
Submitted:
31 August 2023
Posted:
04 September 2023
You are already at the latest version
Abstract
Pt-coated Ni layer supported on Ni foam catalyst (denoted PtNi/Nifoam) was investigated for the oxidation of the formic acid (FAO) in acidic media. The prepared PtNi/Ni foam catalyst was studied as a function of the formic acid (FA) concentration at bare Pt and PtNi/Nifoam catalysts. The catalytic activity of the PtNi/Nifoam catalysts, studied on the basis of the ratio of the direct and indirect current peaks (jd)/(jnd) for the FAO reaction, showed values about 10 times higher compared to those on bare Pt, particularly at a low formic acid concentrations, reflecting the superiority of the former catalysts for the oxidation of FA to CO2. Ni foams provide a large surface area for the FOR while synergistic effects between Pt nanoparticles and Ni-oxy species layer on Ni foams contribute significantly to the enhanced oxidation of FA via the direct pathway, making it almost equal to the indirect pathway, particularly at low formic acid concentrations.
Keywords:
platinum
; nickel foam
; electroless deposition
; formic acid
; oxidation
1. Introduction
Currently, both formic acid (FA) and formate are of considerable interest for their possible direct production from CO2 as a green feedstock [1,2,3,4]. Formic acid or formate oxidation pertain to the most important model electrocatalytic reactions of small organic molecules that have been studied extensively [5,6] due their relevance as fuel for fuel cell applications including direct methanol fuel cell [7], direct formic acid or formate fuel cells (DFAFCs or DFFCs) [8,9]. High energy density, facile storage, operation and transportation make formic acid-based fuel cells rather promising for next-generation power sources, especially for small devices and portable applications [15].
The viability of DFAFCs intensely relies on the efficient formic acid or formate oxidation reactions. In general, Pt or Pd-based materials are considered to be the most suitable and advanced catalysts for the efficiency of these reactions [8,16,17]. It is well known that FAO in acidic media occurs via two different reaction pathways on Pt [10,19,20]. FA can be oxidized to CO2: (i) directly via a reactive intermediate (HCOOH + A → CO2 + 2H+ + 2e−) - dehydrogenation; or (ii) indirectly via an adsorbed COads species produced by dissociation of formic acid (HCOOH→B→COads +H2O→ CO2 + 2H+ + 2e−) – dehydration. Recently, a new reaction pathway has been proposed, including hydrogen oxidation reaction (HOR), where the H2 produced is supposed to exist as a new intermediate product, which is rapidly electro-oxidised to H+, contributing to the overall process [21].
Typically, COads species are identified as poisoning intermediates, but the nature of the active intermediate is still a matter of debate. A formyl СООН– [22] or adsorbed formate HCOO– [23,24,25,26,27] are supposed to be the rate-determining species, nevertheless in some cases the latter is concluded to be a spectator species rather than the active intermediate [5,29,30,31,32]. In some studies it is considered formate to be a common key intermediate in both direct and indirect pathways [25,26,27,28]. Meanwhile, a three-pathway mechanism has been proposed in the ref. [5], in which weakly adsorbed HCOOHads molecules are considered to be the active FAO intermediate, with their direct oxidation to CO2 being the predominant pathway. FAO reaction is very sensitive to pH of the solution [34,35,36,37,38,39,40,41], composition of formic acid/formate [40,42,43,44], temperature [45], and nature of the electrode or surface structure [6,26,46,47].
The successful commercialisation of DFAFCs is largely determined by the selection of the appropriate anode catalyst. Although Pt and Pt-based materials are widely used in commercial applications and are the most promising electrochemical catalysts, they are rare and still suffer from high cost, insufficient durability and low performance due to rapid deactivation of in situ generated carbon monoxide intermediates. For this reason, the development of an efficient, stable and low-cost anode catalyst is of paramount importance. To address these requirements, several strategies have been pursued to reduce carbonaceous poisoning effects and improve Pt-based catalyst performance. They either resist CO adsorption on the Pt surface and/or facilitate oxidative removal of adsorbed CO from the Pt surface. The first approach is realised by coupling Pt with other metals such as Ni [48,49,50,51], Bi [52,53,54], Sb [55], Rh [56] through so-called ensemble and/or electronic effects. Another approach is based on the enrichment of the surface with oxygen-containing species via the so-called bifunctional mechanism by alloying Pt e.g. with metal oxides such as NiOx [57,58,59,60], CoOx [57], Cu2O [61], FeOx [62], MnOx [63,64], which are characterised by their ability to allow the electrochemical dissociation of water at potentials more negative than that of bare Pt [65,66].
In order to reduce the use of Pt nanoparticles while minimising the cost of electrocatalysts for commercial applications, emerging materials with large specific areas such as porous carbon, carbon nanotubes, carbon black, doped graphene or graphene nanosheets [48,53,66,68,69,70] are used as supports. Recently, conductive substrates such as conductive polymers have been successfully used as catalyst supports for FAO due to their porous structures and high surface area [71,72]. Alternatively, careful engineering of nanocatalysts from solid dimensions to porous nanostructures, e.g. by a simple dealloying process, could achieve large specific areas [49,68,73]. The porous structure and alloy synergy was found to provide a significant gain in the preferred dehydrogenation pathway. The use of porous structures is of interest as they can not only shift [74,76,79] but even change [73,77,78] the reaction pathway from the undesirable indirect to the preferred direct oxidation pathway of formic acid.
In this context, the use of three-dimensional Ni foam with its unique architecture as a catalyst support has attracted particular attention due to its low density, high thermal and mechanical stability, high electrical conductivity, large specific surface area and ease of reactant and product diffusion.
Recently, a Pt-modified Ni layer coated on Nifoam (PtNi/Nifoam) has been proposed for efficient formate oxidation in an alkaline medium [80]. It showed an enhanced electrocatalytic activity towards formate oxidation via the direct pathway in alkaline medium, in contrast to the pure Pt electrode. As a follow-up to our previous studies [80], the behaviour of the prepared PtNi/Nifoam catalyst in acidic media is presented in this study.
2. Materials and Methods
The Ni foam with 20 pores/cm, a bulk density of 0.45 g cm-3, and a thickness of 1.6 mm was purchased from the supplier GoodFellow GmbH (Hamburg, Germany). The thin Ni layer was deposited on Ni foam substrate by the use of sodium hypophosphite as a reducing agent. The Pt thin layer was electroplated on Ni/Nifoam using the electrolyte containing PtCl2(NH3)2, NH4NO3, NH4OH, and NaNO2 (pH 8) at a current density of 1 A dm-2 for 40 min. The electrolyte temperature was kept at 95 oC.
The oxidation of formate (FOR) was investigated using a Zennium electrochemical workstation (ZAHNER-Elektrik GmbH & Co.KG, Kronach, Germany). A conventional three-electrode cell was used for electrochemical measurements. The Ni/Nifoam and PtNi/Nifoam catalysts with a geometric area of 2,45 cm2 were employed as working electrodes. An Ag/AgCl/KCl (3 M KCl) electrode was used as a reference, and a Pt sheet with a geometric area of 4 cm2 was used as a counter electrode. The bulk Pt bulk electrode with a geometric area of 1 cm2 was used for comparison. Cyclic voltammograms (CVs) were recorded at a potential scan rate of 50 mV s-1 from the open-circuit potential value in the anodic voltammetric scan up to +1.4 V unless otherwise stated in a 0.5 M H2SO4 solution containing FA concentration in the range of 0.05–0.7 M at a temperature of 25 oC. All potential values given are referred to as "Ag/AgCl".
Before each measurement of the electrochemical CV curves, the Pt and PtNi/Nifoam electrodes were pretreated in 0.5 M H2SO4 solution in a potential window of -0.2 to 1.3 V, at a potential scan rate of 50 mV s-1. The electrochemically active surface areas (ECSA) of the prepared catalysts were determined by calculating the charge associated with hydrogen adsorption (210 μC cm–2) [87]. CVs for the oxidative CO stripping from the surface of Pt and PtNi/Nifoam catalysts were performed in 0.5 M H2SO4, in N2 saturated solution, at 50 mV/s. CO was adsorbed in 0.5 M H2SO4 at a potential of -0.2 V for 15 min.
3. Results and Disscusion
X-ray photoelectron spectroscopy (XPS) was used to analyse the electronic state of the surface composition of the prepared Pt-modified Ni layer deposited on a Ni foam substrate (PtNi/Nifoam), as described in our previous work [80]. The data obtained are briefly presented below. The determined Pt 4f spectra gave a doublet of a high energy band (Pt 4f5/2) and a low energy band (Pt 4f7/2). Deconvolution of the latter revealed two peaks centered at 70.9 and 72.4 eV showing that Pt is present in two different oxidation states, Pt (0) and Pt (II), indicating that the Pt species grown on the Ni/Nifoam are in the metallic state and PtO or Pt (OH)2, respectively [81]. The Ni 2p3/2 XPS spectrum split into three resolved peaks centered at 852.3 eV, 853.9 and 855.8 eV, corresponding to the presence of Ni, NiO and Ni(OH)2 species on the Nifoam surface, respectively. The resulting XPS spectrum of O 1s split into three resolved peaks centered at 529.8, 531.3 and 532.8 eV. The lowest energy contributions at 529.8, 531.3 eV were assigned to the oxide/hydroxide species such as NiO and Ni(OH)2, respectively [82]. Meanwhile, the highest BE value at 532.8 eV is generally associated with physically adsorbed water molecules [83,84].
The electrochemical behaviour of bare Pt and PtNi/Nifoam electrodes towards the oxidation of formic acid in an acidic medium was evaluated using cyclic voltammetry. The cyclic voltammograms (CVs) of the bare Pt and PtNi/Nifoam electrodes in 0.5 M H2SO4 solution, measured at a potential scan rate of 50 mV s-1, are shown in Figure 1. The typical behaviour of bare Pt in acidic media is characterised by three clearly identifiable peak pairs, labelled I/I', II/II' and III/III'. The first two pairs in the negative potential region correspond to the adsorption/desorption of hydrogen. The third, at more positive potentials, corresponds to the surface redox transition associated with the Pt/PtO transformation.
In the case of the Ni/Nifoam electrode modified with Pt nanoparticles, an enormous increase in current is observed compared to the current values obtained for the bare Pt substrate (Figure 1). The dissolution of Ni in sulphuric acid takes place when anodic potentials are applied. Meanwhile, on the catalyst surface, (NiOH)ads species are being formed. The reaction sequence in acidic media is as follows [85]:
Ni+H2O → (NiOH)ads + H+ + e–
(NiOH)ads → Ni(OH+)ads + e–
Ni(OH+)ads +2H+ → Ni(OH)2+ H+
Ni(OH)2+ 2H+ → Ni2+ +H2O
Ni(OH)2 → NiO(Ni2O3)
Net reaction:
Ni→ Ni2+ +2e–
It should be noted that although Ni species are very susceptible to dissolution in acidic media, the mesoporous Ni-Pt films appear to be more corrosion resistant, especially with increasing Pt content, as discussed in ref. [86]. Moreover, the latter simultaneously show very high activity in the redox reaction of Ni(OH)2⇔NiOOH in sulfuric acid [86].
The enormous increase in current on the PtNi/Nifoam electrode indicates that it has a much larger surface area than the bare Pt substrate. The electrochemically active surface areas (ECSA) of the prepared catalysts were determined from the CVs of the Pt and PtNi/ Nifoam catalysts recorded in a deaerated 0.5 M H2SO4 solution at a scan rate of 50 mV s–1 by calculating the charge associated with hydrogen adsorption (210 μC cm–2) [87]. For the bare Pt substrate, this value is 1.5 cm2, while for the PtNi/ Nifoam electrode the average value is 71 cm2. Before each measurement of the electrochemical CV curve, the PtNi/Nifoam electrode was pre-treated in 0.5 M H2SO4 solution (as specified in the experimental part) and the ECSA was then re-evaluated.This section may be divided by subheadings. It should provide a concise and precise description of the experimental results, their interpretation, as well as the experimental conclusions that can be drawn.
Representative CV curves as a function of the formic acid concentration of 0.3, 0.5 and 0.7 M in 0.5 M H2SO4 solution for the bare Pt and of 0.05, 0.07, 0.1, 0.3, 0.5 and 0.7 M for PtNi/ Nifoam catalysts are plotted in Figure 2a, b, respectively. They show three oxidation peaks labelled Peak (I), Peak (II) and Peak (III) in the positive-going potential scan and a peak labelled Peak IV followed by a relatively well-developed shoulder labelled Peak (V) in the reverse negative potential scan with the latter being pronounced at lower concentrations of formic acid (0.05, 0.07 and 0.1 M FA) for the PtNi/Nifoam catalyst (Figure 2c). The voltammograms determined do not undergo radical transformations with the formic acid concentration and are similar in shape to those typically found for the bare Pt electrode [30,46,88].
The first peak current (jd) for FAO on Pt and PtNi/Nifoam catalysts is in the potential region of about 0.34 V and about 0.43-0.55 V, respectively, depending on the FA concentration, the values of which are summarised in Table 1 and Table 2. It is attributed to the direct oxidation of FA via a reactive intermediate (formate) to CO2 according to the following reaction sequence [51]:
Direct pathway (dehydrogenation pathway):
HCOOH + Pt → Hads + COOHads/HCOOads + H+ + e– + Pt→CO2 + 2H+ + e– + Pt
The value of the direct current peak (jd) generated under the potential region of the anodic peak (I) show the catalytic activity of the surface for the direct oxidation of FA. Whereas, the second oxidation peak (II) at more positive potentials mainly corresponds to the indirect oxidation of FA via adsorbed COad oxidation to CO2, which is realised through the following reactions [51]:
Indirect pathway (dehydration pathway):
HCOOH + Pt → Pt-CO + H2O (non-faradaic, poisoning)
Pt + H2O →Pt-OH +H+ + e– (Pt hydroxylation)
Pt-OH + Pt-CO → CO2 + 2P t + H+ + e– (CO oxidation)
The value of the indirect current peak (jind) generated under the potential region of the anodic peak (II) characterizes the surface poisoning by the CO adsorption process that effectively blocks the Pt surface required for the formation of OHad (via Eq. 9), which in turn is consumed in oxidising COads to complete FAO (via Eq. 10). In general, the insufficient availability of OHad leads to the accumulation of COads and limits the conversion efficiency of FA to CO2. It should be noted that direct FAO is not completely excluded and could occur in this potential region of peak (II) [5,29]. Meanwhile, the last peak (III) at the most positive potentials during the anodic potential scan of FAO is related to the formation of surface oxides.
During the negative potential scan, are assumed electrochemical reactions to take place simultaneously, including the reductive dehydroxylation of the Pt surface, as well as the oxidation of the FA by both direct and possibly indirect routes. Peak (IV) on the negative-going potential scan represents the oxidation of carbonaceous species on a clean and real catalytic activity containing Pt surface after partial reduction of the irreversibly formed surface oxides. Whereas the oxidation process at the shoulder marked as peak (V) at about 0.3 V, particularly on the PtNi/Nifoam catalyst (Figure 2c), is influenced by COad and the contribution of its oxidation [89].
The CVs presented in Figure 2 as well as the corresponding values of the current peaks in different potential regions for different concentrations of FA for Pt and PtNi/Nifoam catalysts listed in Table 1 and Table 2, respectively, show that increasing FA concentration results in higher current values defined in the potential regions of peak (II) for both catalysts and is followed by a potential shift of the current peaks to a more positive potential region, indicating that the electrode process is irreversible. In the case of the PtNi/Nifoam electrode, an enormous increase in current emerges compared to the current values observed on the bare Pt substrate in 0.5 M H2SO4 solutions (Figure 2b). It is approximately 0.44, 53.1 and 72.3 times higher for 0.3, 0.5 and 0.7 M FA, respectively. Such an efficient enhancement is attributed to the volumetric mesoporous structure of the PtNi/Nifoam catalyst, possessing a large specific surface area containing numerous active sites for the FAO reaction to proceed, and not only on the top of the surface, but in the vicinity of the substrate also.
A similar increase in current is observed in the peak potential region (I) of both catalysts. However, for the Pt catalyst it is relatively negligible, whereas for the PtNi/Nifoam catalyst it is detected only at lower FA concentrations of 0,05, 0,07, 0,1 and 0,3 M with the peak potential being shifted towards a more positive potential region. Further increase in FA concentration results in a decrease in the current of peak (I), indicating that FAO via the indirect pathway starts to dominate. The analysis of the ratio of the two oxidation current peaks (jd)/(jnd) determined for the Pt electrode shows a decrease in value from 0.18 to 0.12 with the change of the formic acid concentration from 0.3 to 0.7 M (Table 1), denoting the gain in poisoning level of the catalyst and indicates a rather low catalytic activity toward FAO via the direct route. A low number of free Pt active sites are available for FAO via the dehydrogenation pathway (Eq. 7). The poor oxidation of FA during the positive potential scan and the susceptibility of the Pt surface to COad poisoning is confirmed by the low value of another ratio of the direct current peak value (jd) on the positive going potential scan to the backward-going current peak value (jb) generated under the potential region of the anodic peak (IV), denoted as (jd)/(jb), which is only about 0.05.
On the contrary upon modifying Ni/Nifoam with Pt particles higher or equal current values are defined for the first current peak (jd) when increasing the FA concentration to 0.3 M (Figure 2b and Table 2), meaning that less CO is formed on the modified surface. The ratio of the current values (jd)/(jind) in the potential region of peaks (I) and (II) equals to 0.98, 1.14, and 1.00, for 0.05, 0.07, and 0.1 M FA, respectively, pointing to the fact that FAO via the direct pathway dominates and exceeds that via the indirect route on PtNi/Nifoam catalysts. This ratio is about 10 times higher for the PtNi/Nifoam catalysts as compared to that determined at the Pt surface. However, with increasing FA concentration from 0.3 to 0.5 and 0.7 M, this ratio decreases from 0.75 to 0.13 or even 0.07. This shows that the level of the PtNi/Nifoam catalyst poisoning increases due to the accumulation of incompletely oxidised carbonaceous species, indicating a change in the dominant pathway of the FAO reaction. Similarly, the ratio value of (jd)/(jb) in the potential region of peaks (I) and (IV) also decreases from 0.65 to even 0.07 for FA concentrations growing up from 0.05 to 0.7 M, implying the cumulative poisoning of the PtNi/Nifoam catalyst. The measurements show a higher electrocatalytic activity of the PtNi/Nifoam electrode towards FAO and a significantly better tolerance of the catalyst to poisoning species, especially at lower FA concentrations, as compared to the catalytic response of the bare Pt electrode indicating, the synergy between the embedded Pt and Ni layer on the porous structure of the Nifoam substrate [48]. The presence of Ni species could avoid the accumulation of carbonaceous species, especially at low FA concentrations, providing more electrochemical active sites of Pt for FAO through the direct pathway.
In order to evaluate the electrocatalytic activity of the investigated catalysts towards FAO, the current density values were normalized with respect to ECSA for each catalyst in acid media (Figure 3). For the sake of simplicity only positive going scans are presented. These values represent the specific activity of the catalysts. The CVs clearly show that the current density values at the both potential peaks (I) and (II) for the PtNi/Nifoam catalyst are significantly increased as compared to the current density values of the bare Pt electrode in the same potential region for all FA concentrations studied. In the case of 0.3 M FA this value for the PtNi/Nifoam catalyst is 5.5 times higher compare to the (jd) for the bare Pt catalyst and is followed by an onset potential shifted to a more negative potential region. Such efficiently improved results are attributed to the synergistic effect between Pt and Ni layer coated porous structure of Nifoam substrate that could avoid the accumulation of incompletely oxidised carbonaceous species (COads), directing FAO reaction towards the dehydrogenation pathway.
A comparison of the electrochemical performance, in terms of (jd)/(jind), of the catalysts included in this study with those of Pt-and Pt-based electrocatalysts used for FAO in acidic media reported in the literature is presented in Table 3. A selection of relevant references, summarised in Table 3, clearly shows that the operating conditions, in particular the acidity of the FAO achieved by applying a suitable amount of sodium hydroxide, leads to a higher value of the (jd)/(jind) ratio. In most cases a pH of 3.5 was used, where a significant amount of FA is ionised to formate anion (about one third), which reduces the polarisation resistance and increases the ionic conductivity of the electrolyte, as well as compressing the thickness of the diffusion layer [58,61,65,66]. Meanwhile, in the present study, the PtNi/Nifoam catalyst in a highly acidic solution at pH 0.3 showed that, under certain conditions, this ratio can be achieved at around 1, indicating the predominance of the FAO direct pathway.
In order to confirm better tolerance to catalysts poisoning by adsorbed carbonaceous species on the PtNi/Nifoam catalyst CO stripping measurements were adjusted. The current values measured for each sample were normalized to the electrochemically active surface area (ECSA), which was determined from the hydrogen adsorption region. Figure 4a reveals an obvious COads oxidation current peak at about 0.60 V during the positive potential on the bare Pt electrode in 0.5 M H2SO4. Meanwhile this peak on the PtNi/Nifoam catalyst in acid solution is shifted to the negative direction by about 0.32 V as compared to that on Pt and is located at 0.28 V (Figure 4b), suggesting that the PtNi/Nifoam catalyst has better CO tolerance than the single-metal Pt catalyst. The promotion in oxidation of carbonaceous species such as CO to CO2 could be attributed to the availability of transition metal oxides such as NiOx, which allow the electrochemical dissociation of water at potentials more negative than that of a bare Pt [65,66].
The enhanced oxidation of FA could be explained by the presence of Ni-oxy species, which are supposed to act as catalytic mediators via above mentioned reaction by facilitating charge transfer during the direct oxidation of FA to CO2 while simultaneously oxidizing CO at a rather low potential through the following reactions [57,65,66]:
and/or
Pt ... HCOOHad + NiOOH → Ptfree + CO2+ Ni(OH)2 + H+ + e–
Pt .... HCOO–ad + NiOOH → Ptfree + CO2 + Ni(OH)2 + e–
Pt ... COad + NiOOH + H2O → Ptfree + Ni(OH)2 + CO2+ H+ + e–
The above mentioned reactions show that the presence of the Ni(OH)2 species could be relatively successful in renewing the free and active Pt sites for further FAO by directing it via the dehydrogenation pathway, especially at lower FA concentrations. However, the dissolution of Ni species in highly acidic solution should be taken into account. In explaining the enhanced oxidation of formic acid on a binary PtNi/Nifoam catalyst, the synergy of the three necessary components, each performing a very specific function, should be outlined: Pt nanoparticles serve as the active site for FAO; Ni-oxy species facilitate the oxidative removal of carbon poisons from adjacent Pt sites, thus avoiding the accumulation of COads; and finally, Nifoam provides the large surface area and high electrical conductivity required for fast electrocatalysis.
4. Conclusions
A novel binary catalyst composed of Pt nanoparticles modified Ni layer coated on Nifoam catalyst has been proposed for efficient FAO in acidic media. The activity of the prepared catalyst towards FAO in acidic media was investigated. It was found that the PtNi/Nifoam catalyst has a significantly higher electrochemically active surface area compared to that of Pt, equal to about 71 cm2 and shows an enhanced electrocatalytic activity towards the FAO via the direct pathway as compared to pure Pt electrodes, particularly at lower FA concentrations. The prepared PtNi/Nifoam catalyst shows better CO tolerance than the single metal Pt in acidic solution. The reason for the enhanced electrocatalytic activity is due to the synergistic effects between the Pt nanoparticles and the porous structure of the Ni-oxy species layer on Nifoam with large ECSA. It is suggested that Ni-oxy species assist in the oxidative removal of accumulated carbonaceous species from the surface and act as catalytic mediators for charge transfer in the oxidation process.
Author Contributions
This study was conducted through the contributions of all authors. Conceptualization, A.N. and D. S.; Methodology, D. U., R. L., V. K. and V. J.; Investigation, A. B., B. S. and A. Z.; Formal analysis, R. L., V. K., B. S. and V. J.; Validation, A. Z., and A. B.; Writing – Original Draft Preparation, A. N. and D. S.; Writing – Review and Editing, L. T.- T. and D. S.; Visualization, A. N., D. S. and D. U.; Supervision, L. T.-T. and E. N.; Project Administration, A. N., L. T.-T. and E. N. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by European Union Structural Funds project “Development of Doctoral Studies” under grant No. 09.3.3.-ESFA-V-711-01-0001.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ma, Z.; Legrand, U.; Pahija, E.; Tavares, J. R.; Boffito D., C. From CO2 to Formic Acid Fuel Cells. Ind. Eng. Chem. Res. 2021, 60, 803–815. [Google Scholar] [CrossRef]
- Artz, J.; Müller, T. E.; Thenert, K.; Kleinekorte, J.; Meys, R.; Sternberg, A.; Bardow, A.; Leitner, W. Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment. Chem. Rev. 2018, 118, 434–504. [Google Scholar] [CrossRef] [PubMed]
- Legrand, U.; Boudreault, R.; Meunier, J. L. Decoration of N-Functionalized Graphene Nanoflakes with Copper-Based Nanoparticles for High Selectivity CO2 Electroreduction towards Formate. Electrochim. Acta. 2019, 318, 142–150. [Google Scholar] [CrossRef]
- Gunasekar, G. H.; Park, K.; Jung, K.-D.; Yoon, S. Recent Developments in the Catalytic Hydrogenation of CO2 to Formic acid/Formate using Heterogeneous Catalysts. Inorg. Chem. Front. 2016, 3, 882–895. [Google Scholar] [CrossRef]
- Chen, Y.; Heinen, M.; Jusys, Z.; Behm, R. Kinetics and mechanism of the electrooxidation of formic acid – Spectroscopical study in a flow cell. Angew. Chem., Int. Ed. 2006, 45, 981–985. [Google Scholar] [CrossRef] [PubMed]
- Scheijen, F. J. E.; Beltramo, G. L.; Hoeppener, S.; Housmans, T. H. M.; Koper, M. T. M. The electrooxidation of small organic molecules on platinum nanoparticles supported on gold: influence of platinum deposition procedure. J. Solid-State Electrochem. 2008, 12, 483–495. [Google Scholar] [CrossRef]
- Tiwari, J. N.; Tiwari, R. N.; Singh, G.; Kim, K. S. Recent progress in the development of anode and cathode catalysts for direct methanol fuel cells. Nano Energy. 2013, 2, 553–578. [Google Scholar] [CrossRef]
- An, L.; Chen, R. Direct formate fuel cells: A review. J.Power Sources. 2016, 320, 127–139. [Google Scholar] [CrossRef]
- Ulas, B.; Kivrak, H. D. Direct Liquid Fuel Cells. Fundamentals, Advances and Future. 2021, 149-176.
- Yu, X.; Pickup, P. G. Recent advances in direct formic acid fuel cells (DFAFC). J. Power Sources. 2008, 182, 124–132. [Google Scholar] [CrossRef]
- Hong, P.; Liao, S.; Zeng, J.; Huang, X. Design, fabrication and performance evaluation of a miniature air breathing direct formic acid fuel cell based on printed circuit board technology. J. of Power Sources. 2010, 195, 7332–7337. [Google Scholar] [CrossRef]
- Miesse C., M.; Jung, W. S.; Jeong, K.-J.; Lee, J. K.; Lee, J.; Han, J.; Yoon, S. P.; Nam, S. W.; Lim, T.-H. ; Hong. S.-A. Direct formic acid fuel cell portable power system for the operation of a laptop computer. J Power Sources. 2006, 162, 532–540. [Google Scholar]
- Ahmad, K. N.; Azam, A. M. I. N.; Isahak, W. N. R. W.; Zainoodin, A. M.; Masdar, M. S. Improving the electrocatalytic activity for formic acid oxidation of bimetallic Ir–Zn nanoparticles decorated on graphene nanoplatelets. Mater. Res. Express. 2020, 7, 015095. [Google Scholar] [CrossRef]
- El-Nagar, G. A.; Delikaya, Ö.; Lauermann, I.; Roth, C. Platinum Nanostructure Tailoring for Fuel Cell Applications Using Levitated Water Droplets as Green. ACS Appl. Mater. Interfaces. 2019, 11, 22398–22407. [Google Scholar] [CrossRef] [PubMed]
- Yu, X. W.; Manthiram, A. Catalyst-selective, scalable membraneless alkaline direct formate fuel cells. Appl. Catal. B Environ. 2015, 165, 63–67. [Google Scholar] [CrossRef]
- Uwitonze, N.; Chen, Y. The study of Pt and Pd based anode catalysis for formic acid fuel cell, Chem. Sci. J. 2017, 8, 167. [Google Scholar]
- Folkman, S.J.; González-Cobos, J.; Giancola, S.; Sánchez-Molina, I.; Galán-Mascarós, J.R. Benchmarking Catalysts for Formic Acid/Formate Electrooxidation. Molecules. 2021, 26, 4756. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Chen, W. Recent advances in formic acid electro-oxidation: from the fundamental mechanism to electrocatalysts. Nanoscale Adv. 2021, 3, 94–105. [Google Scholar] [CrossRef]
- Wang, H. F.; Liu, Z. P. Formic acid oxidation at Pt/H2O interface from periodic DFT calculations integrated with a continuum solvation model. J. Phys. Chem. C. 2009, 113, 17502–17508. [Google Scholar] [CrossRef]
- Petrii, O. A. The Progress in Understanding the Mechanisms of Methanol and Formic Acid Electrooxidation on Platinum Group Metals (a Review). Russian Journal of Electrochem. 2019, 55, 1–33. [Google Scholar] [CrossRef]
- Yang, X.; Meng, Q.; Wang, X.; Jin, Z.; Liu, C.; Ge, J.; Xing, X. A new pathway for formic acid electro-oxidation: The electro-chemically decomposed hydrogen as a reaction intermediate. Journal of Energy Chemistry. 2022, 71, 188–191. [Google Scholar] [CrossRef]
- Ross, P. N.; Lipkowski, J. Electrocatalysis. New York: Wiley-VCH, 1998.
- Miki, A.; Ye, S.; Osawa, M. Surface-enhanced IR absorption on platinum nanoparticles: an application to real-time monitoring of electrocatalytic reactions. Chem. Commun. 2002, 150, 1500–1501. [Google Scholar] [CrossRef]
- Columbia, M. R.; Thiel, P. A. The interaction of formic acid with transition metal surfaces, studied in ultrahigh vacuum. J. Electroanal. Chem. 1994, 369, 1–14. [Google Scholar] [CrossRef]
- Cuesta, A.; Cabello, G.; Gutierrez, C.; Osawa, M. Adsorbed formate: the key intermediate of the oxidation of formic acid on platinum electrodes. Phys. Chem. Chem. Phys. 2011, 13, 20091–20095. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, A.; Cabello, G.; Osawa, M.; Gutierrez, C. Mechanism of the electrocatalytic oxidation of formic acid on metals. ACS Catal. 2012, 2, 728–738. [Google Scholar] [CrossRef]
- Salamon, M. J.; Briega-Martos, V.; Cuesta, A.; Herrero, E. , Insight into the role of adsorbed formate in the oxidation of formic acid from pH-dependent experiments with Pt single-crystal electrodes. J. Electroanl. Chem. 2022, 925, 116886. [Google Scholar] [CrossRef]
- Betts, A.; Briega-Martos, V.; Cuesta, A.; Herrero, E. Adsorbed Formate is the Last Common Intermediate in the Dual-Path Mechanism of the Electrooxidation of Formic Acid. ACS Catal. 2020, 10, 8120–8130. [Google Scholar] [CrossRef]
- Chen, Y.-X.; Heinen, M.; Jusys, Z.; Behm, R. J. Bridge-bonded formate: active intermediate or spectator species in formic acid oxidation on a Pt film electrode. Langmuir. 2006, 22, 10399–10408. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y. X.; Ye, S.; Heinen, M.; Jusys, Z.; Osawa, M.; Behm, R. J. Application of in-situ attenuated total reflection-Fourier transform infrared spectroscopy for the understanding of complex reaction mechanism and kinetics: formic acid oxidation on a Pt film elektrode at elevated temperatures. J. Phys. Chem. B. 2006, 110, 9534–9544. [Google Scholar] [CrossRef]
- Chen, Y.-X.; Heinen, M.; Jusys, Z.; Behm, R. J. Kinetic isotope effects in complex reaction networks: formic acid electrooxidation. Chem., Phys. Chem. 2007, 8, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Yuan, D. F.; Yang, F.; Mei, D.; Zhang, Z.; Chen, Y.-X. On the mechanism of the direct path way formic acid oxidation at Pt(111) electrodes. Phys. Chem. Chem. Phys. 2013, 15, 4367–4376. [Google Scholar] [CrossRef]
- Okamoto, H.; Numata, Y.; Gojuki, T.; Mukouyama, Y. Different behavior of adsorbed bridgebonded formate from that of current in the oxidation of formic acid on platinum. Electrochim. Acta. 2014, 116, 263–270. [Google Scholar] [CrossRef]
- Haan, J. L.; . Masel, R. T. The influence of solution pH on rates of an electrocatalytic reactions: Formic acid electrooxidation on platinum and palladium. Electrochim. Acta. 2009, 54, 4073–4078. [Google Scholar] [CrossRef]
- Joo, J.; Uchida, T.; Cuesta, A.; Koper, M. T. M.; Osawa, M. Importance of acid-base equilibrium in electrocatalytic oxidation of formic acid on platinum. J. Am. Chem. Soc. 2013, 135, 9991–9994. [Google Scholar] [CrossRef] [PubMed]
- Perales-Rondon, J. V.; Herrero, E.; Feliu, J. M. Effects of the anion adsorption and pH on the formic acid oxidation reaction on Pt(111) electrodes. Electrochim. Acta. 2014, 140, 511–517. [Google Scholar] [CrossRef]
- Perales-Rondon, J. V.; Brimaud, S.; Solla-Gullon, J.; Herrero, E.; Behm, R. J.; Feliu, J. M. Further insights into the formic acid oxidation mechanism on platinum: pH and anion adsorption effects. Electrochim. Acta. 2015, 180, 479–485. [Google Scholar] [CrossRef]
- Joo, J.; Choun, N.; Jeong, J.; Lee, J. Influence of solution pH on Pt anodic catalyst in direct formic acid fuel cells. ACS Catal. 2015, 5, 6848–6851. [Google Scholar] [CrossRef]
- McCrum, I. T.; Janik, M. J. pH and alkali cation effects in the Pt cyclic voltammogram explained using density functional theory. J. Phys. Chem. C. 2016, 120, 457–471. [Google Scholar] [CrossRef]
- Abdelrahman, A.; Hermann, J. M.; Kibler, L. A. Electrocatalytic oxidation of formate and formic acid on platinum and gold: study of pH dependence with phosphate buffers. Electrocatalysis. 2017, 8, 509–517. [Google Scholar] [CrossRef]
- Zhang, M.-K.; Wei, Z.; Chen, W.; Xu, M.-L.; Cai, J.; Chen, Y.-X. Bell shape vs volcano shape pH dependent kinetics of the electrochemical oxidation of formic acid and formate, intrinsic kinetics or local pH shift? Electrochim. Acta. 2020, 363, 13716. [Google Scholar] [CrossRef]
- Joo, J.; Uchida, T.; Cuesta, A.; Koper, M. T. M.; Osawa, M. The effect of pH on the electrocatalytic oxidation of formic acid/formate on platinum: A mechanistic study by surface-enhanced infrared spectroscopy coupled with cyclic voltammetry. Electrochim. Acta. 2014, 129, 127–136. [Google Scholar] [CrossRef]
- Wei, Y.; Zuo, K. Q.; Zhengda, H.; Chen, W.; Lin, C. H.; Cai, J.; Sartin, M.; Chen, Y.-X. The mechanisms of HCOOH/HCOO– oxidation on Pt electrodes: Implication from pH effect and H/D kinetic izotope effect. Electrochem. Commun. 2017, 81, 1–4. [Google Scholar] [CrossRef]
- Buso-Rogero, C.; Ferre-Vilaplana, A.; Herrero, E.; Feliu, J. M. The role of formic acid/formate equilibria in the oxidation of formic acid on Pt (111). Electrochem. Commun. 2019, 98, 10–14. [Google Scholar] [CrossRef]
- Jiang, J.; Scott, J.; Wieckowski, A. Direct evidence of triple-path mechanism of formate electrooxidation on Pt black in alkaline media at varying temperature. Part I: The electrochemical studies. Electrochim. Acta. 2013, 104, 124–133. [Google Scholar]
- Brimaud, S.; Solla-Gullon, J.; Weber, I.; Feliu, J. M.; Behm, R. J. Formic acid electrooxidation on noble-metal electrodes: role and mechanistic implication of pH, surface structure, and anion adsorption. ChemElectroChem, 2014, 1, 1075–1083. [Google Scholar] [CrossRef]
- Ferre-Vilaplana, A.; Perales-Rondóon, J. V.; Buso-Rogero, C.; Feliu, J. M.; Herrero, E. Formic acid oxidation on platinum electrodes:a detailed mechanism supported by experiments and calculations on well-defined surfaces. J. Mater. Chem. A, 2017, 5, 21773–21784. [Google Scholar] [CrossRef]
- Themsirimongkon, S.; Pongpichayakul, N.; Fang, L.; Jakmunee, J.; Saipanya, S. New catalytic designs of Pt on carbon nanotube-nickel-carbon black for enhancement of methanol and formic acid oxidation. J. of Electroanalyt Chem. 2020, 876, 114518. [Google Scholar] [CrossRef]
- Kiani, M.; Zhang, J.; Luo, Y.; Chen, Y.; Chen, Ji.; Fan, J.; Wang, G.; Wang, R. Facile synthesis and enhanced catalytic activity of electrochemically dealloyed platinum–nickel nanoparticles towards formic acid electro-oxidation. J. Energy Chem. 2019, 35, 9–16. [Google Scholar] [CrossRef]
- Zhang, B.-W.; Zhang, Z.-C.; Liao, H.-G.; Gong, Y.; Gu, L.; Qu, X.-M.; You, L.-X.; Liu, S.; Huang, L.; Tian, X.-C.; Huang, R.; Zhu, F.-C.; Liu, T.; Jiang, Y.-X.; Zhou, Z.-Y.; Sun, S.-G. Tuning Pt-skin to Ni-rich surface of Pt3Ni catalysts supported on porous carbon for enhanced oxygenreduction reaction and formic electro-oxidation. Nano Energy. 2016, 19, 198–209. [Google Scholar] [CrossRef]
- Pei, A.; Ruan, L.; Liu, B.; Chen, W.; Lin, S.; Chen, B.; Liu, Y.; Zhu, L. H.; Chen, B. H. Ultra-low Au decorated PtNi alloy nanoparticles on carbon for high-efficiency electro-oxidation of methanol and formic acid. Int. J. Hydrogen Energy. 2020, 45, 22893–22905. [Google Scholar] [CrossRef]
- Perales-Rondón, J. V.; Ferre-Vilaplana, A.; Feliu, J. M.; Herrero, E. Oxidation mechanism of formic acid on the bismuth adatom-modified Pt (111) surface. J. Amer. Chem. Soc. 2014, 136, 13110–13113. [Google Scholar] [CrossRef]
- Choi, M.; Ahn, C.-Y.; Lee, H.; Kim, J. K.; Oh, S.-H.; Hwang, W.; Yang, S.; Kim, J.; Kim, O.-H.; Choi, I.; Sung, Y.-E.; Cho, Y.-H.; Rhee, C. K.; Shin, W. Bi-modified Pt supported on carbon black as electro-oxidation catalyst for 300 W formic acid fuel cell stack. Applied Catalysis B: Environmental. 2019, 253, 187–195. [Google Scholar] [CrossRef]
- Wang, C. Y.; Yu, Z.-Y.; Li, G.; Song, Q.-T.; Li, G.; Luo, C.-X.; Yin, S.-H.; Lu, B.-A.; Xiao, C.; Xu, B.-B.; Zhou, Z.-Y.; Tian, T.; Sun, S.-G. ; Intermetallic PtBi Nanoplates with High Catalytic Activity towards Electrooxidation of Formic Acid and Glycerol. ChemElectroChem. 2020, 7, 239–245. [Google Scholar] [CrossRef]
- Zhang, Y.; Qiao, M.; Huang, Y.; Zou, Y.; Liu, Z.; Tao, L.; Li, Y.; Dong, C. L.; Wang, S. In Situ Exfoliation and Pt Deposition of Antimonene for Formic Acid Oxidation via a Predominant Dehydrogenation Pathway. Research. 2020, 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sawy, E. N. E.; Pickup, P. G. Carbon monoxide and formic acid oxidation at Rh@Pt nanoparticles. Electrochimi. Acta. 2019, 302, 234–240. [Google Scholar] [CrossRef]
- El-Nagar, G. A.; Mohammad, A. M.; El-Deab, M. S.; El-Anadouli, B. E. Electrocatalysis of Formic Acid Electro-Oxidation at Platinum Nanoparticles Modified Surfaces with Nickel and Cobalt Oxides Nanostructured. Progress in Clean Energy. 2015, 1, 577–594. [Google Scholar]
- El-Nagar, G. A.; Mohammad, A. M. Enhanced electrocatalytic activity and stability ofplatinum, gold, and nickel oxide nanoparticlesbased ternary catalyst for formic acid electro-oxidation. Int. J. Hydrogen Energy. 2014, 39, 11955–11962. [Google Scholar] [CrossRef]
- Mohammad, A. M.; El-Nagar, G. A.; Al-Akraa, I. M; El-Deab, M. S.; El-Anadouli, B. E. Towards improving the catalytic activity and stability of platinum-based anodes in direct formic acid fuel cells. Hydrogen Energy. 2015, 40, 7808–7816. [Google Scholar] [CrossRef]
- El-Refaei, S. M.; El-Nagar, G. A.; Mohammad, A. M.; El-Deab, M. S.; El-Anadouli, B. E. Electrocatalytic Activity of NiOx Nanostructured Modified Electrodes Towards Oxidation of Small Organic Molecules. In book: 2nd International Congress on Energy Efficiency and Energy Related Materials (ENEFM2014). 2015, 1-7.
- El-Nagar, G. A Mohammad, A. M.; El-Deab, M. S.; El-Anadouli, B. E. Propitious Dendritic Cu2O−Pt Nanostructured Anodes for Direct Formic Acid Fuel Cells. ACS Appl. Mater. Interfaces. 2017, 9, 19766–19772. [Google Scholar]
- Ali Al-Qodami, B.; Farrag, H. H.; Sayed, S. Y.; Allam, N. K.; El-Anadouli, B. E.; Mohammad, A. M. Bifunctional Tailoring of Platinum Surfaces with Earth Abundant Iron Oxide Nanowires for Boosted Formic Acid Electro-Oxidation. Journal of Nanotechnology. 2018, 2018, 1–10. [Google Scholar] [CrossRef]
- Mohammad, A. M.; Al-Akraa, I. M.; El-Deab, M. S. Superior electrocatalysis of formic acid electrooxidation on a platinum, gold and manganese oxide nanoparticle-based ternary catalyst. Int J Hydrogen Energy. 2018, 43, 139–149. [Google Scholar] [CrossRef]
- Asal, Y. M.; Al-Akraa, I. M.; Mohammad, A. M.; El-Deab, M. S. A competent simultaneously co-electrodeposited Pt-MnOx nanocatalyst for enhanced formic acid electro-oxidation. J. Taiwan Inst. Chem. Engineers. 2019, 96, 169–175. [Google Scholar] [CrossRef]
- El-Nagar, G. A.; Mohammad, A. M.; El-Deab, M. S.; El-Anadouli, B. E. Facilitated electrooxidation of formic acid at nickel oxide nanoparticles modified electrodes. J Electrochem Soc. 2012, 159, F249–F254. [Google Scholar] [CrossRef]
- El-Nagar, G. A.; Mohammad, A. M.; El-Deab, M. S.; El-Anadouli, B. E. Electrocatalysis by design: enhanced electrooxidation of formic acid at platinum nanoparticles–nickel oxide nanoparticles binary catalysts. Electrochim Acta. 2013, 94, 62–71. [Google Scholar] [CrossRef]
- Caglar, A.; Cogenli, M. S.; Yurtcan, A. B.; Kivrak, H. Effective carbon nanotube supported metal (M=Au, Ag, Co, Mn, Ni, V, Zn) core Pd shell bimetallic anode catalysts for formic acid fuel cells. Renewable Energy. 2020, 150, 78–90. [Google Scholar] [CrossRef]
- Xu, H.; Yan, B.; Li, S.; Wang, J.; Wang, C.; Guo, J.; Du, Y. N-doped graphene supported PtAu/Pt intermetallic core/dendritic shell nanocrystals for efficient electrocatalytic oxidation of formic acid. Chem Eng J. 2018, 334, 2638–2646. [Google Scholar] [CrossRef]
- Al-Akraa, I. M.; Salama, A. E.; Asal, Y. M.; Mohammad, A. M. Boosted performance of NiOx/Pt nanocatalyst for the electro-oxidation of formic acid: A substrate’s functionalization with multi-walled carbon nanotubes. Arabian Journal of Chemistry. 2021, 14, 103383. [Google Scholar] [CrossRef]
- Saquib, M.; Halder, A. Ensemble Effect of Ni in Bimetallic PtNi on Reduced Graphene Oxide Support for Temperature-Dependent Formic Acid Oxidation. ChemistrySelect. 2018, 3, 3909–3917. [Google Scholar] [CrossRef]
- Shatla, A. S.; Hassan, K. M.; Abd-El-Latif, A. A.; Hathoot, A. A.; Baltruschat, H.; Abdel-Azzem, M. Poly 1,5 diaminonaphthalene supported Pt, Pd, Pt/Pd and Pd/Pt nanoparticles for direct formic acid oxidation. J. Electroanalyt. Chemistry. 2019, 833, 231–241. [Google Scholar] [CrossRef]
- El-Moghny, M. G. A.; Alalawy, H. H.; Mohammad, A. M.; Mazhar, A. A.; El-Deab, M. S.; El-Anadouli, B. E. Conducting polymers inducing catalysis: Enhanced formic acid electro-oxidation at a Pt/polyaniline nanocatalyst. Int J Hydrogen Energy. 2017, 42, 11166–11176. [Google Scholar] [CrossRef]
- Xie, Y.; Dimitrov, N. Ultralow Pt loading nanoporous Au-Cu-Pt thin film as highly active and durable catalyst for formic acid oxidation. Applied Catalysis B: Environmental. 2020, 263, 118366. [Google Scholar] [CrossRef]
- Qiu, H.-J.; Xu, H. T.; Li, X.; Wang, J. Q.; Wang, Y. Core–shell-structured nanoporous PtCu with high Cu content and enhanced catalytic performance. J. Mater. Chem. A. 2015, 3, 7939–7944. [Google Scholar] [CrossRef]
- Jiang, X.; Yan, X.; Ren, W.; Jia, Y.; Chen, J.; Sun, D.; Xu, L.; Tang, Y. Porous AgPt@Pt Nanooctahedra as an Efficient Catalyst toward Formic Acid Oxidation with Predominant Dehydrogenation Pathway. ACS Appl Mater Interfaces. 2016, 8, 31076–31082. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, J.; Zhou, F.; Zhang, J.; Wei, X.; Luo, R.; Wang, G.; Wang, R. Dealloyed platinum-copper with isolated Pt atom surface: Facile synthesis and promoted dehydrogenation pathway of formic acid electro-oxidation. J Electroanal Chem. 2017, 799, 78–83. [Google Scholar] [CrossRef]
- Li, D.; Meng, F.; Wang, H.; Jiang, X.; Zhu, Y. Nanoporous AuPt alloy with low Pt content: a remarkable electrocatalyst with enhanced activity towards formic acid electro-oxidation. Electrochim. Acta. 2016, 190, 852–861. [Google Scholar] [CrossRef]
- Han, S.-H.; Liu, H.-M.; Bai, J.; Tian, X. L.; Xia, B. Y.; Zeng, J.-H.; Jiang, J.-X.; Chen, Y. Platinum-silver alloy nanoballoon nanoassemblies with super catalytic activity for the formate electrooxidation. ACS Appl Energy Mater. 2018, 1, 1252–1258. [Google Scholar] [CrossRef]
- Wang, J.; Chen, F.; Jin, Y.; Guo, L.; Gong, X.; Wanga, X.; Johnston, R. L. In situ high-potential-driven surface restructuring of ternary AgPd–Ptdilute aerogels with record-high performance improvement for formate oxidation electrocatalysis. Nanoscale. 2019, 11, 14174–14185. [Google Scholar] [CrossRef] [PubMed]
- Nacys, A.; Balčiūnaitė, A.; Zabielaitė, A.; Upskuvienė, D.; Šebeka, B.; Jasulaitienė, V.; Kovalevskij, V.; Šimkūnaitė, D.; Norkus, E.; Tamašauskaitė-Tamašiūunaitė, L. An Enhanced Oxidation of Formate on PtNi/Ni Foam Catalyst in an Alkaline Medium. Crystals, 2022, 12, 362. [Google Scholar] [CrossRef]
- Briggs, D.; Seah, M. P. Practical Surface Analysis, John Wiley & Sons. 1993, 1.
- Kim, K. S.; Winograd, N. X-ray photoelectron spectroscopic studies of nickel oxygen surfaces using oxygen and argon ion-bombardment. Surf. Sci. 1974, 43, 625–643. [Google Scholar] [CrossRef]
- Wittstok, G.; Strübing, A.; Szargan, R.; Werner, G. Glucose oxidation at bismuth-modified platinum electrodes. J. Electroanal. Chem. 1998, 444, 61–73. [Google Scholar] [CrossRef]
- Blasini, D. R.; Rochefort, D.; Fachini, E.; Aldena, L. R.; DiSalvo, F. J.; Cabrera, C. R.; Abruna, H. D. Surface composition of ordered intermetallic compounds PtBi and PtPb. Surf. Sci., 2006, 600, 2670–2680. [Google Scholar] [CrossRef]
- Sanli, A. E.; Aytaç, A. Electrochemistry of The Nickel Electrode as a Cathode Catalyst In The Media Of Acidic Peroxide For Application of The Peroxide Fuel Cell. ECS Transactions. 2012, 42, 3–22. [Google Scholar] [CrossRef]
- Eiler, K.; Krawiec, H.; Kozina, I.; Sort, J.; Pellicer, E. Electrochemical characterisation of multifunctional electrocatalytic mesoporous Ni-Pt thin films in alkaline and acidic media. Electroch. Acta, 2020, 359, 136952. [Google Scholar] [CrossRef]
- Trasatti, S.; Petrii, O. A. Real surface area measurements in electrochemistry. Pure Appl. Chem. 1991, 63, 711–734. [Google Scholar] [CrossRef]
- Samjeské, G.; Miki, A.; Ye, S.; Osawa, M. Mechanistic Study of Electrocatalytic Oxidation of Formic Acid at Platinum in Acidic Solution by Time-Resolved Surface-Enhanced Infrared Absorption Spectroscopy. J. Phys. Chem. B. 2006, 110, 16559–16566. [Google Scholar] [CrossRef] [PubMed]
- Lemos, S. G.; Oliveira, R. T. S.; Santos, M. C.; Nascente, P. A. P.; Bulhões, L. O. S.; Pereira, E. C. Electrocatalysis of methanol, ethanol and formic acid using a Ru/Pt metallic bilayer. J. Power Sources. 2007, 163, 695–701. [Google Scholar] [CrossRef]
- Nicholson, R. S.; Shain, I. Theory of Stationary Electrode Polarography. Single Scan and Cyclic Methods Applied to Reversible, Irreversible, and Kinetic Systems. Analytical Chemistry. 1964, 36, 706–723. [Google Scholar]
- Çögenli, M.S.; Yurtcan, A.B. Catalytic activity, stability and impedance behavior of PtRu/C, PtPd/C and PtSn/C bimetallic catalysts toward methanol and formic acid oxidation. Int. J. Hydrog. Energy. 2018, 43, 10698–10709. [Google Scholar] [CrossRef]
- Al-Akraa, I.M.; Mohammad, A.M. A spin-coated TiOx/Pt nanolayered anodic catalyst for the direct formic acid fuel cells. Arab. J. Chem. 2020, 13, 4703–4711. [Google Scholar] [CrossRef]
- Al-Akraa, I.M.; Asal, Y.M.; Darwish, S.A. A simple and effective way to overcome carbon monoxide poisoning of platinum surfaces in direct formic acid fuel cells. Int. J. Electrochem. Sci. 2019, 14, 8267–8275. [Google Scholar] [CrossRef]
- Sun, Y.; Huang, B.; Li, Y.; Qin, Y.; Fu, Z.; Sun, M.; Wang, L.; Guo, S. Segmented Au/PtCo heterojunction nanowires for efficient formic acid oxidation catalysis. Fun. Res. 2021, 1, 453–460. [Google Scholar] [CrossRef]
Figure 1.
Typical stabilized CVs of Pt (dotted red line) and PtNi/Nifoam (solid blue line) recorded in a 0.5 M H2SO4 solution at a scan rate of 50 mV s-1.
Figure 1.
Typical stabilized CVs of Pt (dotted red line) and PtNi/Nifoam (solid blue line) recorded in a 0.5 M H2SO4 solution at a scan rate of 50 mV s-1.
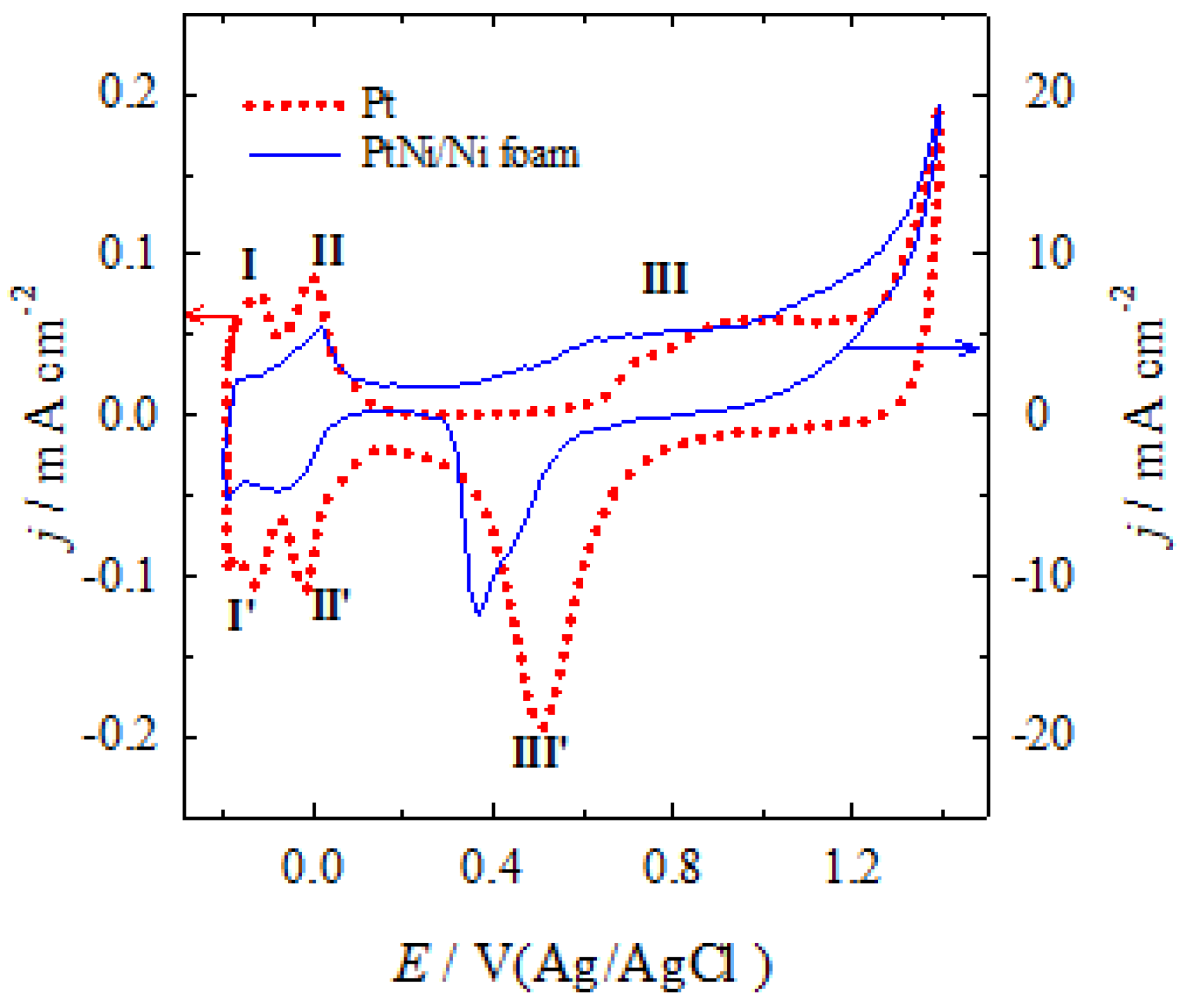
Figure 2.
CVs of Pt (a) and PtNi/Nifoam (b, c) recorded in a 0.5 M H2SO4 solution, containing 0.3, 0.5, 0.7 M FA (a), 0.05, 0.07, 0.1, 0.3, 0.5, 0.7 M FA (b) and 0.05, 0.07, 0.1 M FA (c) at a scan rate of 50 mV s-1. (Positively going potential scan - solid lines, negatively going potential scan - dashed lines).
Figure 2.
CVs of Pt (a) and PtNi/Nifoam (b, c) recorded in a 0.5 M H2SO4 solution, containing 0.3, 0.5, 0.7 M FA (a), 0.05, 0.07, 0.1, 0.3, 0.5, 0.7 M FA (b) and 0.05, 0.07, 0.1 M FA (c) at a scan rate of 50 mV s-1. (Positively going potential scan - solid lines, negatively going potential scan - dashed lines).
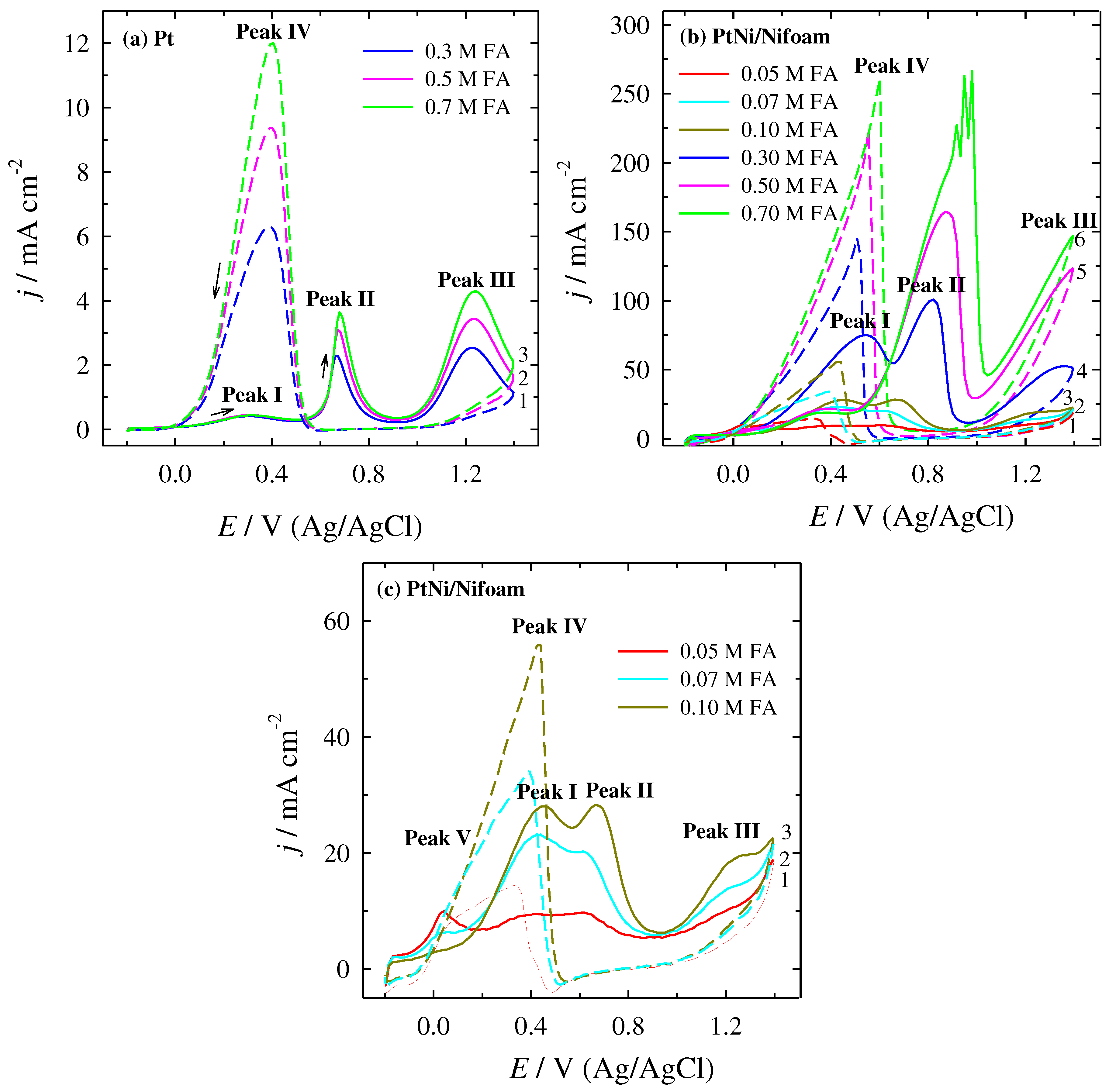
Figure 3.
ECSA normalised positive-going potential scans for the bare Pt catalyst, containing 0.3, 0.5, 0.7 M FA (1-3 dotted lines) and the PtNi/Nifoam catalyst, containing 0.05, 0.07, 0.1, 0.3, 0.5, 0.7 M FA (4-9 solid lines) recorded in 0.5 M H2SO4 solution, at a scan rate of 50 mV s-1.
Figure 3.
ECSA normalised positive-going potential scans for the bare Pt catalyst, containing 0.3, 0.5, 0.7 M FA (1-3 dotted lines) and the PtNi/Nifoam catalyst, containing 0.05, 0.07, 0.1, 0.3, 0.5, 0.7 M FA (4-9 solid lines) recorded in 0.5 M H2SO4 solution, at a scan rate of 50 mV s-1.
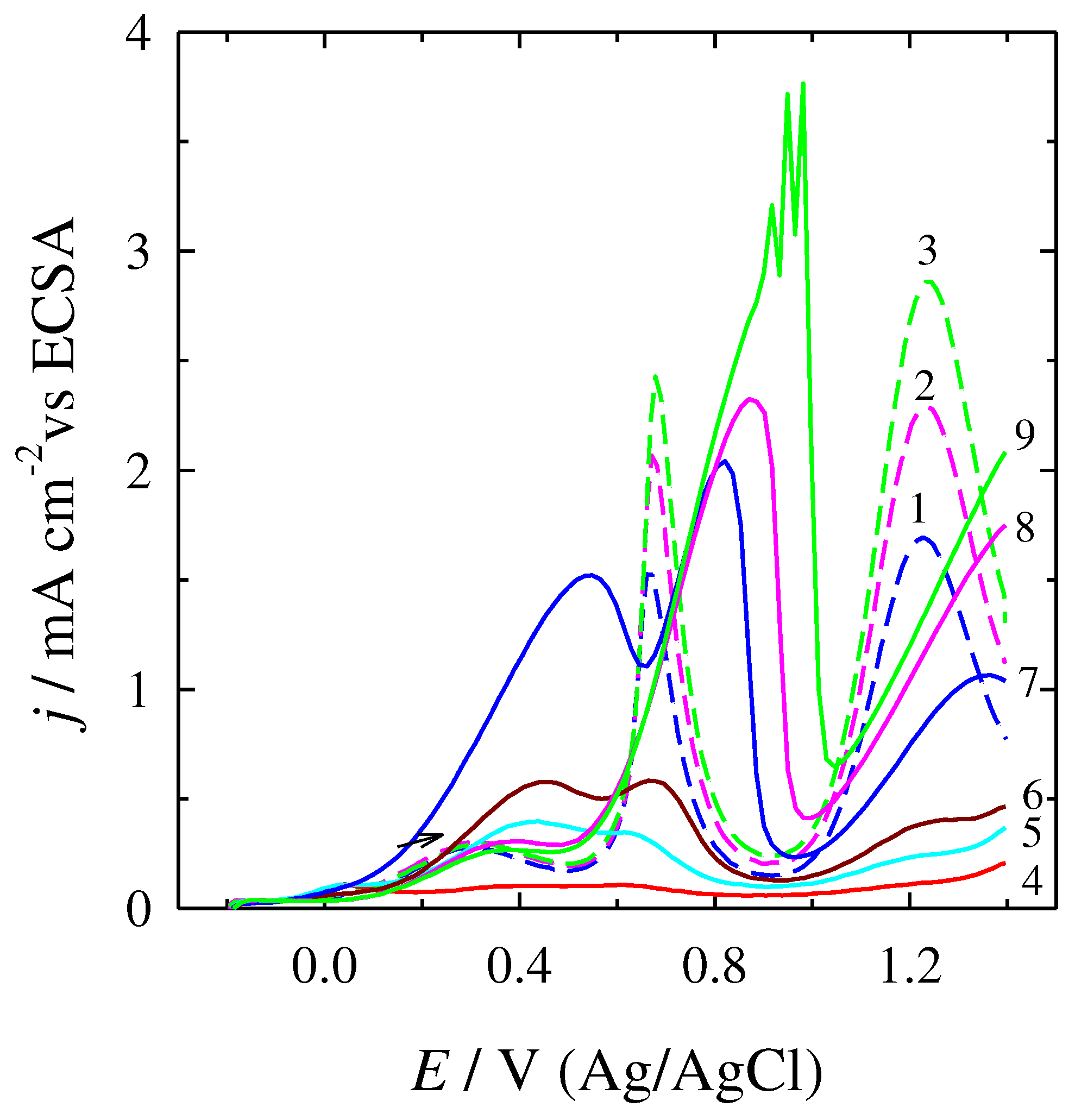
Figure 4.
CVs for the oxidative CO stripping from the surface of Pt (a,) and PtNi/Nifoam catalysts (b) in 5 M H2SO4, at 50 mV/s. CO was adsorbed at -0.2 V in 0.5 M H2SO4 for 15 min; potential sweep was carried out in N2 saturated solution.
Figure 4.
CVs for the oxidative CO stripping from the surface of Pt (a,) and PtNi/Nifoam catalysts (b) in 5 M H2SO4, at 50 mV/s. CO was adsorbed at -0.2 V in 0.5 M H2SO4 for 15 min; potential sweep was carried out in N2 saturated solution.

Table 1.
Summary of electrochemical measurements at the Pt catalyst for the data in Figure 2a.
Table 1.
Summary of electrochemical measurements at the Pt catalyst for the data in Figure 2a.
| cFA, M | Peak I | Peak II | Peak III | Peak IV | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Epc, V |
jd, mA cm-2 |
Epc, V |
jind, mA cm-2 |
jd/jind | Epc, V |
jd, mA cm-2 |
Epc, V |
jb, mA cm-2 |
(jd)/(jb) | |
| 0.3 0.5 0.7 |
0.320 | 0.42 | 0.661 | 2.29 | 0.18 | 1.227 | 2.54 | 0.390 | 6.30 | 0.07 |
| 0.319 | 0.45 | 0.671 | 3.10 | 0.14 | 1.236 | 3.43 | 0.391 | 9.37 | 0.05 | |
| 0.327 | 0.44 | 0.679 | 3.64 | 0.12 | 1.234 | 4.29 | 0.405 | 12.00 | 0.04 | |
Table 2.
Summary of electrochemical measurements at the PtNi/Nifoam catalyst for the data in Figure 2b.
Table 2.
Summary of electrochemical measurements at the PtNi/Nifoam catalyst for the data in Figure 2b.
| cFA, M | Peak I | Peak II | Peak IV | Peak V | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Epc, V |
jd, mA cm-2 |
Epc, V |
jind, mA cm-2 |
jd/jind | Epc, V |
jd, mA cm-2 |
Epc, V |
jb, mA cm-2 |
(jd)/(jb) | |
| 0.05 0.07 0.1 0.3 0.5 0.7 |
0.426 | 9.47 | 0.618 | 9.70 | 0.98 | 0.326 | 14.52 | 0.086 | 8.865 | 0.65 |
| 0.439 | 23.18 | 0.615 | 20.27 | 1.14 | 0.393 | 34.14 | 0.249 | 25.740 | 0.68 | |
| 0.456 0.549 0.390 0.390 |
28.01 75.10 21.71 18.90 |
0.680 0.821 0.870 0.949 |
28.14 100.80 164.55 263.10 |
1.00 0.75 0.13 0.07 |
0.441 0.508 0.556 0.603 |
55.75 145.30 223.55 261.50 |
0.297 |
38.565 |
0.50 0.52 0.10 0.07 |
|
Table 3.
A comparison of electrochemical performance, in terms of (jd)/(jind), of the electrodes included in this investigation with those of Pt-and Pt-based electrocatalysts used for FAO in acidic media reported in the literature.
Table 3.
A comparison of electrochemical performance, in terms of (jd)/(jind), of the electrodes included in this investigation with those of Pt-and Pt-based electrocatalysts used for FAO in acidic media reported in the literature.
| Catalyst | I d /Iind | Conditions of Experiment | pH | Reference |
|---|---|---|---|---|
| NiOx/Pt/GC | 0.33 | 0.3 M FA,pH 3.5, 100 mV/s |
3.5 | [69] |
| NiOx/Pt/CNTs/GC | ∝ | 0.3 M FA, pH 3.5, 100 mV/s |
3.5 | [69] |
| Pt/GC | 0.69 | 0.3 M FA, pH 3.5, 100 mV/s |
3.5 | [69] |
| Commercial Pt/C | 0.16 | 0.5 M FA + 0.1M HClO4, pH ≈1.0 50 mV/s |
1.0 | [51] |
| Pt11.1Ni88.9/C | 0.33 | 0.5 M FA + 0.1M HClO4, pH ≈1.0 50 mV/s |
1.0 | [51] |
| Pt10.9 Au0.2Ni88.9/C | 0.34 | 0.5 M FA + 0.1M HClO4, pH ≈1.0 50 mV/s |
1.0 | [51] |
| Pt/C | 0.29 | 0. 5 M FA + 0.5 M H2SO4 , pH 0.3 50 mV s |
0.3 | [91] |
| Pt black | 0.24 | 0. 5 M FA + 0.5 M H2SO4, pH 0.3, 50 mV s |
0.3 | [91] |
| PtPd/C | 0.87 | 0. 5 M FA + 0.5 M H2SO4, pH 0.3, 20 mV s |
0.3 | [91] |
| Pt-TiOx (700 C) | 10.00 | 0.3 M FA, pH = 3.5, 100 mV/s |
3.5 | [92] |
| Pt/MWCNTs-GC | 7.50 | 0.3 M FA, pH = 3.5, 100 mV/s |
3.5 | [93] |
| MnOx/Au/Pt/GC | 30.20 | 0.3 M FA , pH ≈ 3.5 + a proper amount of NaOH, 100 mV/s |
3.5 | [63] |
| NiOx/Au/Pt/GC | ∝ | 0.3 M FA , pH = 3.5 +a proper amount of NaOH, 100 mV/s |
3.5 | [59] |
| nano-NiOx/Pt | 50.00 | 0.3 M FA , pH = 3.5 +a proper amount of NaOH, 100 mV/s |
3.5 | [65] |
| nano-NiOx/Pt/GC | 17.00 | 0.3 M FA , pH = 3.5 +a proper amount of NaOH, 100 mV/s |
3.5 | [66] |
| Au23/Pt63Co14 | 3.60 | 0.5 M FA + 0.1 M HClO4 pH ≈ 1.0, 50 mV/s |
1.0 | [94] |
| PtNi/Nifoam | 1.14 | 0.07 M FA + 0.5 M H2SO4 , pH 0.3, 50 mV/s |
0.3 | This work |
| PtNi/Nifoam | 1.00 | 0.1 M FA + 0.5 M H2SO4 , pH 0.3, 50 mV/s |
0.3 | This work |
| PtNi/Nifoam | 0.70 | 0.3 M FA + 0.5 M H2SO4 , pH 0.3, 50 mV/s |
0.3 | This work |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



