Submitted:
31 August 2023
Posted:
01 September 2023
You are already at the latest version
Abstract
The MET proto-oncogene encodes a pivotal tyrosine kinase receptor, binding the hepatocyte growth factor (HGF, also known as scatter factor, SF) and governing essential biological processes such as organogenesis, tissue repair, and angiogenesis. The pleiotropic physiological functions of MET explain its diverse role in cancer progression in a broad range of tumors; genetic/epigenetic alterations of MET drive tumor cell dissemination, metastasis, and acquired resistance to conventional and targeted therapies. Therefore, targeting MET emerged as a promising strategy, and many efforts were devoted to identifying the optimal way of hampering MET signaling. Despite encouraging results, however, the complexity of MET's functions in oncogenesis yields intriguing observations, fostering a humbler stance on our comprehension. This review explores recent discoveries concerning MET alterations in cancer, elucidating their biological repercussions, discussing therapeutic avenues, and outlining future directions. By contextualizing the research question and articulating the study's purpose, this work navigates MET biology's intricacies in cancer, offering a comprehensive perspective.
Keywords:
MET alterations
; cancer progression
; invasive growth
; therapeutic targeting.
1. Introduction
The human MET protooncogene encodes the tyrosine kinase receptor for the hepatocyte growth factor / scatter factor (HGF/SF) [1,2,3]. It is expressed in a broad range of epithelial cells, and the resulting receptor is a 170kDa transmembrane protein organized in two disulfide bond-linked 𝛼 and β subunits of respectively 50kDa and 145kDa [4,5]. The ligand, SF is produced and secreted in physiologic conditions by mesenchymal cells close to MET-expressing epithelial cells [6,7]. This paracrine effect leads to MET activation by autophosphorylation of the cytoplasmic catalytic domain and recruitment of adaptor proteins, promoting signal transduction. MAPK pathway, PI3K/AKT signaling, and STAT3 constitute the primary signal transducers [8,9,10]. MET plays an essential role during embryogenesis (e.g., epithelial to mesenchymal transition, organ development) and the postnatal period (e.g., angiogenesis, organ regeneration, and wound healing (reviewed in [11]).
The broad range of critical biological responses induced by MET awards the latter as a crucial oncogene in tumor progression, enabling cancer cells to survive and escape the hostile primary tumor microenvironment and form distal metastases [12]. MET is altered in multiple cancer types and behaves as a pivotal regulator of invasive growth, a complex and intertwined sequence of events including epithelial-to-mesenchymal transition (EMT), scattering, migration, and growth [2,13,14,15,16,17]. Tumor cells harboring such alterations become ‘addicted’ to MET (described in [2]), therefore instituting a tumor cell-specific vulnerability point and justifying targeted therapies. Accordingly, MET inhibition reduces tumor size and impedes metastases in rodent models [12,18,19]. Therefore, MET was recently proposed to stand among the top five proteins to prioritize in the targeted therapies [20].
However, it should be noted that the path to efficient MET-targeted therapy for patients is long and covered with complications. Despite the presumed ‘addiction’ to MET, many patients do not respond to therapy [12,21,22,23]. Moreover, acquired resistance to MET inhibitors might arise [24,25,26,27].
This review reminds the overall structure of MET and summarizes the observed MET alterations in cancer, their impact on invasive growth, and their therapeutic potential. The lessons from the disappointing results of targeted therapies will be investigated to propose more accurate strategies to extend disease-free survival time.
Structure and function of the MET kinase
MET is the tyrosine kinase receptor (RTK) for SF. The translated precursor protein of 175kDa matures through proteolytic cleavage by FURIN in the Golgi apparatus (Figure 1, left panel and [28]). The mature protein forms a heterodimer of an extracellular 𝛼 subunit (50kDa) linked via disulfide bonds to the 145kDa transmembrane 𝛽 subunit. The extracellular portion of the 𝛽 chain comprises a SEMA (semaphorin) domain, a Plexin-Semaphorin-Integrin (PSI) homology domain, and four immunoglobulin-like IPT domains [29,30]. SEMA and IPT domains are crucial for ligand binding and receptor dimerization, while the PSI domain is essential for the proper maturation of the receptor through its recently described disulfide isomerase activity [31]. The intracellular part of MET is composed of a short juxtamembrane domain (JM), followed by the catalytic site and the docking site for signal transducers.
Upon SF binding, the catalytic domain of MET gets auto-phosphorylated on Tyrosine 1234 and Tyrosine 1235 [32]. The activation of the catalytic site triggers the phosphorylation of two tyrosine on the docking site (Y1349 and Y1356), priming the interaction with Src homology 2 (SH2) domain-containing proteins [10,33,34]. MET docking site is multifunctional; Y1349 phosphorylation leads to the activation of PI3K/AKT pathway (migration/survival) while Y1356 phosphorylation activates the RAS/MAPK pathway (proliferation/cell cycle progression) [8,10]. STAT3 transcription factor is also activated by MET [9]. Like other RTKs, MET transmits the information from the extracellular space to the cytoplasm, generating a multilayered network that activates various biological processes, including migration, growth, and differentiation/stemness (Figure 2 and [35]).
SF/MET interaction induces the phosphorylation of Y1003 within the juxtamembrane domain, allowing the recruitment of the E3 ubiquitin ligase casitas B-lineage lymphoma (CBL), promoting MET monoubiquitylation [36], receptor internalization and lysosomal degradation (Figure 1, right panel and [37]). Accordingly, experimental evidence has shown that Y1003F mutation stabilizes the receptor [38]. The juxtamembrane domain is therefore accepted as a negative regulator of the MET/SF axis [36,39,40,41,42].
MET alterations in tumors and their biological significance
Recent advances in next-generation sequencing technologies allowed a significant reduction in their cost, ultimately leading to an inflation of publicly available ‘OMICS’ data. Besides, improvements in the standardization of data curation, analyses, and presentation offered researchers an unprecedented quantity of comprehensive information regarding genetic alterations during cancer onset and progression. In line with this, we developed an auto-updatable ‘MET observatory’ to catalogue genetic alterations of the MET gene in cancer. Here, we present some of the features of this observatory. Briefly, the catalog of alterations results from data collection from TCGA, COSMIC, and ClinVar datasets.
MET amplification
An oncogene can be defined as an entity that can transform cells by conferring some attributes of a cancer cell. MET is an excellent example of an oncogene by its critical involvement in cell migration, metastasis, and cell survival. Unsurprisingly, MET is altered in many cancers, including non-small cell lung cancer (NSCLC), lung squamous carcinoma, gastric cancer, colorectal adenocarcinoma, melanoma, gliomas, and renal cancer [44,45,46,47,48,49]. The most observed MET alteration is gene amplification, with a prevalence of 4% (Figure 3A and [50,51,52,53,54,55]). MET amplification is observed in a broad range of tumors. In some tumors, the incidence increases by nearly 5-fold, reaching almost 20% of all kidney papillary cell carcinomas (KIRP, Figure 3B). Besides, MET amplification sustains secondary resistance to EGFR-targeted therapies in NSCLC and colorectal cancers, showing its importance in cell survival and acquired resistance to EGFR-targeting therapies [54,56,57].
Exon14 ‘skipping’, the predominant MET alteration
It was thus proposed that other mutations might drive ligand-independent activation of MET. Surprisingly for an RTK (e.g., EGFR, BCL-ABL, FGFR, SRC in cancer, for review, see [58]), very few patients exhibited MET mutations in the kinase domain or regulatory regions (Figure 3C and [17,59]). Splice site mutations spanning the exon14 were by far the most common MET mutations. These mutations (complex or simple) are a consequence of the loss of acceptor or donor sites, resulting in exon 14 ‘skipping’. Indeed, the latter was described in a significant number of patients: 13% in pulmonary sarcomatoid carcinoma, 6% in adenosquamous carcinoma, 3% in lung adenocarcinomas, 2% in lung squamous cell carcinomas, 0.4% in gliomas, and 0.4% in cancers of unknown primary origin (CUP) [16,60]. MET Exon14 encodes for the juxtamembrane domain. Because of the regulatory role of the JM described above, it was thought that its loss would lead to increased receptors and subsequent ligand-independent, uncontrolled activation of MET, driving invasive growth. Accordingly, exon 14 re-insertion into the oncogenic gene fusion (TPR)–MET, which consists of the MET sequence downstream from the juxtamembrane domain fused to the dimerization motif of TPR, resulted in decreased oncogenic potential [61]. Therefore, targeted therapies against MET constituted appealing strategies for cancer patients carrying MET exon14 deletion (MET∆14) [21,23,62,63,64,65,66,67,68,69,70,71,72,73,74]. However, only half of the patients harboring MET∆14 benefited from MET-targeted therapies, suggesting that the critical aspects of MET∆14 remain to be elucidated [75,76]. Recently, two independent studies demonstrated that deletion of exon 14 does not result in constitutive activation of the kinase. MET∆14 activity requires SF and drives a robust and selective AKT activation, rendering cancer cells more prone to survival and migration [17,77]. Therefore, it is proposed that cancer cells expressing MET∆14 choose the astute strategy to ‘fly’ the local hostile micro milieu to form distal metastases instead of ‘fighting’ to proliferate locally. The absence of SF in the tumor microenvironment or PI3K/AKT axis mutations may explain the insensitivity to targeted therapies. Fittingly, PI3K/AKT activating mutations co-occur with MET exon14 ‘skipping’ in 14% of cancer patients [78,79]. Based on these results, a better stratification of patients might lead to a better response to MET-targeted therapies.
Point mutations within the MET coding sequence
As shown in Figure 3C, mutations affecting the catalytic site or regulatory sites of MET are sporadic but exist. The first activating mutations of MET were identified in hereditary papillary renal carcinoma (HPRC), and the authors suggested that the mutations affecting the kinase domain of MET (M1149T, V1206L, V1238I, D1246N, and Y1248C) were causal in HPRC [80]. Similar MET mutations (D1246H, Y1228C, and M1268T) spanning the critical Y1234 and Y1235 were described in the sporadic renal carcinoma [81]. Moreover, cytogenetic analyses showed non-random chromosome 7 trisomy, which affected the mutated MET allele [82]. All these mutations have the common feature of inducing the constitutive activation of the kinase [81,83,84], leading to oncogene ‘addiction’ [2]. Experiments in transgenic murine demonstrated the oncogenic potential of these activating mutants.
Interestingly, tumors formed in mice were not restricted to the kidney; animals developed lymphomas, carcinomas, or aggressive mammary tumors [85,86]. Importantly, these independent studies demonstrate that activating mutations affecting the MET catalytic site drive tumorigenesis in multiple tissues. Researchers attempted to identify activating mutations in other human cancers in the following years. Somatic or germline mutations were described in hepatocellular carcinoma, head and neck cancers, oropharynx squamous cell cancer, gastric cancer, CUP, and colorectal cancer [87,88,89,90,91,92,93].
A growing number of point mutations were described in the SEMA domain (both in 𝛼 and 𝛽 chains, Figure 3C) responsible for the ligand binding [94]. Six non-small cell lung cancer patients out of 127 harbored mutations (L229F, N375S, E168D, N375S, S323G, N375S) within the exon 2 encoding for the SEMA domain [95]. SEMA domain mutations are not restricted to lung cancer. In an independent study, MET mutations were detected in 9% of advanced breast cancer (8/88 patients). Six of eight MET mutations affected the SEMA domain (N375S in five patients, M362T in one patient, [96]). Despite some evidence showing that SEMA domain mutations are oncogenic [92,97], our knowledge of the biological significance of MET SEMA mutations remains poor. They likely affect the ligand-binding domain’s structure, promoting a constitutively active or ligand hypersensitive kinase. Accordingly, in CUP – owning the unique ability of self-renewal in the absence of any exogenous growth factor [98] – MET mutations clustered within the SEMA domain; in a total of 23 CUP patients, five out of seven MET alterations localized in the SEMA domain (H150Y, Q142X, C385Y, and two patients with E168D [92]).
Point mutations, although rare, should not be overlooked. With recent advancements in genomic screening technologies, we might find a growing number of these mutations. By combining in silico, in vitro, and in vivo findings, we aim to have a sharper picture of MET alterations in cancer and their biological significance, opening new avenues for targeted therapies.
For RTKs, it is generally accepted that gene amplification leads to a higher number of receptors at the cell surface, priming kinase activation in the presence of small amounts of ligands. MET challenges this concept; overexpression of human MET in mouse liver induces hepatocellular carcinomas [99]. Since human MET cannot interact with murine SF [100], these results demonstrate the ligand-independent activation of the receptor when over-expressed.
Fusion partners of MET drive oncogene ‘addiction’
For decades the only known MET gene rearrangement in human tumors has been TPR-MET, mostly occurring in gastric cancers [101]. Recently, the thorough analyses of the vast TCGA tumor collection uncovered new hybrid proteins [102]: the MET intracellular domain fused at the N-terminus with several partners, some of them encompass the dimerization ‘coiled-coil’ (CC) motif (i.e., C8orf34, BAIAP2L1, TFG, and KIF5B). Consequently, the chimeric MET dimerizes in a ligand-independent fashion, driving constitutive kinase activity and tumorigenesis. Although occurring at low frequencies, these fusions have been found in lung adenocarcinomas, hepatocellular carcinomas, papillary renal carcinomas, and thyroid carcinomas; thus, they cannot be ignored [102].
Another recurrent gene rearrangement involves MET and the PTPRZ1 gene, encoding a tyrosine phosphatase [103]. PTPRZ1-MET fusions include almost the entire MET sequence fused at its N-terminus with a variable number of exons of the PTPRZ1 gene [104]. PTPRZ1-MET fusions have been found in brain tumors, such as low-grade gliomas, secondary glioblastomas arising in adults from the progression of lower-grade gliomas, and pediatric glioblastomas at a remarkably high frequency (~10%, [104]). Notably, the chromosomal rearrangement between PTPRZ1 and MET leads to fusion protein overexpression and enhanced kinase activation [105]. The mechanism explaining enhanced MET activity in tumors expressing the fusion protein remains to be determined; the highly active PTPRZ1 promoter fused to the MET gene [104] and the coiled-coil domain of PTPRZ1 fused to MET [106] are two mutually non-exclusive hypotheses.
Experiments show that MET fusion proteins respond to anti-MET monotherapy: PTPRZ1-MET in a pediatric glioma [104] and KIF5B-MET in lung cancers [107]. MET gene fusions also happen in melanomas, where six different N-terminal partners fused in-frame with the intracellular MET domain have been described (23).
Different MET genetic alterations can induce either ligand-independent (or hypersensitive) or SF-dependent (MET∆14) activation of the kinase (Figure 4). They have a common denominator for driving invasive growth. Tumor cells thus become addicted to MET and vulnerable to targeted therapies.
Genetic alterations of MET: the peak of the iceberg?
MET-amplified tumors represent 4%, where MET overexpression is observed in more than 50% of cancers (Figure 3D). The discrepancy is too large and deserves the attention of the scientific community. It should be noted that large dataset often restricts their analyses to protein-coding regions, overlooking important regulatory regions crucial for gene expression. The inducible nature of the MET promoter was previously described and its importance in tumor biology was established [108,109,110]. Thus, alterations affecting the MET promoter should not be disregarded: re-analyses of TCGA dataset showed a remarkable decrease in promoter methylation in cancer patients, a synonym for transcription activation and MET overexpression (Figure 3E). This observation suggests that central regulatory mechanisms remain to be elucidated. Gene expression is not only regulated at the transcription level. Post-transcription control mechanisms affecting the translation efficiency and the messenger’s stability are hubs for deregulations observed in cancer. Indeed, we observed a small but significant fraction of patients harboring mutations within untranslated regions of the mRNA (Figure 3F). However, their biological significance is unknown but their exploration might reveal an unsuspected ‘dark energy’ for tumor cells. Besides, we have recently revealed MET translational regulation by the PI3K/AKT/mTOR axis and its relevance in therapy resistance. This aspect is detailed below.
MET-targeted therapies
In line with the statements above, several MET-targeting agents have been developed (summarized in Table 1). As with other targeted therapies, several questions must be addressed before trying to quench MET signaling – and tumor growth – in patients. We can encapsulate these matters in three main topics: 1) how to quench MET signaling in patients; 2) who should benefit from MET-targeting agents. 3) how to face the inevitable problem in targeted therapy: drug resistance.
Different MET-blocking agents: advantages and pitfalls
Three main strategies were employed to extinguish the MET signaling (schematized in Figure 5): small kinase inhibitors and monoclonal antibodies targeting SF or its receptor [2,12,111]. Small kinase inhibitors are chemical compounds that pass through the plasma membrane and interact with the receptor kinase domain. They can target a large panel of receptors (multitarget tyrosine kinase inhibitors) or, specifically MET. The latter has the advantage of reducing off-target effects. On the other hand, because of the crosstalk of receptors [43,112,113,114], targeting a broader number of RTKs might induce a better clinical outcome. Tyrosine kinase inhibitors act as ATP mimetics, hampering receptor phosphorylation and subsequent kinase activity [115]. However, as discussed in the first chapter, MET phosphorylation is indispensable for its degradation [36,37]. In the chronic treatment setting, small molecules can thus potentially increase the number of receptors at the cell surface, suffocating the treatment efficacy. Besides, an acquired, or existing mutation in the ATP binding pocket can engender resistance, as previously observed for small molecule inhibitors of EGFR, KIT, and BCR-ABL [116,117,118,119].
An alternative approach is to target the extracellular moiety of MET using antibodies. They are more specific by nature than chemical inhibitors. Importantly, antibodies are insensitive to multidrug resistance, a feature of aggressive cancer cells that augments the drug efflux, reduces its influx, or increases the drug catabolism [54,120,121,122,123]. Furthermore, antibodies recognize MET even if the intracellular part is mutated in cancer cells (METΔ14 or activating mutations in the catalytic site). However, antibodies can induce receptor dimerization and activation. Different strategies have been employed to circumvent this issue. One-armed monoclonal antibodies (MetMab, also known as Onartuzumab, [124]) and antibodies inducing CBL-independent degradation of MET (SAIT301, [125,126]) have been developed and tested in clinics (Table 1).
Another innovative strategy is taking advantage of the shedding of the plasma membrane to maintain homeostasis [127]; DN30 antibodies interact with the IPT domain of MET with subnanomolar affinities and induce shedding through the cleavage of the extracellular moiety of MET [128,129,130]. The proteasome subsequently degrades the intracellular moiety [131,132]. Additionally, since the extracellular part of MET is trimmed, it can potentially sequestrate the circulating SF, further hampering the SF/MET axis. Furthermore, based on the straightforward elimination of MET from the cell surface, independently of receptor activation (phosphorylated vs. unphosphorylated state), DN30 has a substantial advantage over other MET antibodies, as it is effective in the full spectrum of MET activation mechanisms, whether SF-dependent or independent (that is, induced by mutations, gene fusions, or amplification). Multiple studies demonstrated its remarkable potential; DN30 hampers cell growth and induces apoptosis in multiple MET-addicted cell lines in vitro and induces an impressive reduction of tumor mass in vivo [130,133]. Importantly, DN30 displayed a favorable pharmacokinetics and safety profile in non-human primates [133], encouraging the design of a clinical trial in MET-addicted cancer patients.
Targeting the ligand is an alternative option to impede MET signaling. The benefit of the SF antibodies AMG 102 (Rilotumumab, [134]) and AV-299 (Ficlatuzumab) was assessed in many clinical trials with disappointing results (Table 1). In one clinical trial, SF antibodies significantly increased mortality, causing the premature ending of the study [135]. As discussed previously, MET alterations in tumors typically promote ligand-independent kinase activity (gene amplification, activating mutations, fusion proteins), partially explaining these frustrating results. Nevertheless, targeting SF should not be dismissed; their ability to sequestrate the ligand could be exploited to inhibit SF-dependent invasion and survival in tumors without MET amplification (e.g., METΔ14). Farther, it should be reminded that SF acts on the tumor micro milieu on cancer-associated fibroblasts and macrophages to foster angiogenesis [136,137,138,139]. A strategy is valid only if employed toward the right target.
Patient stratification: a key for success in targeted therapies
Identifying the target population for treatment might look self-evident, but it is laborious and critical for setting the scene for success in a clinical trial. More than three decades of basic research guided drawing an overall picture of patients that can benefit from MET inhibition. Studies using cell lines or patient-derived xenografts have shown that only tumors harboring MET alterations (mostly amplification) respond to MET blockade. The cell cycle is arrested, and/or apoptosis is induced in vitro [140], and complete inhibition of tumor growth (and even tumor shrinkage) is observed in vivo [57]. MET alterations must be assessed in patients before assigning them to the group receiving targeted therapies. This is indeed a golden rule to follow: targeted therapies can only benefit patients presenting MET alterations. Nevertheless, their unambiguous identification is challenging. In many clinical trials, MET levels were assessed by immunohistochemistry [141,142], a strategy being far from objective and hardly reproducible. Moreover, the increased protein intensity is not necessarily a synonym for MET amplification and ‘addiction’. Because of the inducible nature of the MET promoter [108,109,110], high MET protein levels may be transient due to changes in the tumor microenvironment (e.g., hypoxia, ionizing radiation, cytotoxic reagents, described as ‘expedience’ in [2]). Indeed, post hoc analyses of some clinical trials illustrate this issue; patients with gene copy number gain of MET >4 exhibited the uppermost progression-free survival [142]. Another study testing the efficiency of MetMab failed to demonstrate any benefit of the treatment. Though, 88% of patients were MET-negative [143], demonstrating abruptly the validity of the golden rule stated above.
For tumors expressing wt MET (the vast majority of patients), a priori ineligible for MET-targeted therapies, it should be noted that hampering the MET signaling reduces migration and metastatic dissemination drastically without affecting the growth of cancer cells [144,145,146]. Using MET-targeted therapy might constitute a promising adjuvant therapy after tumor resection with curative intent, an ideal setting to eradicate the persistence and dissemination of subclinical tumor foci.
With the progress in next-generation sequencing technologies (lesser quantity of material required for acceptable coverage), genomic interrogations of liquid biopsies (circulating tumor DNA and circulating tumor cells) are largely feasible [147,148] to adequately and objectively stratify patients. Ideally, MET alterations must be assessed before the initiation of the clinical trial. Liquid biopsies also enable longitudinal evaluation of tumor evolution in a non-invasive manner, crucial for understanding mechanisms hidebound to drug resistance, the major challenge in targeted therapy.
Understanding and overcoming drug resistance
Targeted therapies were a breakthrough in cancer research, emphasizing our cutting-edge understanding of cancer cells’ vulnerabilities. The design of appropriate treatment strategies was thus achievable. However, they suffer a significant limitation: resistance frequently occurs [179,180,181] after an initial response. Understanding how cancer cells evade targeted therapies constitutes a substantial challenge in the clinic.
Tumors can be defined as pseudo-organs constituted of heterogeneous clones presenting highly diverse genotypes and phenotypes. Some might prosper while others regress depending on their ability to face microenvironmental selection pressures [181]. MET genetic alterations might dominate the majority of tumor cells and dictate drug sensitivity; however, minor subclones harboring other mutations that confer resistance to MET blockade may coexist and be positively selected under drug pressure. Resistant subclones must be promptly detected (e.g., liquid biopsies at regular intervals). In line with this idea, many efforts have been made to characterize the molecular profile of emerging resistant clones [182,183,184]. These forerunner studies have undeniable value for discovering new targetable biomarkers in persistent clones that limit their propagation with proper and timely therapeutic interventions.
The flare effect: mTOR pathway comes to scene
In current clinical practice, the line of treatment is dismissed when resistance arises. In many patients, discontinuation of kinase inhibitors results in rapid tumor regrowth: this phenomenon is known as disease ‘flare’ or tumor ‘rebound’ [185,186,187,188], characterized by an unknown incidence and a cumbersome prognosis [189]. The occurrence of tumor ‘rebound’ complicates disease management and has led to the idea of continuing the therapy beyond the progression [189]. Notably, treatment with a MET therapeutic antibody that induces ‘shedding’ (proteolytic cleavage of the receptor at the cell surface) substantially prevents this effect, providing a rationale to combine, or alternate, MET-targeted drugs with different mechanisms of action [190].
We recently presented a mechanistic elucidation of the ‘flare’ effect [191]. Within MET-amplified cells, halting the administration of the small molecule JNJ-38877618 (MET inhibitor) triggered activation within the AKT/mTOR pathway. Notably, mTOR orchestrates cell growth by governing mRNA translation, ribosome biogenesis, and metabolic processes [192,193,194,195]. Noteworthy transformations have been observed due to the overexpression of mTOR’s target, eIF4E, in rodent fibroblasts [196], consequently prompting extensive investigations into mTOR’s role in cancer [192]. Efforts towards inhibiting mTOR with rapalogs (including rapamycin and analogs) have been pursued in cancer therapeutics; however, the exclusive utilization of rapalogs displayed suboptimal efficacy [197].
A captivating observation emerged as mTOR swiftly augmented MET translation upon MET inhibitor withdrawal [191], uncovering a novel aspect of the AKT/mTOR axis in conferring resistance against therapies. Beyond de novo protein synthesis, the influence of the AKT/mTOR pathway on MET extends further. This pathway’s Ser/Thr kinase activity phosphorylates and incapacitates the protein phosphatase PTP1B, resulting in elevated phosphorylated (active) MET at the cell surface [191]. Consequently, the AKT/mTOR pathway emerges as a promising candidate for targeted interventions due to its paramount role in driving tumor rebound. These mechanistic insights hold the potential to guide the formulation of metronomic treatment strategies, incorporating alternating MET inhibitors and mTOR inhibitors with minimal washout periods.
In a broader context, the intricate interplay between the mTOR axis and MET biology underscores the pivotal role of translational regulation in governing MET expression within the realm of cancer. Translational control, distinct from the transcriptional regulations, operates on pre-existing mRNA, facilitating a swift adaptability to the dynamic shifts within the tumor microenvironment. While a comprehensive comprehension of the relevance of MET’s translational control in the context of cancer demands further exploration, it might provide insight into the apparent paradox between sporadic MET amplifications (accounting for 3-5% of cases) and the nearly ubiquitous MET overexpression observed in cancer [198,199,200,201].
Discussion
MET is a potent oncogene driving invasive growth [2,13,14,15,16,17]. Decades of fundamental research led to the understanding of the different roles of MET in human tumors. New studies are ongoing to fine-tune our understanding of the role of varying MET mutations in cancer [17,77,97]. The biological knowledge of MET needs to be translated into clinical applications. Recent failures of clinical trials can be primarily explained by the lack of consideration of MET genetic alterations before patient selection. On the contrary, when patient stratification was appropriately performed a priori or during post hoc analyses, a general picture of the target patients’ genomic profile can be pictured: MET alterations are an absolute prerequisite for the success of targeted therapies.
These lessons witness the crucial importance of functional preclinical insights to guide clinical practice. Indeed, it was already clear from studies in cell lines and animal models that substantial reduction of cancer cell viability in vitro and tumor shrinkage in vivo occur only in settings where stable and heritable genetic alterations of MET sustain oncogene ‘addiction’. For the large majority of cancer patients, where MET is not mutated, preclinical comprehension of the role of MET can be directly translated to the clinic: MET has anti-apoptotic and pro-invasive functions [145,146]. Adjuvant treatment with MET inhibitors after tumor resection with curative intent could be a viable strategy.
Another unexplained phenomenon is the relatively poor frequency of MET-amplified tumors [16,50,51,52,53,54,55,60] compared to many MET-overexpressing tumors [198,199,200,201]. This apparent paradox can be partially explained by the inducible nature of the MET promoter: ionizing radiations, hypoxia, and cytotoxic reagents enhance MET expression [108,109,110], previously described as oncogene-mediated ‘expedience’ [2]. Translational control mechanisms (e.g., mTOR) might add another layer of regulation of the receptor expression [191]. Another possibility is the existence of other mutations, either in the coding sequence or regulatory regions of the MET gene. The number of previously unidentified MET mutations is constantly increasing thanks to the advances in next-generation sequencing techniques and comprehensive publicly available databases (TCGA, cBioPortal, ClinVar). Thus, efforts aiming at the identification of new variants in cancer must be encouraged. The primary task should be to functionally characterize these mutations to broaden the panel of MET ‘addicted’ tumors and increase the number of patients eligible for next-generation precision medicine.
Author Contributions
Conceptualization, D.M.A. and P.M.C.; writing—original draft preparation, D.M.A and P.M.C.; writing—review and editing, D.M.A. and P.M.C.; visualization, D.M.A.; supervision, P.M.C.; funding acquisition, P.M.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the grant from Associazione Italiana per la Ricerca sul Cancro AIRC-IG-19 Grant (Number 23820 to P.M.C.) and the post-doctoral fellowship from Fondazione Umberto Veronesi to D.M.A (Ref 5109).
Data Availability Statement
Not applicable.
Acknowledgments
We acknowledge Dr. Carla Boccaccio for her thoughtful comments. Illustrations were performed using Biorender.com.
Conflicts of Interest
P.M.C. is a Vertical Bio AG consultant (Basel, Switzerland). Other authors declare no competing interests.
References
- Weidner, K.M.; Hartmann, G.; Naldini, L.; Comoglio, P.M.; Sachs, M.; Fonatsch, C.; Rieder, H.; Birchmeier, W. Molecular characteristics of HGF-SF and its role in cell motility and invasion. EXS 1993, 65, 311–328. [Google Scholar] [PubMed]
- Comoglio, P.M.; Trusolino, L.; Boccaccio, C. Known and novel roles of the MET oncogene in cancer: a coherent approach to targeted therapy. Nat Rev Cancer 2018, 18, 341–358. [Google Scholar] [CrossRef] [PubMed]
- Graveel, C.R.; Tolbert, D.; Vande Woude, G.F. MET: a critical player in tumorigenesis and therapeutic target. Cold Spring Harb Perspect Biol 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Di Renzo, M.F.; Narsimhan, R.P.; Cooper, C.S.; Rosa, C.; Comoglio, P.M. Biosynthesis of the protein encoded by the c-met proto-oncogene. Oncogene 1989, 4, 1383–1388. [Google Scholar]
- Uchikawa, E.; Chen, Z.; Xiao, G.Y.; Zhang, X.; Bai, X.C. Structural basis of the activation of c-MET receptor. Nat Commun 2021, 12, 4074. [Google Scholar] [CrossRef]
- Nakamura, T. Structure and function of hepatocyte growth factor. Prog Growth Factor Res 1991, 3, 67–85. [Google Scholar] [CrossRef]
- Tajima, H.; Matsumoto, K.; Nakamura, T. Regulation of cell growth and motility by hepatocyte growth factor and receptor expression in various cell species. Exp Cell Res 1992, 202, 423–431. [Google Scholar] [CrossRef]
- Ponzetto, C.; Bardelli, A.; Maina, F.; Longati, P.; Panayotou, G.; Dhand, R.; Waterfield, M.D.; Comoglio, P.M. A novel recognition motif for phosphatidylinositol 3-kinase binding mediates its association with the hepatocyte growth factor/scatter factor receptor. Mol Cell Biol 1993, 13, 4600–4608. [Google Scholar] [CrossRef]
- Boccaccio, C.; Ando, M.; Tamagnone, L.; Bardelli, A.; Michieli, P.; Battistini, C.; Comoglio, P.M. Induction of epithelial tubules by growth factor HGF depends on the STAT pathway. Nature 1998, 391, 285–288. [Google Scholar] [CrossRef]
- Ponzetto, C.; Bardelli, A.; Zhen, Z.; Maina, F.; dalla Zonca, P.; Giordano, S.; Graziani, A.; Panayotou, G.; Comoglio, P.M. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 1994, 77, 261–271. [Google Scholar] [CrossRef]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol 2010, 11, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; Vande Woude, G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Boccaccio, C.; Comoglio, P.M. Invasive growth: a MET-driven genetic programme for cancer and stem cells. Nat Rev Cancer 2006, 6, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L.; Comoglio, P.M. Scatter-factor and semaphorin receptors: cell signalling for invasive growth. Nat Rev Cancer 2002, 2, 289–300. [Google Scholar] [CrossRef]
- Christofori, G. New signals from the invasive front. Nature 2006, 441, 444–450. [Google Scholar] [CrossRef]
- Frampton, G.M.; Ali, S.M.; Rosenzweig, M.; Chmielecki, J.; Lu, X.; Bauer, T.M.; Akimov, M.; Bufill, J.A.; Lee, C.; Jentz, D.; et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov 2015, 5, 850–859. [Google Scholar] [CrossRef]
- Cerqua, M.; Botti, O.; Arigoni, M.; Gioelli, N.; Serini, G.; Calogero, R.; Boccaccio, C.; Comoglio, P.M.; Altintas, D.M. MET14 promotes a ligand-dependent, AKT-driven invasive growth. Life Sci Alliance 2022, 5. [Google Scholar] [CrossRef]
- Comoglio, P.M.; Giordano, S.; Trusolino, L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov 2008, 7, 504–516. [Google Scholar] [CrossRef]
- Corso, S.; Migliore, C.; Ghiso, E.; De Rosa, G.; Comoglio, P.M.; Giordano, S. Silencing the MET oncogene leads to regression of experimental tumors and metastases. Oncogene 2008, 27, 684–693. [Google Scholar] [CrossRef]
- Behan, F.M.; Iorio, F.; Picco, G.; Goncalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef]
- Drilon, A.; Clark, J.W.; Weiss, J.; Ou, S.I.; Camidge, D.R.; Solomon, B.J.; Otterson, G.A.; Villaruz, L.C.; Riely, G.J.; Heist, R.S.; et al. Antitumor activity of crizotinib in lung cancers harboring a MET exon 14 alteration. Nat Med 2020, 26, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Seto, T.; Han, J.-Y.; Reguart, N.; Garon, E.B.; Groen, H.J.M.; Tan, D.S.-W.; Hida, T.; Jonge, M.J.D.; Orlov, S.V.; et al. Capmatinib (INC280) in METΔex14-mutated advanced non-small cell lung cancer (NSCLC): Efficacy data from the phase II GEOMETRY mono-1 study. Journal of Clinical Oncology 2019, 37, 9004–9004. [Google Scholar] [CrossRef]
- Landi, L.; Chiari, R.; Tiseo, M.; D’Inca, F.; Dazzi, C.; Chella, A.; Delmonte, A.; Bonanno, L.; Giannarelli, D.; Cortinovis, D.L.; et al. Crizotinib in MET-Deregulated or ROS1-Rearranged Pretreated Non-Small Cell Lung Cancer (METROS): A Phase II, Prospective, Multicenter, Two-Arms Trial. Clin Cancer Res 2019, 25, 7312–7319. [Google Scholar] [CrossRef] [PubMed]
- Cruickshanks, N.; Zhang, Y.; Hine, S.; Gibert, M.; Yuan, F.; Oxford, M.; Grello, C.; Pahuski, M.; Dube, C.; Guessous, F.; et al. Discovery and Therapeutic Exploitation of Mechanisms of Resistance to MET Inhibitors in Glioblastoma. Clin Cancer Res 2019, 25, 663–673. [Google Scholar] [CrossRef]
- Kim, S.; Kim, T.M.; Kim, D.W.; Kim, S.; Kim, M.; Ahn, Y.O.; Keam, B.; Heo, D.S. Acquired Resistance of MET-Amplified Non-small Cell Lung Cancer Cells to the MET Inhibitor Capmatinib. Cancer Res Treat 2019, 51, 951–962. [Google Scholar] [CrossRef]
- Suzawa, K.; Offin, M.; Lu, D.; Kurzatkowski, C.; Vojnic, M.; Smith, R.S.; Sabari, J.K.; Tai, H.; Mattar, M.; Khodos, I.; et al. Activation of KRAS Mediates Resistance to Targeted Therapy in MET Exon 14-mutant Non-small Cell Lung Cancer. Clin Cancer Res 2019, 25, 1248–1260. [Google Scholar] [CrossRef]
- Virzi, A.R.; Gentile, A.; Benvenuti, S.; Comoglio, P.M. Reviving oncogenic addiction to MET bypassed by BRAF (G469A) mutation. Proc Natl Acad Sci U S A 2018, 115, 10058–10063. [Google Scholar] [CrossRef]
- Komada, M.; Hatsuzawa, K.; Shibamoto, S.; Ito, F.; Nakayama, K.; Kitamura, N. Proteolytic processing of the hepatocyte growth factor/scatter factor receptor by furin. FEBS Lett 1993, 328, 25–29. [Google Scholar] [CrossRef]
- Gherardi, E.; Youles, M.E.; Miguel, R.N.; Blundell, T.L.; Iamele, L.; Gough, J.; Bandyopadhyay, A.; Hartmann, G.; Butler, P.J. Functional map and domain structure of MET, the product of the c-met protooncogene and receptor for hepatocyte growth factor/scatter factor. Proc Natl Acad Sci U S A 2003, 100, 12039–12044. [Google Scholar] [CrossRef]
- Zhen, Z.; Giordano, S.; Longati, P.; Medico, E.; Campiglio, M.; Comoglio, P. Structural and functional domains critical for constitutive activation of the HGF-receptor (Met). Oncogene 1994, 9, 1691–1697. [Google Scholar]
- Altintas, D.M.; Gallo, S.; Basilico, C.; Cerqua, M.; Bocedi, A.; Vitacolonna, A.; Botti, O.; Casanova, E.; Rancati, I.; Milanese, C.; et al. The PSI Domain of the MET Oncogene Encodes a Functional Disulfide Isomerase Essential for the Maturation of the Receptor Precursor. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Longati, P.; Bardelli, A.; Ponzetto, C.; Naldini, L.; Comoglio, P.M. Tyrosines1234-1235 are critical for activation of the tyrosine kinase encoded by the MET proto-oncogene (HGF receptor). Oncogene 1994, 9, 49–57. [Google Scholar] [PubMed]
- Giordano, S.; Bardelli, A.; Zhen, Z.; Menard, S.; Ponzetto, C.; Comoglio, P.M. A point mutation in the MET oncogene abrogates metastasis without affecting transformation. Proc Natl Acad Sci U S A 1997, 94, 13868–13872. [Google Scholar] [CrossRef] [PubMed]
- Ponzetto, C.; Zhen, Z.; Audero, E.; Maina, F.; Bardelli, A.; Basile, M.L.; Giordano, S.; Narsimhan, R.; Comoglio, P. Specific uncoupling of GRB2 from the Met receptor. Differential effects on transformation and motility. J Biol Chem 1996, 271, 14119–14123. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Peschard, P.; Fournier, T.M.; Lamorte, L.; Naujokas, M.A.; Band, H.; Langdon, W.Y.; Park, M. Mutation of the c-Cbl TKB domain binding site on the Met receptor tyrosine kinase converts it into a transforming protein. Mol Cell 2001, 8, 995–1004. [Google Scholar] [CrossRef]
- Lefebvre, J.; Ancot, F.; Leroy, C.; Muharram, G.; Lemiere, A.; Tulasne, D. Met degradation: more than one stone to shoot a receptor down. FASEB J 2012, 26, 1387–1399. [Google Scholar] [CrossRef]
- Abella, J.V.; Peschard, P.; Naujokas, M.A.; Lin, T.; Saucier, C.; Urbe, S.; Park, M. Met/Hepatocyte growth factor receptor ubiquitination suppresses transformation and is required for Hrs phosphorylation. Mol Cell Biol 2005, 25, 9632–9645. [Google Scholar] [CrossRef]
- Tan, Y.H.; Krishnaswamy, S.; Nandi, S.; Kanteti, R.; Vora, S.; Onel, K.; Hasina, R.; Lo, F.Y.; El-Hashani, E.; Cervantes, G.; et al. CBL is frequently altered in lung cancers: its relationship to mutations in MET and EGFR tyrosine kinases. PLoS One 2010, 5, e8972. [Google Scholar] [CrossRef]
- Villa-Moruzzi, E.; Puntoni, F.; Bardelli, A.; Vigna, E.; De Rosa, S.; Comoglio, P.M. Protein tyrosine phosphatase PTP-S binds to the juxtamembrane region of the hepatocyte growth factor receptor Met. Biochem J 1998, 336 Pt 1, 235–239. [Google Scholar] [CrossRef]
- Lee, C.C.; Yamada, K.M. Identification of a novel type of alternative splicing of a tyrosine kinase receptor. Juxtamembrane deletion of the c-met protein kinase C serine phosphorylation regulatory site. J Biol Chem 1994, 269, 19457–19461. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Gao, C.F.; Lee, C.C.; Kim, M.D.; Vande Woude, G.F. An alternatively spliced form of Met receptor is tumorigenic. Exp Mol Med 2006, 38, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. A signaling adapter function for alpha6beta4 integrin in the control of HGF-dependent invasive growth. Cell 2001, 107, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Go, H.; Jeon, Y.K.; Park, H.J.; Sung, S.W.; Seo, J.W.; Chung, D.H. High MET gene copy number leads to shorter survival in patients with non-small cell lung cancer. J Thorac Oncol 2010, 5, 305–313. [Google Scholar] [CrossRef]
- Graziano, F.; Galluccio, N.; Lorenzini, P.; Ruzzo, A.; Canestrari, E.; D’Emidio, S.; Catalano, V.; Sisti, V.; Ligorio, C.; Andreoni, F.; et al. Genetic activation of the MET pathway and prognosis of patients with high-risk, radically resected gastric cancer. J Clin Oncol 2011, 29, 4789–4795. [Google Scholar] [CrossRef]
- Zhang, M.; Li, G.; Sun, X.; Ni, S.; Tan, C.; Xu, M.; Huang, D.; Ren, F.; Li, D.; Wei, P.; et al. MET amplification, expression, and exon 14 mutations in colorectal adenocarcinoma. Hum Pathol 2018, 77, 108–115. [Google Scholar] [CrossRef]
- Pal, S.K.; Ali, S.M.; Yakirevich, E.; Geynisman, D.M.; Karam, J.A.; Elvin, J.A.; Frampton, G.M.; Huang, X.; Lin, D.I.; Rosenzweig, M.; et al. Characterization of Clinical Cases of Advanced Papillary Renal Cell Carcinoma via Comprehensive Genomic Profiling. Eur Urol 2018, 73, 71–78. [Google Scholar] [CrossRef]
- Cheng, F.; Guo, D. MET in glioma: signaling pathways and targeted therapies. J Exp Clin Cancer Res 2019, 38, 270. [Google Scholar] [CrossRef]
- Zhou, Y.; Song, K.Y.; Giubellino, A. The Role of MET in Melanoma and Melanocytic Lesions. Am J Pathol 2019, 189, 2138–2148. [Google Scholar] [CrossRef]
- Houldsworth, J.; Cordon-Cardo, C.; Ladanyi, M.; Kelsen, D.P.; Chaganti, R.S. Gene amplification in gastric and esophageal adenocarcinomas. Cancer Res 1990, 50, 6417–6422. [Google Scholar]
- Kuniyasu, H.; Yasui, W.; Kitadai, Y.; Yokozaki, H.; Ito, H.; Tahara, E. Frequent amplification of the c-met gene in scirrhous type stomach cancer. Biochem Biophys Res Commun 1992, 189, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Ooi, A.; Kobayashi, M.; Mai, M.; Yanagihara, K.; Nakanishi, I. Amplification of c-myc, K-sam, and c-met in gastric cancers: detection by fluorescence in situ hybridization. Lab Invest 1998, 78, 1143–1153. [Google Scholar] [PubMed]
- Miller, C.T.; Lin, L.; Casper, A.M.; Lim, J.; Thomas, D.G.; Orringer, M.B.; Chang, A.C.; Chambers, A.F.; Giordano, T.J.; Glover, T.W.; et al. Genomic amplification of MET with boundaries within fragile site FRA7G and upregulation of MET pathways in esophageal adenocarcinoma. Oncogene 2006, 25, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Bubendorf, L.; Dafni, U.; Schobel, M.; Finn, S.P.; Tischler, V.; Sejda, A.; Marchetti, A.; Thunnissen, E.; Verbeken, E.K.; Warth, A.; et al. Prevalence and clinical association of MET gene overexpression and amplification in patients with NSCLC: Results from the European Thoracic Oncology Platform (ETOP) Lungscape project. Lung Cancer 2017, 111, 143–149. [Google Scholar] [CrossRef]
- Bean, J.; Brennan, C.; Shih, J.Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A 2007, 104, 20932–20937. [Google Scholar] [CrossRef]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov 2013, 3, 658–673. [Google Scholar] [CrossRef]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol Cancer 2018, 17, 58. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Vuong, H.G.; Ho, A.T.N.; Altibi, A.M.A.; Nakazawa, T.; Katoh, R.; Kondo, T. Clinicopathological implications of MET exon 14 mutations in non-small cell lung cancer - A systematic review and meta-analysis. Lung Cancer 2018, 123, 76–82. [Google Scholar] [CrossRef]
- Vigna, E.; Gramaglia, D.; Longati, P.; Bardelli, A.; Comoglio, P.M. Loss of the exon encoding the juxtamembrane domain is essential for the oncogenic activation of TPR-MET. Oncogene 1999, 18, 4275–4281. [Google Scholar] [CrossRef] [PubMed]
- Food, U.; Administration, D. TABRECTA (capmatinib) Prescribing Information. 2022.
- Wolf, J.; Seto, T.; Han, J.-Y.; Reguart, N.; Garon, E.B.; Groen, H.J.; Tan, D.S.; Hida, T.; de Jonge, M.; Orlov, S.V. Capmatinib in MET exon 14–mutated or MET-amplified non–small-cell lung cancer. New England Journal of Medicine 2020, 383, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Leonardi, G.C.; Kravets, S.; Dahlberg, S.E.; Drilon, A.; Noonan, S.A.; Camidge, D.R.; Ou, S.-H.I.; Costa, D.B.; Gadgeel, S.M. Impact of MET inhibitors on survival among patients with non-small cell lung cancer harboring MET exon 14 mutations: a retrospective analysis. Lung Cancer 2019, 133, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Clark, J.W.; Weiss, J.; Ou, S.-H.I.; Camidge, D.R.; Solomon, B.J.; Otterson, G.A.; Villaruz, L.C.; Riely, G.J.; Heist, R.S. Antitumor activity of crizotinib in lung cancers harboring a MET exon 14 alteration. Nature medicine 2020, 26, 47–51. [Google Scholar] [CrossRef]
- Paik, P.K.; Drilon, A.; Fan, P.-D.; Yu, H.; Rekhtman, N.; Ginsberg, M.S.; Borsu, L.; Schultz, N.; Berger, M.F.; Rudin, C.M. Response to MET Inhibitors in Patients with Stage IV Lung Adenocarcinomas Harboring MET Mutations Causing Exon 14 SkippingMET Inhibitors in MET Exon 14 Splice Variant Lung Cancer. Cancer discovery 2015, 5, 842–849. [Google Scholar] [CrossRef]
- Lu, S.; Fang, J.; Li, X.; Cao, L.; Zhou, J.; Guo, Q.; Liang, Z.; Cheng, Y.; Jiang, L.; Yang, N. Phase II study of savolitinib in patients (pts) with pulmonary sarcomatoid carcinoma (PSC) and other types of non-small cell lung cancer (NSCLC) harboring MET exon 14 skipping mutations (METex14+). 2020.
- Engstrom, L.D.; Aranda, R.; Lee, M.; Tovar, E.A.; Essenburg, C.J.; Madaj, Z.; Chiang, H.; Briere, D.; Hallin, J.; Lopez-Casas, P.P.; et al. Glesatinib Exhibits Antitumor Activity in Lung Cancer Models and Patients Harboring MET Exon 14 Mutations and Overcomes Mutation-mediated Resistance to Type I MET Inhibitors in Nonclinical Models. Clin Cancer Res 2017, 23, 6661–6672. [Google Scholar] [CrossRef]
- Paik, P.K.; Drilon, A.; Fan, P.D.; Yu, H.; Rekhtman, N.; Ginsberg, M.S.; Borsu, L.; Schultz, N.; Berger, M.F.; Rudin, C.M.; et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov 2015, 5, 842–849. [Google Scholar] [CrossRef]
- Paik, P.K.; Felip, E.; Veillon, R.; Sakai, H.; Cortot, A.B.; Garassino, M.C.; Mazieres, J.; Viteri, S.; Senellart, H.; Van Meerbeeck, J.; et al. Tepotinib in Non-Small-Cell Lung Cancer with MET Exon 14 Skipping Mutations. N Engl J Med 2020, 383, 931–943. [Google Scholar] [CrossRef]
- Wolf, J.; Seto, T.; Han, J.Y.; Reguart, N.; Garon, E.B.; Groen, H.J.M.; Tan, D.S.W.; Hida, T.; de Jonge, M.; Orlov, S.V.; et al. Capmatinib in MET Exon 14-Mutated or MET-Amplified Non-Small-Cell Lung Cancer. N Engl J Med 2020, 383, 944–957. [Google Scholar] [CrossRef]
- Hu, H.; Mu, Q.; Bao, Z.; Chen, Y.; Liu, Y.; Chen, J.; Wang, K.; Wang, Z.; Nam, Y.; Jiang, B.; et al. Mutational Landscape of Secondary Glioblastoma Guides MET-Targeted Trial in Brain Tumor. Cell 2018, 175, 1665–1678.e1618. [Google Scholar] [CrossRef]
- Klempner, S.J.; Borghei, A.; Hakimian, B.; Ali, S.M.; Ou, S.I. Intracranial Activity of Cabozantinib in MET Exon 14-Positive NSCLC with Brain Metastases. J Thorac Oncol 2017, 12, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Moro-Sibilot, D.; Cozic, N.; Perol, M.; Mazieres, J.; Otto, J.; Souquet, P.J.; Bahleda, R.; Wislez, M.; Zalcman, G.; Guibert, S.D.; et al. Crizotinib in c-MET- or ROS1-positive NSCLC: results of the AcSe phase II trial. Ann Oncol 2019, 30, 1985–1991. [Google Scholar] [CrossRef] [PubMed]
- Salgia, R.; Sattler, M.; Scheele, J.; Stroh, C.; Felip, E. The promise of selective MET inhibitors in non-small cell lung cancer with MET exon 14 skipping. Cancer Treat Rev 2020, 87, 102022. [Google Scholar] [CrossRef] [PubMed]
- Fujino, T.; Suda, K.; Mitsudomi, T. Lung Cancer with MET exon 14 Skipping Mutation: Genetic Feature, Current Treatments, and Future Challenges. Lung Cancer (Auckl) 2021, 12, 35–50. [Google Scholar] [CrossRef]
- Wang, F.; Liu, Y.; Qiu, W.; Shum, E.; Feng, M.; Zhao, D.; Zheng, D.; Borczuk, A.; Cheng, H.; Halmos, B. Functional Analysis of MET Exon 14 Skipping Alteration in Cancer Invasion and Metastatic Dissemination. Cancer Res 2022, 82, 1365–1379. [Google Scholar] [CrossRef] [PubMed]
- Rotow, J.K.; Gui, P.; Wu, W.; Raymond, V.M.; Lanman, R.B.; Kaye, F.J.; Peled, N.; Fece de la Cruz, F.; Nadres, B.; Corcoran, R.B.; et al. Co-occurring Alterations in the RAS-MAPK Pathway Limit Response to MET Inhibitor Treatment in MET Exon 14 Skipping Mutation-Positive Lung Cancer. Clin Cancer Res 2020, 26, 439–449. [Google Scholar] [CrossRef]
- Jamme, P.; Fernandes, M.; Copin, M.C.; Descarpentries, C.; Escande, F.; Morabito, A.; Gregoire, V.; Jamme, M.; Baldacci, S.; Tulasne, D.; et al. Alterations in the PI3K Pathway Drive Resistance to MET Inhibitors in NSCLC Harboring MET Exon 14 Skipping Mutations. J Thorac Oncol 2020, 15, 741–751. [Google Scholar] [CrossRef]
- Schmidt, L.; Duh, F.M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; Scherer, S.W.; Zhuang, Z.; Lubensky, I.; Dean, M.; et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet 1997, 16, 68–73. [Google Scholar] [CrossRef]
- Schmidt, L.; Junker, K.; Nakaigawa, N.; Kinjerski, T.; Weirich, G.; Miller, M.; Lubensky, I.; Neumann, H.P.; Brauch, H.; Decker, J. Novel mutations of the MET proto-oncogene in papillary renal carcinomas. Oncogene 1999, 18, 2343–2350. [Google Scholar] [CrossRef]
- Zhuang, Z.; Park, W.-S.; Pack, S.; Schmidt, L.; Vortmeyer, A.O.; Pak, E.; Pham, T.; Weil, R.J.; Candidus, S.; Lubensky, I.A. Trisomy 7-harbouring non-random duplication of the mutant MET allele in hereditary papillary renal carcinomas. Nature genetics 1998, 20, 66–69. [Google Scholar] [CrossRef]
- Jeffers, M.; Schmidt, L.; Nakaigawa, N.; Webb, C.P.; Weirich, G.; Kishida, T.; Zbar, B.; Vande Woude, G.F. Activating mutations for the met tyrosine kinase receptor in human cancer. Proceedings of the National Academy of Sciences 1997, 94, 11445–11450. [Google Scholar] [CrossRef] [PubMed]
- Jeffers, M.; Fiscella, M.; Webb, C.P.; Anver, M.; Koochekpour, S.; Vande Woude, G.F. The mutationally activated Met receptor mediates motility and metastasis. Proceedings of the National Academy of Sciences 1998, 95, 14417–14422. [Google Scholar] [CrossRef]
- Graveel, C.; Su, Y.; Koeman, J.; Wang, L.-M.; Tessarollo, L.; Fiscella, M.; Birchmeier, C.; Swiatek, P.; Bronson, R.; Vande Woude, G. Activating Met mutations produce unique tumor profiles in mice with selective duplication of the mutant allele. Proceedings of the National Academy of Sciences 2004, 101, 17198–17203. [Google Scholar] [CrossRef] [PubMed]
- Graveel, C.R.; London, C.A.; Woude, G.F.V. A mouse model of activating Met mutations. Cell cycle 2005, 4, 518–520. [Google Scholar] [CrossRef]
- Park, W.S.; Dong, S.M.; Kim, S.Y.; Na, E.Y.; Shin, M.S.; Pi, J.H.; Kim, B.J.; Bae, J.H.; Hong, Y.K.; Lee, K.S. Somatic mutations in the kinase domain of the Met/hepatocyte growth factor receptor gene in childhood hepatocellular carcinomas. Cancer research 1999, 59, 307–310. [Google Scholar] [PubMed]
- Di Renzo, M.F.; Olivero, M.; Martone, T.; Maffe, A.; Maggiora, P.; De Stefani, A.; Valente, G.; Giordano, S.; Cortesina, G.; Comoglio, P. Somatic mutations of the MET oncogene are selected during metastatic spread of human HNSC carcinomas. Oncogene 2000, 19, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, D.M.; Landt, O.; Berthou, S.; Gruber, G.; Beer, K.T.; Greiner, R.H.; Zimmer, Y. Prevalence and clinical impact of Met Y1253D-activating point mutation in radiotherapy-treated squamous cell cancer of the oropharynx. Oncogene 2003, 22, 8519–8523. [Google Scholar] [CrossRef]
- Lee, J.-H.; Han, S.-U.; Cho, H.; Jennings, B.; Gerrard, B.; Dean, M.; Schmidt, L.; Zbar, B.; Vande Woude, G.F. A novel germ line juxtamembrane Met mutation in human gastric cancer. Oncogene 2000, 19, 4947–4953. [Google Scholar] [CrossRef]
- Ghadjar, P.; Blank-Liss, W.; Simcock, M.; Hegyi, I.; Beer, K.T.; Moch, H.; Aebersold, D.M.; Zimmer, Y. MET Y1253D-activating point mutation and development of distant metastasis in advanced head and neck cancers. Clinical & experimental metastasis 2009, 26, 809–815. [Google Scholar]
- Stella, G.M.; Benvenuti, S.; Gramaglia, D.; Scarpa, A.; Tomezzoli, A.; Cassoni, P.; Senetta, R.; Venesio, T.; Pozzi, E.; Bardelli, A. MET mutations in cancers of unknown primary origin (CUPs). Human mutation 2011, 32, 44–50. [Google Scholar] [CrossRef]
- Neklason, D.W.; Done, M.W.; Sargent, N.R.; Schwartz, A.G.; Anton-Culver, H.; Griffin, C.A.; Ahnen, D.J.; Schildkraut, J.M.; Tomlinson, G.E.; Strong, L.C. Activating mutation in MET oncogene in familial colorectal cancer. BMC cancer 2011, 11, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kong-Beltran, M.; Stamos, J.; Wickramasinghe, D. The Sema domain of Met is necessary for receptor dimerization and activation. Cancer cell 2004, 6, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.C.; Jagadeeswaran, R.; Jagadeesh, S.; Tretiakova, M.S.; Nallasura, V.; Fox, E.A.; Hansen, M.; Schaefer, E.; Naoki, K.; Lader, A. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non–small cell lung cancer. Cancer research 2005, 65, 1479–1488. [Google Scholar] [CrossRef] [PubMed]
- de Melo Gagliato, D.; Jardim, D.L.F.; Falchook, G.; Tang, C.; Zinner, R.; Wheler, J.J.; Janku, F.; Subbiah, V.; Piha-Paul, S.A.; Fu, S. Analysis of MET genetic aberrations in patients with breast cancer at MD Anderson Phase I unit. Clinical breast cancer 2014, 14, 468–474. [Google Scholar] [CrossRef]
- Sebai, M.; Tulasne, D.; Caputo, S.M.; Verkarre, V.; Fernandes, M.; Guerin, C.; Reinhart, F.; Adams, S.; Maugard, C.; Caron, O.; et al. Novel germline MET pathogenic variants in French patients with papillary renal cell carcinomas type I. Hum Mutat 2022, 43, 316–327. [Google Scholar] [CrossRef]
- Verginelli, F.; Pisacane, A.; Gambardella, G.; D’Ambrosio, A.; Candiello, E.; Ferrio, M.; Panero, M.; Casorzo, L.; Benvenuti, S.; Cascardi, E.; et al. Cancer of unknown primary stem-like cells model multi-organ metastasis and unveil liability to MEK inhibition. Nat Commun 2021, 12, 2498. [Google Scholar] [CrossRef]
- Wang, R.; Ferrell, L.D.; Faouzi, S.; Maher, J.J.; Bishop, J.M. Activation of the Met receptor by cell attachment induces and sustains hepatocellular carcinomas in transgenic mice. J Cell Biol 2001, 153, 1023–1034. [Google Scholar] [CrossRef]
- Jeffers, M.; Rong, S.; Vande Woude, G.F. Hepatocyte growth factor/scatter factor-Met signaling in tumorigenicity and invasion/metastasis. J Mol Med (Berl) 1996, 74, 505–513. [Google Scholar] [CrossRef]
- Yu, J.; Miehlke, S.; Ebert, M.P.; Hoffmann, J.; Breidert, M.; Alpen, B.; Starzynska, T.; Stolte Prof, M.; Malfertheiner, P.; Bayerdorffer, E. Frequency of TPR-MET rearrangement in patients with gastric carcinoma and in first-degree relatives. Cancer 2000, 88, 1801–1806. [Google Scholar] [CrossRef]
- Stransky, N.; Cerami, E.; Schalm, S.; Kim, J.L.; Lengauer, C. The landscape of kinase fusions in cancer. Nat Commun 2014, 5, 4846. [Google Scholar] [CrossRef]
- Bao, Z.S.; Chen, H.M.; Yang, M.Y.; Zhang, C.B.; Yu, K.; Ye, W.L.; Hu, B.Q.; Yan, W.; Zhang, W.; Akers, J.; et al. RNA-seq of 272 gliomas revealed a novel, recurrent PTPRZ1-MET fusion transcript in secondary glioblastomas. Genome Res 2014, 24, 1765–1773. [Google Scholar] [CrossRef] [PubMed]
- International Cancer Genome Consortium PedBrain Tumor, P. Recurrent MET fusion genes represent a drug target in pediatric glioblastoma. Nat Med 2016, 22, 1314–1320. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.M.; Yu, K.; Tang, X.Y.; Bao, Z.S.; Jiang, T.; Fan, X.L.; Chen, X.W.; Su, X.D. Enhanced expression and phosphorylation of the MET oncoprotein by glioma-specific PTPRZ1-MET fusions. FEBS Lett 2015, 589, 1437–1443. [Google Scholar] [CrossRef]
- Huang, R.; Liu, Y.; Wang, K.; Wang, Z.; Zhang, C.; Zhang, W.; Zhao, Z.; Li, G.; Huang, L.; Chang, Y.; et al. High-sensitive clinical diagnostic method for PTPRZ1-MET and the characteristic protein structure contributing to ligand-independent MET activation. CNS Neurosci Ther 2021, 27, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Plenker, D.; Bertrand, M.; de Langen, A.J.; Riedel, R.; Lorenz, C.; Scheel, A.H.; Muller, J.; Bragelmann, J.; Dassler-Plenker, J.; Kobe, C.; et al. Structural Alterations of MET Trigger Response to MET Kinase Inhibition in Lung Adenocarcinoma Patients. Clin Cancer Res 2018, 24, 1337–1343. [Google Scholar] [CrossRef]
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer cell 2003, 3, 347–361. [Google Scholar] [CrossRef]
- De Bacco, F.; Luraghi, P.; Medico, E.; Reato, G.; Girolami, F.; Perera, T.; Gabriele, P.; Comoglio, P.M.; Boccaccio, C. Induction of MET by ionizing radiation and its role in radioresistance and invasive growth of cancer. JNCI: Journal of the National Cancer Institute 2011, 103, 645–661. [Google Scholar] [CrossRef] [PubMed]
- Gambarotta, G.; Pistoi, S.; Giordano, S.; Comoglio, P.M.; Santoro, C. Structure and inducible regulation of the human MET promoter. Journal of Biological Chemistry 1994, 269, 12852–12857. [Google Scholar] [CrossRef]
- Peters, S.; Adjei, A.A. MET: a promising anticancer therapeutic target. Nature reviews Clinical oncology 2012, 9, 314–326. [Google Scholar] [CrossRef]
- Linklater, E.S.; Tovar, E.A.; Essenburg, C.J.; Turner, L.; Madaj, Z.; Winn, M.E.; Melnik, M.K.; Korkaya, H.; Maroun, C.R.; Christensen, J.G.; et al. Targeting MET and EGFR crosstalk signaling in triple-negative breast cancers. Oncotarget 2016, 7, 69903–69915. [Google Scholar] [CrossRef]
- Jo, M.; Stolz, D.B.; Esplen, J.E.; Dorko, K.; Michalopoulos, G.K.; Strom, S.C. Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. J Biol Chem 2000, 275, 8806–8811. [Google Scholar] [CrossRef] [PubMed]
- K, K.B.; Balagopal, A.; Vizeacoumar, F.S.; Vizeacoumar, F.J.; Freywald, A.; Giambra, V. Protein Tyrosine Kinases: Their Roles and Their Targeting in Leukemia. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef]
- Toschi, L.; Janne, P.A. Single-agent and combination therapeutic strategies to inhibit hepatocyte growth factor/MET signaling in cancer. Clinical Cancer Research 2008, 14, 5941–5946. [Google Scholar] [CrossRef]
- Bell, D.W.; Gore, I.; Okimoto, R.A.; Godin-Heymann, N.; Sordella, R.; Mulloy, R.; Sharma, S.V.; Brannigan, B.W.; Mohapatra, G.; Settleman, J. Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nature genetics 2005, 37, 1315–1316. [Google Scholar] [CrossRef]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non–small-cell lung cancer to gefitinib. New England Journal of Medicine 2005, 352, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L.; Trent, J.C.; Wu, E.F.; Fuller, G.N.; Ramdas, L.; Zhang, W.; Raymond, A.K.; Prieto, V.G.; Oyedeji, C.O.; Hunt, K.K. A missense mutation in KIT kinase domain 1 correlates with imatinib resistance in gastrointestinal stromal tumors. Cancer research 2004, 64, 5913–5919. [Google Scholar] [CrossRef]
- Gorre, M.E.; Mohammed, M.; Ellwood, K.; Hsu, N.; Paquette, R.; Rao, P.N.; Sawyers, C.L. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001, 293, 876–880. [Google Scholar] [CrossRef]
- Chun, S.-Y.; Kwon, Y.-S.; Nam, K.-S.; Kim, S. Lapatinib enhances the cytotoxic effects of doxorubicin in MCF-7 tumorspheres by inhibiting the drug efflux function of ABC transporters. Biomedicine & Pharmacotherapy 2015, 72, 37–43. [Google Scholar]
- Brouwer, K.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.; Hoffmaster, K.; Ishikawa, T.; Keppler, D.; Kim, R. Membrane transporters in drug development. Nature 2010. [Google Scholar]
- Filomeni, G.; Turella, P.; Dupuis, M.L.; Forini, O.; Ciriolo, M.R.; Cianfriglia, M.; Pezzola, S.; Federici, G.; Caccuri, A.M. 6-(7-Nitro-2, 1, 3-benzoxadiazol-4-ylthio) hexanol, a specific glutathione S-transferase inhibitor, overcomes the multidrug resistance (MDR)-associated protein 1–mediated MDR in small cell lung cancer. Molecular cancer therapeutics 2008, 7, 371–379. [Google Scholar] [CrossRef]
- Rodriguez-Antona, C.; Ingelman-Sundberg, M. Cytochrome P450 pharmacogenetics and cancer. Oncogene 2006, 25, 1679–1691. [Google Scholar] [CrossRef] [PubMed]
- Martens, T.; Schmidt, N.-O.; Eckerich, C.; Fillbrandt, R.; Merchant, M.; Schwall, R.; Westphal, M.; Lamszus, K. A novel one-armed anti-c-Met antibody inhibits glioblastoma growth in vivo. Clinical cancer research 2006, 12, 6144–6152. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.-S.; Kang, S.; Kim, K.-A.; Song, Y.-J.; Cheong, K.H.; Cha, H.-Y.; Kim, C.H. Met degradation by SAIT301, a Met monoclonal antibody, reduces the invasion and migration of nasopharyngeal cancer cells via inhibition of EGR-1 expression. Cell death & disease 2014, 5, e1159–e1159. [Google Scholar]
- Lee, J.M.; Kim, B.; Lee, S.; Jeong, Y.; Oh, Y.; Song, Y.; Jung, S.; Choi, J.; Lee, S.; Cheong, K. Cbl-independent degradation of Met: ways to avoid agonism of bivalent Met-targeting antibody. Oncogene 2014, 33, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.R.; Handsley, M.M.; Pennington, C.J. The ADAM metalloproteinases. Molecular aspects of medicine 2008, 29, 258–289. [Google Scholar] [CrossRef]
- Vigna, E.; Pacchiana, G.; Mazzone, M.; Chiriaco, C.; Fontani, L.; Basilico, C.; Pennacchietti, S.; Comoglio, P.M. “Active” cancer immunotherapy by anti-Met antibody gene transfer. Cancer research 2008, 68, 9176–9183. [Google Scholar] [CrossRef] [PubMed]
- Prat, M.; Crepaldi, T.; Pennacchietti, S.; Bussolino, F.; Comoglio, P.M. Agonistic monoclonal antibodies against the Met receptor dissect the biological responses to HGF. Journal of cell science 1998, 111, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, A.; Circosta, P.; Granziero, L.; Mazzone, M.; Pisacane, A.; Fenoglio, S.; Comoglio, P.M.; Giordano, S. Ab-induced ectodomain shedding mediates hepatocyte growth factor receptor down-regulation and hampers biological activity. Proceedings of the National Academy of Sciences 2006, 103, 5090–5095. [Google Scholar] [CrossRef]
- Schelter, F.; Kobuch, J.; Moss, M.L.; Becherer, J.D.; Comoglio, P.M.; Boccaccio, C.; Krüger, A. A disintegrin and metalloproteinase-10 (ADAM-10) mediates DN30 antibody-induced shedding of the met surface receptor. Journal of Biological Chemistry 2010, 285, 26335–26340. [Google Scholar] [CrossRef]
- Foveau, B.; Ancot, F.; Leroy, C.; Petrelli, A.; Reiss, K.; Vingtdeux, V.; Giordano, S.; Fafeur, V.; Tulasne, D. Down-regulation of the met receptor tyrosine kinase by presenilin-dependent regulated intramembrane proteolysis. Molecular biology of the cell 2009, 20, 2495–2507. [Google Scholar] [CrossRef]
- Martinelli, I.; Modica, C.; Chiriaco, C.; Basilico, C.; Hughes, J.M.; Corso, S.; Giordano, S.; Comoglio, P.M.; Vigna, E. hOA-DN30: a highly effective humanized single-arm MET antibody inducing remission of ‘MET-addicted’ cancers. J Exp Clin Cancer Res 2022, 41, 112. [Google Scholar] [CrossRef]
- Burgess, T.L.; Sun, J.; Meyer, S.; Tsuruda, T.S.; Sun, J.; Elliott, G.; Chen, Q.; Haniu, M.; Barron, W.F.; Juan, T. Biochemical Characterization of AMG 102: A Neutralizing, Fully Human Monoclonal Antibody to Human and Nonhuman Primate Hepatocyte Growth FactorAMG 102 Neutralizes Hepatocyte Growth Factor. Molecular cancer therapeutics 2010, 9, 400–409. [Google Scholar] [CrossRef]
- Catenacci, D.V.T.; Tebbutt, N.C.; Davidenko, I.; Murad, A.M.; Al-Batran, S.E.; Ilson, D.H.; Tjulandin, S.; Gotovkin, E.; Karaszewska, B.; Bondarenko, I.; et al. Rilotumumab plus epirubicin, cisplatin, and capecitabine as first-line therapy in advanced MET-positive gastric or gastro-oesophageal junction cancer (RILOMET-1): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2017, 18, 1467–1482. [Google Scholar] [CrossRef]
- Bussolino, F.; Di Renzo, M.F.; Ziche, M.; Bocchietto, E.; Olivero, M.; Naldini, L.; Gaudino, G.; Tamagnone, L.; Coffer, A.; Comoglio, P. Hepatocyte growth factor is a potent angiogenic factor which stimulates endothelial cell motility and growth. The Journal of cell biology 1992, 119, 629–641. [Google Scholar] [CrossRef]
- Wu, X.; Chen, X.; Zhou, Q.; Li, P.; Yu, B.; Li, J.; Qu, Y.; Yan, J.; Yu, Y.; Yan, M. Hepatocyte growth factor activates tumor stromal fibroblasts to promote tumorigenesis in gastric cancer. Cancer letters 2013, 335, 128–135. [Google Scholar] [CrossRef]
- Galimi, F.; Cottone, E.; Vigna, E.; Arena, N.; Boccaccio, C.; Giordano, S.; Naldini, L.; Comoglio, P.M. Hepatocyte growth factor is a regulator of monocyte-macrophage function. The Journal of Immunology 2001, 166, 1241–1247. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, J.; Chen, S.; Lu, M.; Luo, X.; Yao, S.; Liu, S.; Qin, Y.; Chen, H. Tumor-associated macrophages provide a suitable microenvironment for non-small lung cancer invasion and progression. Lung cancer 2011, 74, 188–196. [Google Scholar] [CrossRef]
- McDermott, U.; Sharma, S.V.; Dowell, L.; Greninger, P.; Montagut, C.; Lamb, J.; Archibald, H.; Raudales, R.; Tam, A.; Lee, D.; et al. Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proc Natl Acad Sci U S A 2007, 104, 19936–19941. [Google Scholar] [CrossRef]
- Grande, E.; Giovannini, M.; Marriere, E.; Pultar, P.; Quinlan, M.; Chen, X.; Rahmanzadeh, G.; Curigliano, G.; Cui, X. Effect of capmatinib on the pharmacokinetics of digoxin and rosuvastatin administered as a 2-drug cocktail in patients with MET-dysregulated advanced solid tumours: A phase I, multicentre, open-label, single-sequence drug-drug interaction study. Br J Clin Pharmacol 2021, 87, 2867–2878. [Google Scholar] [CrossRef]
- Scagliotti, G.; Moro-Sibilot, D.; Kollmeier, J.; Favaretto, A.; Cho, E.K.; Grosch, H.; Kimmich, M.; Girard, N.; Tsai, C.M.; Hsia, T.C.; et al. A Randomized-Controlled Phase 2 Study of the MET Antibody Emibetuzumab in Combination with Erlotinib as First-Line Treatment for EGFR Mutation-Positive NSCLC Patients. J Thorac Oncol 2020, 15, 80–90. [Google Scholar] [CrossRef]
- Dieras, V.; Campone, M.; Yardley, D.A.; Romieu, G.; Valero, V.; Isakoff, S.J.; Koeppen, H.; Wilson, T.R.; Xiao, Y.; Shames, D.S.; et al. Randomized, phase II, placebo-controlled trial of onartuzumab and/or bevacizumab in combination with weekly paclitaxel in patients with metastatic triple-negative breast cancer. Ann Oncol 2015, 26, 1904–1910. [Google Scholar] [CrossRef] [PubMed]
- Benvenuti, S.; Gentile, A.; Lazzari, L.; Arnesano, A.; Trusolino, L.; Comoglio, P.M. An ‘in-cell trial’ to assess the efficacy of a monovalent anti-MET antibody as monotherapy and in association with standard cytotoxics. Mol Oncol 2014, 8, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Michieli, P.; Mazzone, M.; Basilico, C.; Cavassa, S.; Sottile, A.; Naldini, L.; Comoglio, P.M. Targeting the tumor and its microenvironment by a dual-function decoy Met receptor. Cancer Cell 2004, 6, 61–73. [Google Scholar] [CrossRef]
- Mazzone, M.; Basilico, C.; Cavassa, S.; Pennacchietti, S.; Risio, M.; Naldini, L.; Comoglio, P.M.; Michieli, P. An uncleavable form of pro-scatter factor suppresses tumor growth and dissemination in mice. J Clin Invest 2004, 114, 1418–1432. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nature reviews Clinical oncology 2017, 14, 531–548. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nature Reviews Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Oxnard, G.R.; Jackman, D.M.; Savukoski, D.O.; Hall, D.; Shivdasani, P.; Heng, J.C.; Dahlberg, S.E.; Janne, P.A.; Verma, S.; et al. MET Exon 14 Mutations in Non-Small-Cell Lung Cancer Are Associated With Advanced Age and Stage-Dependent MET Genomic Amplification and c-Met Overexpression. J Clin Oncol 2016, 34, 721–730. [Google Scholar] [CrossRef]
- Lennerz, J.K.; Kwak, E.L.; Ackerman, A.; Michael, M.; Fox, S.B.; Bergethon, K.; Lauwers, G.Y.; Christensen, J.G.; Wilner, K.D.; Haber, D.A. MET amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinib. Journal of clinical oncology 2011, 29, 4803. [Google Scholar] [CrossRef]
- Mo, H.N.; Liu, P. Targeting MET in cancer therapy. Chronic Dis Transl Med 2017, 3, 148–153. [Google Scholar] [CrossRef]
- Rosen, E.Y.; Johnson, M.L.; Clifford, S.E.; Somwar, R.; Kherani, J.F.; Son, J.; Bertram, A.A.; Davare, M.A.; Gladstone, E.; Ivanova, E.V.; et al. Overcoming MET-Dependent Resistance to Selective RET Inhibition in Patients with RET Fusion-Positive Lung Cancer by Combining Selpercatinib with Crizotinib. Clin Cancer Res 2021, 27, 34–42. [Google Scholar] [CrossRef]
- Pal, S.K.; Tangen, C.; Thompson, I.M., Jr.; Balzer-Haas, N.; George, D.J.; Heng, D.Y.C.; Shuch, B.; Stein, M.; Tretiakova, M.; Humphrey, P.; et al. A comparison of sunitinib with cabozantinib, crizotinib, and savolitinib for treatment of advanced papillary renal cell carcinoma: a randomised, open-label, phase 2 trial. Lancet 2021, 397, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Daud, A.; Kluger, H.M.; Kurzrock, R.; Schimmoller, F.; Weitzman, A.L.; Samuel, T.A.; Moussa, A.H.; Gordon, M.S.; Shapiro, G.I. Phase II randomised discontinuation trial of the MET/VEGF receptor inhibitor cabozantinib in metastatic melanoma. Br J Cancer 2017, 116, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Elisei, R.; Schlumberger, M.J.; Muller, S.P.; Schoffski, P.; Brose, M.S.; Shah, M.H.; Licitra, L.; Jarzab, B.; Medvedev, V.; Kreissl, M.C.; et al. Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol 2013, 31, 3639–3646. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.A.; Wainberg, Z.A.; Catenacci, D.V.; Hochster, H.S.; Ford, J.; Kunz, P.; Lee, F.C.; Kallender, H.; Cecchi, F.; Rabe, D.C.; et al. Phase II study evaluating 2 dosing schedules of oral foretinib (GSK1363089), cMET/VEGFR2 inhibitor, in patients with metastatic gastric cancer. PLoS One 2013, 8, e54014. [Google Scholar] [CrossRef]
- Patnaik, A.; Gadgeel, S.; Papadopoulos, K.P.; Rasco, D.W.; Haas, N.B.; Der-Torossian, H.; Faltaos, D.; Potvin, D.; Tassell, V.; Tawashi, M.; et al. Phase I Study of Glesatinib (MGCD265) in Combination with Erlotinib or Docetaxel in Patients with Advanced Solid Tumors. Target Oncol 2022, 17, 125–138. [Google Scholar] [CrossRef]
- Kudo, M.; Morimoto, M.; Moriguchi, M.; Izumi, N.; Takayama, T.; Yoshiji, H.; Hino, K.; Oikawa, T.; Chiba, T.; Motomura, K.; et al. A randomized, double-blind, placebo-controlled, phase 3 study of tivantinib in Japanese patients with MET-high hepatocellular carcinoma. Cancer Sci 2020, 111, 3759–3769. [Google Scholar] [CrossRef]
- Basilico, C.; Pennacchietti, S.; Vigna, E.; Chiriaco, C.; Arena, S.; Bardelli, A.; Valdembri, D.; Serini, G.; Michieli, P. Tivantinib (ARQ197) displays cytotoxic activity that is independent of its ability to bind MET. Clinical Cancer Research 2013, 19, 2381–2392. [Google Scholar] [CrossRef]
- Calles, A.; Kwiatkowski, N.; Cammarata, B.K.; Ercan, D.; Gray, N.S.; Jänne, P.A. Tivantinib (ARQ 197) efficacy is independent of MET inhibition in non-small-cell lung cancer cell lines. Molecular oncology 2015, 9, 260–269. [Google Scholar] [CrossRef]
- Wu, Y.L.; Zhang, L.; Kim, D.W.; Liu, X.; Lee, D.H.; Yang, J.C.; Ahn, M.J.; Vansteenkiste, J.F.; Su, W.C.; Felip, E.; et al. Phase Ib/II Study of Capmatinib (INC280) Plus Gefitinib After Failure of Epidermal Growth Factor Receptor (EGFR) Inhibitor Therapy in Patients With EGFR-Mutated, MET Factor-Dysregulated Non-Small-Cell Lung Cancer. J Clin Oncol 2018, 36, 3101–3109. [Google Scholar] [CrossRef]
- Schuler, M.; Berardi, R.; Lim, W.T.; de Jonge, M.; Bauer, T.M.; Azaro, A.; Gottfried, M.; Han, J.Y.; Lee, D.H.; Wollner, M.; et al. Molecular correlates of response to capmatinib in advanced non-small-cell lung cancer: clinical and biomarker results from a phase I trial. Ann Oncol 2020, 31, 789–797. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Moonsamy, P.; Gainor, J.F.; Lennerz, J.K.; Piotrowska, Z.; Lin, J.J.; Lennes, I.T.; Sequist, L.V.; Shaw, A.T.; Goodwin, K.; et al. A Phase 2 Study of Capmatinib in Patients With MET-Altered Lung Cancer Previously Treated With a MET Inhibitor. J Thorac Oncol 2021, 16, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Han, J.Y.; Ahn, M.J.; Cho, B.C.; Yu, H.; Kim, S.W.; Yang, J.C.; Lee, J.S.; Su, W.C.; Kowalski, D.; et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: interim results from a multicentre, open-label, phase 1b study. Lancet Oncol 2020, 21, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Heng, D.Y.C.; Lee, J.L.; Cancel, M.; Verheijen, R.B.; Mellemgaard, A.; Ottesen, L.H.; Frigault, M.M.; L’Hernault, A.; Szijgyarto, Z.; et al. Efficacy of Savolitinib vs Sunitinib in Patients With MET-Driven Papillary Renal Cell Carcinoma: The SAVOIR Phase 3 Randomized Clinical Trial. JAMA Oncol 2020, 6, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Cheng, Y.; Zhou, J.; Lu, S.; Zhang, Y.; Zhao, J.; Kim, D.W.; Soo, R.A.; Kim, S.W.; Pan, H.; et al. Tepotinib plus gefitinib in patients with EGFR-mutant non-small-cell lung cancer with MET overexpression or MET amplification and acquired resistance to previous EGFR inhibitor (INSIGHT study): an open-label, phase 1b/2, multicentre, randomised trial. Lancet Respir Med 2020, 8, 1132–1143. [Google Scholar] [CrossRef] [PubMed]
- Decaens, T.; Barone, C.; Assenat, E.; Wermke, M.; Fasolo, A.; Merle, P.; Blanc, J.F.; Grando, V.; Iacobellis, A.; Villa, E.; et al. Phase 1b/2 trial of tepotinib in sorafenib pretreated advanced hepatocellular carcinoma with MET overexpression. Br J Cancer 2021, 125, 190–199. [Google Scholar] [CrossRef]
- Shah, M.A.; Bang, Y.J.; Lordick, F.; Alsina, M.; Chen, M.; Hack, S.P.; Bruey, J.M.; Smith, D.; McCaffery, I.; Shames, D.S.; et al. Effect of Fluorouracil, Leucovorin, and Oxaliplatin With or Without Onartuzumab in HER2-Negative, MET-Positive Gastroesophageal Adenocarcinoma: The METGastric Randomized Clinical Trial. JAMA Oncol 2017, 3, 620–627. [Google Scholar] [CrossRef]
- Kishi, K.; Sakai, H.; Seto, T.; Kozuki, T.; Nishio, M.; Imamura, F.; Nokihara, H.; Satouchi, M.; Nakagawa, S.; Tahata, T.; et al. First-line onartuzumab plus erlotinib treatment for patients with MET-positive and EGFR mutation-positive non-small-cell lung cancer. Cancer Treat Res Commun 2019, 18, 100113. [Google Scholar] [CrossRef]
- Harding, J.J.; Zhu, A.X.; Bauer, T.M.; Choueiri, T.K.; Drilon, A.; Voss, M.H.; Fuchs, C.S.; Abou-Alfa, G.K.; Wijayawardana, S.R.; Wang, X.A.; et al. A Phase Ib/II Study of Ramucirumab in Combination with Emibetuzumab in Patients with Advanced Cancer. Clin Cancer Res 2019, 25, 5202–5211. [Google Scholar] [CrossRef]
- Camidge, D.R.; Moran, T.; Demedts, I.; Grosch, H.; Mileham, K.; Molina, J.; Juan-Vidal, O.; Bepler, G.; Goldman, J.W.; Park, K.; et al. A Randomized, Open-Label Phase II Study Evaluating Emibetuzumab Plus Erlotinib and Emibetuzumab Monotherapy in MET Immunohistochemistry Positive NSCLC Patients with Acquired Resistance to Erlotinib. Clin Lung Cancer 2022, 23, 300–310. [Google Scholar] [CrossRef]
- Liu, L.; Zeng, W.; Wortinger, M.A.; Yan, S.B.; Cornwell, P.; Peek, V.L.; Stephens, J.R.; Tetreault, J.W.; Xia, J.; Manro, J.R. LY2875358, a neutralizing and internalizing anti-MET bivalent antibody, inhibits HGF-dependent and HGF-independent MET activation and tumor growth. Clinical Cancer Research 2014, 20, 6059–6070. [Google Scholar] [CrossRef]
- Hultberg, A.; Morello, V.; Huyghe, L.; De Jonge, N.; Blanchetot, C.; Hanssens, V.; De Boeck, G.; Silence, K.; Festjens, E.; Heukers, R. Depleting MET-Expressing Tumor Cells by ADCC Provides a Therapeutic Advantage over Inhibiting HGF/MET SignalingARGX-111 Kills MET-Expressing Cancer Cells by ADCC. Cancer research 2015, 75, 3373–3383. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, S.T.; Park, S.; Lee, S.; Park, S.H.; Park, J.O.; Lim, H.Y.; Ahn, H.; Bok, H.; Kim, K.M.; et al. Phase I Trial of Anti-MET Monoclonal Antibody in MET-Overexpressed Refractory Cancer. Clin Colorectal Cancer 2018, 17, 140–146. [Google Scholar] [CrossRef]
- Affronti, M.L.; Jackman, J.G.; McSherry, F.; Herndon, J.E., 2nd; Massey, E.C., Jr.; Lipp, E.; Desjardins, A.; Friedman, H.S.; Vlahovic, G.; Vredenburgh, J.; et al. Phase II Study to Evaluate the Efficacy and Safety of Rilotumumab and Bevacizumab in Subjects with Recurrent Malignant Glioma. Oncologist 2018, 23, 889–e898. [Google Scholar] [CrossRef]
- Mok, T.S.; Geater, S.L.; Su, W.C.; Tan, E.H.; Yang, J.C.; Chang, G.C.; Han, M.; Komarnitsky, P.; Payumo, F.; Garrus, J.E.; et al. A Randomized Phase 2 Study Comparing the Combination of Ficlatuzumab and Gefitinib with Gefitinib Alone in Asian Patients with Advanced Stage Pulmonary Adenocarcinoma. J Thorac Oncol 2016, 11, 1736–1744. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Weiss, G.J.; Papadopoulos, K.P.; Hofmeister, C.C.; Tibes, R.; Tolcher, A.; Isaacs, R.; Jac, J.; Han, M.; Payumo, F.C.; et al. Phase I ficlatuzumab monotherapy or with erlotinib for refractory advanced solid tumours and multiple myeloma. Br J Cancer 2014, 111, 272–280. [Google Scholar] [CrossRef] [PubMed]
- D’Arcangelo, M.; Cappuzzo, F. Focus on the potential role of ficlatuzumab in the treatment of non-small cell lung cancer. Biologics: targets & therapy 2013, 7, 61. [Google Scholar]
- Wagle, N.; Emery, C.; Berger, M.F.; Davis, M.J.; Sawyer, A.; Pochanard, P.; Kehoe, S.M.; Johannessen, C.M.; MacConaill, L.E.; Hahn, W.C. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. Journal of clinical oncology 2011, 29, 3085. [Google Scholar] [CrossRef]
- Russo, M.; Siravegna, G.; Blaszkowsky, L.S.; Corti, G.; Crisafulli, G.; Ahronian, L.G.; Mussolin, B.; Kwak, E.L.; Buscarino, M.; Lazzari, L. Tumor Heterogeneity and Lesion-Specific Response to Targeted Therapy in Colorectal CancerLesion-Specific Response to Therapy in Colorectal Cancer. Cancer discovery 2016, 6, 147–153. [Google Scholar] [CrossRef]
- Tabassum, D.P.; Polyak, K. Tumorigenesis: it takes a village. Nature Reviews Cancer 2015, 15, 473–483. [Google Scholar] [CrossRef]
- Russo, M.; Pompei, S.; Sogari, A.; Corigliano, M.; Crisafulli, G.; Puliafito, A.; Lamba, S.; Erriquez, J.; Bertotti, A.; Gherardi, M.; et al. A modified fluctuation-test framework characterizes the population dynamics and mutation rate of colorectal cancer persister cells. Nature Genetics 2022, 54, 976–984. [Google Scholar] [CrossRef]
- Cipponi, A.; Goode, D.L.; Bedo, J.; McCabe, M.J.; Pajic, M.; Croucher, D.R.; Rajal, A.G.; Junankar, S.R.; Saunders, D.N.; Lobachevsky, P. MTOR signaling orchestrates stress-induced mutagenesis, facilitating adaptive evolution in cancer. Science 2020, 368, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Crisafulli, G.; Sogari, A.; Reilly, N.M.; Arena, S.; Lamba, S.; Bartolini, A.; Amodio, V.; Magrì, A.; Novara, L. Adaptive mutability of colorectal cancers in response to targeted therapies. Science 2019, 366, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Chaft, J.E.; Oxnard, G.R.; Sima, C.S.; Kris, M.G.; Miller, V.A.; Riely, G.J. Disease flare after tyrosine kinase inhibitor discontinuation in patients with EGFR-mutant lung cancer and acquired resistance to erlotinib or gefitinib: implications for clinical trial design. Clin Cancer Res 2011, 17, 6298–6303. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, R.M.; Torcuator, R.; Jain, R.; Anderson, J.; Doyle, T.; Schultz, L.; Mikkelsen, T. Rebound tumour progression after the cessation of bevacizumab therapy in patients with recurrent high-grade glioma. J Neurooncol 2010, 99, 237–242. [Google Scholar] [CrossRef]
- West, H.; Oxnard, G.R.; Doebele, R.C. Acquired resistance to targeted therapies in advanced non-small cell lung cancer: new strategies and new agents. Am Soc Clin Oncol Educ Book 2013. [Google Scholar] [CrossRef]
- Pop, O.; Pirvu, A.; Toffart, A.C.; Moro-Sibilot, D. Disease flare after treatment discontinuation in a patient with EML4-ALK lung cancer and acquired resistance to crizotinib. J Thorac Oncol 2012, 7, e1–2. [Google Scholar] [CrossRef]
- Yap, T.A.; Macklin-Doherty, A.; Popat, S. Continuing EGFR inhibition beyond progression in advanced non-small cell lung cancer. Eur J Cancer 2017, 70, 12–21. [Google Scholar] [CrossRef]
- Pupo, E.; Ducano, N.; Lupo, B.; Vigna, E.; Avanzato, D.; Perera, T.; Trusolino, L.; Lanzetti, L.; Comoglio, P.M. Rebound Effects Caused by Withdrawal of MET Kinase Inhibitor Are Quenched by a MET Therapeutic Antibody. Cancer Res 2016, 76, 5019–5029. [Google Scholar] [CrossRef]
- Altintas, D.M.; Cerqua, M.; De Laurentiis, A.; Trusolino, L.; Boccaccio, C.; Comoglio, P.M. An mTOR feedback loop mediates the ‘flare’ (‘rebound’) response to MET tyrosine kinase inhibition. Sci Rep 2023, 13, 1378. [Google Scholar] [CrossRef]
- Guertin, D.A.; Sabatini, D.M. Defining the Role of mTOR in Cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef]
- Mamane, Y.; Petroulakis, E.; LeBacquer, O.; Sonenberg, N. mTOR, translation initiation and cancer. Oncogene 2006, 25, 6416–6422. [Google Scholar] [CrossRef]
- Huber, A.; Bodenmiller, B.; Uotila, A.; Stahl, M.; Wanka, S.; Gerrits, B.; Aebersold, R.; Loewith, R. Characterization of the rapamycin-sensitive phosphoproteome reveals that Sch9 is a central coordinator of protein synthesis. Genes Dev 2009, 23, 1929–1943. [Google Scholar] [CrossRef]
- Thoreen, C.C.; Chantranupong, L.; Keys, H.R.; Wang, T.; Gray, N.S.; Sabatini, D.M. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 2012, 485, 109–113. [Google Scholar] [CrossRef]
- Lazaris-Karatzas, A.; Montine, K.S.; Sonenberg, N. Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5’ cap. Nature 1990, 345, 544–547. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Rapalogs in cancer prevention: anti-aging or anticancer? Cancer Biol Ther 2012, 13, 1349–1354. [Google Scholar] [CrossRef]
- Wei, K.; Li, M.; Zöller, M.; Wang, M.; Mehrabi, A.; Hoffmann, K. Targeting c-MET by Tivantinib through synergistic activation of JNK/c-jun pathway in cholangiocarcinoma. Cell Death Dis 2019, 10, 231. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Han, J.-Y.; Lee, G.K.; Shin, J.; Yun, S.A.; Oh, J.Y.; Lee, S.; Kim, H.T.; Lee, J.S. C-MET overexpression as a resistance biomarker to epidermal growth factor receptor tyrosine kinase inhibitors in EGFR-mutant non-small cell lung cancer. Journal of Clinical Oncology 2016, 34, e20660–e20660. [Google Scholar] [CrossRef]
- Danilkovitch-Miagkova, A.; Zbar, B. Dysregulation of Met receptor tyrosine kinase activity in invasive tumors. The Journal of clinical investigation 2002, 109, 863–867. [Google Scholar] [CrossRef]
- Blumenschein Jr, G.R.; Mills, G.B.; Gonzalez-Angulo, A.M. Targeting the hepatocyte growth factor–cMET axis in cancer therapy. Journal of clinical oncology 2012, 30, 3287. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Biosynthesis (left, blue box) and degradation (right, pink box) of MET. ER: Endoplasmic reticulum; SEMA: semaphorin domain; PSI: plexin-semaphoring-integrin homology domain; IPT: immunoglobulin-like domain; JM: juxtamembrane domain; CS: catalytic site; DS: docking site.
Figure 1.
Biosynthesis (left, blue box) and degradation (right, pink box) of MET. ER: Endoplasmic reticulum; SEMA: semaphorin domain; PSI: plexin-semaphoring-integrin homology domain; IPT: immunoglobulin-like domain; JM: juxtamembrane domain; CS: catalytic site; DS: docking site.
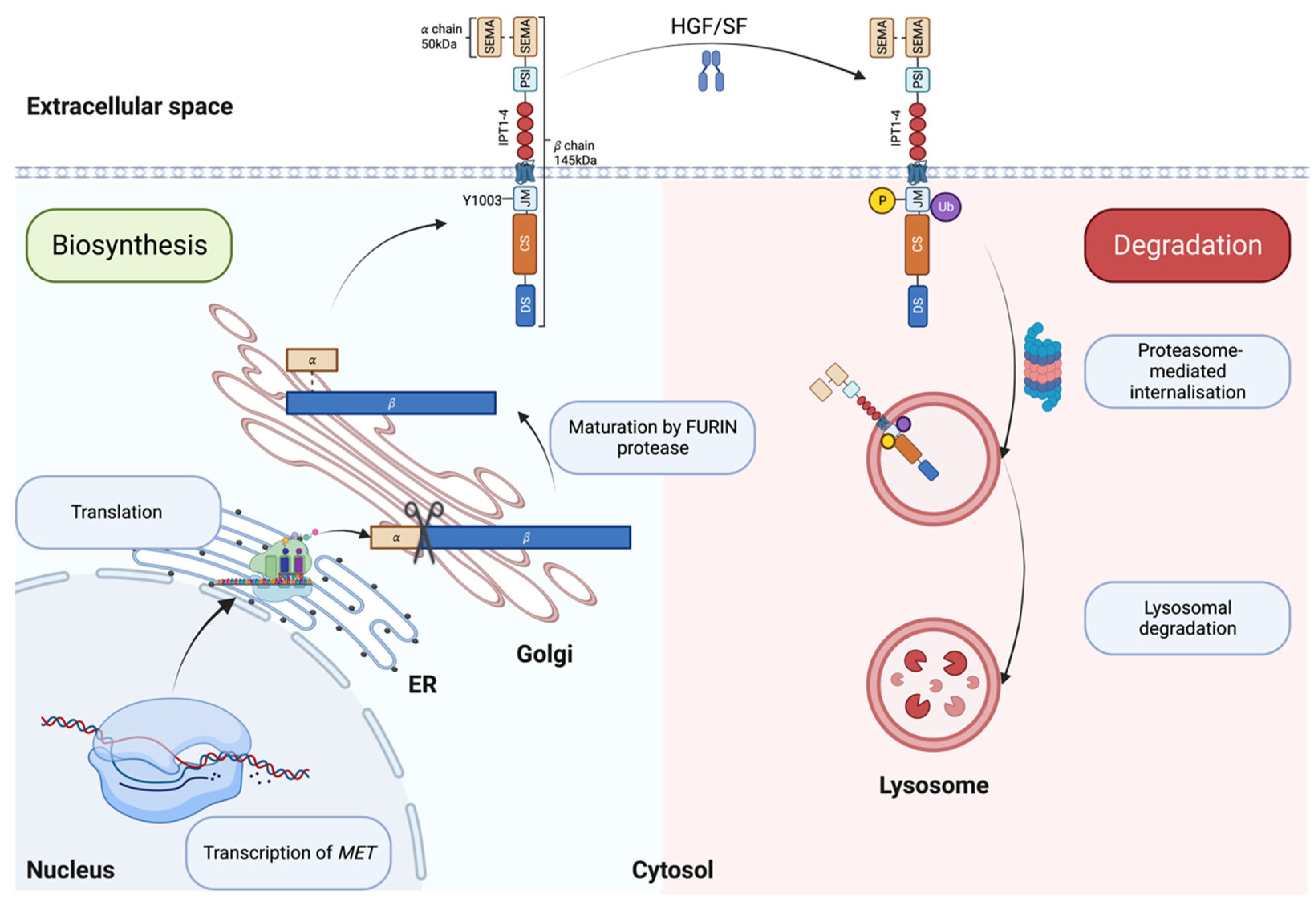
Figure 2.
SF-dependent MET activation. Upon ligand binding (HGF/SF), MET heterodimer forms a tetramer [5]. The extracellular signal is transmitted into the cytoplasm, leading to the recruitment of adaptor proteins (GRB2 for MAPK pathway and p85/p110 for AKT) and signal transduction.
Figure 2.
SF-dependent MET activation. Upon ligand binding (HGF/SF), MET heterodimer forms a tetramer [5]. The extracellular signal is transmitted into the cytoplasm, leading to the recruitment of adaptor proteins (GRB2 for MAPK pathway and p85/p110 for AKT) and signal transduction.
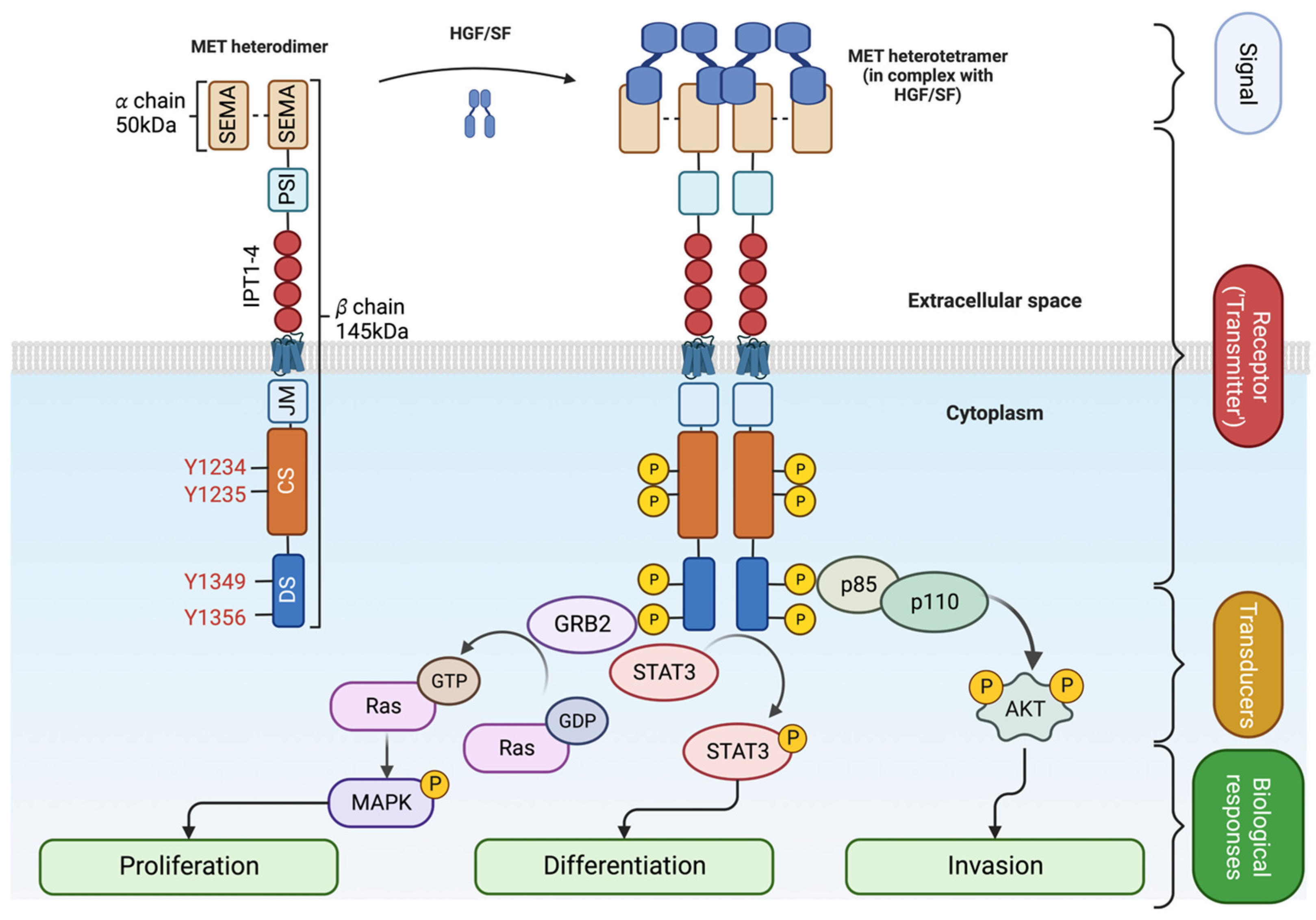
Figure 3.
The MET Observatory. (A) Copy number variation (CNV) in human tumors (TCGA pan cancer atlas). Dashed lines correspond to a copy lumber variation equal to 2 in absolute values. (B) MET CNV across tumour types. The dashed line corresponds to the average percentage of CNV in all types of cancer (ACC= Adrenocortical carcinoma; KIRP= Kidney renal papillary cell carcinoma; GBM=Glioblastoma multiform; KICH= Kidney Chromophobe; SKCM= Skin Cutaneous Melanoma; OV= Ovarian serous cystadenocarcinoma; SARC=Sarcoma; ESCA= Esophageal carcinoma; LGG=Low grade glioma; STAD= Stomach adenocarcinoma). (C) MET mutations in the protein-coding region and distribution over the MET protein domains (SEMA=semaphoring; PSI= plexin-semaphorin-integrin homology; IPT=immunoglobulin-like; TM=transmembrane; JM=juxtamembrane; CS=catalytic site; DS=docking site for transducers). (D) Comparison between the incidence of MET amplification, mutations, and overexpression. (E) B-value of CpG methylation of the MET promoter using the cg22116492 probe in cancer and in non-tumoral tissues. Percentage of mutations affecting protein-coding and regulatory regions of MET mRNA.
Figure 3.
The MET Observatory. (A) Copy number variation (CNV) in human tumors (TCGA pan cancer atlas). Dashed lines correspond to a copy lumber variation equal to 2 in absolute values. (B) MET CNV across tumour types. The dashed line corresponds to the average percentage of CNV in all types of cancer (ACC= Adrenocortical carcinoma; KIRP= Kidney renal papillary cell carcinoma; GBM=Glioblastoma multiform; KICH= Kidney Chromophobe; SKCM= Skin Cutaneous Melanoma; OV= Ovarian serous cystadenocarcinoma; SARC=Sarcoma; ESCA= Esophageal carcinoma; LGG=Low grade glioma; STAD= Stomach adenocarcinoma). (C) MET mutations in the protein-coding region and distribution over the MET protein domains (SEMA=semaphoring; PSI= plexin-semaphorin-integrin homology; IPT=immunoglobulin-like; TM=transmembrane; JM=juxtamembrane; CS=catalytic site; DS=docking site for transducers). (D) Comparison between the incidence of MET amplification, mutations, and overexpression. (E) B-value of CpG methylation of the MET promoter using the cg22116492 probe in cancer and in non-tumoral tissues. Percentage of mutations affecting protein-coding and regulatory regions of MET mRNA.
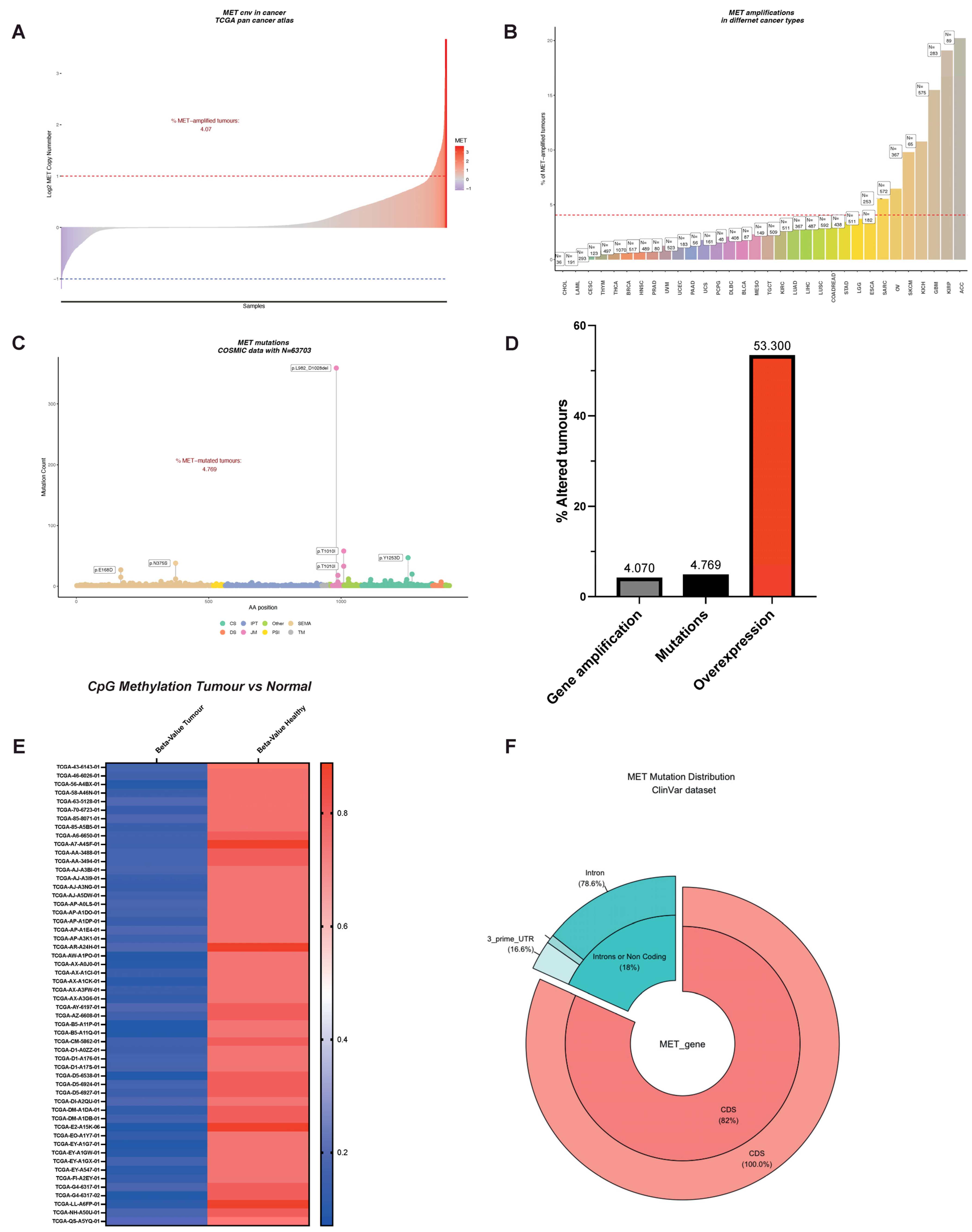
Figure 4.
Overview of MET alterations in cancer that drives invasive growth. Tumors become ‘addicted’ to MET and candidate for targeted therapies.
Figure 4.
Overview of MET alterations in cancer that drives invasive growth. Tumors become ‘addicted’ to MET and candidate for targeted therapies.
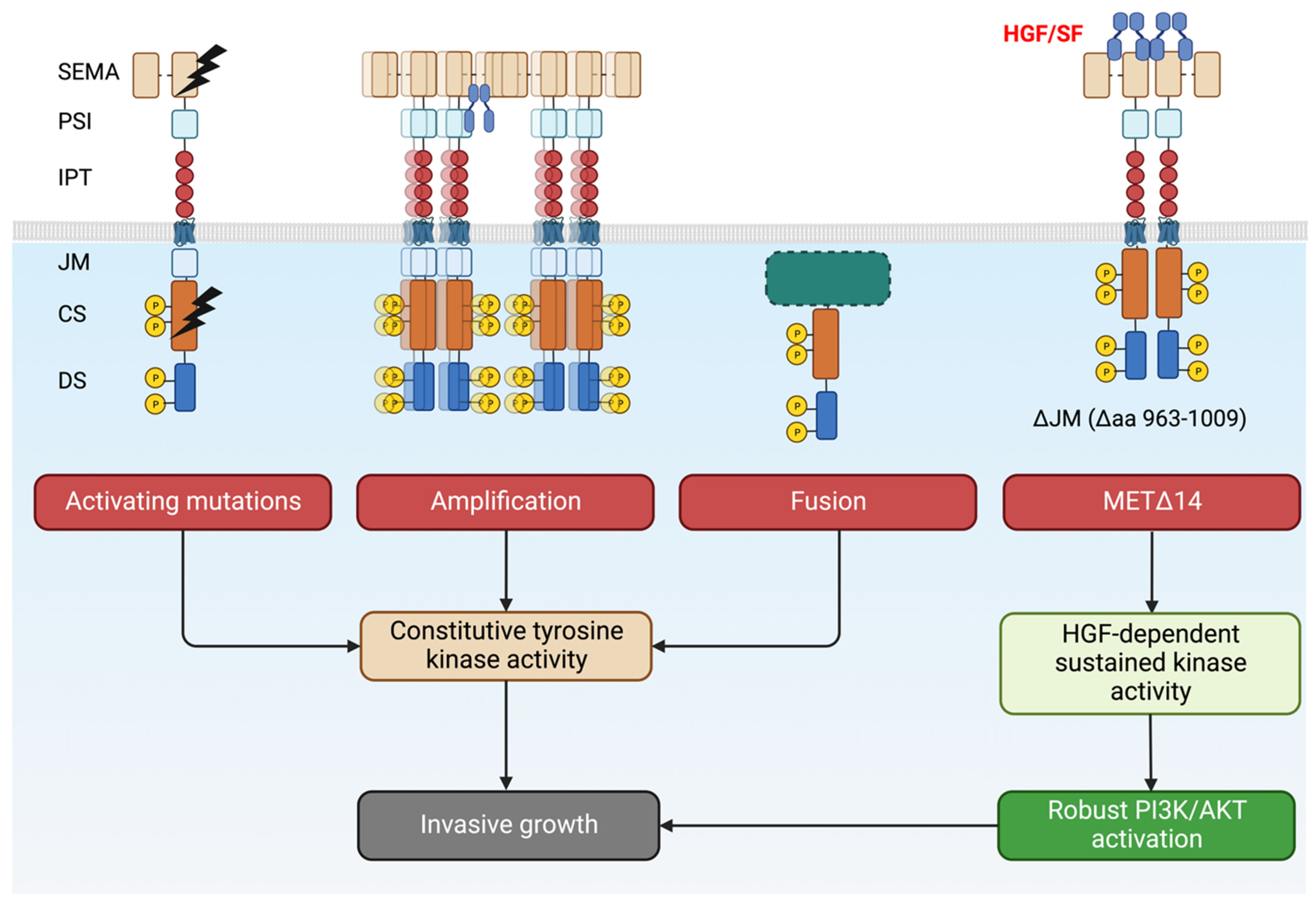
Figure 5.
Targeting MET/SF axis in cancer. Tyrosine kinase inhibitors (TKI) act as ATP mimetics to impede receptor phosphorylation. MET/SF interaction using can be prevented using either SF antibodies or MET antibodies. Some MET antibodies can induce receptor degradation in CBL-independent manner (SAIT301) or by shedding (DN30). Besides, in the case of DN30, the extracellular part of the trimmed receptor has the potential to sequestrate circulating SF.
Figure 5.
Targeting MET/SF axis in cancer. Tyrosine kinase inhibitors (TKI) act as ATP mimetics to impede receptor phosphorylation. MET/SF interaction using can be prevented using either SF antibodies or MET antibodies. Some MET antibodies can induce receptor degradation in CBL-independent manner (SAIT301) or by shedding (DN30). Besides, in the case of DN30, the extracellular part of the trimmed receptor has the potential to sequestrate circulating SF.
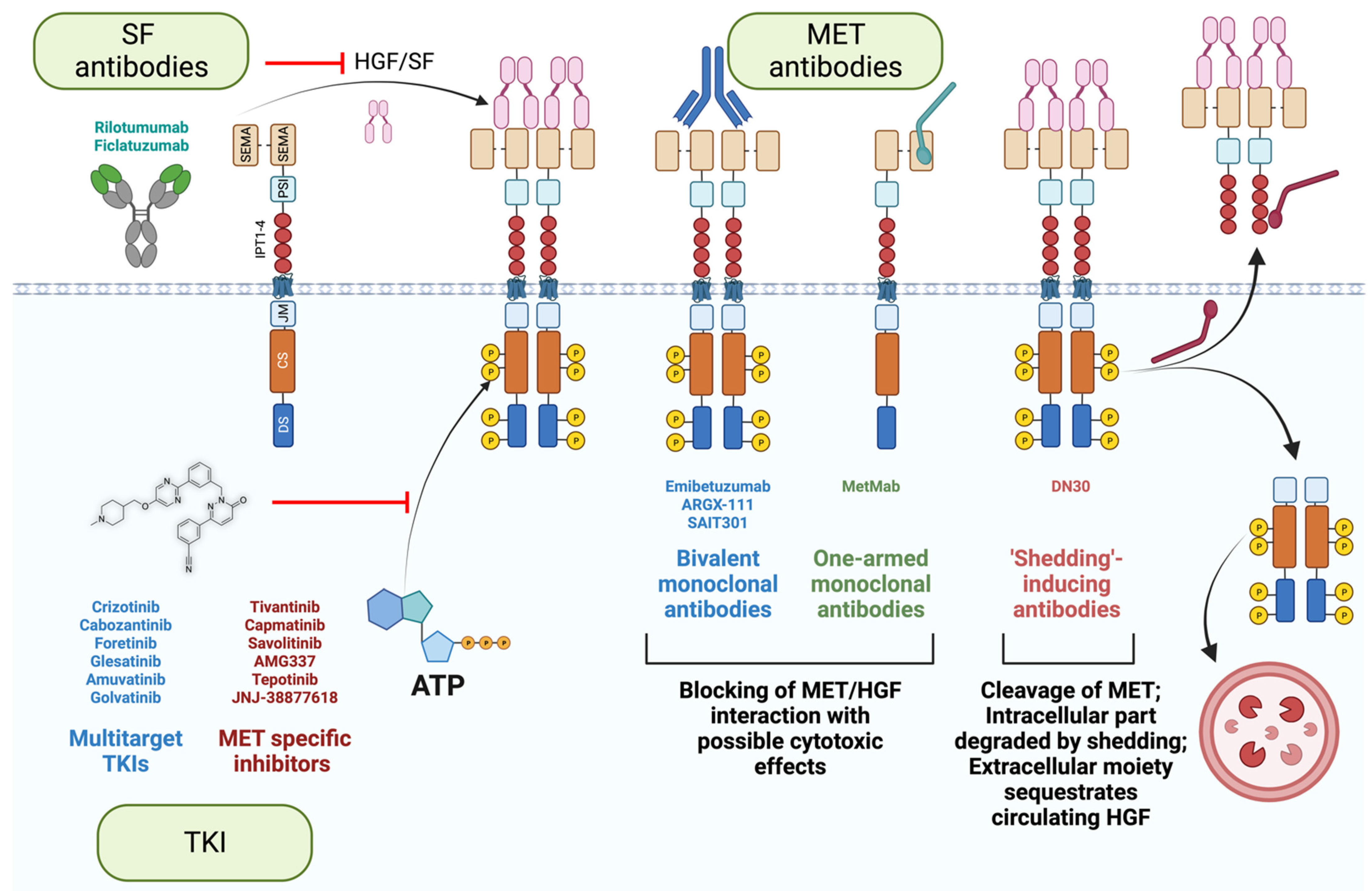

Table 1.
*As described in August 2022 in clinicaltrials.gov. Early phase I: exploratory trials before phase I. NA: trials without FDA-defined phases. **Advanced cancers of various origins; ADCC: antibody-dependent cell-mediated cytotoxicity; AML: acute myeloid lymphoma; HCC: hepatocellular carcinoma; NSCLC: non-small cell lung cancer; PRCC: papillary renal cell carcinoma; SCLC: small cell lung cancer.
Table 1.
*As described in August 2022 in clinicaltrials.gov. Early phase I: exploratory trials before phase I. NA: trials without FDA-defined phases. **Advanced cancers of various origins; ADCC: antibody-dependent cell-mediated cytotoxicity; AML: acute myeloid lymphoma; HCC: hepatocellular carcinoma; NSCLC: non-small cell lung cancer; PRCC: papillary renal cell carcinoma; SCLC: small cell lung cancer.
| Drug | Number of trials (phase)* | Cancer types | Principal outcome | Notes |
|---|---|---|---|---|
| Multitarget tyrosine kinase inhibitors | ||||
| PF02341066 (Crizotinib) | 51 (early I/I); 62 (II); 18 (III); 5 (IV); 7 (NA) | Breast cancer, renal clear cell cancer, glioblastoma, inflammatory myofibroblastic tumors, lymphoma, papillary renal cancers, MET+ gastric adenocarcinoma, MET+ or RON+ metastatic urothelial cancer and NSCLC | Substantial antitumor activity in patients with MET amplification and/or METΔ14 [149,150,151]. Crizotinib overcomes resistance to selpercatinib in RET-fusion positive NSCLC patients [152]. | • Targets: MET, ROS1, and ALK • Approved for treating NSCLC with EML4–ALK in 2011 and NSCLC with CD74–ROS1 in 2016 [150]. |
| XL184 (Cabozantinib) | 54 (early I/I); 157 (II); 19 (III); 2 (IV); 7 (NA) | Breast cancer, glioblastoma, HCC, kidney cancer, medullary thyroid cancer, melanoma, NSCLC, ovarian cancer, and prostate cancer | Cabozantinib significantly improved progression-free survival in patients with metastatic PRCC and melanoma [153,154]. Complete response was reported in one patient with METΔ14 [66]. | • Targets: MET, RET, and others. • Approved for the treatment of medullary thyroid cancer [155]. |
| GSK1363089 (Foretinib) | 6 (early I/I); 7 (II) | Mixed cancer, breast cancer, gastric cancer, head and neck cancer, liver cancer, NSCLC, and PRCC | No activity in unselected patients [156]. | Targets: MET, RET, and others. |
| MGCD265 (Glesatinib) | 4 (early I/I); 2 (II) | Mixed cancer and NSCLC | Results are pending. The safety profile is acceptable in non-genetically selected patients with advanced solid tumors [157]. | Targets: MET and AXL |
| MP470 (Amuvatinib) | 2 (early I/I); 1 (II); 1 (NA) | Mixed cancer, gastric cancer, glioblastoma, pancreatic cancer, and SCLC | Results are pending. | Targets: MET, RET, FLT3, and PDGFRA |
| E7050 (Golvatinib) | 8 (early I/I) | Mixed cancer, gastric cancer, head and neck cancer, and HCC | Results are pending. | Targets: MET and VEGFR2 |
| Specific MET inhibitors (small molecules) | ||||
| ARQ197 (Tivantinib) | 25 (early I/I); 21 (II); 4 (III) | Mixed cancer**, colorectal cancer, HCC, liver cancer, mesothelioma, NSCLC, SCLC, and stomach cancer | Tivantinib treatment did not demonstrate efficacy in a Phase III trial for HCC patients with high MET levels (based on staining intensity) [158]. | Specificity to MET is controversial [159,160]. |
| INCB28060 (Capmatinib) | 18 (early I/I); 27 (II); 3 (III); 1 (IV) | Mixed cancer, colorectal cancer, glioblastoma, head and neck cancer, HCC, NSCLC, and PRCC | Capmatinib showed substantial antitumor activity in patients with MET amplification or METΔ14 [71,161,162]. Capmatinib induces potentially similar resistance mechanisms as Crizotinib [163] but is a promising option in MET-amplified, EGFR-inhibitor-resistant tumors [161]. | Approved to treat adult patients with metastatic NSCLC with METΔ14 [141]. |
| AZD6094 (Savolitinib or Volitinib) | 7 (early I/I); 8 (II); 3 (III) | Mixed cancer, colorectal cancer, gastric cancer, NSCLC, kidney cancer, and PRCC | Encouraging results in EGFR-mutated, MET-amplified tumors [164,165]. | NA |
| AMG337 | 3 (early I/I); 5 (II) | Mixed cancer, renal clear cell cancer, esophageal cancer, and stomach cancer | Results are pending. | NA |
| MSC2156119J (Tepotinib) | 5 (early I/I); 5 (II) | Mixed cancer, lung cancer, and NSCLC | Partial response in NSCLC patients with METΔ14 [70]. Promising results in patients with MET amplification [166,167]. | NA |
| OMO-1 (JNJ-38877618) | 1 (early I/I) | Mixed cancer, lung cancer, and NSCLC | Results are pending. | NA |
| MET antibodies | ||||
| MetMab (Onartuzumab) | 6 (early I/I); 8 (II); 5 (III) | Mixed cancer, breast cancer, colorectal cancer, glioblastoma, HER2- and MET+ gastric cancer, HCC, and MET+ NSCLC | Some clinical trials were inconclusive due to poor patient selection or the premature termination of the study [143,168,169]. Other results are pending. | One-armed monoclonal antibody [124]. |
| LY2875358 (Emibetuzumab) | 1 (early I/I); 2 (II) | Mixed cancer, gastric cancer, and NSCLC | Cytostatic antitumor activity [170]. Significant increase in median progression-free survival for patients with the highest MET expression [142]. It cannot reverse acquired resistance to Erlotinib, an EGFR inhibitor [171]. | Humanized IgG4 bivalent monoclonal antibody [172]. |
| ARGX-111 | 1 (early I/I) | Mixed cancer, gastric cancer, glioblastoma, liver cancer, and renal cancer | Results are pending. | Bivalent monoclonal antibody with the property to activate ADCC [173]. |
| SAIT301 | 1 (early I/I) | Mixed cancer | Phase I completed, and the recommended dose for phase II was established [174]. | Bivalent monoclonal antibodies targeted against the MET 𝛼 chain, inducing CBL-independent degradation of MET to circumvent the detrimental agonist effect of other bivalent antibodies [125,126]. |
| SF antibodies | ||||
| AMG 102 (Rilotumumab) | 6 (early I/I); 13 (II); 3 (III) | Mixed cancer, gastric cancer, glioblastoma, lung cancer, mesothelioma, and prostate cancer | No improvement in the clinical outcome in patients with MET+ gastric cancer [135]. Toxicity issue [175]. | Humanized IgG2 monoclonal antibody [134]. |
| AV-299 (Ficlatuzumab) | 6 (early I/I); 4 (II); 3 (III) | AML, head and neck cancer, liver cancer, and NSCLC | Low benefit compared to other drugs [176,177]. | Humanized IgG1 monoclonal antibody [178]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



