
Submitted:
31 August 2023
Posted:
31 August 2023
You are already at the latest version
Abstract
The presence of high concentrations of flammable gases and volatile organic compounds in the atmosphere has been widely reported to be detrimental to human survival. A lot of research effort has been put towards finding an efficient means of quick detection of these gases below their ‘immediately dangerous to life or health’ concentrations. Detecting these gases in an oxygen-deficient environment is a crucial task to consider and has been overlooked. In this research, double substitution spinel with chemical formula Co1-2xNixMnxFe2-yCeyO4, where 0 ≤ x = y ≤ 0.3, were prepared via the glycol-thermal technique. The final products following the appropriate substitution are CoFe2O4 (dried naturally), CoFe2O4 (dried with Infrared lamp), Co0.8Ni0.1Mn0.1Fe1.9Ce0.1O4, Co0.6Ni0.2Mn0.2Fe1.8Ce0.2O4, and Co0.4Ni0.3Mn0.3Fe1.7Ce0.3O4 spinel ferrites. The X-ray diffractometry (XRD), High resolution transmission electron micrographs (HRTEM), and X-ray photoelectron spectroscopy (XPS) of the samples confirm the formation of the spinel. The gas sensing performance of this samples were tested at operating temperature of 225 ºC towards liquefied petroleum gas (LPG), ammonia, ethanol, and propanol. The Co0.8Ni0.1Mn0.1Fe1.9Ce0.1O4-based sensor was selective to LPG, with a high response of 116.43 toward 6 000 ppm of LPG when helium was used as carrier gas, 3.35 when dry-air was the carrier gas, 4.4 when nitrogen was the carrier gas, but was not sensitive when argon was used as a carrier gas.
Keywords:
Gas sensor
; Spinel nanoferrite
; Oxygen-deficiency
; Carrier gas
; LPG-sensing
1. INTRODUCTION
The ever-growing importance of chemical gas sensing is due to the necessity for environmental air quality monitoring, fire hazard prevention, food security and improved clinical diagnosis [1,2,3,4]. Expansion of industrialization and urbanization has been accompanied with tremendous impact on the environment owing to the release of harmful gases and volatile organic compounds (VOC’s), posing life-threatening conditions on human survival [5,6]. In addition, explosion of highly flammable and toxic gases such as liquefied petroleum gas (LPG), H2S, CO2, and SO2 with accompanied risk of casualty and loss of properties has caused a lot of attention towards the detection and monitoring of these gases, to ensure safe environment for human existence [7,8]. Gas sensors based on semiconducting metal oxides have been widely investigated for gas detection owing to their ease of fabrication, low production cost, low power consumption, and scalability [1,2,9]. Diverse binary and ternary metal oxides have been explored for gas sensing application. Cobalt based ferrites, of the widely explored ternary materials, possess interesting characteristics for gas sensing [10]. The cobalt-based ferrite belongs to the spinel group with formula AB2O4, where A and B are divalent and trivalent metal, respectively. This group of metal oxides is suitable as gas sensor owing to its tolerance for substitution in its structure, oxygen vacancies and adjustable conductivity type [11]. MgFe2O4 [12], CoFe2O4 [13], NiFe2O4 [14], ZnFe2O4 [15], CuFe2O4 [16], and NiCo2O4 [17] among other spinels have attracted substantial attention in gas sensing field.
The greatest concern in gas sensing research is the improvement of the sensitivity and selectivity of sensor materials to a target gas. One effective method of achieving this is by introducing a moderate amount of dopant to the structure of a host material [11] causing a change in electrical properties of the material. Study reveals that researchers have leveraged this characteristic tunability of the spinel to enhance its sensing performance [11,18]. The cobalt ferrite (CoFe2O4) has an inverse cubic spinel structure of which the tetrahedral A site is occupied by half of Fe3+ ions, the octahedral B site is occupied by the remaining half of Fe3+ and all Co2+ ions. Many of the previous works have reported the altered sensing performance of the spinel upon substitution into the site A or B of the structure. This study considers a cobalt ferrite material with both A and B sites cations partly replaced with Ni and Mn divalent metal cations (on A site) and Ce trivalent cations (on B site). It is worth mentioning that the double substitution of the spinel is normal and has been reported previously elsewhere [19]. However, the gas sensing performance of these new class of spinel has never been reported. The understanding is that the atmospheric oxygen is chemisorbed on the surface of the sensor when the dry-air is used as a carrier gas, whereas when an inert gas such as helium, argon, or nitrogen is used as carrier-gas an oxygen-deficient ambience is created resulting in the chemisorption of a fewer atmospheric oxygen species on the surface of the sensor. In the oxygen-deficient ambience, we expect the sensor to perform poorly as compared to the normal situation when the atmospheric oxygen is abundant (i.e., in dry-air). This conclusion arises from the long-held notion of the active participation of atmospheric oxygen in gas sensing mechanism of electrochemical gas sensors, as real-time gas sensing takes place in the atmosphere where there is an abundance of oxygen.
Metal oxide semiconductor-based sensors operate on the principle of chemoresistivity, i.e., change in electrical conductivity or resistivity of thin films on exposure to and reaction with analyte gas. In air, at a particular temperature atmospheric oxygen get adsorbed on the sensing surface of a chemoresistive sensor, capturing electrons in conduction band and changes the resistance of the sensor. On exposure of a sensor to an analyte gas, the gas molecules interact with the sensing surface and act as either electron donor or acceptor [20,21]. A reducing gas reacts with chemisorbed oxygen species and causes the release of captured electron back to the conduction band, thereby increasing the conductivity of an n-type sensor (because the majority carriers of n-type materials are electrons). If the analyte gas is an oxidizing type, its reaction with an n-type sensor will lead to increased resistivity as more electron in the conduction band will be captured by further adsorption of oxygen on the sensor surface. Generally, gas sensing is believed to be largely due to oxygen adsorption and desorption on the surface of metal oxide semiconductor gas sensor [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22] and by implication seems to take place favourably in the presence of oxygen. In addition, the real-life detection of gases occurs in air where atmospheric oxygen acts as a medium to dilute and carry these gases. This may suggest that chemical gas sensing will only take place or be more supported in an environment with abundant oxygen than in an oxygen-deficient environment such as in outer deep space or moon South Pole, where the lack of atmospheric oxygen is common [23].
NASA had found that metal oxide-based sensors are suitable for deep space exploration [23]. The drawback would be the low temperatures and insufficient atmospheric oxygen in deep space. In this work, we investigate the chemical gas-sensing performance of Co1-2xNixMnxFe2-yCeyO4 in abundant oxygen molecules and deficient oxygen atmosphere in order to re-create the outer and deep space situations. These carrier gases, dry-air, nitrogen, argon and helium, were used to carry the analyte gas and to dilute it into different concentrations. The idea to challenge the widely accepted chemical gas sensing mechanism is novel. The Co1-2xNixMnxFe2-yCeyO4 based sensor exhibits high sensitivity and selectivity towards LPG in helium gas atmosphere when x = y = 0.1. However, the sensitivity and selectivity shifted towards NH3 gas in dry air atmosphere when x = y = 0. In a nitrogen gas atmosphere (i.e., nitrogen-dominant chamber), sensitivity, and selectivity favour propanol at x = y = 0.3. The sensors’ performance in argon gas atmosphere (i.e., argon-dominant chamber) was generally poor towards the tested gases. All these gas-sensing measurements were conducted at 225 °C optimal operating temperature. The idea of testing our sensors’ performance on different ambient environments, as well as the application of double-substitution CoFe2O4 spinel nanoferrites in gas sensing experiment is unprecedented.
2. EXPERIMENTAL DETAILS
2.1. Synthesis Procedure
Co1-2xNixMnxFe2-yCeyO4, where x = y = 0.0, 0.1, 0.2 and 0.3, were prepared via the glycol-thermal technique. Stoichiometric amounts of NiCl2.6H2O (98 %), FeCl3.6H2O (98 %), MnCl2.4H2O (99 %), CoCl2.6H2O (97 %) and CeCl3.7H2O (99.9 %) obtained were dissolved in 100 mL of distilled water. Ammonium hydroxide solution was added dropwise and mixed under vigorous stirring using a magnetic stirrer for one hour to adjust the pH to 10. Deionized water was used several times to wash the resultant precipitate until it is free of chloride ions. The precipitate was then added to 300 mL of ethylene glycol for reaction on a PARR 4843 Watlow series stirred pressure reactor at 200 °C for 6hrs. The reaction pressures were between 50 to 100 psi and the stirring speed was 300 rpm. After cooling, the glycol-reacted precipitate was washed using deionized water and heated rapidly to 200 °C for 4 hrs in the open air. The substance was finally cooled to obtain the as-prepared samples. Some portion of the sample for which x = y = 0 (i.e., CoFe2O4) was left to dry overnight under 250 W infrared lamp. The powder samples were then annealed for 3 hrs at 500 °C to obtain the final product and were labelled S1 (IR lamb dried CoFe2O4), S2 (natural-dried CoFe2O4), S3 (Co0.8Ni0.1Mn0.1Fe1.9Ce0.1O4), S4 (Co0.6Ni0.2Mn0.2Fe1.8Ce0.2O4), and S5 (Co0.4Ni0.3Mn0.3Fe1.7Ce0.3O4).
2.2. Characterization
The crystal structure of each powder sample was studied using a Bruker D8 Advance X-ray diffractometer having Cu-Kα (λ = 0.1541 nm) radiation source with 0.3° per minute scan rate. Scanning electron microscope (SEM, Carl ZEISS Sigma VP-03-67) was used to capture the images of the particle size and surface morphology of the powder samples. A Micrometrics TRISTAR II (USA) surface area analyser was used for nitrogen adsorption-desorption study and for Brunauer-Emmett-Teller (BET) surface area study. The pore size, pore volume and surface area were measured using nitrogen at 77 K and degassing the samples at 150 °C for 3 hrs. High-resolution transmission electron microscopy (HR-TEM) images were captured by a JEOL 1400 system. PHI 5000 Scanning ESCA Microprobe was used to examine the chemical state of the samples by X-ray photoelectron spectroscopy (XPS) analysis using A 100 μm diameter monochromatic Al Kα (1486.6 eV) X-ray beam at a pressure of 9.3 ᵡ 10-10 Torr. Ar+ with energy of 2 kV and 2 µA was used for sputter-etching the samples for 1 minute at the sputter rate of 15 nm per minute. Surface charging was minimized by using a low energy Ar+ ion gun and low energy neutralizer electron gun. The low energy peak of Au 4f7 at 83.96 eV and the high energy peak of Cu 2p3 at 932.62 eV were used for the binding energy calibration. The retard linearity was set in order to keep the difference between these two peaks constant at 848.66 eV. To keep the Ag3d5 peak at 368.27 ± 0.1 eV, the work function of the analyser was set to 3.7 eV.
2.3. Sensor Fabrication and Measurements
To fabricate the gas sensors, each powder sample was uniformly distributed in ethanol and sonicated for 2 h and the solution was drop-casted onto alumina substrate screen-printed with gold electrode. The other surface of the substrate was heated to 90 °C to remove the organic solvent and leave a dry well adhering sensing layer. For sensing measurement, each of the sensors was simultaneously placed in an airtight chamber having electrical and gas feeds. The measurements of the target gases (LPG, NH3, ethanol, and propanol) at different ppm concentrations, were conducted using a KS026K16 (KENOSISTEC model, Italy) gas testing system. These measurements were first performed at operating temperatures of 175 °C and 225 °C and constant applied voltage of 5.0 V across the sensor to determine the optimal operating temperature. The responses of the sensors were very low at the operating temperature of 175 °C as compared to that of 225 °C. Once the optimal operating temperature was determined to be 225 °C, the remaining measurements were conducted at 225 °C. Dry air (79% N2 and 21% O2) was used to dilute and carry the gases into the chamber for measurements. For gas sensing measurement in oxygen-deficient ambient, helium, argon, and nitrogen were used as carrier gases at different times. The gas sensing measurements were conducted in 0.1 %RH dry ambient. Keithley 6487/E picoammeter/voltage source meter was used to quantify the change in the sensor resistance of the device. The sensor response, S, was calculated using:
where Ig is the current in the presence of gaseous species and Ia is the current in air.
3. RESULTS AND DISCUSSION
3.1. X-ray diffraction, surface morphology and Chemical Composition
Figure 1a depicts the X-ray diffraction patterns of the pure CoFe2O4 that was dried using infrared (IR) lamp, dried with oven at 200 °C (natural dried), and the Co1-2xNixMnxFe2-yCeyO4 ferrites spinels resulting from the substitution of tetrahedral A-sites and octahedral B-sites of CoFe2O4 with Ni, Mn, and Ce atoms in a linearly increasing molar amount.
The substitution was done at equal molar amount of x and y, where 0 ≤ x = y ≤ 0.3. These substituted spinel nanoferrites were dried naturally at 200 °C for 4 hours and subjected to annealing as described in the experimental section. The final products of the appropriate substitution of Ni, Mn and Ce atoms are Co0.8Ni0.1Mn0.1Fe1.9Ce0.1O4, Co0.6Ni0.2Mn0.2Fe1.8Ce0.2O4, and Co0.8Ni0.1Mn0.1Fe1.9Ce0.1O4, including the unsubstituted CoFe2O4 spinel ferrites. The samples are labelled S1 (IR lamb dried dried CoFe2O4), S2 (natural-dried CoFe2O4), S3 (Co0.8Ni0.1Mn0.1Fe1.9Ce0.1O4), S4 (Co0.6Ni0.2Mn0.2Fe1.8Ce0.2O4), and S5 (Co0.4Ni0.3Mn0.3Fe1.7Ce0.3O4).
The diffraction patterns of the samples having peaks at 2θ positions can be indexed as follows: 30.18° (220), 35.58° (311), 43.39° (400), 53.82° (422), 57.23° (511) and 62.91° (440). The position of the XRD peaks for the samples matched well with the standard XRD pattern for CoFe2O4 spinel ferrites with JCP2 card No. 96-154-0974, and it is in good agreement with the XRD results previously reported elsewhere [19,24,25,26,27]. It is noteworthy to mention that at x = y ≥ 0.2, additional peaks at 2θ positions, 28.63, 33.10 and 47.87° were observed. These peaks correspond respectively to (111), (200) and (220) planes of CeO2 with PDF card no. 96-721-7888 [19,28,29,30]. This indicates the formation of CeO2 secondary phase in the spinel structure. No other phase attributable to Ni and Mn oxides was observed; this is owing to the fact that ionic radii of Ni2+, Mn2+ and Co2+ are very close to each other with 0.083, 0.081, and 0.079 nm, respectively. However, the ionic radius of Ce3+ (0.101 nm) is greater than that of Fe3+ (0.064 nm). As a results, cerium dioxides secondary phase was formed as Ce content increased in the sample. Mkwae et al. [31] reported the formation of the CeO2 as a secondary phase at higher Ce dopant content during the synthesis of MgCexFe2-xO4 by hydrothermal synthesis method. Many other researchers have previously observed the CeO2 secondary phase when Ce3+ was substituted in their spinel nanoferrites [19,32,33,34]. In addition, the crystallinity of the sample with x = y = 0.1 is less than that of the other samples as some peaks corresponding to the spinel appears to be absent or inconspicuous as observed in their low intensity. Table 1 shows the crystallite sizes of the samples, calculated using Debye-Scherrer’s equation [35]. Figure 1b–f shows the scanning electron microscope (SEM) images of the spinel nanoferrites samples. The two CoFe2O4 samples- infrared lamb dried and natural-dried in Figure 1b,c, possess similar surface morphology with predominantly fine particles and few agglomerates. Upon addition of impurities to the spinel structure, difference in surface morphology becomes distinct especially on the x = y = 0.1 shown in Figure 1d. This sample contains more agglomerates than fine nanoparticles as compared to the previous two samples with no impurities- Ni, Mn and Ce cations. The formation of these closely packed agglomerates could be due to the introduction of the Ni, Mn, and Ce dopants into the spinel, which might have led to the complication of the spinel structure. At the substitution of 0.1 molar concentration of the impurities, i.e., when x = y = 0.1, the crystallinity of the spinel decreased as observed earlier in the x-ray diffraction (XRD) analysis. At x = y = 0.2, the spinel looks like solidified magma sparsely covered by agglomerates (see Figure 1e). This is more crystalline than the sample with x = y = 0.1 as can be observed in the high XRD peak intensities in Figure 1a with secondary phases corresponding to the CeO2. The sample depicted in Figure 1f has the appearance of tightly bound particles.
Pore sizes and pore size distribution were estimated by conducting nitrogen adsorption-desorption measurement on the sample. The samples are generally mesoporous, having pore size p, where 2 nm < p < 50 nm. Figure 2 depicts the nitrogen adsorption-desorption isotherm of the Co0.8Ni0.1Mn0.1Fe1.9Ce0.1O4 double-substitution spinel. It is a type IV isotherm [36] which is characteristic of mesoporous materials. The inset is the pore size distribution.
Table 1 presents the samples’ crystallite sizes of the samples, calculated from the most prominent peak located at (311) plane of the spinel, the pore sizes, and the specific surface areas. The crystallite sizes are observed to vary with dopant amount, while the pore sizes and specific surface areas do not follow a particular order as regards the change in their values upon dopant addition.
Figure 3a–e show the High-Resolution Transmission Electron Microscope (HRTEM) images of the spinel nanoferrites. These spinels are generally crystalline nanoparticles. This is evident in the clear fringes observed on the HRTEM micrographs. The interplanar d-spacing (dhkl) fringes of the crystalline plane (Figure 3a) can be attributed to the most prominent ring indexed (311) in the corresponding selected area electron diffraction (SAED) pattern and was calculated to be d311 = 0.255 nm. This value varies slightly between samples due to changes in lattice parameters resulting from sites loading. The cubic spinel structure was highly expected to undergo both expansion and shrinkage between 0 ≤ x = y ≤ 0.3. The SAED pattern observed for these nanoferrites show bright crystalline spotted rings with the (311) exhibiting prominence. The SAED ring corresponding to the CeO2 can also be observed very close to the most prominent ring of 311 peak. This agrees well with XRD result and the ring corresponds to (111) plane of CeO2 fluorite structure [37] whose inter-planar distance (d-spacing) is 0.32 nm. It is noteworthy that the secondary phase of CeO2 was only formed as Ce3+ content increased in the spinel structure.
Figure 4a–h depicts the x-ray photoelectron spectroscopy (XPS) of the Co1-2xNixMnxFe2-yCeyO4 nanoferrites with 0 ≤ x = y ≤ 0.3. The survey spectra shown in Figure 4a,b before and after sputtering the surface with Ar+ ions were identical with peaks confirming the presence of Co, Ni, Mn, Fe, Ce, O, and C elements in the Co1-2xNixMnxFe2-yCeyO4. The sputtering took place for a minute at the sputtering rate of 15 nm per minute. The similarity of the survey spectra suggests the homogeneity of these nanoferrites. Observing from the survey, Ni 2p peak appears between the edge and trough of a peak. This may suggest the scarcity or total absence of Ni atoms in the sample composition. Furthermore, Ni 2p scan (Figure 4d) confirms the scarcity of Ni atoms as the peak situated at 848.66 eV and 886.12 eV could not be ascribed to Ni. Figure 4c shows Co 2p core level spectrum. The fitting peaks at 779.96 eV ascribed to B-site Co2+ and 781.62 eV ascribed to A-site Co2+ correspond to Co 2p3/2 with its satellites at 785.11 eV, while the peak at 796.06 eV corresponds to Co 2p1/2 with its satellite at 803.25 eV. It is obvious that there are no Co3+ cations in the sample since they normally exhibit peaks centred at 798.5 eV and 783.3 eV while occupying tetrahedral and octahedral sites respectively [38]. Figure 4e shows the Mn 2p core level spectrum consisting of spin-orbit doublets with their respective satellites.
The Mn 2p3/2 peak at 640.68 eV ascribed to Mn2+ cations and 642.06 eV ascribed to Mn3+ cations has apparently combined satellites located at 645.75 eV. In addition, the peak at 652.96 eV is attributed to Mn 2p1/2 with its satellite at 657.54 eV. The Ce 3d core level spectrum depicted in Figure 4f exhibits peaks at 882.72 eV and 888.47 eV which are respectively attributable to Ce3+ and Ce4+ cations of Ce 3d5/2. The peak at 901.88 eV is ascribed to Ce 3d3/2 with its satellite peak at 916.21 eV. The Fe 2p spectrum shown in Figure 4g comprises two spin-orbit doublets situated as follows: Fe 2p3/2 (710.43 eV, Fe3+ in B-sites and 712.45 eV, Fe3+ in A-sites) with its combined shake-up satellites at 718.20 eV and Fe 2p1/2 (724.10 eV, Fe3+ in B-sites and 725.61 eV, Fe3+ in A-sites) with its satellites at 732.32 eV. The positions of the binding energy of Co 2p and Fe 2p peaks and their satellites confirms the oxidation state of Co and Fe (Co2+ and Fe3+) in the sample. The spectra also indicate the presence of Co and Fe at the octahedral and tetrahedral sites [19]. The O 1s spectrum (Figure 4h) consists of three peaks ascribed to OL (centred at 529.00 eV), OV (centred at 529.9 eV) and OC (centred at 531.23 eV). OL is the oxygen inherent in the sample, OV signifies the oxygen deficient sites in the sample. OC is connected to surface oxygen on the sample owing to its exposure to air.
Various deconvolutions of O 1s peaks are presented in Table 2. Chemisorbed oxygen is only present in two of the samples while the existence of oxygen vacancy and lattice oxygen is generally observed.
3.2. Gas sensing properties
The sensors were exposed to ethanol, propanol, ammonia, and LPG at operating temperature of 225 ºC. The concentration range for each of the target gases is based on the recommended exposure limits. Dry-air, helium, nitrogen, and argon were used as carrier-gases and for analyte gas dilution to produce different concentrations.
Figure 5 depicts the selectivity plots of Co1-2xNixMnxFe2-yCeyO4 ,0 ≤ x=y ≤ 0.1, double-substitution spinel (i.e., S3) towards ethanol, propanol, ammonia, and LPG using dry-air and helium gas as carrier gases. It should be mentioned that the nitrogen and argon carrier-gas driven results are not included here as these did not yield good gas sensing performance as expected. These results are shown in Figure S5 of the ESI. Figure 5a shows that when dry air was used as a carrier and to dilute the analytes, sensors S1 and S3 were favourable to ethanol vapour. However, propanol vapour could be highly detected by S1 sensor. Moreover, sensor S3 is highly sensitive to ammonia. Furthermore, LPG could be detected by S1. Overall, sensor S3 gave the highest response of 7.95 to ammonia. The whole situation changed unexpectedly when helium is used as a carrier gas and to dilute the analytes. The expectation was that helium is an inert gas and will create an oxygen deficient atmosphere inside the sensing chamber resulting in the decrease in sensors’ sensitivity towards the analyte gas. The opposite was observed in Figure 5b. The sensor S3 was extremely sensitive and selective to LPG at 6000 ppm concentration with a response of 116. Interestingly, these experiments that was designed to demonstrate the performance of semiconductor gas sensors in different atmospheres, operated with some accompanied anomalies. Figure 5c depicts the transient current plot with time of the LPG concentrations on S3 sensor. The results show an increasing response with LPG concentration until 6000 ppm is reached thereafter the response begins to drop with further increase in concentration. However, on the 10000 ppm concentration the response is slightly higher than that towards the 9000 ppm. This can be attributed to the fact that on the 10000 ppm, the carrier gas is not present, and this is a maximum concentration. All the maximum gas analyte concentrations should look the same regardless of the carrier gas. The reason for dropping of the response after 6000 ppm is not clear as well as the high response when helium was used compared to dry air.
In Figure 5d the transient current plot of the sensor toward LPG in dry air atmosphere, i.e., using dry air as carrier gas is shown. The sensor exhibits n-type characteristics towards the first three concentration cycles of LPG: 1000, 2000, and 3000 ppm. On the peak corresponding to the 3000 ppm concentration, a large split is observed, which is attributed to fluctuation in operating temperature as once observed by Mkwae et al. [31]. However, from 4000 ppm onward, charge carrier transition began to occur in the sensor upon LPG sensing. The response of this sensor towards 3000 ppm of LPG is 3.35.
The LPG sensing of the sensor S3 in dry air atmosphere was characterized by two distinct phenomena, namely: current oscillation resulting from fluctuation in operating temperature, and charge carrier transition, from n- to p- type. The response, response time, and the recovery time of the sensor are 5.87 (10000ppm), 5.83 minutes, and 4.00 minutes respectively. The arrow at the bottom of the curve indicates the moment LPG was allowed into the chamber, while the arrow at the top of the curve indicates the moment LPG was stopped from entering the chamber. In helium gas atmosphere, there was no charge carrier transition observed in the sensors upon LPG detection, rather, there was a decrease in sensors’ response from 7000 ppm concentration of LPG in addition to the observed current oscillation which occurred at the peak as well as at the sensors’ relaxation. Generally, the sensors behaved abnormally upon LPG detection when helium was used as a carrier gas (i.e., in helium dominant chamber or atmosphere). The response increased from 1000 ppm to 6000 ppm then decreased the same way from 7000 ppm to 10000 ppm. This made the response of S3 at 10000 ppm (81.73) less than at 6000 ppm (116.43). The response could have been much greater than 116.43 had the abnormal decrease in response not occurred. Moreover, while the sensors’ response pattern towards other gases was normal, it was generally abnormal towards LPG (and occasionally, towards NH3) irrespective of the carrier gas.
Figure 6 shows the response patterns of the sensors towards LPG in dry air atmosphere and in helium gas atmosphere. The peaks labelled 1-10 corresponds to 1000-10000 ppm LPG concentration with 10 equal steps. Figure 6a is the response pattern of the naturally dried pristine CoFe2O4-based sensor towards LPG in dry air atmosphere. This pattern is like that of the IR-dried sample. The pristine samples did not undergo charge carrier transition, rather, their LPG sensing performance was accompanied by current oscillation which becomes prominent with gas concentration. At concentrations of 5000 to 7000 ppm, the frequency of the oscillation was high, while the amplitude was higher from 8000 to 10000 ppm concentrations as seen in Figure 6a. This current oscillation is caused by the fluctuating operating temperature during the LPG sensing. This anomaly was discussed elsewhere [39]. The response pattern of the sensor (S5), fabricated from the sample with the chemical formula Co0.4Ni0.3Mn0.3Fe1.7Ce0.3O4 is depicted in Figure 6b. This pattern is similar for all the doped samples. They exhibited charge carrier transition (n- to p- type) upon LPG detection. Figure 6c,d show the response patterns of S2 and S5 respectively towards LPG in helium gas atmosphere. S1 and S2 were fabricated from the pure samples, and they exhibit similar sensitivity pattern, while S3, S4, and S5 are doped samples, and they also exhibit similar sensitivity pattern. In nitrogen gas atmosphere, the sensitivity pattern of the sensors towards LPG is similar to that of the dry air atmosphere - there was charge carrier transition of the sensors from n- to p-type. Also, there were series of current oscillation at the peaks of the response. In argon gas atmosphere, the performance of the sensors was generally poor and unquantifiable. The gas sensing performance of the sensor (S3) towards LPG in dry air atmosphere, nitrogen gas atmosphere, helium gas atmosphere, and argon gas atmosphere are compared in Table 3. The resistance values presented in Table 3 are the sensor resistance in the carrier gas before the target gas, i.e., LPG was introduced into the chamber. The idea of gas sensing using different carrier gases (as carried out in this work) is unprecedented. The outstanding response of the Co0.8Ni0.1Mn0.1Fe1.9Ce0.1O4 -based sensor (S3) towards LPG in helium gas atmosphere is noteworthy and could be suggested that helium gas created a more conduction-enhancing condition for the sensor than the dry air, nitrogen gas, and argon gas did. It probably was acting as a catalyst in the reaction between the sensing layer and the gas. Moreover, the optimal resistance for best performance appears to be attained in helium gas as can be observed in Table 3. This could be a reason for higher conductivity in the sensor when the LPG was allowed into the chamber with helium as carrier gas. The responses presented in Table 3 are towards 3000 ppm of LPG in dry air atmosphere, 6000 ppm of LPG in helium gas atmosphere, and 3000 ppm of LPG in nitrogen gas atmosphere because of the charge carrier transition that was observed on the sensor during LPG sensing in dry air atmosphere, and the decrease in response observed starting from 7000 ppm of LPG in helium gas atmosphere. The highest response recorded when dry air was used as a carrier gas was towards ammonia on the sensor S2. The response is 9 towards 1000 ppm; the transient current curve of the sensor is shown in Figure 6e.
3.2.1. Gas sensing Mechanism
Figure 7 illustrates the gas sensing mechanism of the Co0.8Ni0.1Mn0.1Fe1.9Ce0.1O4-based sensor in the different ambient conditions (oxygen abundant and oxygen deficient) it was operated while determining its gas sensing performance. At the operating temperature of 225 °C, the atmospheric oxygen is chemisorbed on the surface of the sensor as O - oxygen species (Figure 7a) as illustrated in the equations 2-5. These oxygen species captured the free electrons at the conduction band, creating a depletion layer on the sensor [40,41], and thus increase the resistivity of the n-type sensor. When the target gas (LPG- a reducing gas) was introduced into the chamber, it reacted with the chemisorbed oxygen on the sensor surface to produce water and carbon dioxide (Figure 7b). The surface reduction of the sensor’s sensing layer led to the release of the captured electrons, causing the thinning of the depletion layer, and resulting in increased conductivity of the sensor [40,41,42]. In helium gas atmosphere (Figure 7c), the sensor resistance (27.95 kΩ) was observed to be far less than observed in the air (see Table 3). This implies the availability of more electrons at the conduction band of the n-type semiconductor-based sensor as a result of a very thin depletion region. This could be owing to the displacement of oxygen from the chamber where, at the operating temperature, it could have been adsorbed again to the sensor surface and reduced the quantity of electrons or impede the mobility of electrons in the conduction band. Nevertheless, it could not be explicitly stated that some chemisorbed species of oxygen are no longer available at all on the sensor surface, but rather that there is limited amount of them, otherwise, the current would have remained the same when LPG was allowed into the chamber, as adsorption/desorption of oxygen is currently understood to be the principle upon which the operation of electrochemical gas sensors is based [20,43,44]. However, the sensor’s response increased with increasing concentration of the target gas (LPG). The effect of helium was more noticeable when used as a carrier gas for LPG (depicted in Figure 7d). The relatively high sensor response towards helium-borne LPG indicates the catalytic effect of helium in the process. While helium did not actually participate in the chemical reaction (being inert as shown in Figure 7c), it is obvious that it facilitated the reaction of LPG with the residual oxygen species on the sensing layer by easily bringing LPG into close contact with the sensing layer, and thus aiding further thinning of the depletion layer as shown in Figure 7d.
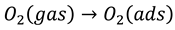
Figure 8a shows the transient current curve resulting from the detection of 3 cycles each of 5000 ppm, 6000 ppm, and 7000 ppm LPG concentrations by the sensor (S3). The sensor could not maintain the same response towards three cycles of 5000 ppm concentration of LPG. Its response towards three cycles of 6000 ppm concentration is consistent, while its response to three cycles of 7000 ppm concentration is as observed for 5000 ppm concentration but with lower response. The decrease in the response that occur towards 7000 ppm, in comparison with the 6000-ppm concentration, is consistent with the result obtained earlier. The test was carried out over 10 months after the initial measurement. The response of the sensor has decreased to 12.39 over this period.
Figure 8b shows the sensitivity plot of the sensor (S3) towards 5000 ppm and 6000 ppm of LPG using helium as carrier gas in the presence of 50% relative humidity. The response of the sensor under this condition is 10.98 as compared to 12.39 under dry ambient condition. The sensor is not much affected by the high humidity; this indicates its potential for LPG detection and monitoring in an atmosphere with high relative humidity.
The long-term stability of the sensor (S3) in helium gas atmosphere is presented in Figure 8c. The sensor retained about 50 % of its initial sensitivity towards LPG in 6 months. Surprisingly, in the following 5 months the response has drastically diminished relative to its initial response. This drastic diminishment in response could be the effect of series of current instability the sensor underwent while detecting LPG over this period. This instability, otherwise called current oscillation, occurred at the peaks (marked with red arrow in Figure 6d) as well as at the relaxation (marked with green arrow in Figure 6d). This could have caused an enormous stress on the sensor, and thereby rapidly reduced its efficiency.
The result of this work in comparison to other works on LPG detection is summarised in Table 4. The performance of the sensor with helium as carrier is quite remarkable.
4. CONCLUSIONS
The gas sensing performance of a double-substitution spinel is reported in this work. Both doped and pure Cobalt ferrite spinels, prepared by glycolthermal method, were exposed to flammable gases using dry air, helium gas, nitrogen gas, and argon gas as carrier gases at different times. The gas sensing performance of S3, i.e., the sample with chemical formula Co0.8Ni0.1Mn0.1Fe1.9Ce0.1O4 towards 6000 ppm concentration of LPG, using helium as carrier gas, is relatively high. In helium atmosphere, the sensor experienced series of oscillating current upon LPG detection, at the peaks as well as at the relaxation. In addition, the sensor’s response declined from 7000 ppm of LPG concentration contrary to expectation. The gas sensing performance of the sensor in dry air atmosphere is low and its LPG sensing was accompanied with current oscillation which was observed to have been triggered by fluctuation of operating temperature. Moreover, the sensors underwent n-type to p-type charge carrier transition from 4000 ppm concentration of LPG. This may suggest the reason for its poor performance in dry air atmosphere. In the nitrogen gas atmosphere, the sensitivity pattern of the sensor (S3) was similar to that in the dry air atmosphere as charge carrier transition of the sensor was also observed. The response of the sensor towards LPG in argon gas atmosphere was poor and unquantifiable. The sensor seems to exhibit preference for operation in oxygen-deficient environment as its response was higher in the helium gas atmosphere than in the dry air atmosphere. The response of the sensor after a period of 11 months was found to have diminished significantly. However, it exhibits very high resistance towards 50 % relative humidity.
Currently, there is not adequate knowledge to explain the phenomena that accompany LPG sensing in both dry air atmosphere and helium gas atmosphere. A more comprehensive study is very crucial for better understanding of the phenomena and to fabricate sensors operable efficiently despite the anomalous phenomena.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Acknowledgments
The authors affiliated to UNIZULU wish to thank the research office for research support. National Research Foundation is also duly acknowledged for its consistent support to carry out research nationwide. The authors wish to acknowledge the University of the Free State for its continuing support in the surface science characterisation and analysis.
References
- Majhi, S.M.; Naik, G.K.; Lee, H.-J.; Song, H.-G.; Lee, C.-R.; Lee, I.-H.; Yu, Y.-T. Au@NiO core-shell nanoparticles as p-type gas sensor: Novel synthesis, characterization, and their gas sensing properties with sensing mechanism. Sensors and Actuators B 2018, 268, 223–231. [Google Scholar] [CrossRef]
- Bhati, V.S.; Kumar, M.; Banerjee, R. Gas sensing performance of 2D nanomaterials/metal oxide nanocomposites: a review. J. Mater. Chem. C 2021, 9, 8776–8808. [Google Scholar] [CrossRef]
- Qin, W.; Yuan, Z.; Gao, H.; Zhang, R.; Meng, F. Perovskite-structured LaCoO3 modified ZnO gas sensor and investigation on its gas sensing mechanism by first principle. Sensors Actuators B: Chem. 2021, 341, 130015. [Google Scholar] [CrossRef]
- Ji, H.; Zeng, W.; Li, Y. Gas sensing mechanisms of metal oxide semiconductors: a focus review. Nanoscale 2019, 11, 22664–22684. [Google Scholar] [CrossRef]
- Vraj Shah, Jaydip Bhaliya, Gautam M. Patel, Priyanka Joshi. Room-Temperature Chemiresistive Gas Sensing of SnO2 Nanowires: A Review. Journal of Inorganic and Organometallic Polymers and Materials 2022, 32, 741–772. [Google Scholar] [CrossRef]
- Zhu, L.; Zeng, W. Room-temperature gas sensing of ZnO-based gas sensor: A review. Sensors and Actuators A 2017, 267, 242–261. [Google Scholar] [CrossRef]
- Malik, R.; Tomer, V.K.; Mishra, Y.K.; Lin, L. Functional gas sensing nanomaterials: A panoramic view. Appl. Phys. Rev. 2020, 7, 021301. [Google Scholar] [CrossRef]
- G. J. Thangamani, S. K. khadheer pasha. Titanium dioxide (TiO2) nanoparticles reinforced polyvinyl formal (PVF) nanocomposites as chemiresistive gas sensor for sulphur dioxide (SO2) monitoring. Chemosphere 2021, 275, 129960. [Google Scholar] [CrossRef]
- Walker, J.M.; Akbar, S.A.; Morris, P.A. Synergistic effects in gas sensing semiconducting oxide nano-heterostructures: A review. Sensors Actuators B: Chem. 2019, 286, 624–640. [Google Scholar] [CrossRef]
- Kumar, E.R.; Kamzin, A.; Janani, K. Effect of annealing on particle size, microstructure and gas sensing properties of Mn substituted CoFe2O4 nanoparticles. J. Magn. Magn. Mater. 2016, 417, 122–129. [Google Scholar] [CrossRef]
- Zhou, T.; Cao, S.; Zhang, R.; Tu, J.; Fei, T.; Zhang, T. Effect of cation substitution on the gas sensing performances of ternary spinel MCo2O4 (M= Mn, Ni, and Zn). Journal of applied Materials Interfaces. 2019, 11, 28023–28032. [Google Scholar] [CrossRef] [PubMed]
- Patil, J.; Nadargi, D.; Mulla, I.S.; Suryavanshi, S. Spinel MgFe2O4 thick films: A colloidal approach for developing gas sensors. Mater. Lett. 2018, 213, 27–30. [Google Scholar] [CrossRef]
- Shozi, N.N.; Kortidis, I.; Mkwae, P.S.; Chonco, N.P.; Leshabane, N.; Jozela, M.; Kroon, R.E.; Swart, H.C.; Nkosi, S.S. Extremely sensitive and selective flammable liquefied hydrocarbon gas sensing and inter-dependence of fluctuating operating temperature and resistance: Perspective of rare-earth doped cobalt nanoferrites. J. Alloy. Compd. 2021, 859, 157846. [Google Scholar] [CrossRef]
- Koli, P.B.; Kapadnis, K.H.; Deshpande, U.G. Nanocrystalline-modified nickel ferrite films: an effective sensor for industrial and environmental gas pollutant detection. J. Nanostructure Chem. 2019, 9, 95–110. [Google Scholar] [CrossRef]
- Nemufulwi, M.I.; Swart, H.C.; Mhlongo, G.H. A comprehensive comparison study on magnetic behaviour, defects-related emission and Ni substitution to clarify the origin of enhanced acetone detection capabilities. Sens. Actuators B Chem. 2021, 339, 129860. [Google Scholar] [CrossRef]
- Manikandan, V.; Singh, M.; Yadav, B.C.; Mane, R.S.; Vigneselvand, S.; Mirzaei, A.; Chandrasekaran, J. Room temperature LPG sensing properties of tin substituted copper ferrite (Sn-CuFe2O4) thin film. Materials Chemistry and Physics 2020, 240, 122265. [Google Scholar] [CrossRef]
- Wu, S.; Li, X.; Xu, Y.; Wu, J.; Wang, Z.; Han, Y.; Zhang, X. Hierarchical spinel NixCo1-xFe2O4 microcubes derived from Fe-based MOF for high-sensitive acetone sensor. Ceramics International 2018, 44, 19390–19396. [Google Scholar] [CrossRef]
- Gonçalves, J.M.; Rocha, D.P.; Silva, M.N.T.; Martins, P.R.; Nossol, E.; Angnes, L.; Rout, C.S.; Munoz, R.A.A. Feasible strategies to promote the sensing performances of spinel MCo2O4 (M= Ni, Fe, Mn, Cu and Zn) based electrochemical sensors: a review. Journal of Materials Chemistry C. 2021, 9, 7852–7887. [Google Scholar] [CrossRef]
- Almessiere, M.A.; Slimani, Y.; Baykal, A. Synthesis and characterization of Co1-2xNixMnxFe2-yCeyO4 nanoparticles. Journal of Rare Earths 2020, 38, 188–194. [Google Scholar] [CrossRef]
- Shankar, P.; Rayappan, J.B.B. Gas sensing mechanism of metal oxide: The role of ambient atmosphere, type of semiconductor and gases- A review. Science Letters Journal 2015, 4, 126. [Google Scholar]
- Zhang, J.; Qin, Z.; Zeng, D.; Xie, C. Metal-oxide-based semiconductor gas sensor: screening, preparation and integration. Journal of Physical Chemistry 2017, 19, 6313–6329. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.M.; Akbar, S.A.; Morris, P.A. Synergistic effects in gas sensing semiconducting oxide nano-heterostructures: A review. Sensors Actuators B: Chem. 2019, 286, 624–640. [Google Scholar] [CrossRef]
- Pi, W.; Chen, X.; Humayun, M.; Yuan, Y.; Dong, W.; Zhang, G.; Chen, B.; Fu, Q.; Lu, Z.; Li, H.; Tang, Z.; Luo, W. Highly Sensitive Chemiresistive H2S Detection at Subzero Temperature over the Sb-Doped SnO2@g-C3N4 Heterojunctions under UV Illumination. Applied Materials and Interfaces 2023, 11, 14979–14989. [Google Scholar] [CrossRef] [PubMed]
- P. Munindra, M. Sai Bhargava Reddy, B. Geeta Rani, N. Jayarambabu, Saraswathi Kailasa, P. Srinivasa Subba Rao, K. Venkateswara Rao. A high-performance low-temperature LPG detection by MgFe2O4/BiVO4 chemiresistive sensor. Journal of Materials Science: Materials in Electronics 2020, 31, 2370–2377. [Google Scholar]
- Almessiere, M.A.; Slimani, Y.; Ali, S.; Baykal, A.; Ercan, I.; Sozeri, H. Nd3+ Ion-Substituted Co1−2xNixMnxFe2−yNdyO4 Nanoparticles: Structural, Morphological, and Magnetic Investigations. Journal of Inorganic and Organometallic Polymers and Materials 2019, 29, 783–791. [Google Scholar] [CrossRef]
- Almessiere, M.A.; Slimani, Y.; Güner, S.; Nawaz, M.; Baykal, A.; Aldakheel, F.; Akhtar, S.; Ercan, I.; Beleli, I.; Ozҫelik, B. magnetic and structural characterization of Nb3+ -substututed CoFe2O4 nanoparticles. Ceramics International 2019, 45, 8222–8232. [Google Scholar] [CrossRef]
- Rani, B.J.; Ravina, M.; Saravanakumar, B.; Ravi, G.; Ganesh, V.; Ravichandran, S.; Yuvakkumar, R. Ferrimagnetism in cobalt ferrite (CoFe 2 O 4 ) nanoparticles. Nano-Structures Nano-Objects 2018, 14, 84–91. [Google Scholar] [CrossRef]
- Motaung, D.E.; Tshabalala, Z.P.; Makgwane, P.R.; Mahmoud, F.A.; Oosthuizen, D.N.; Cummings, F.R.; Leshabane, N.; Hintsho-Mbita, N.; Li, X.; Ray, S.S.; et al. Multi-functioning of CeO2-SnO2 heterostructure as room temperature ferromagnetism and chemiresistive sensors. J. Alloy. Compd. 2021, 906, 164317. [Google Scholar] [CrossRef]
- Kirankumar, V.S.; Mayank, N.; Sumathi, S. Photocatalytic performance of cerium doped copper aluminate nanoparticles under visible light irradiation. Journal of the Taiwan Institute of Chemical Engineers 2019, 95, 602–615. [Google Scholar] [CrossRef]
- Nurhasanah, I.; Safitri, W.; Arifin, Z.; Subagio, A.; Windarti, T. Antioxidant activity and dose enhancement factor of CeO2 nanoparticles synthesized by precipitation method. IOP Conf. Series: Mater. Sci. Eng. 2018, 432, 012031. [Google Scholar] [CrossRef]
- Mkwae, P.S.; Ogundipe, S.A.; Jozela, M.; Revaprasadu, N.; Nkosi, S.S. The heat rate kinetics on the liquefied hydrocarbon gases sensing and food quality control detecting strategy. Mater. Chem. Phys. 2022, 277. [Google Scholar] [CrossRef]
- Chandamma, N.; Santhosh Kumar, M.V.; Shankarmurthy, G.J.; Melagiriyappa, E.; Nagaraja, K.K. Effect of gamma irradiation on some electrical and dielectric properties of Ce3+ substituted Ni–Zn nano ferrites. Chin. J. Phys. 2017, 55, 1729–1738. [Google Scholar] [CrossRef]
- Wang, J.; Sun, A.; Jiang, Y.; Huang, X.; Shao, L.; Zhang, Y. Structural and magnetic properties of Ce3+doped Mg-Co ferrite prepared by sol–gel method. J. Mater. Sci. Mater. Electron. 2022, 33, 11881–11895. [Google Scholar] [CrossRef]
- Li, L.-Z.; Zhong, X.-X.; Wang, R.; Tu, X.Q.; He, L.; Wang, F.-H. Effects of Ce substitution on structural and electromagnetic properties of NiZn nano ferrite. Journal of Magnetism and Magnetic Materials 2019, 475, 1–4. [Google Scholar] [CrossRef]
- Mustapha, S.; Tijani, J.O.; Ndamitso, M.M.; Abbdulkareem, A.S.; Shuahib, D.T.; Amigun, A.T.; Abubakar, H.L. Facile synthesis and characterisation of TiO2 nanoparticles: X-ray peak profile analysis using Williamson-Hall and Debye-Scherrer methods. International Nano Letters 2021, 11, 241–261. [Google Scholar] [CrossRef]
- Khalfaoui, M.; Knani, S.; Hachicha, M.A.; Lamine, A.B. New theoretical expreesions for the five adsorption isotherms classifield by BET based on statistical physics treatment. Journal of colloid and interface science 2003, 263, 350–356. [Google Scholar] [CrossRef]
- Hailstone, R.K.; DiFrancesco, A.G.; Leong, J.G.; Allston, T.D.; Reed, K.J. A Study of Lattice Expansion in CeO2 Nanoparticles by Transmission Electron Microscopy. J. Phys. Chem. C 2009, 113, 15155–15159. [Google Scholar] [CrossRef]
- Tiberio Magno de Lima Alves, Bruno Ferreira Amorim, Marco Antonio Morales Torres, Claudionor Gomes Bezerra, Suzana Nobrega de Medeiros, Pedro Lana Gastelois, Luis Eugenio Fernandez Outon, Waldemar Augusto de Almeida Macedo. Wasp-waisted behavior in magnetic hysteresis curves of CoFe2O4 Nano powder at a low temperature: experimental evidence and theoretical approach. Journal of Royal Society Chemistry Advance 2017, 7, 22187. [Google Scholar]
- Gumbi, S.W.; Mkwae, P.S.; Kortidis, I.; Kroon, R.E.; Swart, H.C.; Moyo, T.; Nkosi, S.S. Electronic and simple oscillatory conduction in ferrite gas sensing: Gas-sensing mechanism, Long-term gas monitoring, Heat transfer, and other anomalies. Journal of Applied Material Interfaces 2020, 12, 43231–43249. [Google Scholar] [CrossRef]
- Mirzaei, A.; Bonyani, M.; Torkian, S.; Feizpour, M.; Bonavita, A.; Leonardi, S.G.; Neri, G. A comparative study on the electrical and gas sensing properties of thick films prepared with synthesized nano-sized and commercial micro-sized Fe2O3 powders. Process. Appl. Ceram. 2017, 11, 265–274. [Google Scholar] [CrossRef]
- Rai, P.; Majhi, S.M.; Yu, Y.-T.; Lee, J.-H. Nobel metal@metal oxide semiconductor core@shell nano-architectures as a new platform for gas sensing applications. RSC Adv. 2015, 5, 76229. [Google Scholar] [CrossRef]
- Nikolic, M.V.; Milovanovic, V.; Vasiljevic, Z.Z.; Stamenkovic, Z. Semiconductor Gas Sensors: Materials, Technology, Design, and Application. Sensors 2020, 20, 6694. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Mojumder, S.; Saha, D.; Pa, M. Influence of major parameters on the sensing mechanism of semiconductor metal oxide based chemiresitive gas sensors: A review focused on personalized healthcare. Sensors & Actuators: B. Chemical 2022, 352, 131066. [Google Scholar]
- Staerz, A.; Weimar, U.; Barsan, N. Current state of knowledge on the metal oxide based gas sensing mechanism. Sensors Actuators B: Chem. 2022, 358, 131531. [Google Scholar] [CrossRef]
- Choudhary, S.; Annapoorni, S.; Malik, R. Evolution and growth mechanism of hexagonal ZnO nanorods and their LPG sensing response at low operating temperature. Sensors Actuators A: Phys. 2019, 293, 207–214. [Google Scholar] [CrossRef]
- Nakate, U.T.; Patil, P.; Ghule, B.; Nakate, Y.T.; Ekar, S.; Ambare, R.C.; Mane, R. Room temperature LPG sensing properties using spray pyrolysis deposited nano-crystalline CdO thin films. Surfaces Interfaces 2019, 17, 100339. [Google Scholar] [CrossRef]
Figure 1.
(a) The XRD patterns of cobalt ferrite spinel doped with Ni & Mn in tetrahedral A- sites, Ce at octahedral B-sites. Scanning electron microscopy micrograms of the Co1-2xNixMnxFe2-yCeyO4 samples with (b) x = y = 0: dried with infrared lamp, (c) x = y = 0: dried naturally, (d) x = y = 0.1, (e) x = y = 0.2 and (f) x = y = 0.3.
Figure 1.
(a) The XRD patterns of cobalt ferrite spinel doped with Ni & Mn in tetrahedral A- sites, Ce at octahedral B-sites. Scanning electron microscopy micrograms of the Co1-2xNixMnxFe2-yCeyO4 samples with (b) x = y = 0: dried with infrared lamp, (c) x = y = 0: dried naturally, (d) x = y = 0.1, (e) x = y = 0.2 and (f) x = y = 0.3.
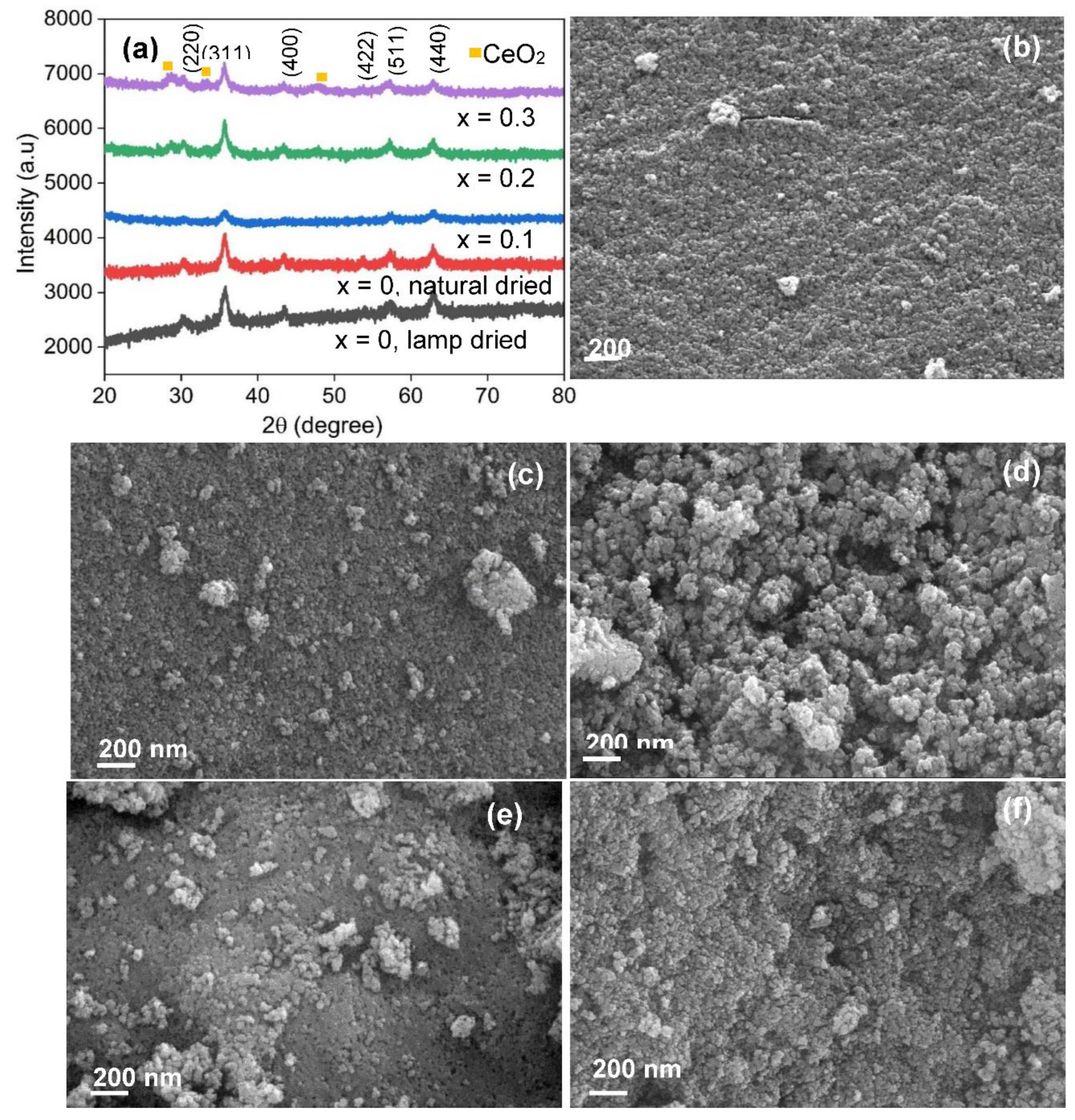
Figure 2.
Nitrogen adsorption-desorption isotherms of the Co0.8Ni0.1Mn0.1Fe1.9Ce0.1O4 double-substitution spinel with Inset: The corresponding pore size distribution.
Figure 2.
Nitrogen adsorption-desorption isotherms of the Co0.8Ni0.1Mn0.1Fe1.9Ce0.1O4 double-substitution spinel with Inset: The corresponding pore size distribution.
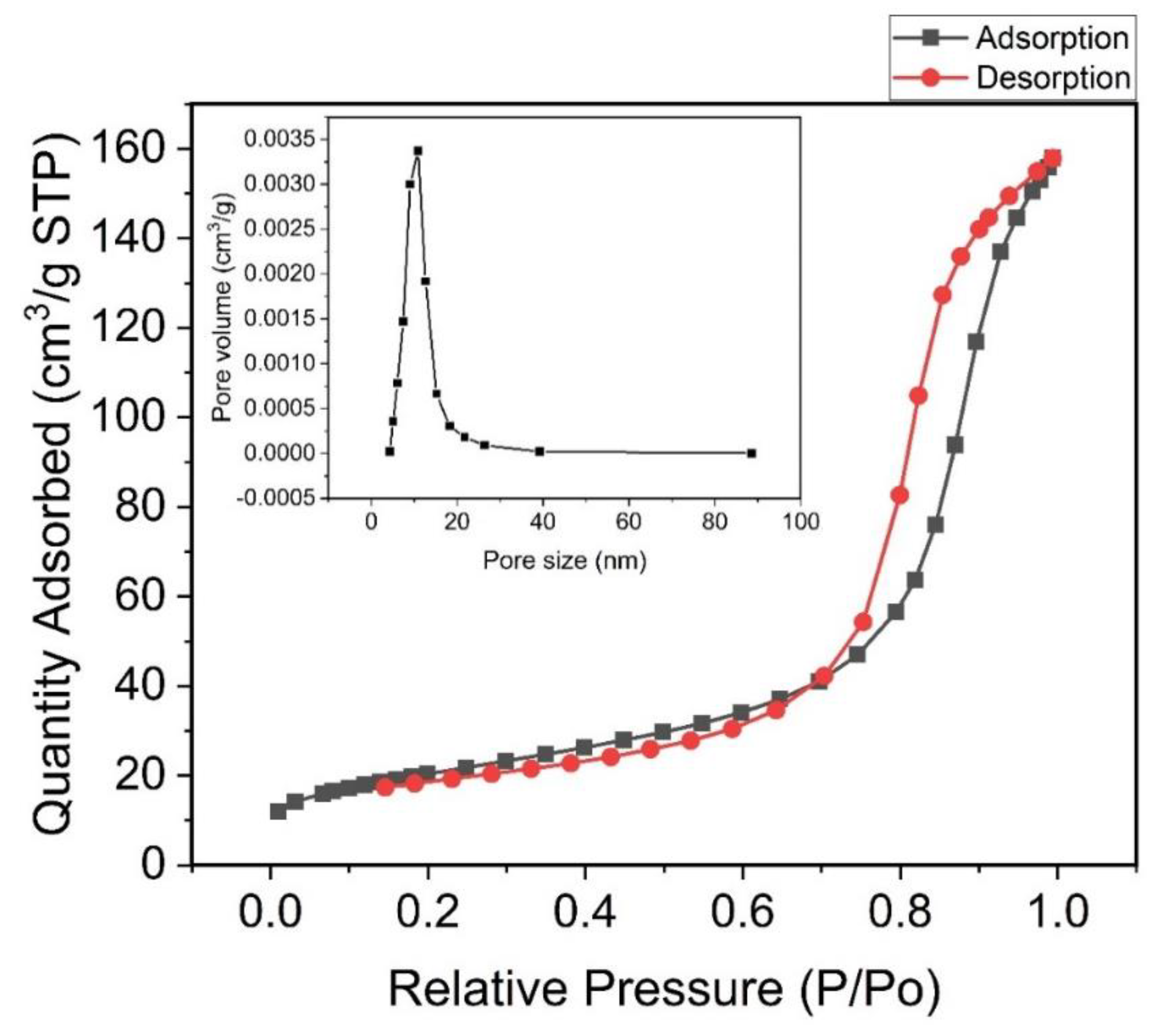
Figure 3.
HRTEM images of the Co1-2xNixMnxFe2-yCeyO4 samples with (a) x = y = 0: dried with UV lamp, (b) x = y = 0: dried naturally, (c) x = y = 0.1, (d) x = y = 0.2 and (e) x = y = 0.3. The insets are the corresponding Selected Area Electron Diffraction patterns.
Figure 3.
HRTEM images of the Co1-2xNixMnxFe2-yCeyO4 samples with (a) x = y = 0: dried with UV lamp, (b) x = y = 0: dried naturally, (c) x = y = 0.1, (d) x = y = 0.2 and (e) x = y = 0.3. The insets are the corresponding Selected Area Electron Diffraction patterns.
Figure 4.
XPS spectra of Co1-2xNixMnxFe2-yCeyO4 sample for which x = y = 0.1: (a) Survey spectrum before sputtering (b) Survey spectrum after sputtering, (c) Co 2p scan, (d) Ni 2p scan, (e) Mn 2p scan (f) Ce 3d scan, (g) Fe 2p scan and (h) O 1s scan.
Figure 4.
XPS spectra of Co1-2xNixMnxFe2-yCeyO4 sample for which x = y = 0.1: (a) Survey spectrum before sputtering (b) Survey spectrum after sputtering, (c) Co 2p scan, (d) Ni 2p scan, (e) Mn 2p scan (f) Ce 3d scan, (g) Fe 2p scan and (h) O 1s scan.
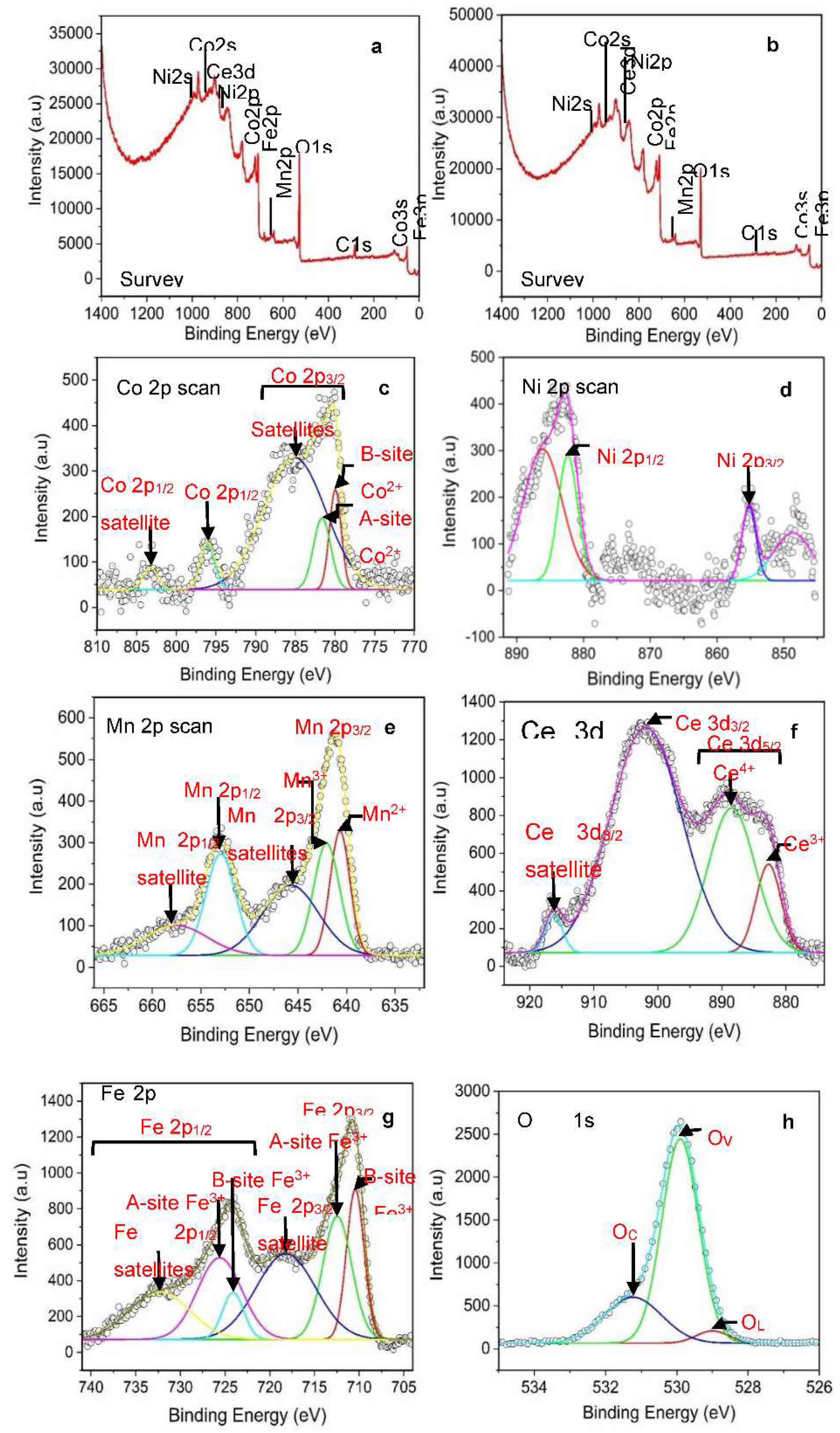
Figure 5.
3D selectivity plots of Co1-2xNixMnxFe2-yCeyO4 double-substitution spinel towards: (a) ethanol, propanol, ammonia in dry air atmosphere, (b) ethanol, propanol, ammonia, and LPG (6000 ppm) in helium gas atmosphere, (c), ethanol, propanol, ammonia, and LPG (10k ppm) in helium gas atmosphere, and (d) transient current plot of S3 towards LPG in dry air atmosphere.
Figure 5.
3D selectivity plots of Co1-2xNixMnxFe2-yCeyO4 double-substitution spinel towards: (a) ethanol, propanol, ammonia in dry air atmosphere, (b) ethanol, propanol, ammonia, and LPG (6000 ppm) in helium gas atmosphere, (c), ethanol, propanol, ammonia, and LPG (10k ppm) in helium gas atmosphere, and (d) transient current plot of S3 towards LPG in dry air atmosphere.
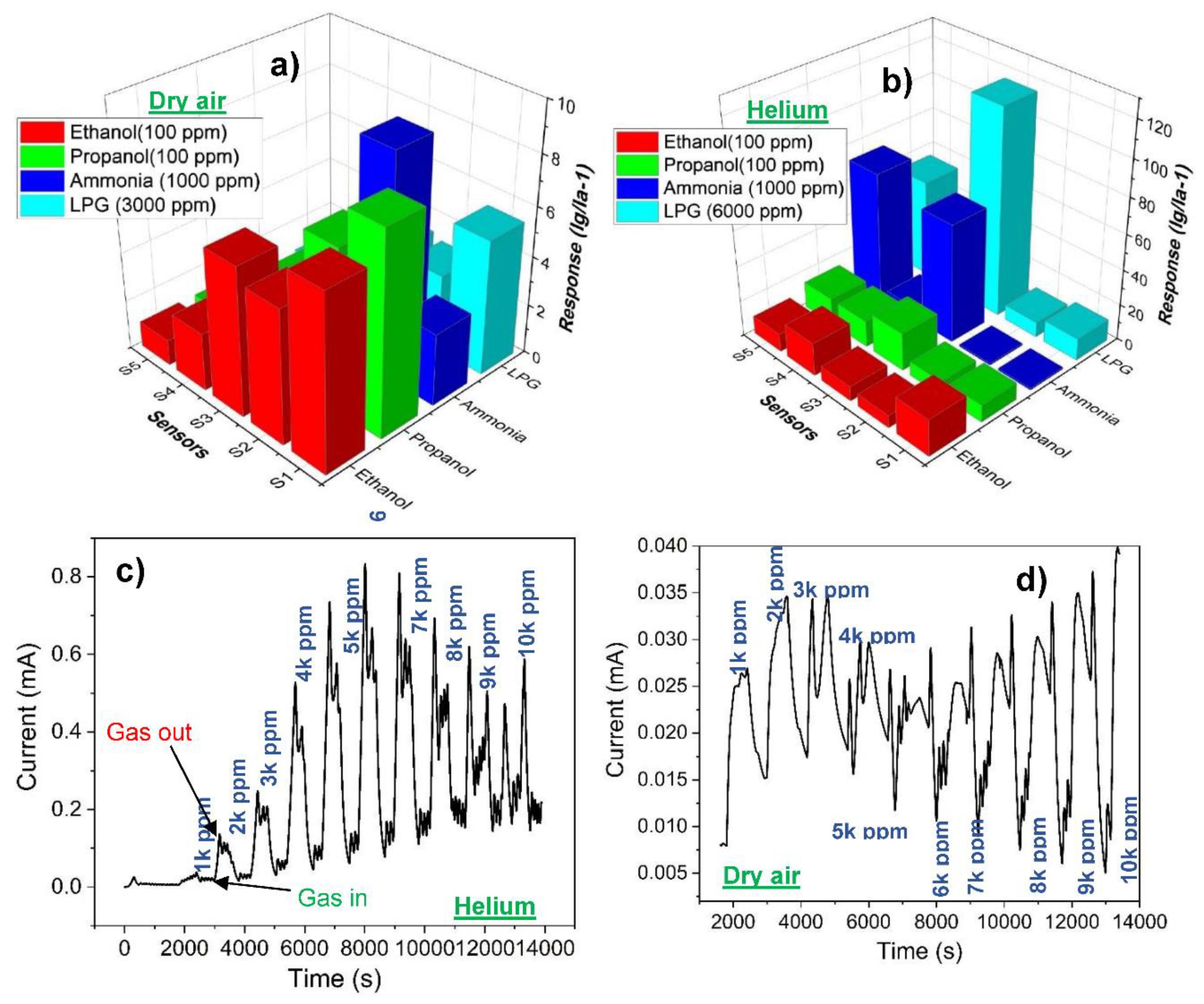
Figure 6.
Gas sensing response pattern of: (a) S2 towards LPG in dry air atmosphere, (b) S5 towards LPG in dry air atmosphere, (c) S2 towards LPG in helium gas atmosphere, (d) S5 towards LPG in helium atmosphere, and (e) transient current curve of S2 towards ammonia gas in dry air atmosphere.
Figure 6.
Gas sensing response pattern of: (a) S2 towards LPG in dry air atmosphere, (b) S5 towards LPG in dry air atmosphere, (c) S2 towards LPG in helium gas atmosphere, (d) S5 towards LPG in helium atmosphere, and (e) transient current curve of S2 towards ammonia gas in dry air atmosphere.
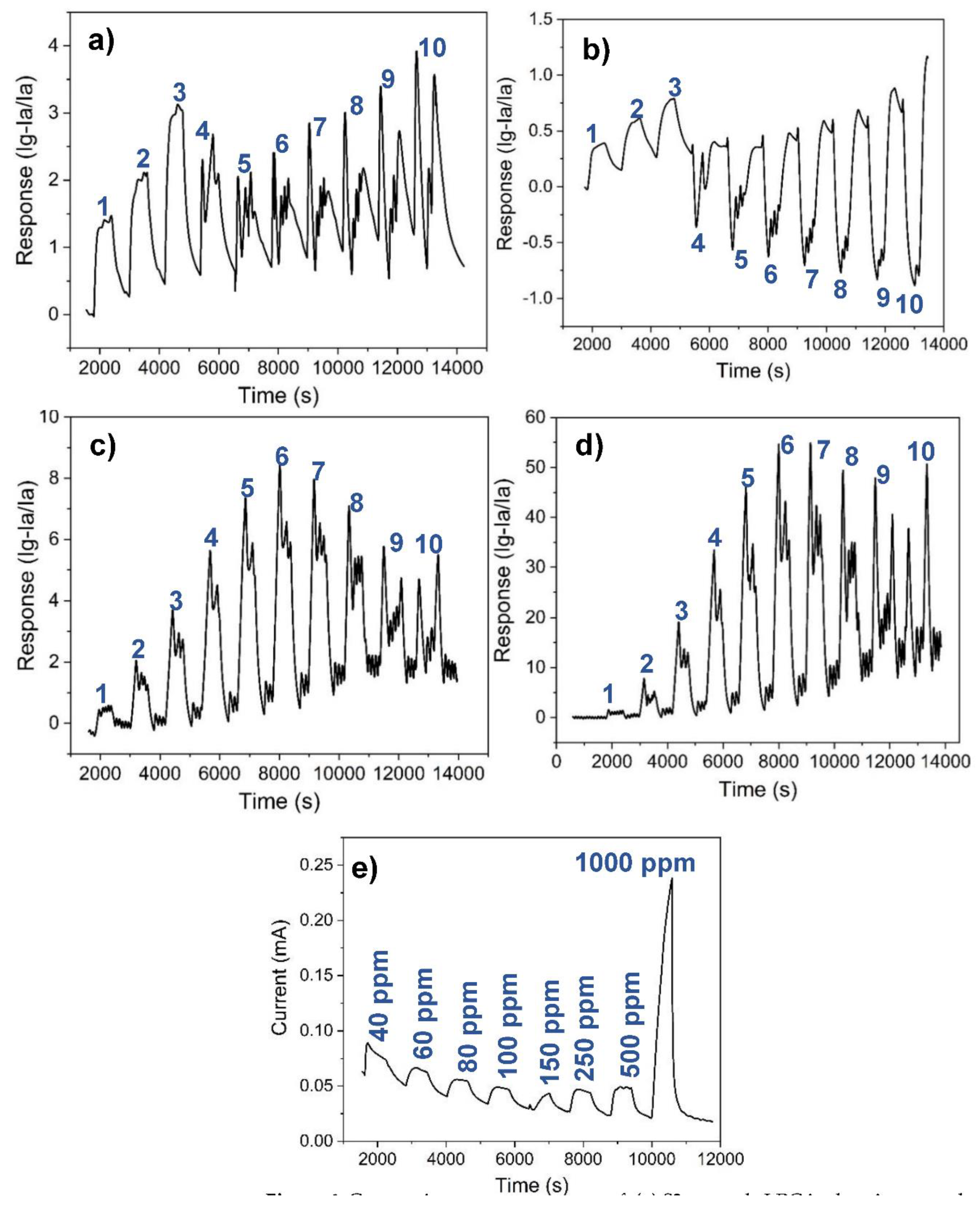
Figure 7.
Schematic illustration of the gas sensing mechanism of the Co0.8Ni0.1Mn0.1Fe1.9Ce0.1O4-based sensor: (a) in air, (b) towards air-borne LPG, (c) in helium gas atmosphere, and (d) towards helium-borne LPG.
Figure 7.
Schematic illustration of the gas sensing mechanism of the Co0.8Ni0.1Mn0.1Fe1.9Ce0.1O4-based sensor: (a) in air, (b) towards air-borne LPG, (c) in helium gas atmosphere, and (d) towards helium-borne LPG.
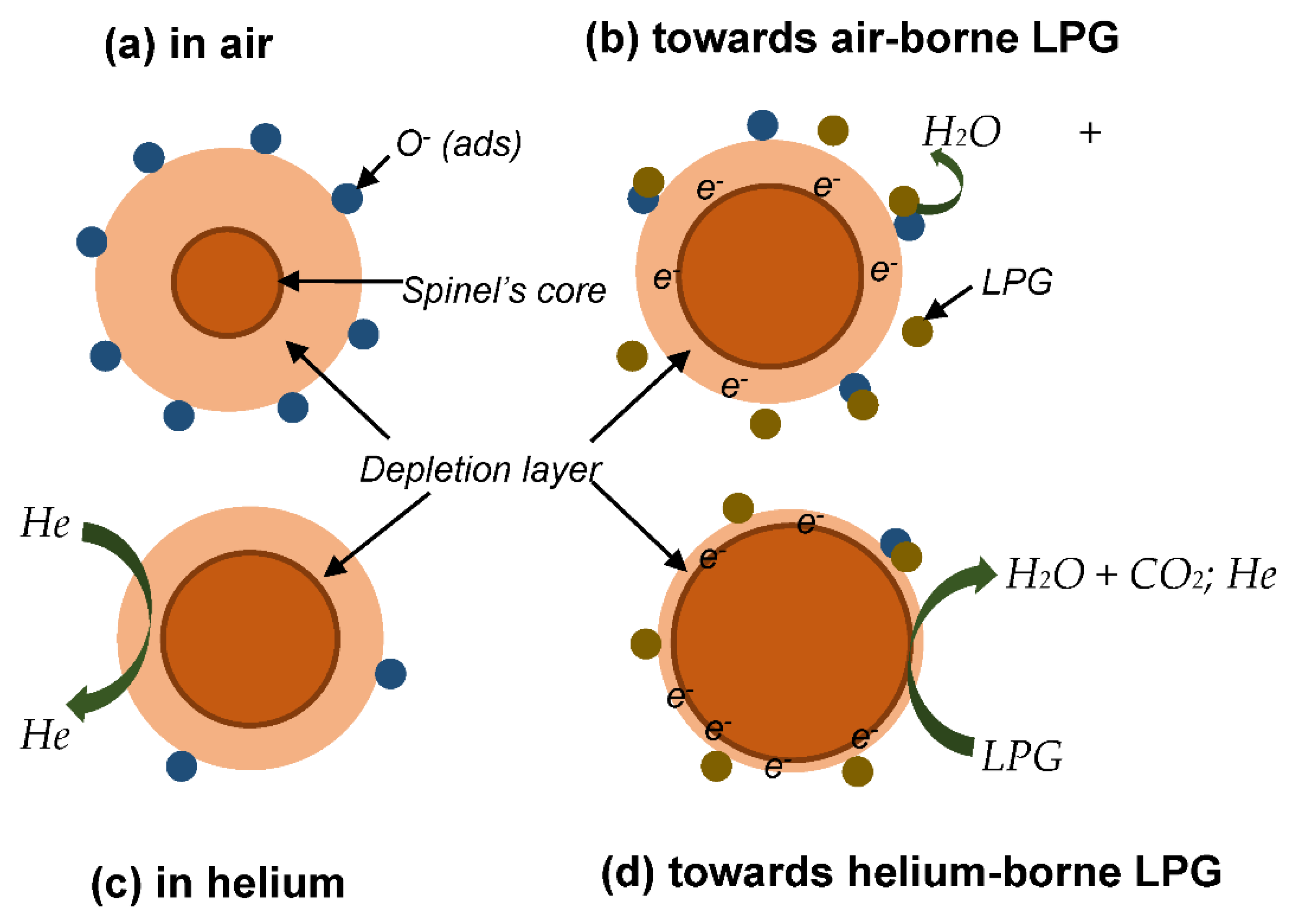
Figure 8.
(a) Transient current and response curve from the sensitivity of the sensor (S3) towards 5000, 6000, and 7000 ppm of LPG in helium gas atmosphere, (b) response of S3 towards 5000 and 6000 ppm of LPG in helium gas atmosphere in the presence of 50% RH, and (c) long-term stability of the sensor (S3) in helium gas atmosphere.
Figure 8.
(a) Transient current and response curve from the sensitivity of the sensor (S3) towards 5000, 6000, and 7000 ppm of LPG in helium gas atmosphere, (b) response of S3 towards 5000 and 6000 ppm of LPG in helium gas atmosphere in the presence of 50% RH, and (c) long-term stability of the sensor (S3) in helium gas atmosphere.
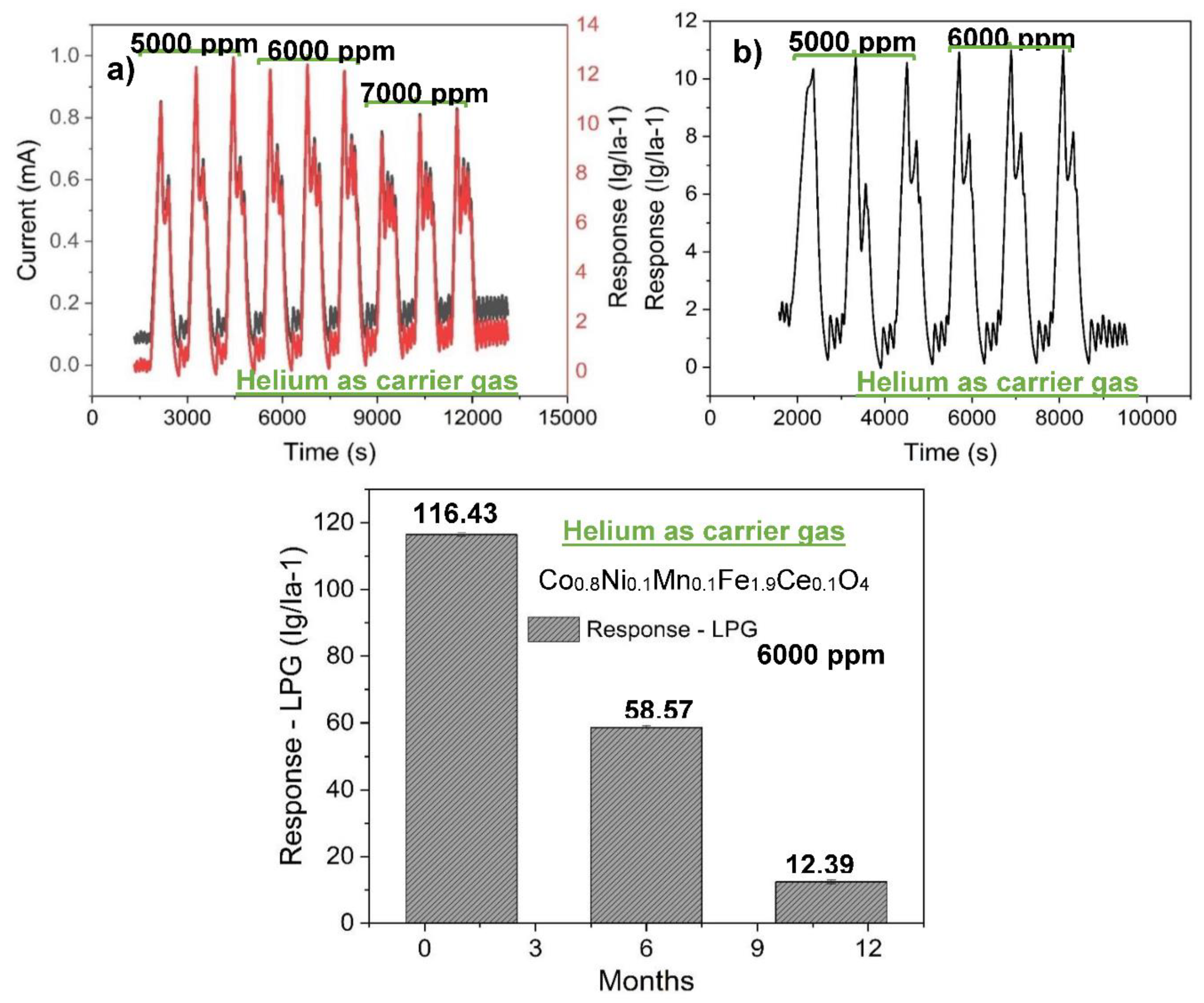

Table 1.
The crystallite sizes, pore size distribution and the BET specific surface areas.
| Co1-2xNixMnxFe2-yCeyO4 | D311 (nm) | Pore Size (nm) | Specific surface Area (m2/g) |
|---|---|---|---|
| x = y = 0, lamp-dried | 9.99 | 11.38 | 66.99 |
| x = y = 0, natural-dried | 11.30 | 11.96 | 62.48 |
| x = y = 0.1 | 9.28 | 12.34 | 72.82 |
| x = y = 0.2 | 12.05 | 10.52 | 51.93 |
| x = y = 0.3 | 12.30 | 13.29 | 60.76 |
Table 2.
Deconvolution of O 1s peaks from XPS analysis.
| Co1-2xNixMnxFe2-yCeyO4 | OL: | (OV+OC): | (OL+OV): |
|---|---|---|---|
| x = y = 0, lamp-dried | 0.55 | 0.45 | - |
| x = y = 0, natural-dried | 0.69 | OV = 0.31 | - |
| x = y = 0.1 | 0.04 | 0.96 | - |
| x = y = 0.2 | 0.84 | OV = 0.16 | - |
| x = y = 0.3 | 0.61 | OV = 0.39 | - |
Table 3.
comparison of gas sensing performance of the sensor (S3) towards LPG, in various carrier gases.
Table 3.
comparison of gas sensing performance of the sensor (S3) towards LPG, in various carrier gases.
| Carrier Gas | Resistance (kΩ) | Response |
|---|---|---|
| Dry air | 641.03 | 5.87 |
| Nitrogen | 28.57 | 5.28 |
| Helium | 769.23 | 116.43 |
| Argon | - | - |
Table 4.
Comparison of the performance of the double-substitution spinel with other sensor materials towards LPG detection.
Table 4.
Comparison of the performance of the double-substitution spinel with other sensor materials towards LPG detection.
| Material | Carrier gas | Operating temp. (°C) | Concentration (ppm) | Response | Ref. |
|---|---|---|---|---|---|
| ZnO | Dry air | 200 | 100 | 49% | [45] |
| CdO | Dry air | 50 | 10 000 | 4.6% | [46] |
| MgFe2O4 | Dry air | 225 | 10 000 | 395.47 | [31] |
| MgFe2O4/BiVO4 | Dry air | 50 | 500 | 58% | [24] |
| Sn-CuFe2O4 | Dry air | 25 | 2 000 | 78% | [16] |
| Co0.8Ni0.1Mn0.1Fe1.9Ce0.1O4 | Dry air | 225 | 3 000 | 3.35 | This work |
| Co0.8Ni0.1Mn0.1Fe1.9Ce0.1O4 | Helium gas | 225 | 6 000 | 116.43 | This work |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



