
Submitted:
30 August 2023
Posted:
31 August 2023
You are already at the latest version
Abstract
Infection with Ebola virus (EBOV) is responsible for haemorrhagic fever in humans with a high mortality rate. Combined efforts of prevention and therapeutic intervention are required to tackle highly variable RNA viruses, whose infections often lead to outbreaks. Here, we have screened the 2P2I3D chemical library using a nanoluc-based protein complementation assay (NPCA) and isolated two compounds disrupting the interaction of the EBOV protein fragment VP35IID with the N terminus of the dsRNA-binding proteins PKR and PACT, involved in IFN response and/or intrinsic immunity, respectively. The two compounds inhibited EBOV infection in cell culture as well as infection by measles virus (MV) independently of IFN induction. Consequently, we propose the compounds are antiviral by restoring intrinsic immunity driven by PACT. Given that PACT is highly conserved across mammals, we support further testing of the compounds in other species, as well as against further negative sense RNA viruses.
Keywords:
Ebola virus
; measles virus
; VP35
; PKR
; PACT
; RIG-I
; drug screen.
1. Introduction
Ebola virus (EBOV) remains a public health concern since periodic outbreaks occur in Africa and several cases have spread into other continents (see [1] for a recent review). In humans, EBOV causes a cytokine storm leading to haemorrhagic fever responsible of its high lethality rate [2]. Current FDA approved treatments are monoclonal antibodies only evaluated for efficacy against Zaire EBOV strain (https://www.cdc.gov/vhf/ebola/treatment/index.html). Therefore, complementary treatments with broad action are required for this rapidly evolving RNA virus [3].
EBOV is an enveloped non-segmented negative sense single stranded RNA ((-)ssRNA) filovirus that contains seven genes arranged from 3’ to 5’ encoding the nucleoprotein (NP), viral proteins VP35, VP40, VP24, VP30, glycoprotein (GP), and the RNA-dependent RNA polymerase [4]. The structure and crosstalk between these proteins have been uncovered through numerous studies aiming at identifying druggable targets [5,6,7,8,9,10]. One of the EBOV proteins in the spotlight is VP35 since it is a cofactor of the viral RNA-polymerase complex required for viral replication [5,7,10]. VP35 structure comprises an N-terminus domain (NTD, aa 180) followed by an oligomerization domain (OD, aa 80-145) and a C-terminus domain (CTD, aa 215-340). VP35 CTD has been also termed interferon inhibitory domain (IID) because of its implication in counteracting multiple antiviral responses; RNA-based viruses as EBOV tend to generate secondary structures consisting of double stranded RNA (dsRNA) which are immunogenic, that is, can be detected by a variety of dsRNA intracellular sensors to trigger an immune response to clear the invading RNA [11]. However, VP35IID binding to viral dsRNA masks its recognition by host dsRNA-sensors to avoid their driven IFN-α/β production [12,13]. More in detail, VP35IID contains a key hydrophobic residue (F239) whose sidechain contacts dsRNA blunt ends, as well as a central basic patch (CBP) where amino acid (aa) residues R312 and R322 provide a charged surface for dsRNA binding, while R305, K309 and K319 increase dsRNA interaction strength [14]. Both main VP35 functions (polymerase cofactor and dsRNA binding) have been targeted in silico [15,16,17,18].
In parallel, VP35IID interacts directly with protein kinase dsRNA dependent (PKR) through paired combinations of CBP residues R305/K309/R312, preventing its activation to bypass its role in translation arrest, subsequently allowing viral translation to occur [19,20,21]. Moreover, VP35IID binding to PKR activator (PACT) [22] prevents its interaction and activation of retinoic acid-inducible gene I (RIG-I), further impairing IFN production [12,23], as well as the PACT-mediated activation of RNAi [22,24]. PKR and PACT complex interplay with other RIG-I-like Receptors (RLRs) as MDA5 and LGP2 is less characterized [25], especially in the context of EBOV infection.
Since VP35IID interaction with PKR and PACT impairs their antiviral functions, we hypothesized that releasing PKR and PACT from VP35IID sequestration may restore their antiviral potential facilitating viral clearance. Accordingly, our goal in this study was to screen for compounds disrupting such interactions. To do so, we set up an in cellula nanoluciferase protein complementation assay (NPCA, [26]) to screen a pre-filtered library of chemical compounds [27,28]. The efficacy of the identified compounds as antiviral agents against EBOV and measles virus was addressed, as well as their effect on the intrinsic and innate immune response.
2. Results
2.1. Generation and assessment of a VP35IID/NtPKR and VP35IID/PACT NPCA.
In order to identify inhibitors of the interaction between VP35IID and PKR or PACT, we established an in cellula nanoluciferase protein complementation assay (NPCA). For this, we constructed plasmids expressing VP35IID, the N-terminus of PKR (NtPKR; aa 1-265) as well as full length PACT (Figure 1a). In addition, to enhance the specificity of this assay, we generated plasmids individually expressing each of their double stranded RNA binding domains (DRBDs) abbreviated as K1 and K2 for PKR, and A1 and A2 for PACT. In the case of PACT, we also generated constructs with A1 and A2 together (NtPACT), and A2 with M3 (A2M3), where M3 represents the CTD of PACT (Figure 1a). Each construct was generated in all possible combinations namely, in fusion with each luciferase moiety inserted in either the N- or C-terminus of each protein. For each assay, reconstituted luciferase activity resulting from the protein interactions was measured 24 h after co-transfection in HEK293T cells. NtPKR/PACT homo- or hetero- dimerization served as positive controls, given their known ability to interact through their DRBDs [29]. Since the strongest luciferase signal was obtained when the luciferase moieties 1 and 2 were fused to the N-terminus of NtPKR or PACT (N1) and VP35IID (N2), respectively (Figure S1), we used these constructs combinations subsequently.
We observed that the luciferase signal was higher when VP35IID interacted with NtPKR than with PACT (Figure 1b), presumably because individual DRBDs from PKR (K1 and K2) interact qualitatively stronger with VP35IID than the ones from PACT (A1 and A2) (Figure 1b). Therefore, the NtPACT and A2M3 constructs of PACT were generated to determine whether the M3 region was somehow masking PACT DRBDs, which would account for the lower luciferase signal in the VP35IID/PACT pair than VP35IID/NtPKR one. However, the luciferase signal resulting from the VP35IID/NtPACT or VP35IID/A2M3 interaction was weaker as compared with that obtained when VP35IID interacts with full length PACT (Figure 1c). Thus, full length PACT and the two DRBDs of PKR (NtPKR) were required for the strongest interaction with VP35IID.
To further assess the specificity of the VP35IID interactions with NtPKR or PACT by NPCA, as well as ensuring this system could display a detectible drop in luciferase signal when protein-protein interactions are compromised, we generated a panel of VP35IID constructs with relevant aa substitutions to alanine (Figure 1d). The interaction of VP35IID with NtPKR was significantly reduced with the K309A/R312A (p-value of 0.0433) and qualitatively decreased in R305A/R312A VP35IID, confirming the role of these residues in the PKR/VP35 interaction [20]. In contrast, only a qualitative decrease could be detected for the interaction with PACT. These results confirm the reported importance of the VP35IID central basic patch in the interactions with PKR and PACT, their interaction strength, and give an indication of the luc signal decrease to expect from inhibitory compounds. Of note, we evidence here that VP35IID interactions with NtPKR or PACT can take place in the absence of viral dsRNA, suggesting for the first time that their functionality may be compromised irrespective of dsRNA binding. This is of importance because other screens [15] have focused on abrogating the specific interaction between VP35 and dsRNA, which would restore IFN induction presumably enhancing a systemic cytokine storm.
In conclusion, since the strongest luciferase signals were obtained when VP35IID was co-expressed with NtPKR or full length PACT all tagged in their N-terminus, we proceeded to use this NPCA as platform towards screening compounds which would abrogate these interactions.
2.2. Identification of compounds targeting the interaction of VP35IID with PACT or NtPKR.
To set up the screening platform abrogating protein interactions between VP35IID and NtPKR or PACT, HEK293T cells were transfected with the plasmid N2-VP35IID and either the plasmids N1-NtPKR or N1-PACT. After 24 h, the cells were trypsinated and distributed into 96-well-plates containing the 2P2I3D library of 1,664 compounds, each at a final concentration of 20 μM [27] as described in Materials and Methods. Luciferase activity measured after another 24 h yielded 287 hits for VP35IID/NtPKR and 262 hits for VP35IID/PACT that could inhibit the luciferase activity by 60-90%. False positives due to direct interference with the luciferase enzymatic activity and/or to cytotoxicity were then discarded through two additional screenings, as well as non-specific hits known to be frequently selected whatever the screening procedure. From these three rounds of screening and analysis (Figure 2a), 44 compounds confirmed their ability to: i) inhibit luciferase activity less than 1%, ii) reduce cell viability to less than 1% and iii) decrease the VP35IID/PACT and/or VP35IID/NtPKR split luc signal in a 60-90% range. Out of these 44 compounds, 41 were available from the suppliers and were further tested at four different concentrations (1, 5, 10 and 20 μM) on HEK293T cells 24 h post-transfection with each pair of plasmids (VP35IID/NtPKR or VP35IID/PACT) (Figure S2a). Compounds 13 and 36 (reference K221-3357/ MolPort-007-903-447 (ChemDiv) and STK283971/ MolPort-002-995-533 (Vitas-M Laboratory), respectively, (Figure S2b) were the most efficient in inhibiting the interaction of VP35IID with either NtPKR or PACT (Figure S2a,b) therefore, they were selected for further study.
To ensure that the compounds were not cytotoxic, an additional MTT assay was performed in another cell line (Huh7.25/CD81) upon 24 h and 72 h treatment. After 24 h, cell viability was preserved for both compounds, while at 72 h only a dose-response toxicity was observed for compound 36 at concentrations around 7 µM; compound 13 rather appeared to be cytostatic, since cell viability did not decrease from 30 % even above 10 µM (Figure S3).
2.3. Compounds 13 and 36 decrease EBOV RNA and infectious virus.
Next, we tested the effect of either compound at 0, 1, 5 and 10 µM on two cell types (HEK293T and Huh7) uninfected or infected with EBOV (MOI 0.1) for 24, 48 and 72 hours. Once more, both cell lines were assessed for viability through the experiment through a different assay (CellTiter-Glo) (Figure S4 a and b); again, cell viability was generally not affected by addition of the compounds, except for compound 13 at 10 µM at 3 dpi in both cell types, independently of EBOV infection.
The effect of the compounds on EBOV was determined by quantifying the amount of intracellular (internal) and extracellular (external) viral RNA at 24 (Day 1) and 72 (Day 3) hours post infection (hpi), respectively, as well as production of infectious virus released in the supernatant at these time points (EBOV titer) (Figure 3a). We evidenced that the presence of both intracellular and extracellular EBOV RNA was reduced by the compounds in a dose-dependent manner already 24 hpi (Figure 3b), and the production of the infectious virus (titer) qualitatively decreased between 0.5-1.5 logs (Figure 3c).
Given the reported ability of VP35 to coat and mask viral dsRNA from dsRNA sensors to prevent IFN induction [14], we wanted to assess whether impairing VP25IID from interacting with two dsRNA sensors (PKR and PACT) would have an impact on IFN induction. Of note, we believe most of the abrogation of IFN induction by VP35 is precisely through its dsRNA binding capability, which is not directly targeted with this NPCA; we designed the NPCA screen to abrogate the interaction between VP35IID with NtPKR or PACT to unlock their antiviral effect against EBOV evidenced in the previous section. Therefore, as expected, upon assessing the RNA expression of the two early (IFNβ, IFNα1) and one late (IFNα2) IFN genes 24 hpi, IFNs were not induced by EBOV infection (Figure S5), neither dose-response changes were observed in the presence of the compounds.
2.4. Compounds 13 and 36 also impair MV independently of IFN.
Given that dsRNA sensors are at the frontline defense against RNA virus infections, these proteins are common targets of RNA viruses (reviewed in [11]). Therefore, we next investigated whether the above anti-EBOV compounds could be antiviral against another (-)ssRNA virus. For this, we used measles virus (MV, family Paramyxoviridae), harboring or not (MVΔV) the non-structural virulence V protein [30]. In agreement with the above observations, the two compounds reduced the intracellular presence of the two MV variants in a dose dependent manner as shown by RT-qPCR and immunoblot through decreased expression of the nucleoprotein (N) (Figure 4a) independently of IFNβ transcription (Figure 4b). Moreover, PKR and PACT total protein levels remained constant throughout the experiments, suggesting that they could be available to perform other functions. Altogether, these data show that compounds 13 and 36 are antiviral agents, at least against negative RNA viruses such as EBOV and MV, and that their effect is independent of the IFN induction pathway.
2.5. Mechanism of action of compounds 13 and 36
We then examined the effect of compounds 13 and 36 on combinatorial associations of the RIG-I/PACT/PKR proteins using NPCA. We corroborated the ability of RIG-I to interact with PACT and NtPKR [31,32], and we showed that VP35IID can also interact with RIG-I. Moreover, all interactions were dose-response inhibited by compound 13 and with less efficacy by compound 36 (Figure 5a). Thus, in addition to interfere with the interaction of VP35IID with PACT or NtPKR, the compounds may interfere with interactions of PACT or NtPKR with other RLRs as RIG-I.
We have previously reported that the association of PKR and PACT is involved in the induction of pro-inflammatory cytokines in response to stress, and this association could be inhibited at the level of their DRBDs by luteolin, a natural compound member of the flavonoid family [33], reviewed in [34]. We therefore compared the action of the compounds 13 and 36 to that of luteolin and observed that they could also inhibit the NtPKR/PACT interaction, as well as their interaction with VP35IID (Figure 5b). Therefore, it is tempting to speculate that by interfering with protein interactions involved in EBOV or MV infections, as well as in IFN and pro-inflammatory cytokines induction, compounds 13 and 36 behave as antiviral agents while controlling the inflammatory response often associated with these viral infections.
3. Discussion
In this study, we have screened a chemical library of 1,664 compounds and identified two compounds disrupting the interaction of the EBOV protein fragment VP35IID with the cellular proteins PACT and NtPKR. K221-3357 and STK283971 (referred here as compounds 13 and 36, respectively) have the ability of inhibiting negative-sense RNA viruses from different families (EBOV from Filoviridae and MV from Paramyxoviridae), therefore these compounds may have broad spectrum antiviral activity. Determining their antiviral extent individually or in combination against other negative-sense RNA viruses such as Crimean–Congo hemorrhagic fever, influenza or rabies viruses, may offer alternative treatments in case of emergency. Both compounds appeared to be nevertheless specific against negative-sense RNA viruses, as we examined their effect on a positive-sense RNA virus as HCV where no antiviral effect was found (not shown).
Since negative-sense RNA viruses have to be transcribed to a positive-sense RNA to translate viral proteins, this process generates dsRNA intermediates unique of negative-sense RNA viruses that may be detected by dsRNA sensors and/or by dsRNA RNAses as Dicer [35]; VP35 is known to interfere with this process [24,36]. Interestingly, the interaction of VP35IID with either NtPKR or PACT was two orders of magnitude stronger than with itself (Figure 1b), suggesting that PKR and PACT may interfere with VP35 di-/tri-/tetramerisation required to act as polymerase cofactor [5] and/or efficiently coat dsRNA [14] however, the interactions strengths may be different with full length proteins. Given that the compounds were selected through a screen platform aiming to disrupt protein-protein interactions, instead of abrogating the dsRNA binding by VP35IID as in [15,37,38], the compounds may not interfere with the dsRNA binding capabilities of VP35IID, NtPKR or PACT. Thus, from our studies, we do not know unequivocally whether the compounds also abrogated dsRNA binding to these proteins therefore, further investigation is warranted. Detailed high resolution microscopy studies are also required to determine whether the compounds interfere with VP35 viral polymerase cofactor.
Additionally, we observed a weak inhibitory effect of the compounds on the interaction between NtPKR or PACT with RIG-I (Figure 5a). Of note, this is the first report showing a direct interaction between PKR and RIG-I, although they have been previously reported to be in close vicinity in virus-induced stress granules or in complex [31,32,39]. Although the significance of this interaction and inhibition by the compounds remains to be further explored, inhibiting the interaction between PACT and RIG-I suggests that IFN synthesis driven by RIG-I would not occur in presence of the compounds. In fact, we evidenced the antiviral effect of the compounds to occur independently of IFN levels during EBOV and MV infections. Therefore, we classify compounds 13 and 36 as boosters of intrinsic immunity. In our proposed model (Figure 6), the two compounds inhibit protein interactions, which may make dsRNA more accessible to host RNAses. Complementarily, since PACT can use any of its dsRNA domains to interact with Dicer [35] its known participation on RNA interference (RNAi) may also decrease viral RNA levels. Further studies are required to determine whether key interactions of PACT with RNAi pathway components as TRBP or DICER1 are not disrupted by the compounds, and that RNAi pathway functionality is indeed restored by the compounds during infection. Additionally, the central role of PACT in the mechanism of action of the compounds could be further confirmed by testing the compounds in PACT deficient backgrounds. In line with this, we would like to emphasize that PACT is a highly conserved protein across several mammalian species relevant to EBOV infection in comparison to PKR (Figure S6a-b). Also, in contrast to PKR and RLRs, PACT and other RNAi components are not ISGs in a range of vertebrate species (Figure 6c), which suggests a translation of our results without species barriers. In view with the current evidence, the functional significance of PACT interactions with IFN-inducible RLS will have to be reassessed. Thereby, we endorse these compounds for animal testing, especially because their lack of effect on IFN induction may not aggravate a typical EBOV systemic storm, avoiding further morbidity and mortality in human. Studies assessing a synergistic effect when the compounds are administered in combination with current monoclonal treatments are also desirable. In addition, we have evidenced luteolin to target combinations of VP35IID, NtPKR or PACT heterodimers offering further mechanistic explanations to its previously reported antiviral effect [40,41,42,43,44,45].
Our screen also identified compounds increasing the interaction of VP35IID with both NtPKR and PACT (#2, MolPort-005-948-404, NAT17-347111 (AnalytiCon Discovery, GmbH); #9, MolPort-001-572-901, 7961700 (ChemBridge Corporation); #16, MolPort-010-967-340, K783-5489 (ChemBridge Corporation); #23, MolPort-002-527-771, STOCK1N-55055 (InterBioScreen Ltd.); #29, MolPort-002-611-996, STL343551 (Vitas-M Laboratory, Ltd.); #35, MolPort-019-950-511, STOCK1N-77683 (InterBioScreen), Figure S1); therefore, it is tempting to speculate that these compounds may fill the dsRNA pocket between VP35IID and NtPKR or PACT in our NPCA, strengthening their interaction when viral dsRNA is absent. Nevertheless, these compounds were not investigated in the context of an infection because our objective was to find and test compounds inhibiting these proteins’ interactions to impair viral infection.
According to the viability tests we performed in different cell lines, ≤ 10 µM treatment for 24 h seems to give an antiviral effect without compromising cell viability, while lower concentrations (e.g., ≤ 5 µM) should be used by 72 h. These orders of magnitude are comparable to drugs already used to inhibit EBOV [46]. Although our viability tests may offer a starting point to choose a concentration range for experimentation, we want to remark that the compounds were pulsed only once and then kept through the assay, without pulsing each day. Therefore, the viability should be re-assessed when translating these results into animal tests since the compounds half-life in plasma and cytotoxicity in multicellular organisms remains to be determined.
Overall, we identified two compounds and validated their antiviral action against two negative sense RNA viruses (EBOV and MV) in two cell types also tested for cell viability. Finally, we propose a model of action of the compounds through PACT-driven RNAi and highlight further areas of research.
4. Materials and Methods
4.1. Cell culture
HEK293T, Huh7 and Huh7.25/CD81 cells were cultured in Dulbecco’s modified Eagle’s Medium (DMEM + GlutaMAX; Gibco laboratories; Grand Island, NY, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Hyclone; GE Healthcare Life Sciences), 1% nonessential amino acids (Gibco), 1000 U/mL penicillin and 0.1 mg/mL streptomycin (Invitrogen; USA) at 37 °C with 5 % CO2. Vero (CRL1586) cells were cultured as above but supplemented with 5% heat-inactivated fetal bovine serum (FBS) instead.
4.2. Antibodies
Anti-PACT polyclonal antibodies were purchased from Santa Cruz Biotechnology (#sc-377103). Mouse monoclonal antibodies anti-Myc and rabbit polyclonal anti-FLAG antibodies were from Santa Cruz. Mouse monoclonal anti-β-actin antibody was from Sigma (#A1978). Goat anti-rabbit IgG (H&L) Secondary Antibody DyLight 800 4X PEG (#SA5-35571) and Goat anti-mouse IgG(H+L) secondary Antibody DyLight 680 (#35518) were from Invitrogen. The anti-measles nucleoprotein mouse monoclonal antibody (#ab9397) was from Abcam.
4.3. Expression vectors
The ORF sequences corresponding to N-terminus PKR 1-265 [49] and full length PACT [29] were cloned by in vitro recombination into pDONR207 (Gateway® BP Clonase™ II Enzyme MIX, Invitrogen™ by Life Technologies™) and transferred by LR recombination into vectors expressing in frame complementary fragments of nanoluciferase, either at their N- (luciferase N1 or N2) or C- (luciferase C1 or C2) terminus [26,50]. A similar procedure was used to generate the different fragments of PKR (K1, K2) and PACT (A1, A2, A2-M3) in frame with the luciferase moieties (Table S1). Because of French regulatory constraints on highly pathogenic micro-organisms to which EBOV belongs, only manipulation of constructs of ≤500 nucleotides (nt) are accepted in laboratories without special authorization. Therefore, the codon optimized version of EBOV VP35 (GenBank: AF086833.2, Ebola Virus-Mayinga, Zaire, 1976) generated in [51] was used to clone the EBOV VP35IID fragment of 378 nt (215-340 aa): aaagcccgacattagtgctaaggacctgcgcaacatcatgtacgatcacctgccaggctttggcaccgcctttcaccagctggtgcaggtcatctgcaagctgggcaaagactcc
aattctctggacatcatccacgccgagttccaggcttccctggccgagggcgattcaccccagtgcgctctgatccagatcaccaa
gagggtgcccattttccaggatgcagctccccctgtgatccacattcgctccaggggcgacatccccagggcttgccagaagtcc
ctgcgaccagtccctccctccccaaagatcgacaggggctgggtctgcgtgtttcagctgcaggacggcaagaccctgggtctg
aagatttga) using the VP35F/R primer pair into Zero Blunt® TOPO® (ThermoFisher); this sequence served as a PCR template for generating all VP35IID-related nanoluciferase moieties constructs using primers without and with stop codon consisting of the primer VP35-B1 paired with either VP35-B2N or VP35-B2S, respectively (Table S1). The PCR products were purified and cloned into pDONR207 by BP reaction to obtain the pDONR207+VP35IID+NoStop and pDONR207+VP35IID+stop constructs, respectively. The VP35IID amino acid changes K319A/R322A (M1), R312 (M2), F239 (M3), R305A/K309A (M4), K309A/R312A (M5) and R305A/R312A (M6) were generated by site directed mutagenesis (QuikChange II Site-directed mutagenesis kit, Agilent) with the primer pairs M1F/M1R, M2F/M2R, M3F/M3R, M4F/M4R, M5F/M5R and M6F/M6R respectively, using WT VP35IID in pDONR207 as a template and transferring the resulting mutants into N2 destination vector by Gateway LR recombination. The pDONR207 vector containing the RIG-I sequence was prepared as described [50] and transferred to vectors expressing in frame the nanoluciferase moiety N2 at their N-t.
4.4. Nanoluc protein complementation assay
The nanoluciferase protein complementation assay (NPCA) was performed as described [26,33]. Briefly, HEK293T cells (32000 cells/well were seeded in 96-well opaque white plates. After 24 h, cells were transfected using PEI (Linear polyethylenimine PEI “MAX”, Polysciences Inc) with 200 ng of the VP35IID, NtPKR or PACT constructs in fusion with the luciferase moieties. At 24 h post-transfection, the cells were incubated in the presence of 40 μL of Nano-Glo luciferase reagent (Promega #N1120). Luciferase enzymatic activity was measured with a Centro XS LB960 luminometer (Berthold) using 100 ms integration time unless otherwise indicated. Luciferase signal given by the luminometer is plotted as absolute Luc signal.
4.5. Compound library screening with NPCA
The 2P2I3D chemical library of 1664 compounds dedicated to orthosteric modulation of protein-protein interactions was acquired (Protisvalor, France) and stored in 384 well plates. This library contains PPI-like compounds corresponding to medicinally important privileged structures identified as core structures in numerous therapeutics, after filtering of 8.3 million compounds representing the main chemical providers commercially available [28,52,53]. The screening was performed on a Freedom EVO® platform (Tecan). 1 µL of each compound (20 mM in DMSO) from the 2P2I3D chemical library was spiked into white, flat bottom, bar-coded tissue culture 96-well plates (Greiner). For each plate, the first four wells of column 1 and the four last wells of column 12 contained only 1 µL of DMSO to act as positive controls of fluorescence signal (cells co-transfected either with the VP35IID/NtPKR pair or the VP35IID/PACT pair). The remaining wells of columns 1 and 12 also contained 1 µL of DMSO but the added cells have been co-transfected with vectors expressing both luciferase moieties (Luc1 or Luc2) to serve as negative controls. HEK293T cells were seeded in thee 10 cm2 round plates and two 484 cm2 square plates (Corning) to have 8.2 x 106 and 40 x 106 cells/plate, respectively. 24 h after seeding, cells of the square plates were co-transfected with a total amount of 125 µg of either N1-PACT or N1-NtPKR with N2-VP35IID while cells of two 10 cm2 plates were co-transfected with 25.6 µg of the vectors expressing only Luc1 and Luc2. On the following day, the cells were trypsinated, washed with DMEM and resuspended at 3.2 x 105 cells/mL in DMEM with 10 % FBS and without antibiotics to load 100 μL/well into the plates containing 1 μL of each compound at 2 mM, so the final concentration was 20 μM. After 24 h, 40 μL of Nano-Glo luciferase reagent (Promega #N1120) were added to measure the luciferase activity on a M1000 Pro (Tecan) using a 100 ms integration time. For each plate, positive and negative controls were used to calculate the Z’-factor [53] following the formula Z' = 1 – 3 × (σ+ + σ-) / (μ+ – μ-), where σ+ and σ- correspond to standard deviations for positive and negative controls, respectively, and μ+ and μ- correspond to means of luminescence signal measured for positive and negative controls respectively.
4.6. Cell viability assays
In parallel to the drug screens, a methyl-thiazol-tetrazolium (MTT) cell proliferation assay (Sigma, CGD-1) was performed following the manufacturer instructions. Briefly, a non-transfected preparation of HEK293T cells was distributed into a further set of 96-well opaque white plates containing the compounds from the 2P2I3D chemical library. The first column only contained 1% DMSO (no compound) serving as positive controls, and the last column was devoid of cells, DMSO and compounds to serve as a blank. After 24 h incubation at 37 °C, the MTT solution was added, and the formation of crystals was monitored. Once they started to appear in the positive control, the MTT solvent was added, and the crystals dissolved by pipetting up and down with the Freedom EVO® platform (Tecan) robot and the signal was immediately measured spectrophotometrically at 570 nm.
Further MTT viability assays were done in Huh7.25-CD81. 1 x 104 cells seeded in 96-well culture plates were incubated for 24 h to reach 60-70% confluence. After incubation for 24 h to reach 60-70% confluence, the medium was replaced by 200 µL of medium containing the compounds 13 or 36 at 1, 5, 10, 20, 30 and 40 µM or DMSO at equivalent concentrations. At 24 h or 72 h post treatment, supernatants were replaced by 100 µL OPTIMEM containing 10% MTT and incubated at 37°C during 4 h. Then, supernatants were replaced by 100 µL of lysis buffer/well and incubated for another 15 min under orbital agitation according to the manufacturer's instructions (Sigma); plates were read at 570 nm. IC50 estimations were done on GraPhad Prism 2.0 upon fitting a non-linear curve. The percentage of viable cells was presented as a percentage of the untreated samples at each time point for each sample.
4.7. Whole luciferase enzymatic assay
A further set of the 2P2I3D chemical library was used to discard false positives resulting from inhibition of an in vitro transcribed whole luciferase enzymatic activity in 96-well opaque white plates. As above, the first column only contained the enzyme in 1% DMSO (no compound) serving as positive control, while the last column was devoid of enzyme to serve as negative control.
4.8. Real-time quantitative PCR and analysis.
For EBOV infection IFN analysis, total cellular RNA was extracted on day one, according to the instructions of the RNeasy kit manufacturer (Qiagen, Hilden, Germany). Potential genomic DNA contamination was minimized by treatment with DNase I (RNase-free DNase set; Qiagen). Reverse transcription was used to generate cDNA from extracted RNA, SuperScript III reverse transcriptase, oligo(dT), a deoxynucleoside triphosphate (dNTP) mixture, RNaseOUT, dithiothreitol (DTT), and 5× RT buffer (all from Invitrogen). cDNA was then quantified, using GAPDH as a reference, by quantitative PCR (qPCR) on an LightCycler480 (Roche) using TaqMan Universal Master Mix and commercial TaqMan primers and probes for GAPDH (VIC probe). Other TaqMan assays were carried out with the primers and probes listed in Table S1. Genomic DNA contamination was checked by testing for amplification in RNA samples without the reverse transcriptase step. The results were normalized to the amount of GAPDH cDNA and are plotted as RNA relative expression.
For MV RNA presence, total cellular RNA was extracted using TRI-Reagent (Sigma), according to the manufacturer’s instruction and reverse transcribed using 1 µg of total RNA with either OligodT18 (ThermoFisher) or MV genome specific primers ([30], Table S1). Host cell mRNAs transcription and MV RNA genome levels were quantified by a two-step quantitative real time PCR (RT-qPCR) assay. RT-qPCR was performed using an AbiPrism 7900HT machine, with a FastStart Universal SYBR Green Master (Roche). The results were normalized to the amount of GAPDH cDNA and are plotted as RNA relative expression.
4.9. Immunoblot
Cells were washed once with PBS and scraped into CHAPS buffer (50 mM Tris-HCl pH 7.5, 140 mM NaCl, 5 mM EDTA, 5% glycerol, 1% CHAPS) containing cOmplete™ EDTA-free protease inhibitor and PhosSTOP™ phosphatase inhibitor (ROCHE). Protein concentrations were determined according to Bradford using Protein Assay Dye Reagent Concentrate (Bio-Rad). Protein electrophoresis was performed in NuPAGE 4-12 % Bis-TRIS gels (Invitrogen). After protein blotting, nitrocellulose membranes (Fisher Bioblock Scientific) were blocked with 2.5 % skimmed milk and probed with specific antibodies as described in the figure legends. Odyssey scanner was used for immunoblot image acquisition with the Odyssey 3.1 software (Li-Cor Biosciences).
4.10. Viruses
The Zaire strain of EBOV was obtained after two passages in Vero E6 cells using the plasma from a fatally infected patient during an outbreak in Gabon in 2001. The titer of the viral stock was 5 x 106 focus-forming Units/mL with confirmed absence of mycoplasma. Infections in experiments were done at MOI of 0.1 and subsequent titer was determined by immunostaining of infectious foci on Vero cells with mouse polyclonal primary antibodies from ascites conjugated to anti-mouse alkaline phosphatase.
4.10. Statistical analysis
All experiments were independently repeated at least twice with triplicate samples, unless otherwise stated. Plotted results are shown as mean ± standard deviation (SD). Differences between more than two groups were assessed by Kruskal-Wallis since sample distributions did not follow a Gaussian distribution. Statistical significance is indicated with asterisks, where one represents a p-value < 0.05, two p-value < 0.01, three when p-value < 0.001, and four when p-value < 0.0001. Although all graphs were assessed for statistical significance, only significant comparisons are indicated for clarity.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, M.C.R, H.M-L, Y.J., A.V.K and E.F.M.; methodology, M.C.R, H.M-L, Y.J., A.V.K and E.F.M.; formal analysis, M.C.R., A.V.K. and E.F.M.; investigation, M.C.R, O.H., P.M, A.J, Y.J. and V.N.; resources, E.F.M., H.M-L., O.H., A.V.K., R-Y.S-D ,Y.J., M.G. and J.Z.; data curation, M.C.R., E.F.M., and A.V.K.; writing—original draft preparation, M.C.R., H.M-L and E.F.M.; writing—review and editing, M.C.R, H.M-L, E.F.M. and A.V.K.; visualization, M.C.R, E.F.M., A.J., and A.V.K.; supervision, E.F.M., Y.J., H.M-L, A.V.K., S.B. and M.C.R; project administration, E.F.M.; funding acquisition, H.M-L and E.F.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by grants raised by Institut Pasteur for the program “Task force Ebola” and by CNRS (UMR 3569).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
This paper contains all relevant data required to support its results.
Acknowledgments
We thank Xavier Morelli, Jean-Claude Guillemot and Philippe Roche for the PPI chemical library and fruitful discussion.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jain, S.; Khaiboullina, S.; Martynova, E.; Morzunov, S.; Baranwal, M. Epidemiology of Ebolaviruses from an Etiological Perspective. Pathogens 2023, 12, 248. [Google Scholar] [CrossRef]
- Falasca, L.; Agrati, C.; Petrosillo, N.; Di Caro, A.; Capobianchi, M.R.; Ippolito, G.; Piacentini, M. Molecular mechanisms of Ebola virus pathogenesis: focus on cell death. Cell Death Differ. 2015, 22, 1250–1259. [Google Scholar] [CrossRef]
- Jain, S.; Khaiboullina, S.F.; Baranwal, M. Immunological Perspective for Ebola Virus Infection and Various Treatment Measures Taken to Fight the Disease. Pathogens 2020, 9, 850. [Google Scholar] [CrossRef]
- Elliott, L.H.; Kiley, M.P.; McCormick, J.B. Descriptive analysis of Ebola virus proteins. Virology 1985, 147, 169–176. [Google Scholar] [CrossRef]
- Yuan, B.; Peng, Q.; Cheng, J.; Wang, M.; Zhong, J.; Qi, J.; Gao, G.F.; Shi, Y. Structure of the Ebola virus polymerase complex. Nature 2022, 610, 394–401. [Google Scholar] [CrossRef]
- Leung, D.W.; Borek, D.; Luthra, P.; Binning, J.M.; Anantpadma, M.; Liu, G.; Harvey, I.B.; Su, Z.; Endlich-Frazier, A.; Pan, J.; et al. An Intrinsically Disordered Peptide from Ebola Virus VP35 Controls Viral RNA Synthesis by Modulating Nucleoprotein-RNA Interactions. Cell Rep. 2015, 11, 376–389. [Google Scholar] [CrossRef]
- Kirchdoerfer, R.N.; Abelson, D.M.; Li, S.; Wood, M.R.; Saphire, E.O. Assembly of the Ebola Virus Nucleoprotein from a Chaperoned VP35 Complex. Cell Rep. 2015, 12, 140–149. [Google Scholar] [CrossRef]
- Dong, S.; Yang, P.; Li, G.; Liu, B.; Wang, W.; Liu, X.; Xia, B.; Yang, C.; Lou, Z.; Guo, Y.; et al. Insight into the Ebola virus nucleocapsid assembly mechanism: crystal structure of Ebola virus nucleoprotein core domain at 1.8 Å resolution. Protein Cell 2015, 6, 351–362. [Google Scholar] [CrossRef]
- Beniac, D.R.; Melito, P.L.; Devarennes, S.L.; Hiebert, S.L.; Rabb, M.J.; Lamboo, L.L.; Jones, S.M.; Booth, T.F. The Organisation of Ebola Virus Reveals a Capacity for Extensive, Modular Polyploidy. PLOS ONE 2012, 7, e29608. [Google Scholar] [CrossRef]
- Prins, K.C.; Delpeut, S.; Leung, D.W.; Reynard, O.; Volchkova, V.A.; Reid, S.P.; Ramanan, P.; Cárdenas, W.B.; Amarasinghe, G.K.; Volchkov, V.E.; et al. Mutations Abrogating VP35 Interaction with Double-Stranded RNA Render Ebola Virus Avirulent in Guinea Pigs. J. Virol. 2010, 84, 3004–3015. [Google Scholar] [CrossRef]
- Zinzula, L.; Tramontano, E. Strategies of highly pathogenic RNA viruses to block dsRNA detection by RIG-I-like receptors: Hide, mask, hit. Antivir. Res. 2013, 100, 615–635. [Google Scholar] [CrossRef]
- Edwards, M.R.; Liu, G.; Mire, C.E.; Sureshchandra, S.; Luthra, P.; Yen, B.; Shabman, R.S.; Leung, D.W.; Messaoudi, I.; Geisbert, T.W.; et al. Differential Regulation of Interferon Responses by Ebola and Marburg Virus VP35 Proteins. Cell Rep. 2016, 14, 1632–1640. [Google Scholar] [CrossRef]
- Bale, S.; Julien, J.-P.; Bornholdt, Z.A.; Krois, A.S.; Wilson, I.A.; Saphire, E.O. Ebolavirus VP35 Coats the Backbone of Double-Stranded RNA for Interferon Antagonism. J. Virol. 2013, 87, 10385–10388. [Google Scholar] [CrossRef]
- Leib, D.; Dw, L.; Kc, P.; Dm, B.; M, F.; Jm, T.; P, R.; Jc, N.; LA, H.; Z, O.; et al. Faculty Opinions recommendation of Structural basis for dsRNA recognition and interferon antagonism by Ebola VP35.. 2010, 17. [CrossRef]
- Daino, G.L.; Frau, A.; Sanna, C.; Rigano, D.; Distinto, S.; Madau, V.; Esposito, F.; Fanunza, E.; Bianco, G.; Taglialatela-Scafati, O.; et al. Identification of Myricetin as an Ebola Virus VP35–Double-Stranded RNA Interaction Inhibitor through a Novel Fluorescence-Based Assay. Biochemistry 2018, 57, 6367–6378. [Google Scholar] [CrossRef]
- Liu, G.; Nash, P.J.; Johnson, B.; Pietzsch, C.; Ilagan, M.X.G.; Bukreyev, A.; Basler, C.F.; Bowlin, T.L.; Moir, D.T.; Leung, D.W.; et al. A Sensitive in Vitro High-Throughput Screen To Identify Pan-filoviral Replication Inhibitors Targeting the VP35–NP Interface. ACS Infect. Dis. 2017, 3, 190–198. [Google Scholar] [CrossRef]
- Ren, J.-X.; Zhang, R.-T.; Zhang, H.; Cao, X.-S.; Liu, L.-K.; Xie, Y. Identification of novel VP35 inhibitors: Virtual screening driven new scaffolds. BioMedicine 2016, 84, 199–207. [Google Scholar] [CrossRef]
- Brown, C.S.; Lee, M.S.; Leung, D.W.; Wang, T.; Xu, W.; Luthra, P.; Anantpadma, M.; Shabman, R.S.; Melito, L.M.; MacMillan, K.S.; et al. In Silico Derived Small Molecules Bind the Filovirus VP35 Protein and Inhibit Its Polymerase Cofactor Activity. J. Mol. Biol. 2014, 426, 2045–2058. [Google Scholar] [CrossRef]
- Banerjee, A.; Mitra, P. Ebola Virus VP35 Protein: Modeling of the Tetrameric Structure and an Analysis of Its Interaction with Human PKR. J. Proteome Res. 2020, 19, 4533–4542. [Google Scholar] [CrossRef]
- Schümann, M.; Gantke, T.; Mühlberger, E. Ebola Virus VP35 Antagonizes PKR Activity through Its C-Terminal Interferon Inhibitory Domain. J. Virol. 2009, 83, 8993–8997. [Google Scholar] [CrossRef]
- Feng, Z.; Cerveny, M.; Yan, Z.; He, B. The VP35 Protein of Ebola Virus Inhibits the Antiviral Effect Mediated by Double-Stranded RNA-Dependent Protein Kinase PKR. J. Virol. 2007, 81, 182–192. [Google Scholar] [CrossRef]
- Fabozzi, G.; Nabel, C.S.; Dolan, M.A.; Sullivan, N.J. Ebolavirus Proteins Suppress the Effects of Small Interfering RNA by Direct Interaction with the Mammalian RNA Interference Pathway. J. Virol. 2011, 85, 2512–2523. [Google Scholar] [CrossRef]
- Luthra, P.; Ramanan, P.; Mire, C.E.; Weisend, C.; Tsuda, Y.; Yen, B.; Liu, G.; Leung, D.W.; Geisbert, T.W.; Ebihara, H.; et al. Mutual Antagonism between the Ebola Virus VP35 Protein and the RIG-I Activator PACT Determines Infection Outcome. Cell Host Microbe 2013, 14, 74–84. [Google Scholar] [CrossRef]
- Haasnoot, J.; de Vries, W.; Geutjes, E.-J.; Prins, M.; de Haan, P.; Berkhout, B. The Ebola Virus VP35 Protein Is a Suppressor of RNA Silencing. PLOS Pathog. 2007, 3, e86–e86. [Google Scholar] [CrossRef]
- David, R.Y.S.; Combredet, C.; Najburg, V.; Millot, G.A.; Beauclair, G.; Schwikowski, B.; Léger, T.; Camadro, J.-M.; Jacob, Y.; Bellalou, J.; et al. LGP2 binds to PACT to regulate RIG-I– and MDA5-mediated antiviral responses. Sci. Signal. 2019, 12, eaar3993. [Google Scholar] [CrossRef]
- Mo, X., et al., AKT1, LKB1, and YAP1 Revealed as MYC Interactors with NanoLuc-Based Protein-Fragment Complementation Assay. Mol Pharmacol, 2017. 91(4): p. 339-347.
- Hamon, V.; Bourgeas, R.; Ducrot, P.; Theret, I.; Xuereb, L.; Basse, M.J.; Brunel, J.M.; Combes, S.; Morelli, X.; Roche, P. 2P2I HUNTER : a tool for filtering orthosteric protein–protein interaction modulators via a dedicated support vector machine. J. R. Soc. Interface 2014, 11, 20130860. [Google Scholar] [CrossRef]
- Basse, M.J.; Betzi, S.; Bourgeas, R.; Bouzidi, S.; Chetrit, B.; Hamon, V.; Morelli, X.; Roche, P. 2P2Idb: a structural database dedicated to orthosteric modulation of protein–protein interactions. Nucleic Acids Res. 2012, 41, D824–D827. [Google Scholar] [CrossRef]
- Singh, M.; Patel, R.C. Increased interaction between PACT molecules in response to stress signals is required for PKR activation. J. Cell. Biochem. 2012, 113, 2754–2764. [Google Scholar] [CrossRef]
- Mura, M.; Combredet, C.; Najburg, V.; David, R.Y.S.; Tangy, F.; Komarova, A.V. Nonencapsidated 5′ Copy-Back Defective Interfering Genomes Produced by Recombinant Measles Viruses Are Recognized by RIG-I and LGP2 but Not MDA5. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Yoo, J.-S.; Takahasi, K.; Ng, C.S.; Ouda, R.; Onomoto, K.; Yoneyama, M.; Lai, J.C.; Lattmann, S.; Nagamine, Y.; Matsui, T.; et al. DHX36 Enhances RIG-I Signaling by Facilitating PKR-Mediated Antiviral Stress Granule Formation. PLOS Pathog. 2014, 10, e1004012. [Google Scholar] [CrossRef]
- Kok, K.-H.; Lui, P.-Y.; Ng, M.-H.J.; Siu, K.-L.; Au, S.W.N.; Jin, D.-Y. The Double-Stranded RNA-Binding Protein PACT Functions as a Cellular Activator of RIG-I to Facilitate Innate Antiviral Response. Cell Host Microbe 2011, 9, 299–309. [Google Scholar] [CrossRef]
- Dabo, S.; Maillard, P.; Rodriguez, M.C.; Hansen, M.D.; Mazouz, S.; Bigot, D.-J.; Tible, M.; Janvier, G.; Helynck, O.; Cassonnet, P.; et al. Inhibition of the inflammatory response to stress by targeting interaction between PKR and its cellular activator PACT. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef]
- Chukwurah, E.; Farabaugh, K.T.; Guan, B.J.; Ramakrishnan, P.; Hatzoglou, M. A tale of two proteins: PACT and PKR and their roles in inflammation. FEBS J, 2021, 288, 6365–6391. [CrossRef]
- Kok, K.H.; Ng, M.-H.J.; Ching, Y.-P.; Jin, D.-Y. Human TRBP and PACT Directly Interact with Each Other and Associate with Dicer to Facilitate the Production of Small Interfering RNA. J. Biol. Chem. 2007, 282, 17649–17657. [Google Scholar] [CrossRef]
- Zhu, Y.; Cherukuri, N.C.; Jackel, J.N.; Wu, Z.; Crary, M.; Buckley, K.J.; Bisaro, D.M.; Parris, D.S. Characterization of the RNA Silencing Suppression Activity of the Ebola Virus VP35 Protein in Plants and Mammalian Cells. J. Virol. 2012, 86, 3038–3049. [Google Scholar] [CrossRef]
- Luthra, P.; Liang, J.; Pietzsch, C.A.; Khadka, S.; Edwards, M.R.; Wei, S.; De, S.; Posner, B.; Bukreyev, A.; Ready, J.M.; et al. A high throughput screen identifies benzoquinoline compounds as inhibitors of Ebola virus replication. Antivir. Res. 2017, 150, 193–201. [Google Scholar] [CrossRef]
- Luthra, P.; Aguirre, S.; Yen, B.C.; Pietzsch, C.A.; Sanchez-Aparicio, M.T.; Tigabu, B.; Morlock, L.K.; García-Sastre, A.; Leung, D.W.; Williams, N.S.; et al. Topoisomerase II Inhibitors Induce DNA Damage-Dependent Interferon Responses Circumventing Ebola Virus Immune Evasion. Mbio 2017, 8, e00368–17. [Google Scholar] [CrossRef]
- Onomoto, K. , et al., Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS One 2012, 7, e43031. [Google Scholar]
- Onomoto, K.; Jogi, M.; Yoo, J.S.; Narita, R.; Morimoto, S.; Takemura, A.; Sambhara, S.; Kawaguchi, A.; Osari, S.; Nagata, K.; Matsumiya, T. Luteolin inhibits respiratory syncytial virus replication by regulating the MiR-155/SOCS1/STAT1 signaling pathway. Virol. J. 2020, 17, 187. [Google Scholar]
- Theerawatanasirikul, S.; Thangthamniyom, N.; Kuo, C.-J.; Semkum, P.; Phecharat, N.; Chankeeree, P.; Lekcharoensuk, P. Natural Phytochemicals, Luteolin and Isoginkgetin, Inhibit 3C Protease and Infection of FMDV, In Silico and In Vitro. Viruses 2021, 13, 2118. [Google Scholar] [CrossRef]
- Peng, M.; Watanabe, S.; Chan, K.W.K.; He, Q.; Zhao, Y.; Zhang, Z.; Lai, X.; Luo, D.; Vasudevan, S.G.; Li, G. Luteolin restricts dengue virus replication through inhibition of the proprotein convertase furin. Antivir. Res. 2017, 143, 176–185. [Google Scholar] [CrossRef]
- Men, X.; Li, S.; Cai, X.; Fu, L.; Shao, Y.; Zhu, Y. Antiviral Activity of Luteolin against Pseudorabies Virus In Vitro and In Vivo. Animals 2023, 13, 761. [Google Scholar] [CrossRef]
- Dai, W.; Bi, J.; Li, F.; Wang, S.; Huang, X.; Meng, X.; Sun, B.; Wang, D.; Kong, W.; Jiang, C.; et al. Antiviral Efficacy of Flavonoids against Enterovirus 71 Infection in Vitro and in Newborn Mice. Viruses 2019, 11, 625. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, Y.; Song, Z.; Chang, H.; Kuang, Q.; Zheng, Z.; Wang, H.; Zhang, G. Luteolin restricts ASFV replication by regulating the NF-kappaB/STAT3/ATF6 signaling pathway. Vet. Microbiol. 2022, 273, 109527. [Google Scholar] [CrossRef]
- Jasenosky, L.D.; Cadena, C.; Mire, C.E.; Borisevich, V.; Haridas, V.; Ranjbar, S.; Nambu, A.; Bavari, S.; Soloveva, V.; Sadukhan, S.; et al. The FDA-Approved Oral Drug Nitazoxanide Amplifies Host Antiviral Responses and Inhibits Ebola Virus. iScience 2019, 19, 1279–1290. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Shaw, A.E.; Hughes, J.; Gu, Q.; Behdenna, A.; Singer, J.B.; Dennis, T.; Orton, R.J.; Varela, M.; Gifford, R.J.; Wilson, S.J.; et al. Fundamental properties of the mammalian innate immune system revealed by multispecies comparison of type I interferon responses. PLOS Biol. 2017, 15, e2004086–e2004086. [Google Scholar] [CrossRef]
- Bonnet, M.C.; Daurat, C.; Ottone, C.; Meurs, E.F. The N-terminus of PKR is responsible for the activation of the NF-kappaB signaling pathway by interacting with the IKK complex. Cell Signal 2006, 18, 1865–1875. [Google Scholar]
- Sanchez David, R.Y.; Combredet, C.; Sismeiro, O.; Dillies, M.A.; Jagla, B.; Coppee, J.Y.; Mura, M.; Guerbois Galla, M.; Despres, P.; Tangy, F.; Komarova, A.V. Comparative analysis of viral RNA signatures on different RIG-I-like receptors. Elife 2016, 5, e11275. [Google Scholar] [CrossRef]
- Tao, W.; Gan, T.; Guo, M.; Xu, Y.; Zhong, J. Novel Stable Ebola Virus Minigenome Replicon Reveals Remarkable Stability of the Viral Genome. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Milhas, S.; Raux, B.; Betzi, S.; Derviaux, C.; Roche, P.; Restouin, A.; Basse, M.-J.; Rebuffet, E.; Lugari, A.; Badol, M.; et al. Protein–Protein Interaction Inhibition (2P2I)-Oriented Chemical Library Accelerates Hit Discovery. ACS Chem. Biol. 2016, 11, 2140–2148. [Google Scholar] [CrossRef]
- Zhang, J.-H.; Chung, T.D.Y.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. SLAS Discov. Adv. Sci. Drug Discov. 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Combredet, C.; Labrousse, V.; Mollet, L.; Lorin, C.; Delebecque, F.; Hurtrel, B.; McClure, H.; Feinberg, M.B.; Brahic, M.; Tangy, F. A Molecularly Cloned Schwarz Strain of Measles Virus Vaccine Induces Strong Immune Responses in Macaques and Transgenic Mice. J. Virol. 2003, 77, 11546–11554. [Google Scholar] [CrossRef]
Figure 1.
Interaction of VP35IID with NtPKR or PACT individual domains in cellula. (a) Schematic representation of Zaire EBOV VP35 and VP35IID constructs with approximate position of amino acid (aa) changes used in (d) indicated with pins, as well as the N-terminus (Nt) aa 1-265 fragment of PKR (NtPKR), full length PACT and the derived constructs for either PKR or PACT, indicating the position of the two double stranded RNA binding domains (DRBDs: K1 and K2 for PKR and A1 and A2 for PACT, respectively), and the M3 domain of PACT. (b) HEK293T cells (30,000 cells/well in 96-well plates) were co-transfected with 100 ng of the VP35IID construct with the luciferase moiety 2 at its N-terminus (N2-VP35IID) in the presence of each of the following constructs with the luciferase moiety 1 at their N-terminus: NtPKR, PACT and each of their individual DRBDs. (c) N2-VP35IID construct transfected in presence of full-length PACT its fragments (NtPACT, A2M3, A1 or A2) each tagged with the luciferase moiety 1 at their N-terminus. (d) Cells were co-transfected with either N1-NtPKR or N1-PACT in the presence of each of the VP35IID, WT or constructs with the indicated aa substitutions. (c-d) experiments were done as in (b); luciferase enzymatic activity was measured for 100 ms (b-c) or 5 s (d). Representative graphs are shown.
Figure 1.
Interaction of VP35IID with NtPKR or PACT individual domains in cellula. (a) Schematic representation of Zaire EBOV VP35 and VP35IID constructs with approximate position of amino acid (aa) changes used in (d) indicated with pins, as well as the N-terminus (Nt) aa 1-265 fragment of PKR (NtPKR), full length PACT and the derived constructs for either PKR or PACT, indicating the position of the two double stranded RNA binding domains (DRBDs: K1 and K2 for PKR and A1 and A2 for PACT, respectively), and the M3 domain of PACT. (b) HEK293T cells (30,000 cells/well in 96-well plates) were co-transfected with 100 ng of the VP35IID construct with the luciferase moiety 2 at its N-terminus (N2-VP35IID) in the presence of each of the following constructs with the luciferase moiety 1 at their N-terminus: NtPKR, PACT and each of their individual DRBDs. (c) N2-VP35IID construct transfected in presence of full-length PACT its fragments (NtPACT, A2M3, A1 or A2) each tagged with the luciferase moiety 1 at their N-terminus. (d) Cells were co-transfected with either N1-NtPKR or N1-PACT in the presence of each of the VP35IID, WT or constructs with the indicated aa substitutions. (c-d) experiments were done as in (b); luciferase enzymatic activity was measured for 100 ms (b-c) or 5 s (d). Representative graphs are shown.
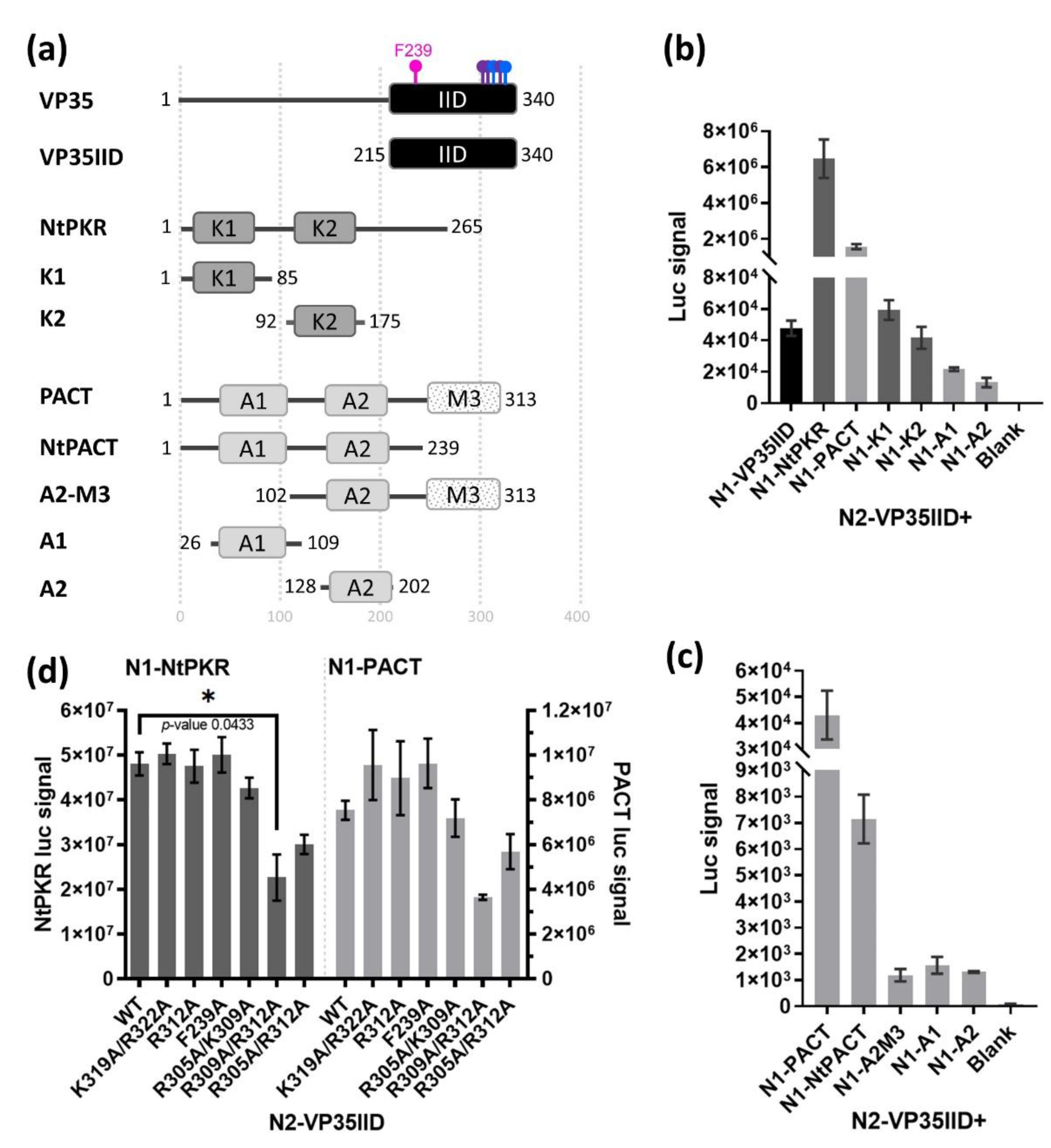
Figure 2.
Selection and testing of compounds. (a) High throughput screening flow-through diagram of a chemical library using NPCA. The 1,664 com-pounds were applied to the split luc in cellula interactions of VP35IID/PACT (1) and VP35IID/NtPKR (2), full length luciferase in vitro (full luc, 3) and to untransfected cells to perform an MTT viability test (4). Hits from (1) and (2) giving 60-90 % luc signal decrease comprised the first selection of hits from which false positives (3; hits also inhibiting full luc <1 %), toxic hits (4; hits reducing cell viability <1%) and frequent hits were subtracted. The final selection resulted in 44 hits. (b) Disruption of the interaction NtPKR/VP35IID and PACT/VP35IID by compounds 13 and 36. Compilation of three independent experiments with three experimental replicates (in cyan and blue dots, respectively) as shown in Figure S3, normalized to the untreated (U) and expressed in percentage to appreciate the effect of increasing concentrations of compounds 13 and 36 (1, 5, 10 and 20 µM) on the interaction NtPKR/VP35IID (left) or PACT/VP35IID (right). U, untreated with DMSO or compounds.
Figure 2.
Selection and testing of compounds. (a) High throughput screening flow-through diagram of a chemical library using NPCA. The 1,664 com-pounds were applied to the split luc in cellula interactions of VP35IID/PACT (1) and VP35IID/NtPKR (2), full length luciferase in vitro (full luc, 3) and to untransfected cells to perform an MTT viability test (4). Hits from (1) and (2) giving 60-90 % luc signal decrease comprised the first selection of hits from which false positives (3; hits also inhibiting full luc <1 %), toxic hits (4; hits reducing cell viability <1%) and frequent hits were subtracted. The final selection resulted in 44 hits. (b) Disruption of the interaction NtPKR/VP35IID and PACT/VP35IID by compounds 13 and 36. Compilation of three independent experiments with three experimental replicates (in cyan and blue dots, respectively) as shown in Figure S3, normalized to the untreated (U) and expressed in percentage to appreciate the effect of increasing concentrations of compounds 13 and 36 (1, 5, 10 and 20 µM) on the interaction NtPKR/VP35IID (left) or PACT/VP35IID (right). U, untreated with DMSO or compounds.
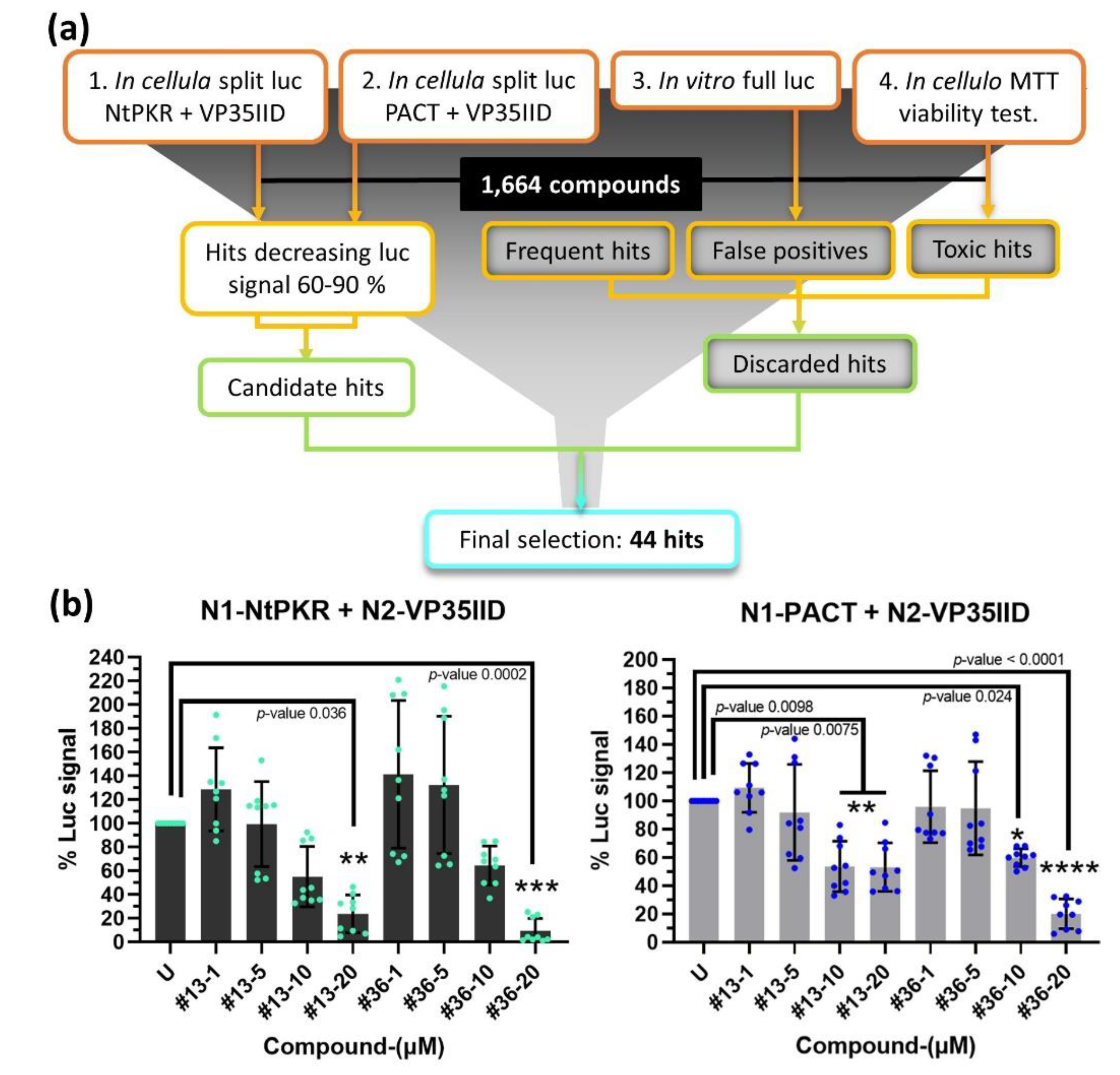
Figure 3.
Effect of compounds 13 and 36 in EBOV infection. (a) Experimental procedure diagram. HEK293T or Huh7 cells were seeded in 12-well or 96-well plates at 4 x 106 cells/plate for HEK293T cells or 3 x 106 cells/plate for Huh7 cells. 24 h after, they were infected with EBOV for 1 h at 37 °C. After adsorption, the medium was replaced with culture medium containing compound 13 or 36 at a final concentrations of 1, 5 or 10 µM; an aliquot of medium was taken at this moment representing time 0 of infection (Day 0). Aliquots of the supernatants were taken at day 1, 2 and 3 for viability assay by CellTiter-Glo (Figure S4), and at day 1 and day 3 for titration of the virus released from the cells (infectious EBOV released). RNA was extracted after 24 h (Day 1) for measuring of endogenous EBOV RNA (EBOV RNA internal) and for expression of cytokines (Figure S5), and after 72 h (Day 3) for measuring of the EBOV RNA secreted by the cells (EBOV RNA external). (b) Internal and external EBOV RNA from infected 293T or Huh7 cells untreated or treated with a concentration range of compounds 13 or 36; all scales are x 106. Graphs display averages of 2 technical replicates. (c) EBOV titer estimated by immunostaining; NI, not infected sample.
Figure 3.
Effect of compounds 13 and 36 in EBOV infection. (a) Experimental procedure diagram. HEK293T or Huh7 cells were seeded in 12-well or 96-well plates at 4 x 106 cells/plate for HEK293T cells or 3 x 106 cells/plate for Huh7 cells. 24 h after, they were infected with EBOV for 1 h at 37 °C. After adsorption, the medium was replaced with culture medium containing compound 13 or 36 at a final concentrations of 1, 5 or 10 µM; an aliquot of medium was taken at this moment representing time 0 of infection (Day 0). Aliquots of the supernatants were taken at day 1, 2 and 3 for viability assay by CellTiter-Glo (Figure S4), and at day 1 and day 3 for titration of the virus released from the cells (infectious EBOV released). RNA was extracted after 24 h (Day 1) for measuring of endogenous EBOV RNA (EBOV RNA internal) and for expression of cytokines (Figure S5), and after 72 h (Day 3) for measuring of the EBOV RNA secreted by the cells (EBOV RNA external). (b) Internal and external EBOV RNA from infected 293T or Huh7 cells untreated or treated with a concentration range of compounds 13 or 36; all scales are x 106. Graphs display averages of 2 technical replicates. (c) EBOV titer estimated by immunostaining; NI, not infected sample.
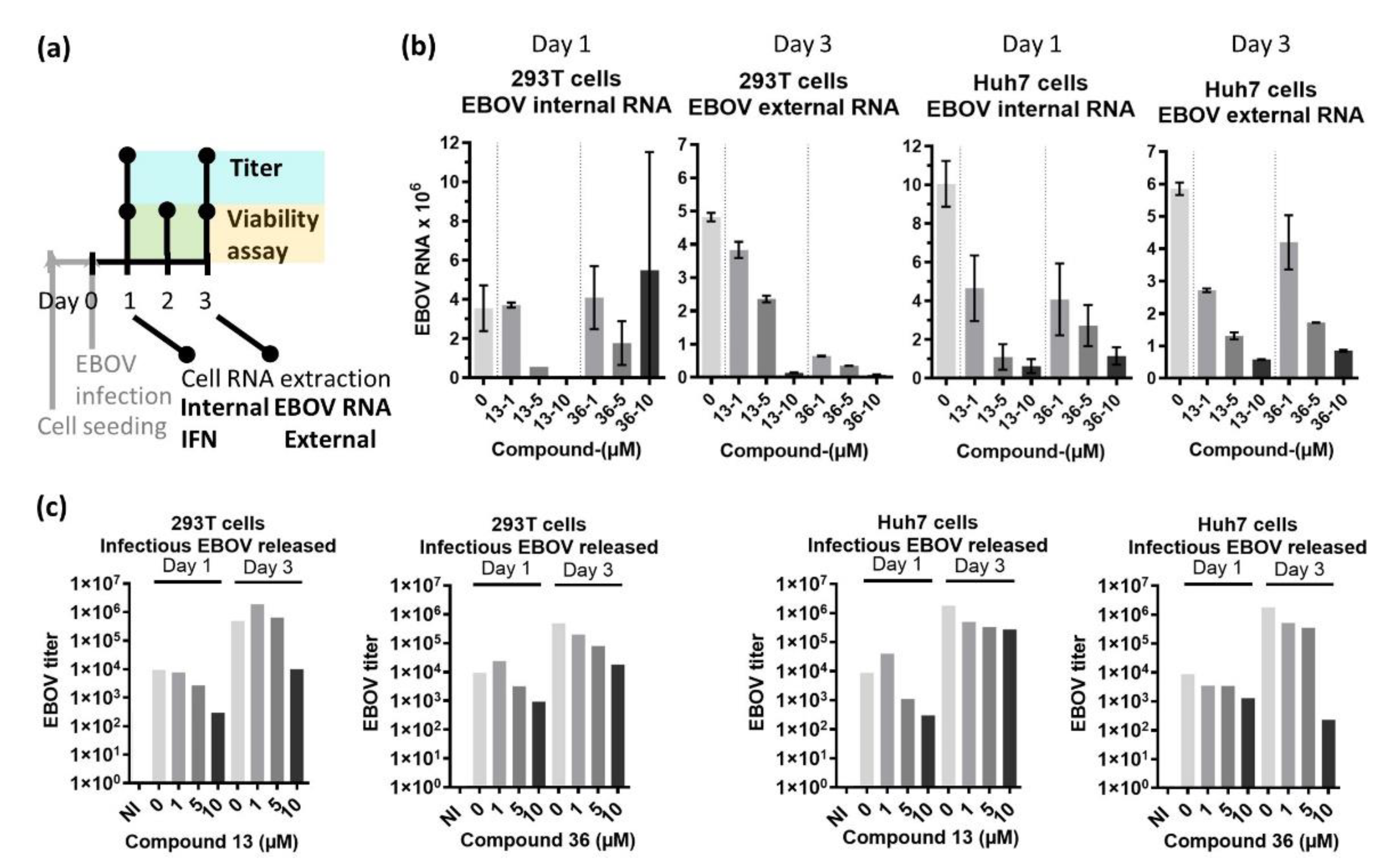
Figure 4.
Effect of compounds 13 and 36 on measles virus (MV) infection. (a) After adsorption of uninfected or infected HEK293T cells with MV (left) or MVΔV (right), the medium was replaced with fresh one containing the indicated concentrations of the compounds. After 24 h, total RNA was extracted for RT-qPCR detection of viral RNA genome as in [30], as well as of (b) IFNβ mRNA. Total protein cell extracts were prepared for immunoblot (representative shown underneath the graph in (a), to analyze the expression of the MV N protein and of endogenous PKR and PACT; detection of β-actin served as loading control. Averages of three technical replicates from two independent experiments are shown.
Figure 4.
Effect of compounds 13 and 36 on measles virus (MV) infection. (a) After adsorption of uninfected or infected HEK293T cells with MV (left) or MVΔV (right), the medium was replaced with fresh one containing the indicated concentrations of the compounds. After 24 h, total RNA was extracted for RT-qPCR detection of viral RNA genome as in [30], as well as of (b) IFNβ mRNA. Total protein cell extracts were prepared for immunoblot (representative shown underneath the graph in (a), to analyze the expression of the MV N protein and of endogenous PKR and PACT; detection of β-actin served as loading control. Averages of three technical replicates from two independent experiments are shown.
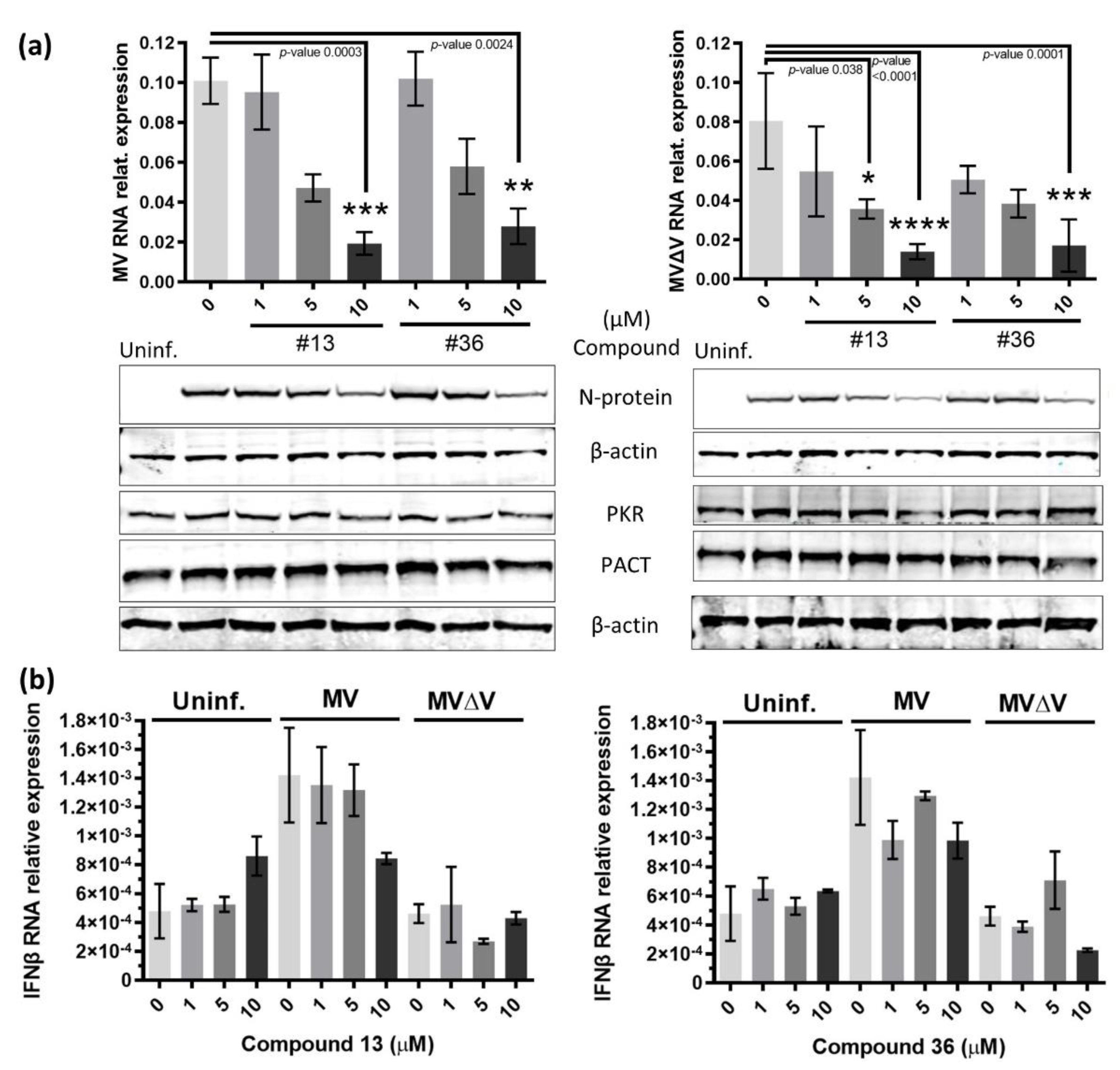
Figure 5.
Effect of compounds 13, 36 or luteolin on different protein pairs’ interactions. HEK293T cells were co-transfected with 100 ng of the different constructs (PACT, RIG-I, NtPKR or VP35IID) bearing the luciferase moiety 1 or moiety 2 at N-terminus; luciferase signal is expressed as percent normalized to untreated (U) with compounds; the horizontal purple line indicates 100 % luciferase signal. Representative experiments are shown. (a) Effect of compounds 13 or 36 at different concentrations on RIG-I interaction with either PACT, NtPKR or VP35IID. (b) Effect of compounds 13, 36 and luteolin on combinations of interacting PACT/NtPKR/VP35IID proteins. Representative graphs are shown.
Figure 5.
Effect of compounds 13, 36 or luteolin on different protein pairs’ interactions. HEK293T cells were co-transfected with 100 ng of the different constructs (PACT, RIG-I, NtPKR or VP35IID) bearing the luciferase moiety 1 or moiety 2 at N-terminus; luciferase signal is expressed as percent normalized to untreated (U) with compounds; the horizontal purple line indicates 100 % luciferase signal. Representative experiments are shown. (a) Effect of compounds 13 or 36 at different concentrations on RIG-I interaction with either PACT, NtPKR or VP35IID. (b) Effect of compounds 13, 36 and luteolin on combinations of interacting PACT/NtPKR/VP35IID proteins. Representative graphs are shown.
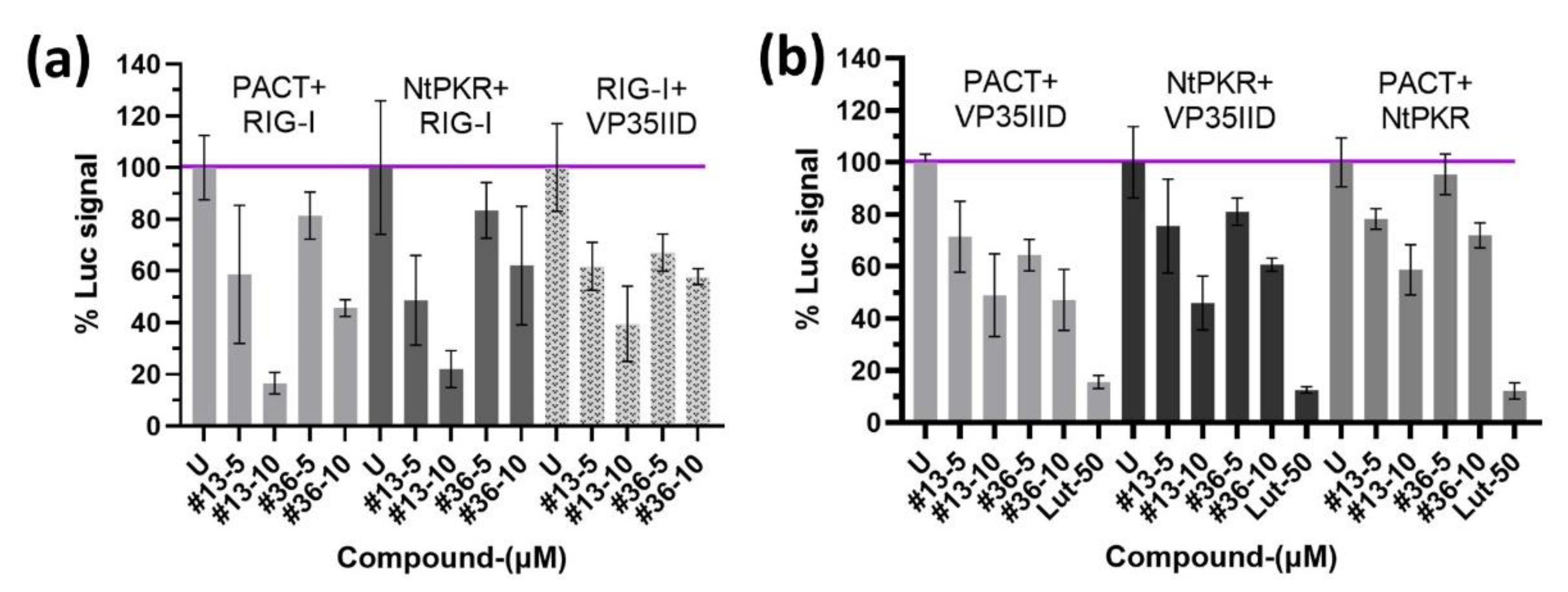
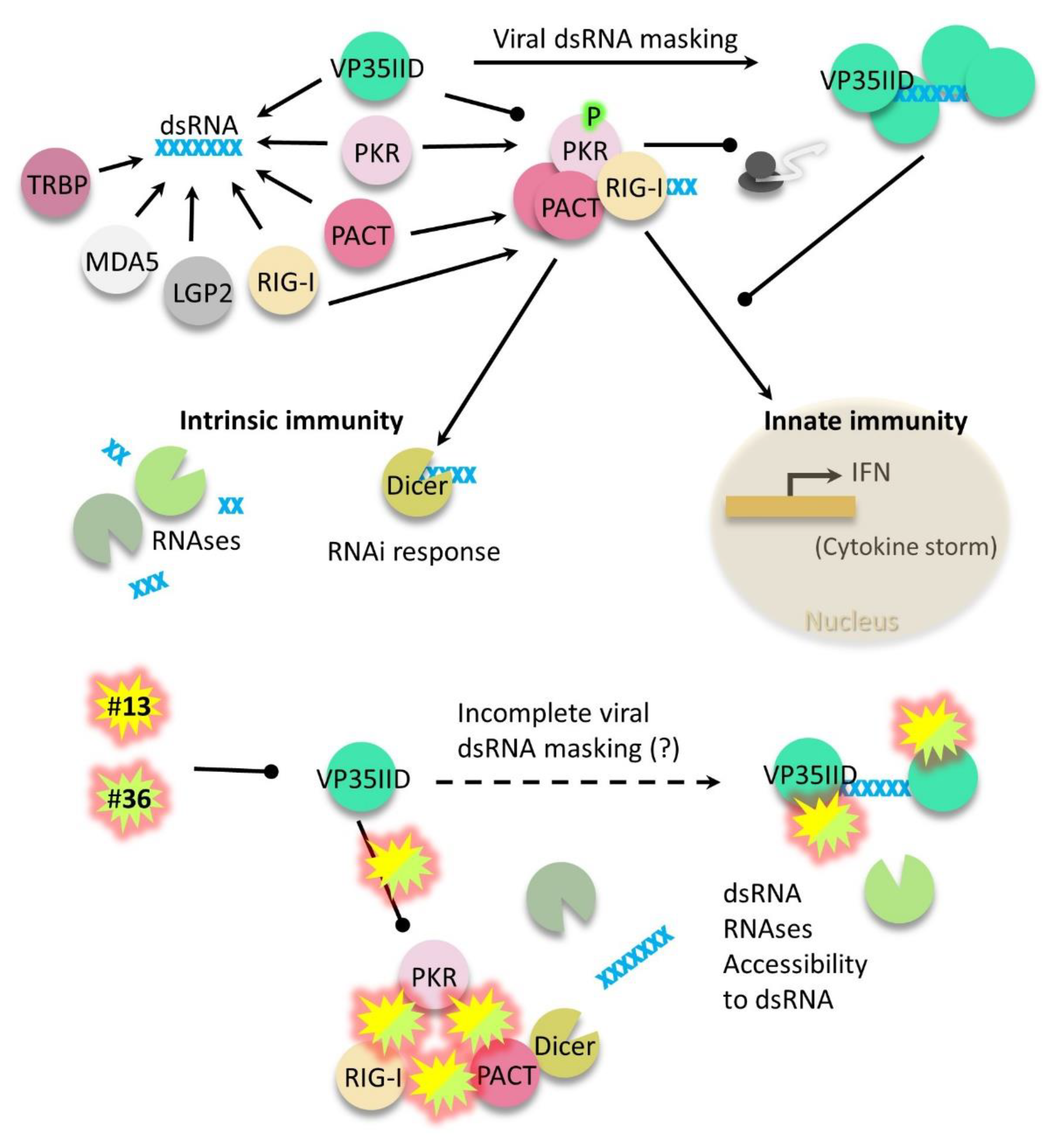
Figure 6.
Summary model of the VP35IID crosstalk with dsRNA cell sensors and the effect of compounds #13 and #36. Overall, these compounds restore intrinsic immunity response (immediate degradation of viral dsRNA by RNAses and RNAi) without affecting the inhibition of innate immunity by VP35IID, therefore preventing a cytokine storm aggravation. Trans-activation response (TAR) RNA binding protein (TRBP); anti-melanoma differentiation-associated gene 5 (MDA5); Laboratory of Genetics and Physiology 2 (LGP2).
Figure 6.
Summary model of the VP35IID crosstalk with dsRNA cell sensors and the effect of compounds #13 and #36. Overall, these compounds restore intrinsic immunity response (immediate degradation of viral dsRNA by RNAses and RNAi) without affecting the inhibition of innate immunity by VP35IID, therefore preventing a cytokine storm aggravation. Trans-activation response (TAR) RNA binding protein (TRBP); anti-melanoma differentiation-associated gene 5 (MDA5); Laboratory of Genetics and Physiology 2 (LGP2).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



