
Submitted:
30 August 2023
Posted:
31 August 2023
You are already at the latest version
Abstract
Vine growth habit (VGH) is a notable property of wild soybean plants that also holds a high degree of importance in the context of domestication as it can preclude the use of these wild cultivars for the breeding and improvement of domesticated soybean. Here, a bulked segregant analysis (BSA) approach was employed to study the genetic etiology of VGH in soybean plants by integrating linkage mapping and population sequencing approaches. To develop a recombinant inbred line (RIL) population, the cultivated Zhongdou41 (ZD41) soybean cultivar was bred with ZYD02787, a wild soybean accession. The VGH status of each line in the resultant population was assessed, ultimately leading to the identification of 6 and 9 QTLs from the BSA sequencing of the F4 population and F6- F8 population sequence mapping, respectively. One QTL shared across these analyzed generations was detected on chromosome 19. Three other QTLs detected by BSA-seq were validated and localized to 90.93 kb, 2.9 Mb, and 602.08 kb regions of chromosomes 6 and 13, respectively harboring 14, 53, and 4 genes. Four consistent VGH-related QTLs located on chromosomes 10, 13, and 19 were detected by both analytical approaches in a minimum of two environments, while an additional five loci on chromosomes 2, 10, and 18 were detected in at least two environments only via ICIM mapping. Of the detected loci, five had been reported previously whereas six represent novel QTLs. Together, these data offer new insight into the genetic basis for VGH in soybean plates, providing a rational basis to inform the use of wild accessions in future breeding efforts.
Keywords:
Glycine soja
; Vine growth habit
; QTL mapping
1. Introduction
The domestication of soybean [Glycine max (L.) Merr.] plants from wild Glycine soja first occurred in East Asia [1,2,3]. At present, wild soybeans represent a potentially invaluable resource that may harbor elite alleles capable of broadening the genetic basis of domesticated soybean plants while improving particular traits of interest. The vine growth habit (VGH) of these wild plants, however, hinders efforts to effectively breed them with cultivated soybean cultivars [2,4]. An important step in the process of soybean domestication was the transition from the twining growth tendency of wild soybeans to the upright growth of widely cultivated varieties [4]. Efforts to fully understand the genetic etiological basis for VGH will aid the effective utilization of wild resources to enable the more effective breeding-based improvement of soybean crops. As such, many research efforts have sought to clarify the genetic regulation of VGH in order to provide an evidence-based foundation for molecular breeding.
The mechanisms responsible for the regulation of VGH are complex, as evidenced by the divergent vining growth of the offspring produced through various crosses [2,4,5,6]. Vining-type growth is a dominant trait over erect growth in soybean plants, but it is also a quantitative trait influenced by many different genes, further complicating efforts to study this trait and to reliably facilitate effective domestication [2,4]. VGH-related quantitative trait loci (QTLs) vary across populations of different backgrounds and developmental stages. For example, one study of an interspecific recombinant inbred line (RIL) identified two QTLs associated with the twining growth of mature soybean plants designated as qTH-D1b and qTH-G that were located on chromosomes 2 and 18 [5]. Genotyping sequencing data derived from two Glycine max × Glycine soja populations also revealed 132 domestication-associated QTLs, 12 of which were associated with growth habit, although only one of these QTLs. qGH-19-2 (PVE = 5 and 10), was identified in both analyzed populations [6]. A further 7 and 5 QTLs were respectively detected in the flowering (R1) and mature (R8) stages when analyzing two RIL populations arising from the crossing of the wild soybean accession PI342618B with two different types of cultivated soybeans. These QTLs were mapped to chromosomes 1, 13, 18, and 19, and major loci included qVGH-18-1, qVGH-18-2, qVGH-19-3, and qVGH-19-4 (R2>10%, detection time ≥ 2), although of these QTLs only qVGH-18-2 was consistently identified in both of these populations across cropping years and growth stages. The gibberellin oxidase (GAox) member of the 2-oxoglutarate-dependent dioxygenase (2-ODD) family known as Glyma18g06870 (VGH1) was identified as a candidate gene within this qVGH-18-2 region, as it exhibited significant divergence between soybean plants with vining and upright growth with an FST > 0.25 [2]. One 2-ODD/GAox family gene was also detected in each of the qVGH-18-1 and qVGH-19-4 regions and respectively designated VGH2 and VGH3 [2]. GAox genes thus appear to be important regulators of the heritability of soybean VGH, suggesting that research focused on the genetic regulation of GAox activity may inform work focused on VGH and other stem-related traits, thereby potentially aiding the genetic improvement of soybean crops. In another study, major growth-related QTLs were identified on chromosome 11 in the W05 soybean cultivar and chromosome 13 in the Wm82 cultivar explaining 16-32% of the variance in a wild W05× cultivar C08 RIL population. The latter of these two genes was also associated with a copy number variation (CNV) in the apical bud-expressed gibberellin 2-oxidase 8A/B(GA2ox8) gene, with a positive correlation between gene copy numbers and expression levels whereas these copy numbers were negatively correlated with trailing growth and shoot length [7].
For vine-type plants, shoot apical meristems (SAMs) are indeterminate such that they can continuously grow from the vegetative to the reproductive state, whereas for plants with erect or semi-erect growth, SAMs determinate such that they cease growing after flowering, suggesting a potentially close genetic link between VGH and stem growth habit. In soybean plants, two genes designated Dt1 and Dt2 have been found to control the stem termination type [8,9]. The Dt1 gene is an ortholog of the Arabidopsis TERMINAL FLOWER 11 (TFL11) gene [8,10,11], and may regulate determinate growth as a result of an earlier drop in the expression of GmTFL1b coinciding with floral induction, despite functioning normally in the non-inductive flowering phase [8]. Dt2 is a gain-of-function of MADS-domain factor gene capable of specifying semi-determinacy, apparently via repressing Dt1 expression in SAMs and thereby promoting early SAM conversion into reproductive inflorescences [9,11].
The present study was developed with the goal of further clarifying the genetic architecture of VGH. To that end, bulked segregant analysis (BSA) sequencing and population resequencing of an RIL population derived from crossing the Zhongdou41 (ZD41) cultivar with the wild ZYD02787 accession. Through these approaches and associated mapping analyses, candidate genes associated with major VGH-related QTLs were screened in different growth environments.
2. Results
2.1. VGH Characterization of Parental and RIL Soybean Plants
To begin exploring the genetic regulation of soybean VGH, an RIL population was generated by crossing the ZD41 cultivar and the wild ZYD02878 accession. The F6-F8 populations were grown in three different environments (19SY, 19JZ, and 20JZ), and the F4 RIL population was grown in Jingzhou in 2018. Vining-type growth was evident for all members of the F1 generation, consistent with vining-type growth being dominant over upright growth. Four phenotypes were isolated in the F2 generation, including erect-, semi-erect-, semi-vining-, and vining-type growth (Figure 1A). In the F6-F8 populations, 3.55%, 35.53%, and 10.92% of lines exhibited erect growth, respectively (Figure 1B–E). This suggests that vining-type growth is a complex trait under the control of more than two genes. Vining-type growth was more common than erect growth for all analyzed generations, with a higher proportion of upright growth having been observed in Sanya (35.51%) relative to Jingzhou, potentially owing to the higher temperatures and lower levels of rainfall in the former region.
2.2. BSA-based Identification of VGH-associated Loci
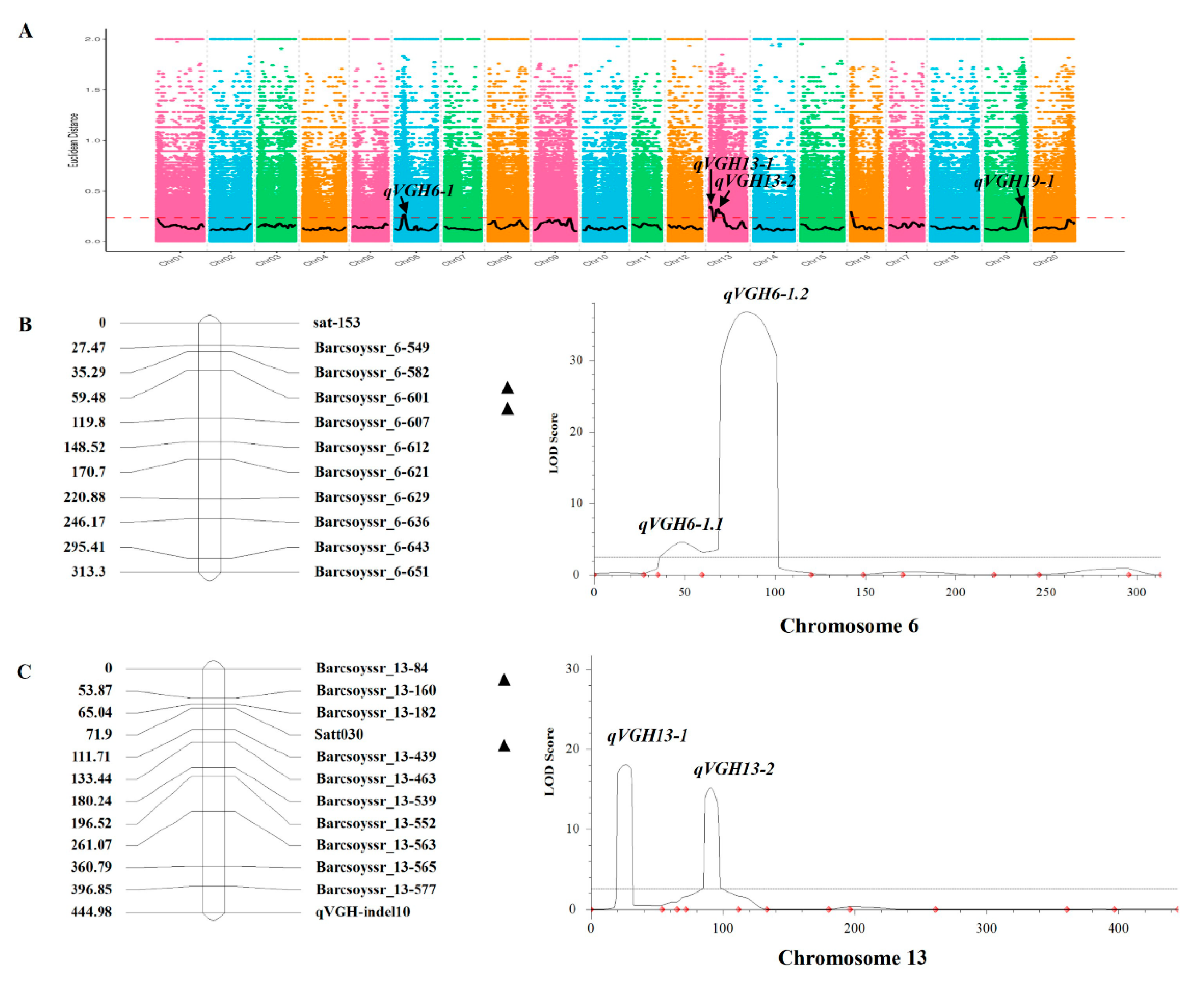
Two bulk DNA samples from the F4 RIL population (Vining-type and Erect-type bulk samples) were used for sequencing with an Illumina instrument, yielding 60.63 Gbp of data at an average sequencing depth of 25.00x. After filtering these reads, 103,459,889 and 97,176,238 clean reads were respectively obtained from the vining-type and erect-type mixed pools, with both pools exhibiting > 97% genomic coverage and > 99% genomic coverage at 1x depth. When comparing genomic variants between these two DNA bulk pools using GATK packages, 1,079,331 single nucleotide polymorphisms (SNPs) and 253,477 small (< 50 bp) insertions/deletions (InDels) were identified.
Based on these identified SNPs and InDels, 6 candidate QTLs were identified on chromosomes 6, 9, 13, 16, and 19, with all of these QTLs other than qVGH-9-1 harboring both SNPs and InDels in these analyses (Table 1). The QTL identified on chromosome 6, designated qVGH6-1, exhibited a physical distance of 2.86 Mb and was found to harbor 585 total genes. Two QTLs were identified on chromosome 13, including the 4.41 Mb QTL qVGH13-1 (Chr13:0...4,410,000) harboring 314 genes and the 10.13 Mb QTL qVGH13-2 (Chr13:8,030,000...18,160,000) harboring 740 genes. The QTL identified on chromosome 16, qVGH16-1 (Chr16:0...1,900,000), exhibited a physical distance of 1.9 Mb and was found to harbor 379 genes. The QTL identified on chromosome 19, qVGH19-1 (Chr19:42,250,000...47,850,000), spans a physical distance of 5.60 Mb and contains a total of 1062 genes.
2.3. Validation of BSA Mapping Results
To validate the candidate regions on chromosomes 6 and 13 identified via BSA mapping but absent in prior studies (qVGH6-1, qVGH13-1, and qVGH13-2), 23 SSR markers in these candidate regions that were polymorphic between the two parental lines were used for genotype identification in the F4 RIL population. The chromosome 6 interval contained two QTLs, of which qVGH6-1.1 was located in a 396.80 kb region between the Barcsoyssr_6-582 and Barcsoyssr_6-601 markers (11,032,521..11,429,318), with a LOD of 4.62 and a PVE of 1.41%. In addition, qVGH6-1.2 was in a 90.93 kb region located between the Barcsoyssr_6-601 and Barcsoyssr_6-607 markers (11,429,373..11,520,306), with a LOD of 36.83 and a PVE of 5.63%. This region on chromosome 6 exhibited an ADD effect of 0.77 and was found to contain 14 genes. The QTLs of interest on chromosome 13 were located between the Barcsoyssr_13-84 and Barcsoyssr_13-160 markers and between the satt030 and Barcsoyssr_13-439 markers. qVGH13-1 was mapped to a 2.9 Mb region (1,641,266 to 3,015,232) containing 53 genes, with a LOD of 18.08 and a PVE of 4.88%, while qVGH13-2 was mapped to a 602.08 kb region (8,722,749.. 9,324,831) containing 4 genes, with a LOD of 15.14 and a PVE of 4.86%.
Figure 2.
VGH-related QTL mapping in the F4 RIL population. (A) QTLs identified through BSA analyses. (B,C) ICIM-ADD mapping on Chr6 (B) and Chr13 (C). Black triangles represent QTLs, while red diamonds indicate SSR marker locations.
Figure 2.
VGH-related QTL mapping in the F4 RIL population. (A) QTLs identified through BSA analyses. (B,C) ICIM-ADD mapping on Chr6 (B) and Chr13 (C). Black triangles represent QTLs, while red diamonds indicate SSR marker locations.
2.4. VGH-related QTL Analyses in the F6-F8 Population Derived from ZD41×ZYD02878
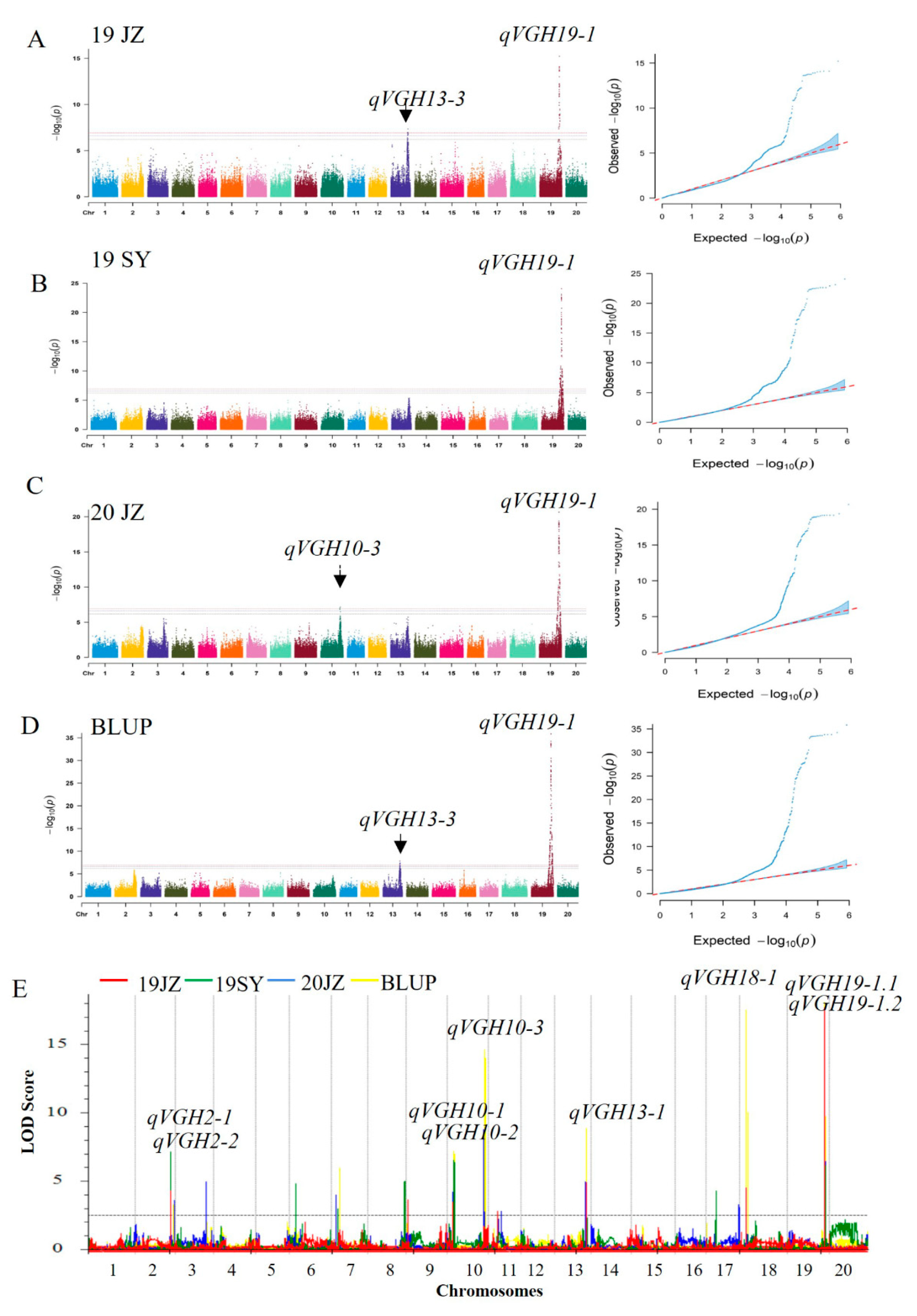
To confirm these VGH-related results, phenotypic data collected across two years, two locations, and three environments were analyzed through EMMAX analyses and ICIM-ADD mapping. To eliminate the effects of different environmental factors on these localization results, BLUP values were calculated for the three test environments and used as phenotypic data to conduct mapping. Genotyping was performed using 8,284 bin markers identified based on the sequencing of the RIL population (ZD41 × ZYD02878), ranging from 297–526 per LG, with an average genetic distance of 479.98 cM [12]. These analyses identified 9 VGH-related loci present in at least 2 environments on chromosomes 2, 10, 13, 18, and 19 (Table 3). The qVGH19-1 QTL was detected in 3 environments and using BLUP data through two methods, and was further subdivided via ICIM mapping into the qVGH19-1.1 and qVGH19-1.2 QTLs, of which qVGH19-1.1 had a higher PVE (5.73-14.51%) in three environments and in BLUP data, while the PVE of qVGH19-1.2 ranged from 4.59-5.20% in two environments and BLUP data (Supplementary Table S1). The qVGH19-1 QTL coincided with BSA-seq results, and the stem growth habit-related gene Dt1 was located in this range. Three loci were detected on chromosome 10, ranging from 200 kb to 910 kb in size, including qVGH10-3, which was detectable using both methods, but only in the 20JZ population with the EMMAX method (Table 3). qVGH13-3 (Chr13:8,196,043...39,755,321) was also detected when analyzing the 19JZ and BLUP data using both methods, while other QTLs were detected only through ICIM analyses, including qVGH2-1, qVGH2-2, qVGH10-1, qVGH10-2 and qVGH18-1.
Figure 3.
VGH-related QTL mapping in the F6-F8 RIL population (A) Manhattan and QQ plots highlighting SNPs associated with VGH for the 19JZ, (B) 19SY, (C) 20JZ, and (D) BLUP populations using the EMMAX approach. (E) Composite interval QTL mapping for the VGH of the 19JZ (red), 19SY (green), 20JZ (blue), and BLUP (yellow) populations using the ICIM-ADD approach.
Figure 3.
VGH-related QTL mapping in the F6-F8 RIL population (A) Manhattan and QQ plots highlighting SNPs associated with VGH for the 19JZ, (B) 19SY, (C) 20JZ, and (D) BLUP populations using the EMMAX approach. (E) Composite interval QTL mapping for the VGH of the 19JZ (red), 19SY (green), 20JZ (blue), and BLUP (yellow) populations using the ICIM-ADD approach.
2.5. Candidate gene analyses
To narrow the scope of candidate gene analyses, QTL regions that were shared across environments and analytical methods were selected, yielding a list of 143 total candidate genes within the qVGH6-1.2, qVGH13-1, qVGH13-2, qVGH13-3, qVGH19-1.1, and qVGH19-1.2 QTLs. Among these, 4 of the candidate genes were only present in the Wm82.a4.v1 version whereas they were absent from the Wm82.a2.v1 version. Owing to the domestication-related nature of VGH, FST values were calculated for the 139 retained genes, leading to the identification of just 23 candidate genes with an FST > 0.6 (Landraces vs. Wild and Improved vs. Wild) in coding sequence (CDS) regions that may be subject to domestication-related selection (Supplementary Table S3).
Six candidate genes of interest were identified in the qVGH6-1.2 region, including Glyma.06G140300 encoding a GroES-like zinc-binding alcohol dehydrogenase family protein, Glyma.06G140600 encoding a RING/U-box superfamily protein, Glyma.06G140700 with unknown function, Glyma.06G140800 encoding a metallo-hydrolase/oxidoreductase superfamily protein, Glyma.06G141100 encoding a leucine-rich repeat protein kinase family protein, and Glyma.06G141300 with unknown function. These 6 genes harbored 11 SNPs, and 4 small InDels were identified in the Glyma.06G140600, Glyma.06G140700, and Glyma.06G141100 genes (Supplementary Table S4). Gene atlas analyses indicated that Glyma.06G141100 is co-expressed with genes in the stem-specific coexpression subnetwork with higher expression levels in the stem, suggesting that this gene may play an important role in shaping VGH phenotypic variability in soybean. Low Glyma.06G140700 expression was also evident in stems, and Glyma.06G140100 was found to encode a calcium-dependent lipid-binding (CaLB) domain family protein.
Three genes of interest were identified in the qVGH19-1.1 region, including Glyma.19G192900 encoding pleiotropic drug resistance protein 11, Glyma.19G193400 encoding a basic-leucine zipper (bZIP) transcription factor family protein, and Glyma.19G194600 encoding F-box/RNI-like superfamily protein. While Dt1 (Glyma.19G194300) is also present within this interval, it was not selected. Two genes exhibited higher expression levels in stem tissues, including Glyma.19G192800, which encodes starch branching enzyme 2.1, as well as Glyma.19G193300, which encodes a calmodulin-binding motif family protein. The growth-regulating factor 4 gene Glyma.19G192700 also has the potential to impact VGH via influencing stem growth.
Six candidate genes were additionally selected in the qVGH19-1.2 region, including Glyma.19G202300 encoding a VQ motif-containing protein, Glyma.19G202800 encoding a protein of unknown function, Glyma.19G203700 and Glyma.19G203800 encoding ubiquitin-specific protease 13, Glyma.19G204200 encoding a cleavage and polyadenylation specificity factor (CPSF) A subunit protein, and Glyma.19G204700 encoding a ubiquitin carboxyl-terminal hydrolase family protein. Of these genes, Glyma.19G202300 and Glyma.19G203800 were not expressed in stem tissues. Additionally, the sterile alpha motif domain-containing protein-coding gene Glyma.19G203100 has the potential to impact VGH through its effects on apical meristem growth. High expression of the peptidase M28 family protein-coding gene Glyma.19G20340 and the low-molecular-weight cysteine-rich 8-coding gene Glyma.19G204900 also exhibited high levels of expression in stem tissues.
Seven genes in the QTL region located on chromosome 13 exhibited a CDS Fst > 0.6, including Glyma.13G302800 encoding a major facilitator superfamily protein, Glyma.13G304000 encoding a GH3 auxin-responsive promoter, Glyma.13G304500 encoding a geranyl diphosphate diphosphatase/geranyl pyrophosphate pyrophosphatase, Glyma.13G304700 and Glyma.13G304800 encoding isoprene synthase, Glyma.13G304900 encoding a GRAS domain family protein, and Glyma.13G305000 encoding MET-1 isoform A. Based on available expression data, Glyma.13G008100 exhibited higher levels of specific expression in stem tissues and encodes a stress-responsive A/B barrel domain protein. Based on functional analyses, Glyma.13G302900 was found to encode a photosynthetic electron transfer C protein, while Glyma.13G304000 encodes a GH3 auxin-responsive promoter and may influence VGH through effects on photosynthetic activity and GH3 auxin hormone activity.

Table 4.
Functional annotation of candidate genes.
| QTL name | Gene | Annotation | Fst>0.6 |
|---|---|---|---|
| qVGH6-1.2 | Glyma.06G141100 | Leucine-rich repeat protein kinase family protein | Yes |
| Glyma.06G140600 | RING/U-box superfamily protein | Yes | |
| Glyma.06G140700 | -- | Yes | |
| qVGH13-1 | Glyma.13G008100 | Stress responsive A/B Barrel Domain | No |
| Glyma.13G006500 | NUDT2,nudix hydrolase homolog 2 | No | |
| qVGH13-2 | Glyma.13G029500 | UDP-glucosyl transferase 85A2 | No |
| qVGH13-3 | Glyma.13G302800 | Major facilitator superfamily protein | Yes |
| Glyma.13G302900 | photosynthetic electron transfer C | No | |
| Glyma.13G304000 | GH3 auxin-responsive promoter (GH3) | Yes | |
| qVGH19-1.1 | Glyma.19G192700 | growth-regulating factor 4 | No |
| Glyma.19G194300 | TFL1,PEBP (phosphatidylethanolamine-binding protein) family protein | No | |
| qVGH19-1.2 | Glyma.19G203100 | Sterile alpha motif (SAM) domain-containing protein | No |
3. Discussion
While domesticated G. max generally exhibits upright bush-like growth, wild G. soja instead exhibits indeterminate vine-like growth [4]. VGH is a typical quantitative trait that is strongly influenced by environmental conditions, with soybean plants growing in shaded field environments exhibiting slender lodging stems, for example [13]. To identify QTLs associated with soybean VGH, RIL populations prepared via crossing G. max × G. soja were analyzed, ultimately leading to the identification of major QTLs that were conserved across environments and analytical methods.
BSA-seq and population resequencing strategies have increasingly been applied in recent years to aid in the rapid identification of QTLs associated with a range of soybean traits of interest [12,14,15,16,17,18,19]. Here, BSA-seq and RIL population resequencing strategies led to the identification of VGH-related regions on chromosomes 2, 6, 10, 13, 18, and 19. Of these loci, qVGH19-1 was detected across multiple environments using both analytical approaches and corresponds to a stable QTL that has repeatedly been mapped in prior reports. The growth habit-related qGH-19-2 locus was mapped to the same position in one past study, with respective PVE values of 10% and 5% in WP468 and WP479. Liu et al. [5] and Lu et al. [4] conducted analyses of VGH based on the number of times the main soybean stem wrapped around a support, leading to the identification of the plant height-related qVG-19 and qVG-19.1 QTLs on chromosome 9 (LG L) in two RIL populations [4]. Three QTLs associated with VGH at maturity were detected on chromosome 19, including a 44-47 Mb region (qVGH19-1) that was separated into two QTLs, with qVGH-19-3 and qVGH-19-4 mapping to similar or identical positions to qVGH-19-1.1 and qVGH-19-1.2 on this chromosome [2]. The previously characterized stem growth-related Dt1 gene was located in the qVGH-19-1.1, and there are phenotypic similarities between vine growth and indeterminate stem growth. It remains uncertain as to whether Dt1 influences VGH, however, as the W82 female parents used in the population for qGH-19-2 and qVGH-19-3 exhibited the same haplotype as the wild male parents [2,6,8]. In this study, Glyma.19G194300 harbored a nonsynonymous SNP (45184804, C-A), while Glyma.19G192700 located within this region was found to encode a growth-regulating factor 4 that may be related to VGH.
The qVGH2-1 QTL was detected in the analyzed populations from at least two environments in this study. Liu et al. previously reported the qTH-D1b QTL at satt546, which exhibited respective PVE values of 20.5% and 10.1% for testing performed in 2004 and 2005 [5]. The growth habit-related QTL qGH-2 has also been mapped to a 44.6 Mb region of chromosome 2 in the WP479 cultivar, exhibiting a PVE value of 10.1% [6]. In a population derived from crossing ZH39 and NY27-38, qVG-2 was mapped to a 43.3-44.3 Mb region with a PVE of 5.80-9.84% [4], while qVGH13-3 was localized to a 39.0-39.5 Mb region, and qVG-13, which was also identified in a population derived from crossing the ZH39 and NY27-38 cultivars, has been mapped to a 36-50 Mb region of the Wm82 genome [4]. Increases in GA2ox8 gene copy number have also been reported to decrease trailing growth [7].
Studies of VGH are relatively rare owing to the strong environmental influence and difficulties associated with phenotypic identification. Similar ‘vining score’ rating systems have previously been reported when discussing the VGH of plants derived from two G. max × G. soja crosses [20]. In several reports, authors have classified these plants as exhibiting growth that was erect (upright growth for the entirety of the plant stem), semi-erect (trailing of the top of the main stem), semi-trailing (trailing of >50% of the main stem), or trailing (the entirety of the main stem was wound around supporting stakes or twine) [2,7]. In other scores, vining tendencies were quantified on a numerical scale from 1 (indeterminate growth) to 5 (prolific vining growth similar to that of G. soja) [6]. There have even been efforts to study VGH based on the quantification of the number of times a main stem wound around its support [4,5]. These methods have enabled the identificaiton of shared loci including qTH-L[5], qGH-19-2[6], qVGH-19-3[2], and qVG-19[4] on chromosome 19, as well as qTH-G[5], qGH-18[6], and qVGH-18-2[2] on chromosome 18. Here, phenotypic identification was performed using four classical types of phenotypic classification. To improve the accuracy of these results, loci were analyzed across four generations and in different environments, ultimately revealing 9 loci that were detected in a minimum of two environments, of which 5 (qVGH2-1, qVGH13-3, qVGH18-1, qVGH19-1.1, qVGH19-1.2) were identical or nearly identical to previously reported QTLs, highlighting the accuracy of this experimental approach.
VGH is a domestication-associated trait that developed through anthropogenic selection as it has important implications for the agricultural cultivation of soybean plants [1,3]. To gain greater insight into the genetic basis for the domestication of wild soybean plants, it is vital that genes and loci associated with domestication-related traits be identified such that these wild resources can be more effectively utilized. FST values are used in population genetics to evaluate the evolution of genetic variation within and among populations [21,22]. Here, Fst analyses of candidate genes of interest were performed based on data derived from 2,214 soybean accessions, leading to the selection of 23 genes with CDS SNPs, revealing no significant domestication of the qVGH19-1 region in line with prior reports [2]. Selection for a GH3 auxin-responsive promoter was also observed. Gibberellin (GA) and other hormones play a central role in regulating plant characteristics, and a GmDW1 mutant has been reported to exhibit lower GA levels associated with a dwarf phenotype [23]. GA2ox8 can reportedly influence both trailing growth and shoot length [7]. GmIAA27 codes for an AUX/IAA protein associated with dwarfing and multi-branched growth, relying on auxin for interactions with TIR1, and with the induction of certain GH3 genes [24]. Brassinosteroids can regulate leaf angle, and brassinolide synthesis-related gene upregulation in maize, wheat, and rice has been reported to promote increased leaf inclination while the downregulation of these genes has the opposite effect [25,26,27,28,29]. The GWAS results for these 2,214 soybean accessions can be used to enable VGH analyses, gene cloning, and the evaluation of soybean cyst nematode resistance.
In the present study, we combined BSA-seq, linkage mapping and population sequence, and identified 6 and 9 QTLs from F4 BSA sequence and population sequence mapping in F6- F8, respectively. A common QTL shared by all generation was located on chromosome 19. Three additional QTL identified(qVGH6-1.2, qVGH13-1 and qVGH13-2) by BSA-seq were also validated and narrow to 90.93 kb, 2.9 Mb, 602.08 kb, on Chromosomes 6 and 13, contain 14, 53, 4 genes, respectively. Four consistency QTLs, qVGH10-3, qVGH13-3, qVGH19-1.1 and qVGH19-1.2 detected by both methods and present in at least two environments for VGH, located on chromosomes 10, 13, 19. Among them, 5 loci were identified in previous studies, and 6 novel QTLs, and the fine mapping of these QTLs is need for the cloning of the underlying genes, which will broaden out understanding of the genetic of VGH, and thus facilitate the utilization of wild resources in breeding.
4. Materials and Methods
4.1. Plant Materials and Phenotypic Analyses
The ZD41 soybean cultivar and the wild ZYD02878 soybean accession were obtained from the Chinese Academy of Agricultural Sciences. ZD41 exhibits upright growth without twining and is widely grown throughout central China, whereas ZYD02878 is a wild variety with typical vining phenotypes originally harvested from Shanxi Province in China. ZD41 (the female parent) and ZYD02878 (the male parent) were originally crossed in 2015, and RIL populations from F2 to F8 were generated through single-seed descent, ultimately yielding a population comprised of 366 lines.
For phenotypic analyses, all experimental lines were cultivated in experimental stations in Sanya and Jingzhou. The RIL-F4, RIL-F6, and RIL-F8 populations were planted in the experimental field of Crop Science of Yangtze University (30.37°N, 112.06°E) with a plant spacing of 30, 50, and 100 cm during the 2018, 2019, and 2020 seasons, respectively, while the RIL-F7 population was planted at the Sanya NanFan Research Center of CAAS (18.25°N,109.51°E) from November–February 2020 with a plant spacing of 30 cm. The F6-F8 populations were respectively designated 19JZ, 19SY, and 20JZ for subsequent analyses. VGH phenotypic analyses were performed in the drum stage of growth, classifying plants into four categories [2,7]: Type 1 (erect-type) plants with upright growth for > 80% of the main stem, as in the prototypical female ZD41 parental variety; Type 2 (semi-erect-type) plants exhibiting upright growth for over 60% of the main stem, with slight thinning and waviness for the upper portion of the main stem but without winding; Type 3 (semi-vining-type) plants with upright growth for over 20% of the main stem but with middle and upper portions consisting of an elongated climbing vine with some winding; or Type 4 (vining-type) plants with a weak stem that cannot stand upright, instead relying on the trunk or other objects for spreading with intense entanglement, as in the case of the paternal wild ZYD02878 accession. The chisq.test.4 function was used to perform chi-square analyses. The R reshape2, lmerTest, lme4, Reshape2, and lsmeans packages were used to compute BLUP values, integrating samples repeatedly detected across multiple environments and multiple years while removing outlier phenotypic data to control for environmental effects on downstream data analyses.
4.2. Genotyping of Individual and DNA Bulk Samples
The cetyltrimethylammonium bromide (CTAB) method was used to extract whole genomic DNA from healthy leaves [30]. To prepare vining and erect bulk DNA samples (VB and EB, respectively), 200 ng of DNA was pooled from 20 vining or 20 erect F4 lines. These pooled DNA samples were then sequenced with an Illumina HiSeq4000 instrument based on standard recommendations for DNA sequencing. The resultant reads were cleaned and mapped to the Glycine max Wm82.a2.v1 reference genome from Phytozome using Burrows-Wheeler Aligner (BWA) with the default parameters [31]. The calling of SNPs and InDels (<50 bp) was performed using GATK (Genome Analysis Toolkit, v 4.2) [32]. Trait-associated regions were evaluated with a Euclidean Distance (ED) algorithm, with ED values being calculated based on the genotype and depth of different mixed pools [33].
An Illumina instrument was used for the 150 bp paired-end read-based sequencing of 366 F7 RIL individuals. Clean reads were generated by filtering raw data in the FASTQ format, after which the BWA software was used to map these data to the reference genome (Wm82.a4.v1) under the default parameters [31]. SAMtools was used to call SNPs/InDels with the following settings: mpileup -m 2 -F 0.002 -d 1000. Only SNPs with a variable position depth > 4 and a mapping quality > 20 were retained for analysis.
4.3. Genome-wide association mapping
Loci with minor allele frequencies < 0.05, a missing rate > 50%, or multiple allele loci were removed from genotyping data using Plink2 [34]. Subsequent analyses were performed using the remaining SNPs. Beagle.21 was used for genotypic predictions for the missing loci. Principal component calculations were performed with the PCA module in the GCTA program to correct for population structure [35]. Kinship among individuals was analyzed using TASSEL (v5) [36]. EMMAX was used to conduct a genome-wide association analysis of 3-year 3-point phenotypic data [37], and the post.GWAS, tidyr, and Cmplot R packages were used to plot the resultant association data and to identify the association interval. The threshold value for calculated SNPs was set to P = 0.05/N, where N represents the total SNP number, for a final P value of 6.05 × 10−8 (N = 825,945).
4.4. Linkage map construction and QTL mapping
After removing anomalous SNPs following SNP calling, polymorphic SNPs were used for the estimation of recombinant points defined based on a difference in a bin of 15 continuous SNPs relative to another individual RIL [38]. Bin markers exhibiting identical genotypes within a 100 kb window were merged into a single bin marker, ultimately leading to the selection of 8284 bin markers across 20 chromosomes [12]. VGH-related QTLs were then identified via complete interval mapping (ICIM-ADD) using QTL IciMapping4.0 [39], with the threshold line LOD of 2.5.
4.5. Candidate genes prioritization
The Phytozome database Gene models within the QTL regions were searched on Phytozome (http://www.phytozome.com) was searched for gene models within QTL regions. Further candidate gene screening was then performed by calculating fixation index (Fst) values based on published genomic sequencing data from 2,214 soybean plants [40] using vcftools (v0.1.13) [41], with a 100 bp window size. A coding region Fst ≥ 0.6 was considered indicative of a possible domestication-associated gene [42]. Spatiotemporal analyses of candidate gene expression were then performed using W82 transcriptomic data (http://www.phytozome.com), yielding results in the Fragments Per kilobase of transcript per Million reads mapped (FPKM) format. Candidate gene functional annotation was performed with Phytozome and the SoyBase database (http://www.soybase.org).
Supplementary Materials
Table S1: The QTL for VGH in 3 environments and Blup by ICIM mapping; Table S2: SNPs identified by EMMAX for VGH in F6~F8 populations; Table S3: The CDS Fst>0.6 genes in major QTL; Table S4: SNP and small InDel in major QTL; Table S5: The FPKM of different tissue sample in Wm82.
Author Contributions
Conceptualization, J.W. and L.Q.; generated the populations, J.W.; K.L.; Y.X.; Q.Y.; J.W. and C.W.; Investigation: X. Y. and K.L.; Geotyping and analysis, X.Y.,Y.C. and J. S.; Writing-Original Draft preparation, J.S. Review & Editing, J.W. and L.Q. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Agricultural Science and Technology Innovation Program (ASTIP) of Chinese Academy of Agricultural Sciences, Key Science and Technology Project of Yunnan (202202AE090014).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data generated and analyzed during the current study are available from the corresponding author on reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cao, D.; Takeshima, R.; Zhao, C.; Liu, B.; Jun, A.; Kong, F. Molecular mechanisms of flowering under long days and stem growth habit in soybean. J. Exp. Bot. 2016, 68, 1873–1884. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Liu, L.; Kong, J.; Xu, Z.; Bhat, J.A.; Zhao, T. QTL architecture of vine growth habit and gibberellin oxidase gene diversity in wild soybean (Glycine soja). Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Wang, Y.; Wei, H.; Hufnagel, D.E.; Wang, Y.; Guo, S.; Li, Y.; Wang, L.; Qiu, L.-J. The prevalence of deleterious mutations during the domestication and improvement of soybean. Crop. J. 2023, 11, 523–530. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, J.; Guo, X.; Chen, J.; Chang, R.; Guan, R.; Qiu, L. Identification of Genomic Regions Associated with Vine Growth and Plant Height of Soybean. Int. J. Mol. Sci. 2022, 23, 5823. [Google Scholar] [CrossRef]
- Liu, B.; Fujita, T.; Yan, Z.-H.; Sakamoto, S.; Xu, D.; Abe, J. QTL Mapping of Domestication-related Traits in Soybean (Glycine max). Ann. Bot. 2007, 100, 1027–1038. [Google Scholar] [CrossRef]
- Swarm, S.A.; Sun, L.; Wang, X.; Wang, W.; Brown, P.J.; Ma, J.; Nelson, R.L. Genetic dissection of domestication-related traits in soybean through genotyping-by-sequencing of two interspecific mapping populations. Theor. Appl. Genet. 2019, 132, 1195–1209. [Google Scholar] [CrossRef]
- Wang, X.; Li, M.; Wong, F.; Luk, C.; Chung, C.Y.; Yung, W.; Wang, Z.; Xie, M.; Song, S.; Chung, G.; et al. Increased copy number of gibberellin 2-oxidase 8 genes reduced trailing growth and shoot length during soybean domestication. Plant J. 2021, 107, 1739–1755. [Google Scholar] [CrossRef]
- Tian, Z.; Wang, X.; Lee, R.; Li, Y.; Specht, J.E.; Nelson, R.L.; McClean, P.E.; Qiu, L.; Ma, J. Artificial selection for determinate growth habit in soybean. Proc. Natl. Acad. Sci. 2010, 107, 8563–8568. [Google Scholar] [CrossRef]
- Ping, J.; Liu, Y.; Sun, L.; Zhao, M.; Li, Y.; She, M.; Sui, Y.; Lin, F.; Liu, X.; Tang, Z.; et al. Dt2Is a Gain-of-Function MADS-Domain Factor Gene That Specifies Semideterminacy in Soybean. Plant Cell 2014, 26, 2831–2842. [Google Scholar] [CrossRef]
- Liu, B.; Watanabe, S.; Uchiyama, T.; Kong, F.; Kanazawa, A.; Xia, Z.; Nagamatsu, A.; Arai, M.; Yamada, T.; Kitamura, K.; et al. The Soybean Stem Growth Habit Gene Dt1 Is an Ortholog of Arabidopsis TERMINAL FLOWER1. Plant Physiol. 2010, 153, 198–210. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, D.; Ping, J.; Li, S.; Chen, Z.; Ma, J. , Innovation of a Regulatory Mechanism Modulating Semi-determinate Stem Growth through Artificial Selection in Soybean. PLoS Genet 2016, 12, e1005818. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xiong, Y.; Hong, H.; Li, G.; Gao, J.; Guo, Q.; Sun, R.; Ren, H.; Zhang, F.; Wang, J.; et al. Genetic dissection of and genomic selection for seed weight, pod length, and pod width in soybean. Crop. J. 2023, 11, 832–841. [Google Scholar] [CrossRef]
- Liu, W.; Zou, J.; Zhang, J.; Yang, F.; Wan, Y.; Yang, W. Evaluation of Soybean (Glycine max) Stem Vining in Maize-Soybean Relay Strip Intercropping System. Plant Prod. Sci. 2015, 18, 69–75. [Google Scholar] [CrossRef]
- Campbell, B.W.; Hofstad, A.N.; Sreekanta, S.; Fu, F.; Kono, T.J.Y.; O’rourke, J.A.; Vance, C.P.; Muehlbauer, G.J.; Stupar, R.M. Fast neutron-induced structural rearrangements at a soybean NAP1 locus result in gnarled trichomes. Theor. Appl. Genet. 2016, 129, 1725–1738. [Google Scholar] [CrossRef]
- Song, J.; Li, Z.; Liu, Z.; Guo, Y.; Qiu, L.-J. Next-Generation Sequencing from Bulked-Segregant Analysis Accelerates the Simultaneous Identification of Two Qualitative Genes in Soybean. Front. Plant Sci. 2017, 8, 919. [Google Scholar] [CrossRef] [PubMed]
- Amin, G. M. A. , Characterization and rapid gene-mapping of leaf lesion mimic phenotype of spl-1 mutant in soybean (Glycine max (L. ) Merr.). International Journal of Molecular Sciences 2019, 20, 2193. [Google Scholar] [CrossRef]
- Li, R.; Jiang, H.; Zhang, Z.; Zhao, Y.; Xie, J.; Wang, Q.; Zheng, H.; Hou, L.; Xiong, X.; Xin, D.; et al. Combined Linkage Mapping and BSA to Identify QTL and Candidate Genes for Plant Height and the Number of Nodes on the Main Stem in Soybean. Int. J. Mol. Sci. 2019, 21, 42. [Google Scholar] [CrossRef]
- Liu, X.-T.; Wu, X.-Y.; Wu, W.-P.; Wu, M.; Chen, J.-Q.; Wang, B. A bean common mosaic virus-resistance gene in the soybean variant V94-5152 was mapped to the Rsv4 locus conferring resistance to soybean mosaic virus. Theor. Appl. Genet. 2021, 134, 2367–2377. [Google Scholar] [CrossRef]
- da Silva, M.P.; Zaccaron, A.Z.; Bluhm, B.H.; Rupe, J.C.; Wood, L.; Mozzoni, L.A.; Mason, R.E.; Yingling, S.; Pereira, A. Bulked segregant analysis using next-generation sequencing for identification of genetic loci for charcoal rot resistance in soybean. Physiol. Mol. Plant Pathol. 2019, 109, 101440. [Google Scholar] [CrossRef]
- Carpenter, J.A.; Fehr, W.R. Genetic Variability for Desirable Agronomic Traits in Populations Containing Glycine soja Germplasm1. Crop. Sci. 1986, 26, 681–686. [Google Scholar] [CrossRef]
- Holsinger, K.E.; Weir, B.S. Genetics in geographically structured populations: defining, estimating and interpreting FST. Nat. Rev. Genet. 2009, 10, 639–650. [Google Scholar] [CrossRef]
- Bhatia, G.; Patterson, N.; Sankararaman, S.; Price, A. L. , Estimating and interpreting FST: The impact of rare variants. Genome Research 2013, 23, (9), 1514–1521. [Google Scholar] [CrossRef]
- Li, Z.-F.; Guo, Y.; Ou, L.; Hong, H.; Wang, J.; Liu, Z.-X.; Guo, B.; Zhang, L.; Qiu, L. Identification of the dwarf gene GmDW1 in soybean (Glycine max L.) by combining mapping-by-sequencing and linkage analysis. Theor. Appl. Genet. 2018, 131, 1001–1016. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Wu, H.; Guo, Y.; Gao, H.; Wei, Z.; Zhao, Y.; Qiu, L. GmIAA27 Encodes an AUX/IAA Protein Involved in Dwarfing and Multi-Branching in Soybean. Int. J. Mol. Sci. 2022, 23, 8643. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A. , The Role of OsBRI1 and Its Homologous Genes, OsBRL1 and OsBRL3, in Rice. Plant Physiology 2006, 140, (2), 580–590. [Google Scholar] [CrossRef]
- Hu, X.; Qian, Q.; Xu, T.; Zhang, Y.; Dong, G.; Gao, T.; Xie, Q.; Xue, Y. The U-Box E3 Ubiquitin Ligase TUD1 Functions with a Heterotrimeric G α Subunit to Regulate Brassinosteroid-Mediated Growth in Rice. PLOS Genet. 2013, 9, e1003391. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Li, X.; Zhou, W.; Ren, Y.; Wang, Z.; Liu, Z.; Tang, J.; Tong, H.; Fang, J.; Bu, Q. Transcription Factor OsWRKY53 Positively Regulates Brassinosteroid Signaling and Plant Architecture. Plant Physiol. 2017, 175, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Cao, J.; Yu, K.; Liu, X.; Gao, Y.; Chen, Q.; Zhang, W.; Peng, H.; Du, J.; Xin, M.; Hu, Z.; Guo, W.; Rossi, V.; Ni, Z.; Sun, Q.; Yao, Y. Y. Wheat TaSPL8 Modulates Leaf Angle Through Auxin and Brassinosteroid Signaling1[OPEN]. Plant Physiology 2019, 181, 179–194. [Google Scholar] [CrossRef]
- Tian, J.; Wang, C.; Xia, J.; Wu, L.; Xu, G.; Wu, W.; Li, D.; Qin, W.; Han, X.; Chen, Q.; et al. Teosinte ligule allele narrows plant architecture and enhances high-density maize yields. Science 2019, 365, 658–664. [Google Scholar] [CrossRef]
- Doyle, J. , A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 1987, 19. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.T.; Demarest, B.L.; Bisgrove, B.W.; Gorsi, B.; Su, Y.-C.; Yost, H.J. MMAPPR: Mutation Mapping Analysis Pipeline for Pooled RNA-seq. Genome Res. 2013, 23, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.; Bender, D.; Maller, J.; Sklar, P.; Bakker, P.; Daly, M. J. , PLINK: a tool set for whole-genome association and population-based linkage analyses. The American Journal of Human Genetics 2007, 81, (3), 559–575. [Google Scholar] [CrossRef]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, Y.-W.; Zhang, Z.-C.; Xiang, Y.; Liu, M.-H.; Zhou, Y.-H.; Zuo, J.-F.; Zhang, H.-Q.; Chen, Y.; Zhang, Y.-M. A compressed variance component mixed model for detecting QTNs and QTN-by-environment and QTN-by-QTN interactions in genome-wide association studies. Mol. Plant 2022, 15, 630–650. [Google Scholar] [CrossRef]
- Huang, X.; Feng, Q.; Qian, Q.; Zhao, Q.; Wang, L.; Wang, A.; Guan, J.; Fan, D.; Weng, Q.; Huang, T.; et al. High-throughput genotyping by whole-genome resequencing. Genome Res. 2009, 19, 1068–1076. [Google Scholar] [CrossRef]
- Meng, L.; Li, H.; Zhang, L.; Wang, J. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J. 2015, 3, 269–283. [Google Scholar] [CrossRef]
- Li, Y.-H.; Qin, C.; Wang, L.; Jiao, C.; Hong, H.; Tian, Y.; Li, Y.; Xing, G.; Wang, J.; Gu, Y.; et al. Genome-wide signatures of the geographic expansion and breeding of soybean. Sci. China Life Sci. 2022, 66, 350–365. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Hyten, D.L.; Jia, G.; Quigley, C.V.; Fickus, E.W.; Nelson, R.L.; Cregan, P.B. Development and Evaluation of SoySNP50K, a High-Density Genotyping Array for Soybean. PLOS ONE 2013, 8, e54985. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
VGH phenotypic and frequency distributions for an RIL population derived from crossing ZD41×ZYD02878 (n = 366). (A) ZD41, ZYD02878, and four VGH phenotypes. (B) VGH data for the F4 RIL population in 2018JZ. (C) VGH data for the F6 RIL population in 2019JZ. (D) VGH data for the F7 RIL population in 2019SY. (E) VGH data for the F8RIL population in 2020JZ.
Figure 1.
VGH phenotypic and frequency distributions for an RIL population derived from crossing ZD41×ZYD02878 (n = 366). (A) ZD41, ZYD02878, and four VGH phenotypes. (B) VGH data for the F4 RIL population in 2018JZ. (C) VGH data for the F6 RIL population in 2019JZ. (D) VGH data for the F7 RIL population in 2019SY. (E) VGH data for the F8RIL population in 2020JZ.
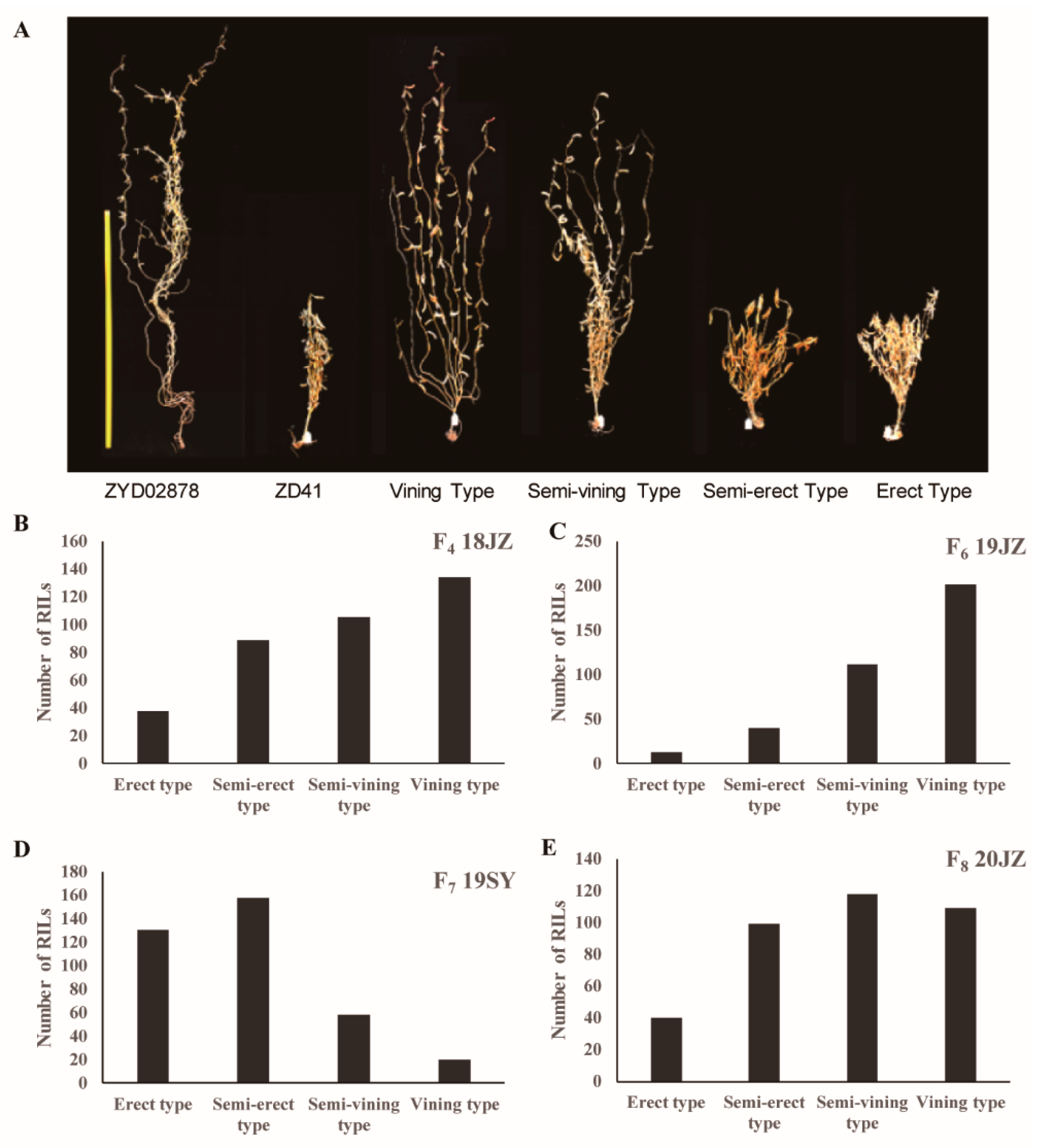
Table 1.
ED association analysis of SNPs and InDels for VGH.
| QTLs | Chr | Start(W82a2) | End(W82a2) | Position (Mb) | Gene_Number | Genotype |
|---|---|---|---|---|---|---|
| qVGH6-1 | 6 | 9,580,000 | 12,440,000 | 2.86 | 585 | SNP, InDel |
| qVGH9-1 | 9 | 41,910,000 | 42,920,000 | 1.01 | 187 | SNP |
| qVGH13-1 | 13 | 0 | 4,410,000 | 4.41 | 314 | SNP, InDel |
| qVGH13-2 | 13 | 8,030,000 | 18,160,000 | 10.13 | 740 | SNP, InDel |
| qVGH16-1 | 16 | 0 | 1,900,000 | 1.90 | 379 | SNP, InDel |
| qVGH19-1 | 19 | 42,250,000 | 47,850,000 | 5.60 | 1062 | SNP, InDel |
Table 3.
Colocalized VGH-related QTLs identified using the ICIM and EMMAX methods for populations grown in three environments and BLUP.
Table 3.
Colocalized VGH-related QTLs identified using the ICIM and EMMAX methods for populations grown in three environments and BLUP.
| QTL Name | Environment | Chr. | Start(W82a4) | End(W82a4) | Position (Mb) | Method |
|---|---|---|---|---|---|---|
| qVGH2-1 | 19JZ,19SY,BLUP | 2 | 44,549,340 | 44,748,366 | 0.2 | ICIM |
| qVGH2-2 | 20JZ,BLUP | 2 | 48,133,595 | 49,091,931 | 0.96 | ICIM |
| qVGH10-1 | 19JZ,20JZ | 10 | 7,448,517 | 7,648,577 | 0.2 | ICIM |
| qVGH10-2 | 19SY,BLUP | 10 | 8,298,686 | 9,212,033 | 0.91 | ICIM |
| qVGH10-3 | 19SY,BLUP | 10 | 45,339,503 | 45,848,959 | 0.51 | ICIM |
| qVGH13-3 | 19JZ,BLUP | 13 | 39,347,224 | 39,548,455 | 0.21 | ICIM |
| qVGH18-1 | 19JZ,BLUP | 18 | 7,843,488 | 8,011,289 | 0.17 | ICIM |
| qVGH19-1.1 | 19JZ,19SY,20JZ,BLUP | 19 | 45,447,240 | 45,839,781 | 0.19 | ICIM |
| qVGH19-1.2 | 19SY,20JZ,BLUP | 19 | 46,449,908 | 46,641,266 | 0.19 | ICIM |
| qVGH10-3 | 20JZ | 10 | 44,008,695 | 45,515,403 | 1.5 | EMMAX |
| qVGH13-3 | 19JZ,BLUP | 13 | 39,070,209 | 39,574,546 | 0.5 | EMMAX |
| qVGH19-1 | 19JZ,19SY,20JZ,BLUP | 19 | 44,047,388 | 46,935,042 | 2.9 | EMMAX |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



