.; Levin, A.; Cherney, D.; Maggioni, A. P.; Pontremoli, R.; Deo, R.; Goto, S.; Rossello, X.; Tuttle, K. R.; Steubl, D.; Petrini, M.; Massey, D.; Eilbracht, J.; Brueckmann, M.; Landray, M. J.; Baigent, C.; Haynes, R. , Empagliflozin in Patients with Chronic Kidney Disease. N Engl J Med 2023, 388, 117–127. [Google Scholar]
- Heerspink, H. J. L.; Stefansson, B. V.; Correa-Rotter, R.; Chertow, G. M.; Greene, T.; Hou, F. F.; Mann, J. F. E.; McMurray, J. J. V.; Lindberg, M.; Rossing, P.; Sjostrom, C. D.; Toto, R. D.; Langkilde, A. M.; Wheeler, D. C.; Committees, D.-C. T. ; Investigators, Dapagliflozin in Patients with Chronic Kidney Disease. N Engl J Med 2020, 383, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Prattichizzo, F.; de Candia, P.; Ceriello, A. , Diabetes and kidney disease: emphasis on treatment with SGLT-2 inhibitors and GLP-1 receptor agonists. Metabolism 2021, 120, 154799. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M.; Takama, H.; Ichikawa, T.; Mukai, K.; Kojima, M.; Suzuki, Y.; Watada, H.; Wada, T.; Ueki, K.; Narita, I.; Kashihara, N.; Kadowaki, T.; Hase, H.; Akizawa, T. , Randomized, double-blind, placebo-controlled phase 3 study of bardoxolone methyl in patients with diabetic kidney disease: design and baseline characteristics of the AYAME study. Nephrol Dial Transplant 2023, 38, 1204–1216. [Google Scholar] [CrossRef] [PubMed]
- Leehey, D. J.; Carlson, K.; Reda, D. J.; Craig, I.; Clise, C.; Conner, T. A.; Agarwal, R.; Kaufman, J. S.; Anderson, R. J.; Lammie, D.; Huminik, J.; Polzin, L.; McBurney, C.; Huang, G. D.; Emanuele, N. V. , Pentoxifylline in diabetic kidney disease (VA PTXRx): protocol for a pragmatic randomised controlled trial. BMJ Open 2021, 11, e053019. [Google Scholar] [CrossRef]
- Chertow, G. M.; Pergola, P. E.; Chen, F.; Kirby, B. J.; Sundy, J. S.; Patel, U. D.; Investigators, G.-U.-. . Effects of Selonsertib in Patients with Diabetic Kidney Disease. J Am Soc Nephrol 2019, 30, 1980–1990. [Google Scholar] [CrossRef]
- Tuttle, K. R.; Brosius, F. C., 3rd; Adler, S. G.; Kretzler, M.; Mehta, R. L.; Tumlin, J. A.; Tanaka, Y.; Haneda, M.; Liu, J.; Silk, M. E.; Cardillo, T. E.; Duffin, K. L.; Haas, J. V.; Macias, W. L.; Nunes, F. P.; Janes, J. M. , JAK1/JAK2 inhibition by baricitinib in diabetic kidney disease: results from a Phase 2 randomized controlled clinical trial. Nephrol Dial Transplant 2018, 33, 1950–1959. [Google Scholar] [CrossRef]
- Lin, C. W.; Mostafa, N. M.; D, L. A.; J, J. B.; Klein, C. E.; Awni, W. M. , Relationship Between Atrasentan Concentrations and Urinary Albumin to Creatinine Ratio in Western and Japanese Patients With Diabetic Nephropathy. Clin Ther 2018, 40, 242–251. [Google Scholar] [CrossRef]
- Toledano, S.; Nir-Zvi, I.; Engelman, R.; Kessler, O.; Neufeld, G. , Class-3 Semaphorins and Their Receptors: Potent Multifunctional Modulators of Tumor Progression. Int J Mol Sci 2019, 20, (3). [Google Scholar] [CrossRef]
- Tamagnone, L.; Artigiani, S.; Chen, H.; He, Z.; Ming, G. I.; Song, H.; Chedotal, A.; Winberg, M. L.; Goodman, C. S.; Poo, M.; Tessier-Lavigne, M.; Comoglio, P. M. , Plexins are a large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell 1999, 99, 71–80. [Google Scholar] [CrossRef]
- Janssen, B. J.; Malinauskas, T.; Weir, G. A.; Cader, M. Z.; Siebold, C.; Jones, E. Y. , Neuropilins lock secreted semaphorins onto plexins in a ternary signaling complex. Nat Struct Mol Biol 2012, 19, 1293–1299. [Google Scholar] [CrossRef]
- Lu, D.; Shang, G.; He, X.; Bai, X. C.; Zhang, X. , Architecture of the Sema3A/PlexinA4/Neuropilin tripartite complex. Nat Commun 2021, 12, 3172. [Google Scholar] [CrossRef]
- Aggarwal, P. K.; Veron, D.; Thomas, D. B.; Siegel, D.; Moeckel, G.; Kashgarian, M.; Tufro, A. , Semaphorin3a promotes advanced diabetic nephropathy. Diabetes 2015, 64, 1743–1759. [Google Scholar] [CrossRef]
- Jayakumar, C.; Ranganathan, P.; Devarajan, P.; Krawczeski, C. D.; Looney, S.; Ramesh, G. , Semaphorin 3A is a new early diagnostic biomarker of experimental and pediatric acute kidney injury. PLoS One 2013, 8, e58446. [Google Scholar] [CrossRef] [PubMed]
- Tapia, R.; Guan, F.; Gershin, I.; Teichman, J.; Villegas, G.; Tufro, A. , Semaphorin3a disrupts podocyte foot processes causing acute proteinuria. Kidney Int 2008, 73, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Gan, H.; Zeng, Y.; Zhao, H.; Tang, R.; Xia, Y. , Inhibition of semaphorin-3a suppresses lipopolysaccharide-induced acute kidney injury. J Mol Med (Berl) 2018, 96, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Sang, Y.; Tsuji, K.; Inoue-Torii, A.; Fukushima, K.; Kitamura, S.; Wada, J. , Semaphorin3A-Inhibitor Ameliorates Doxorubicin-Induced Podocyte Injury. Int J Mol Sci 2020, 21, (11). [Google Scholar] [CrossRef]
- Tufro, A. , Semaphorin3a signaling, podocyte shape, and glomerular disease. Pediatr Nephrol 2014, 29, 751–755. [Google Scholar] [CrossRef]
- Reidy, K. J.; Villegas, G.; Teichman, J.; Veron, D.; Shen, W.; Jimenez, J.; Thomas, D.; Tufro, A. , Semaphorin3a regulates endothelial cell number and podocyte differentiation during glomerular development. Development 2009, 136, 3979–3989. [Google Scholar] [CrossRef]
- Guan, F.; Villegas, G.; Teichman, J.; Mundel, P.; Tufro, A. , Autocrine class 3 semaphorin system regulates slit diaphragm proteins and podocyte survival. Kidney Int 2006, 69, 1564–1569. [Google Scholar] [CrossRef]
- Wu, H.; Kirita, Y.; Donnelly, E. L.; Humphreys, B. D. , Advantages of Single-Nucleus over Single-Cell RNA Sequencing of Adult Kidney: Rare Cell Types and Novel Cell States Revealed in Fibrosis. J Am Soc Nephrol 2019, 30, 23–32. [Google Scholar] [CrossRef]
- Kolodkin, A. L.; Levengood, D. V.; Rowe, E. G.; Tai, Y. T.; Giger, R. J.; Ginty, D. D. , Neuropilin is a semaphorin III receptor. Cell 1997, 90, 753–762. [Google Scholar] [CrossRef]
- Omoto, M.; Yoshida, S.; Miyashita, H.; Kawakita, T.; Yoshida, K.; Kishino, A.; Kimura, T.; Shibata, S.; Tsubota, K.; Okano, H.; Shimmura, S. , The semaphorin 3A inhibitor SM-345431 accelerates peripheral nerve regeneration and sensitivity in a murine corneal transplantation model. PLoS One 2012, 7, e47716. [Google Scholar] [CrossRef] [PubMed]
- Ivakhnitskaia, E.; Chin, M. R.; Siegel, D.; Guaiquil, V. H. , Vinaxanthone inhibits Semaphorin3A induced axonal growth cone collapse in embryonic neurons but fails to block its growth promoting effects on adult neurons. Sci Rep 2021, 11, 13019. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, R.; Yamazoe, K.; Yoshida, S.; Hatou, S.; Inagaki, E.; Okano, H.; Tsubota, K.; Shimmura, S. , The Semaphorin 3A inhibitor SM-345431 preserves corneal nerve and epithelial integrity in a murine dry eye model. Sci Rep 2017, 7, 15584. [Google Scholar] [CrossRef] [PubMed]
- Kretschmer, K.; Stichel, J.; Bellmann-Sickert, K.; Baumann, L.; Bierer, D.; Riedl, B.; Beck-Sickinger, A. G. , Pinpointing the interaction site between semaphorin-3A and its inhibitory peptide. J Pept Sci 2023, 29, e3460. [Google Scholar] [CrossRef]
- Tran, T. S.; Kolodkin, A. L.; Bharadwaj, R. , Semaphorin regulation of cellular morphology. Annu Rev Cell Dev Biol 2007, 23, 263–292. [Google Scholar] [CrossRef]
- Reidy, K.; Tufro, A. , Semaphorins in kidney development and disease: modulators of ureteric bud branching, vascular morphogenesis, and podocyte-endothelial crosstalk. Pediatr Nephrol 2011, 26, 1407–1412. [Google Scholar] [CrossRef]
- Tufro, A.; Teichman, J.; Woda, C.; Villegas, G. , Semaphorin3a inhibits ureteric bud branching morphogenesis. Mech Dev 2008, 125, (5–6), 558. [Google Scholar] [CrossRef]
- Inoue-Torii, A.; Kitamura, S.; Wada, J.; Tsuji, K.; Makino, H. , The level of urinary semaphorin3A is associated with disease activity in patients with minimal change nephrotic syndrome. Int J Nephrol Renovasc Dis 2017, 10, 167–174. [Google Scholar] [CrossRef]
- Mohamed, R.; Ranganathan, P.; Jayakumar, C.; Nauta, F. L.; Gansevoort, R. T.; Weintraub, N. L.; Brands, M.; Ramesh, G. , Urinary semaphorin 3A correlates with diabetic proteinuria and mediates diabetic nephropathy and associated inflammation in mice. J Mol Med (Berl) 2014, 92, 1245–1256. [Google Scholar] [CrossRef]
- Veron, D.; Bertuccio, C. A.; Marlier, A.; Reidy, K.; Garcia, A. M.; Jimenez, J.; Velazquez, H.; Kashgarian, M.; Moeckel, G. W.; Tufro, A. , Podocyte vascular endothelial growth factor (Vegf(1)(6)(4)) overexpression causes severe nodular glomerulosclerosis in a mouse model of type 1 diabetes. Diabetologia 2011, 54, 1227–1241. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, P.; Jayakumar, C.; Mohamed, R.; Weintraub, N. L.; Ramesh, G. , Semaphorin 3A inactivation suppresses ischemia-reperfusion-induced inflammation and acute kidney injury. Am J Physiol Renal Physiol 2014, 307, F183–F194. [Google Scholar] [CrossRef] [PubMed]
- Ning, L.; Li, Z.; Wei, D.; Chen, H.; Yang, C.; Wu, D.; Wang, Y.; Zhang, J. , Urinary semaphorin 3A as an early biomarker to predict contrast-induced acute kidney injury in patients undergoing percutaneous coronary intervention. Braz J Med Biol Res 2018, 51, e6487. [Google Scholar] [CrossRef] [PubMed]
- Sang, Y.; Tsuji, K.; Fukushima, K.; Takahashi, K.; Kitamura, S.; Wada, J. , Semaporin3A inhibitor ameliorates renal fibrosis through the regulation of JNK signaling. Am J Physiol Renal Physiol 2021, 321, F740–F756. [Google Scholar] [CrossRef] [PubMed]
- Vadasz, Z.; Ben-Izhak, O.; Bejar, J.; Sabo, E.; Kessel, A.; Storch, S.; Toubi, E. , The involvement of immune semaphorins and neuropilin-1 in lupus nephritis. Lupus 2011, 20, 1466–1473. [Google Scholar] [CrossRef]
- Vadasz, Z.; Toubi, E. , Semaphorin 3A - a marker for disease activity and a potential putative disease-modifying treatment in systemic lupus erythematosus. Lupus 2012, 21, 1266–1270. [Google Scholar] [CrossRef]
- Koehler, S.; Miner, J. H.; Staruschenko, A. , Call for papers: podocyte physiology and pathophysiology. Am J Physiol Renal Physiol 2023, 324, F505–F510. [Google Scholar] [CrossRef]
- Tufro, A. , Podocyte Shape Regulation by Semaphorin 3A and MICAL-1. Methods Mol Biol 2017, 1493, 393–399. [Google Scholar]
- Reidy, K. J.; Aggarwal, P. K.; Jimenez, J. J.; Thomas, D. B.; Veron, D.; Tufro, A. , Excess podocyte semaphorin-3A leads to glomerular disease involving plexinA1-nephrin interaction. Am J Pathol 2013, 183, 1156–1168. [Google Scholar] [CrossRef]
- Kumagai, K.; Hosotani, N.; Kikuchi, K.; Kimura, T.; Saji, I. , Xanthofulvin, a novel semaphorin inhibitor produced by a strain of Penicillium. J Antibiot (Tokyo) 2003, 56, 610–616. [Google Scholar] [CrossRef]
- Samsu, N. , Diabetic Nephropathy: Challenges in Pathogenesis, Diagnosis, and Treatment. Biomed Res Int 2021, 2021, 1497449. [Google Scholar] [CrossRef] [PubMed]
- Valencia, W. M.; Florez, H. , How to prevent the microvascular complications of type 2 diabetes beyond glucose control. BMJ 2017, 356, i6505. [Google Scholar] [CrossRef] [PubMed]
- Burrows, N. R.; Hora, I.; Geiss, L. S.; Gregg, E. W.; Albright, A. , Incidence of End-Stage Renal Disease Attributed to Diabetes Among Persons with Diagnosed Diabetes - United States and Puerto Rico, 2000-2014. MMWR Morb Mortal Wkly Rep 2017, 66, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Lane, P. H.; Steffes, M. W.; Mauer, S. M. , Renal histologic changes in diabetes mellitus. Semin Nephrol 1990, 10, 254–259. [Google Scholar]
- Maezawa, Y.; Takemoto, M.; Yokote, K. , Cell biology of diabetic nephropathy: Roles of endothelial cells, tubulointerstitial cells and podocytes. J Diabetes Investig 2015, 6, 3–15. [Google Scholar] [CrossRef]
- Cao, Z.; Cooper, M. E. , Pathogenesis of diabetic nephropathy. J Diabetes Investig 2011, 2, 243–247. [Google Scholar] [CrossRef]
- Kumar Pasupulati, A.; Chitra, P. S.; Reddy, G. B. , Advanced glycation end products mediated cellular and molecular events in the pathology of diabetic nephropathy. Biomol Concepts 2016, 7, (5–6), 293. [Google Scholar] [CrossRef]
- Fu, Y.; Wang, C.; Zhang, D.; Chu, X.; Zhang, Y.; Li, J. , miR-15b-5p ameliorated high glucose-induced podocyte injury through repressing apoptosis, oxidative stress, and inflammatory responses by targeting Sema3A. J Cell Physiol 2019, 234, 20869–20878. [Google Scholar] [CrossRef]
- Fei, B.; Zhou, H.; He, Z.; Wang, S. , KCNQ1OT1 inhibition alleviates high glucose-induced podocyte injury by adsorbing miR-23b-3p and regulating Sema3A. Clin Exp Nephrol 2022, 26, 385–397. [Google Scholar] [CrossRef]
- Jiang, Z.; Qian, L.; Yang, R.; Wu, Y.; Guo, Y.; Chen, T. , LncRNA TCF7 contributes to high glucose-induced damage in human podocytes by up-regulating SEMA3A via sponging miR-16-5p. J Diabetes Investig 2023, 14, 193–204. [Google Scholar] [CrossRef]
- Singbartl, K.; Kellum, J. A. , AKI in the ICU: definition, epidemiology, risk stratification, and outcomes. Kidney Int 2012, 81, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Jang, H. R.; Rabb, H. , Immune cells in experimental acute kidney injury. Nat Rev Nephrol 2015, 11, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Kellum, J. A.; Romagnani, P.; Ashuntantang, G.; Ronco, C.; Zarbock, A.; Anders, H. J. , Acute kidney injury. Nat Rev Dis Primers 2021, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Lewandowska, L.; Matuszkiewicz-Rowinska, J.; Jayakumar, C.; Oldakowska-Jedynak, U.; Looney, S.; Galas, M.; Dutkiewicz, M.; Krawczyk, M.; Ramesh, G. , Netrin-1 and semaphorin 3A predict the development of acute kidney injury in liver transplant patients. PLoS One 2014, 9, e107898. [Google Scholar] [CrossRef]
- Parikh, C. R.; Mishra, J.; Thiessen-Philbrook, H.; Dursun, B.; Ma, Q.; Kelly, C.; Dent, C.; Devarajan, P.; Edelstein, C. L. , Urinary IL-18 is an early predictive biomarker of acute kidney injury after cardiac surgery. Kidney Int 2006, 70, 199–203. [Google Scholar] [CrossRef]
- Doi, K.; Negishi, K.; Ishizu, T.; Katagiri, D.; Fujita, T.; Matsubara, T.; Yahagi, N.; Sugaya, T.; Noiri, E. , Evaluation of new acute kidney injury biomarkers in a mixed intensive care unit. Crit Care Med 2011, 39, 2464–2469. [Google Scholar] [CrossRef]
- Bennett, M.; Dent, C. L.; Ma, Q.; Dastrala, S.; Grenier, F.; Workman, R.; Syed, H.; Ali, S.; Barasch, J.; Devarajan, P. , Urine NGAL predicts severity of acute kidney injury after cardiac surgery: a prospective study. Clin J Am Soc Nephrol 2008, 3, 665–673. [Google Scholar] [CrossRef]
- Tian, M.; Zhao, S. , [The early diagnostic value of urinary Sema3A for ICU adult patients with acute kidney injury]. Zhonghua Yi Xue Za Zhi 2015, 95, 1457–1462. [Google Scholar]
- Doi, K.; Noiri, E.; Nangaku, M.; Yahagi, N.; Jayakumar, C.; Ramesh, G. , Repulsive guidance cue semaphorin 3A in urine predicts the progression of acute kidney injury in adult patients from a mixed intensive care unit. Nephrol Dial Transplant 2014, 29, 73–80. [Google Scholar] [CrossRef]
- Fang, W.; Wang, Z.; Li, Q.; Wang, X.; Zhang, Y.; Sun, Y.; Tang, W.; Ma, C.; Sun, J.; Li, N.; Yi, F. , Gpr97 Exacerbates AKI by Mediating Sema3A Signaling. J Am Soc Nephrol 2018, 29, 1475–1489. [Google Scholar] [CrossRef]
- Kar, F.; Hacioglu, C.; Senturk, H.; Donmez, D. B.; Kanbak, G.; Uslu, S. , Curcumin and LOXblock-1 ameliorate ischemia-reperfusion induced inflammation and acute kidney injury by suppressing the semaphorin-plexin pathway. Life Sci 2020, 256, 118016. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Pei, L.; Lin, F.; Yin, H.; Li, X.; He, W.; Liu, N.; Gou, X. , Exosomes from human-bone-marrow-derived mesenchymal stem cells protect against renal ischemia/reperfusion injury via transferring miR-199a-3p. J Cell Physiol 2019, 234, 23736–23749. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ortega, M.; Rayego-Mateos, S.; Lamas, S.; Ortiz, A.; Rodrigues-Diez, R. R. , Targeting the progression of chronic kidney disease. Nat Rev Nephrol 2020, 16, 269–288. [Google Scholar] [CrossRef] [PubMed]
- Viazzi, F.; Ramesh, G.; Jayakumar, C.; Leoncini, G.; Garneri, D.; Pontremoli, R. , Increased urine semaphorin-3A is associated with renal damage in hypertensive patients with chronic kidney disease: a nested case-control study. J Nephrol 2015, 28, 315–320. [Google Scholar] [CrossRef]
- Iwano, M.; Plieth, D.; Danoff, T. M.; Xue, C.; Okada, H.; Neilson, E. G. , Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 2002, 110, 341–350. [Google Scholar] [CrossRef]
- Takagawa, S.; Nakamura, F.; Kumagai, K.; Nagashima, Y.; Goshima, Y.; Saito, T. , Decreased semaphorin3A expression correlates with disease activity and histological features of rheumatoid arthritis. BMC Musculoskelet Disord 2013, 14, 40. [Google Scholar] [CrossRef]
- Rimar, D.; Nov, Y.; Rosner, I.; Slobodin, G.; Rozenbaum, M.; Halasz, K.; Haj, T.; Jiries, N.; Kaly, L.; Boulman, N.; Vadasz, Z. , Semaphorin 3A: an immunoregulator in systemic sclerosis. Rheumatol Int 2015, 35, 1625–1630. [Google Scholar] [CrossRef]
- Kiriakidou, M.; Ching, C. L. , Systemic Lupus Erythematosus. Ann Intern Med 2020, 172, ITC81–ITC96. [Google Scholar] [CrossRef]
- Kessel, A.; Haj, T.; Peri, R.; Snir, A.; Melamed, D.; Sabo, E.; Toubi, E. , Human CD19(+)CD25(high) B regulatory cells suppress proliferation of CD4(+) T cells and enhance Foxp3 and CTLA-4 expression in T-regulatory cells. Autoimmun Rev 2012, 11, 670–677. [Google Scholar] [CrossRef]
- Luft, F. C. , Semaphorin-3A is a repulsive but attractive renal guidance cue to therapy. J Mol Med (Berl) 2014, 92, 1225–1227. [Google Scholar] [CrossRef]
- Summers, S. A.; Hoi, A.; Steinmetz, O. M.; O’Sullivan, K. M.; Ooi, J. D.; Odobasic, D.; Akira, S.; Kitching, A. R.; Holdsworth, S. R. , TLR9 and TLR4 are required for the development of autoimmunity and lupus nephritis in pristane nephropathy. J Autoimmun 2010, 35, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Capolunghi, F.; Rosado, M. M.; Cascioli, S.; Girolami, E.; Bordasco, S.; Vivarelli, M.; Ruggiero, B.; Cortis, E.; Insalaco, A.; Fanto, N.; Gallo, G.; Nucera, E.; Loiarro, M.; Sette, C.; De Santis, R.; Carsetti, R.; Ruggiero, V. , Pharmacological inhibition of TLR9 activation blocks autoantibody production in human B cells from SLE patients. Rheumatology (Oxford) 2010, 49, 2281–2289. [Google Scholar] [CrossRef] [PubMed]
- Doron, R.; Merav, L.; Nasrin, E.; Adi, S. D.; Elias, T.; Gleb, S.; Itzhak, R.; Michael, R.; Zahava, V. , Low Urine Secretion of Semaphorin3A in Lupus Patients with Proteinuria. Inflammation 2022, 45, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Bejar, J.; Kessler, O.; Sabag, A. D.; Sabo, E.; Itzhak, O. B.; Neufeld, G.; Vadasz, Z. , Semaphorin3A: A Potential Therapeutic Tool for Lupus Nephritis. Front Immunol 2018, 9, 634. [Google Scholar] [CrossRef]
Figure 1.
The structure of SEMA3A and its receptors, NRP1 and PlexinA. SEMA3A: semaphorin3A; SEMA3A-I: semaphorin3A inhibitor; NRP1: neuropirin-1; N: N-terminal; SEMA: SEMA domain; PSI: cysteine-rich plexin-semaphorin-integrin domain; Ig: immunoglobulin-like domain; Basic: basic domain; C: C-terminal; a1: complement binding domain a1; a2: complement binding domain a2; b1: coagulation factor V homology domain; b2: coagulation factor VIII homology domain; C: c-domain; IPT: Ig-like, plexin and transcription factor domain; GAP domain: GAP domain GTPase activating protein.
Figure 1.
The structure of SEMA3A and its receptors, NRP1 and PlexinA. SEMA3A: semaphorin3A; SEMA3A-I: semaphorin3A inhibitor; NRP1: neuropirin-1; N: N-terminal; SEMA: SEMA domain; PSI: cysteine-rich plexin-semaphorin-integrin domain; Ig: immunoglobulin-like domain; Basic: basic domain; C: C-terminal; a1: complement binding domain a1; a2: complement binding domain a2; b1: coagulation factor V homology domain; b2: coagulation factor VIII homology domain; C: c-domain; IPT: Ig-like, plexin and transcription factor domain; GAP domain: GAP domain GTPase activating protein.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



