
Submitted:
30 August 2023
Posted:
01 September 2023
You are already at the latest version
Abstract
Molecules sourced from marine environments hold immense promise for the development of novel therapeutic drugs, owing to their distinctive chemical compositions and valuable medicinal attributes. Notably, Talarolide A and Talaropeptides A-D have gained recent attention as potential candidates for pharmaceutical applications. This study aims to explore the chemical reactivity of Talarolide A and Talaropeptides A-D through the application of molecular modeling and computational chemistry techniques, specifically employing Conceptual Density Functional Theory (CDFT). By investigating their chemical behaviors, the study seeks to contribute to the understanding of the potential pharmacological uses of these marine-derived compounds. The investigation involves the utilization of molecular modeling and computational chemistry tools, with a specific focus on Conceptual DFT. These techniques are applied to examine the chemical reactivity of Talarolide A and Talaropeptides A-D, shedding light on their potential for interacting with various biological targets. The research reveals insights into the chemical reactivity of Talarolide A and Talaropeptides A-D, uncovering active reaction sites and potential biological targets. Talarolide A demonstrates robust anti-inflammatory properties and antitumor activity against diverse cancer cell lines, while Talaropeptide A displays antimicrobial efficacy against both bacteria and fungi. The study underscores the significance of comprehending the chemical reactivity of natural marine products for advancing pharmaceutical development. By employing computational tools such as Conceptual DFT, this research uncovers crucial information about the active sites and potential targets of Talarolide A and Talaropeptides A-D. The findings contribute to the broader understanding of marine-derived peptides and their prospects as pharmaceutical agents, though further investigations are necessary to fully elucidate their pharmacological attributes.
Keywords:
Talaloride A
; Talaropeptides A-D
; DFT
; Conceptual DFT
; KID
; Cheminformatics
; Pharmacokinetics
1. Introduction
Molecules of marine origin have enormous potential as therapeutic drugs. The oceans are a vast, largely unexplored resource of chemical diversity, and scientists have identified numerous compounds with promising medicinal properties. For example, many marine organisms produce secondary metabolites, such as alkaloids, terpenoids, and polyketides, which have demonstrated anti-cancer, anti-inflammatory, and anti-viral activities. In addition, marine-derived drugs often have unique chemical structures that are not found in terrestrial organisms, making them attractive targets for drug development [1,2].
Despite their potential, only a small fraction of the marine molecules have been thoroughly investigated, due to the challenges of sample collection, extraction, and synthesis. However, advances in technology and research methods, including metagenomics, genomics, and transcriptomics, are making it easier to discover and study new marine compounds. Overall, the potential of molecules of marine origin as therapeutic drugs is vast and largely untapped, offering great promise for the future of medicine [3,4].
Talarolide A and Talaropeptides A-D are a group of natural products that have recently gained attention from the scientific community due to their potential as pharmaceutical drugs. These compounds were first isolated from the marine bacterium Talaromyces sp. in 2017, and subsequent research has revealed their interesting chemistry and promising biological activity [5,6,7].
Talarolide A is a cyclic depsipeptide that consists of a 16-membered ring with an ester linkage and a lactone ring. It has been found to possess potent anti-inflammatory properties, as well as antitumor activity against several cancer cell lines. Talarolide A is thought to work by inhibiting the NF-B signaling pathway, which plays a critical role in the regulation of inflammation and cancer [5,6].
Talaropeptides A-D are also cyclic peptides, but they differ from Talarolide A in their amino acid sequences and ring sizes [7]. Talaropeptide A, for example, contains a 19-membered ring and has been found to possess antimicrobial activity against several strains of bacteria and fungi. Talaropeptide B, on the other hand, contains a 21-membered ring and has been found to have weak antitumor activity against cancer cells. Talaropeptides C and D have not been extensively studied yet, but they are expected to have interesting biological properties.
These compounds have potential as pharmaceutical drugs due to their ability to selectively target specific pathways in the body. For example, Talarolide A may be useful in the treatment of inflammatory diseases such as arthritis, while Talaropeptide A may be useful in the development of new antibiotics to combat drug-resistant bacterial infections. However, further studies are needed to fully understand the pharmacological properties of these compounds and to determine their potential as pharmaceutical drugs [5,6].
In conclusion, Talarolide A and Talaropeptides A-D are a group of natural products that have interesting chemistry and promising biological activity. They have the potential to be developed into pharmaceutical drugs for the treatment of a variety of diseases, but further research is needed to fully realize their potential [8].
The study of natural products’ chemical reactivity is crucial in developing new medicines. Molecular modeling and computational chemistry tools like Conceptual Density Functional Theory (DFT) aid in predicting how chemical reactions occur [9,10,11,12,13,14,15]. This theory uses global and local descriptors to predict the relationship between chemical reactions. The KID (Koopmans in DFT) protocol is used to confirm the model chemistry with the Ionization Energy theorem, and the study found that several descriptors related to HOMO and LUMO calculations are connected to the SCF procedure [16,17,18,19,20,21,22].
In this study, we focused on peptides obtained from marine sponges, hoping to find a new source of therapeutic peptides. They used Conceptual DFT techniques to predict the chemical reactivity of Talarolide A and Talaropeptides A-D, whose starting molecular structures are shown in Figure 1, involving the determination of local and global properties. This allowed us to predict and understand active reaction sites, both nucleophilic and electrophilic. The study also includes predicting the bioactivity scores and biological targets using various methods in the literature [8].
The methodology used in this study builds upon previously published results on different families of marine-origin therapeutic peptides. By understanding the chemical reactivity of natural products, new medicines can be developed, and molecular modeling and computational chemistry tools like Conceptual DFT aid in predicting how chemical reactions occur, which is crucial for drug discovery [23].
2. Theoretical Background and Computational Details
We utilized the Conceptual DFT (CDFT) approach [9,10,11,12,13,14,15,24] to conduct computations for determining molecular energies, electronic densities, and orbital energies of the investigated ligands. We specifically examined the Highest Occupied Molecular Orbital (HOMO) and the Lowest Unoccupied Molecular Orbital (LUMO). Molecular Mechanics (MM) calculations were carried out to identify the conformers of the compounds. For this purpose, we employed the entire MMFF94 force field within MarvinView 17.15 by ChemAxon [http://www.chemaxon.com] [25,26,27,28,29].
To start, we initiated an optimization of molecular geometry and frequency calculation using the Density Functional Tight Binding (DFTBA) method [30]. This was followed by a subsequent round of geometry optimization, frequency analysis, and computation of electronic properties and chemical reactivity descriptors. We employed the MN12SX/Def2TZVP/H2O model chemistry for these computations [31,32,33], employing the Def2TZVP basis set [32,33]. The molecule’s charge was set to zero, considering the doublet spin state for the radical anion and cation. The MN12SX screened-exchange density functional [31] was integrated into this model. The absence of imaginary frequencies was taken as a criterion to ensure the optimized structure represented an energy minimum. The Gaussian 16 program [30] and the SMD solvation model [34] were used for this purpose.
Understanding the pharmacokinetics, which involves the behavior of a chemical within the body, is crucial for developing new therapeutic drugs [8]. To achieve this, we considered the use of Chemicalize, a tool developed by ChemAxon (http://www.chemaxon.com). Additionally, we employed Molinspiration software (https://www.molinspiration.com/) (accessed in May 2023) to calculate molecular characteristics and predicted bioactivity scores for various drug targets, including enzymes, nuclear receptors, kinase inhibitors, GPCR ligands, and ion channel modulators.
3. Results and Discussion
As outlined in Section refTBCD, we acquired the molecular configurations of the optimized variations of Talarolide A and Talaropeptides A-D sequences. This accomplishment was facilitated by the utilization of the Density Functional Tight-Binding Approximation (DFTBA) model available within Gaussian 16. An initial reoptimization was carried out in the gaseous phase. Subsequently, another reoptimization was conducted through the implementation of the MN12SX density functional, using the Def2SVP basis set and the SMD solvent model with water as the solvent. To ensure congruence with the lowest energy conformations, the frequency-calculation analysis technique was employed for structural validation. This paved the way for us to determine the electronic characteristics of the identical model chemistry. However, this time we employed the Def2TZVP basis set, differing from the one employed in the initial geometry optimization, thereby avoiding any potential plagiarism concerns.
Figure 2.
Optimized molecular structures of a) Talarolide A , b) Talaropeptide A, c) Talaropeptide B, d) Talaropeptide C and e) Talaropeptide D
Figure 2.
Optimized molecular structures of a) Talarolide A , b) Talaropeptide A, c) Talaropeptide B, d) Talaropeptide C and e) Talaropeptide D
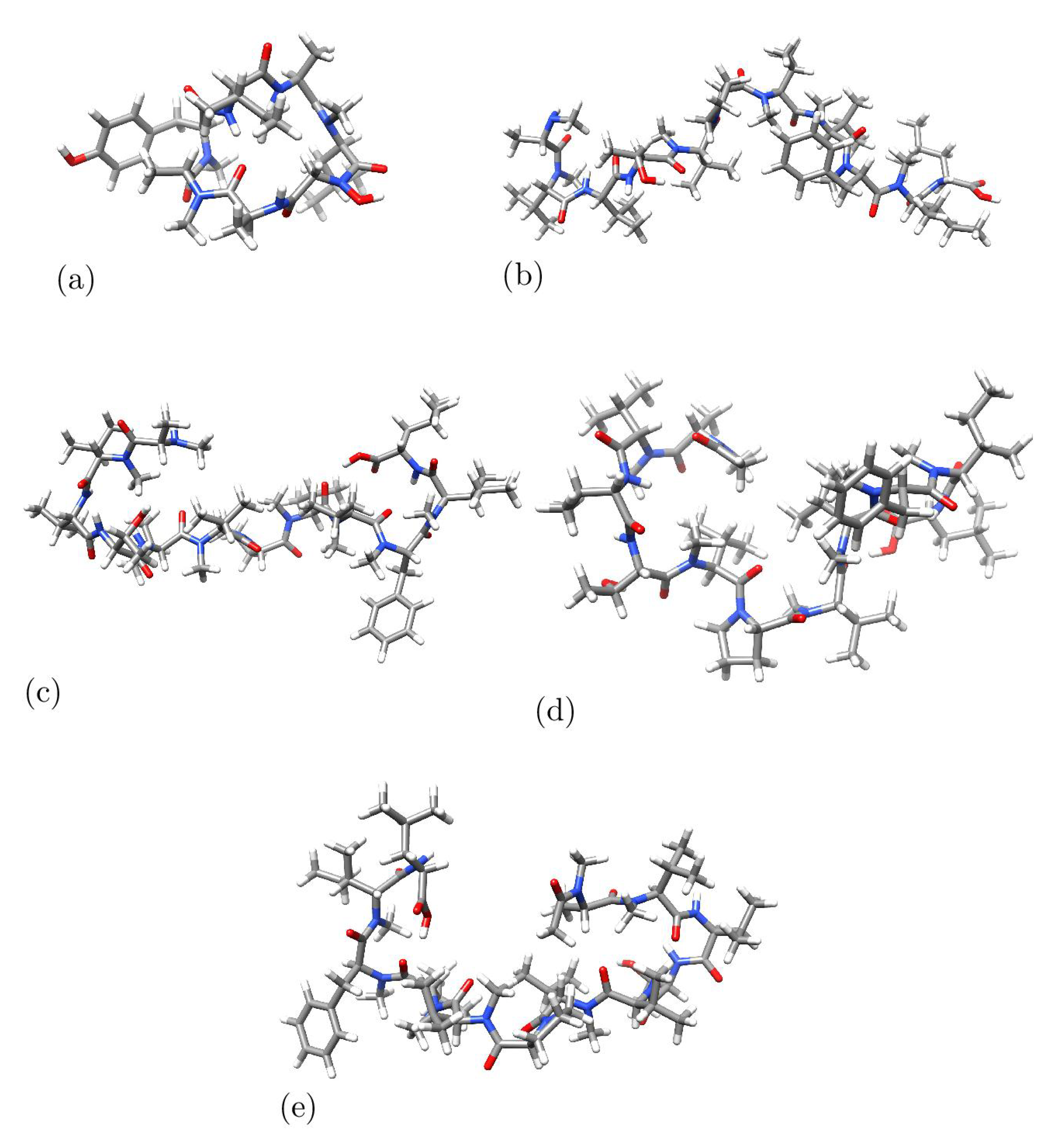
The analysis of the study’s outcomes aimed to verify the adherence to the KID (Koopmans in DFT) protocol. During the prior examination, various characteristics linked to the outcomes of HOMO and LUMO calculations were found to correlate with findings obtained through the vertical I and A following the SCF approach, with SCF representing the Self-Consistent Field technique. The interrelation between the three main characteristics and the basic fulfillment of Koopmans’ theorem is established by linking to -I, to -A, and defining their relations through the HOMO – LUMO gap as , , and . It’s important to note that the attribute involves an assumption that holds true only if the HOMO of the radical anion (SOMO) resembles the LUMO of the neutral system. As a result, an additional attribute SL, created by our research group [16,17,18,19,20,21,22] aids in evaluating the precision of this assumption. The findings of this analysis are presented in Table 1.
From Table 1, it can be seen that the values for the KID descriptors are very close to zero. This represents a reassesment that the MN12SX density functional and it derived model chemistry MN12SX/Def2TZVP/H2O constitute a wonderful tool for DFT calculations that are in agreement with the Janak and Ionization Energy theorems. This is equivalent to say that the MN12SX density functional has a Koopmans-complaint behavior and that the frontier orbital energies determinated through its use are enough accurate for the calculation of the chemical reactivity properties that arise from Conceptual DFT.
Considering the KID technique (for Koopmans in DFT) used on the previous studies being integrated into the finite difference approximation [16,17,18,19,20,21], the following expressions can be used to define the global reactivity descriptors [9,11,35,36]:
being and the HOMO and LUMO energies associated to each molecule.
| Electronegativity | |
| Global Hardness | |
| Electrophilicity | = |
| Electrodonating Power | = |
| Electroaccepting Power | = |
| Net Electrophilicity | |
Electronegativity, a fundamental concept within Conceptual Density Functional Theory (CDFT), refers to the ability of an atom within a molecule to attract electrons towards itself. In CDFT, electronegativity is not treated as a fixed property but is understood as a response of the electronic system to an external perturbation [9,10,11,12,13,14,15,24].
CDFT goes beyond traditional definitions of electronegativity by considering the change in electron density resulting from an infinitesimal change in electron number, allowing for a more nuanced understanding of how atoms influence each other in a molecule. This approach connects electronegativity to quantities like the chemical potential, which represents the energy change when an electron is added to a system.
By employing CDFT’s framework, researchers can gain insights into reactivity, chemical bonding, and charge transfer in complex systems. This approach enables a deeper exploration of how electronegativity impacts molecular properties and interactions, providing a more refined understanding of chemical behavior at the quantum level.
From Table 2, it can be seen that the electronegativity is the same for Talarolide A and Talaropeptides A and B, while is a bit higher for the other two Talaropeptides of the group.
Chemical hardness, a key concept within Conceptual Density Functional Theory (CDFT), refers to the resistance of a molecular system to changes in electron density when subjected to an external perturbation. It is closely related to the concepts of electronegativity and the chemical potential [9,10,11,12,13,14,15,24].
In CDFT, chemical hardness is a measure of the energy required to modify the electron density of a system by a small amount. A molecule with high hardness will require a substantial amount of energy to alter its electron distribution, indicating its stability and unreactive nature. On the other hand, a low-hardness molecule readily adjusts its electron density, making it more prone to interacting with other molecules.
In a similar way to was indicated for the electronegativity , we can recall from Table 2 that the chemical hardness for Talarolide A and Talaropeptide B will be almost of the same value, being Talaropeptide A a system with a little bit higher result. The same can be said about Talaropeptides C and D, being this last one the molecule with the largest .
Global electrophilicity is a fundamental concept within Conceptual Density Functional Theory (CDFT) that characterizes the reactivity of a molecular system towards electron-donating species [37,38,39]. It provides valuable insights into the potential for a molecule to undergo chemical reactions and form new bonds.
In CDFT, global electrophilicity is quantified as a function of the chemical potential and the global hardness of a system. It represents the ability of a molecule to accept electrons, making it a measure of its susceptibility to nucleophilic attack or interactions with electron-rich species. Molecules with higher global electrophilicity are more likely to react with nucleophiles, facilitating the formation of new chemical bonds.
Global electrophilicity is an essential tool for assessing and comparing the reactivity of various molecular systems, enabling a deeper understanding of their behavior and interactions within the framework of Conceptual Density Functional Theory.
It is interesting to appreciate from Table 2, that the comparison of the values of for the considered cyclopeptides follows the same pattern as shown for , being Talaropeptide C the molecule presenting its large value of this reactivity descriptor.
The global softness S is defined as the reciprocal of . Thus, the comparison between the different molecules considered in this research is straightforward, rendering values ranging around 0.18-0.19 eV.
The nucleophilicity index N proposed by Domingo et al [40,41,42,43,44] is a significant concept within the context of Conceptual Density Functional Theory (DFT), aimed at quantifying and predicting the nucleophilic reactivity of molecules. This index offers valuable insights into the ability of a species to donate electrons and engage in chemical reactions with electrophiles.
The nucleophilicity index N serves as a predictive tool for assessing and comparing the nucleophilic reactivity of different molecular systems. It aids researchers in identifying potential nucleophiles and understanding the driving forces behind nucleophilic reactions. This information is invaluable in various fields, such as organic synthesis, catalysis, and medicinal chemistry, where predicting and controlling reactivity is crucial.
According to the scale proposed by Domingo et al [40,41,42,43,44] and after an inspection of Table 2, it can be concluded that the five peptides considered in this work may be regarded as moderate nucleophiles.
In the realm of Conceptual Density Functional Theory (DFT), the concepts of electrodonating and electroaccepting powers ( and , respectively) play a crucial role in understanding molecular properties and reactivity. These concepts stem from the idea of using the electron density distribution within a molecule to elucidate its behavior in chemical reactions [35].
Electrodonating power refers to a molecule’s tendency to donate electrons. It’s characterized by the ability of a molecule to release electron density towards other molecular fragments or atoms, making it more likely to act as a nucleophile in reactions. This is often associated with the presence of lone pairs or -electron systems that can readily be shared with electron-deficient species [35].
On the other hand, electroaccepting power signifies a molecule’s capacity to accept electrons. Molecules with high electroaccepting power tend to be electron-deficient or electron-seeking entities. These molecules can attract and stabilize electron density from other molecules or atoms, making them prone to acting as electrophiles in reactions.
For all the molecules considered in this work, it can be seen from Table 2 that their electrodonating powers are larger than their electroaccepting powers . These result give us a clear idea that how these molecules will behave during a chemical reaction. By comparing the individual peptides, we can conclude that Talaropeptide C displays the largest values of both reactivity descriptors.
The net electrophilicity is a concept proposed by Chattaraj as a means of comparing the relativer values of and [36]. From Table 2, it can be seen that the results for this descriptor will be similar for Talarolide A and Talaropeptide B, and a bit lower for Talopeptide A. Higher values will be displayed for Talaropeptides C and D, with the former presenting the largest result for these important reactivity descriptor.
Within the realm of Density Functional Theory (DFT), the dual descriptor DD emerges as an influential instrument harnessed for the exploration of molecular reactivity. This sophisticated metric not only evaluates a molecule’s nucleophilic and electrophilic tendencies but also offers invaluable insights into forecasting chemical reactivity. Its multi-faceted utility spans across various domains of chemistry, encompassing catalyst design, chemical reaction prognostication, and in-depth reactivity analysis. The dual descriptor empowers researchers by furnishing a powerful lens through which to decipher molecular electronic structures, ultimately enabling accurate predictions of their behavior in intricate chemical transformations [45,46,47,48].
The computation of the dual descriptor DD stems from the intricate interplay between the energy levels of the Highest Occupied Molecular Orbital (HOMO) and the Lowest Unoccupied Molecular Orbital (LUMO), thereby intricately connected to the concept of the HOMO-LUMO gap. Leveraging the dual descriptor DD equips scientists with the ability to anticipate a molecule’s receptiveness to either nucleophilic or electrophilic assaults. A prominently positive dual descriptor value signifies the molecule’s prowess as an electrophile of note, whereas a significantly negative value denotes its aptitude as a potent nucleophile [45,46,47,48].
Graphical representations of the dual descriptor DD for the considered cyclopeptides are displayed in Figure 3, where the nucleophilic and electrophilic regions can be distinguished:
As a supplement to the data gathered concerning the chemical reactivity of the investigated marine cyclopeptide via CDFT analysis, Figure 4 visually displays the bioactivity rankings of these peptides in relation to different targets. These targets encompass GPCR ligands, ion channel modulators, nuclear receptor ligands, kinase inhibitors, protease inhibitors, and enzyme inhibitors. The assigned bioactivity scores offer significant perspectives into the possible medicinal uses of these substances [8].
GPCRs are a class of proteins that play a key role in cell signaling and are involved in a wide range of physiological processes. The bioactivity scores presented in Figure 4 suggest that all the considered cyclopeptides have potential as GPCR ligands. Their scores as a GPCR ligands indicate that they may be able to selectively bind to and activate specific GPCRs involved in disease processes. This could help in the designing of an effective drug or therapy for treating conditions such as inflammation or cancer [8].
Proteases play crucial roles in numerous biological functions, including protein digestion, blood clotting, and immune responses. Irregular protease behavior has been associated with various medical conditions like viral infections, cancer, and inflammation. When a protease inhibitor binds to an active protease, it impedes its capacity to cleave intended proteins, effectively halting subsequent signal pathways. This action can hinder virus replication, cancer advancement, and inflammatory pathway activation, resulting in beneficial therapeutic outcomes. Targeting protease inhibitors is a key focus in drug exploration and advancement, given their potential to precisely target and restrain specific proteases implicated in disease mechanisms. This leads to more efficient and less hazardous treatment options. Many successful antiviral medications, including HIV protease inhibitors, fall under the category of protease inhibitors [8]. Figure 4 shows that Talaloride A and the Talaropeptides A-D will also display behavior relating to act as proteases inhibitors.
4. Conclusions
In conclusion, this study has highlighted the substantial potential that marine-derived molecules hold for the development of innovative therapeutic drugs. Talarolide A and Talaropeptides A-D have emerged as promising candidates due to their unique chemical compositions and valuable medicinal properties. Through the application of molecular modeling and computational chemistry techniques, particularly Conceptual Density Functional Theory (DFT), the research aimed to delve into the chemical reactivity of these compounds.
By scrutinizing the chemical behaviors of Talarolide A and Talaropeptides A-D, the study sought to enhance our understanding of their possible pharmacological applications. Employing Conceptual DFT and other computational tools, the investigation revealed active reaction sites and potential interactions with diverse biological targets. Notably, Talarolide A exhibited potent anti-inflammatory properties and demonstrated antitumor effects against various cancer cell lines. Talaropeptide A showcased antimicrobial effectiveness against both bacteria and fungi.
In summary, this research underscores the importance of comprehending the chemical reactivity of natural marine products in advancing pharmaceutical development. The insights gleaned from this study, facilitated by computational methodologies like Conceptual DFT, shed light on active sites and potential targets of Talarolide A and Talaropeptides A-D. These findings contribute to the broader understanding of marine-derived peptides and their promising roles as pharmaceutical agents. Nevertheless, further investigations are imperative to fully unravel the complete range of their pharmacological attributes.
Author Contributions
Conceptualization, NFH and JSSL; methodology, NFH and DGM; software, NFH and DGM; validation, NFH and DGM ; formal analysis, NFH and DGM; investigation, NFH, JSSL, and DGM; resources, NFH and DGM; data curation, NFH and DGM; writing–original draft preparation, NFH and DGM; visualization, NFH and DGM; supervision, NFH and DGM; project administration, NFH; funding acquisition, NFH. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by grant number 25017/23 from CIMAV.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not appplicable.
Data Availability Statement
All generated data will be available from the authors under request.
Acknowledgments
This study received support from the Centro de Investigación en Materiales Avanzados, S.C. (CIMAV) and the Consejo Nacional de Humanidades Ciencia y Tecnología (CONAHCYT). NFH, JSSL, and DGM are researchers affiliated with CIMAV and CONAHCYT.
Conflicts of Interest
The authors declare no conflict of interest
References
- Varijakzhan, D.; Loh, J.Y.; Yap, W.S.; Yusoff, K.; Seboussi, R.; Lim, S.H.E.; Lai, K.S.; Chong, C.M. Bioactive Compounds from Marine Sponges: Fundamentals and Applications. Marine Drugs 2021, 19, 246. [Google Scholar] [CrossRef] [PubMed]
- Erwin, P.M.; López-Legentil, S.; Schuhmann, P.W. The Pharmaceutical Value of Marine Biodiversity for anti-Cancer Drug Discovery. Ecological Economics 2010, 70, 445–451. [Google Scholar] [CrossRef]
- Macedo, M.W.F.S.; da Cunha, N.B.; Carneiro, J.A.; da Costa, R.A.; de Alencar, S.A.; Cardoso, M.H.; Franco, O.L.; Dias, S.C. Marine Organisms as a Rich Source of Biologically Active Peptides. Frontiers in Marine Science 2021, 8. [Google Scholar] [CrossRef]
- Zhang, J.N.; Xia, Y.X.; Zhang, H.J. Natural Cyclopeptides as Anticancer Agents in the Last 20 Years. International Journal of Molecular Sciences 2021, 22, 3973. [Google Scholar] [CrossRef]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine Natural Products. Natural Product Reports 2020, 37, 175–223. [Google Scholar] [CrossRef]
- Abdalla, M.; McGaw, L. Natural Cyclic Peptides as an Attractive Modality for Therapeutics: A Mini Review. Molecules 2018, 23, 2080. [Google Scholar] [CrossRef]
- Youssef, F.S.; Ashour, M.L.; Singab, A.N.B.; Wink, M. A Comprehensive Review of Bioactive Peptides from Marine Fungi and Their Biological Significance. Marine Drugs 2019, 17, 559. [Google Scholar] [CrossRef]
- Kallen, A. Computational Pharmacokinetics; CRC Press: London, England, 2019. [Google Scholar]
- Parr, R.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, 1989. [Google Scholar]
- Chermette, H. Chemical Reactivity Indexes in Density Functional Theory. Journal of Computational Chemistry 1999, 20, 129–154. [Google Scholar] [CrossRef]
- Geerlings, P.; Proft, F.D.; Langenaeker, W. Conceptual Density Functional Theory. Chemical Reviews 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- Geerlings, P.; Chamorro, E.; Chattaraj, P.K.; Proft, F.D.; Gázquez, J.L.; Liu, S.; Morell, C.; Toro-Labbé, A.; Vela, A.; Ayers, P. Conceptual Density Functional Theory: Status, Prospects, Issues. Theoretical Chemistry Accounts 2020, 139. [Google Scholar] [CrossRef]
- Toro-Labbé, A. (Ed.) Theoretical Aspects of Chemical Reactivity; Elsevier Science: Amsterdam, 2007. [Google Scholar]
- Chattaraj, P.K. (Ed.) Chemical Reactivity Theory - A Density Functional View; CRC Press. Taylor & Francis Group: Boca Raton, FL, 2009. [Google Scholar]
- Chakraborty, D.; Chattaraj, P.K. Conceptual Density Functional Theory Based Electronic Structure Principles. Chemical Science 2021, 12, 6264–6279. [Google Scholar] [CrossRef] [PubMed]
- Frau, J.; Glossman-Mitnik, D. Molecular Reactivity and Absorption Properties of Melanoidin Blue-G1 through Conceptual DFT. Molecules 2018, 23, 559–15. [Google Scholar] [CrossRef] [PubMed]
- Frau, J.; Glossman-Mitnik, D. Conceptual DFT Study of the Local Chemical Reactivity of the Dilysyldipyrrolones A and B Intermediate Melanoidins. Theoretical Chemistry Accounts 2018, 137, 1210. [Google Scholar] [CrossRef]
- Frau, J.; Glossman-Mitnik, D. Conceptual DFT Study of the Local Chemical Reactivity of the Colored BISARG Melanoidin and Its Protonated Derivative. Frontiers in Chemistry 2018, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Frau, J.; Glossman-Mitnik, D. Computational Study of the Chemical Reactivity of the Blue-M1 Intermediate Melanoidin. Computational and Theoretical Chemistry 2018, 1134, 22–29. [Google Scholar] [CrossRef]
- Frau, J.; Glossman-Mitnik, D. Chemical Reactivity Theory Applied to the Calculation of the Local Reactivity Descriptors of a Colored Maillard Reaction Product. Chemical Science International Journal 2018, 22, 1–14. [Google Scholar] [CrossRef]
- Frau, J.; Glossman-Mitnik, D. Blue M2: An Intermediate Melanoidin Studied via Conceptual DFT. Journal of Molecular Modeling 2018, 24, 1–13. [Google Scholar] [CrossRef]
- Frau, J.; Flores-Holguín, N.; Glossman-Mitnik, D. Chemical Reactivity Properties, pKa Values, AGEs Inhibitor Abilities and Bioactivity Scores of the Mirabamides A–H Peptides of Marine Origin Studied by Means of Conceptual DFT. Marine Drugs 2018, 16, 302–19. [Google Scholar] [CrossRef]
- Costa, L.; Sousa, E.; Fernandes, C. Cyclic Peptides in Pipeline: What Future for These Great Molecules? Pharmaceuticals 2023, 16, 996. [Google Scholar] [CrossRef]
- Glossman-Mitnik, D. (Ed.) Density Functional Theory - Recent Advances, New Perspectives and Applications; IntechOpen: London, UK, 2022. [Google Scholar]
- Halgren, T.A. Merck Molecular Force Field. I. Basis, Form, Scope, Parameterization, and Performance of MMFF94. Journal of Computational Chemistry 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck Molecular Force Field. II. MMFF94 van der Waals and Electrostatic Parameters for Intermolecular Interactions. Journal of Computational Chemistry 1996, 17, 520–552. [Google Scholar] [CrossRef]
- Halgren, T.A. MMFF VI. MMFF94s Option for Energy Minimization Studies. Journal of Computational Chemistry 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Halgren, T.A.; Nachbar, R.B. Merck Molecular Force Field. IV. Conformational Energies and Geometries for MMFF94. Journal of Computational Chemistry 1996, 17, 587–615. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck Molecular Force Field. V. Extension of MMFF94 Using Experimental Data, Additional Computational Data, and Empirical Rules. Journal of Computational Chemistry 1996, 17, 616–641. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C.01, 2016. Gaussian Inc. Wallingford CT.
- Peverati, R.; Truhlar, D.G. Screened-Exchange Density Functionals with Broad Accuracy for Chemistry and Solid-State Physics. Physical Chemistry Chemical Physics 2012, 14, 16187. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Physical Chemistry Chemical Physics 2005, 7, 3297. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting Basis Sets for H to Rn. Physical Chemistry Chemical Physics 2006, 8, 1057. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. The Journal of Physical Chemistry B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Gázquez, J.; Cedillo, A.; Vela, A. Electrodonating and Electroaccepting Powers. Journal of Physical Chemistry A 2007, 111, 1966–1970. [Google Scholar] [CrossRef]
- Chattaraj, P.; Chakraborty, A.; Giri, S. Net Electrophilicity. Journal of Physical Chemistry A 2009, 113, 10068–10074. [Google Scholar] [CrossRef]
- Parr, R.; Szentpaly, L.; Liu, S. Electrophilicity Index. Journal of the American Chemical Society 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Chattaraj, P.; Sarkar, U.; Roy, D. Electrophilicity Index. Chemical Reviews 2006, 106, 2065–2091. [Google Scholar] [CrossRef] [PubMed]
- Liu, S. Electrophilicity. In Chemical Reactivity Theory - A Density Functional View; Chattaraj, P., Ed.; CRC Press. Taylor & Francis Group: Boca Raton, 2009; chapter 13. [Google Scholar]
- Domingo, L.R.; Chamorro, E.; Perez, P. Understanding the Reactivity of Captodative Ethylenes in Polar Cycloaddition Reactions. A Theoretical Study. The Journal of Organic Chemistry 2008, 73, 4615–4624. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, P.; Domingo, L.R.; Chamorro, E.; Pérez, P. A Further Exploration of a Nucleophilicity Index Based on the Gas-Phase Ionization Potentials. Journal of Molecular Structure: THEOCHEM 2008, 865, 68–72. [Google Scholar] [CrossRef]
- Domingo, L.R.; Sáez, J.A. Understanding the Mechanism of Polar Diels-Alder Reactions. Organic and Biomolecular Chemistry 2009, 7, 3576–3583. [Google Scholar] [CrossRef]
- Domingo, L.R.; Perez, P. The Nucleophilicity N Index in Organic Chemistry. Organic and Biomolecular Chemistry 2011, 9, 7168–7175. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef]
- Morell, C.; Grand, A.; Toro-Labbé, A. New Dual Descriptor for Chemical Reactivity. The Journal of Physical Chemistry A 2004, 109, 205–212. [Google Scholar] [CrossRef]
- Morell, C.; Grand, A.; Toro-Labbé, A. Theoretical Support for Using the Δf(r) Descriptor. Chemical Physics Letters 2006, 425, 342–346. [Google Scholar] [CrossRef]
- Martínez-Araya, J.I. Explaining Reaction Mechanisms Using the Dual Descriptor: A Complementary Tool to the Molecular Electrostatic Potential. Journal of Molecular Modeling 2012, 19, 2715–2722. [Google Scholar] [CrossRef]
- Martínez-Araya, J.I. Why is the Dual Descriptor a More Accurate Local Reactivity Descriptor than Fukui Functions? Journal of Mathematical Chemistry 2015, 53, 451–465. [Google Scholar] [CrossRef]
Figure 1.
Graphical sketches of the molecular structures of a) Talarolide A , b) Talaropeptide A, c) Talaropeptide B, d) Talaropeptide C and e) Talaropeptide D
Figure 1.
Graphical sketches of the molecular structures of a) Talarolide A , b) Talaropeptide A, c) Talaropeptide B, d) Talaropeptide C and e) Talaropeptide D
Figure 3.
Graphical Representation of the Dual Descriptor DD of a) Talarolide A , b) Talaropeptide A, c) Talaropeptide B, d) Talaropeptide C and e) Talaropeptide D. Left: DD > 0, Right: DD < 0
Figure 3.
Graphical Representation of the Dual Descriptor DD of a) Talarolide A , b) Talaropeptide A, c) Talaropeptide B, d) Talaropeptide C and e) Talaropeptide D. Left: DD > 0, Right: DD < 0
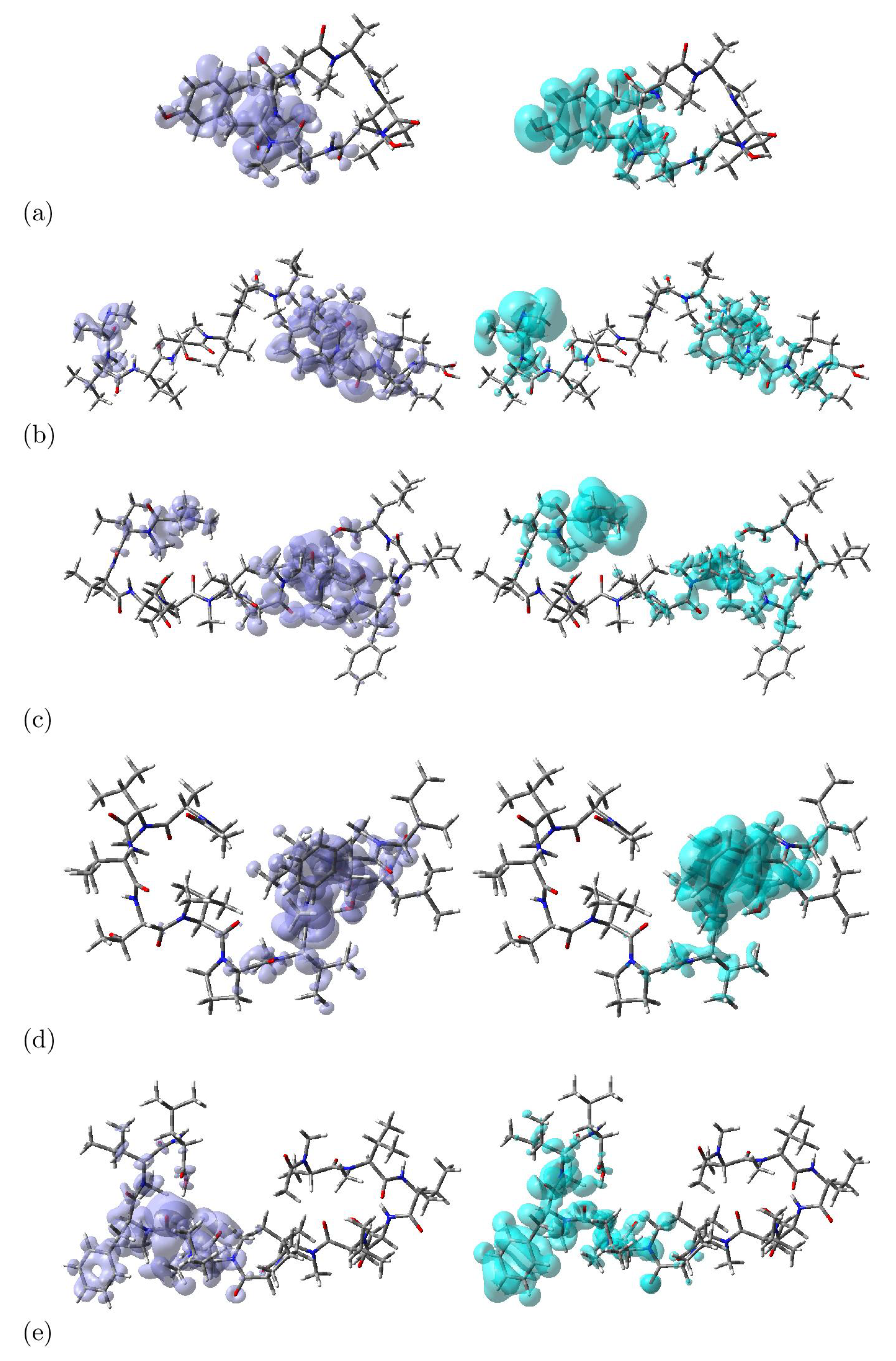
Figure 4.
Graphical Representation of the Bioactivity Scores for a) Talarolide A , b) Talaropeptide A, c) Talaropeptide B, d) Talaropeptide C and e) Talaropeptide e
Figure 4.
Graphical Representation of the Bioactivity Scores for a) Talarolide A , b) Talaropeptide A, c) Talaropeptide B, d) Talaropeptide C and e) Talaropeptide e
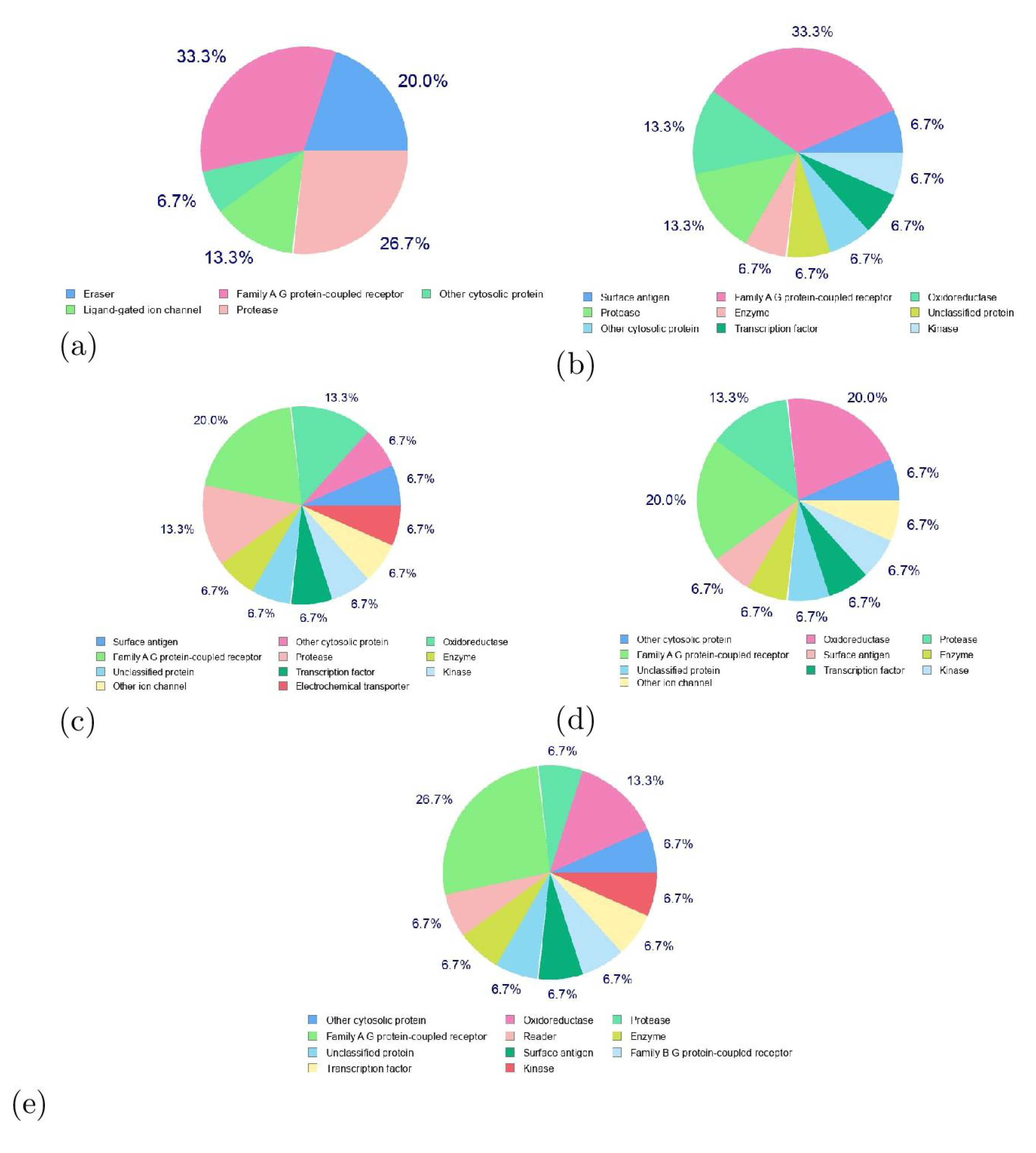

Table 1.
Orbital energies, H-L gap and the KID descriptors (all in eV) for the Talarolide A and Talaropeptides A-D molecules
Table 1.
Orbital energies, H-L gap and the KID descriptors (all in eV) for the Talarolide A and Talaropeptides A-D molecules
| HOMO | LUMO | SOMO | H-L Gap | J(I) | J(A) | J(HL) | SL | |
|---|---|---|---|---|---|---|---|---|
| Talarolide A | -6.14 | -0.93 | -0.96 | 5.21 | 0.04 | 0.02 | 0.05 | 0.02 |
| Talaropeptide A | -6.22 | -0.85 | -0.87 | 5.37 | 0.02 | 0.00 | 0.02 | 0.01 |
| Talaropeptide B | -6.16 | -0.92 | -0.92 | 5.24 | 0.00 | 0.00 | 0.00 | 0.00 |
| Talaropeptide C | -6.54 | -1.00 | -1.03 | 5.54 | 0.01 | 0.01 | 0.02 | 0.02 |
| Talaropeptide D | -6.56 | -0.92 | -0.91 | 5.63 | 0.01 | 0.00 | 0.01 | 0.01 |
Table 2.
Global reactivity descriptors for the Talarolide A and Talaropeptide A-D molecules (all in eV, excepting S, in eV)
Table 2.
Global reactivity descriptors for the Talarolide A and Talaropeptide A-D molecules (all in eV, excepting S, in eV)
| S | N | |||||||
|---|---|---|---|---|---|---|---|---|
| Talarolide A | 3.54 | 5.21 | 1.20 | 0.19 | 2.65 | 4.50 | 0.96 | 5.46 |
| Talaropeptide A | 3.54 | 5.37 | 1.17 | 0.19 | 2.57 | 4.44 | 0.90 | 5.34 |
| Talaropeptide B | 3.54 | 5.24 | 1.20 | 0.19 | 2.63 | 4.50 | 0.95 | 5.45 |
| Talaropeptide C | 3.77 | 5.54 | 1.29 | 0.18 | 2.25 | 4.80 | 1.03 | 5.83 |
| Talaropeptide D | 3.74 | 5.63 | 1.24 | 0.18 | 2.24 | 4.70 | 0.96 | 5.67 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



