
Submitted:
01 September 2023
Posted:
04 September 2023
You are already at the latest version
Abstract
Skin cancer is a prevalent and heterogenous disease with several subtypes, such as melanoma, basal cell carcinoma, and squamous cell carcinoma. Among them, melanoma is the most aggressive subtype, with a higher propensity to spread compared to most solid tumors. The application of OMICS approaches has revolutionized the field of melanoma research by providing comprehensive insights into the molecular alterations and biological processes underlying melanoma development and progression. This review aims to offer an overview of melanoma biology, covering its transition from primary to malignant melanoma, as well as the key genes and pathways involved in the initiation and progression of this disease. Utilizing online databases, we extensively explored the general expression profile of genes, identified the most frequently altered genes, and gene mutations, and examined genetic alterations responsible for drug resistance. Additionally, we studied the mechanisms responsible for immune checkpoint inhibitors resistance in melanoma.
Keywords:
Melanoma
; BRAF mutations
; BRAF inhibitors
; MEK inhibitors
; Immunotherapy
1. Introduction
Skin cancer is a major challenge to public health globally and the growing burden of the disease might have a significant impact on the global economy and manpower [1]. The epidermis and dermis are the two main layers that make up the skin. Melanocytes, Keratinocytes, Merkel cells, and Langerhans cells are found in the epidermis, which is the top layer of skin. Any irregularity in this layer can result in several skin injuries, including cancer.
The etiology of skin cancer is complex and heterogenous due to the involvement of environmental, phenotypic, and genetic risk factors. Ultraviolet radiations (UVR) are the most common environmental risk factor for skin cancers which induces breaks in the DNA causing its damage. This could happen due to oxidative stress, pyrimidine dimer production, gene mutations, photoproducts, inflammation, and immunosuppression, all of which favor the carcinogenic process [2].
Skin cancers are commonly divided into 2 main types: melanoma (cancers resulting from melanocyte malfunction) and non-melanoma skin cancers (NMSC) (from cells generated from the epidermis) [3]. According to GLOBOCAN 2020, NMSC accounts for over one million new cases and 64,000 deaths globally and has an incidence that is approximately twice as high for men as for women [4]. NMSC also referred to as keratinocyte carcinoma, are the most common, comprising about 95% of the total skin cancers. They are classified into 2 types: Basal Cell Carcinoma (BCC) and Squamous Cell Carcinoma (SCC). On the contrary, melanoma accounts for over 1.7% of all cases and an anticipated 0.57% of deaths [4]. Melanoma, the most aggressive type of skin cancer, has a higher propensity to spread than most solid tumors. Studies have revealed that UV is the causative agent for nearly 65% of melanoma and 90% of NMSC. The World Health Organization (WHO) 2018 classified melanoma into different types based on sun exposure or sun damage (Figure 1 and Table 1) [5].
The American Joint Committee on Cancer (AJCC) developed a set of guidelines that are used to grade melanoma after diagnosis to direct patient care and prognosis. Patients with melanoma can be divided into five separate stages, ranging from 0 to IV, with the prognosis getting worse as the stage rises [6]. Melanoma in situ is referred to as stage 0 and metastatic melanoma is referred to as stage IV. To categorize early-stage melanoma from late-stage, AJCC criteria uses many permutations of the TNM (Tumor, Node, Metastasis) system. It provides a standardized way to describe the extent of the cancer based on tumor size, lymph node involvement, and the presence of distant metastasis. Early in situ diagnosis of melanoma is crucial for its prognosis and survival as the 5-year survival rate of primary melanoma is 99% while that of metastatic melanoma is only 27% [7].
The "era" of personalised medicine has seen a significant evolution in the treatment and care of individuals with metastatic melanoma. Patients with advanced-stage melanoma have benefited from immunotherapy and have shown improved life expectancy [8]. However, considering the (still) high proportion of patients who do not respond to treatments or experience adverse side effects, there are still a lot of advancements to be achieved. In this scenario, the significance of precision medicine becomes apparent as it plays a crucial role in identifying the most suitable treatment approach for individual patients and exerting an impact on the decisions made. In the recent years, OMICS approach has gained prominence in health care and research as it involves the study of various biological components on a large scale, such as genomics, transcriptomics, proteomics, and metabolomics. By integrating data from various OMICS-based approaches, researchers aim to gain a comprehensive understanding of the molecular mechanisms underlying skin cancer. This knowledge can lead to the development of personalized treatment strategies and the discovery of novel therapeutic targets for improved patient outcomes.
This article provides an overview of the key OMICS approaches and their applications in advancing our understanding of melanoma, with major focus on the genes involved and the mutation spectrum, associated pathways, gene mutations or signatures unique to certain geographic areas and the immunotherapy of melanoma.
2. Genetics of melanoma
Melanoma has the maximum mutational burden of all cancers due to UV-induced DNA damage and/or errors in DNA replication [7]. All these variants accelerate the transition from primary to metastatic melanoma. In the breakthrough phase or the initial phase, a normal melanocyte develops an initial driver mutation causing melanocyte hyperplasia and production of a melanocytic nevi. BRAF and NRAS mutations are the most common mutations found in melanocyte nevi with the latter found mostly in congenital nevi [9,10,11]. In the subsequent step called the expansion phase, certain melanocytic nevi advance into intermediate lesions that develop TERT promoter mutations and eventually develop into melanoma in situ [12]. Once the primary melanoma has accumulated several mutations in CDKN2A, TP53, PTEN, and other genes, it enters the invasive phase and transforms into malignant melanoma [11,13]. This is the phase with a high number of genetic variants and a high mutational burden.
Specific genomic alterations play a crucial role in the initiation and progression of melanoma. These alterations could be either somatic or germline. Somatic mutations, undergo erratic cell division and proliferation and can be the causative of melanoma. The hereditary or family melanomas are caused by germline mutations, which are less frequent and occur within genes associated with an increased susceptibility to melanoma in the germline [14].
A single incidence of melanoma can have many genes altered, but only a small number of these mutations-either gain-of-function (GOF)/activating or loss-of-function (LOF)/deleterious mutations – are true “drivers” of the tumor. Melanoma may exhibit mutations in reported oncogenes, which make melanoma cells hyperactive thus enabling uncontrolled tumor growth. Similarly, tumor suppressor genes, which regulate cell development, are also susceptible to mutations, which when altered, cease to function. As a result, their inactivation may activate the downstream growth pathways, enabling unregulated tumor growth [15].
In the recent years, many genetic variants have been catalogued comprising both GOF and LOF variants which includes copy number variants (CNVs) and single nucleotide variants (SNVs) which could be either somatic or germline mutations [14]. GOF mutations usually occur in the oncogenes. By exploring UALCAN, the expression pattern of the genes in melanoma is obtained. We found that the most frequently altered oncogenes include BRAF and NRAS. Furthermore, GOF mutations have also been identified in CDKN2A, CDK4, RAC1, KIT, TERT, MAP2K1, MAP2K2, GNAQ, GNA11, IDH1, ERBB2/4, KRAS, SF3B1. LOF/deleterious mutations occur in the tumor suppressor genes such as NF1, PTEN, TP53, and RASA2 (Figure 2) [16,17].
The most recurrent somatic mutations occur in genes that regulate central cellular processes, such as proliferation, growth and metabolism, resistance to apoptosis, and cell cycle control genes. These genetic variations lead to the abnormal activation of their respective signaling pathways. A summary of the most frequently altered genes in somatic and germline mutations in melanoma and the pathway involved is listed in Table 2 [15,18].
According to the estimates, the occurrence of cutaneous melanoma is higher in Caucasians when compared to Hispanic, African American, Indo-American, and Asian population groups [19]. Based on the evaluation of various literature sources it was found that the following genetic mutations or gene signatures have been found specifically in certain geographic areas. The presence of NRAS (12%) and KIT gene mutations is associated with melanoma subtype and location in Australian patients [20]. Furthermore, it has been discovered that there is an association between the prevalence of BRAF gene mutations in melanoma patients and the populations of Brazil, Australia, Italy, the United States, Sweden, and Japan, with prevelance being 70%, 48%, 46%, 47%, 71%, and 68% respectively [20]. Several studies have also focused on the co-occurrence of the BRAF and NRAS mutations suggesting that the concurrent presence of these mutations may complicate treatment options, as they may have distinct responses to targeted therapies [21,22]. Recent study focused on the positional identity linked to the anatomical location is a determining factor for transformation potential for oncogenes in Melanoma. Compared to melanocytes from other anatomic regions, acral melanocytes were more vulnerbale to CRKL transformation [23].
3. Understanding the Genetic Changes that fuel treatment resistance: The Melanoma Code
RAS functions as a molecular switch that becomes activated by extracellular signals, subsequently triggering downstream signaling through MAP kinase pathways (Supplementary Figure S1) [24]. The RAS family of proteins comprises NRAS and BRAF, both integral in governing cell development, differentiation, and survival processes. Controlling cell survival, differentiation, and proliferation depends on the functionality of MAPK pathway, which interfaces multiple transcription factors and is triggered by receptor tyrosine kinases (RTKs) to turn on transcription of several genes (Figure 3). In melanoma, the MAPK pathway frequently activated due to mutations in genes like BRAF and NRAS. BRAF mutations cause the pathway to be permanently activated, leading to unchecked cell growth and proliferation. MAPK inhibitors, like BRAF inhibitors, block BRAF activity, blocking downstream kinases, causing cell death and suppressing growth and proliferation.
There are several gene mutation which drive the progression of melanoma post treatment. These encompass BRAF (V600E) mutations, NRAS mutations (G12, G13, Q61) and NF1 LOF mutations. These mutations contribute to the development of melanoma and trigger the activation of MAPK signalling pathway [25]. One of the distinguishing characteristics of melanoma is the activation of the BRAF mutation, prominently seen in cases of malignant melanoma. A recurrent missense mutation, c.1796T>A in exon 15 of BRAF gene causes a change in valine (V) to glutamic acid (E) – V600E, thereby resulting in a non-functional protein. Assocaition between BRAF missense mutation and early resistance to the treatment in melanoma is depicted below (Figure 4).
A classification scheme for cutaneous melanomas was created by the cancer genome atlas (TCGA) based on the existence of NF1, RAS (N/H/KRAS), or BRAF oncogenic mutations. MAPK co-alterations were seen in 30% of NF1 mutants, most frequently BRAF non-V600, despite the fact that NF1 was classified as a genomically distinct subgroup. BRAF alterations have been divided into three classes in more recent functional analyses, on the basis of class 1 (all V600 variations) is dimer- and RAS-independent; class 2 is dimer-dependent; and class 3 is dimer- and RAS-dependent, albeit it frequently necessitates the activation of RAS via co-alteration before MAPK activation can occur [26]. These molecular information imply that the classification of the TCGA driver subgroups may require improvement. There are additional melanoma-related genes, including NF, NRAS, and CDKN2A that may be influenced (either of these genes gets primarily mutated). These melanomas frequently exhibit distinct gene alterations from melanomas that arise in sun-exposed regions, such as variations in the gene C-KIT (commonly KIT). Changes in tumour suppressor genes like CDKN2A (p16) or CDK4 which prevents from performing their usual function. This could also inevitably cause cancer [27].
The primary resistance of melanoma to targeted therapy mainly stems from the heterogeneity and plasticity of the tumor. This diversity in cell populations is largely attributed to the presence of numerous UV-induced mutations in melanoma cells. Apart from the genetic diversity inherent to melanoma, the resistance to BRAF inhibitors can also arise due to influences from the tumor microenvironment and epigenetic alterations [28,29,30,31,32,33].
Understanding these mutations opens up new opportunities, improves melanoma management, and offers new insights for tailored medicines. Using cBioPortal, we observed non significant correlation between fraction of genome alteration and mutation count in melaonoma (Figure 5).
By exploring UALCAN, BRAF expression in melanoma metastasized to lymph node was monitored. We observed that the BRAF expression in the normal cells is negligible when compared to those of nodal metastases (Figure 6). BRAF expression in N0 and N1 are matching. While N0 represents no metastasis to the lymph nodes, N1 represents melanoma being metastasized to 1 to 3 axillary lymph nodes. BRAF expression was high in N2 nodal metastasis, where the melanoma was metastasized to 4 to 9 axillary lymph nodes. However, the expression is reduced in the N3 nodal metastasis where 3-10 axillary lymph nodes have the metastatic melanoma (Figure 6).
Gene effect scores derived from CRISPR knockout screens were released by the broad's achilles and sanger's SCORE projects [34]. We monitored the gene effect score for BRAF in skin cancer cell lines. Negative results suggest gene knockout-induced cell growth inhibition and/or death of cell. The scores are normalised so that independently determined common requirements have a median value of -1 and nonessential genes have a median score of 0 (Supplementary Figure S2).
3.1. Acquired gene mutations
Gene mutations associated with melanoma typically occur within one's lifetime and are not handed on to one's descendants. On rare occasions, these acquired mutations in a cell seem to develop randomly and for no apparent reason. There are several mutations such as missense mutations, truncating mutation which acts as a putative driver alteration that takes place in gene which alters its functions (Supplementary Figure S3). Among all the different mutation BRAF mutation is predominant mutations (Supplementary Figure S3).
We generated a cascade diagram to illustrate the activation of the specific pathway, particularly the RTK-RAS pathway using cBioPortal database's analysis of data from the TCGA Pan-cancer Atlas. We found that melanoma resistance is significantly influenced by the RTK-RAS pathway. Importanly, RTK or N-RAS activation was observed to confer resistance to B-RAF (V600E) suppression in melanomas (Figure 7). Upon inhibition of the ERK pathway, the regulation of RAS activity is halted, resulting in partial RAS activity, which subsequently causes RAS hyperactivation and the development of melanoma. Receptor tyrosine kinases (RTKs) play a crucial role in the evasion of cancer cells from BRAF inhibition, constituing a crucial mechanism in this RTK-mediated process. The most important mechanism contributing to resistance against BRAF/MEK inhibition is the reactivation of MAPK pathway [35].
Because of its prevalent NRAS and BRAF mutations, the system exhibits an overactive RAS-ERK1 and ERK2 pathway, which promotes cell growth and survival. This leads to translocation of RAF to the plasma membrane, where it triggers the activatation of serine/threonine kinases within the RAF family. Consequenlty, this activation cascades to MEK and subsequently activates ERK1 and ERK2. Mutations in BRAF and NRAS are not UVR signature variations but rather mutational pathways, regardless of their significance in melanoma. The activation of ERK1 and ERK2 results in cell-cycle arrest and senescence. While the interaction between BRAF and NRAS affects optimal results, but BRAF inhibitors triggers a paradoxical activation of ERK1 and ERK2. The progression of melanomas is initiated by an accumulation of mutations synergizing with factors like BRAF-V600E8 [36]. In the context of BRAF-mutant or NRAS-mutant cells, ERK1 and ERK2 are hyperactivated leading to growth suppression.
3.2. Drugs that attack cells with altered BRAF gene
The mutations in the BRAF gene affect half of all melanoma cases, resulting in altered BRAF proteins that aid growth. Among thes mutations, the V600E mutation stands out, as it results in the constant activation of the BRAF protein within the MAPK pathway [37]. This particular mutation is found in approximately 40-50% of melanomas. Drugs that trap the BRAF or MEK proteins (BRAF inhibitors or MEK inhibitors) are unlikely to be effective against melanomas with a unaltered BRAF gene. The combined application of both these drugs generally more effective compared to using either one in isolation. As a part of targeted therapy for patient with a BRAF mutation, it's customary to administer both a BRAF and a MEK inhibitor, given their complementary effects.
Three specific medications directly target the BRAF protein: encorafenib (Braftovi), dabrafenib (Tafinlar), and vemurafenib (Zelboraf) (Figure 8). Some patients with advanced or incurable melanoma may benefit from these medications because they can minimize or halt the growth of tumors [38]. In melanoma patients (stage III), who have had surgery, dabrafenib is administered (in addition to trametinib, an MEK inhibitor) to help minimize the probability of the tumour returning [39]. In a study, researchersidentified a subsequent BRAF L505H mutation in melanoma, which conferred resistance to vemurafenib treatment. This suggest that the BRAF L505H mutation may play a substantial role in therapy resistance [40]. Patients with metastatic BRAF(V600)-mutant melanoma develop resistance to selective RAF kinase inhibitors, which results in changes to the MAPK pathway that confer resistance. Additionally, the RAF inhibitor therapy results in a variety of genetic resistance mechanisms, most notably the reactivation of the MAPK pathway [41]. There are several mechanisms by which BRAF inhibitors can acquire resistance. There are multiple ways that BRAF inhibitors can develop resistance. Additional mutations in the MAPK pathway, specifically in the MEK1 and MEK2 genes, are one of the most frequent. Even in the presence of the BRAF inhibitor, these alterations can result in reactivation of the pathway, enabling the cancer cells to proliferate and advance [42].
In addition to MAPK pathway, PI3K signalling cascade is activated by the activation of RTKs, attracts PI3K either directly or through adaptor proteins. PI3K then phosphorylates PIP2 to produce PIP3, which activates AKT and a host of other effectors that control important cellular processes in cancer cells. PTEN controls this process adversely by dephosphorylating PIP3 (Figure 9) [43]. All the significant components of this signalling axis gets frequently altered in cancer. Tumor growth and development are encouraged by the PI3K/Akt pathway, which is abnormally active in malignancies and crucial for many cellular processes. By examining the upstream and downstream nodes of this pathway (Supplementary Figure S4), it may be feasible to fully comprehend its function [44]. Furthermore, the emergence of treatment resistance occurs along with this pathway activation [45]. Abnormal PI3K/AKT/NF-B and PI3K/AKT/mTOR pathways are linked to MDR activation. According to a number of studies, the network with the highest frequency of mutations in human malignancies was the PI3K/AKT route. The two main mutant pathways associated with MDR are PI3K/AKT/NF-B and PI3K/AKT/mTOR, which have been linked to cancer and apoptosis, respectively [46].
MEK inhibitors
Given the collaborative nature of the MEK and BRAF genes, drugs that hinder MEK proteins can be beneficial in addressing BRAF gene alterations present in melanomas. Notably, MEK inhibitors such as Trametinib (Mekinist), cobimetinib (Cotellic), and binimetinib (Mektovi) have been developed for this purpose.
These medications are effective in treating melanoma that has spread or that cannot be entirely removed. Stage III melanoma patients can also take trametinib in addition to dabrafenib following surgery to lessen the chance of the tumour recurring. An MEK inhibitor and a BRAF inhibitor together is once more the most typical strategy compared to using either type of medicine alone, this appears to decrease tumours for a longer period of time. With the combination, some negative effects—like the emergence of further skin cancers—are actually less frequent [47,48].
A major barrier in the therapy of melanoma is the emergence of drug resistance. Melanoma cells can become resistance to therapy through a number of methods, including:
Activation of alternative signalling pathways: Melanoma cells can activate alternative signalling pathways to bypass the inhibition of the primary pathway targeted by the treatment. The MAPK pathway is routinely targeted while treating melanoma, however if Resistance may develop if other mechanisms, such as the PI3K/AKT pathway, are active [33,49].
- Genetic mutations: Melanoma cells may gain mutations in genes that impact drug metabolism or the treatment's intended target. For instance, BRAF gene alterations result in resistance to BRAF inhibitors.
- Epigenetic changes: Epigenetic changes that alter gene expression and lead to treatment resistance may occur in melanoma cells. Modifications to histone or DNA methylation may affect, for example, the expression of genes related to how the body reacts to treatment or how drugs are metabolised.
- Tumour microenvironment: The tumour microenvironment may promote immune evasion and cell survival, which could aid in the formation of resistance. For instance, immune cells that target melanoma cells may become dormant in the presence of immunosuppressive cells or cytokines in the tumour microenvironment [50]
In clinical trials, BRAF and MEK inhibitors increased overall and progression-free survival rates in patients with advanced melanoma. The majority of patients do, however, eventually become resistant. A few of the processes that could cause this resistance include the development of additional MAPK pathway mutations and the upregulation of genes necessary for cell survival and proliferation. PI3K/AKT pathway activation can result in resistance to MAPK inhibitors. The PI3K/AKT pathway is essential for a different processes, including cell growth, metabolism, and survival. Resistance to MAPK inhibitors has been associated with activation of this pathway. Upregulation of anti-apoptotic proteins, which enhance cell survival and shield cells from death brought on by MAPK inhibitors, is one way the PI3K/AKT pathway can result in resistance (Table 4).
A general view of the RAS signalling pathway which regulates membrane trafficking, survival, calcium signalling, and cell apoptosis (Figure 10). The activation of compensatory signalling pathways that avoid the MAPK pathway's inactivation constitutes a second mechanism. For instance, the MAPK pathway's reactivation and activation of the PI3K/AKT pathway may lead to the development of resistance to MAPK inhibitors, which can elevate the expression of the RAS/RAF/MEK/ERK network's genes. However, the activation of other signalling pathways, such as the PI3K/AKT pathway, might result in resistance to MAPK inhibitors. Various cytokines and growth factors that activate the PI3K/AKT pathway are crucial for regulating cell survival and growth. The MAPK pathway can be reactivated and become resistant to MAPK inhibitors if the PI3K/AKT pathway is engaged, which can promote the production of genes implicated in the process. The MAPK pathway is upstream of the RAS/RAF/MEK/ERK pathway is capable of being stimulated by activation of the PI3K/AKT pathway. For RAF, MEK, and ERK to be activated, one of these genes, RAS, must be present. RAS and RAF activity can override the MAPK pathway's inhibition by BRAF inhibitors and revive it, leading to resistance [51].
The Figure 11. depicts the PI3K/Akt, RAS/MAPK, JAK/STAT signalling pathways, govering pivotal processes like cell growth, angiogenesis, and cell migration. Activation of the PI3K/AKT pathway not only regulates gene expression that promote cell survival but also protect cell death induced by MAPK inhibitors [46]. In melanoma cells, the activation of the RAS pathway encourages uncontrolled cell proliferation. Dysregulated signalling through oncogenic BRAF, a crucial constituent of the RAS pathway, causes ongoing activation of downstream effectors involved in cell cycle progression, leading to an increase in cell division and tumour growth. Moreover, this RAS activation promotes enhanced cell survival and heightened resistance to apoptosis. Melanoma cells can avoid the cell death mechanism owing to the dysregulated signalling through the MAPK pathway, which enables them to endure and develop. Additionally, induction of angiogenesis further feeds the developing tumour with nutrition and oxygen, thereby aid in its survival and growth. Furthermore, the increased invasion and metastasis observed in melanoma cells may be caused by dysregulated RAS signalling. Small GTPases can be turned against themselves to encourage invasion and metastasis. The epidermal growth factor receptors (EGFR) which is closely associated in melanoma reoccurrence & progression. Further, by elevating protein synthesis and glucose metabolism, the PI3K/AKT pathway can support cancer cells' survival and development [52].

Table 4.
Genetic mutations causing resistance mechanism to BRAF inhibitors.
| Mutation | Mechanism |
| BRAF gene amplification and splicing | The BRAF gene was amplified, which significantly increased the expression (BRAF protein) and prompted the reactivation of ERK when BRAF inhibitors were present. The production of shortened BRAF proteins, which contain the kinase domain but lack the RAS-binding N-terminus region, can result through alternative splicing and form homodimers that are resistant to BRAF inhibitors [53]. |
| BRAF secondary mutations | Patients who were resistant to BRAF inhibitors showed secondary mutations in L505H or the single-nucleotide alteration V600E. The V600E mutation raises BRAF kinase activity and results in MEK inhibitor cross-resistance [33]. |
| MEK1/2 mutations | Without BRAF activation, MEK1/2 mutations could restart downstream ERK signaling [33,53]. |
| Receptor interaction proteins, RTKs, or membrane receptors are upregulated | Through the stimulation of parallel pathways or by directly inducing the RAS pathway, overexpression or hyperactivation of membrane receptors/RTKs, which is partially mediated by MITF copy gain, may promote acquired resistance [33]. |
| Inconsistencies with in PI3K-AKT cascade | PI3K and AKT-activating mutations enhance AKT signalling by promoting anti-apoptotic signals and elevating expression of essential proliferative genes, enabling survival signals independent of BRAF [33]. |
4. Immune Therapy/ Checkpoint Inhibitors
Cancer cells can manipulate the PD-1/PD-L1 pathway to evade immune recognition. CTLA-4 (cytotoxic T lymphocyte–associated antigen-4) and PD-1 (anti-programmed cell death protein 1) are T cell surface receptors associated with immune suppression and dysfunction. Currently, 7 immune checkpoint inhibitors (ICIs) have been approved by U.S. FDA (1 CTLA-4 inhibitor (ipilimumab), 3 PD-1 inhibitors (nivolumab, pembrolizumab, and cemiplimab) and 3 PD-L1 (ligand for PD-1 receptor) inhibitors (atezolizumab, durvalumab, and avelumab) have received U.S. Food and Drug Administration approval. ICIs have changed the treatment of numerous cancer types. Particularly in certain melanoma patients, ICIs produce a good response, however, the most of patients show no response to the treatment. The underlying mechanism for the resistance remains elusive [54,55,56].
4.1. Mechanisms of Resistance
The major concern with ICIs is to decipher the intricate resistance mechanisms and to develop novel drug combinations to optimize treatment approaches to overcome the resistance. The resistance mechanism can be primary or acquired. Primary resistance is an inherent lack of response to the treatment and acquired resistance emerges during the course of the treatment. The mechanism of resistance is categorized as intrinsic or extrinsic to tumor cells. Intrinsic resistances are related to the mechanism that is specific to the tumor cells involved in immune responses, cell signaling, gene expression, and DNA damage response. Extrinsic resistances are associated externally to the tumor cells throughout the T-cell activation [54].
Transcriptomic analysis from the melanoma patients reveals that responsiveness to the pretreatment with anti-CTLA-4 showed a positive correlation with increased tumor mutational burden (TMB), increased expression of neoantigen and cytolytic markers in the immune microenvironment [57]. In the case of anti-PD-1 pretreatment, responsive melanoma tumor exhibited elevated levels of CD8+ T-cell infiltration, and expression of PD-L1 on tumor cells or immune cells, thus these particular signatures might act as a potential biomarker for the treatment responsiveness [58,59,60]. In the melanoma mice model, more infiltration of intratumoral follicular Treg cells reduced responsiveness to anti-PD1 treatment [61,62]. In melanoma patients, MHC-II expression on tumor cells correlates with a more favorable response to anti-PD1/PDL1 treatment [63]. In certain individuals due to immunoediting, the immune system selects subclones of tumor cells lacking expression of neoantigens causing poor immunogenicity and resistance to ICIs [54]. Altogether, high TMB, increased expression of MHC-II and depleted levels of Tregs improve the efficacy of anti-PD1 treatment [61].
4.2. Clinical predictors of CTLA-4 blockade in metastatic Melanoma
For multi-institutional retrospective analysis from 229 melanoma patients, 60 patients (26%) had NRASG12/G13/Q61 mutations, 53 patients (23%) had BRAFV600 mutations and 116 (51%) had neither NRAS/BRAF mutations were chosen. In response to first-line immune therapy (IL2, Ipilimumab, and anti-PD-1/PD-L1), 28% of NRAS-mutant cohort showed complete response/partial response (CR/PR) whereas NRAS/BRAF wild type cohort exhibited 16% response (28% vs. 16%, P=0.04) and best response to any line of immunotherapy is 32% and 20% response respectively (32% vs. 20%, P=0.07). The group of patients with NRAS-mutant melanoma exhibited a heightened response rate and experienced clinical benefit from immune therapy (Table 5). This retrospective study indicates that advanced melanoma with NRAS mutations exhibit better outcomes in immune-based treatment compared with non-NRAS mutations [64].
We evaluated OS and PFS for patients with BRAF mutations from ICI-cohort (Miao_Melanoma-OS and Miao_Melanoma-PFS datasets) to generate survival curves using Kaplan-Meier analysis. We observed an improved OS and PFS trend for patients with BRAF mutations (Figure 12).
Besides somatic mutations, copy number variations (CNVs) might also aid in selective response to ICIs. Data from small cohorts of melanoma patients treated with ICIs suggest that the integrity of the IFN-γ pathway is essential for the responsiveness to anti-PD1 and anti-CTLA-4 treatment. It indicates that loss of IFN-γ signaling in tumor cells may promote resistance to immune checkpoint therapies [65,66].
High mutational load (nonsynonymous mutations per exome) also exhibited better clinical benefit from Ipilimumab treatment. However, mutational load alone is not an effective indicator of CTLA-4 blockade therapy response. The therapeutic advantages of ipilimumab were observed in correlation with the presence of tumor-specific neoantigens. The tumor-specific expression of somatic neoepitopes increased the overall antigenicity trend. Patients with sustained clinical benefits demonstrated the expression of a tetrapeptide neoantigen signature. Similarly, the presence of this tetrapeptide signature correlated strongly with survival. Mutations resulting in the presentation of specific neoepitopes enhance MHC class I binding, eliciting an intensified antitumor response augmented by CTLA-4 blockade [67].
In a parallel study, transcriptomic analysis of tumor biopsies from 40 melanoma patients reveals a connection between improved immune therapy response and factors such as higher mutational load, increased neoantigen load, and elevated expression of cytolytic markers within the immune microenvironment [57]. Single-cell RNA sequencing and computational analyses on 33 melanoma tumors identified a distinct resistance program unique to tumor cells. This program is linked to T-cell exclusion and immune evasion. CDK4 is one of the key master regulators involved in the resistance program. Counteracting this program through CDK4/6-inhibition enhances the responsiveness of melanoma tumors to ICIs in mouse models [68].
4.3. Predictive features of response to immune checkpoint blockade (ICB)
Auslander et al., developed an immuno-predictive score (IMPRES), a predictor of ICB response in melanoma patients. IMPRES is constructed based on pair-wise relations between the expressions of 28 checkpoint genes with co-stimulatory or co-inhibitory effects. In the above study, seven immune-related consistently differentially expressed pathways (termed CDPs) that are common in all anti-PD-1 datasets and 4 CDPs common across all anti-CTLA-4 datasets were identified. The correlation between each IMPRES feature and the expression of each of the CDPs was computed. Subsequently, IMPRES was used to predict the response to ICB among melanoma patients. While IMPRES can predict all the true responders but miss half of the nonresponders. Additionally, elevated IMPRES scores correlate with enhanced OS and PFS in melanoma patients treated with ICB [52].
Analysis of copy number variations using whole exome sequences (WES) from 469 melanoma tumors did not identify any specific recurrent variation to either responders or non-responders to immune therapy treatment. BRAC2 with nsSNVs (6 of 21 tumors) are better responders. These BRAC2 loss-of-function mutations might lead to a defect in homologous recombination and double-strand DNA break repair or some unknown effects that add to responsiveness to anti-PD-1 treatment. Transcriptomic analysis was performed on anti-PD-1 responding and non-responding tumors to analyse the differentially expressed genes (DEGs). 693 genes were differentially expressed, and relative gene up-expression events were more in non-responding tumors compared to the responding tumors. DEGs that are expressed in higher levels in pre-treatment tumors that do not respond encompass genes linked to mesenchymal transition, immunosuppression, chemotaxis of monocytes and macrophages, and as well as genes associated with wound healing and angiogenesis. Transcriptomic signatures derived from perturbation-based analysis displayed co-enrichment patterns (9 of 13 non-responding vs 1 of 15 responding pretreated anti-PD-1 tumors). These collective signatures are termed as the innate anti-PD-1 resistance (IPRES) signature. Innately resistant tumors exhibit the IPRES, indicating upregulation of events involved in the regulation of mesenchymal transition, cell adhesion, remodelling of the extracellular matrix (ECM), wound healing, and angiogenesis. Notably, treatment with mitogen-activated protein kinase (MAPK) inhibitors causes comparable alterations in residual melanoma tumors. This observation implies that these signatures might negatively impact the responsiveness to anti-PD-1 therapy [69].
5. Discussion
Skin cancer is a potentially life-threatening disease and is responsible for causing significant loss to human health across the globe. As discussed, the aetiology of skin cancer is complex and heterogenous due to the involvement of environmental, phenotypic, and genetic risk factors however the prime factor causing skin cancer is UV radiation which causes breaks in DNA leading to its damage.
Among all malignancies, melanoma has the largest mutational burden because of UV-induced DNA damage and/or replication mistakes. All these accelerate the transition of primary to metastatic melanoma. Numerous somatic and germline mutations including SNVs and CNVs contribute to the formation of melanoma. During the process, the oncogenes attain a GOF mutation while the tumor suppressor genes undergo silencing by LOF mutations. UALCAN-UAB is one of the best freely available OMICS tools to study the expression of these oncogenes and tumor suppressor genes.
Various approaches, including immunotherapy, cryotherapy, radiotherapy, and photodynamic therapy are used for the management of skin cancer. Owing to various limitations of skin cancer treatment and increased severity, a cost-effective, novel, and efficient treatment for skin cancer is required. In the past decade, the systemic treatment landscape for metastatic melanoma has experienced a profound revival, marked by the emergence of two primary strategies: targeted therapy and immunotherapy.
Notably, the resurgence of interest in this field has been driven by the revelation that BRAF mutations are prevalent in approximately 40–50% of melanoma cases. This genetic alteration triggers the constitutive activation of the downstream MAPK pathway (RAS/RAF/MEK/ERK proteins), providing indispensable support for the proliferation of melanoma tumor cells. The primary mutations encompass BRAF (V600E), responsible for approximately 80% of all BRAF mutations. This mutation involves a singular nucleotide alteration (GTG to GAG), leading to the substitution of valine with glutamate. Furthermore, V600K mutations constitute roughly 16% of BRAF mutations, entailing a two-fold nucleotide modification (GTG to AAG), resulting in the replacement of valine with lysine.
These oncogenic mutations play a pivotal role in initiating and promoting tumor formation and metastasis. In response to this understanding, innovative BRAF small molecule inhibitors like vemurafenib, dabrafenib, and encorafenib have been developed as therapeutic interventions for melanoma treatment. These inhibitors target the aberrant BRAF signaling, offering a promising approach to combat this challenging disease.
The immune system plays a vital role in immune therapy responses in cancer. Importantly, in melanomas, ICI treatment exhibited widespread success. However, a large proportion of patients exhibit resistance to the treatment. Several transcriptomic studies from melanoma patients reveal that differential expression of certain transcripts involved in various cellular processes such as increased expression of neoantigen, cytolytic markers, PD-L1, and MHC-II might contribute to resistance to ICI treatment.
Multi-institutional retrospective analysis involving small-cohort suggests that NRAS mutations in advanced melanoma exhibit improved response to first-line immunotherapy. In a similar kind of analysis, BRAF mutations from ICI-cohort exhibited a better response to the treatment. This indicates besides DEGs of certain targets involved in immune responses, mutational load and mutations in NRAS and BRAF can serve as a predictive marker for the treatment.
Recent studies developed methods to predict features that are responsible for improved treatment outcomes. One such predictor is IMPRES, a higher IMPRES score indicates improved OS and PFS in ICI-treated melanoma patients. IPRES signatures also suggest the impact of treatment resistance in melanoma patients. However, due to the complex mechanisms involved in resistance, identifying predictive markers with certainty would be challenging. Nonetheless, the identified predictive markers need to be validated using large cohorts.
A better understanding of the tumor microenvironment, its components, biochemical composition, and metabolic profile of constituent cells would lead to the evaluation of new treatment strategies.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: The figure depicts the downstream effect of the RAS signaling pathway; Figure S2: Gene effect score for BRAF in skin cancer cell lines; Figure S3: Genetic alterations in the context of early resistance; Figure S4. Actions of Akt/PKB pathway.
Author Contributions
Conceptualization, D.M.; writing—original draft preparation, P.V.S., R.N. and K.K.K.; writing—review and editing, P.V.S., R.N., K.K.K., Y.M., S.M.V., R.A. and D.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding
Institutional Review Board Statement
Not applicable.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Data sharing not applicable.
Acknowledgments
We would like to acknowledge the team of Global Cancer Consortium (GCC) for organizing the Cancer OMICS Training Workshop. We would also like to acknowledge Dr. Sooryanarayana Varambally for providing comprehensive insight on UALCAN-UAB. PVS has received Intra Mural Funding (IMF) from Manipal Academy of Higher Education.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Khan NH, Mir M, Qian L, Baloch M, Ali Khan MF, Rehman A-U-, Ngowi EE, Wu D-D & Ji X-Y Skin cancer biology and barriers to treatment: Recent applications of polymeric micro/nanostructures. J Adv Res 2022, 36, 223–247.
- Vink AA & Roza L Biological consequences of cyclobutane pyrimidine dimers. J Photochem Photobiol B 2001, 65, 101–104.
- Esteva A, Kuprel B, Novoa RA, Ko J, Swetter SM, Blau HM & Thrun S Dermatologist-level classification of skin cancer with deep neural networks. Nature 2017, 542, 115–118.
- 4. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A & Bray F Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin, 2021; 71, 209–249.
- Ferrara G & Argenziano G The WHO 2018 Classification of Cutaneous Melanocytic Neoplasms: Suggestions From Routine Practice. Front Oncol 2021, 11, 675296.
- Gershenwald JE, Scolyer RA, Hess KR, Sondak VK, Long G V, Ross MI, Lazar AJ, Faries MB, Kirkwood JM, McArthur GA, Haydu LE, Eggermont AMM, Flaherty KT, Balch CM & Thompson JF Melanoma staging: Evidence-based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J Clin 2017, 67, 472–492.
- Eddy K, Shah R & Chen S Decoding Melanoma Development and Progression: Identification of Therapeutic Vulnerabilities. Front Oncol 2020, 10, 626129.
- 8. Valenti F, Falcone I, Ungania S, Desiderio F, Giacomini P, Bazzichetto C, Conciatori F, Gallo E, Cognetti F, Ciliberto G, Morrone A & Guerrisi A (2021) Precision Medicine and Melanoma: Multi-Omics Approaches to Monitoring the Immunotherapy Response. Int J Mol Sci, 22.
- Pollock PM, Harper UL, Hansen KS, Yudt LM, Stark M, Robbins CM, Moses TY, Hostetter G, Wagner U, Kakareka J, Salem G, Pohida T, Heenan P, Duray P, Kallioniemi O, Hayward NK, Trent JM & Meltzer PS High frequency of BRAF mutations in nevi. Nat Genet 2003, 33, 19–20.
- Poynter JN, Elder JT, Fullen DR, Nair RP, Soengas MS, Johnson TM, Redman B, Thomas NE & Gruber SB BRAF and NRAS mutations in melanoma and melanocytic nevi. Melanoma Res 2006, 16, 267–273.
- Sensi M, Nicolini G, Petti C, Bersani I, Lozupone F, Molla A, Vegetti C, Nonaka D, Mortarini R, Parmiani G, Fais S & Anichini A Mutually exclusive NRASQ61R and BRAFV600E mutations at the single-cell level in the same human melanoma. Oncogene 2006, 25, 3357–3364.
- Chiba K, Lorbeer FK, Shain AH, McSwiggen DT, Schruf E, Oh A, Ryu J, Darzacq X, Bastian BC & Hockemeyer D Mutations in the promoter of the telomerase gene TERT contribute to tumorigenesis by a two-step mechanism. Science 2017, 357, 1416–1420.
- Shain AH & Bastian BC From melanocytes to melanomas. Nat Rev Cancer 2016, 16, 345–358.
- Bruno W, Martinuzzi C, Dalmasso B, Andreotti V, Pastorino L, Cabiddu F, Gualco M, Spagnolo F, Ballestrero A, Queirolo P, Grillo F, Mastracci L & Ghiorzo P Combining molecular and immunohistochemical analyses of key drivers in primary melanomas: interplay between germline and somatic variations. Oncotarget 2018, 9, 5691–5702.
- Scatena C, Murtas D & Tomei S Cutaneous Melanoma Classification: The Importance of High-Throughput Genomic Technologies. Front Oncol 2021, 11, 635488.
- Davis EJ, Johnson DB, Sosman JA & Chandra S Melanoma: What do all the mutations mean? Cancer 2018, 124, 3490–3499.
- Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVSK & Varambally S UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658.
- Leonardi GC, Falzone L, Salemi R, Zanghì A, Spandidos DA, Mccubrey JA, Candido S & Libra M Cutaneous melanoma: From pathogenesis to therapy (Review). Int J Oncol 2018, 52, 1071–1080.
- Cormier JN, Xing Y, Ding M, Lee JE, Mansfield PF, Gershenwald JE, Ross MI & Du XL Ethnic differences among patients with cutaneous melanoma. Arch Intern Med 2006, 166, 1907–1914.
- Devitt B, Liu W, Salemi R, Wolfe R, Kelly J, Tzen C-Y, Dobrovic A & McArthur G Clinical outcome and pathological features associated with NRAS mutation in cutaneous melanoma. Pigment Cell Melanoma Res 2011, 24, 666–672.
- Raaijmakers MIG, Widmer DS, Narechania A, Eichhoff O, Freiberger SN, Wenzina J, Cheng PF, Mihic-Probst D, Desalle R, Dummer R & Levesque MP Co-existence of BRAF and NRAS driver mutations in the same melanoma cells results in heterogeneity of targeted therapy resistance. Oncotarget 2016, 7, 77163–77174.
- Gutiérrez-Castañeda LD, Gamboa M, Nova JA, Pulido L & Tovar-Parra JD Mutations in the BRAF, NRAS, and C-KIT Genes of Patients Diagnosed with Melanoma in Colombia Population. Biomed Res Int 2020, 2020, 2046947.
- Weiss JM, Hunter M V, Cruz NM, Baggiolini A, Tagore M, Ma Y, Misale S, Marasco M, Simon-Vermot T, Campbell NR, Newell F, Wilmott JS, Johansson PA, Thompson JF, Long G V, Pearson J V, Mann GJ, Scolyer RA, Waddell N, Montal ED, Huang T-H, Jonsson P, Donoghue MTA, Harris CC, Taylor BS, Xu T, Chaligné R, Shliaha P V, Hendrickson R, Jungbluth AA, Lezcano C, Koche R, Studer L, Ariyan CE, Solit DB, Wolchok JD, Merghoub T, Rosen N, Hayward NK & White RM Anatomic position determines oncogenic specificity in melanoma. Nature 2022, 604, 354–361.
- Fox DB, Ebright RY, Hong X, Russell HC, Guo H, LaSalle TJ, Wittner BS, Poux N, Vuille JA, Toner M, Hacohen N, Boland GM, Sen DR, Sullivan RJ, Maheswaran S & Haber DA Downregulation of KEAP1 in melanoma promotes resistance to immune checkpoint blockade. NPJ Precis Oncol 2023, 7, 25.
- Loftus SK, Baxter LL, Cronin JC, Fufa TD & Pavan WJ Hypoxia-induced HIF1α targets in melanocytes reveal a molecular profile associated with poor melanoma prognosis. Pigment Cell Melanoma Res 2017, 30, 339–352.
- Shoushtari AN, Chatila WK, Arora A, Sanchez-Vega F, Kantheti HS, Rojas Zamalloa JA, Krieger P, Callahan MK, Betof Warner A, Postow MA, Momtaz P, Nair S, Ariyan CE, Barker CA, Brady MS, Coit DG, Rosen N, Chapman PB, Busam KJ, Solit DB, Panageas KS, Wolchok JD & Schultz N Therapeutic Implications of Detecting MAPK-Activating Alterations in Cutaneous and Unknown Primary Melanomas. Clin cancer Res an Off J Am Assoc Cancer Res 2021, 27, 2226–2235.
- O’Bryan JP Pharmacological targeting of RAS: Recent success with direct inhibitors. Pharmacol Res 2019, 139, 503–511. [CrossRef]
- 28. Olbryt M (2022) Potential Biomarkers of Skin Melanoma Resistance to Targeted Therapy—Present State and Perspectives. Cancers (Basel), 14.
- Hendrix MJC, Seftor EA, Hess AR & Seftor REB Molecular plasticity of human melanoma cells. Oncogene 2003, 22, 3070–3075.
- 30. Ahmed F & Haass NK (2018) Microenvironment-Driven Dynamic Heterogeneity and Phenotypic Plasticity as a Mechanism of Melanoma Therapy Resistance. Front Oncol, 8.
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin A V, Bignell GR, Bolli N, Borg A, Børresen-Dale A-L, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjörd JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Jäger N, Jones DTW, Jones D, Knappskog S, Kool M, Lakhani SR, López-Otín C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson J V, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt ANJ, Valdés-Mas R, van Buuren MM, van ’t Veer L, Vincent-Salomon A, Waddell N, Yates LR, Zucman-Rossi J, Andrew Futreal P, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR, Initiative APCG, Consortium IBC, Consortium IM-S & PedBrain I Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421.
- Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, Patch A-M, Kakavand H, Alexandrov LB, Burke H, Jakrot V, Kazakoff S, Holmes O, Leonard C, Sabarinathan R, Mularoni L, Wood S, Xu Q, Waddell N, Tembe V, Pupo GM, De Paoli-Iseppi R, Vilain RE, Shang P, Lau LMS, Dagg RA, Schramm S-J, Pritchard A, Dutton-Regester K, Newell F, Fitzgerald A, Shang CA, Grimmond SM, Pickett HA, Yang JY, Stretch JR, Behren A, Kefford RF, Hersey P, Long G V, Cebon J, Shackleton M, Spillane AJ, Saw RPM, López-Bigas N, Pearson J V, Thompson JF, Scolyer RA & Mann GJ Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180.
- 33. Proietti I, Skroza N, Bernardini N, Tolino E, Balduzzi V, Marchesiello A, Michelini S, Volpe S, Mambrin A, Mangino G, Romeo G, Maddalena P, Rees C & Potenza C (2020) Mechanisms of Acquired BRAF Inhibitor Resistance in Melanoma: A Systematic Review. Cancers (Basel), 12.
- Dempster JM, Boyle I, Vazquez F, Root DE, Boehm JS, Hahn WC, Tsherniak A & McFarland JM Chronos: a cell population dynamics model of CRISPR experiments that improves inference of gene fitness effects. Genome Biol 2021, 22, 343.
- 35. Al Mahi A & Ablain J (2022) RAS pathway regulation in melanoma. Dis Model Mech, 15.
- 36. Centeno PP, Pavet V & Marais R (2023) The journey from melanocytes to melanoma. Nat Rev Cancer.
- Guo W, Wang H & Li C Signal pathways of melanoma and targeted therapy. Signal Transduct Target Ther 2021, 6, 424.
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O’Day SJ, Sosman JA, Kirkwood JM, Eggermont AMM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT & McArthur GA Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011, 364, 2507–2516.
- Ribas A, Lawrence D, Atkinson V, Agarwal S, Miller WHJ, Carlino MS, Fisher R, Long G V, Hodi FS, Tsoi J, Grasso CS, Mookerjee B, Zhao Q, Ghori R, Moreno BH, Ibrahim N & Hamid O Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat Med 2019, 25, 936–940.
- Hoogstraat M, Gadellaa-van Hooijdonk CG, Ubink I, Besselink NJM, Pieterse M, Veldhuis W, van Stralen M, Meijer EFJ, Willems SM, Hadders MA, Kuilman T, Krijgsman O, Peeper DS, Koudijs MJ, Cuppen E, Voest EE & Lolkema MP Detailed imaging and genetic analysis reveal a secondary BRAF(L505H) resistance mutation and extensive intrapatient heterogeneity in metastatic BRAF mutant melanoma patients treated with vemurafenib. Pigment Cell Melanoma Res 2015, 28, 318–323.
- Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, Place CS, Taylor-Weiner A, Whittaker S & Kryukov G V The Genetic Landscape of Clinical Resistance to RAF Inhibition in Metastatic MelanomaGenetic Resistance Mechanisms to RAF Inhibition in Melanoma. Cancer Discov 2014, 4, 94–109.
- Gerosa L, Chidley C, Fröhlich F, Sanchez G, Lim SK, Muhlich J, Chen J-Y, Vallabhaneni S, Baker GJ, Schapiro D, Atanasova MI, Chylek LA, Shi T, Yi L, Nicora CD, Claas A, Ng TSC, Kohler RH, Lauffenburger DA, Weissleder R, Miller MA, Qian W-J, Wiley HS & Sorger PK Receptor-Driven ERK Pulses Reconfigure MAPK Signaling and Enable Persistence of Drug-Adapted BRAF-Mutant Melanoma Cells. Cell Syst 2020, 11, 478–494.e9.
- Yuan TL & Cantley LC PI3K pathway alterations in cancer: variations on a theme. Oncogene 2008, 27, 5497–5510.
- 44. Parkman GL, Turapov T, Kircher DA, Burnett WJ, Stehn CM, O’Toole K, Flaherty R, Elmer RC, Culver KM, Foth M, Andtbacka RHI, Lum DH, Judson-Torres R, VanBrocklin MW, Holmen SL & McMahon M (2022) Genetic silencing of AKT induces melanoma cell death. bioRxiv, 2022.08.15.504039.
- He Y, Sun MM, Zhang GG, Yang J, Chen KS, Xu WW & Li B Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct Target Ther 2021, 6, 425.
- Liu R, Chen Y, Liu G, Li C, Song Y, Cao Z, Li W, Hu J, Lu C & Liu Y PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis 2020, 11, 797.
- Eroglu Z & Ribas A Combination therapy with BRAF and MEK inhibitors for melanoma: latest evidence and place in therapy. Ther Adv Med Oncol 2016, 8, 48–56.
- Sanchez JN, Wang T & Cohen MS BRAF and MEK Inhibitors: Use and Resistance in BRAF-Mutated Cancers. Drugs 2018, 78, 549–566.
- Carè A, Del Bufalo D & Facchiano A (2022) Editorial on Special Issue “Advances and Novel Treatment Options in Metastatic Melanoma” Cancers (Basel) 14.
- Lim SY, Shklovskaya E, Lee JH, Pedersen B, Stewart A, Ming Z, Irvine M, Shivalingam B, Saw RPM, Menzies AM, Carlino MS, Scolyer RA, Long G V & Rizos H The molecular and functional landscape of resistance to immune checkpoint blockade in melanoma. Nat Commun 2023, 14, 1516.
- Patterson A & Auslander N Mutated processes predict immune checkpoint inhibitor therapy benefit in metastatic melanoma. Nat Commun 2022, 13, 5151.
- Auslander N, Zhang G, Lee JS, Frederick DT, Miao B, Moll T, Tian T, Wei Z, Madan S, Sullivan RJ, Boland G, Flaherty K, Herlyn M & Ruppin E Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma. Nat Med 2018, 24, 1545–1549.
- Carlino MS, Fung C, Shahheydari H, Todd JR, Boyd SC, Irvine M, Nagrial AM, Scolyer RA, Kefford RF, Long G V & Rizos H Preexisting MEK1P124 mutations diminish response to BRAF inhibitors in metastatic melanoma patients. Clin cancer Res an Off J Am Assoc Cancer Res 2015, 21, 98–105.
- Fares CM, Van Allen EM, Drake CG, Allison JP & Hu-Lieskovan S Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am Soc Clin Oncol Educ book Am Soc Clin Oncol Annu Meet 2019, 39, 147–164.
- Sharma P & Allison JP The future of immune checkpoint therapy. Science 2015, 348, 56–61.
- 56. Seidel JA, Otsuka A & Kabashima K (2018) Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front Oncol, 8.
- Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MHG, Goldinger SM, Utikal J, Hassel JC, Weide B, Kaehler KC, Loquai C, Mohr P, Gutzmer R, Dummer R, Gabriel S, Wu CJ, Schadendorf D & Garraway LA Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211.
- Weiss SA, Wolchok JD & Sznol M Immunotherapy of Melanoma: Facts and Hopes. Clin cancer Res an Off J Am Assoc Cancer Res 2019, 25, 5191–5201.
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, Seja E, Cherry G, Gutierrez AJ, Grogan TR, Mateus C, Tomasic G, Glaspy JA, Emerson RO, Robins H, Pierce RH, Elashoff DA, Robert C & Ribas A PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571.
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM & Sznol M Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012, 366, 2443–2454.
- 61. Machiraju D, Schäfer S & Hassel JC (2021) Potential Reasons for Unresponsiveness to Anti-PD1 Immunotherapy in Young Patients with Advanced Melanoma. Life (Basel, Switzerland), 11.
- Eschweiler S, Clarke J, Ramírez-Suástegui C, Panwar B, Madrigal A, Chee SJ, Karydis I, Woo E, Alzetani A, Elsheikh S, Hanley CJ, Thomas GJ, Friedmann PS, Sanchez-Elsner T, Ay F, Ottensmeier CH & Vijayanand P Intratumoral follicular regulatory T cells curtail anti-PD-1 treatment efficacy. Nat Immunol 2021, 22, 1052–1063.
- Johnson DB, Estrada M V, Salgado R, Sanchez V, Doxie DB, Opalenik SR, Vilgelm AE, Feld E, Johnson AS, Greenplate AR, Sanders ME, Lovly CM, Frederick DT, Kelley MC, Richmond A, Irish JM, Shyr Y, Sullivan RJ, Puzanov I, Sosman JA & Balko JM Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat Commun 2016, 7, 10582.
- Johnson DB, Lovly CM, Flavin M, Panageas KS, Ayers GD, Zhao Z, Iams WT, Colgan M, DeNoble S, Terry CR, Berry EG, Iafrate AJ, Sullivan RJ, Carvajal RD & Sosman JA Impact of NRAS mutations for patients with advanced melanoma treated with immune therapies. Cancer Immunol Res 2015, 3, 288–295.
- Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, Chen P-L, Hwu P, Allison JP, Futreal A, Wargo JA & Sharma P Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404.
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, Saco J, Homet Moreno B, Mezzadra R, Chmielowski B, Ruchalski K, Shintaku IP, Sanchez PJ, Puig-Saus C, Cherry G, Seja E, Kong X, Pang J, Berent-Maoz B, Comin-Anduix B, Graeber TG, Tumeh PC, Schumacher TNM, Lo RS & Ribas A Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med 2016, 375, 819–829.
- Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, Hollmann TJ, Bruggeman C, Kannan K, Li Y, Elipenahli C, Liu C, Harbison CT, Wang L, Ribas A, Wolchok JD & Chan TA Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014, 371, 2189–2199.
- Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su M-J, Melms JC, Leeson R, Kanodia A, Mei S, Lin J-R, Wang S, Rabasha B, Liu D, Zhang G, Margolais C, Ashenberg O, Ott PA, Buchbinder EI, Haq R, Hodi FS, Boland GM, Sullivan RJ, Frederick DT, Miao B, Moll T, Flaherty KT, Herlyn M, Jenkins RW, Thummalapalli R, Kowalczyk MS, Cañadas I, Schilling B, Cartwright ANR, Luoma AM, Malu S, Hwu P, Bernatchez C, Forget M-A, Barbie DA, Shalek AK, Tirosh I, Sorger PK, Wucherpfennig K, Van Allen EM, Schadendorf D, Johnson BE, Rotem A, Rozenblatt-Rosen O, Garraway LA, Yoon CH, Izar B & Regev A A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 2018, 175, 984–997.e24.
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, Seja E, Lomeli S, Kong X, Kelley MC, Sosman JA, Johnson DB, Ribas A & Lo RS Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44.
Figure 1.
Classification of melanoma in association with sun-exposure as per WHO 2018. The 2018 classification by WHO has categorized melanoma into different types based on sun exposure or sun damage.
Figure 1.
Classification of melanoma in association with sun-exposure as per WHO 2018. The 2018 classification by WHO has categorized melanoma into different types based on sun exposure or sun damage.
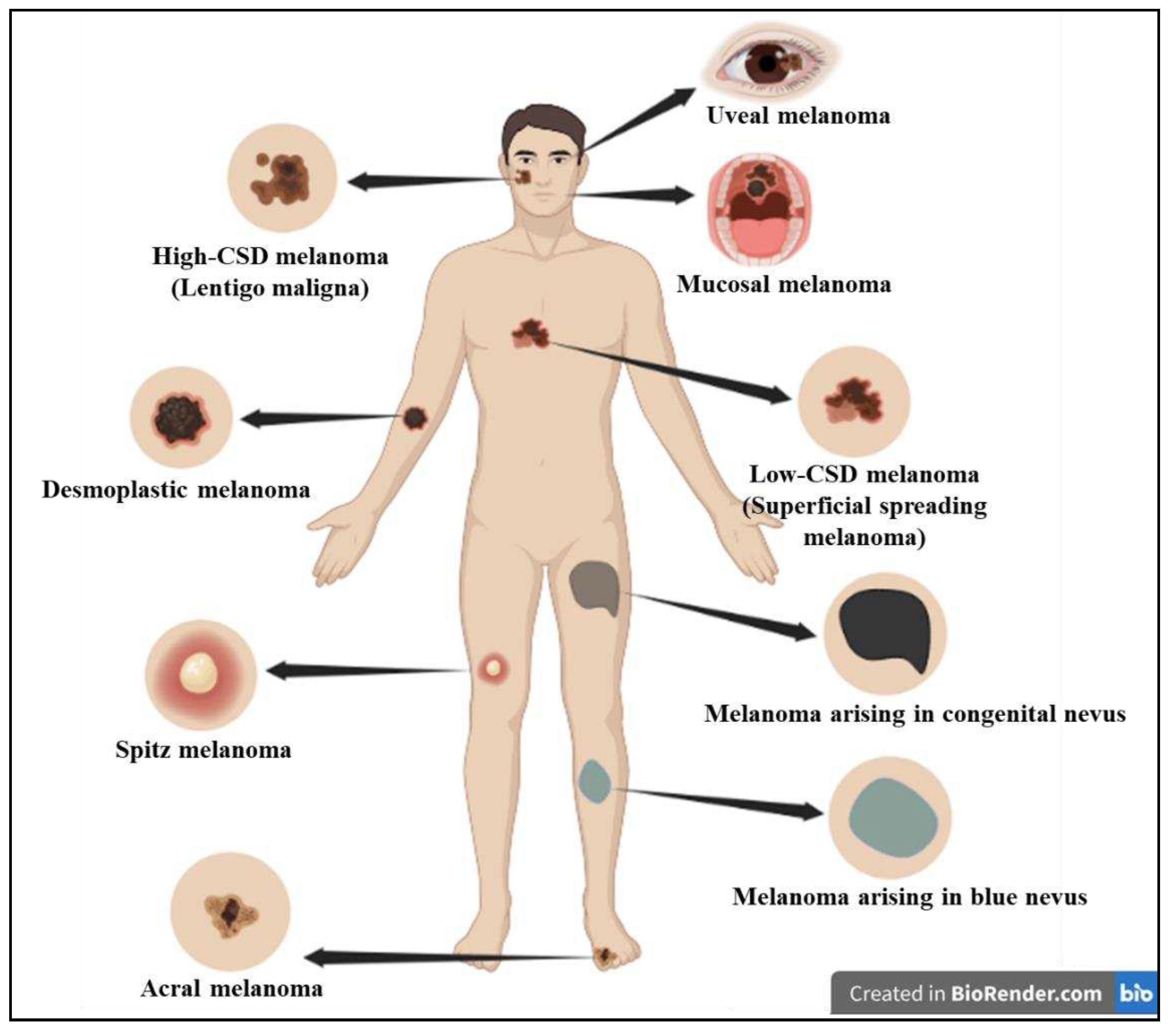
Figure 2.
Expression profile of genes associated with melanoma in melanoma patients obtained from UALCAN-UAB database [17]. The Y-axis represents the major genes associated with melanoma and the X-axis represents the tumor type – Primary tumor or Metastatic tumor. The blue colour represents low expression while the red colour represents high expression. From the above plot, it is observed that RAC1, MAP2K2 and CDK4 are consistently showing high expression in all the patients, while TERT and ERBB4 are showing low expression. The expression profile of the other genes is varying in each patient.
Figure 2.
Expression profile of genes associated with melanoma in melanoma patients obtained from UALCAN-UAB database [17]. The Y-axis represents the major genes associated with melanoma and the X-axis represents the tumor type – Primary tumor or Metastatic tumor. The blue colour represents low expression while the red colour represents high expression. From the above plot, it is observed that RAC1, MAP2K2 and CDK4 are consistently showing high expression in all the patients, while TERT and ERBB4 are showing low expression. The expression profile of the other genes is varying in each patient.
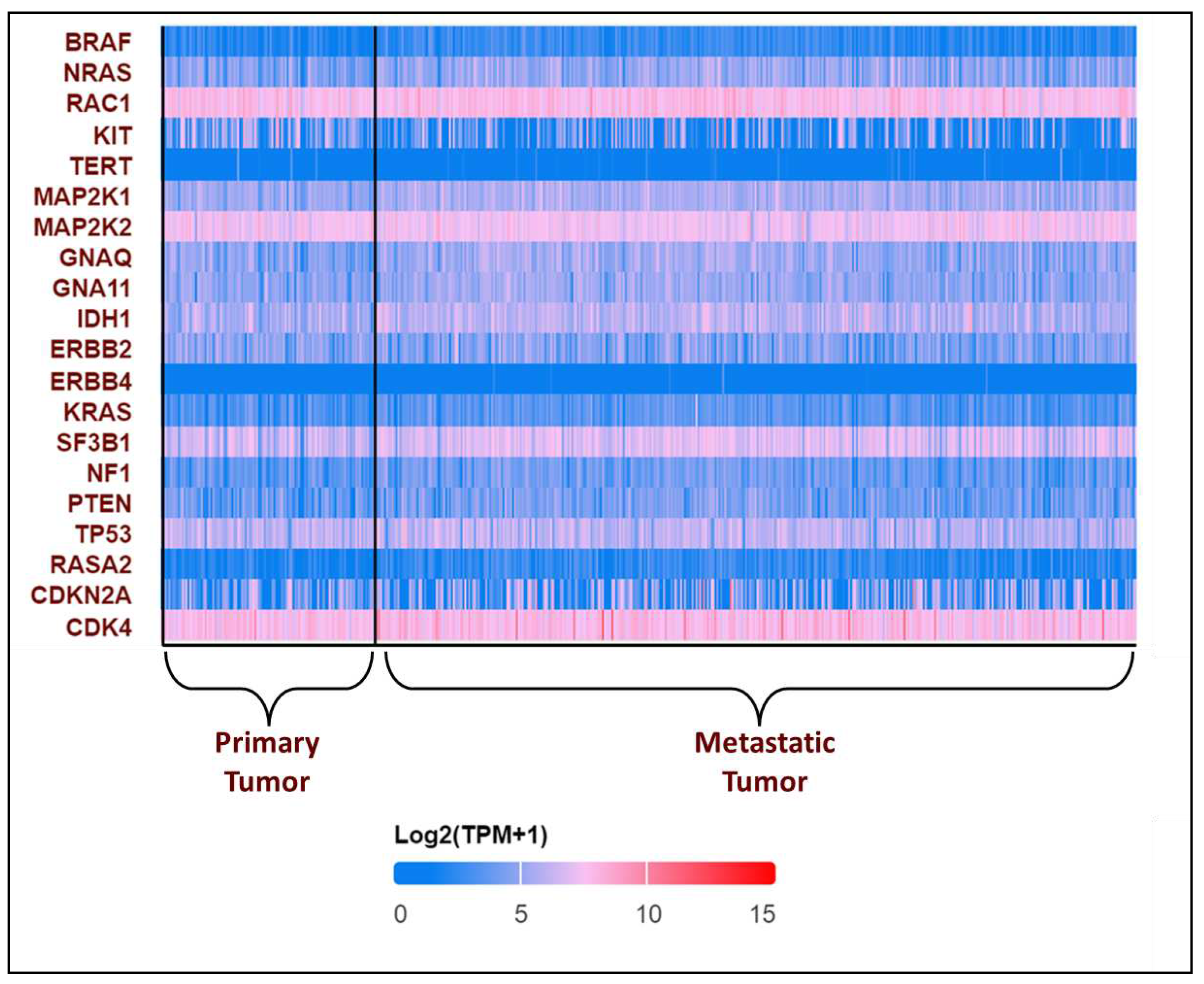
Figure 3.
Ras activation mechanism: of the RAS pathway. This figure demonstrates downstream effect of RAS signaling effector pathway. Ras activation involves the conversion of cytosolic GDP to GTP, thereby initiating a shift from an inactive GDP-bound state to an active GTP-bound state. This interaction leads to the activation of Ras. Subsequently, Ras triggers downstream signaling pathways, crucial for the proliferation and metastasis of melanoma. (Created with BioRender.com).
Figure 3.
Ras activation mechanism: of the RAS pathway. This figure demonstrates downstream effect of RAS signaling effector pathway. Ras activation involves the conversion of cytosolic GDP to GTP, thereby initiating a shift from an inactive GDP-bound state to an active GTP-bound state. This interaction leads to the activation of Ras. Subsequently, Ras triggers downstream signaling pathways, crucial for the proliferation and metastasis of melanoma. (Created with BioRender.com).
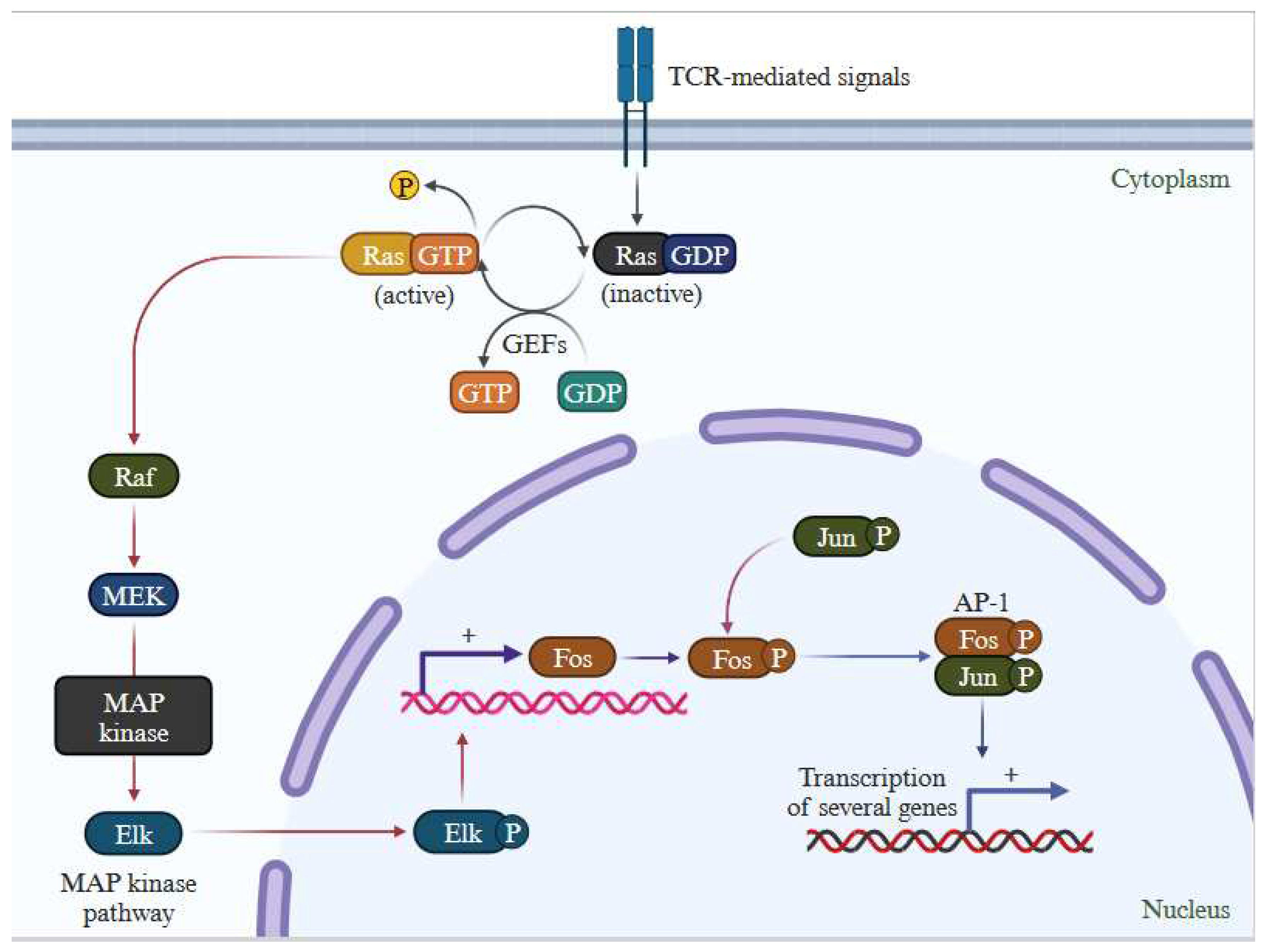
Figure 4.
Association between BRAF missense mutation and early drug resistance in melanoma. The dot plot showing BRAF mutation triggering early resistance. The early resistance is higher post treatments leading to subsequent disease. These mutations are oncogenic drivers that cause progression and metastasis. The data represented from the TCGA dataset using c-BioPortal.
Figure 4.
Association between BRAF missense mutation and early drug resistance in melanoma. The dot plot showing BRAF mutation triggering early resistance. The early resistance is higher post treatments leading to subsequent disease. These mutations are oncogenic drivers that cause progression and metastasis. The data represented from the TCGA dataset using c-BioPortal.

Figure 5.
The plot describes the correlation between set of fraction genome altered (x-axis) and the mutation count (y-axis). No significant correlation between fraction of genome alteration and mutation count is observed. The data represented from the TCGA dataset using c-BioPortal.
Figure 5.
The plot describes the correlation between set of fraction genome altered (x-axis) and the mutation count (y-axis). No significant correlation between fraction of genome alteration and mutation count is observed. The data represented from the TCGA dataset using c-BioPortal.
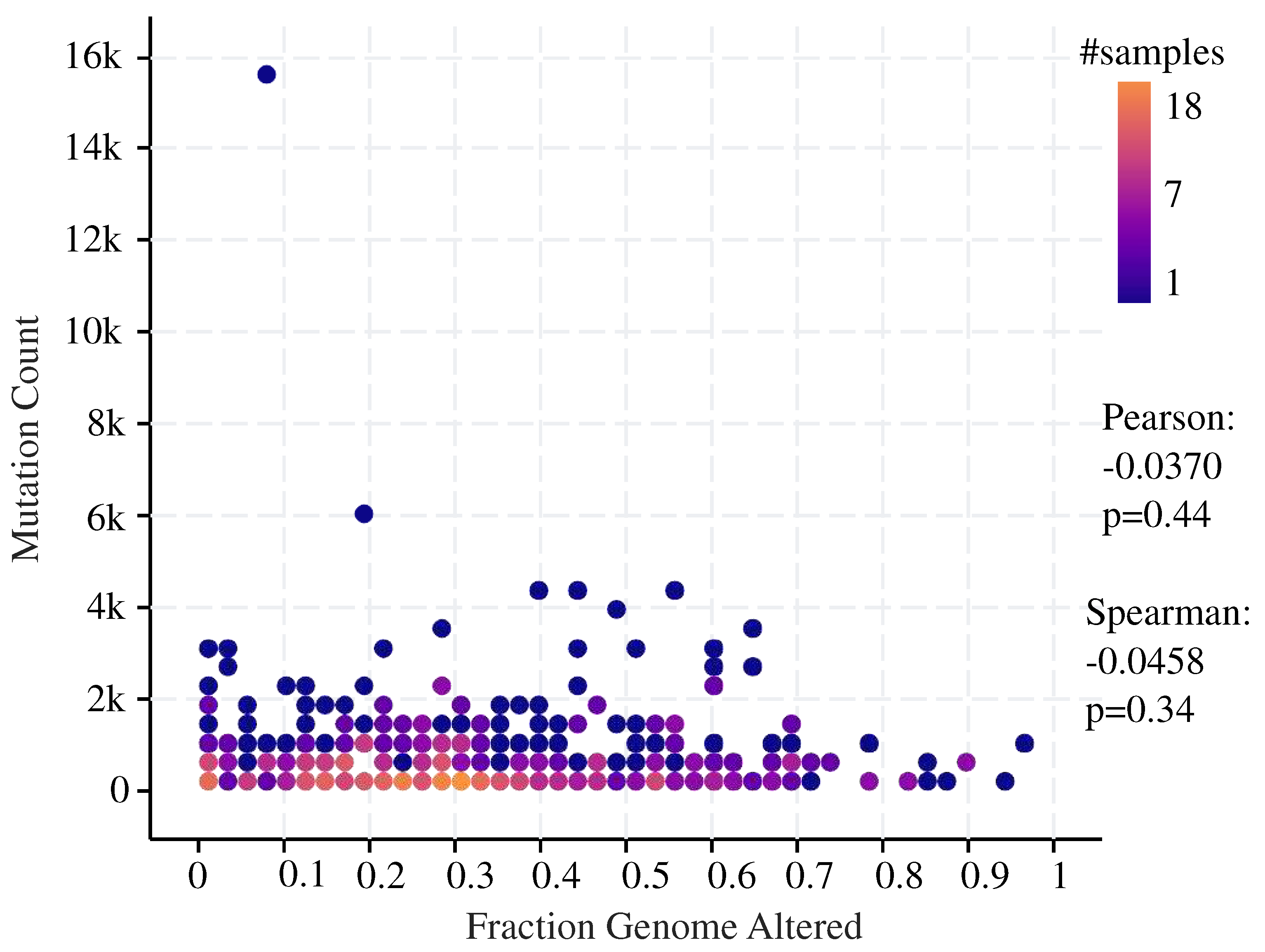
Figure 6.
BRAF expression profile status of nodal metastasis in skin cutaneous melanoma. The BRAF expression profiles data is obtained using UALCAN portal. It is evident from the above figure that the BRAF expression is high in N2, while the expression is low in N3 nodal metastasis. The statistical analysis for the differential expression is represented in the Table 3.
Figure 6.
BRAF expression profile status of nodal metastasis in skin cutaneous melanoma. The BRAF expression profiles data is obtained using UALCAN portal. It is evident from the above figure that the BRAF expression is high in N2, while the expression is low in N3 nodal metastasis. The statistical analysis for the differential expression is represented in the Table 3.
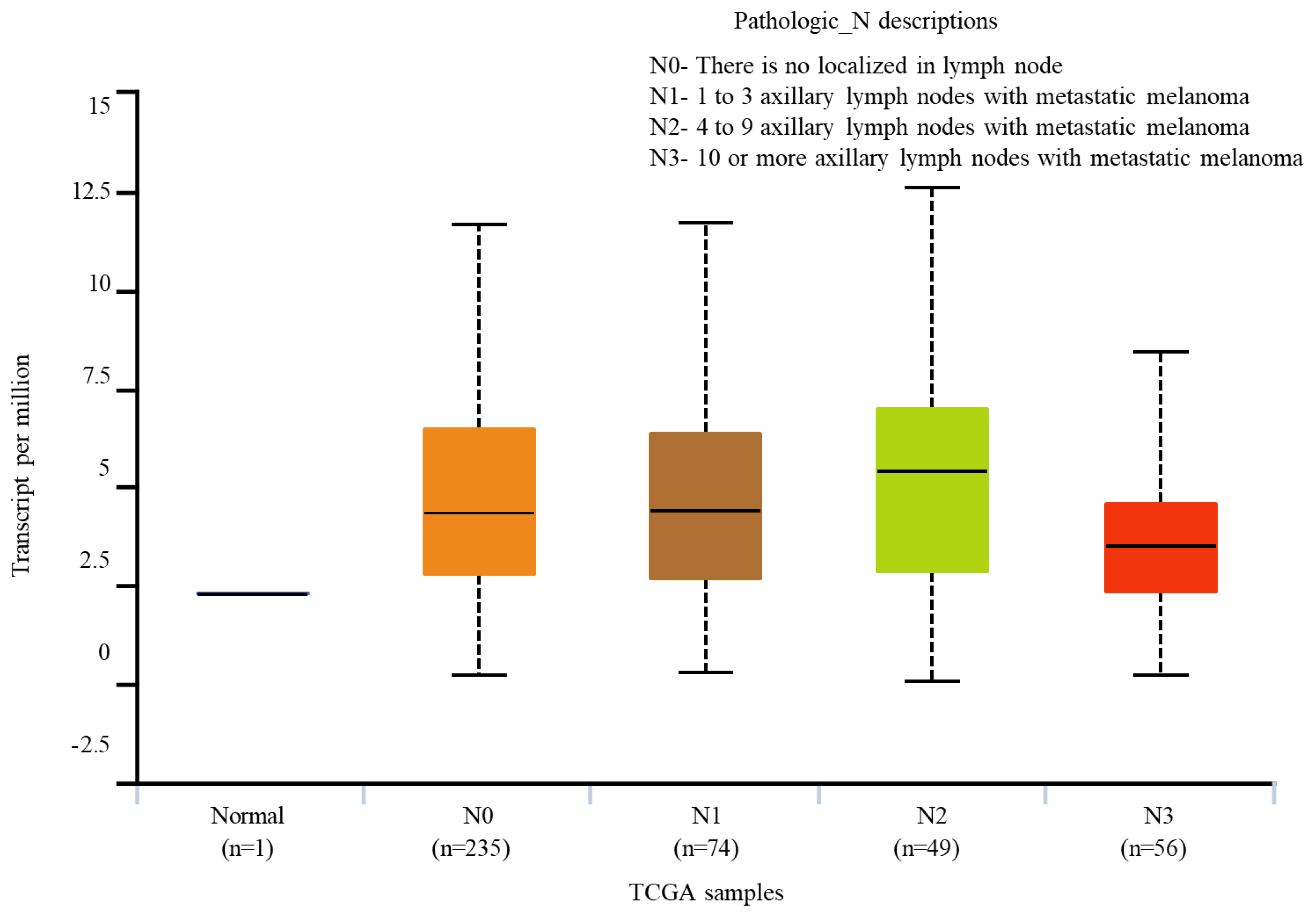
Figure 7.
RTK-RAS Pathway regulates the MAPK signalling in melanoma by activating RAS, RAF and MEK leading to melanoma development. The data represented from the TCGA dataset using c-BioPortal.
Figure 7.
RTK-RAS Pathway regulates the MAPK signalling in melanoma by activating RAS, RAF and MEK leading to melanoma development. The data represented from the TCGA dataset using c-BioPortal.
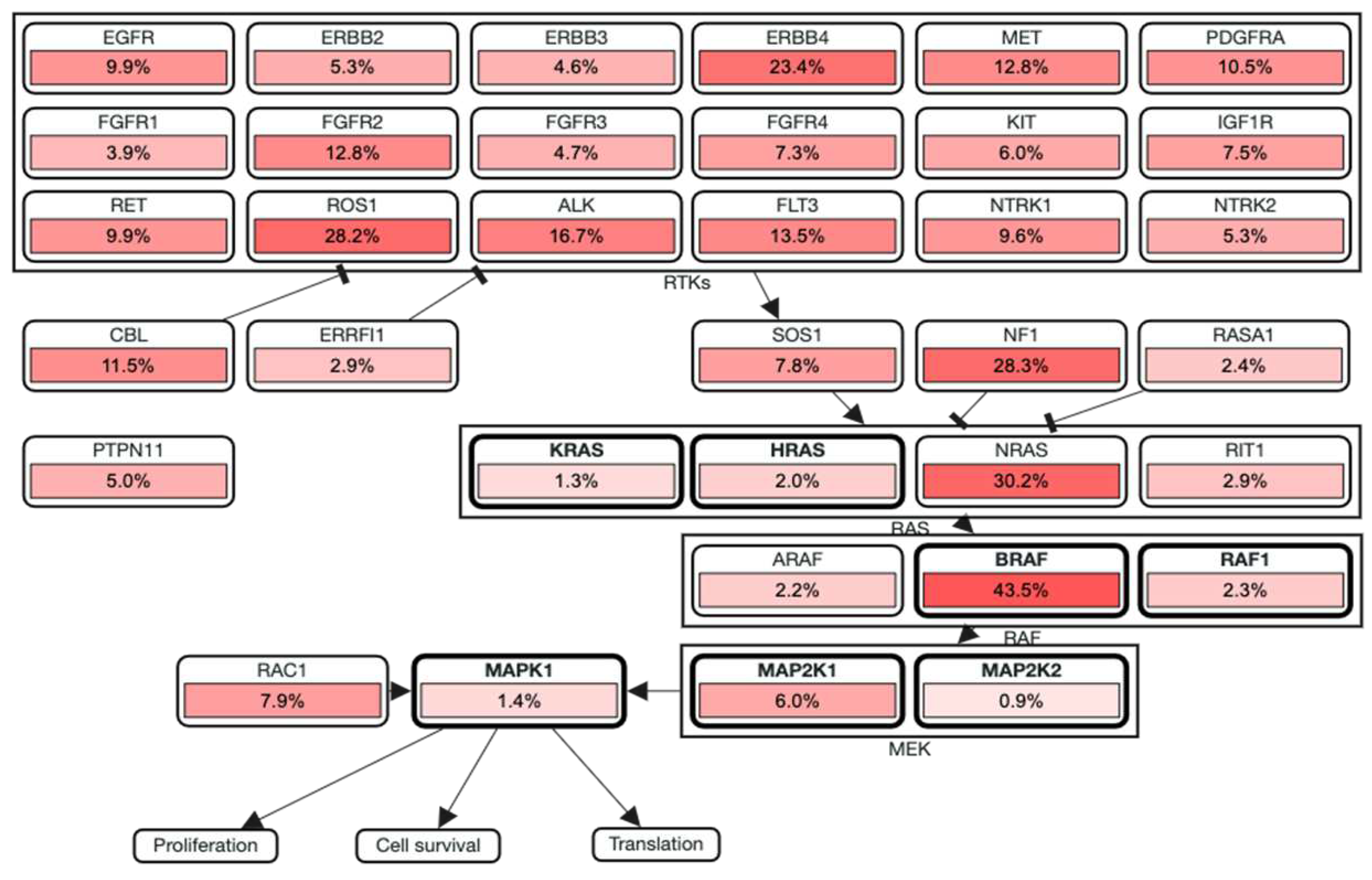
Figure 8.
The oncogenic BRAF signalling pathway in melanoma is targeted using specific drugs that blocks MAPK pathway. Diagram illustartes influcence of oncogenic BRAF signaling on cell proliferation and survival and the effect of BRAF inhbitors on oncogenic BRAF signaling (Created with BioRender.com).
Figure 8.
The oncogenic BRAF signalling pathway in melanoma is targeted using specific drugs that blocks MAPK pathway. Diagram illustartes influcence of oncogenic BRAF signaling on cell proliferation and survival and the effect of BRAF inhbitors on oncogenic BRAF signaling (Created with BioRender.com).
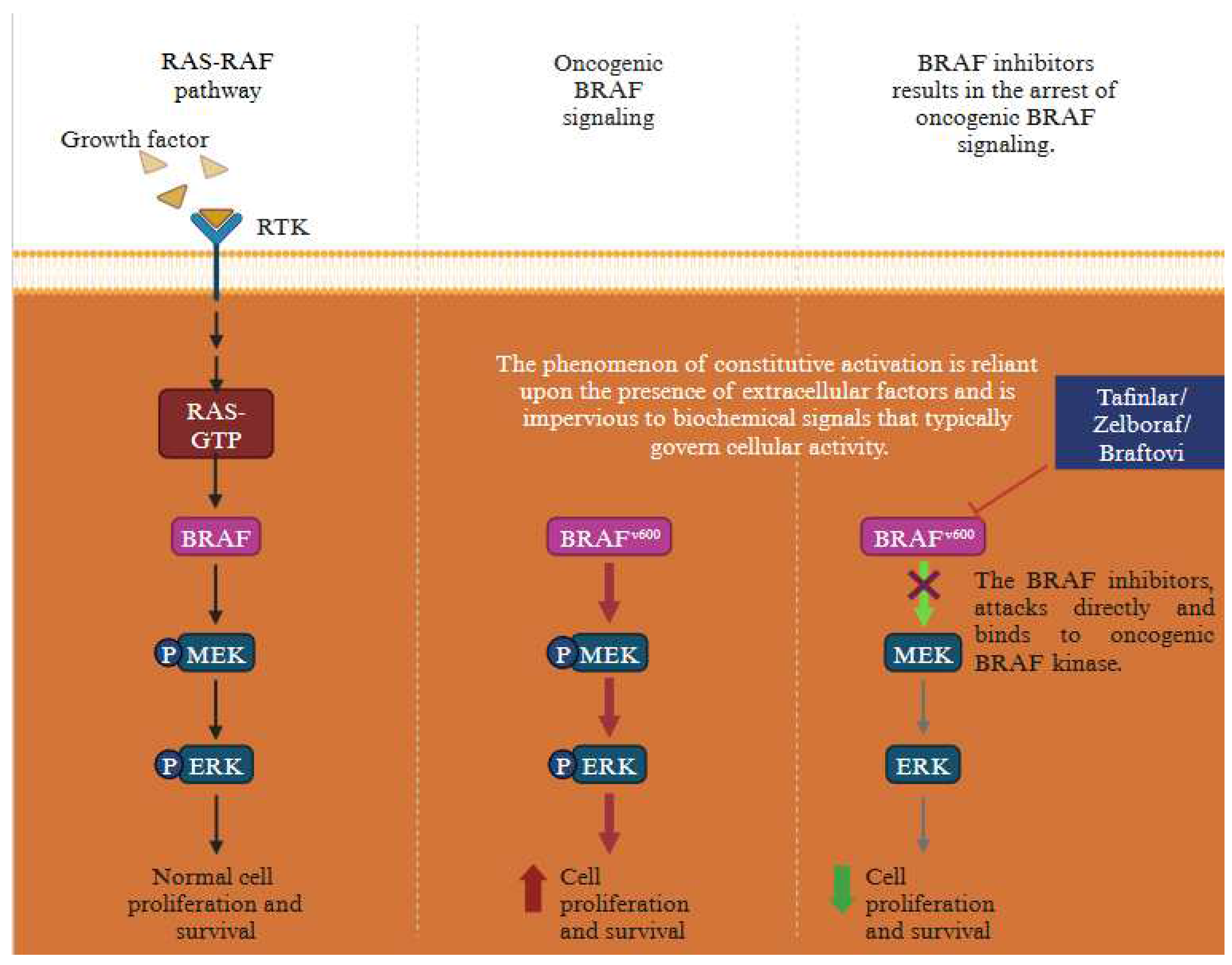
Figure 9.
Tumor growth and development supported by the PI3K/Akt pathway. Figure depicts PI3K pathway in cancer cell growth. The BRAF gene encodes member of the RAF family proteins (serine/threonine kinase). These proteins are a component of the RAF/MEK/ERK serine threonine kinase cascade, which controls a number of cellular processes. RAF inhibitor resistance is connected to numerous of activation methods of the PI3K pathway, including as PTEN loss and AKT activation. RTKs are receptor tyrosine kinases, while PI3K is phosphoinositide 3-kinase (Designed with BioRender).
Figure 9.
Tumor growth and development supported by the PI3K/Akt pathway. Figure depicts PI3K pathway in cancer cell growth. The BRAF gene encodes member of the RAF family proteins (serine/threonine kinase). These proteins are a component of the RAF/MEK/ERK serine threonine kinase cascade, which controls a number of cellular processes. RAF inhibitor resistance is connected to numerous of activation methods of the PI3K pathway, including as PTEN loss and AKT activation. RTKs are receptor tyrosine kinases, while PI3K is phosphoinositide 3-kinase (Designed with BioRender).
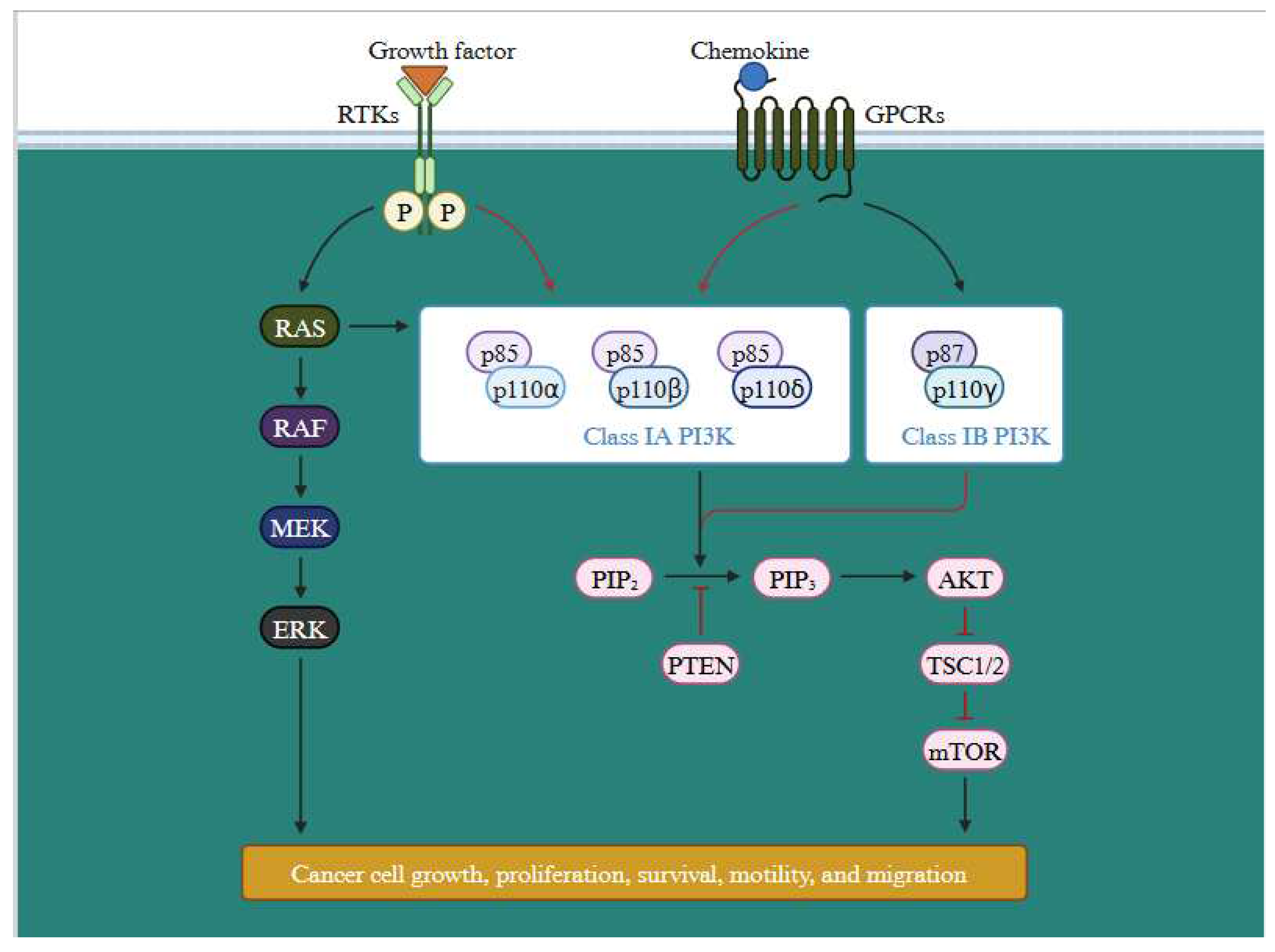
Figure 10.
Overview of the RAS Signaling Pathway: The RAS signaling pathway, encompassing the RAS superfamily of small GTPases, such as RAS itself, governs crucial cellular processes, including membrane trafficking and signaling. Notably, cancer cells exploit the inherent functions of these small GTPases to promote invasion, metastasis, and propagation (Created using Biorender).
Figure 10.
Overview of the RAS Signaling Pathway: The RAS signaling pathway, encompassing the RAS superfamily of small GTPases, such as RAS itself, governs crucial cellular processes, including membrane trafficking and signaling. Notably, cancer cells exploit the inherent functions of these small GTPases to promote invasion, metastasis, and propagation (Created using Biorender).
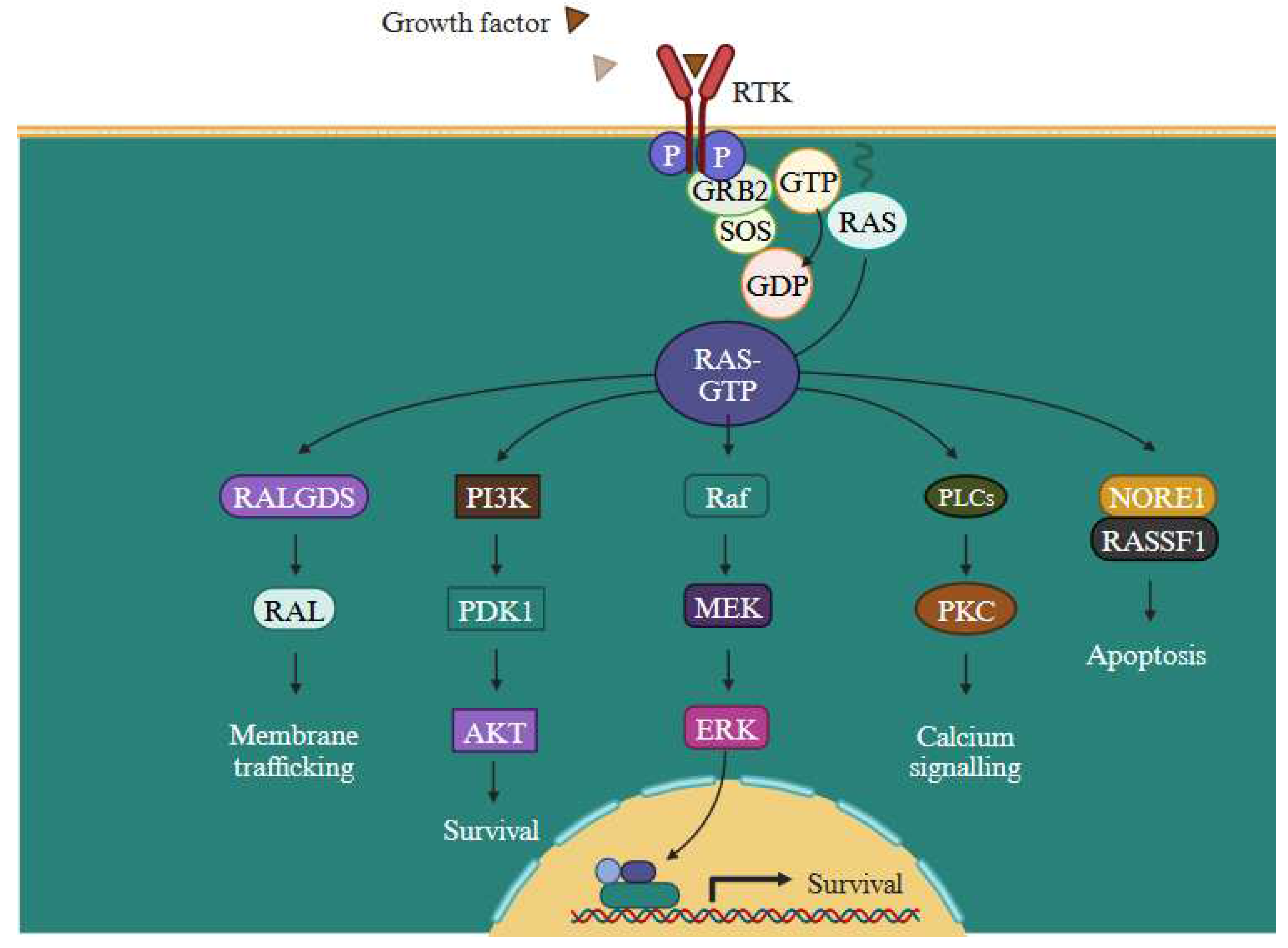
Figure 11.
Overview of PI3K/Akt, RAS/MAPK, and JAK/STAT signalling pathways. In melanoma cells, activation of the RAS pathway encourages unchecked cell proliferation, cell survival and apoptosis resistance (Designed using BioRender). .
Figure 11.
Overview of PI3K/Akt, RAS/MAPK, and JAK/STAT signalling pathways. In melanoma cells, activation of the RAS pathway encourages unchecked cell proliferation, cell survival and apoptosis resistance (Designed using BioRender). .
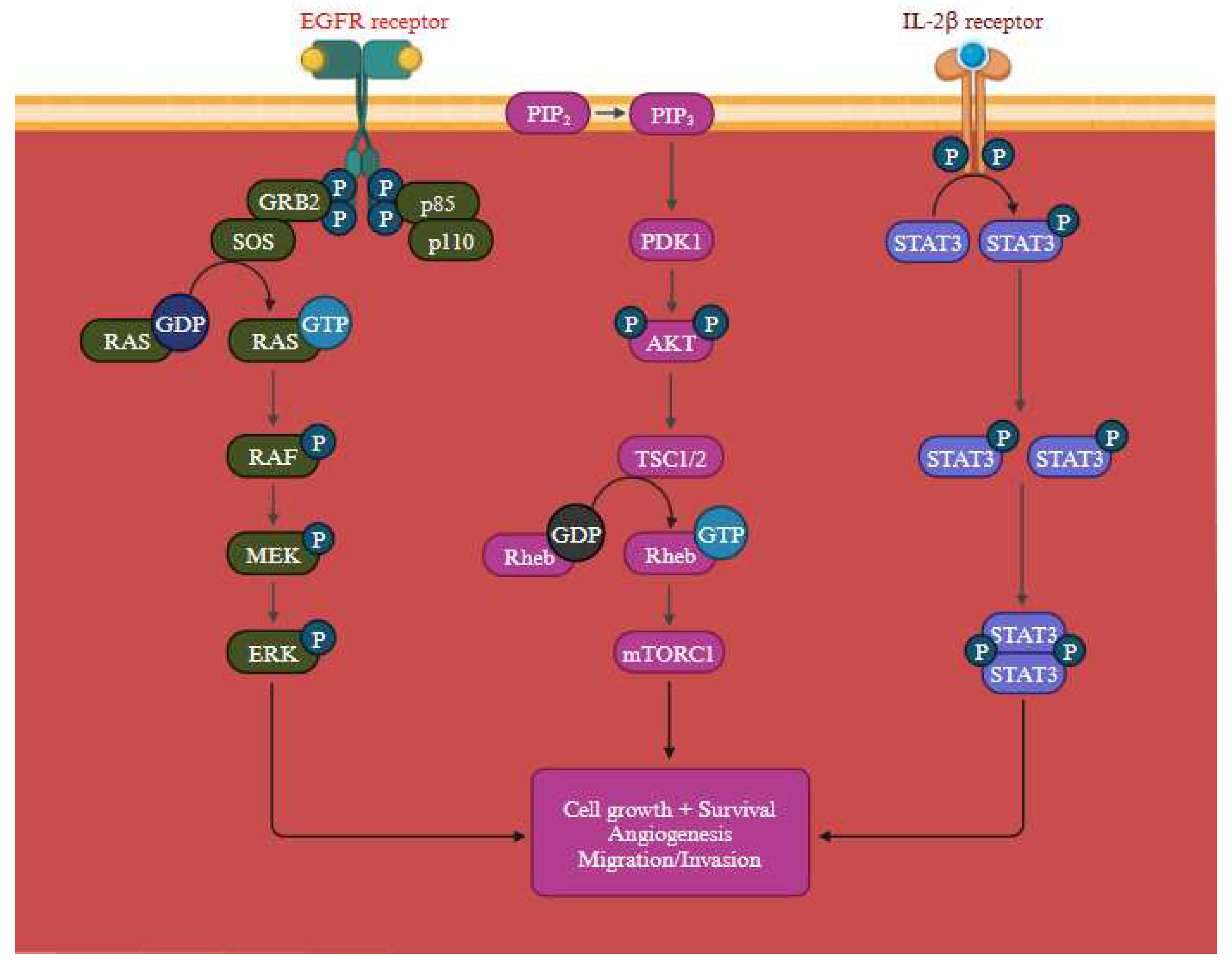
Figure 12.
Kaplan–Meier survival curves for OS and PFS. (A) OS and (B) PFS from ICI-Cohort for BRAF (WT) and BRAF-mutant cohorts. Improved OS and PFS trend for patients with BRAF mutations were depicted. Data was generated using the CAMOIP tool.
Figure 12.
Kaplan–Meier survival curves for OS and PFS. (A) OS and (B) PFS from ICI-Cohort for BRAF (WT) and BRAF-mutant cohorts. Improved OS and PFS trend for patients with BRAF mutations were depicted. Data was generated using the CAMOIP tool.
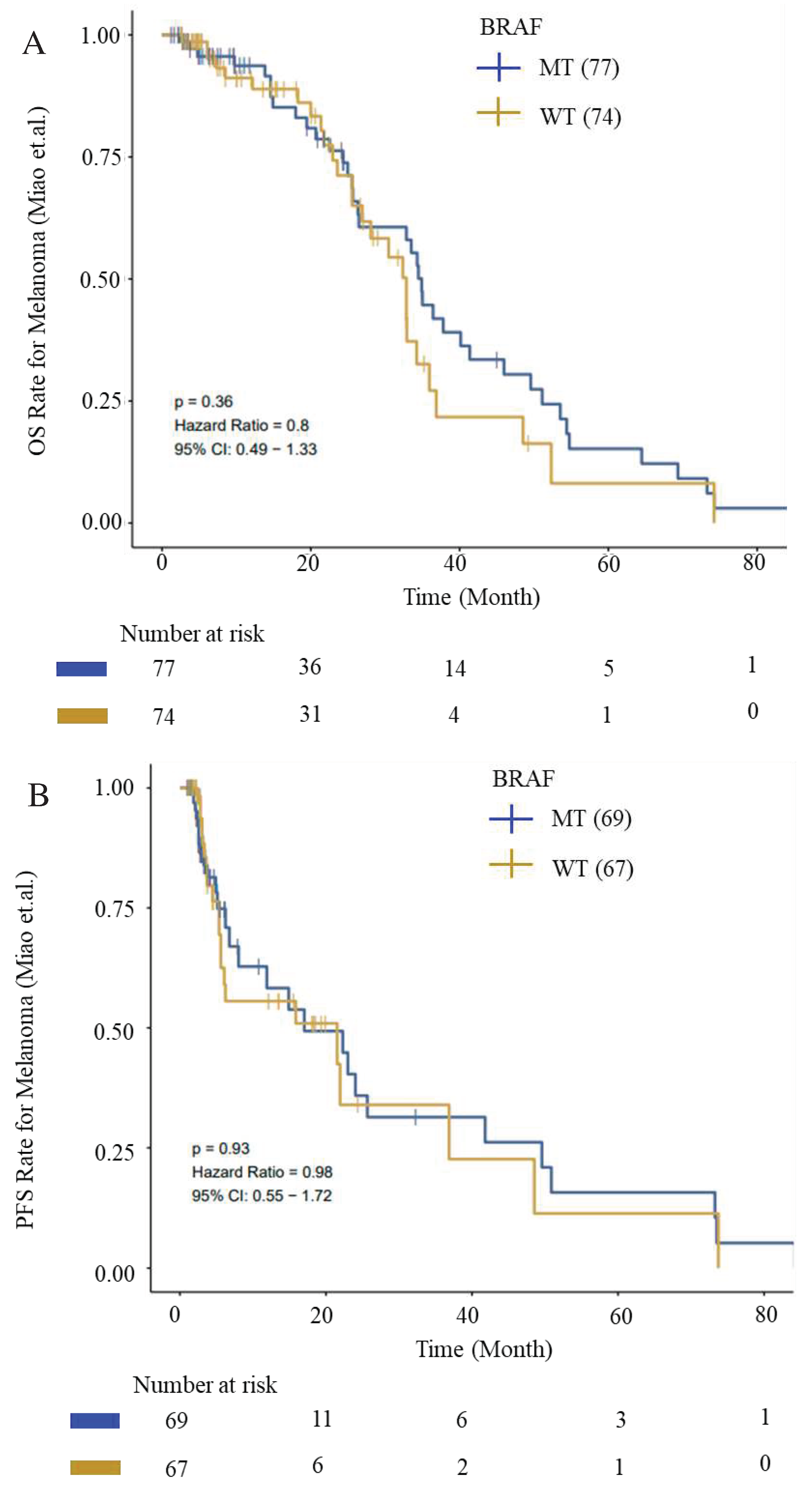
Table 1.
WHO 2018 classification of melanoma [5].
Table 1.
WHO 2018 classification of melanoma [5].
| Melanoma Subtype | Location |
| Melanomas arising in sun exposed skin | |
| Low-CSD* melanoma/ Superficial spreading melanoma | trunk or extremities |
| High-CSD* melanoma (including lentigo maligna melanoma and high-CSD* nodular melanoma) | Head and neck region |
| Desmoplastic melanoma | Head and neck, trunk, or extremities |
| Melanomas developing at sun shielded areas or without known etiological associations with UV radiation exposure | |
| Malignant Spitz tumor (Spitz melanoma) | Head and neck, trunk, or extremities |
| Acral melanoma | Acral sites |
| Mucosal melanoma | Mucosae |
| Melanoma arising in congenital nevus | Trunk and proximal parts of the limbs, scalp, or neck |
| Melanoma arising in blue nevus | Scalp, extremities, or trunk |
| Uveal melanoma | Eyes |
*Cumulative sun damage.
| Gene Name | Gene Symbol | Frequency (%) | Pathway | Function |
| Somatic GOF/Activating mutations | ||||
| Braf Proto-Oncogene, Serine/Threonine Kinase | BRAF | 50-70 | MAPK signaling | Cell proliferation and survival |
| Neuroblastoma RAS Viral Oncogene Homolog | NRAS | 15-30 | MAPK/PI3K signaling | Cell proliferation, differentiation, and survival |
| Ras-related C3 Botulinum Toxin Substrate 1 | RAC1 | ~9 | MAPK signaling | Cell proliferation and migration |
| KIT proto-oncogene receptor tyrosine kinase | KIT | 5-15 | MAPK/PI3K and JAK/STAT signaling | Cell proliferation and survival |
| Telomerase reverse transcriptase | TERT | 14 | Telomerase activity | Cell survival |
| Mitogen-Activated Protein Kinase 1 and 2 | MAP2K1/2 | ~8 | MAPK signaling | Cell proliferation |
| G Protein Subunit Alpha Q and 11 | GNAQ/11 | Rare | MAPK signaling | Cell proliferation |
| Isocitrate Dehydrogenase 1 | IDH1 | ~5 | Metabolism of isocitrate | Cell proliferation and impaired differentiation |
| Erb-b2 Receptor Tyrosine Kinase 2/4 | ERBB2/4 | 1/19 | Tyrosine kinases signaling | Cell proliferation and survival |
| Kirsten Rat Sarcoma Viral Oncogene Homolog, GTPase | KRAS | ~2 | GTPase activity | Cell proliferation and survival |
| Splicing Factor 3b Subunit 1 | SF3B1 | 33 | Alternative splicing | Tumorigenesis |
| Somatic LOF/deleterious mutations | ||||
| Neurofibromin 1 | NF1 | 10-15 | MAPK/PI3K signaling | Cell proliferation, differentiation, and survival |
| Phosphatase and tensin homolog | PTEN | 14 | PI3K signaling | Apoptosis, cell survival and immune evasion |
| Tumor protein p53 | TP53 | 15 | Caspase3, FAS and CTL mediated apoptotic pathways | Cell-cycle progression, DNA repair and apoptosis |
| RAS P21 Protein Activator 2 | RASA2 | ~5 | RAS signaling | Cell proliferation and migration |
| Germline LOF/deleterious mutations | ||||
| Cyclin-dependent kinase inhibitor 2A | CDKN2A | 20-40 | RB pathway | Apoptosis and cell survival |
| Cyclin-dependent kinase 4 | CDK4 | NA | G1/S phase cell cycle checkpoint | Cell-cycle progression |
Table 3.
Statistical analysis based on the BRAF expression on nodal metastasis status.
| Comparison | Statistical Significance |
| N0 vs N1 | 7.717400E-01 |
| N0 vs N2 | 2.674800E-01 |
| N0 vs N3 | 7.441600E-02 |
| N1 vs N2 | 2.294600E-01 |
| N1 vs N3 | 1.707820E-01 |
| N2-vs-N3 | 2.955700E-02 |
Table 5.
Overall response rate and clinical benefit to immune therapy [64].
Table 5.
Overall response rate and clinical benefit to immune therapy [64].
| NRAS mutation | Non-NRAS mutations | |
| Anti-PD-1/PD-L1 (n=48) ORR CBR |
64% 73% |
30% 35% |
| Ipilimumab (n=169) ORR CBR |
19% 42% |
11% 19% |
| first-line immune therapy (Kaplan-Meier analysis)-median duration PFS OS |
4.1 months 19.5 months |
2.9 months 15.2 months |
Overall response rate (ORR), CBR; response rate plus stable disease for 24 weeks, overall survival (OS), progression-free survival (PFS), first-line immune therapy (IL2, Ipilimumab, and anti-PD-1/PD-L1).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



