
Submitted:
29 August 2023
Posted:
31 August 2023
You are already at the latest version
Abstract
New antibiotics are unquestionably needed in the fight to the emergence and spread of multidrug-resistant bacteria. So far, antibiotics targeting bacterial central metabolism have been poorly investigated. By determining the minimal inhibitory concentration (MIC) of desmethylphosphinothricin (Glu-γ-PH), an analogue of glutamate with a phosphinic moiety re-placing the γ-carboxyl group, we previously showed its promising antibacterial activity on Escherichia coli. Herein we synthetized and determined the growth inhibition exerted on E. coli by an L-Leu dipeptide derivative of Glu-γ-PH (L-Leu-D,L-Glu-γ-PH). Furthermore, we compared the growth inhibition obtained with this dipeptide with that exerted by the free amino acid, i.e. Glu-γ-PH, and by their phosphonic and non-desmethylated analogues. All tested compounds were more effective when assayed in chemically defined minimal medium. The dipeptide L-Leu-D,L-Glu-γ-PH had a significantly improved antibacterial activity (2 μg/mL), at a concentra-tion between the non-desmethytaled (0.1 μg/mL) and the phosphonic (80 μg/mL) analogues. Also in Bacillus subtilis the dipeptide L-Leu-D,L-Glu-γ-PH displayed an activity comparable to that of the antibiotic amoxicillin. This work highlights the antibacterial relevance of the phosphinic pharmacophore and proposes new avenues to the development of novel antimicrobial drugs containing the phosphinic moiety.
Keywords:
antibacterial
; glutamate metabolism
; antimicrobial resistance
; central metabolism
; dipeptide permeases
; phosphorus-containing glutamate analogues
1. Introduction
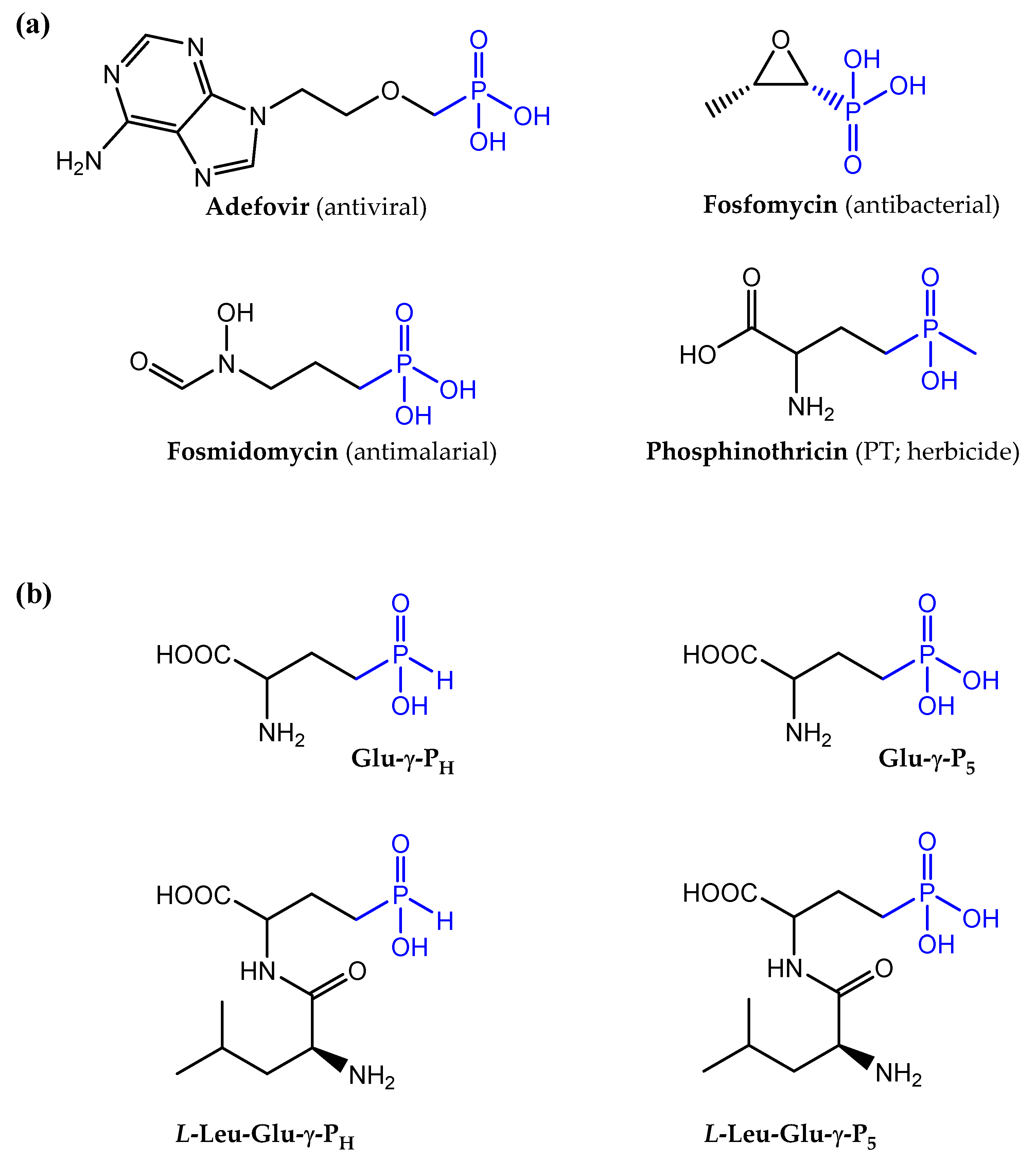
Organic molecules containing phosphonic and phosphinic acidic groups are unusual, though naturally occurring compounds [1-6]. They are unusual because they characterized by the presence of a carbon-phosphorous (C-P) bond. In particular, phosphonates contain a single C-P bond, whereas phosphinates contain either two such bonds (i.e. C-P-C) or C-P-H bonds. For both types of compounds the rest of the valences on the phosphorous atom are engaged in bonding oxygen. The C-P bond, unlike the C-O-P bonds (such as those occurring in the more common organic phosphate esters and anhydrides) has unique features, including a remarkable stability against enzymatic (i.e., cannot be cleaved by hydrolyses) or chemical cleavage, such as acid/base hydrolysis. Notably, the phosphinic and phosphonic moieties structurally mimic phosphate esters, carboxylates and tetrahedral intermediates occurring during carboxyl group transformations [3,4]. This also explains why many of these compounds act as substrates or competitive inhibitors of enzymes the natural substrates of which possess the above functional groups. Prominent examples of these class of molecules include compounds of natural origin, such as the antibiotics fosfomycin [6], dehydrophos and plumbemycin, the antimalarial compounds fosmidomycin and FR-900098, the antifungals rhizocticins, the herbicide phosphinothricin (PT; also known as glufosinate) [3,4], as well as chemically synthesized antivirals Adefovir and Tenofovir, which are successfully employed to treat hepatitis B infections [7]. Examples of such molecules are shown in Figure 1a.
Given that some of the natural compounds mentioned above may be toxic also to the producing micro-organism, they are naturally synthetized as di- or tri-peptide precursors, which then enter the target cell through dipeptide or oligopeptide permeases, Dpp and Opp, respectively [3,4,8,9]. Once internalized, they are cleaved by cytosolic peptidases that cause the release of the active C-P-containing amino acid. This mechanism has been referred as a “Trojan horse” or pro-drug strategy [3,4,8,9]. A remarkable example is phosphinothricin (PT; glufosinate; Fig. 1a), a non-proteinogenic amino acid, which was initially isolated as a bioactive component of the tripeptide Bialaphos, phosphinothricyl-L-alanyl-L-alanine. PT is a phosphinic analogue of L-glutamate, with a phosphinic moiety (C-PO2HCH3) replacing the glutamate γ-carboxyl group. PT was demonstrated to act as a competitive inhibitor of the enzyme glutamine synthetase (GS), which catalyzes the ATP-dependent formation of glutamine starting from glutamate and ammonia, by mimicking the γ-phosphorylated intermediate of glutamate formed during the first step of the reaction [10,11]. In plants, the inhibition of GS leads to a rapid accumulation of intracellular ammonium ion (NH4+), which perturbs pH homeostasis and leads to cell death. Hence, PT and the PT-containing tripeptides, Bialaphos or Phosalacine (i.e. L-alanyl-L-leucyl-PT), exhibit strong herbicidal activity [3,5,12,13]. In addition to the well-established herbicidal effect, Bialaphos and a PT-containing dipeptide, L-Leu-L-PT, were remarkably effective on clinical isolates of Klebsiella pneumoniae, which displayed resistance to more than 20 commercial antibiotics belonging to different classes [14].
Less investigated than PT and Bialaphos is the PT desmethylated on the phosphinic moiety (L-2-amino-4-(hydroxy)-phosphinylbutyric acid; hereafter L-Glu-γ-PH; Fig. 1b), which is also an analogue of L-glutamate, that carries the more rarely occurring H-phosphinic group, i.e. C-P(O)(OH)H. L-Glu-γ-PH was originally isolated as an intermediate in the biosynthesis of Bialaphos in Streptomyces hygroscopicus [15] and more recently in the free form in Nonomureae sp. NRRL B-24552 [1]. In S. hygroscopicus L-Glu-γ-PH was shown to accumulate and inhibit growth in the mutant form of this micro-organism where the Bialaphos biosynthetic pathway was blocked [16]. To protect themselves from the action of the antibiotics they synthetize, the producer organisms (i.e. Streptomyces) inactivate PT by acetylation [17] and then incorporation into Bialaphos, which is then released in the environment. To date the mechanism of antibacterial activity of L-Glu-γ-PH remains unknown. However, we demonstrated that the H-phosphinic group of L-Glu-γ-PH is a bioisostere of carboxylates, and that the desmethylated phosphinic compounds derived from it (i.e. the H-phosphinic analogues of GABA and succinate) can be recognized and metabolized just as the substrate by the relevant enzymes [18]. We also found that only the L-isomer of Glu-γ-PH displays an antibacterial activity, which implies that the compound is indeed metabolized and leads to the formation of intermediate(s) that are eventually responsible for the observed antibacterial activity [18].
To the best of our knowledge, peptides containing amino acids with a H-phosphinic group in distal position from the carboxyl group have never been investigated as antibacterials. Herein, to evaluate the potential of Glu-γ-PH as an effective antibacterial, we studied the inhibition of growth caused by the dipeptide L-Leu-D,L-Glu-γ-PH on both Escherichia coli and Bacillus subtilis, as representatives of Gram-negative and Gram-positive bacteria, respectively. We compared its activity with that of D,L-PT and L-Leu-D,L-PT, its dipeptide derivative, as well as with the corresponding phosphonic analogues of glutamate (i.e., Glu-γ-P5 and L-Leu-D,L-Glu-γ-P5; Fig. 1b). Our data suggest that the incorporation of Glu-γ-PH in a dipeptide significantly improves the penetration of the molecule, thus enhancing its antibacterial activity and potential use for treating bacterial infections caused by different microorganisms. This work represents another piece of evidence that phosphinic compounds can be regarded as interesting molecules with antibacterial activity, as recently proposed for the PT-derived dipeptide on multidrug resistant clinical isolates of K. pneumoniae [14].
2. Materials and Methods
2.1. Materials
D,L-Glu-γ-PH was synthesized as described in [19]; N-(benzyloxycarbonyl)-L-leucine N-hydroxysuccinimide ester (Z-L-Leu-OSu) was prepared according to [20] and was recrystallized from i-PrOH before use. L-2-Amino-4-phosphonobutyric acid (L-AP4) was obtained from Santa Cruz Biotechnology and was recrystallized from H2O-EtOH before use; Amoxicillin (2.0 μg per disk) – from Becton, Dickinson & Co (USA).
Agar agar powder No. 1 for bacteriology was from LobaChemie. All other reagents, salts and solvents were of the highest purity and used as supplied by Sigma-Aldrich and Acros.
TLC was carried out on plastic sheet Cellulose F254 (Merck, Germany) in i-PrOH‒25% NH4OH‒H2O = 7:1:2. L-Leu-D,L-Glu-γ-PH and L-Leu-D,L-Glu-γ-P5 were detected on TLC plates following staining with ninhydrin (0.4% in acetone).
Ion-exchange chromatography was carried out on Dowex 50WX8, H+-form, 100-200 mesh (BioRad) using water for elution.
NMR spectra were recorded on a Bruker AM-300 (300.13 MHz for 1H, 75.43 MHz for 13C, and 121.44 MHz for 31P) using D2O as a solvent with sodium 3-trimethyl-1-propanesulfonate (DSS) as internal, or 85% H3PO4 as external standards. Chemical shifts are given in parts per million (ppm), the letter “J” indicates spin-spin coupling constants which are given in Hertz (Hz).
2.2. Synthesis of L-Leucyl-D,L-Glu-γ-PH
To a solution containing D,L-Glu-γ-PH (500 mg, 3.0 mmol), NaHCO3 (127 mg, 1.5 mmol), Na2CO3 (158 mg, 1.5 mmol) in 1.0 M NaOH (6 mL), water (1 mL), and 1,2-dimethoxyethane (1 mL), a solution of N-Cbz-L-Leu-OSu (1.08 g, 3.0 mmol) in 1,2-dimethoxyethane (5 mL) was added, and the reaction mixture was stirred overnight at 20°C. The reaction mixture was concentrated in vacuo, the residue was then dissolved in water (15 mL), acidified with 37% HCl to pH=1.0, and the separated oil was extracted with EtOAc (3 x 7 mL). The combined EtOAc extracts were washed with water (3.0 mL), brine (2 x 5 mL) and dried (MgSO4). The solvent was removed in vacuo and the residue was dried in vacuo at 1.0 Torr at 40°C for 1 h. The obtained foam was dissolved in glacial AcOH (3 mL), then anisole (0.2 mL) and 35% HBr/AcOH (2.2 mL) were added. The reaction mixture was incubated at 20°C for 1.5 h (until the end of the evolution of CO2), pooled into abs. Et2O (60 mL) and left overnight at -20°C. Solvents were decantated, the residual oil was co-evaporated in vacuo with water (2 x 10 mL), the residue was dissolved in water (10 mL) and applied on a Dowex 50WX8 column (V= 12 mL). Column was eluted with water (600 mL), collecting 10 mL fractions, and then ninhydrin-positive fractions (from 15 to 50) were combined, evaporated to dryness in vacuo, and the residue was dried in vacuo over P2O5 to give L-Leu-D,L-Glu-γ-PH (640 mg, yield 76% for two steps) as a colorless solid, Rf 0.66. 1H NMR (300.13 MHz, D2O): δ = 7.02 (dm, 1H, 1JHP 514.1 Hz, H-P), 4.57-4.47 (m, 1H, CH-COOH), 4.17-4.09 (m, 1H, CH-NH2), 2.24-2.12 (m, 1H, CHa-P), 2.09-1.95 (m, 1H, CHb-P), 1.90-1.62 (m, 5H, CH2-CH2-P, CH2-CH-NH2, CH-(CH3)2), 1.10-1.00 (m, 6H, CH-(CH3)2). 13C NMR (75.43 MHz, D2O): δ = 177.81 and 177.31 (2×s, COOH), 173.31 and 173.20 (2×s, CONH), 56.52 and 56.26 (2×d, 3JPC 16.5 Hz and 3JPC 16.4 Hz, CH-COOH), 54.79 and 54.61 (2×s, CH-NH2), 42.59 (s, CH2-CH-NH2), 30.25 and 30.05 (2×d, 1JPC 89.3 Hz, 1JPC 89.4 Hz, CH2-P), 26.71 and 26.50 (2×s, CH2-CH2-P), 25.82 and 25.79 (2×s, CH-(CH3)2), 24.39 and 24.35 and 23.94 and 23.91 (4×s, CH3).31P NMR (121.44 MHz, D2O): δ = 29.34 and 29.17 (2×s). Symbol “×” indicate differences of the same signals and coupling constants of L,L- and L,D-diastereomers. HRMS (ESI-MS): found m/z 281.1261; calc. forC10H21N2O5P [M+H]+ 281.1266.
2.3. Synthesis of L-Leucyl-D,L-Glu-γ-P5
This dipeptide was prepared as described for L-Leu-D,L-Glu-γ-PH (see section 2.2) starting from D,L-AP4 (366 mg, 2.0 mmol) and N-Cbz-L-Leu-OSu (716 mg, 2.0 mmol) in H2O-1,2-dimethoxyethane mixture. After the deprotection of the crude N-Cbz-dipeptide with 35% HBr/AcOH and the removal of the access of HBr/AcOH as descibed in section 2.2, the residue was dissolved in H2O (10 mL) and applied on a Dowex 50WX8 column (V= 12 mL). Column was eluted with water (700 mL), collecting 10 mL fractions, and then ninhydrin-positive fractions (from 25 to 60) were combined, evaporated to dryness in vacuo and the residue was dried in vacuo over P2O5 to afford L-Leucyl-D,L-Glu-γ-P5 (320 mg, yield 54% for two steps): Rf 0.29. 1Н NMR (300.13 MHz, D2O): δ = 4.44-4.32 (m, 1H, CH-COOH), 4.00 (dd, 1H, 3JHHa 7.5 & 7.4 Hz, 3JHHb 7.4 & 6.7 Hz, CH-NH2), 2.15-2.01 (m, 1H, CHa-P), 2.00-1.85 (m, 1H, CHb-P), 1.77-1.54 (m, 5H, CH2-CH2-P, CH2-CH-NH2, CH-(CH3)2), 0.98-0.85 (m, 6H, CH-(CH3)2). 13С NMR (75.43 MHz, D2O): δ = 178.12 and 177.55 (2×s, COOH), 173.30 and 173.19 (2×s, CONH), 56.84 and 56.50 (2×d, 3JPC 17.5 Hz and 3JPC 17.3 Hz, CH-COOH), 54.81 and 54.62 (2×s, CH-NH2), 42.58 (s, CH2-CH-NH2), 27.90 and 27.88 (2×s, CH-(CH3)2), 26.94 and 26.68 (2×d, 1JPC 134.5 Hz and 1JPC 134.7 Hz, CH2-P), 26.70 and 26.50 (2×s, CH2-CH2-P), 24.39 and 24.35 and 23.93 and 23.91 (4×s, CH3). 31P NMR (121.44 MHz, D2O): δ = 24.93. HRMS (ESI-MS): found m/z 297.1210; calc. for C10H21N2O6P [M+H]+ 297.1215.
2.4. The microdilution method to determine the antimicrobial activity of tested compounds against Escherichia coli
The minimum inhibitory concentration able to inhibit 90% (MIC90) of the the growth of the bacterial population of the test strain E. coli K12 MG1655 was calculated using the broth microdilution method in the minimal medium EG containing MgSO4•7H2O (0.2 g), citric acid•H2O (2.0 g), anhydrous K2HPO4 (10.0 g), NaNH4HPO4•H2O (3.5 g), and glucose (4.0 g), milliQ water (1.0 L), at final pH 7.0 as described elsewhere [18]. Briefly,overnight cultures (2 mL) of E. coli K12 strain MG1655 grown in LB (lysigeny broth) medium were centrifuged at 3500 rpm for 15 min at 15°C and the bacterial cellular pellets resuspended in an isovolume of saline solution (9 g/L NaCl). The OD600 then brought to 1.0. The resuspension of the bacterial cells was used to inoculated (1:25) 2 mL of fresh minimal medium EG and bacteria allowed to grow for 6-7 hours at 37°C from a starting OD600 = 0.04 to a final OD600 = 0.5 (corresponding to 2.5 x 108colony forming units, cfu/mL), then diluted (1:25) to a final OD600 = 0.02 (corresponding to 1.0 x 107 cfu/mL) in the same minimal medium. This dilution was the one used to set up the 96-well microplate containing geometrically increasing concentration of the compounds to be tested. In the microplate, 20 µL of bacterial culture (OD600 = 0.02) were added to a final volume of 200 µL. Thus, a 1:10 dilution was made and the starting OD600 in the microplate reader was 0.002, corresponding toa number of cfu/mL at time zero, as assessed by plating on LB-agar, between 0.5-1.0 x 106/mL, which corresponds to the optimum starting number of cfu/mL to perform a MIC experiment. The microplate was incubated at 37°C for 24 hours in the microplate reader Varioskan Lux (Thermo Scientific). The OD600 was recorded automatically every hour. Before each reading, the microplate was set to shake vigorously 10 seconds, to ensure an even distribution of the bacteria in solution. MIC90 was calculated at 22 hour from the inoculum using the equation: % inhibition = [1-(OD600treated/OD600untreated)]×100.
Unless otherwise specified, all the tested compounds were dissolved in Milli-Q water, pH-adjusted to 7-8 by adding 5.0 N NaOH, filtered, dispensed in aliquots and stored at -20°C.
2.5. The agar diffusion method to analyze antimicrobial activity of L-Leucyl-D,L-Glu-γ-PH against Bacillus subtilis АТСС 6633
Different amounts of L-Leu-D,L-Glu-γ-PH were applied to paper disks, the discs were dried in air and placed on the surface of an agar medium for B. subtilis[21], containing Gibco potato starch (25.0 g), glycerol (2.5 g), L-Asp (2.0 g), D,L-Met (0.4 g), K2HPO4 (6.0 g), KH2PO4 (2.0 g), NH4Cl (1.0 g), NH4NO3 (0.2 g), Na2SO4 (0.2 g), MgSO4•7H2O (0.04 g), MnSO4•4H2O (0.002 g), FeSO4•7H2O (0.002 g), CaCl2 (0.001 g), agar (15 g), and milliQ water (1.0 L), final pH 6.8, with a seeded lawn of B. subtilis АТСС 6633 strain with a seeding density of 106 bacteria per 1 cm2 of agar surface. Dishes were incubated for 20 h at 37℃. Disk with Amoxicillin (2.0 μg per disk) was used as a control. The antibiotic activity was determined by agar diffusion method based on the presence and size of non-growth zones around the disks [22].
3. Results
3.1. Synthesis of L-Leu-D,L-Glu-γ-PH and L-Leu-D,L-Glu-γ-P5
L-Leu-D,L-Glu-γ-PH and L-Leu-D,L-Glu-γ-P5 were synthesized by the condensation of N-hydroxysuccinimide ester of the N-Cbz-L-Leu (N-Cbz-L-Leu-OSu) with Glu-γ-PH or Glu-γ-P5 in water/1,2-dimethoxyethane mixture in the presence of NaHCO3/Na2CO3 followed by one-pot removal of Cbz-protecting group with HBr/AcOH (Scheme 1). Each dipeptide was isolated by ion-exchange chromatography on sulfocationite Dowex-50X8 (H+-form), eluting the resin with a large volume of water. However, water elution did not allow to separate the diastereomeric dipeptides, in contrast to L-Leu-PT [14]. L-Leu-D,L-Glu-γ-PH was obtained with 76% overall yield, while L-Leu-D,L-Glu-γ-P5 – with 54% overall yield. Thus, compared with Glu-γ-P5, the H-phosphinic Glu-γ-PH proved to be more reactive. It should be noted that some hydrogen and carbon atoms of the L,D- and L,L-diastereomers of L-Leu-D,L-Glu-γ-PH and L-Leu-D,L-Glu-γ-P5 have different chemical shifts in the 1H- and 13C-NMR spectra (original spectra are provided in the Supplement). However, only for L-Leu-D,L-Glu-γ-PH but not for L-Leu-D,L-Glu-γ-P5 two characteristic signals corresponding to L,D- and L,L-diastereomers were detected in 31P-NMR spectra (original spectra are provided in the Supplement). This finding was unexpected and to exclude an artifact, we prepared L-Ala-D-Ala-P5 and L-Ala-L-Ala-P5 (description of the synthesis is presented in the Supplement) and compared their 1H- and 31P-NMR spectra. In this case, L,D- and L,L-diastereomers could be easily distinguished by 1H-NMR, but again only one signal was observed in the 31P-NMR spectrum (original spectra are depicted in the Supplement).
3.2. Continuous monitoring during MIC assays in minimal medium EG showed unusual growth behaviour in stationary phase
Since in our lab we started to use a new microplate reader, Varioskan Lux (Thermo Scientific), which allowed the concomitant incubation, growth and monitoring of the growth behaviour for 24 hours, we observed that the E. coli strain MG1655 was showing a growth profile resembling somehow a “diauxic growth” (Figure 2). This initially was not noticed because the monitoring was starting at the 13th hour and ending at the 20th hour, thus causing to miss the first peak at the 10th hour (indeed we were mostly observing a slight decline of the stationary phase OD600). Furthermore, previously the measurements were taken hourly by manually transferring the microplate from the incubator at 37°C to the microplate reader (Sunrise Tecan). All these technical differences and the more stable readings performed in the Variskan Luxmicroplate reader can explain why this growth behaviour had originally escaped our observation.
Basically, following a lag phase, the strain was exponentially growing and then entered the stationary phase after which a decrease of the OD600 was followed by a restart of the growth to attain an OD600 value close to that the first maximum. According to reports in the literature [23,24], E. coli MG1655 strain might display this behaviour in minimal medium as a consequence of a diauxic growth during which glucose (in this work 0.4%, corresponding to approx. 22 mM) is consumed first, followed by acetate consumption. Acetate in fact is produced and released (approx. 6 mM) in the medium by the bacteria starting from glucose, but is then used as carbon source at a later stage following glucose depletion [23,24]. In our opinion this explained why we observed a re-growth phase which anyway reached the stationary phase within few hours, being acetate concentration lower than that of glucose. Starting from this finding, we decided to calculate the MIC90 at 22 h in order to be sure to refer to a more stabilized OD600 in the untreated MG1655 strain, as exemplified in Figure 2b.
3.3. L-Leu-Glu-γ-PH is a more potent inhibitor of the growth of E. coli K12 than L-Leu-Glu-γ-P5.
It is known that aminophosphonic acids poorly penetrate in bacteria, if at all [9], and D,L-Glu-γ-P5 is not an exception, i.e, it did not inhibit the growth of E. coli in a chemically defined minimal medium when tested at in a range of concentrations up to 2.0 mg/mL (Table 1). On the contrary, D,L-Glu-γ-PH displayed already an excellent antimicrobial activity against E. coli under the same assay conditions (MIC90 20 µg/mL;Table 1; [18]). As reported previously, the L-isomer is almost twice as active as the racemic compound (Table 1; [18]), whereas the D-isomer is totally inactive under the testing conditions.
Notably, the dipeptide L-Leu-D,L-Glu-γ-PH was found to be 10 times more active than D,L-Glu-γ-PH against E. coli in the same medium, displaying a MIC90 2.0 µg/mL(Table 1). These differences in the activities of D,L-Glu-γ-PH and L-Leu-D,L-Glu-γ-PH can be most likely explained by the ability of the dipeptide to more easily penetrate into the cell via the dipeptide permease Dpp. Then, the cytoplasmic cleavage of L-Leu-D,L-Glu-γ-PH likely releases L-Glu-γ-PH, which can either be a substrate for some enzymes of glutamate metabolism, thereby producing a number of new biologically active phosphorus-containing compounds, or act as an inhibitor in some enzymatic transformations involving glutamate as a substrate (see Discussion).
When D,L-Glu-γ-P5 was tested, it did not display any antibacterial activity in our assay conditions, whereas its dipeptide derivative, L-Leu-D,L-Glu-γ-P5, exhibited antibacterial activity in minimal medium (MIC90 80 µg/mL, Table 1). This clearly indicated that also in the case of the phosphonic compound, its incorporation in a dipeptide with L-Leu improved its penetration in the microbial cell and made possible to exert an inhibitory effect on the growth, otherwise not datable, i.e. when using the free amino acid form.
It should be noted here that both L-Leu-D,L-Glu-γ-PH and L-Leu-D,L-Glu-γ-P5 were poorly effective when tested in liquid rich medium (Mueller-Hinton broth). In this medium, the MIC90 values for both dipeptides were >>2000 μg/mL (data not shown), which is in agreement with the known literature data on poor antibacterial activity of phosphonopeptides in rich medium, likely due to the large content of dipeptides and oligopeptides present in the growth medium [9]. Our results also show that although at high concentration, the dipeptides activity was somehow measurable, whereas that of the active compounds in the free amino acid form was not detectable at all (data not shown). This again points to an improved efficacy of the tested compounds when used as dipeptides.
Strikingly, the PT containing dipeptide, the detailed synthesis was recently published [14], was >1000 times more effective than racemate PT. This is in line with our previous study where we demonstrated that the L-isomer of PT was more potent on E. coli and multidrug resistant strains of K. pneumoniae when incorporated in a dipeptide with L-Leu [14].
3.4. L-Leu-D,L-Glu-γ-PH effectively inhibits the growth of Bacillus subtilis АТСС 6633.
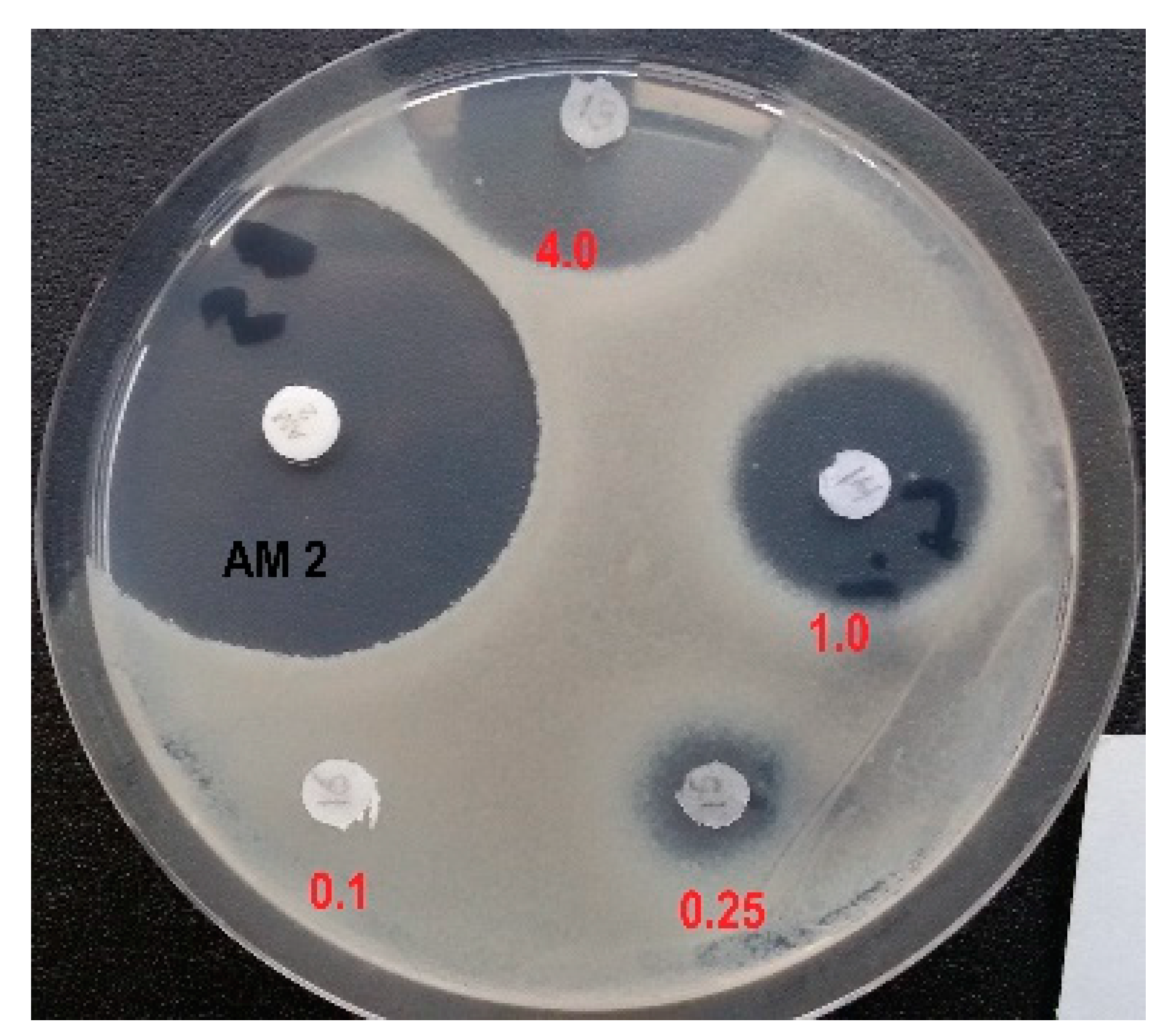
Next, we examined the effects of D,L-Glu-γ-PH and L-Leu-D,L-Glu-γ-PH on the growth of B. subtilis АТСС 6633 using the agar-diffusion test. Using this Gram-positive microorganism D,L-Glu-γ-PH was not active when used even at 100 µg/disk (data not shown). In striking contrast, the dipeptide L-Leu-D,L-Glu-γ-PH inhibited the growth of B. subtilis АТСС 6633 in a dose-dependent manner, starting from 0.25 µg/disk (Fig. 3). In agreement with the results of the previous paragraph, these differences might be attributed to the differences in the transport of D,L-Glu-γ-PH and L-Leu-D,L-Glu-γ-PH in bacteria. Alike in E. coli, the dipeptide most likely actively penetrates in B. subtilis АТСС 6633 using the dipeptide permease Dpp and releases L-Glu-γ-PH upon cleavage in the cytoplasm. When compared with Amoxicillin (2.0 μg/disk, Fig. 3), a β-lactam antibiotic that interferes with the synthesis of the peptidoglycan, the antibacterial activity of L-Leu-D,L-Glu-γ-PH was only slightly lower (4 µg/disk; Fig. 3). However, it is important to note here that the cytoplasmic cleavage of L-Leu-D,L-Glu-γ-PH releases both enantiomers in the cytoplasm, but only the L-enantiomer was reported to possess antimicrobial activity [18]. Therefore, the amount of active diastereomer L-Leu-L-Glu-γ-PH was calculated to be 2.0 μg/disk which confirmed that the dipeptide L-Leu-L-Glu-γ-PH exhibited an antimicrobial activity comparable to that of Amoxicillin.
Figure 3.
Inhibition of the growth of B. subtilisАТСС 6633with L-Leu-D,L-Glu-γ-PH (tested from 4.0 μg to 0.1 μg/disk) and Amoxicillin (2.0 μg/disk).
Figure 3.
Inhibition of the growth of B. subtilisАТСС 6633with L-Leu-D,L-Glu-γ-PH (tested from 4.0 μg to 0.1 μg/disk) and Amoxicillin (2.0 μg/disk).
4. Discussion
The substitution of the carboxyl group of amino acids with a phosphorus-containing group leads to two main families of analogues, the phosphinic and the phosphonic organophosphorous compounds. The latter -P(O)(OH)2 group has a tetrahedral spatial organization, with a double negative charged at neutral pH, unable to mimic the planar single-charged carboxyl group of the amino acids, as we already demonstrated by modelling studies [18]. In agreement, in most of the cases aminophosphonic acids are poor inhibitors of the enzymes of amino acid metabolism [25]. However, among the derivatives of aminophosphonic acids, i.e. esters, amides and the compounds with C-P-C backbone, which are stable mimics of tetrahedral intermediates (or reaction transition states) of the carboxyl group transformations, are not only potent enzyme inhibitors, but even commercial drugs [9,25]. Notable examples are the compounds listed in the Introduction (with some depicted in Fig. 1a) including PT which inhibits GS [10,11]. Another notable example is a peptidomimetic containing a phosphonate moiety in place of the peptide bond, Fosinopril®, which acts as an inhibitor of the zinc-dependent angiotensin converting enzyme and is used to treat hypertension [26].
On the contrary, the substitution of one hydroxyl group of aminophosphonic acids with a hydrogen atom eliminates one negative charge as well a bulky atom (i.e. oxygen) and confers to the H-phosphinic group a flattered tetrahedral geometry as suggested by crystallographic data for β-H-phosphinic analogue of aspartate [27]. Respectively, the H-phosphinic group can be considered as a bioisostere of the carboxyl group, that is confirmed by several substrate-like transformations of α-amino-H-phosphinic acids ([18] and ref. therein).
Demethylphosphinothricin, Glu-γ-PH (Fig. 1b), was first discovered and isolated from Streptomyces hygroscopicus and S. viridochromogenes [15] as a key intermediate of the biosynthetic pathway leading to commercial herbicide Bialaphos (a tripeptide containing phosphinothricin, i.e. PT, and two alanyl residues) [28]The key biochemical peculiarity of Glu-γ-PH is the presence of two pharmacophores in its molecule, i.e. the H-phosphinic group replacing the γ-carboxyl group and the α-amino acid moiety (Figure 4). This explains the ability of Glu-γ-PH to undergo some substrate-like transformations via α-amino acid moiety leading to metabolites containing unusual С-Р-Н bonds (see blue pathways in Figure 4), as recently demonstrated [18]. Some of the de novo synthesized metabolites may eventually be those responsible for the observed antibacterial activity.
It is known that L-Glu-γ-PH is a substrate of the PLP-dependent enzyme aspartate aminotransferase, giving rise to H-phosphinic analogue of α-ketoglutarate, i.e. 2-oxo-4-phosphinobutyric acid (hereafter α-KG-γ-PH, Figure 4, reaction 1) [29]. L-Glu-γ-PH is a substrate of E. coli GABA transaminase producing α-KG-γ-PH [30]. According to our preliminary observations (unpublished), α-KG-γ-PH is also formed in NAD-dependent glutamate dehydrogenase reaction (Fig. 4, reaction 2). As a consequence of one of the above reactions, α-KG-γ-PH may enter the TCA cycle and thereby cause antibacterial activity at some level by acting as an inhibitor of central metabolism. L-Glu-γ-PH was also found to be the substrate of the PLP-dependent enzyme glutamate decarboxylase from E. coli, yielding the H-phosphinic analogue of GABA, GABA-PH (Fig. 4, reaction 3), which then undergoes transamination by GABA-transaminase and the resulting 3-phosphinopropionic aldehyde (the H-phosphinic analogue of succinic semi-aldehyde) is then oxidized in a NAD-dependent reaction to the H-phosphinic analogue of succinate by succinic semialdehyde dehydrogenase [18]. Since the H-phosphinic group is a bioisostere of the carboxyl group, it cannot be excluded that L-Glu-γ-PH may be a substrate of glutamyl-tRNA synthetase (Fig. 4, reaction 4) with a subsequent formation of peptides carrying a few L-Glu-γ-PH residues (though a high incorporation of L-Glu-γ-PH is not expected to occur because of the competition with the much more abundant L-glutamate in the glutamyl-tRNA synthetase reaction). Finally, it is plausible that L-Glu-γ-PH will undergo PLP-dependent racemization to give D-Glu-γ-PH (Fig. 4, reaction 5), which will be not involved in the biosynthesis of peptidoglycan (murein), an important component of the bacterial cell wall, due to the different chemistry of the carboxyl and H-phosphinic groups.
When considering the antibacterial activity of L-Glu-γ-PH, the contribution of the second pharmacophore, the distal H-phosphinic group, must be taken into consideration. Transformations of γ-carboxyl group of glutamate lead to the formation of glutamine (nitrogen assimilation), glutathione (essential antioxidant), dihydrofolate (essential in one-carbon reactions), and are involved in the biosynthesis of proline and ornithine. All these reactions lead to the formation of γ-glutamyl phosphate, or γ-glutamyl adenylate, intermediate through ATP-dependent ligations, key steps in the biosynthesis of the above important metabolites. The intermediate formation of such activated L-Glu-γ-PH derivatives is in principle possible, since the H-phosphinic analogues of methionine and valine were substrates of the ATP-PPi exchange reaction catalyzed by Met- and Val-aminoacyl-tRNA synthetases [31]. However, the transfer of Met- and Val H-phosphinic analogues to the 3’-end of tRNA was not observed and is biochemically impossible because the enzymes are highly complementary to the tetrahedral transition state (intermediate compound) of carboxyl group, while the transition state of the H-phosphinic group is a trigonal bipyramid. These considerations a priori restrict substrate-like transformations of L-Glu-γ-PH via the H-phosphinic group (Fig. 4, pathways 6-10). Therefore, L-Glu-γ-PH may be expected to inhibit glutamine, glutathione, dihydrofolate, proline, and ornithine biosynthetic pathways, but it is difficult to predict how efficient this inhibition would be.
Based on the above, the antibacterial activity of L-Leu-D,L-Glu-γ-PH, which penetrates in bacteria via the peptidyl permease system and upon cleavage via peptidases, releases the antibacterial L-Glu-γ-PH, may be due either to biochemical transformation of its α-amino acid moiety (Fig. 3), giving new biologically active metabolites containing a C-P-H bond, or due to the restriction of metabolically significant transformations at the γ-position of this glutamate analogue – in this case L-Glu-γ-PH would acts as an inhibitor.
In this work we observed that phosphonic dipeptide L-Leu-D,L-Glu-γ-P5 was significantly less active against E. coli when compared to L-Leu-D,L-Glu-γ-PH (Table 1). It is possible that such a difference may be due to the differences in bioavailability, although phosphonopeptides are known to effectively penetrate bacteria using peptidyl permeases [9,30,32]. More p in our opinion is the assumption that these differences are more likely due to the inability of Glu-γ-P5, released in the cytosol after dipeptide cleavage, to undergo substrate-like transformations (reactions 1-5 in Fig. 3) that lead to new biologically active phosphonic compounds, unlike what is very likely to occur with L-Glu-γ-PH (Fig. 3). This is in line with the inability of D,L-Glu-γ-P5 to act as either a substrate or an inhibitor of E. coli glutamate decarboxylase [18,33] and porcine heart aspartate aminotransferase [29]. On the other hand, the phosphonic group is a doubly-charged tetrahedral group which mimic the tetrahedral intermediates (or reaction transition states) of the carboxyl group [9,25] and this explains the rather high competitive inhibition (KI 50 µM) of E. coli glutamine synthetase (GS) with Glu-γ-P5 [34]. However, PT (see Introduction and Fig. 1a), a naturally occurring inhibitor of GS, can undergo ATP-dependent ligation, with the formed pyrophosphonate that mimics the phosphorylated intermediate of glutamate occurring in the GS-catalyzed reaction [10,11]. Notably, PT has KI 0.6 µM against the E. coli GS [35]. These differences in the inhibitory activities of Glu-γ-P5 and PT may partly explain the differences observed in the activities of L-Leu-D,L-Glu-γ-P5 and L-Leu-D,L-PT against E. coli: the first dipeptide has a MIC90 = 80 µg/mL, while the second has a MIC90 = 0.1 µg/mL(Table 1).
5. Conclusions
Antibiotics are chemical substances used to treat bacterial infections in human and veterinary health. It is generally agreed that antibiotics discovery in the 20th century has revolutionized modern medicine by enabling the treatment of life-threating infectious diseases and allowing major advances in modern medicine, such as surgery and chemotherapy. In the last decades however the over- and mis-use of antibiotics, along with the lack of development and innovation in this field exacerbated the phenomenon of the emergence and spreading of antibiotic resistant bacteria. To avoid a return to the pre-antibiotic era, we urgently need original antibiotics with new pharmacophores and mechanisms of action.
Given the abundance and the many key roles played by glutamate in microbial metabolism, Glu-γ-PH may affect one or more of the metabolic pathways in which glutamate is involved. For this multi-target potential, which would significantly delay the appearance of resistance mechanisms, Glu-γ-PH is a new promising antibacterial. Notably, acute toxicity studies conducted with Glu-γ-PH on rats and mouse showed very low toxicity [36]. To the best of our knowledge, amino acids derivatives containing a γ-phosphinic group have never been tested as antibacterial. Our findings and the discussion with literature data strongly suggest that the antibacterial activity of L-Glu-γ-PH can likely be attributed to the formation of H-phosphinic intermediates through substrate-like transformations involving the α-amino acid moiety. The mechanism of action of L-Glu-γ-PH is still unknown and its antibacterial activity may be not conclusively established to arise from one of its pharmacophores, i.e. the α-carbon moiety and the γ-phosphinic moiety (Fig. 4). However, the present work provides evidence of H-phosphinic compounds are attractive molecules with antibacterial activity on novel metabolic target, with an important fallout on treating multidrug resistant pathogenic microorganisms.
6. Patents
A patent IT 102016000098005 has been granted, which includes L-Leu-Glu-γ-PH, one of the compound investigated in detail in this study. (https://www.uniroma1.it/en/brevetto/102016000098005 ).
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, D.D.B.,A.R.K,,O.V.E; methodology, M.A.K., F.G., L.O., M.V.D., B.F.V., A.I.S.; discussed data and gave conceptual advice, D.D.B., F.G., S.N.K.; writing, original draft preparation, D.D.B. O.V.E., A.R.K.; writing, review and editing – all the authors; supervision, O.V.E., D.D.B., A.R.K.; funding acquisition, A.R.K., D.D.B.
Funding
This research was funded by Russian Science Foundation (grant No 22-14-00291), and partially funded by the Sapienza University of Rome (Progetti Medi di Ateneo n. RM11916B861B9985 and RM120172B6587496). The APC was funded by RM120172B6587496.
Data Availability Statement
Additional data are available in Supplementary Materials. If not present there, they can be asked to the authors upon reasonable and motivated request.
Acknowledgments
This work is dedicated to the memory of Dr. Alessandra Occhialini, who passed away in August 2023.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ju, K.-S.; Gao, J.; Doroghazi, J.R.; Wang, K.-K.A.; Thibodeaux, C.J.; Li, S.; Metzger, E.; Fudala, J.; Su, J.; Zhang, J.K.; et al. Discovery of phosphonic acid natural products by mining the genomes of 10,000 actinomycetes. Proceedings of the National Academy of Sciences 2015, 112, 12175–12180. [Google Scholar] [CrossRef]
- Kayrouz, C.M.; Zhang, Y.; Pham, T.M.; Ju, K.-S. Genome Mining Reveals the Phosphonoalamide Natural Products and a New Route in Phosphonic Acid Biosynthesis. ACS Chemical Biology 2020, 15, 1921–1929. [Google Scholar] [CrossRef]
- Metcalf, W.W.; van der Donk, W.A. Biosynthesis of phosphonic and phosphinic acid natural products. Annual review of biochemistry 2009, 78, 65–94. [Google Scholar] [CrossRef]
- Peck, S.C.; Gao, J.; van der Donk, W.A. Discovery and biosynthesis of phosphonate and phosphinate natural products. Methods in enzymology 2012, 516, 101–123. [Google Scholar] [CrossRef]
- Seto, H.; Kuzuyama, T. Bioactive natural products with carbon-phosphorus bonds and their biosynthesis. Natural product reports 1999, 16, 589–596. [Google Scholar] [CrossRef]
- Shiraishi, T.; Kuzuyama, T. Biosynthetic pathways and enzymes involved in the production of phosphonic acid natural products. Bioscience, Biotechnology, and Biochemistry 2021, 85, 42–52. [Google Scholar] [CrossRef]
- Song, B.-C. Switch to tenofovir-based therapy or to continue adefovir-based therapy in CHB patients with suboptimal response to adefovir-based combination? Clin Mol Hepatol 2016, 22, 439–442. [Google Scholar] [CrossRef]
- Circello, B.T.; Miller, C.G.; Lee, J.H.; van der Donk, W.A.; Metcalf, W.W. The antibiotic dehydrophos is converted to a toxic pyruvate analog by peptide bond cleavage in Salmonella enterica. Antimicrobial agents and chemotherapy 2011, 55, 3357–3362. [Google Scholar] [CrossRef]
- Kafarski, P. Phosphonopeptides containing free phosphonic groups: recent advances. RSC advances 2020, 10, 25898–25910. [Google Scholar] [CrossRef]
- Abell, L.M.; Villafranca, J.J. Investigation of the mechanism of phosphinothricin inactivation of Escherichia coli glutamine synthetase using rapid quench kinetic technique. Biochemistry 1991, 30, 6135–6141. [Google Scholar] [CrossRef]
- Gill, H.S.; Eisenberg, D. The crystal structure of phosphinothricin in the active site of glutamine synthetase illuminates the mechanism of enzymatic inhibition. Biochemistry 2001, 40, 1903–1912. [Google Scholar] [CrossRef]
- Omura, S.; Hinotozawa, K.; Imamura, N.; Murata, M. The structure of phosalacine, a new herbicidal antibiotic containing phosphinothricin. The Journal of antibiotics 1984, 37, 939–940. [Google Scholar] [CrossRef]
- Omura, S.; Murata, M.; Hanaki, H.; Hinotozawa, K.; Oiwa, R.; Tanaka, H. Phosalacine, a new herbicidal antibiotic containing phosphinothricin. Fermentation, isolation, biological activity and mechanism of action. The Journal of antibiotics 1984, 37, 829–835. [Google Scholar] [CrossRef]
- Demiankova, M.V.; Giovannercole, F.; Khomutov, M.A.; Salikhov, A.I.; Onillon, L.; Valuev-Elliston, V.T.; Vasilieva, B.F.; Khurs, E.N.; Gabrielyan, N.I.; Kochetkov, S.N.; et al. Antibacterial Activity of Peptide Derivatives of Phosphinothricin against Multidrug-Resistant Klebsiella pneumoniae. Molecules 2023, 28. [Google Scholar] [CrossRef]
- Seto, H.; Sasaki, T.; Imai, S.; Tsuruoka, T.; Ogawa, H.; Satoh, A.; Inouye, S.; Niida, T.; Otake, N. Studies on the biosynthesis of bialaphos (SF-1293). 2. Isolation of the first natural products with a C-P-H bond and their involvement in the C-P-C bond formation. The Journal of antibiotics 1983, 36, 96–98. [Google Scholar] [CrossRef]
- Imai, S.; Seto, H.; Sasaki, T.; Tsuruoka, T.; Ogawa, H.; Satoh, A.; Inouye, S.; Niida, T.; Otake, N. Studies on the biosynthesis of bialaphos (SF-1293). 6. Production of N-acetyl-demethylphosphinothricin and N-acetylbialaphos by blocked mutants of Streptomyces hygroscopicus SF-1293 and their roles in the biosynthesis of bialaphos. The Journal of antibiotics 1985, 38, 687–690. [Google Scholar] [CrossRef]
- Thompson, C.J.; Movva, N.R.; Tizard, R.; Crameri, R.; Davies, J.E.; Lauwereys, M.; Botterman, J. Characterization of the herbicide-resistance gene bar from Streptomyces hygroscopicus. The EMBO journal 1987, 6, 2519–2523. [Google Scholar] [CrossRef]
- De Biase, D.; Cappadocio, F.; Pennacchietti, E.; Giovannercole, F.; Coluccia, A.; Vepsalainen, J.; Khomutov, A. Enzymatic kinetic resolution of desmethylphosphinothricin indicates that phosphinic group is a bioisostere of carboxyl group. Communications chemistry 2020, 3, 121. [Google Scholar] [CrossRef]
- Khomutov, M.A.; Formanovsky, A.A.; Mikhura, I.V.; Vepsalainen, J.; Kochetkov, S.N.; De Biase, D.; Khomutov, A.R. Convenient syntheses of phosphinic analogues of γ-aminobutyric- and glutamic acids. Russian Journal of Bioorganic Chemistry 2016, 42, 672–676. [Google Scholar] [CrossRef]
- Anderson, G.W.; Zimmerman, J.E.; Callahan, F.M. N-Hydroxysuccinimide Esters in Peptide Synthesis. Journal of the American Chemical Society 1963, 85, 3039–3039. [Google Scholar] [CrossRef]
- Fang, A.; Demain, A.L. A new chemically-defined medium for RAC-certified and other strains of Bacillus subtilis. Applied Microbiology and Biotechnology 1989, 30, 144–147. [Google Scholar] [CrossRef]
- Testing, E.C.o.A.S. Antimicrobial susceptibility testing - EUCAST disk diffusion method. version 11.0 2023. [Google Scholar]
- Enjalbert, B.; Letisse, F.; Portais, J.C. Physiological and Molecular Timing of the Glucose to Acetate Transition in Escherichia coli. Metabolites 2013, 3, 820–837. [Google Scholar] [CrossRef]
- Mahadevan, R.; Edwards, J.S.; Doyle, F.J. , 3rd. Dynamic flux balance analysis of diauxic growth in Escherichia coli. Biophysical journal 2002, 83, 1331–1340. [Google Scholar] [CrossRef]
- Horsman, G.P.; Zechel, D.L. Phosphonate Biochemistry. Chemical reviews 2017, 117, 5704–5783. [Google Scholar] [CrossRef]
- Pilote, L.; Abrahamowicz, M.; Eisenberg, M.; Humphries, K.; Behlouli, H.; Tu, J.V. Effect of different angiotensin-converting-enzyme inhibitors on mortality among elderly patients with congestive heart failure. CMAJ : Canadian Medical Association journal = journal de l'Association medicale canadienne 2008, 178, 1303–1311. [Google Scholar] [CrossRef]
- Schwalbe, C.H.W.; Freeman, S.; DasGupta, M. 2-Amino-2-carboxyethylphosphinic acid monohydrate. Acta Crystallographica Section C 1993, 49, 1826–1828. [Google Scholar] [CrossRef]
- Blodgett, J.A.; Thomas, P.M.; Li, G.; Velasquez, J.E.; van der Donk, W.A.; Kelleher, N.L.; Metcalf, W.W. Unusual transformations in the biosynthesis of the antibiotic phosphinothricin tripeptide. Nature chemical biology 2007, 3, 480–485. [Google Scholar] [CrossRef]
- Khurs, E.N.; Osipova, T.I.; Khomutov, R.M. [Enzyme reamination of phosphororganic analogs of aspartate and glutamate]. Bioorganicheskaia khimiia 1989, 15, 552–555. [Google Scholar]
- Schulz, A.; Taggeselle, P.; Tripier, D.; Bartsch, K. Stereospecific production of the herbicide phosphinothricin (glufosinate) by transamination: isolation and characterization of a phosphinothricin-specific transaminase from Escherichia coli. Applied and environmental microbiology 1990, 56, 1–6. [Google Scholar] [CrossRef]
- Biryukov, A.I.; Osipova, T.I.; Khomutov, R.M. alpha-Aminophosphonous acids: the substrates of ATP--PPi exchange reaction, catalysed by aminoacyl-tRNA synthetases. FEBS letters 1978, 91, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.G.; Atherton, F.R.; Hall, M.J.; Hassall, C.H.; Holmes, S.W.; Lambert, R.W.; Nisbet, L.J.; Ringrose, P.S. Phosphonopeptides, a new class of synthetic antibacterial agents. Nature 1978, 272, 56–58. [Google Scholar] [CrossRef] [PubMed]
- Lacoste, A.M.; Mansour, S.; Cassaigne, A.; Neuzil, E. Effect of phosphonic analogues of glutamic acid on glutamate decarboxylase. Experientia 1985, 41, 643–644. [Google Scholar] [CrossRef]
- Meek, T.D.; Villafranca, J.J. Kinetic mechanism of Escherichia coli glutamine synthetase. Biochemistry 1980, 19, 5513–5519. [Google Scholar] [CrossRef]
- Logusch, E.W.; Walker, D.M.; McDonald, J.F.; Franz, J.E.; Villafranca, J.J.; DiIanni, C.L.; Colanduoni, J.A.; Li, B.; Schineller, J.B. Inhibition of Escherichia coli glutamine synthetase by alpha- and gamma-substituted phosphinothricins. Biochemistry 1990, 29, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Yonaga, T.; Akabori, Y.; Fujino, Y. Acute Toxicity Studies of MP101. Pharmacology and therapy (Japan) 1982, 10, 43–51. [Google Scholar]
Figure 1.
Chemical structures of molecules containing phosphonic and phosphinic groups. (a) Notable examples of molecules containing phosphonic and phosphinic groups (in blue) with a pharmacological activity. Only phoshinothricin (PT) belongs to the class of the phosphinic compounds, less frequently present in nature. (b) Chemical structures of H-phosphinic (PH) and phosphonic (P5) analogues of glutamate studied here, Glu-γ-PH and Glu-γ-P5, respectively, and, below them, the two corresponding leucine-containing dipeptides.
Figure 1.
Chemical structures of molecules containing phosphonic and phosphinic groups. (a) Notable examples of molecules containing phosphonic and phosphinic groups (in blue) with a pharmacological activity. Only phoshinothricin (PT) belongs to the class of the phosphinic compounds, less frequently present in nature. (b) Chemical structures of H-phosphinic (PH) and phosphonic (P5) analogues of glutamate studied here, Glu-γ-PH and Glu-γ-P5, respectively, and, below them, the two corresponding leucine-containing dipeptides.
Scheme 1.
i- Cbz-L-Leu-OSu/1,2-dimethoxyethane/H2O/NaHCO3; ii- HBr/АсОН; iii- Dowex 50X8 (H+), elution with H2O.
Scheme 1.
i- Cbz-L-Leu-OSu/1,2-dimethoxyethane/H2O/NaHCO3; ii- HBr/АсОН; iii- Dowex 50X8 (H+), elution with H2O.
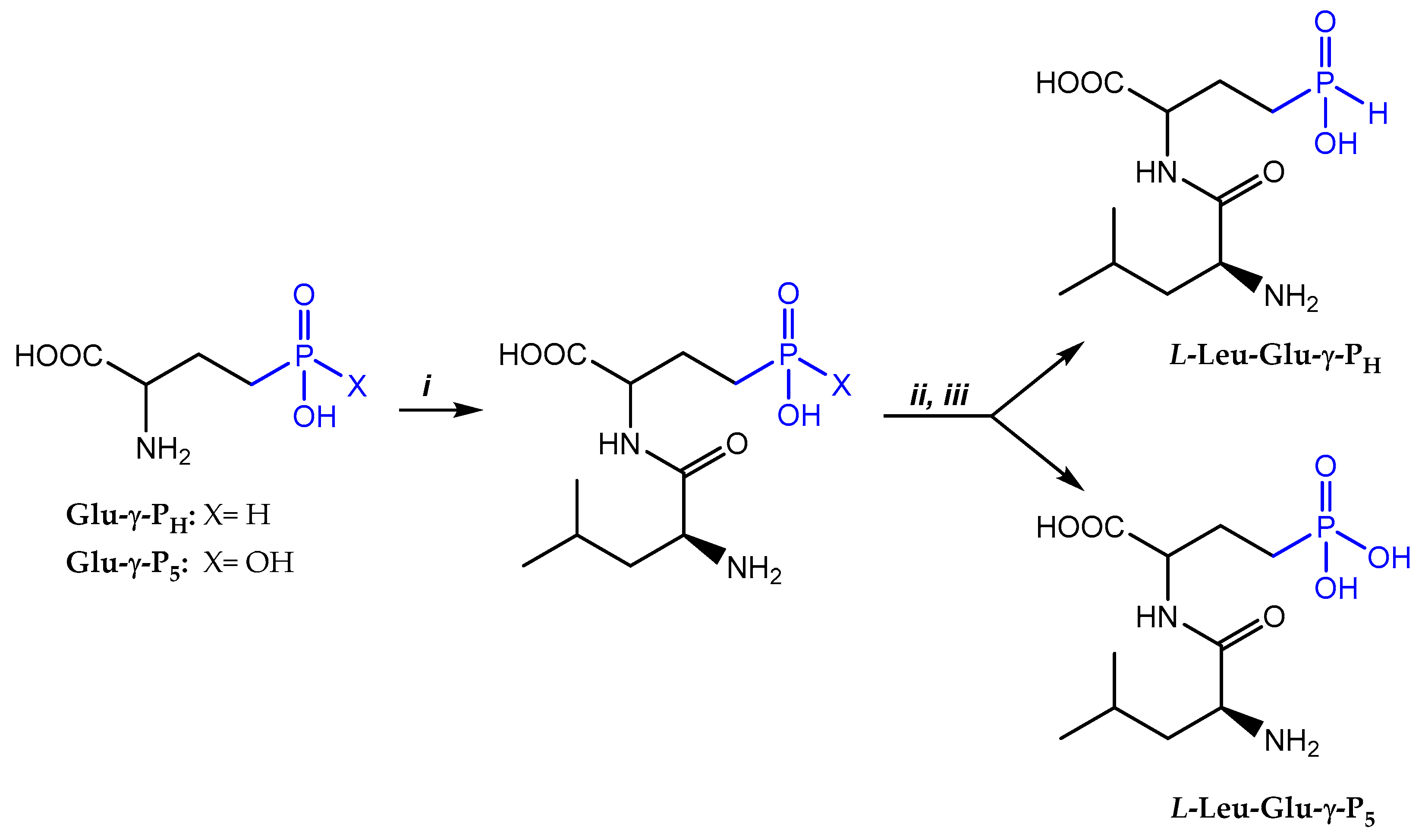
Figure 2.
Growth of E. coli MG1655 in chemically-defined EG medium. (a) curve obtained by averaging 25 growth curves of the control condition (i.e. MG1655 in the absence of treatment); (b) example of the experiments (three biological replicates) with one of the tested dipeptides (see 3.3). Data are shown as mean ± sd.
Figure 2.
Growth of E. coli MG1655 in chemically-defined EG medium. (a) curve obtained by averaging 25 growth curves of the control condition (i.e. MG1655 in the absence of treatment); (b) example of the experiments (three biological replicates) with one of the tested dipeptides (see 3.3). Data are shown as mean ± sd.
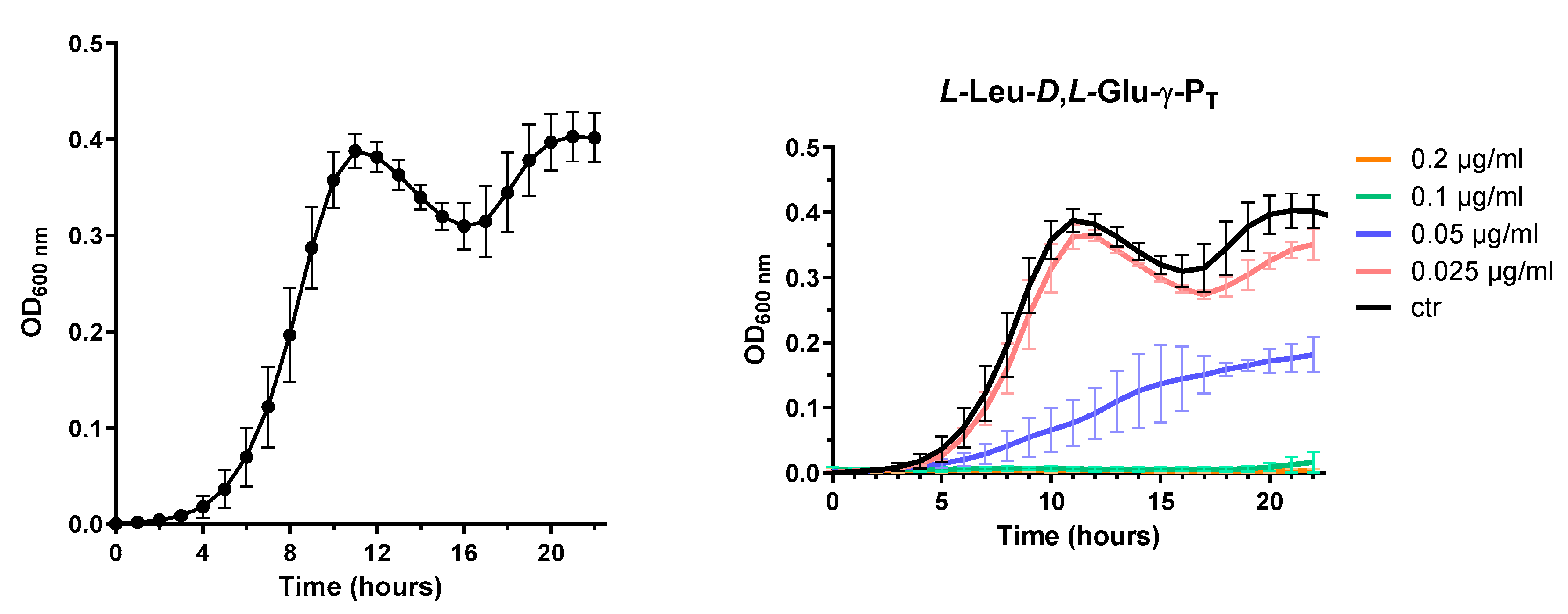
Figure 4.
Some enzymatic transformations of L-Glu which are potentially affected by L-Glu-γ-PH. The two pharmacophores(C-α and phosphinic group) are delimited by an open circle. Substrate-like transformations of L-Glu-γ-PH (in blue): 1) formation of 2-oxo-4-phosphinobutyric acid (α-KG-γ-PH) via PLP-dependent transamination (L-Glu-γ-PH as amino group donor); 2) formation of α-KG-γ-PH in glutamate dehydrogenase reaction (oxidative deamination); 3) H+-dependent decarboxylation of L-Glu-γ-PH, yielding H-phosphinic analogue of GABA; 4) formation of L-Glu-γ-PH-tRNAGlu in glutamyl-tRNA synthetase reaction; 5) PLP-dependent racemization of L-Glu-γ-PH yielding D-Glu-γ-PH. Reactions in which L-Glu-γ-PH cannot be a substrate, but may be an inhibitor (in red): 6) biosynthesis of glutamine; 7) proline; 8) ornithine; 9) glutathione; and 10) dihydrofolate. Reactions 1) and 2) can be reversible. Transformations 6)-10) include the step of ATP-dependent ligation of Glu and subsequent transformations of the glutamyl phosphate into the product(s) that is biochemically impossible for L-Glu-γ-PH.
Figure 4.
Some enzymatic transformations of L-Glu which are potentially affected by L-Glu-γ-PH. The two pharmacophores(C-α and phosphinic group) are delimited by an open circle. Substrate-like transformations of L-Glu-γ-PH (in blue): 1) formation of 2-oxo-4-phosphinobutyric acid (α-KG-γ-PH) via PLP-dependent transamination (L-Glu-γ-PH as amino group donor); 2) formation of α-KG-γ-PH in glutamate dehydrogenase reaction (oxidative deamination); 3) H+-dependent decarboxylation of L-Glu-γ-PH, yielding H-phosphinic analogue of GABA; 4) formation of L-Glu-γ-PH-tRNAGlu in glutamyl-tRNA synthetase reaction; 5) PLP-dependent racemization of L-Glu-γ-PH yielding D-Glu-γ-PH. Reactions in which L-Glu-γ-PH cannot be a substrate, but may be an inhibitor (in red): 6) biosynthesis of glutamine; 7) proline; 8) ornithine; 9) glutathione; and 10) dihydrofolate. Reactions 1) and 2) can be reversible. Transformations 6)-10) include the step of ATP-dependent ligation of Glu and subsequent transformations of the glutamyl phosphate into the product(s) that is biochemically impossible for L-Glu-γ-PH.
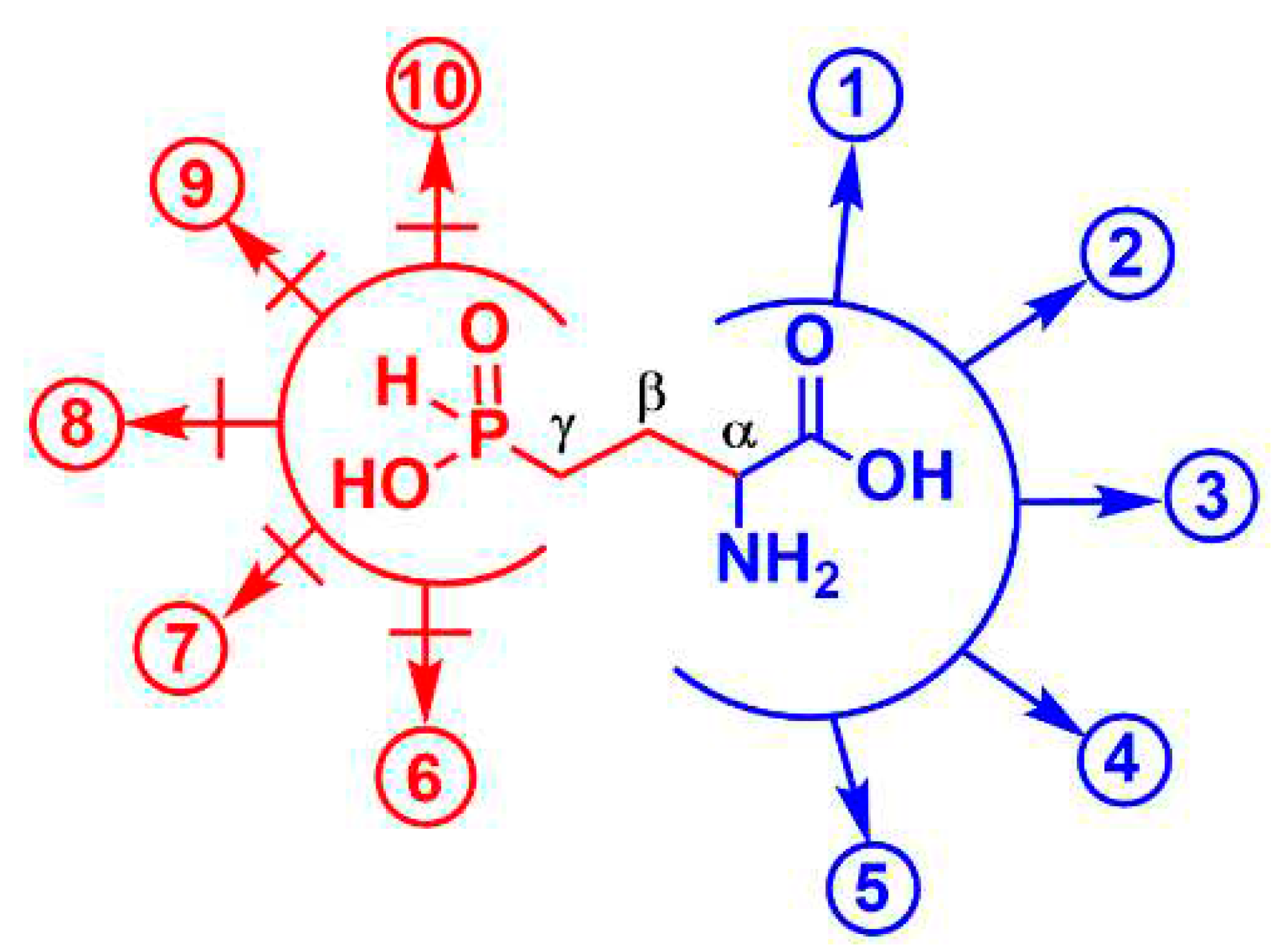

Table 1.
Minimum Inhibitory Concentration (MIC) of the compounds tested on E. coli K12 MG1655 determined by broth microdilution method in chemically-defined minimal medium (EG).
Table 1.
Minimum Inhibitory Concentration (MIC) of the compounds tested on E. coli K12 MG1655 determined by broth microdilution method in chemically-defined minimal medium (EG).
| Compound | MIC90 (µg/mL) | |
|---|---|---|
| Amino acids | D,L-Glu-γ-PH | 20 |
| D,L-PT | 266-3041 | |
| D,L-Glu-γ-P5 | n.d.2 | |
| Dipeptides | L-Leu-D,L-Glu-γ-PH | 2 |
| L-Leu-D,L-PT | 0.1 | |
| L-Leu-D,L-Glu-γ-P5 | 80 | |
1 from [14]; 2 n.d. not detectable when assayed at up to 2 mg/mL.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



