
Submitted:
29 August 2023
Posted:
31 August 2023
You are already at the latest version
Abstract
Some of the most recent advances in enzymatic glycosylation processes focused on the production of important bioactive compounds have been analyzed. The applicability of different enzymes such as glycosidases, glycosyltransferases, glycophosphorylases and glycosynthases have been de-scribed around the review article considering their advantages and disadvantages of these biocat-alysts in the stereoselective and regioselective synthesis of different types of glycosylated mole-cules, phenolic and aliphatic alcohols, oligosaccharides, polysaccharides, glycoderivatives, glyco-peptides and glycoproteins with a clear focus on food and pharmaceutical chemistry. Therefore, these approaches combining the use of different catalytic systems, the improvement of tools such as immobilization technology, or chemical or genetic modification to improve the synthesization in the glycosylation process could be a very useful tool in a continuous biotechnological advance.
Keywords:
enzymes
; glycosylation
; oligosaccharides
; glycoderivatives
1. Introduction
There are many compounds described in the bibliography with good biological activity such as antioxidants, anti-inflammatories or different drugs used in different processes and with good applicability in different fields such as pharmacology, sweeteners, etc. However, in many cases these compounds are absorbed in the gastrointestinal tract and metabolized very quickly in the bloodstream and especially in the liver. In other cases, the problem is that since most of them are highly hydrophobic, their solubility in aqueous medium is very low, so the concentrations in blood are very small, causing a decrease in the biological activity [1]. Thus, in Nature, most of the compounds with biological activity are associated with sugar residues. This helps in different biological processes such as the maintenance of cell integrity, in cell recognition, or in defense mechanisms at the molecular level [2,3,4,5].
Imitating the nature glycosylation is a very effective process to transform natural products, which in many cases, enhances or modifies their biological activity after altering their physical, chemical, and biological properties [6]. Considering this, in many occasions it is interesting the derivatization of the hydroxylated compounds with simple sugars or with oligosaccharide chains. Depending on the type of linkage formed, in many cases these compounds are less hydrolysable and their metabolism occurs in the colon and caused by the gut microbiota. In addition, the sugar derivatization makes the compounds more soluble in aqueous medium increasing the concentration in blood.
At the center of any enzymatic glycoderivatization strategy is a glycosidic bond-forming enzyme. A number of factors must be taken into account when selecting a suitable catalyst, in particular:
- (i)
- Practical access to the corresponding substrates (and catalyst).
- (ii)
- Ability of the catalyst to act on a wide range of substrates.
- (iii)
- Efficiency of glycosidic bond formation.
In organic chemistry, natural glycosidases can be used to catalyze different synthetic processes. Although glycosidases use to be classified according to the type of glycosidic bond formed or hydrolyzed by their catalytic activity (galactosidases, glucosidases, etc.), they are usually grouped into two main groups as glycosidases and transglycosidases. Glycosidases are able to form glycosidic bonds by reverse hydrolysis, with equilibrium shift; and glycosyltransferases (GT) catalyze the transglycosylation, transferring an active glycosyl group to a hydroxyl acceptor to form a new glycoside. Both classes of enzymes are highly widespread in nature and there are different subclasses depending on the type of substrates they accept for catalysis. Among others are thioglycolyases, which catalyze the condensation of activated glycosides and different acceptors [7].
In addition to the multitude of enzymes that exist in nature, in recent years, different mutant enzymes have been developed that have had their hydrolytic capacity eliminated, greatly improving their synthesis yields [8].
Finally, in some cases there are described processes in which different cell lines have been used to catalyze reactions involving glycosidase enzymes [9].
In the present review, different processes of great interest of synthesis using the different catalytic systems described above will be addressed with a special emphasis on the synthesis of derivatives of phenolic compounds because of their high interest due to their, in many cases, good biological activity and their low solubility. This makes them excellent candidates for their glycosylation due to the advantages provided by such treatment as mentioned above.
2. Enzymatic Glycosylation of phenolic compounds
Phenolic compounds are an important class of chemicals containing one or more hydroxyl groups bounded directly to aromatic hydrocarbon group [10]. Considering their chemical structures, phenolic compounds can be divided into several classes such as flavonoids, phenolic acids, tannins, coumarins, quinones, stilbens, and lignans (Figure 1) [11]. They possess various beneficial bioactivities including antioxidant, antiatherogenic, anti-inflammatory and antimicrobial properties, which make them generally interesting in various scientific areas [12,13,14]. However, despite numerous biological activities, their wider application is limited due to their low solubility and stability in water, since they are easily degraded by light irradiation in aqueous solution [10]. Glycosylation of phenolic compound seems to be the useful tool to enhance their solubility and stability in water, as well as to improve their biological and pharmacological properties, such as tyrosinase inhibitory activity, browning-resistant activity and antitumor properties, by increasing their bioavailability or, sometimes, by decreasing the toxicity and side effects [10,15,16].
Extraction of phenolic glycosides from natural sources is complex and uneconomical, while chemical approach for their synthesis requires too many steps of protection, activation, coupling, and deprotection [10]. On the other hand, due to the enzyme’s regio- and stereoselectivity and mild reaction conditions, enzymatic glycosylation is considered very attractive, environmentally friendly strategy for obtaining these valuable compounds [17]. To date, it has been proven that enzymes such as glycosyltransferases (EC2.4) and glycosidases (EC3.2.1) can catalyze the glycosylation of phenolic compounds [10]. In this review, the most representative recent researches of biotransformation of phenolic compounds, using pure enzymes or whole microbial cells conferring the glycosylation potential, as biocatalysts, will be discussed.
2.1. Glycosyltransferase
Glycosyltransferases (GTs) are a subclass of enzymes that catalyze the transfer of a sugar residue from various sugar donors to a variety of important biomolecules as acceptors including mono-, di-, or oligo-carbohydrates, glycans, lipids, peptides, and numerous other small molecules [18]. Therefore, they serve critical roles in oligosaccharide, polysaccharide, and glycoconjugate biosynthesis, as well as protein glycosylation and the synthesis of valuable natural products [18]. Based on their catalytic properties, glycosyltransferases are classified into two groups: Leloir and non-Leloir enzymes [10].
2.1.1. Leloir glycosyltransferase
In vivo synthesis and modification of glycans is catalyzed by Leloir glycosyltransferases. Due to a strict specificity and high efficiency in glycosylation of various natural acceptors using activated glycosyl donors (e.g. nucleotide-sugar conjugates, lipid phosphate sugars and phosphate sugars), these enzymes are widely used for the synthesis of phenolic glycoconjugates [10].
Feng and co-workers identified phenolic glycosyltransferase MhGT1 from the filamentous fungus Mucor hiemalis which exhibited promising capability for regio- and stereospecific O-glycosylation of different phenolic compounds [19]. By examining 93 compound library of phenols from Traditional Chinese Medicinal herbs as substrates, they revealed that MhGT1 exhibited very broad substrate specificity and catalyzed glycosylation of 72 structurally diverse drug-like scaffolds and sterols using uridine diphosphate (UDP) glucose as a sugar donor. Interestingly, MhGT1 showed high regiospecificity towards compounds bearing prenyl moieties and were more active with prenylated phenols as substrates comparing to their non-prenylated analogues which was explained by large active cavity of MhGT1 consisting of hydrophobic and charged amino acid residues [19]. This is particularly important having in mind that prenylated phenolics show better bioactivities comparing to their phenolic precursors, including cardio-, neuro- and osteoprotection, as well as antioxidant and anti-diabetic activity [20].
In another study, Xie et al. identified two novel phenolic UDP glycosyltransferases (PUGTs) in fungi, membrane-bound proteins UGT58A1 (from Absidia coerulea) and UGT59A1 (from Rhizopus japonicas), which can transfer sugar moieties from active donors to phenolic acceptors and form corresponding glycosides [21]. Whole cells of these two fungi were successfully applied for glycosylation of two phenolic compounds, magnolol and honokiol to obtain the corresponding bioactive glycosides with high yields [22], hence the authors used recombinant enzymes from these two fungus and investigated their catalytic promiscuity, regioselectivity and stereoselectivity in formation of different phenolic glycosides. Activities of both enzymes are strongly influenced by presence of various cations - Mg2+, Mn2+, Ca2+, Ba2+ increase activity, Ni2+, Co2+, and Cu2+, decrease activity, while with Zn2+ the glycosylation activity was diminished [21]. As potential substrates, 13 different phenolic acceptors with diverse structures were tested, including lignans, flavonoids, anthraquinon, stilbene, benzophenone, curcuminoid, xanthones, and coumarin. Both enzymes were able to catalyze the glucosylation of magnolol, honokiol, chrysin, emodin, curcumin, and three xanthones (1,3,6-trihydroxyxanthone, 1,3,7-trihydroxyxanthone and 1,3,6,8-tetrahydroxyxanthone). UGT58A1 enabled higher conversion rates of all suitable substrates comparing to UGT59A1. Additionally, several phenolic substrates - fisetin, resveratrol, 2,4-dihydroxy bezophenone, 1,3,8-trihydroxylxanthone, and 7-hydroxy-4-methylcoumarin - were suitable glycosyl acceptors only for UGT58A1 indicating that this enzyme possess particularly high potential to be used as catalyst in the synthesis of bioactive glycosides. By using substrates that contain multiple nucleophiles, single monoglucosylated products were obtained, suggesting regioselectivity of UGT58A1. For example, fisetin possesses four phenolic hydroxyl groups, however single product, fisetin-3-O-β- glucopyranoside, was detected and identified, with a conversion degree of 94%. Regarding tolerance to a sugar donors, out of three tested (UDP-Glc, UDP-GlcA and UDP-Gal) UGT58A1 was able to recognize UDP-Glc and UDP-GlcA with magnolol as an acceptor [21].
Another phenolic UDP glycosyltransferase, Bs-PUGT from Bacillus subtilis PI18, was cloned and expressed in Escherichia coli BL21 (DE3) [23]. Purified enzyme was tested with several naturally occurring phenolics such as tyrosol, cinnamic alcohol, vanillin, ferulic acid, thymol, cinnamic acid, and caffeic acid. Obtained results proved its high activity towards all examined substrates except of thymol and cinnamic acid. By using whole-cell E. coli/Bs-PUGT as a catalyst and coffeic acid as a model substrate in a process assisted by 10 mM Ca2+ ions, 78.3% molar yield of caffeic acid glucosides was accomplished at optimized conditions. Addition of metal ions proved to be valuable tool for increasing the molar yield of caffeic acid glucosides and shortening the reaction time.
A novel glycosyltransferase gene (BbGT) from Beauveria bassiana ATCC 7159 was discovered and heterologously expressed in E. coli by Ren et al. [24]. After functional characterization through in vitro reactions it was shown that purified enzyme was UDP-glucosyltransferase which could convert quercetin into 5 monoglucosylated derivatives and one diglucoside. Metal ions such as Ca2+, Mg2+, and Mn2+ stimulate the activity of BbGT, while Zn2+ show inhibition effect on the enzymatic activity of BbGT. Additionally, the authors reported the broad substrate specificity of BbGT, since it was shown that, besides quercetin, this enzyme could catalyse glucosylation of resveratrol, curcumin and zearalenone, as well. Moreover, considering that whole-cell biotransformation can provide UDP-glucose in situ, the in vivo glucosylation of quercetin by BbGT was also investigated. In addition to E. coli, different other microbial expression host, including Saccharomyces cerevisiae, Pseudomonas putida, and Pichia pastoris, were used. Interestingly, it was shown that BbGT expressed in E. coli showed different product profiles in vivo and in vitro, since the main product of in vivo glucosylation of quercetin was quercetin-7-O-β-D-glucoside, while diglucoside was not detected. On the other hand, the products of quercetin glucosylation depended also on microbial host used, since the major product in P. putida, and P. pastoris was the same as in E. coli, quercetin-7-O-β-D-glucoside, while the enzyme dominantly produced quercetin-3-O-β-D-glucoside in S. cerevisiae.
2.1.2. Non-Leloir glycosyltransferase
Compared to Leloir glycosyltransferases, which require expensive glycosyl donors and generally tolerate natural acceptors, non-Leloir glycosyltransferases are compatible with low-cost donors and a wide range of acceptors, which is very important in terms of production process scale-up [10].
Enzymatic glycosylation of isoquercitrin (IQ) using different glycosyltransferases, comercial cyclodextrin glucanotransferase (CGTase) and amylosucrase from Deinococcus geothermalis (DGAS), was investigated by Rha et al. [25]. Both enzymes showed significant transglycosylation activity and, as a product, reaction mixture of isoquecitrin derivatives, including IQ-glucoside, IQ-diglucoside and IQ-triglucoside, was synthesized. However, several advantages of DGAS over CGTase was observed, including higher productivity, simpler process, and fewer by-products. Higher bioconversion was achieved with DGAS as biocatalyst, 97% conversion of IQ compared to 75% with CGTase [25]. The most important merit of DGAS application is the higher yield of specific isoquercitrin-triglucoside, which is the most bioavailable form among α-isoquercitrin glucosides. In following study, same group investigated the DGAS catalyzed site-specific α-glycosylation of 15 different flavonoids, various mono-, di- and tri-hydroxyflavones (HFVO) and -hydroxyflavanones (HFVA) [26]. The regioselectivity of used biocatalyst was confirmed, since it was shown that in 3–OH and 7–OH positions transglycosylation of flavonoids is negligible (Figure 2), while DGAS showed strong transglycosylation reactivity towards 6–OH and 4ʹ–OH positions of the mono-HFVOs and -HFVAs with glucose released from sucrose as sugar donor. It was hypothesized by the authors that such kind of -OH selectivity is due to the specific interaction between the enzyme and substrate, since diphenyl propane backbone of flavonoids fit well into the active pocket of enzyme, so 4ʹ−OH and 6−OH axial positions in the flavonoid molecules were readily available for transglycosylation, while 3-OH and 7-OH equatorial positions were inaccessible for transglycosylation with DGAS.
Efficient synthesis of novel epigallocatechin gallate-glucosides (EGCG-Gs) was performed by using dextransucrase from Leuconostoc mesenteroides B-1299CB4 expressed in E. coli as a catalyst and sucrose as sugar residue donor [15]. Response surface methodologically optimized process enabled epigallocatechin gallate (EGCG) conversion degree of 91.43%. Nine different reaction products were purified and five of them were identified as novel compounds by NMR spectroscopy. Novel compounds had 45 to 368-fold higher water solubility comparing to parent molecule EGCG. All derivatives had lower antioxidant activity comparing to EGCG and DPPH radical scavenging capacity decrease was higher in molecules with more glucosyl units attached. Furthermore, 7-OH group glucosylation was proven to have a functional role in mushroom tyrosinase inhibitory activity as well as increasing browning-resistant activities [15].
In another work, the synthesis of various α-glucosyl derivatives of EGCG was performed in aqueous reaction medium at 50 °C by a transglycosylation reaction catalyzed by cyclodextrin glycosyltransferase from Thermoanaerobacter sp. using hydrolysed potato starch as glucosyl donor [27]. Two monoglucosides were synthesized as main reaction products, EGCG-3′-O-α-d-glucopyranoside and EGCG-7-O-α-d-glucopyranoside, with conversion degrees of 58% and 13%, respectively (Figure 3.). The same research group investigated the stereo- and regioselectivity of cyclodextrin glycosyltransferase (CGTase) from Thermoanaerobacter sp. in a glycosylation of hesperetin using the soluble starch as glucosyl donor [28]. As products, several glucosides were synthesized. The main product was purified and structural analysis has shown that glucosylation of the hesperetin takes place at position O-7 of the A ring. Under optimized reaction condition, which included the presence of 30% of bis(2-methoxyethyl) ether as cosolvent, the maximum concentration of monoglucoside was approximately 2 mM, obtained after 24 h of reaction. Although the conversion yield of hesperetin was only 4.1%, this is the first report of using the free enzymes for α-glucosylation of hesperetin instead of whole cells.
2.1.3. C-glycosyltransferases
Due to their potential benefits to human health and their better resistance to hydrolysis compared to other type of glycosides, flavonoid C-glycosides have recently attracted increased attention [30]. C-glycosyltransferases (CGT) are the most important biocatalyst used for the enzymatic synthesis of flavonoid C-glycosides, since they have shown high efficiency and regiospecificity [31].
He et al. found a novel C-glycosyltransferase TcCGT1 from medicinal plant Trollius chinensis, which represents the first CGT to catalyze 8-C-glycosylation of flavones (Figure 4) [31]. Moreover, this enzyme could catalyze C-glycosylation of 36 structurally different flavonoids and for 12 substrates conversion yield was above 77%. In addition to C-glycosylation, the authors reported that the TcCGT1 showed high catalytic capabilities to O-glycosylation since it catalyzed the O-glycosylation of 44 substrates of which 31 were flavonoids, with conversion yield of more than 80% for 8 substrates. The most interestingly, TcCGT1 exhibited both C- and O-glycosylation activity towards 17 substrates, including various flavonoids. After investigation of the enzyme crystal structure and the catalytic multifunctional structural mechanism it was revealed that large substrate diversity of TcCGT1 is enabled by spacious sugar acceptor binding pocket. Moreover, conduction of the site-directed mutagenesis at I94E and G284K enabled the O-glycosylation activity of TcCGT1 while suppressing the C-glycosylation activity [31].
Pei et al. developed a one-pot two-enzyme system, which provided an efficient method for the production of isoorientin and isovitexin, flavonoid glycosides, involving the cycling and regeneration of costly UDP-glucose in the reaction [32]. Considering the unsatisfactory productivity of isoorientin synthesis in constructed recombinant E.coli [33], the authors developed environmentally safe efficient enzymatic in vitro glycosylation [32]. They have used C-glucosyltransferase (Gt6CGT) from Gentiana triflora expressed in E. coli BL21 combined with Glycine max sucrose synthase (GmSUS) for the production of isoorientin and isovitexin. GtCGT was used for regioselective glycosylation, since it was reported that this enzyme could glycosylate certain flavonoids at the C-6 position, while GmSUS was utilized to construct regeneration system of UDP-glucose. By optimizing coupled reaction conditions, high molar conversions of precursor flavonoids, luteolin (94.7%) and apigenin (97.1%), were obtained in synthesis of isoorientin and isovitexin, respectively.
2.2. Glycosidases
Glycosyl hydrolases (GHs) or glycosidases are enzymes with primary function of glycosidic linkages hydrolysis, however they can also produce new glycosidic bonds through a transglycosylation reaction when alternative nucleophiles participate as acceptors. Various sugar units could be transferred to a range of different nucleophilic acceptors, including phenolic compounds, with a complete stereoselectivity and very high regioselectivity.
Méndez-Líter and coworkers investigated transglycosylation profiles of Talaromyces amestolkiae β-glucosidases BGL-2, BGL-2T and BGL-3 from the family GH3 using different acceptors, including several phenolic compounds known for their high bioactivities, and cellobiose as sugar donor [16]. BGL-2 was most perspective for investigation with selected acceptors – vanillyl alcohol and hydroxytyrosol. In both cases, anomeric position is attached to the phenolic residue through the aliphatic chain, leaving free phenolic hydroxyl groups, which are responsible for the antioxidant and free radical-scavenging activity of phenolic compounds. Mono-glucosylated derivatives of two phenolics had increased water solubility comparing to parent molecules - for hydroxytyrosol glucoside it was 2.9 times higher, while for the vanillyl alcohol derivative, it was 10.3 times higher. Two glucosides were tested as potential antitumor reagents in preclinical models of cancer and both glycosylated derivatives showed higher effectiveness and/or safer profiles than hydroxytyrosol and vanillin when added to breast cancer cell cultures. With hydroxyltyrosol and its glucoside, significant morphological changes in the MCF-7 cells were observed, while no morphological difference could be detected in case of vanillin and vanillyl glucoside, suggesting that these compounds may be acting as inhibitors of MCF-7 cell proliferation rather than apoptotic stimuli [16].
β-Galactosidase from Aspergillus oryzae was applied as a catalyst in a study focused on enzymatic synthesis of salicin galactoside [34]. This compound is structurally similar to galectin inhibitors, hence potentially implicated in the disease process of different inflammatory conditions. HPLC analyses and ion mobility mass spectrometry proved high selectivity of enzymatic synthesis since only one isomer was formed in which galactosyl moiety was transferred to primary hydroxyl group of glucosyl residue in salicin molecule. Response surface methodological approach in optimization of key experimental factors led to salicin β-galactoside concentrations of up to 30.8 mM after 12 h of reaction, at 40 mM of lactose, 110 mM of salicin and enzyme amount of 360 IU [34].
A recombinant rBxTW1 β-xylosidase from the ascomycete Talaromyces amestolkiae and derived mutant obtained by replacement of the catalytic glutamic acid in position 495 for alanine (rBxTW1-E495A) with dominant synthetic activity were recently examined [35]. The crude extracts of wild-type enzyme and multi-glycoligases, as well as pure multi-glycoligases have been immobilized as magnetic cross-linked enzyme aggregates (mCLEAs) and tested in different hydrolytic and transxylosylation reactions, including examination of several phenolic acceptors with xylose as sugar donor. In a reaction of vanillin and (−)-EGCG xylosides synthesis mCLEAs of the mutant retained the catalytic properties of the purified free enzyme and were successfully reused in a 4 consecutive reaction cycles [35]. Interestingly, no vanillin xyloside was obtained with the soluble crude enzymes suggesting that in crude extracts there may be some component disabling transxylosylation reaction, which was inactivated or removed either by immobilizing or by purifying the enzyme.
In another study, Gonzalez-Alfonso et al. investigated the application of mutant of sucrose phosphorylase from Thermoanaerobacterium thermosaccharolyticum engineered to accept large polyphenols (variant TtSPP_R134A) to synthesize phloretin glucosides [36]. Reaction was performed with 10% (v/v) of acetone as co-solvent and with an excess of glucosyl donor, sucrose. As products of reaction, three compounds were detected, mono-, di- and tri-glucosylated phloretin. In initial stage of reaction, the major product was phloretin glucoside (53% of conversion yield), while later the phloretin diglucoside takes the primate reaching the maximum concentration of 8 mg/ml (73% of conversion yield). The chemical characterization revealed that monoglucosylated product is phloretin 4’-O-α-D-glucopyranoside, while diglucosylated product is phloretin 4’-O-[α-D-glucopyranosyl-(1→3)-O-α-D-glucopyranoside]. Synthesized phloretin diglucoside is novel compound in which the glucoses are bound by an unusual α (1→3) linkage. The authors reported also that aqueous solubility of phloretin was 71-fold and 1200-fold increased by the α-monoglucosylation and diglucosylation, respectively. On the other hand, α-glucosylation led to decrease of the antioxidant capacity of phloretin.
2.2.1. Microbial glycosylation
Microbial transformation of natural phenolic compounds may be an interesting way for obtaining their glycosylated derivatives since such processes have potential industrial application, due to their relatively low costs and mild reaction conditions [37]. Moreover, the glycosides derived by microbial transformation are categorized as natural compounds, allowing them to be used as dietary supplements or ingredients of cosmetics and pharmaceuticals.
The microbial glycosylation of flavonoids was thoroughly investigated by Sordon et al. [38,39]. In one study, three selected filamentous fungi, Beauveria bassiana, Absidia coerulea and Absidia glauca, were used for biotransformation of isoflavones, daidzein, genistein and biochanin A and it was found that these fungi can regioselectively catalyze O-glycosylation of flavonoids (Figure 5) [38].
Fungus B. bassiana AM 278 converted all tested isoflavones to 4''-O-methyl-7-O-glucosyl derivatives, whereas A. coerulea AM 93 and A. glauca AM 177 were able to transform genistein and Biochanin A to genistein-7-O-β-glucoside (genistin) and Biochanin A-7-O-β-glucoside (sissotrin), respectively. In addition to sissotrin, in the culture of A. coerulea AM 93, the presence of second synthesized compound, Biochanin A-5-O-β-glucoside, was noticed. Moreover, two of the obtained compounds (4''-O-methyl-7-O-glucosyl Biochanin A and 5-O-glucosyl Biochanin A) are reported for the first time in the literature [38]. In following investigation, the authors broadened group of potential substrates, so the catalytic activity of fungi using various flavones and flavanones as sugar acceptors was examined [39]. The regioselectivity was confirmed, since it was shown that preferable position of glycosylation is a hydroxyl group bound to C-7 in a flavonoid molecule. Moreover, it was shown that type of flavonoid skeleton and substituents present in aromatic rings strongly influence glycosylation process [39]. Thus, the presence of methoxyl group at C4′ position in ring B of flavonoids, led to an additional glycosylated product in reaction catalyzed by Beauveria bassiana AM 278. Beside the 4''-O-methyl-7-O-glucosyl derivative, another product, in which 4''-O-methylglucose molecule is attached to C-3' position of flavonoid molecule, is synthesized. On the other hand, the lack of double bond between the C-2 and C-3 carbon atoms in flavanone molecules, led to synthesis of two products in reaction catalyzed by Absidia species, in which glucosyl moiety is attached to C-7 and C-3'position of parent molecule [39].
Other group of authors investigated the use of Isaria fungi in biotransformations of flavonoid compounds. They examined the catalytic capacity of filamentous fungi Isaria fumosorosea KCH J2, I. farinosa J1.4, I. farinosa J1.6 and I. farinosa KW1.2 to carry out biotransformations of flavone, 5-hydroxyflavone, 6-hydroxyflavone, 7-hydroxyflavone, daidzein, and 7-aminoflavone [40]. The formation of O-methylglucosides was observed in all cases except in the reaction with 7-aminoflavone which was transformed into two acetamido derivatives. Seven of the synthesized products were not previously reported in the literature. In other study, one step glycosylation of flavonoids was investigated [41]. One strain of Isaria fumosorosea KCH J2 and two strains of I. farinosa (J1.4 and J1.6) were used as biocatalysts for glycosylation of 3-hydroxyflavone, 3-methoxyflavone, quercetin (3,3',4',5,7-pentahydroxyflavone), and baicalein (5,6,7-trihydroxyflavone) [41]. For all the substrates that were used in this study, 4-O-methylglucopyranosides were obtained and the authors suggested that the attachment of 4-O-methylglucopyranose occurs in one step. Moreover, three obtained derivatives have not been described in the literature so far.
3. Enzymatic glycosylation of other alcohols
There are many reports of products of interest that improve their properties after being glycosylated by the different types of glycosidases. The catalytic activity of glycosidases was utilized for the synthesis of β-galactosyl xylitol derivatives using the enzyme LacA β-galactosidase from Lactobacillus plantarum WCFS1. Xylitol is one of the most used sweeteners because of good characteristics, such as low calorie content and the anti-cavities activity. The synthesis was performed with good selectivity being obtained two main derivatives (3-O-β-D-galactopyranosyl-xylitol and (2S-1-O-β-D-galactopyranosyl-xylitol) (Figure 6). This process has the potential to obtain new sweetener with different properties and a very low digestibility. This case constitutes a clear example where the properties of xylitol are improved by glycosylation. The synthesis was performed using lactose as donor and good yields of all the interesting compounds were obtained. The in vitro intestinal digestibility of the main β-galactosyl xylitol derivatives was lower than that of lactose, being around 6 and 15% for the galacto-xylitol derivatives compared to 55% of lactose after 120 min of digestion [42].
Another interesting example has been described in the stereoselective glycosylation of epothilones. Epothilones are polipeptidic macrolides produced by some myxo-bacterium as Sorangium cellulosum [43]. Epothilones are macrolactones with epoxy and keto groups in the lactone ring and a side chain with a thiazole ring. In spite that epothilones were discovered in 1987 as antifungal drugs [44], different analogues have been described with potential in therapy due the high cytotoxic effect in tumour cells [45].
In this way, different isolated derivatives have been recognized as chemotherapeutic drugs against different tumor cell lines. This has been demonstrated in preclinical and clinical trials [46,47].
The reaction was catalyzed by a glycosyl transferase produced by the gene E. coli BL21(DE3) harboring pET28 (a)-YjiC. Different sugar donors allowed the synthesis of different sugar derivatives (Figure 7). The time dependent conversion study of epothilone A to epothilone A 7-O-β-D-glucoside found that maximum (~26%) was achieved between 3 h to 5 h incubation [48].
3.1. Glycan phosphorylases (GP)
The use of glycan phosphorylases alone or in combination is a tool of great interest in the development and production of activated sugars with phosphate groups, as well as in the production of traditional oligosaccharides and other oligosaccharides and polysaccharides composed of non-natural sugars. Glycan phosphorylases (GP) are Leloir GT enzymes that catalyze the formation of sugar-1-phosphates through the phosphorylation of saccharides. These catalysts can be exploited for regio- and stereoselective glycosidic bond formation. Compared to glucosidase-catalyzed transglycosylation, a notable advantage of glucan phosphorylase consists in having less competition considering that hydrolysis is reported to be almost two orders of magnitude slower than the phosphorylitic reaction. In general, glycoside phosphorylases use an inorganic phosphate to break glycosidic bonds, releasing the corresponding sugar-1-phosphate and acceptor (Figure 8) [49,50,51].
Phosphorylases have been classified into different families depending on the sequence, being generally divided between retaining and inverting phosphorylases.
Furthermore, in most cases these enzymes can be combined with other different phosphorylases to produce phosphorylated sugars with different chain lengths or rare sugars that do not occur naturally [52].
It is possible to expand the amount of sugar-1-phosphates beyond Glc-1-phosphate by coupling sucrose phosphorylase (SP) with α-Glc-1-phosphatase (AGP) either sequentially or simultaneously. This allowed the phosphorylation of different residues as D-Mannose (D-Man), N-Acetyl-D-glucosamine (D-GlcNAc), D-Galactosidase (D-Gal) and L-Fucose (L-Fuc) at the anomeric hydroxyl group with axial selectivity (Figure 9) [53].
4. Enzymatic synthetic glycosylation of oligosaccharides
Most of specific enzymes in glycosylation methods goes through the natural capacity to catalyze glycosylation process in aqueous media. In particular, the use of glycosidases for synthetic purpose present some limitation and thus they have been extensively subjected to protein engineering strategies to improve or change catalytic activity and substrate specificity, and to change optimal reaction conditions. [54,55]
In this term, genetically engineered glycoside hydrolases (the so called glycosynthases), in which the catalytic residue (usually an aspartate or glutamate) responsible for promoting nucleophilic attack on the substituted anomeric carbon (Figure 10) is replaced by a non-functional residue (usually alanine or glycine) has been created. As a result of that, they become hydrolytically incompetent. [54,55,56] Then the enzymatic ability of the enzyme goes through the transfer of an activated glycosyl donor (typically a glycosyl fluoride in anomeric position) to a suitable acceptor to catalyse the formation of a glycosidic bond at high yields. [54]
This synthetic potential has enormous impact in the oligosaccharide synthesis, production of therapeutic glycoproteins, the synthesis of carbohydrate-based drugs, and the creation of new biomaterials. [57]
4.1. Glucosynthases
The first glycosynthase was described in 1998 and was derived from β-glycosidase Abg, from Agrobacterium sp., by mutation of the enzyme nucleophile, E358, from glutamate to alanine. [58] This enzyme is a genetically engineered exo-glycosidase capable of synthesising oligosaccharides through utilisation of an activated glycosyl donor, like α-D-glucosyl fluoride (GlcF) with a range of carbohydrate alcohols, in an anomeric configuration opposite to the natural substrate (Figure 11). The glycosynthase is unable to hydrolyse reaction products due to mutation of the catalytic residue, resulting in an inability to form the requisite R-glycosyl-enzyme intermediate (Figure 11). [56,58]
Other notable glycosynthases are β-1,2 glycosynthases, which improved the bioavailability of various phenolic compounds that are known to have positive impacts on human health.
One interesting example in the glucotolerant β-glucosidase (BGL-1) from the ascomycete fungus Talaromyces amestolkiae, heterologously expressed in Pichia pastoris [59]. This was genetically modified to produce a synthase (BGL-1-E521G) with high regioselectivity to β-1,2 transglycosylation. Using as acceptors epigallocatechin gallate (EGCC) compounds, the synthase was able to synthesize mono- and di-glucoside molecules (Figure 12) [59].
Another interesting glycosynthases are focused on a β-1,3 or β-1,4 specificity, for example in glucan synthesis [56,60,61]. A Hordeum vulgare E231G synthase mediated self-condensation of α-laminaribiosyl fluoride and 3-thio-α-laminaribiosyl fluoride to polymers with different polymerization degree. Also, production of mixed-linked 1,3-1,4 β-glucans from di-, tri- and tetra- saccharide donors has been achieved, whereby tuning of the β-1,3 and β-1,4 linkage ratio produced glucans that do not occur in nature.
Recently, progress has been made towards the development of β-1,3-glucan synthases employing thermo-resistant β-glucosidases as native enzymes, with the in situ formation of glycosyl formate donors allowed the use of both the fluoride donor or an exogenous formate nucleophile to produce a β-1,3 disaccharide. [60,61]
On the other hand, there are also glycosynthases derived from endo-glycosidases. These types of enzymes enable the use of oligosaccharides of different degree of polymerizations to act as glycosyl donors. The first glycosynthase reported to efficiently promote the self-condensation of oligosaccharide donors into polysaccharides was constructed by generating the E197A mutant of the retaining cellulase, Cel7B, of Humicola insolens (HiCel7B) (Figure 13) [62]. This powerful glycosynthase catalyses the transfer of α-cellobiosyl and α-lactosyl fluorides (CelF and LacF, respectively) to a variety of substrates, resulting in the formation of a β-1,4 glycosidic linkage (β-1,4 glycosynthases).
4.2. Galactosynthases
Functional oligosaccharides and glycans such as galacto-N-biose (GNB) and lacto-N-biose (LNB) glycoconjugates are important carbohydrates derivatives that are present in a wide scope of bioactive compounds. Thus, the straightforward access to this type of scaffolds is crucial, with versatile applications in medicinal chemistry and biology [63,64,65,66,67]. So, Y-W. Kim et al [68] studied a new route to access D-Lacto- and D-Galacto-N-bioside glycans (D-LNB and D-GNB, respectively) involving an enzymatic pathway. In particular, glycosynthases have revealed to be useful in the preparation of several oligosaccharides and other glycoconjugates [69,70,71,72,73,74], since they possess relevant transglycosylation activity with glycosyl fluorides or glycosyl azides with no appearing hydrolysis, giving opportunity for the synthesis of galactosyl β-1,3-linked transfer products, such as D-LNB and D-GNB. The authors described the synthesis of a galactosynthase derived from glycoside hydrolase (GH) family 35 β-galactosidase. For that, a β-galactosidase from Bacillus circulans mutant (BgaC), where Ala, Gly, and Ser were inserted as substitution of catalytic nucleophiles, was used to explore the potential catalytic activity when α-D-galactopyranosyl fluoride (αGF) and 4-nitrophenyl β-d-glucopyranoside (pNβG) were used as the sugar donor and acceptor, respectively.
After the reaction completion, BgaC synthase bearing Ala and Ser did not yield the desired transfer products, leading only to hydrolysis of the sugar donor (αGF). The one bearing Gly, indeed generated a transfer product and after careful LC-MS analysis by the authors, the observed product was a disaccharide bearing different glycosidic linkages from pNβG. [75]
To further expand the selectivity studies using this BgaC bearing Gly, eighteen different aryl sugar acceptors were employed using αGF as the sugar donor at 25ºC for 5h, with 5 glycosides being identified as acceptors for this BgaC. Thus, these five acceptors were then used to perform the transfer reaction leading to ten different products (1a-1e and 2a-2e) bearing the β-1,3-linkage, which was confirmed by 13C-NMR (Figure 14). Analysis by HPLC and LC-MS also overruled the formation of trisaccharide. [75]
Regarding catalytic efficiency, BgaC-Gly revealed to perform better when α-configured glycosides were used. Thus, the authors were able to develop an unprecedented synthesis of a galactosynthase derived from glycoside hydrolase (GH) family 35 β-galactosidase, which was able to generate transfer products bearing the desired bearing the β-1,3-linkage. Furthermore, para-nitrophenol-αLNB and para-nitrophenol-αGNB were obtained in up to 98% yield. [75]
Given the versatility of β-galactosidase from Bacillus circulans mutant (BgaC), P. Bojarová et al [76] reported the synthetic application of this mutant enzymes for the transformation of α-galactosyl fluoride (αGF) and β-galactosyl azide (βGN3) α-galactosyl, to test the glycosynthase activity. For those three different mutants were obtained using selective mutagenesis, by active site modification to glycine, alanine and threonine, and then applied in the transformation. Still, these donors were not successful and instead, two mutants (bearing glycine and threonine) were employed in the selective synthesis of azido-functionalized N-acetyllactosamine using the p-nitrophenyl β-d-galactoside as a galactosyl donor (Figure 15).
Furthermore, unexpectedly the prepared mutants still retained a minor part of their hydrolytic activity, which can justify why these (αGF and βGN3) were not successful donors. The authors studied this behavior by molecular docking. Thus, the results published by the authors highlight that the catalytic nucleophile may not be entirely valid to all glycosidases but instead structural interactions in the active site should be judged. [76]
In order to achieve the synthesis of valuable α-galactosyl oligosaccharides, [77,78] a glycosynthase from Bacteroides thetaiotaomicron glycoside hydrolase family (GH) 97 (BT synthase) was used in combination with β-galactosyl azide (βGN3) and a-galactosyl fluoride (αGF) as donors, and lactose as acceptor with assistance of external anions. In this case as previously observed, formate proved to be the best assistant for attaining the desired oligosaccharides in the transglycosylation. Even though, inhibition of the donor cleavage reaction is better performed by the azide that by the formate, an accumulation of βGN3 was observed when this was used, with a low yield of transfer product. To justify these results, the authors performed kinetic studies that suggested the formation of a complex between the enzyme, βGN3 and lactose, which limited the transfer reaction in the azide-rescued reaction. [78]
In this way, GT synthase was able to produce α-galactosides via formate-rescued transglycosylation was achieved in 90% yield using xylose or lactose as acceptor with galactosyl fluoride as donor.
4.3. Fucosynthases
Asparagine-linked glycosylation, also known as N-glycosylation, is one of the most prevalent protein post-translational modifications in mammals and plays a key role in regulating the intrinsic properties and biological functions of basic proteins. [79,80] In particular, core fucosylation linking 16-linked fucose to the deepest asparagine-linked N-acetylglucosamine (GlcNAc) moiety in N-glycans is an important modification of N-glycoproteins. Intriguing evidence suggests that core glycoprotein fucosylation regulates diverse cellular functions. For instance, several studies have shown that increased association with core fucosylation is often associated with the development of cancer. [81,82,83]
However, the synthesis of a well-defined fucosylated core glycoprotein structure remains a challenging task due to the complexity in multiphase chemical synthesis or the inability of the biosynthetic 16-fucosyltransferase (FUT8) to directly fucosylate full-size mature N-glycans. [84,85,86,87] For this reason a method for direct fucosylation of intact glycopeptides and glycoproteins is highly desirable.
Following this approach, the investigation group formed by Wang and coworkers [88], describes the design and generation of potential α1,6-fucosynthase and fucoligase for direct core fucosylation of intact N-glycoproteins without product hydrolysis by using novel mutants derived from Lactobacillus casei α-fucosidase.
Firstly, they created several mutants of the L. casei 1,6-fucosidase glycosynthase and glycoligase and they assessed those enzymes' capacity to core fucosylate a range of acceptor substrates (Figure 16).
Following the glycosynthase concept proposed by Withers and co-workers,[89] they performed site-directed mutagenesis at the identified nucleophile in the AlfC α1,6-fucosidase, D200, to generate selected mutants, including D200G, D200S, D200A, and D200T. Similar to this, specific mutants at the putative generic acid/base residue, E274, such as E274A, E274S, E274G, and E274D, were created to provide potential glycoligases. Except for E274D, none of these mutants showed more than traces of residual hydrolysis activity due to mutations at the critical residues. In addition, this study confirmed that the D200 residue is the nucleophile and that the E274 residue is most likely the general acid/base.
Therefore, they assessed the synthesized mutants as potential glycosynthases or glycoligases. Thus, the potential glycosylation activity of the nucleophilic mutants, including D200G, D200S, D200A, and D200T, was tested using both the β-glycosyl azide and the β-glycosyl fluoride as the donor substrates and the Fmoc-Asn(GlcNAc)-OH as the acceptor substrate (Figure 17). No glycosylation products were observed in any of the cases studied, and this finding indicated that the nucleophilic mutants tested did not act as a glycosynthase. Then, they tested the use of α-fucosyl fluoride as the donor substrate (Figure 17). Interestingly, the E274A mutant displayed good enzymatic activity to transfer a fucose residue to the GlcNAc moiety of acceptor, resulting in the disaccharide Fucα1,6GlcNAc-Asn with regio- and stereospecificity (Figure 17). Similar outcomes were produced by the other two mutants, E274G and E274S, which were likewise effective α1,6-fucosylation catalysts.
In addition, they found that the AlfC mutants (E274A, E274G, and E274S) only displayed reduced activity in the absence of the GlcNAc acceptor. While the wild-type AlfC could promptly hydrolyze the donor substrate, it hydrolyzed -fucosyl fluoride slowly. These findings collectively suggested that the AlfC mutations represented a class of distinct O-fucoligase for core fucosylation, able to utilise inexpensive synthetic -fucosyl fluoride as the donor substrate rather than the pricey GDP-fucose as required by the α1,6-fucosyltransferase (FUT8).
Simultaneously, they tested if the mutants could also fucosylate the GlcNAc moiety in the setting of different peptides sequences. The Endo-F3 D165A glycosynthase may use the Fucα1,6GlcNAc-peptides as excellent acceptor substrates to produce core-fucosylated complex N-glycopeptides (Figure 17). Finally, authors demonstrate the potential applicability of the fucoligase E274A to site-specific incorporate a core fucose to complex oligosaccharide moiety in N-glycopeptides (Figure 18). Furthermore, experimental evidence revealed that in the glycoligase-catalyzed fucosylation, the fucose moiety was in fact added particularly to the innermost Asn-linked GlcNAc moiety of the glycopeptide
These strategy was successfully extended in the selective glycosylation of proteins and antibodies [89].
4.4. Chitinases
Chitinases are glycoside hydrolases (GH) that catalyze the hydrolysis of chitin generating chito-oligosaccharides (COS). Chitin and chitosans, its partially deacetylated derivates, are present in most living organisms, including bacteria, fungi, plants and animals [90,91] and exhibit immunostimulant activities in mammals and plants [92,93]. In addition, several studies have shown that their breakdown products, COS, have antimicrobial and antitumor activities in animals, immunoenhancing effects in humans as dietary supplements [94,95,96,97,98], and disease protective responses in plants [99,100], which makes them suitable for agriculture and medicine applications [101,102,103,104,105]. Most of their biological activities require degrees of polymerization larger than the tetrasaccharide [92]. However, this is difficult to produce in a chemical synthesis due to the water insolubility of the products is higher as the degree of polymerization (DP) increases. Thus, in the search for bioactive COS production, enzymatic synthesis represents a potential strategy through the transglycosylation activity of chitinases [106].
For this purpose, several research groups have studied the glycosynthase activity of various chitinases to obtain long-chain oligomers with potential biological applications.
Alsina et al. [92] have studied the glycosynthase-like activity of six chitinases of glycosyl hydrolases family 18 (GH18) to obtain larger oligomers or polymers which could be more resistant. They selected four and two endo-chitinases from bacterial and archaeal origin, respectively, and then mutated the catalytic assisting residue to alanin. Thus, the hydrolase activity would be reduced and an oxazoline derivate could be provided, which would act as a donor substrate for a condensation with an acceptor, catalyzing the polymerization reaction (Figure 19).
The enzymes selected were Bacillus circulans, Serratia proteamaculans ChiD, Laceyella putida ChiA, Serratia marcescens ChiC, Pyrococcus furiosus ChiB and Thermococcus kodakaraensis ChiA. Among them, LpChiA, SmChiC and PfChiB have not reported TG activity on chitooligosaccharide (COS) substrates.
In relation to hydrolase activity, two types of tetrasaccharides were used as substrates, finding that all enzymes showed activity, especially BcChiA, LpChiA and SmChiC. Regarding to glycosynthase activity, the substrate used was pentaacetylchitopentaose oxazoline (DP5ox). After two hours of reaction, the authors found that a mixture of COS was generated for all mutant enzymes, from DP5 to DP15, being DP10, DP7 and DP8 the main products. However, due to residual hydrolase activity, after 18 h of incubation lower than expected yields for the DP>10 products were obtained as a result of hydrolysis and transglycosylation reactions. The best result was obtained for TkChiA, with a yield of 55% for DP10 after 18 hours.
The method followed by the authors performing a single mutation at the assistant residue in GH18 chitinases does not seem adequate to obtain COS with larger DP due to the residual presence of hydrolase activity in the mutant enzymes. Further mutations would be needed to avoid the hydrolyzation and enhance the glycosynthase-like activity.
In a new approach, the group of Alsina et al. [107] focused on the study of bacterial enzyme SpChiD. It had been shown that a single mutation in the assistant residue did not eliminate the hydrolase activity, so a different strategy was tested. For this purpose, different active sites were mutated to achieve a greater reduction in the hydrolase activity. In addition to mutation D151A already performed, others were added based on previous studies and simulations model that would enhance TG and decrease hydrolase activities.
A second generation of 4 mutants with an additional mutation in each of them (S110G/D151A, G113S/D151A, F119A/D151A and D149A/D151A) was performed. In one of the mutants (D149A/D151A), significant improvements in the wanted activity were achieved, obtaining DP10 as the majority product with the best ratio and an insoluble product yield of 30%. This enzyme was selected to design a third generation of new mutants. The triple mutants had reduced hydrolase activity compared to the previous mutants. Glycosynthase activity also improved, except in two of them. After 18 h, precipitate yields of 22 to 68% were obtained, with DP10 as the major product and getting the best results for Y154W/D149A/D151A and Y28A/D149A/D151A.
The chitinases engineered in this third generation have better oligomerization yields than those of a single mutation and also than those of any reported GH18 transglycosylating chitinase.
Thus, through a series of targeted mutations, it has been possible to design hybrids with better glycosyl synthase-like activity, but a further reduction in hydrolase activity would still be necessary for the designed mutants to have truly applicable activity.
Following a similar strategy, Ohnuma et al. [108], mutated GH19 chitinases from Bryum coronatum (BcChi-A) to obtain chitin oligosaccharides using successfully 4,6-dimethoxy-1,3,5-triazin-2-yl α-chitobioside [DMT- α-(GlcNAc)2] as a donor substrate (Figure 20).
Single and double mutants were created by changing the catalytic base (Glu70) and the residue Ser102, which acts fixing a water molecule. In a previous study, the authors had demonstrated that the single mutants showed glycosynthase activity with α-(GlcNAc)2 fluoride as donor substrate. In this paper, the authors tested these single mutants and new double mutants with DMT- α-(GlcNAc)2 to obtain (GlcNAc)4, which were designed to improve the activity of the single ones.
Single mutants did not show glycosynthase activity with the substrate after 48 h of reaction. However, the double mutants (E70G/S102A and E70G/S102C) showed activity and (GlcNAc)4 was obtained as the mayor product. For E70G/S102A, the production yield of (GlcNAc)4 was 22.4%. Small-chain oligosaccharides were also obtained as secondary products, showing the presence of residual hydrolase activity in the mutants, although much lower compared to wild-type enzyme.
Thus, the introduction of well-studied combined mutations in different glycosyl hydrolases, such as the chitinases used in the described studies, can strongly reduce the hydrolase activity, and generate glycosynthase-like activity in the new mutants, which allows to obtain oligosaccharides and long-chain glycoconjugates from different substrates.
5. Conclusions
This review article summarizes some of the most recent advances on the enzymatic glycosylation processes focused on the fabrication of important bioactive compounds. Applicability to glycosylation process by glycosidases, glycosyltransferases, or the so-called glycosynthases have been described with the advantages or disadvantages of these enzymes in each process. Glycosylation of important alcohols such as aromatic as flavonoids or aliphatic such as xylitol have been discussed focus on the fabrication of different products for food technology or pharmaceutical applications. Important advantages in the genetic approaches to create enzymes with full glycosylation power, being able to synthesize tailor-made oligosaccharides or complex glycopeptides have been also presented here. Thus all these different approaches in the glycosylation of molecules can be applied in the future to follow the discovery of more new kind of compounds with improved biological activities with antiviral, antitumoral or anti-inflammatory properties.
Acknowledgments
The authors thank the support of the Spanish National Research Council (CSIC) and Ministry of Science, Technological Development and Innovations of the Republic of Serbia (Contract No. 451-03-47/2023-01/200135). The authors also wish to acknowledge the funding from European Commission, project “Twinning for intensified enzymatic processes for production of prebiotic-containing functional food and bioactive cosmetics “grant no. 101060130, HORIZON-WIDERA-2021-ACCESS-02-01 and Science Fund of the Republic of Serbia, programme IDEAS, project no. 7750109 (PrIntPrEnzy). The Authors would like to acknowledge networking support by the COST Action CA18132 (GlycoNanoProbes).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gantt, R. W.; Pauline Peltier-Pain, P; Thorson, J. S. Enzymatic methods for glyco(diversification/randomization) of drugs and small molecules. Nat. Prod. Rep. 2011, 28, 1811. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Phillips, G. N. Jr.; Thorson, J. S. The structural biology of enzymes involved in natural product glycosylation. Nat Prod Rep. 2012, 29, 1201–1237. [Google Scholar] [CrossRef] [PubMed]
- Song, M. C.; Kim, E.; Ban, Y. H.; Yoo, Y. J.; Kim, E. J.; Park, S. R.; Pandey, R. P.; Sohng, J. K.; Yoon, Y. J. Achievements and impacts of glycosylation reactions involved in natural product biosynthesis in prokaryotes. Appl Microbiol Biotechnol 2013, 97, 5691–5704. [Google Scholar] [CrossRef] [PubMed]
- D. J. Newman, D. J.; Cragg, G. M. Natural products as sources of new drugs over the last 25 years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef]
- Butler, M. S. Natural products to drugs: natural product-derived compounds in clinical trials. Nat. Prod. Rep. 2008, 25, 475–516. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Nguyen, T. T. H.; Kim, N. M.; Moon, Y.-H.; Ha, J.-M.; Park, N.; Lee, D.-G.; Hwang, K.-H.; Park, J.-S.; Kim, D. Functional properties of novel epigallocatechin gallate glucosides synthesized by using dextransucrase from Leuconostoc mesenteroides B-1299CB4. J. Agricul. Food Chem 2016, 64, 9203–9213. [Google Scholar] [CrossRef]
- Gantt, R. W.; Peltier-Pain, P.; Thorson, J. S. Enzymatic methods for glyco(diversification/randomization) of drugs and small molecules. Nat. Prod. Rep. 2011, 28, 1811. [Google Scholar] [CrossRef]
- Lyu, J.; Zhang, J.; Zhu, J.; Chen, S.; Han, T.; Zhang, Y.; Gao, R.; Xie, G.; Guo, Z. Molecular dynamics simulation guided distal mutation of Thermotoga naphthophila β-glucosidase for significantly enhanced synthesis of galactooligosaccharides and expanded product scope. Inter.J. Biol.l Macromol. 2022, 210, 21–32. [Google Scholar] [CrossRef]
- Paliya, B. S.; Sharma, V. K.; Tuohy, M. G.; Singh, H. B.; Koffas, N.; Benhida, R.; Tiwari, B. K.; Kalaskar, D. M.; Singh, B. N.; Gupta, V. K. Bacterial glycobiotechnology: A biosynthetic route for the production of biopharmaceutical glycans. Biotechnol. Adv.s, 2023; 67, 108180. [Google Scholar]
- Xu, L.; Qi, T.; Xu, L.; Lu, L.; Xiao, M. Recent progress in the enzymatic glycosylation of phenolic compounds. Journal of Carbohydrate Chemistry 2016, 35, 1–23. [Google Scholar] [CrossRef]
- Saranraj, P.; Behera, S.S.; Ray, R.C. ; Traditional foods from tropical root and tuber crops: innovations and challenges, Innovations in traditional foods. Elsevier 2019, 159–191. [Google Scholar]
- Sun, Q.; Heilmann, J.; König, B. Natural phenolic metabolites with anti-angiogenic properties–a review from the chemical point of view. Beilstein journal of organic chemistry 2015, 11, 249–264. [Google Scholar] [CrossRef]
- Orhan, D. D.; Özçelik, B.; Özgen, S.; Ergun, F. Antibacterial, antifungal, and antiviral activities of some flavonoids. Microbiological research 2010, 165, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Taamalli, A.; Arráez-Román, D.; Zarrouk, M.; Segura-Carretero, A.; Fernández-Gutiérrez, A. The Occurrence and Bioactivity of Polyphenols in Tunisian Olive Products and by-Products: A Review. Journal of food science 2012, 77, R83–R92. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Nguyen, T. T. H.; Kim, N. M.; Moon, Y.-H.; Ha, J.-M.; Park, N.; Lee, D.-G.; Hwang, K.-H.; Park, J.-S.; Kim, D. Functional properties of novel epigallocatechin gallate glucosides synthesized by using dextransucrase from Leuconostoc mesenteroides B-1299CB4, Journal of agricultural and food chemistry 2016, 64, 9203–9213.
- Méndez-Líter, J.A.; Tundidor, I.; Nieto-Domínguez, M.; de Toro, B. F.; González Santana, A.; de Eugenio, L.I.; Prieto, A.; Asensio, J.L.; Sánchez, C.; Martínez, M.J. Transglycosylation products generated by Talaromyces amestolkiae GH3 β-glucosidases: Effect of hydroxytyrosol, vanillin and its glucosides on breast cancer cells. Microbial cell factories 2019, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Li, B.; Qiao, M.; Li, J.; Xu, H.; Zhang, L.; Zhang, X. Advances on the in vivo and in vitro glycosylations of flavonoids. Applied Microbiology and Biotechnology 2020, 104, 6587–6600. [Google Scholar] [CrossRef]
- Schmid, J.; Heider, D.; Wendel, N. J.; Sperl, N.; Sieber, V. Bacterial glycosyltransferases: challenges and opportunities of a highly diverse enzyme class toward tailoring natural products. Frontiers in microbiology 2016, 7, 182. [Google Scholar] [CrossRef]
- Feng, J.; Zhang, P.; Cui, Y.; Li, K.; Qiao, X.; Zhang, Y. T.; Li, S. M.; Cox, R. J.; Wu, B.; Ye, M. Regio-and Stereospecific O-Glycosylation of Phenolic Compounds Catalyzed by a Fungal Glycosyltransferase from Mucor hiemalis. Advanced Synthesis & Catalysis 2017, 359, 995–1006. [Google Scholar]
- Chang, S.K.; Jiang, Y.; Yang, B. An update of prenylated phenolics: Food sources, chemistry and health benefits. Trends in Food Science & Technology 2021, 108, 197–213. [Google Scholar]
- Xie, K.; Dou, X.; Chen, R.; Chen, D.; Fang, C.; Xiao, Z.; Dai, J. Two novel fungal phenolic UDP glycosyltransferases from Absidia coerulea and Rhizopus japonicus. Applied and environmental microbiology 2017, 83, e03103–e03116. [Google Scholar] [CrossRef]
- Yang, L.; Wang, Z.; Lei, H.; Chen, R.; Wang, X.; Peng, Y.; Dai, J. Neuroprotective glucosides of magnolol and honokiol from microbial-specific glycosylation. Tetrahedron 2014, 70, 8244–8251. [Google Scholar] [CrossRef]
- Li, B.; Chang, S.; Jin, D.; Zhang, S.; Chen, T.; Pan, X.; Fan, B.; Lv, K.; He, X. Ca2+ assisted glycosylation of phenolic compounds by phenolic-UDP-glycosyltransferase from Bacillus subtilis PI18. International journal of biological macromolecules 2019, 135, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Tang, W.; Barton, C. D.; Price, O. M.; Mortensen, M. W.; Phillips, A.; Wald, B.; Hulme, S. E.; Stanley, L. P.; Hevel, J. A highly versatile fungal glucosyltransferase for specific production of quercetin-7-O-β-d-glucoside and quercetin-3-O-β-d-glucoside in different hosts. Applied microbiology and biotechnology 2021, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Rha, C.-S.; Choi, J.-M.; Jung, Y. S.; Kim, E.-R.; Ko, M. J.; Seo, D.-H.; Kim, D.-O.; Park, C.-S. High-efficiency enzymatic production of α-isoquercitrin glucosides by amylosucrase from Deinococcus geothermalis. Enzyme and microbial technology 2019, 120, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Rha, C.-S.; Jung, Y.S.; Seo, D.-H.; Kim, D.-O.; Park, C.-S. Site-specific α-glycosylation of hydroxyflavones and hydroxyflavanones by amylosucrase from Deinococcus geothermalis. Enzyme and microbial technology 2019, 129, 109361. [Google Scholar] [CrossRef]
- Gonzalez-Alfonso, J.L.; Leemans, L.; Poveda, A.; Jimenez-Barbero, J.s.; Ballesteros, A. O.; Plou, F. J. Efficient α-glucosylation of epigallocatechin gallate catalyzed by cyclodextrin glucanotransferase from Thermoanaerobacter species. Journal of agricultural and food chemistry 2018, 66, 7402–7408. [Google Scholar] [CrossRef]
- González-Alfonso, J.L.; Míguez, N.; Padilla, J.D.; Leemans, L.; Poveda, A.; Jiménez-Barbero, J.; Ballesteros, A. O.; Sandoval, G.; Plou, F.J. Optimization of regioselective α-glucosylation of hesperetin catalyzed by cyclodextrin glucanotransferase. Molecules 2018, 23, 2885. [Google Scholar] [CrossRef]
- Méndez-Líter, J. A.; Nieto-Domínguez, M.; de Toro, B. F.; Santana, A. G.; Prieto, A.; Asensio, J. L.; de Eugenio, L. I.; Martínez, M. J. A glucotolerant β-glucosidase from the fungus Talaromyces amestolkiae and its conversion into a glycosynthase for glycosylation of phenolic compounds. Microbial cell factories 2020, 19, 1–13. [Google Scholar] [CrossRef]
- Yang, B.; Liu, H.; Yang, J.; Gupta, V. K.; Jiang, Y. New insights on bioactivities and biosynthesis of flavonoid glycosides. Trends in food science & technology, 2018; 79, 116–124. [Google Scholar]
- He, J. B.; Zhao, P.; Hu, Z. M.; Liu, S.; Kuang, Y.; Zhang, M.; Li, B.; Yun, C. H.; Qiao, X.; Ye, M. Molecular and Structural Characterization of a Promiscuous C-Glycosyltransferase from Trollius chinensis. Angewandte Chemie 2019, 131, 11637–11644. [Google Scholar] [CrossRef]
- Pei, J.; Sun, Q.; Gu, N.; Zhao, L.; Fang, X.; Tang, F.; Cao, F. Production of isoorientin and isovitexin from luteolin and apigenin using coupled catalysis of glycosyltransferase and sucrose synthase. Applied biochemistry and biotechnology 2020, 190, 601–615. [Google Scholar] [CrossRef]
- J. Pei, Q. Sun, L. Zhao, H. Shi, F. Tang, F. Cao, Efficient biotransformation of luteolin to isoorientin through adjusting induction strategy, controlling acetic acid, and increasing UDP-glucose supply in Escherichia coli, Journal of agricultural and food chemistry, 67 (2018) 331-340.
- Carević, M.; Veličković, D.; Stojanović, M.; Milosavić, N.; Rogniaux, H.; Ropartz, D.; Bezbradica, D. Insight in the regioselective enzymatic transgalactosylation of salicin catalyzed by β-galactosidase from Aspergillus oryzae. Process Biochemistry 2015, 50, 782–788. [Google Scholar] [CrossRef]
- Murguiondo, C.; Mestre, A.; Méndez-Líter, J. A.; Nieto-Domínguez, M.; de Eugenio, L. I.; Molina-Gutiérrez, M.; Martínez, M. J.; Prieto, A. Enzymatic glycosylation of bioactive acceptors catalyzed by an immobilized fungal β-xylosidase and its multi-glycoligase variant. International Journal of Biological Macromolecules 2021, 167, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Alfonso, J.L.; Ubiparip, Z.; Jimenez-Ortega, E.; Poveda, A.; Alonso, C.; Coderch, L.; Jimenez-Barbero, J.; Sanz-Aparicio, J.; Ballesteros, A. O.; Desmet, T. Enzymatic Synthesis of Phloretin α-Glucosides Using a Sucrose Phosphorylase Mutant and its Effect on Solubility, Antioxidant Properties and Skin Absorption. Advanced Synthesis & Catalysis, 2021. [Google Scholar]
- Sordon, S.; Popłoński, J.; Huszcza, E. Microbial glycosylation of flavonoids, Polish journal of microbiology 2016, 65, 7. 65.
- Sordon, S.; Popłoński, J.; Tronina, T.; Huszcza, E. Microbial glycosylation of daidzein, genistein and biochanin A: Two new glucosides of biochanin A. Molecules 2017, 22, 81. [Google Scholar] [CrossRef]
- Sordon, S.; Popłoński, J.; Tronina, T.; Huszcza, E. Regioselective O-glycosylation of flavonoids by fungi Beauveria bassiana, Absidia coerulea and Absidia glauca. Bioorganic chemistry 2019, 93, 102750. [Google Scholar] [CrossRef] [PubMed]
- Dymarska, M.; Janeczko, T.; Kostrzewa-Susłow, E. Biotransformations of flavones and an isoflavone (daidzein) in cultures of entomopathogenic filamentous fungi. Molecules 2018, 23, 1356. [Google Scholar] [CrossRef]
- Dymarska, M.; Janeczko, T.; Kostrzewa-Susłow, E. Glycosylation of 3-hydroxyflavone, 3-methoxyflavone, quercetin and baicalein in fungal cultures of the genus Isaria. Molecules, 2018, 23, 2477. [Google Scholar] [CrossRef] [PubMed]
- Rosado, E.; Delgado-Fernandez, P.; de las Rivas, B.; Muñoz, R.; Moreno, J.; Corzo, N.; Mateo. C. Production of β-Galactosyl Xylitol Derivatives Using Heterogeneous Catalysts of LacA β-Galactosidase From Lactobacillus plantarum WCFS1. Molecules 2022, 27, 1235. [Google Scholar] [CrossRef]
- Cheng, K. L.; Bradely, T.; Budman, D. R. Novel microtubule-targeting agents- the epothilones. Biologics 2008, 2, 789–811. [Google Scholar]
- Hofle, G.; Reichenbach, H. Epothilone, a myxobacterial metabolite with promising antitumor activity. In: Cragg GM, Kingston DGI, Newman DJ (eds). Anticancer agents from natural products. CRC Press, 2005; 413–450. [Google Scholar]
- Hardt, I.H.; Steinmetz, H.; Gerth, K.; Sasse, F.; Reichenbach, H.; Hofle, G. New natural epothilones from Sorangium cellulosum, strains So ce 90/B2 and So ce90/D13:isolation, structure elucidation, and SAR studies. J Nat Prod 2001, 64, 847–856. [Google Scholar] [CrossRef]
- Lee, F. Y.; Borzilleri, R.; Fairchild, C. R.; Kim, S. H.; Long, B. H.; Reventos-Suarez, C.; Vite, G. D.; Rose, W. C.; Kramer, R. A. BMS-247550: a novel epothilone analog with a mode of action similar to paclitaxel but possessing superior antitumor efficacy. Clin Cancer Res 2001, 7, 1429–1437. [Google Scholar]
- Thomas, E.; Tabemero, J.; Fornier, M.; Conte, P.; Fumoleau, P.; Liuch, A.; Vahdat, L. T.; Bunnell, C. A.; Burris, H. A.; Viens, P.; Baselqa, J.; Rivera, E.; Guameri, V.; Poulart, V.; Klimovsky, J.; Lebwohl, D.; Martin, M. Phase II clinical trial of ixabepilone (BMS-247550), an epothilone B analog, in patients with taxane-resistant metastatic breast cancer. J Clin Oncol 2007, 25, 3399–3406. [Google Scholar] [CrossRef]
- Parajuli, P.; Pandey, R. P.; Koirala, N. et al. Enzymatic synthesis of epothilone A glycosides. AMB Expr 2014, 4, 31. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, E. C.; Field, R. A. Enzymatic synthesis using glycoside phosphorylases. Carbohydr. Res. 2015, 403, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Puchart, V. Glycoside phosphorylases: Structure, catalytic properties and biotechnological potential. Biotechnol. Adv. 2015, 33, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Nakai, H.; Kitaoka, M.; Svensson, B.; Ohtsubo, K.I. Recent development of phosphorylases possessing large potential for oligosaccharide synthesis. Curr. Opin. Chem. Biol. 2013, 17, 301–309. [Google Scholar] [CrossRef]
- Pergolizzia, G.; Kuhaudomlarpa, S.; Kalitaa, E.; Fielda, R. A. Glycan Phosphorylases in Multi-Enzyme Synthetic Processes. Protein & Peptide Letters 2017, 24, 696–709. [Google Scholar]
- Wildberger, P.; Pfeiffer, M.; Brecker, L.; Nidetzky, B. Diastereoselective synthesis of glycosyl phosphates by using a phosphorylase-phosphatase combination catalyst. Angew. Chem. Int. Ed. 2015, 54, 15867–15871. [Google Scholar] [CrossRef]
- Hancock, S. M.; Vaughan, M. D.; Withers, S. G. Engineering of glycosidases and glycosyltransferases. Current Opinion in Chemical Biology. 2006, 10, 509–519. [Google Scholar] [CrossRef]
- Schmölzer, K.; Weingarten, M.; Baldenius, K.; Nidetzky, B. Glycosynthase Principle Transformed into Biocatalytic Process Technology: Lacto N triose II Production with Engineered exo-Hexosaminidase. ACS Catal. 2019, 9, 5503–5514. [Google Scholar] [CrossRef]
- Bulmer, G. S.; de Andrade, P.; Field, R. A.; van Munster, J. M. Recent advances in enzymatic synthesis of β-glucan and cellulose. Carbohydrate Research. 2021, 508, 108411. [Google Scholar]
- Mészáros, Z.; Nekvasilová, P.; Bojarová, P.; Kren, V.; Fernández Slámová, K. Advanced glycosidases as ingenious biosynthetic instruments. Biotechnology Advances. 2021, 49, 107733. [Google Scholar] [CrossRef]
- Williams, S. J.; Withers, S. G. Glycosynthases: Mutant Glycosidases for Glycoside Synthesis. Australian Journal of Chemistry. 2002, 55, 3–12. [Google Scholar]
- Méndez-Líter, J.A.; Nieto-Domínguez, M.; de Toro, B. et al. A glucotolerant β-glucosidase from the fungus Talaromyces amestolkiae and its conversion into a glycosynthase for glycosylation of phenolic compounds. Microb Cell Fact. 2020, 19, 127. [Google Scholar] [CrossRef] [PubMed]
- Hrmova, M.; Imai, T.; Rutten, S. J.; Fairweather, J. K.; Pelosi, L.; Bulone, V.; Driguez, H.; Fincher, G. B. Mutated Barley (1,3)-β-d -Glucan Endohydrolases Synthesize Crystalline (1,3)-β-d -Glucans*. Journal of Biological Chemistry. 2002, 277, 30102–30111. [Google Scholar] [CrossRef] [PubMed]
- Pérez, X.; Faijes, M.; Planas, A. Artificial Mixed-Linked β-Glucans Produced by Glycosynthase-Catalyzed Polymerization: Tuning Morphology and Degree of Polymerization. Biomacromolecules. 2011, 12, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Smith, P. J.; Ortiz-Soto, M. E.; Roth, C.; Barnes, W. J.; Seibel, J.; Urbanowicz, B. R.; Pfrengle, F. Enzymatic Synthesis of Artificial Polysaccharides. ACS Sustainable Chem. Eng. 2020, 8, 11853–11871. [Google Scholar] [CrossRef]
- E. Steensma, W. G. J. Hol, Protein Sci. 1996, 5, 1184–1188;
- R. Bukowski, L. M. Morris, R. J. Woods, T. Weimar, Eur. J. Org. Chem. 2001, 2697 – 2705.
- Mocchetti, Cell. Mol. Life Sci. 2005, 62, 2283–2294.
- Brockhausen, EMBO Rep. 2006, 7, 599 –604.
- T. Urashima, S. Asakuma, F. Leo, K. Fukuda, M. Messer, O. T. Oftedal, Adv. Nutr. 2012, 3, 473S–482S. [Google Scholar]
- ChemBioChem 2014, 15, 522 – 526.
- K. Yamamoto, Biotechnol. Lett. 2013, 35, 1733–1743. [Google Scholar]
- L.-X. Wang, W. Huang, Curr. Opin. Chem. Biol. 2009, 13, 592 –600; R. Kittl, S. G. Withers,.
- Carbohydr. Res. 2010, 345, 1272 –1279; Z. Armstrong, S. G. Withers, Biopolymers 2013, 99, 666 –674;
- X. P rez, M. Faijes, A. Planas, Biomacromolecules 2011, 12, 494–501.
- B. Cobucci-Ponzano, A. Strazzulli, M. Rossi, M. Moracci, Adv. Synth. Catal. 2011, 353, 2284–2300. [Google Scholar]
- L. F. Mackenzie, Q. Wang, R. A. J. Warren, S. G. Withers, J. Am. Chem. Soc. 1998, 120, 5583–5584. [Google Scholar]
- ChemBioChem 2014, 15, 522 – 526.
- Hovorková, M.; Dr. Kulik, N.; Konvalinková, D.; Dr. Petrásková, L.; Prof. Křen, V.; Prof. Bojarová, P. Mutagenesis of Catalytic Nucleophile of β-Galactosidase Retains Residual Hydrolytic Activity and Affords a Transgalactosidase. ChemCatChem. 2021, 13, 4532–4542. [Google Scholar] [CrossRef]
- Mészáros, Z.; Nekvasilová, P.; Bojarová, P.; Křen, V.; Slámová, K. Reprint of: Advanced glycosidases as ingenious biosynthetic instruments. Biotechnology Advances. 2021, 51, 107820. [Google Scholar] [CrossRef] [PubMed]
- The FEBS Journal ,2017, 284, 766–783.
- Dwek, R. A. A. Chem. Rev. 1996, 96, 683. [Google Scholar] [CrossRef]
- Helenius, A.; Aebi, M. Science. 2001, 291, 2364.
- Taniguchi, N.; Kizuka, Y. Adv. Cancer Res. 2015, 126, 11. [Google Scholar]
- Pinho, S. S.; Reis, C. A. Nat. Rev. Cancer 2015, 15, 540. [Google Scholar] [CrossRef]
- Chen, C. Y.; Jan, Y. H.; Juan, Y. H.; Yang, C. J.; Huang, M. S.; Yu, C. J.; Yang, P. C.; Hsiao, M.; Hsu, T. L.; Wong, C. H. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 630. [CrossRef]
- Li, L.; Liu, Y.; Ma, C.; Qu, J.; Calderon, A. D.; Wu, B.; Wei, N.; Wang, X.; Guo, Y.; Xiao, Z.; Song, J.; Sugiarto, G.; Li, Y.; Yu, H.; Chen, X.; Wang, P. G. Chem. Sci. 2015, 6, 5652. [CrossRef]
- Brzezicka, K.; Echeverria, B.; Serna, S.; van Diepen, A.; Hokke, C. H.; Reichardt, N. C. ACS Chem. Biol. 2015, 10, 1290.
- Calderon, A. D.; Liu, Y.; Li, X.; Wang, X.; Chen, X.; Li, L.; Wang, P. G. Org. Biomol. Chem. 2016, 14, 4027.
- Tseng, T. H.; Lin, T. W.; Chen, C. Y.; Chen, C. H.; Lin, J. L.; Hsu, T. L.; Wong, C. H. J. Am. J. Am. Chem. Soc. 2017, 139, 9431. [CrossRef] [PubMed]
- Chao Li, C.; Shilei Zhu, S.; Christopher Ma, C.; Wan, L.-X. Designer α1,6-Fucosidase Mutants Enable Direct Core Fucosylation of Intact N-Glycopeptides and N-Glycoproteins. J. Am. Chem. Soc. 2017, 139, 15074–15087. [Google Scholar]
- MacKenzie, L. F.; Wang, Q.; Warren, R. A. J.; Withers, S. G. J. Am. J. Am. Chem. Soc. 1998, 120, 5583. [CrossRef]
- Armand, S.; Tomita, H.; Heyraud, A.; Gey, C.; Watanabe, T.; Henrissat, B. Stereochemical course of the hydrolysis reaction catalyzed by chitinases Al and D from Bacillus circulans WL-12. FEBS Lett. 1994, 343, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Henrissat, B.; Davies, G. Structural and sequence-based classification of glycoside hydrolases. Curr. Opin. Struct. Biol. 1997, 7, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Alsina, C.; Faijes, M.; Planas, A. Glycosynthase-type GH18 mutant chitinases at the assisting catalytic residue for polymerization of chitooligosaccharides. Carbohydrate Research 2019, 478, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Elieh Ali Komi, D.; Sharma, L.; Dela Cruz, C. S. Chitin and its effects on inflammatory and immune responses Clin. Rev. Allergy Immunol. 2018, 54, 213–223.
- Yoon, H. J.; Moon, M. E.; Park, H. S.; Im, S. Y.; Kim, Y. H. Chitosan oligosaccharide (COS) inhibits LPS-induced inflammatory effects in RAW 264.7 macrophage cells. Biochem. Biophys. Res. Commun. 2007, 358, 954–959. [Google Scholar] [CrossRef] [PubMed]
- Yousef, M.; Pichyangkura, R.; Soodvilai, S.; Chatsudthipong, V.; Muanprasat, C. Chitosan oligosaccharide as potential therapy of inflammatory bowel disease: therapeutic efficacy and possible mechanisms of action. Pharmacol. Res. 2012, 66, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Chung, M. J.; Park, J. K.; Park, Y. II. Anti-inflammatory effects of low-molecular weight chitosan oligosaccharides in IgE-antigen complex-stimulated RBL-2H3 cells and asthma model mice. Int. Immunopharmacol. 2012, 12, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.-T.; Chen, M.-H.; Chan, H.-Y.; Jeng, J.-H.; Wang, Y.-J. Inhibitory effects of chitooligosaccharides on tumor growth and metastasis. Food Chem. Toxicol. 2009, 47 () 1864–1871.
- Park, J. K.; Chung, M. J.; Choi, H. N.; Park, Y. II. Effects of the molecular weight and the degree of deacetylation of chitosan oligosaccharides on antitumor activity. Int. J. Mol. Sci. 2011, 12, 266–277.
- Hadwiger, L. A. Plant science review: multiple effects of chitosan on plant systems: solid science or hype. Plant Sci. 2013, 208, 42–49.
- Katiyar, D.; Hemantaranjan, A.; Singh, B.; Bhanu, A. N. A future perspective in crop protection: chitosan and its oligosaccharides. Adv Plants Agric Res 2014, 1, 4–11. [Google Scholar]
- Azuma, K.; Ifuku, S.; Osaki, T.; Okamoto, Y.; Minami, S. Preparation and biomedical applications of chitin and chitosan nanofibers. J. Biomed. Nanotechnol. 2014, 10, 2891–2920. [Google Scholar] [CrossRef]
- Ravi Kumar, M. N. A review of chitin and chitosan applications. React. Funct Polym. 2000, 46, 1–27. [Google Scholar]
- Philibert, T.; Lee, B. H.; Fabien, N. Current status and new perspectives on chitin and chitosan as functional biopolymers. Appl. Biochem. Biotechnol. 2017, 181, 1314–1337. [Google Scholar] [CrossRef]
- Rinaudo, M. Chitin and chitosan: properties and applications. Prog. Polym. Sci. 2006, 31, 603–632. [Google Scholar] [CrossRef]
- Xia, W.; Liu, P.; Zhang, J.; Chen, J. Biological activities of chitosan and chitooligosaccharides. Food Hydrocolloids 2011, 25, 170–179. [Google Scholar] [CrossRef]
- Martinez, E. A.; Boer, H.; Koivula, A.; Samain, E.; Driguez, H.; Armand, S.; Cottaz, S. Engineering chitinases for the synthesis of chitin oligosaccharides: catalytic aminoacid mutations convert the GH-18 family glycoside hydrolases into transglycosylases. J. Mol. Catal. B Enzym. 2012, 74, 89–96. [Google Scholar] [CrossRef]
- Alsina, C.; Sancho-Vaello, E.; Aranda-Martínez, A.; Faijes, M.; Planas, A. Auxiliary active site mutations enhance the glycosynthase activity of a GH18 chitinase for polymerization of chitooligosaccharides. Carbohydrate Polymers 2021, 252, 117121. [Google Scholar] [CrossRef] [PubMed]
- Ohnuma, T.; Tanaka, T.; Urasaki, A.; Dozen, S.; Fukamizo, T. A novel method for chemo-enzymatic synthesis of chitin oligosaccharide catalyzed by the mutant of inverting family GH19 chitinase using 4,6-dimethoxy-1,3,5-triazin-2-yl α-chitobioside as a glycosyl donor. J. Biochem. 2019, 165, 497–503. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Chemical structures of the main classes of phenolic compounds.
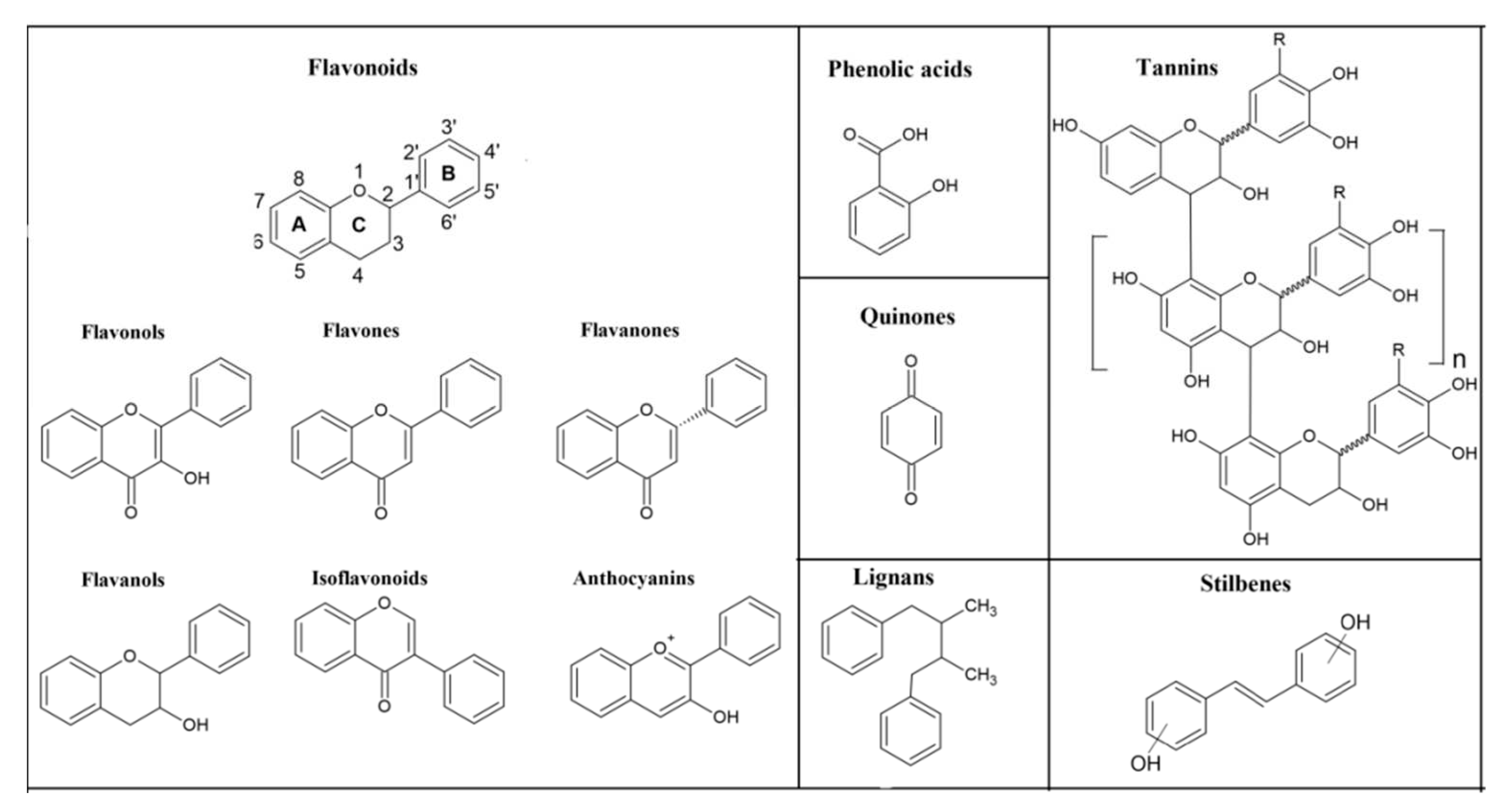
Figure 2.
Regioselectivity of amylosucrase from Deinococcus geothermalis (DGAS) in reaction of hydroxyflavones and hydroxyflavanones α-glycosylation [26].
Figure 2.
Regioselectivity of amylosucrase from Deinococcus geothermalis (DGAS) in reaction of hydroxyflavones and hydroxyflavanones α-glycosylation [26].
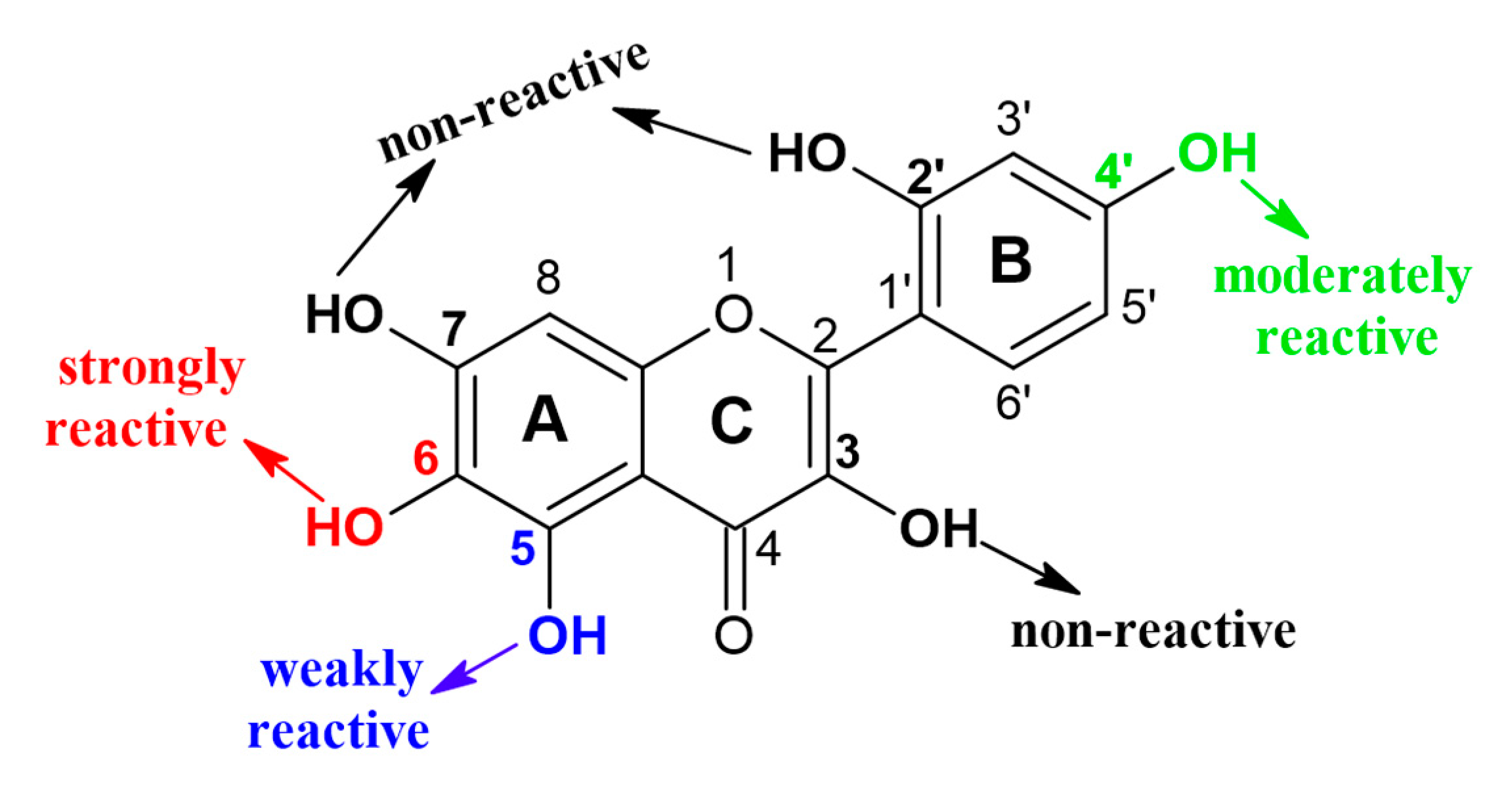
Figure 3.
The illustration of enzyme selection effect on EGCG glycosylation regioselectivity. Reaction catalyzed by cyclodextrin glycosyltransferase from Thermoanaerobacter sp. [27].
Figure 3.
The illustration of enzyme selection effect on EGCG glycosylation regioselectivity. Reaction catalyzed by cyclodextrin glycosyltransferase from Thermoanaerobacter sp. [27].
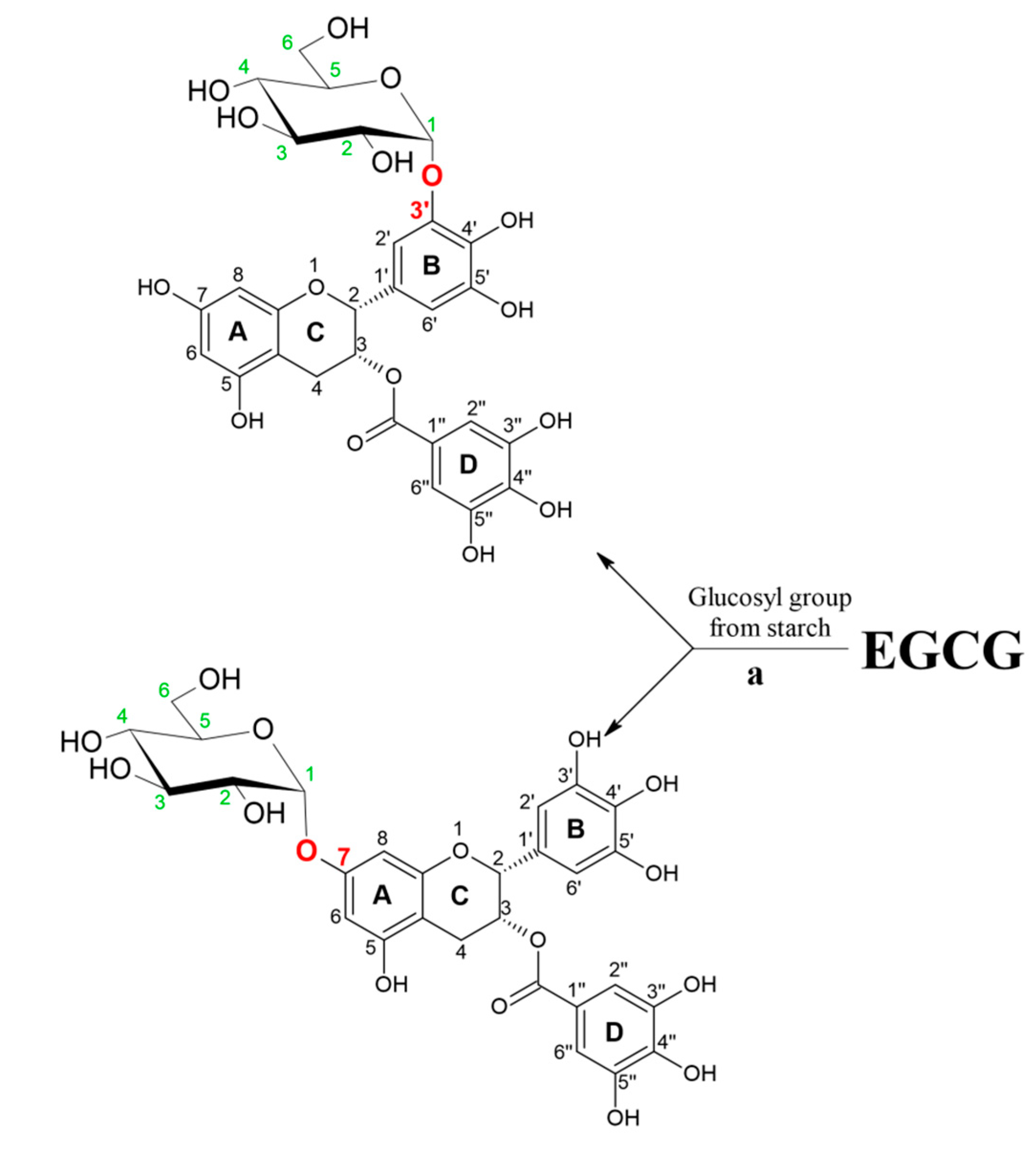
Figure 4.
Regioselective glycosylation of apigenin and luteolin catalyzed by: a) C-glycosyltransferase from medicinal plant Trollius chinensis (TcCGT1) [31] and b) C-glycosyltransferase from Gentiana triflora (Gt6CGT) [32].
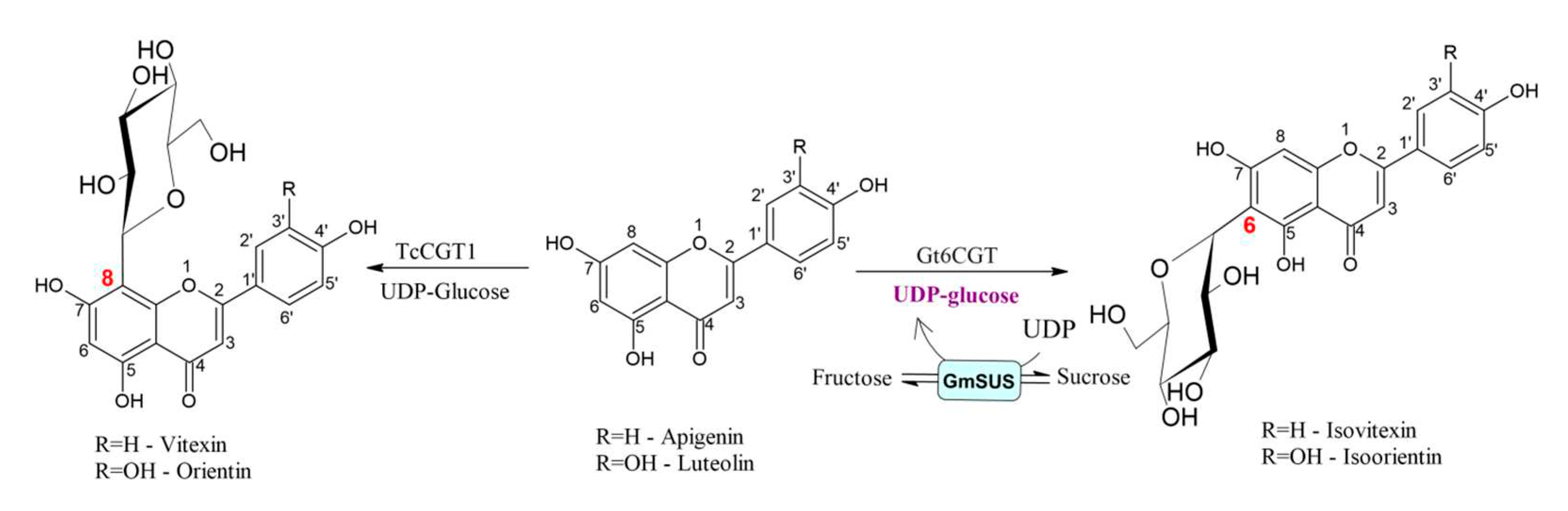
Figure 5.
Different flavonoids chemical structure and glycosylated derivative.
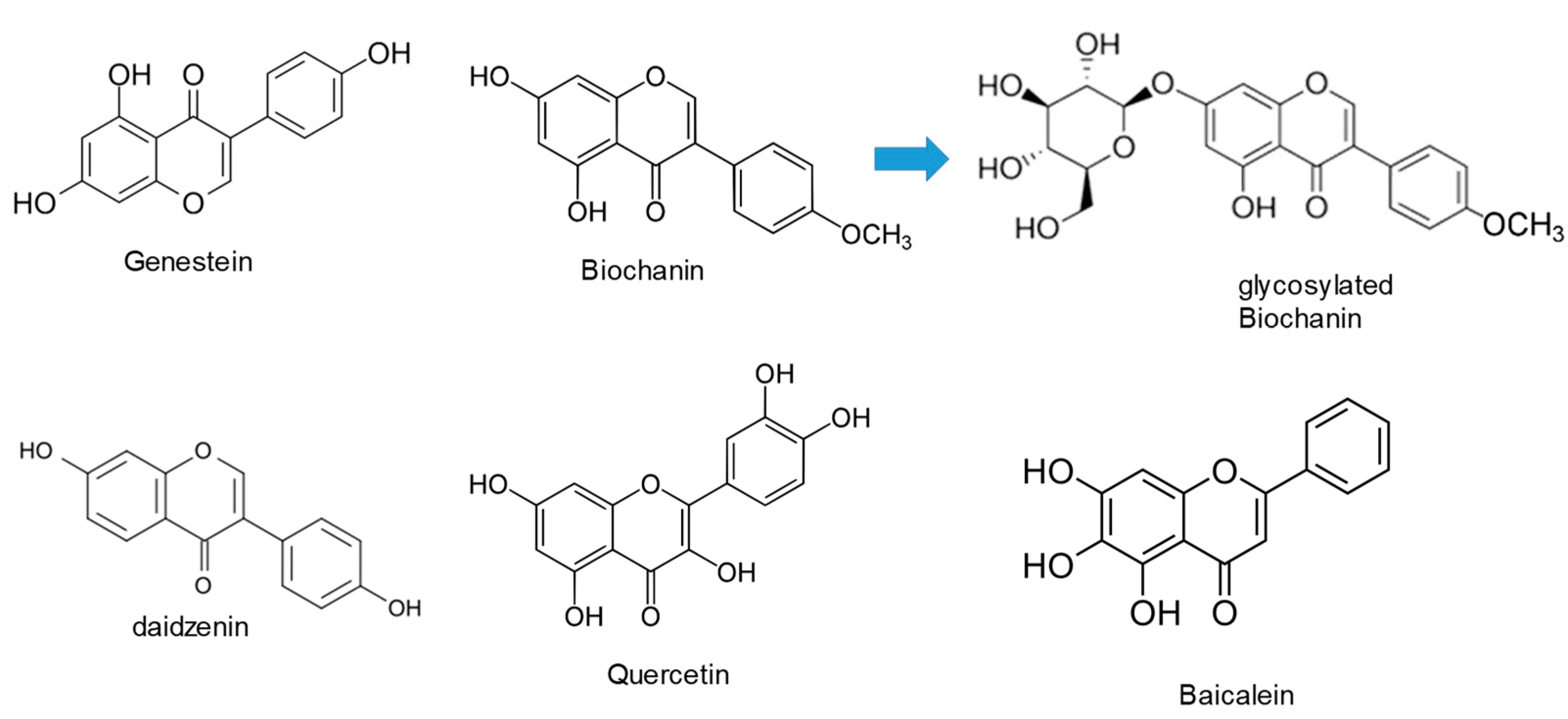
Figure 6.
Enzymatic synthesis of β-galactose-xylitol derivatives using the enzyme β-galactosidase from Lactobacillus plantarum WCFS1.
Figure 6.
Enzymatic synthesis of β-galactose-xylitol derivatives using the enzyme β-galactosidase from Lactobacillus plantarum WCFS1.
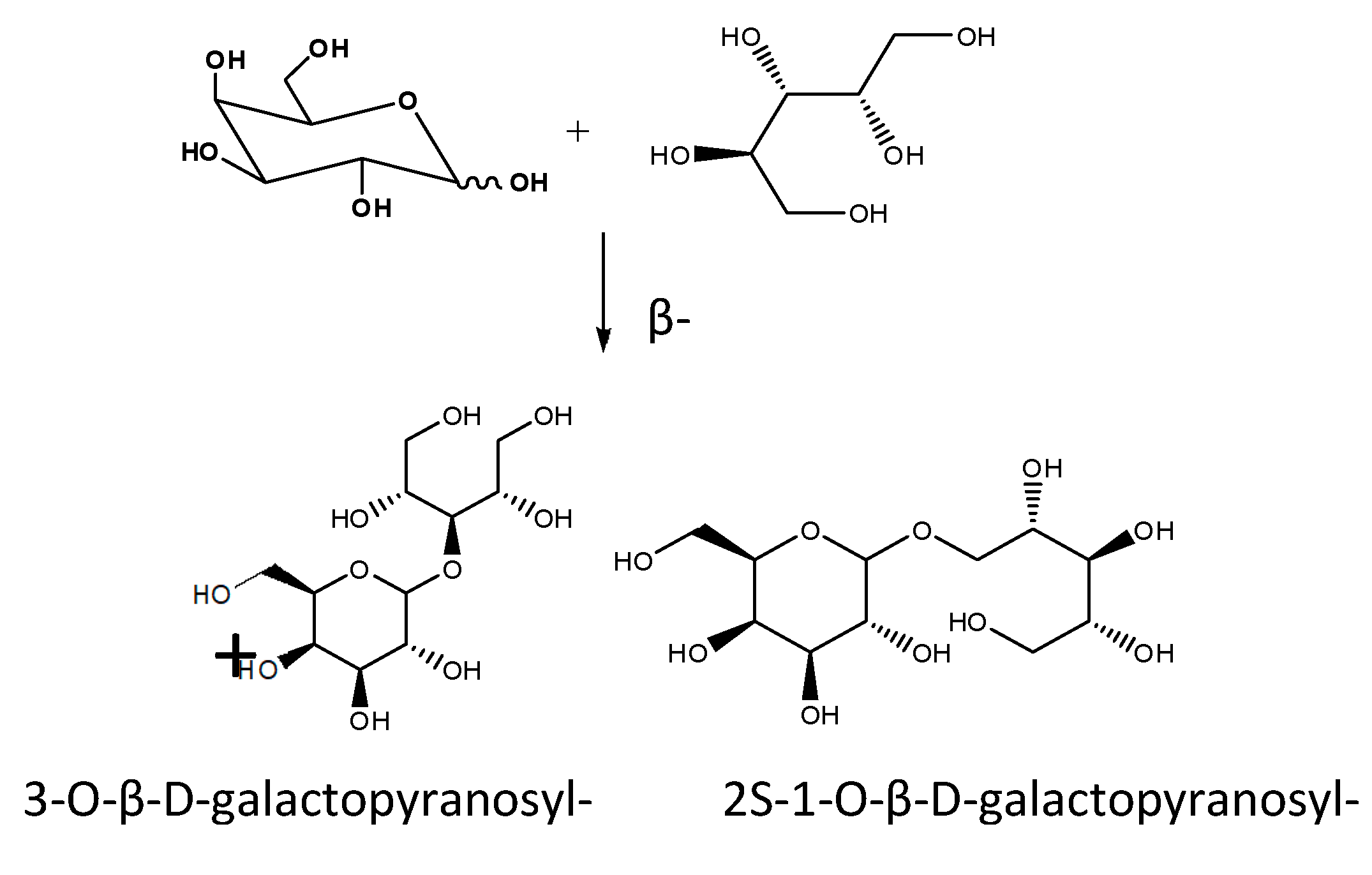
Figure 7.
Glycosylation reaction of epothilone A catalyzed by the YjiC enzyme with diverse NDP-D/L-sugars [48].
Figure 7.
Glycosylation reaction of epothilone A catalyzed by the YjiC enzyme with diverse NDP-D/L-sugars [48].
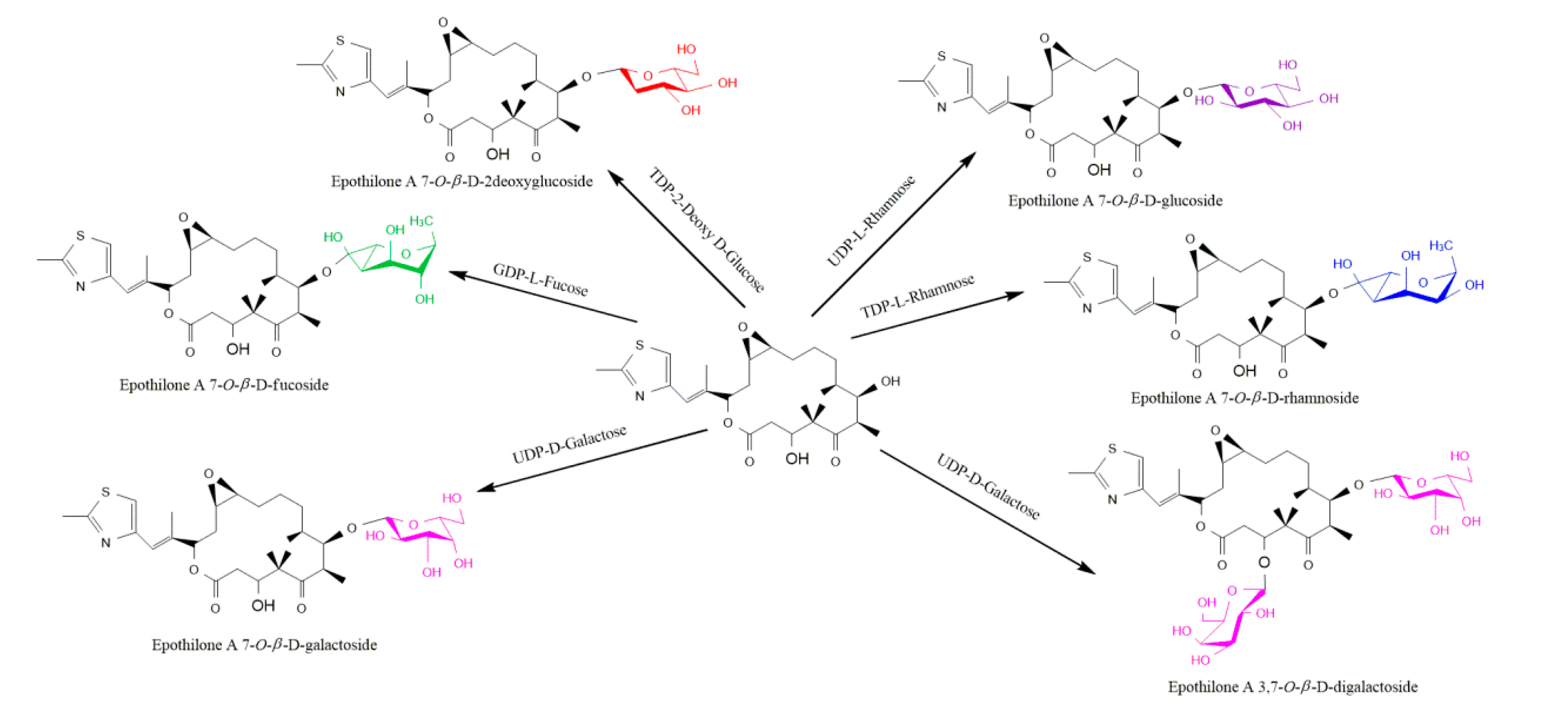
Figure 8.
Figure 8. Reaction scheme of phosphorylation of sugars catalyzed by glycan phosphorylases.
Figure 8.
Figure 8. Reaction scheme of phosphorylation of sugars catalyzed by glycan phosphorylases.
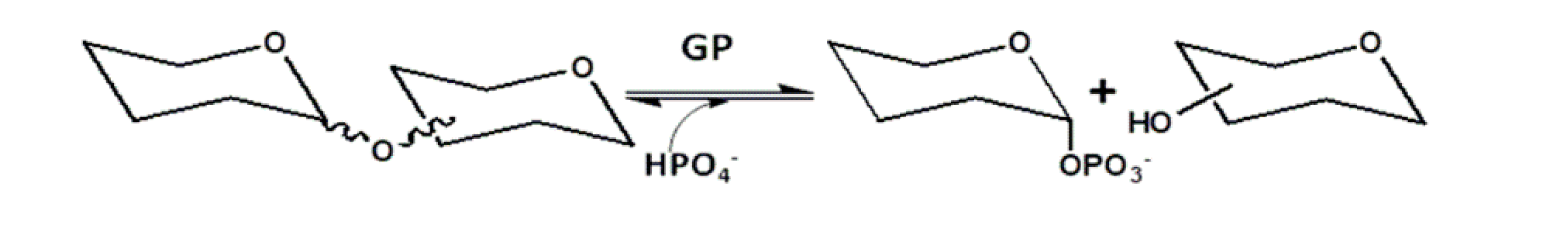
Figure 9.
Coupling SP with AGP for the enzymatic production of a variety of sugar-1-phosphate with axial selectivity at the anomeric position [53]. Sucrose was used by SP to generate α-D-Glc-1-phosphate, which was subsequently or simultaneously used in combination with 4 different acceptors, namely D-Man, D-GlcNAc, D-Gal or L-Fuc, by AGP for the production of the corresponding sugar-1-phosphates. SP = sucrose phosphorylase and AGP = α-glucose-1-phosphatase.
Figure 9.
Coupling SP with AGP for the enzymatic production of a variety of sugar-1-phosphate with axial selectivity at the anomeric position [53]. Sucrose was used by SP to generate α-D-Glc-1-phosphate, which was subsequently or simultaneously used in combination with 4 different acceptors, namely D-Man, D-GlcNAc, D-Gal or L-Fuc, by AGP for the production of the corresponding sugar-1-phosphates. SP = sucrose phosphorylase and AGP = α-glucose-1-phosphatase.
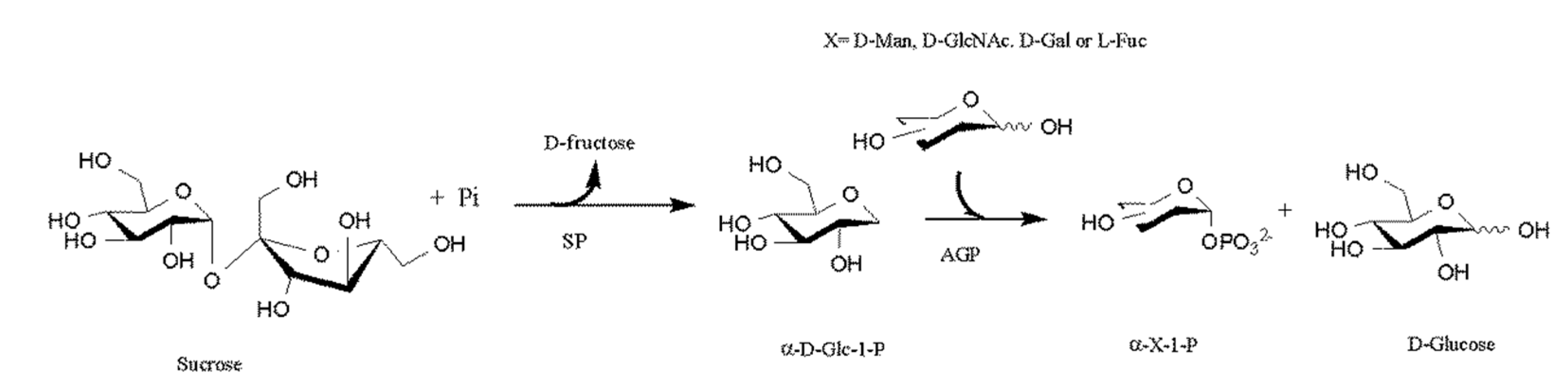
Figure 10.
Trans-Glycosylation process catalysed by β-Glycosynthases.
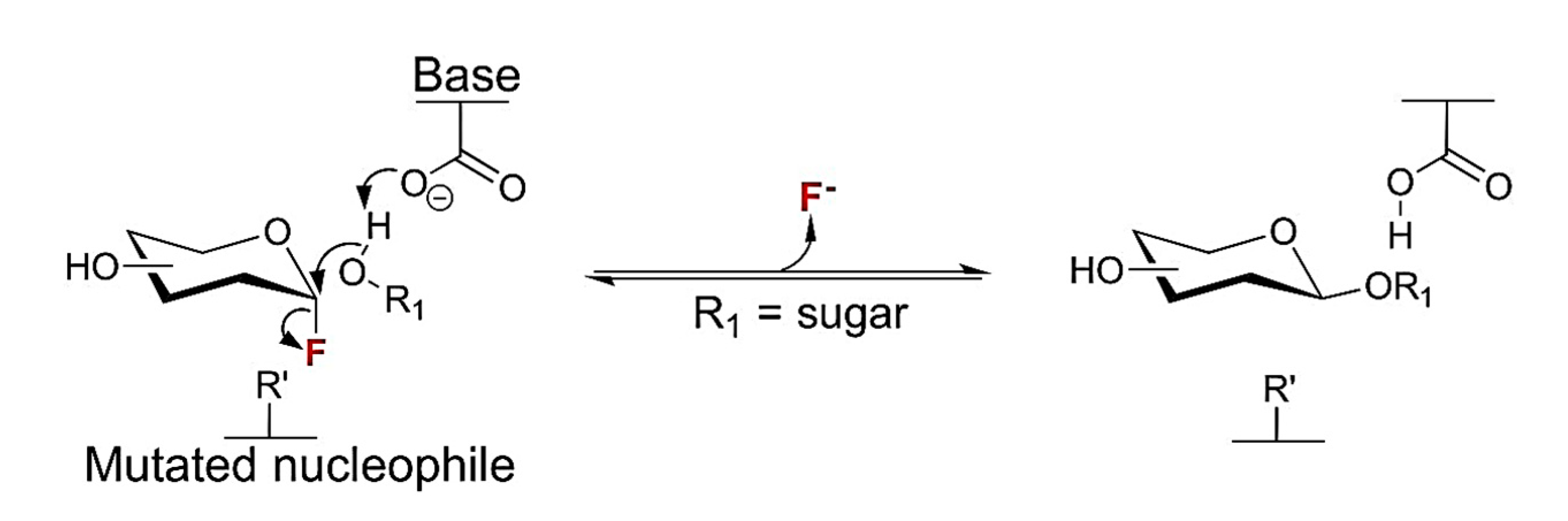
Figure 11.
Example of the synthesis of a mixture of oligosaccharides (β-1,4 Glycosynthase) from the condensation of GlcF with 2-nitrophenyl β-D-glucopyranoside (2NPGlc). [58].
Figure 11.
Example of the synthesis of a mixture of oligosaccharides (β-1,4 Glycosynthase) from the condensation of GlcF with 2-nitrophenyl β-D-glucopyranoside (2NPGlc). [58].
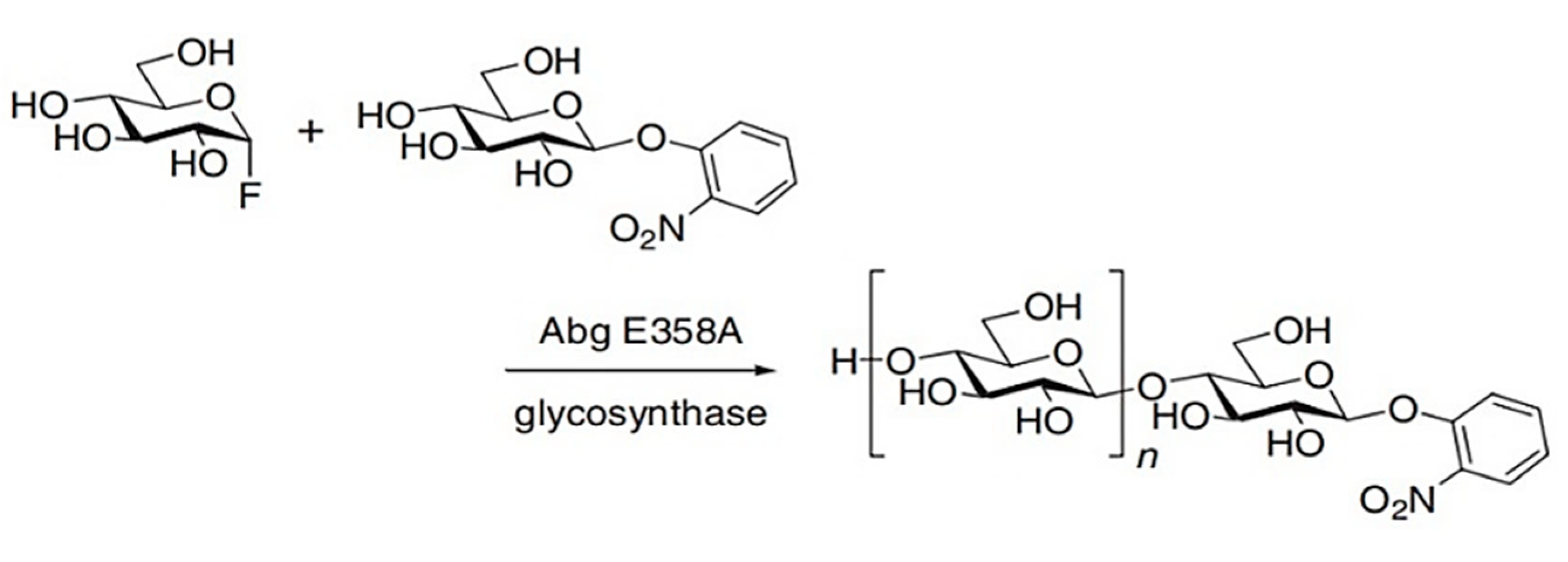
Figure 12.
Glucosides produced by transglycosylation of EGCG with the synthase BGL-1-E521G.[59].
Figure 12.
Glucosides produced by transglycosylation of EGCG with the synthase BGL-1-E521G.[59].
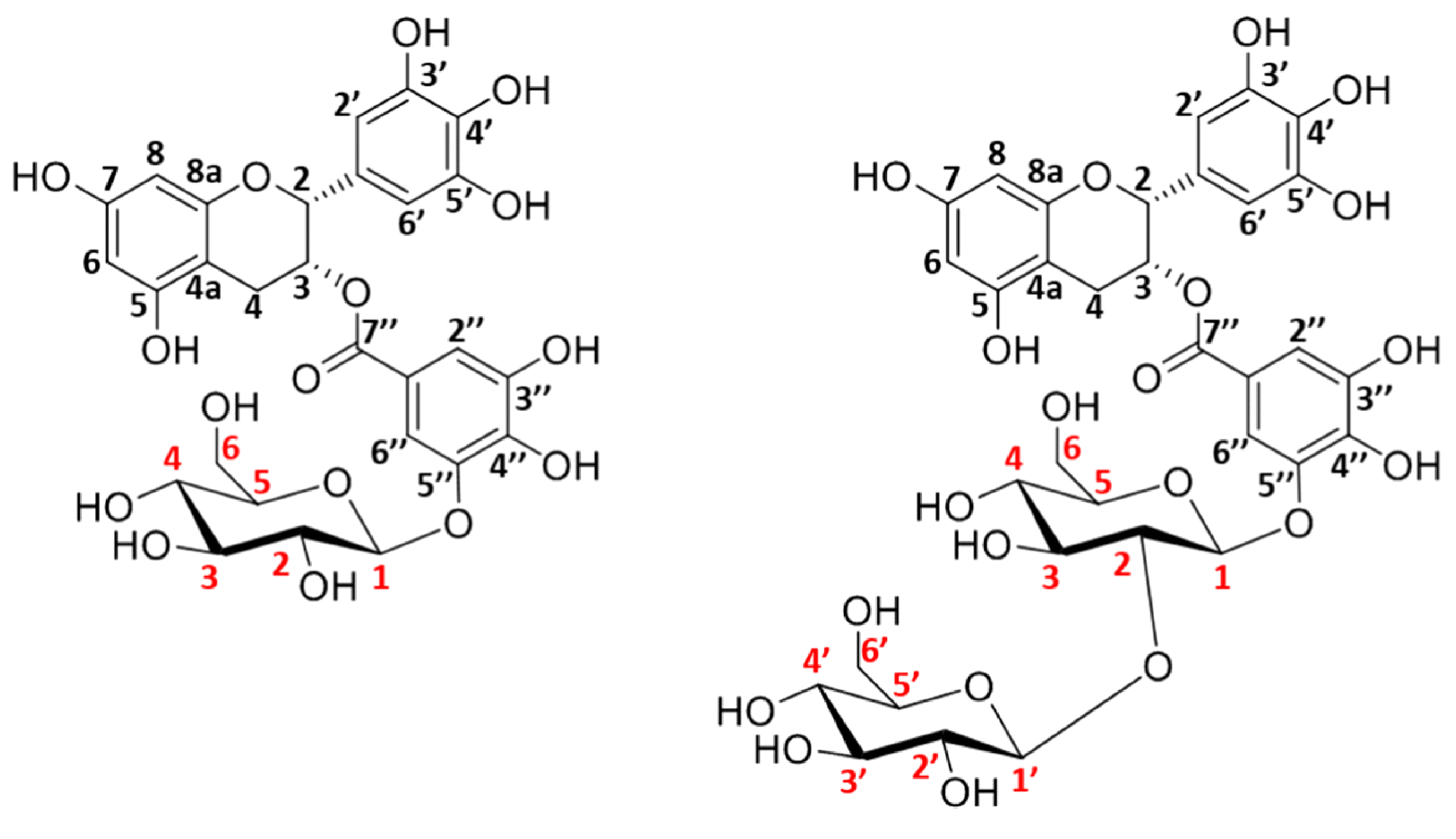
Figure 13.
Polysaccharides synthesized catalyzed by glycosynthases.
Figure 14.
Synthesis of glycoderivatives catalyzed by a BgaC-Gly galactosynthase.
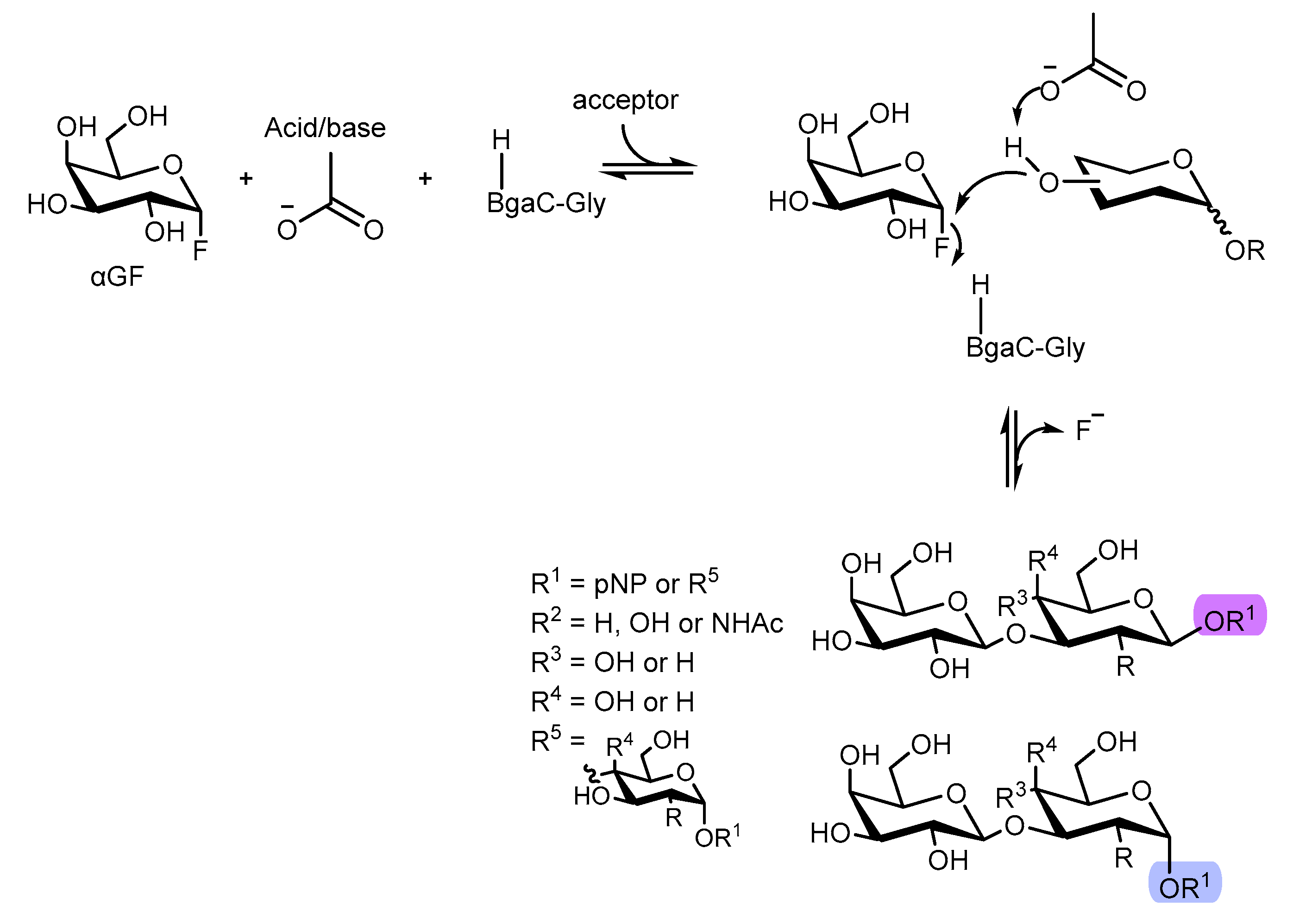
Figure 15.
Synthesis of azido-functionalized N-acetyllactosamine.
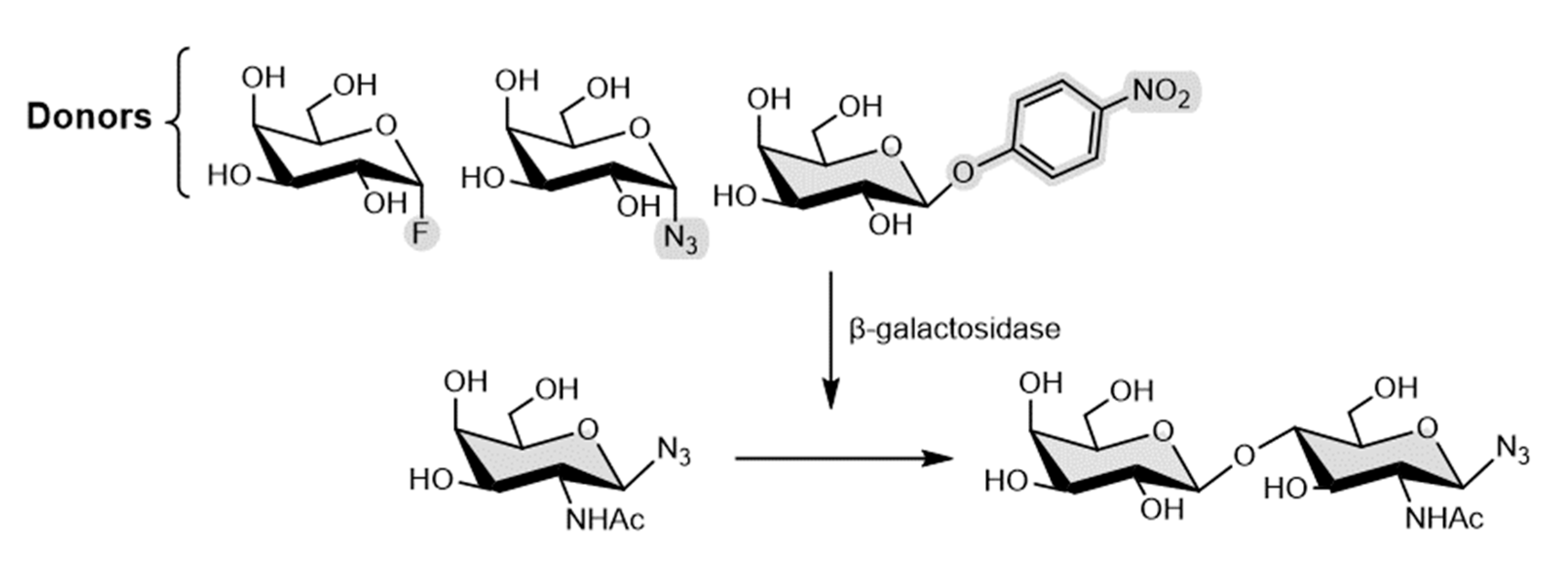
Figure 16.
Evaluation of fucosidase mutants for direct core fucosylation of N-glycans. Reprinted with permission from ref 88. Copyright 2017 American Chemical Society.
Figure 16.
Evaluation of fucosidase mutants for direct core fucosylation of N-glycans. Reprinted with permission from ref 88. Copyright 2017 American Chemical Society.
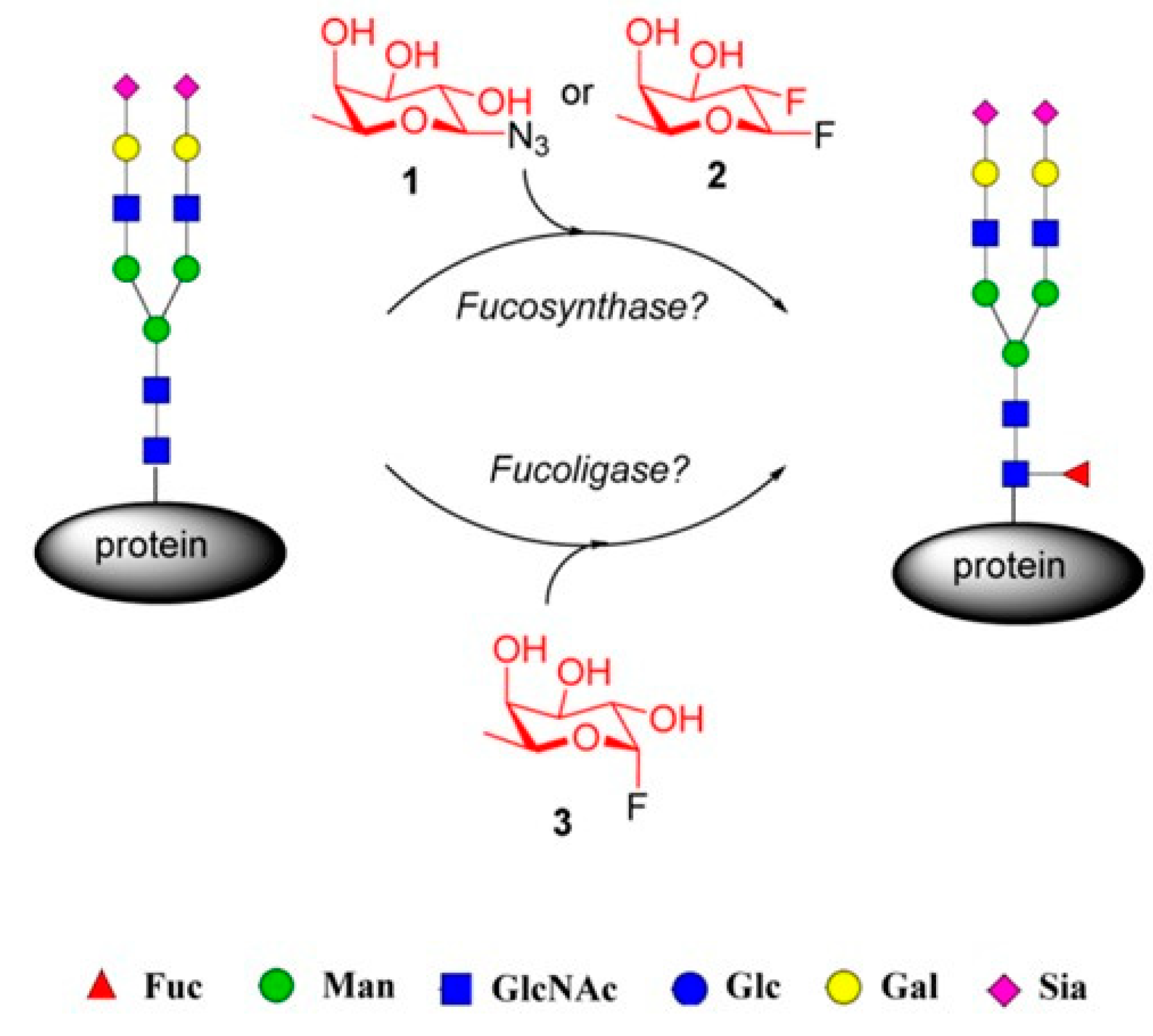
Figure 17.
Transglycosylation with Potential α-Fucosynthase and α-Fucoligase. Adapted figure from ref 89.
Figure 17.
Transglycosylation with Potential α-Fucosynthase and α-Fucoligase. Adapted figure from ref 89.
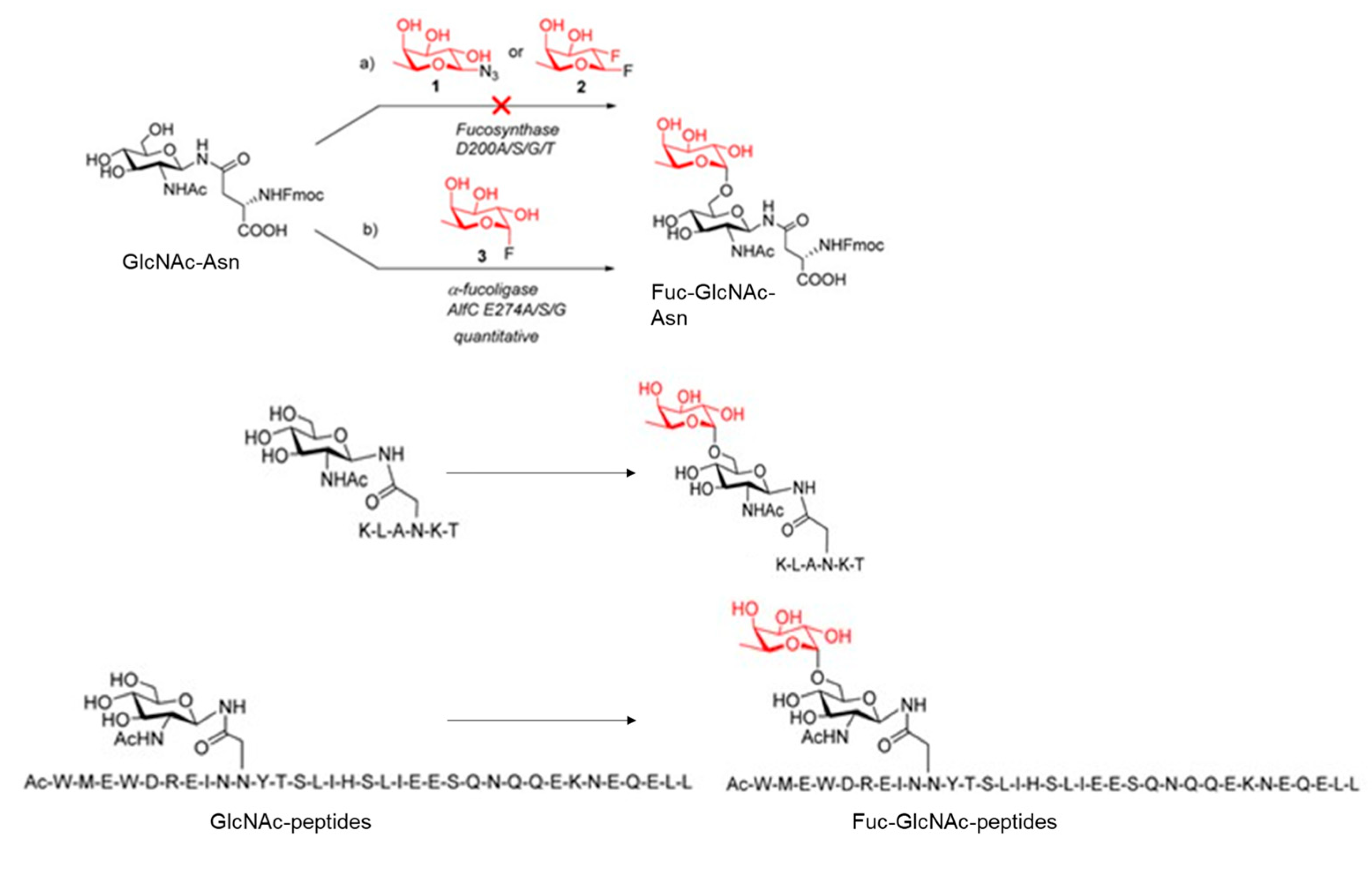
Figure 18.
Fucoligase-Catalyzed Direct Core Fucosylation of N-Glycopeptides.Adapted fig. from ref 8.
Figure 18.
Fucoligase-Catalyzed Direct Core Fucosylation of N-Glycopeptides.Adapted fig. from ref 8.
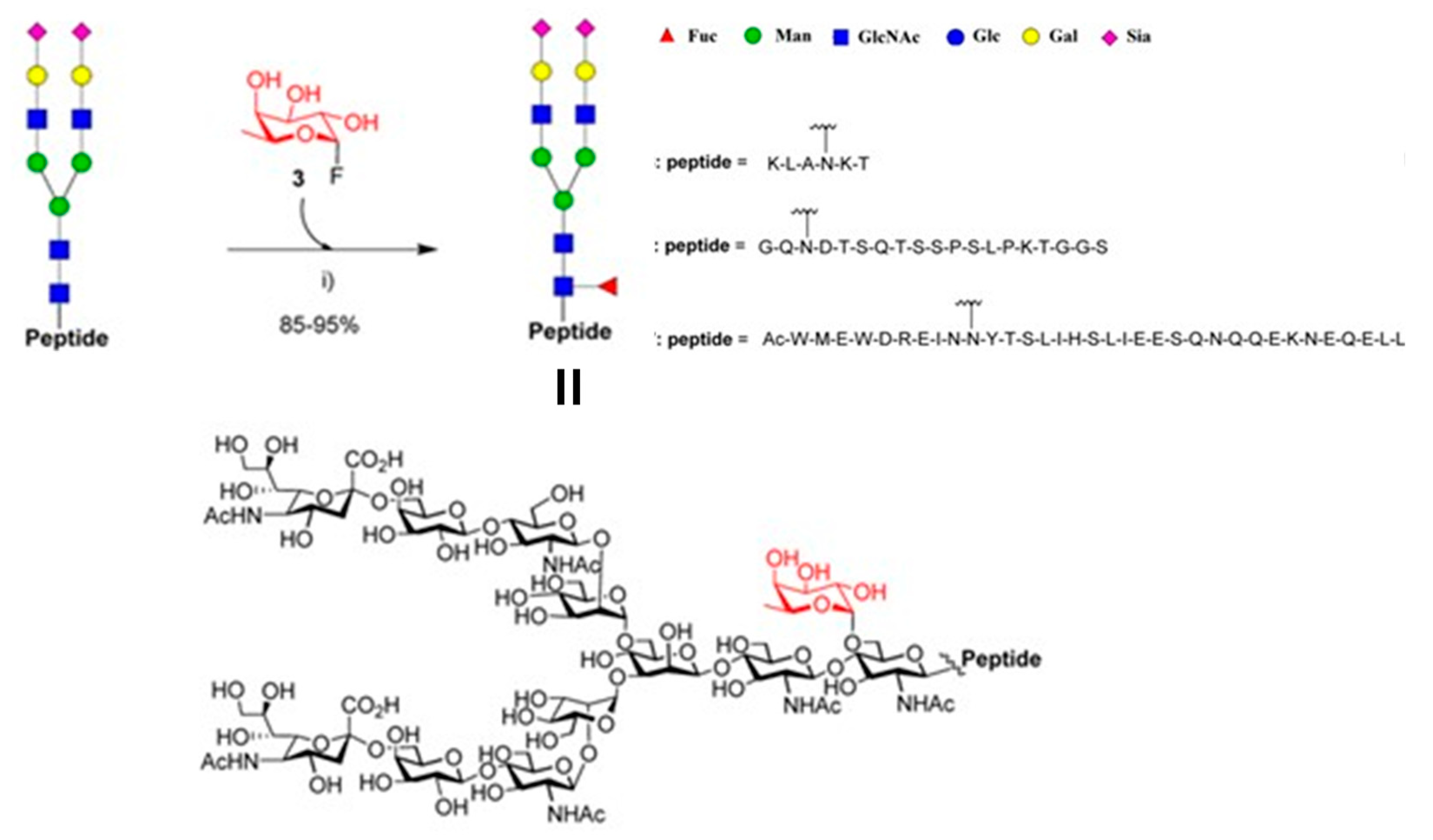
Figure 19.
Scheme illustrating the glycosynthase-like activity of GH18 mutant chitinases to obtain macrooligomers of chitosan using DP5ox as substrate. Reprinted with permission from ref 92. Copyright 2019 Elsevier.
Figure 19.
Scheme illustrating the glycosynthase-like activity of GH18 mutant chitinases to obtain macrooligomers of chitosan using DP5ox as substrate. Reprinted with permission from ref 92. Copyright 2019 Elsevier.
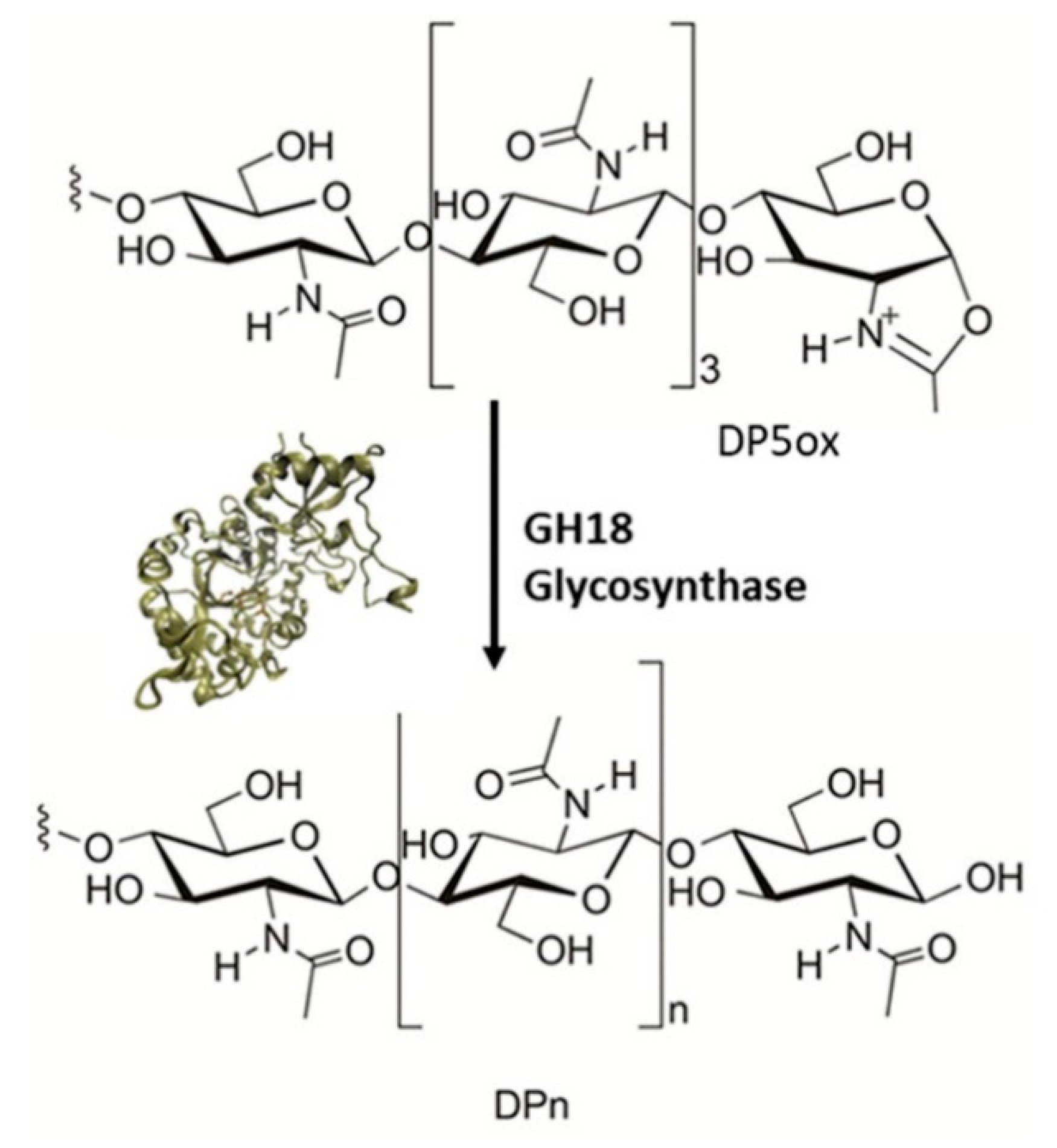
Figure 20.
(GlcNAc)4 synthesis catalyzed by glycosynthase derived from BcChi-A . Reprinted with permission from ref 108. Copyright © 2018, Oxford University Press.
Figure 20.
(GlcNAc)4 synthesis catalyzed by glycosynthase derived from BcChi-A . Reprinted with permission from ref 108. Copyright © 2018, Oxford University Press.
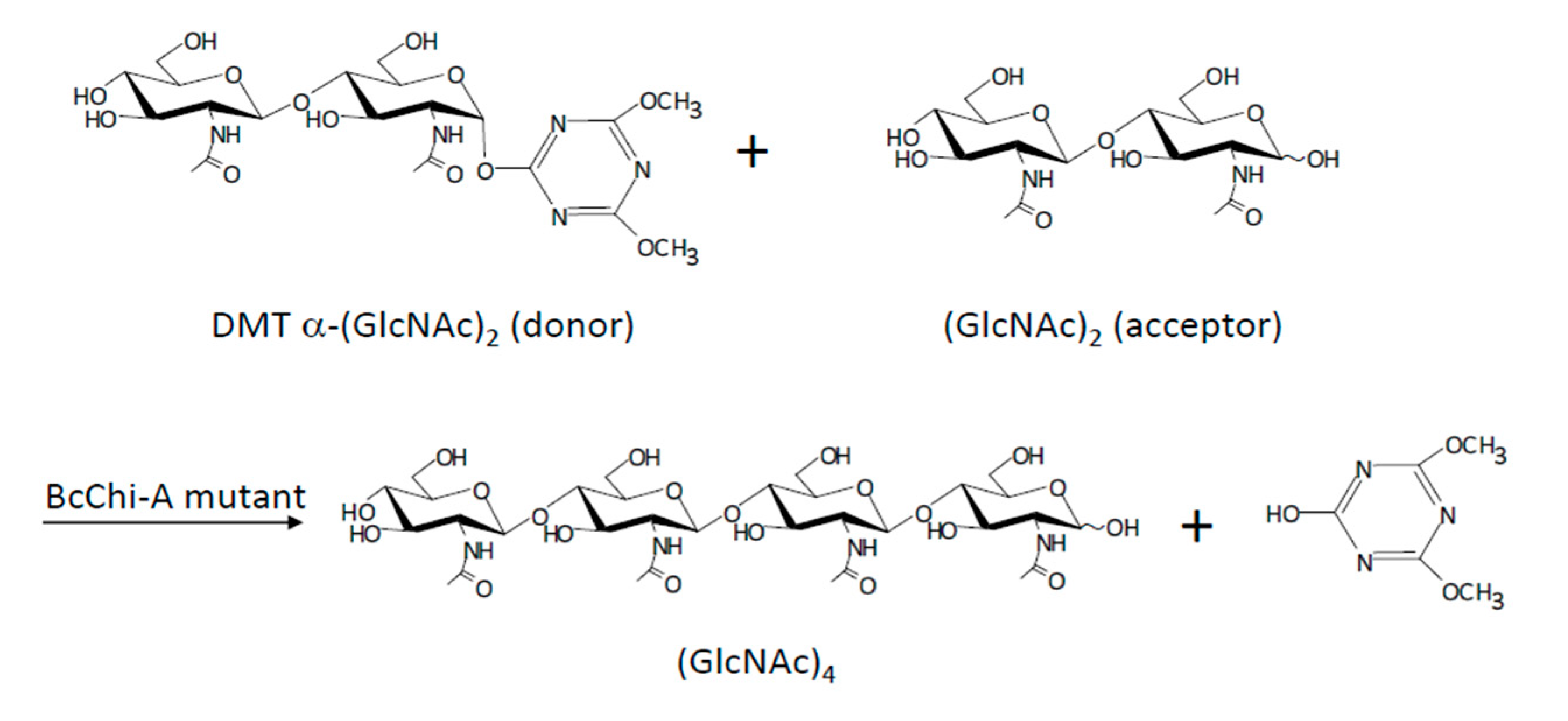
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



