
Submitted:
01 September 2023
Posted:
04 September 2023
You are already at the latest version
Abstract
Regulation at the level of translation plays an essential role in modulating gene expression. Given the importance of knowledge about translationally active mRNAs as an indicator of translation efficiency, it is crucial for researchers to have at their disposal organised sources of functional information on translation - plant translatomic maps similar to transcription maps. The development of a general experimental and computational methodology for constructing translatome maps is essential to address this challenge. In this manuscript, we discuss what researchers can do to design plant translatome maps, namely, we review: (i) experimental methods to obtain translationally active mRNA fractions, considering their advantages and limitations; (ii) additional methodological approaches to obtain complete pools of translationally active mRNAs; (iii) sequencing methods to qualitatively and quantitatively assess different mRNA fractions; and (iv) multivariate analyses of sequencing results by bioinformatics methods, including metrics for assessing mRNA translational activity.
Keywords:
efficiency of translation
; experimental approaches
; computational algorithms
; translatome maps
1. Introduction
The importance of studying the mechanisms of genetic information realization in eukaryotic cells, including plants, was realized by researchers at the earliest stages of research in this field. The first steps in the research of this problem were made through a deep study of the mechanisms of transcription control, as the first stage of gene expression and the easiest for experimental execution and study
Over the past two decades, key mechanisms of transcription regulation have been largely elucidated, and this has greatly expanded researchers' knowledge of the transcriptional control of gene(s) [1]. The results of transcriptional profiling (using high-throughput technologies - microarrays, RNA-Seq) allowed the construction of transcriptome maps - powerful organised sources of functional information on full-genome regulation of gene transcription for a large sample of plant tissues and/or organs, different developmental stages and/or environmental conditions (Figure 1). Many plants that have been the subjects of such transcriptome maps are model systems or economically important crops, and organs or tissues are studied at high morphological or temporal resolution. In plant science, applications of transcriptome maps range widely from inference about gene regulatory networks to evolutionary studies [1].
Note that transcription is only the beginning of the gene expression pathway. According to the current opinion, regulation at the level of translation plays an essential role in modulating gene expression (Figure 1). This became more evident when analysing the quantitative correlation between mRNA levels and protein abundance in different species of living organisms. Thus, comparative omic studies of eukaryotes, including plants, have provided compelling experimental evidence for an extremely modest correlation between levels of transcription (abundance of individual mRNAs) and translation (levels of relevant proteins in the proteome). In this case, the observed fluctuations in transcript level do not necessarily lead to the expected change in the level of the corresponding protein [2,3,4,5,6] (Figure 1). Across a wide range of species, more and more studies are incorporating translation data to better assess how gene regulation occurs at the level of protein synthesis. The inclusion of translation regulation data improves, and has been shown to be more accurate than transcription regulation studies alone [7] (Figure 1). Thus, it has already been convincingly demonstrated that the global association of mRNAs with polysomes changes dramatically both during growth and development and under the action of environmental factors. It should be noted that the genome-wide mechanisms of specific translation control during the development of plants, including the model plant Arabidopsis thaliana, have been studied fragmentarily, even though translation control mechanisms for many specific genes have been described. The question of tissue- and organ-specific control of translation remains open as well.
Modern technological advances have increased knowledge of the modulatory role of translation in gene expression, and have allowed the development and use of new experimental approaches to investigate the translational efficiency of mRNAs on a global scale, i.e., to study populations of translationally active mRNAs associated with ribosomes [5,8]. There is a view, based on current experimental data, that in some cases, assessment of ribosome-associated active mRNA populations can partially replace proteome quantification [4].
Given the importance of knowledge about translationally active mRNAs as an indicator of translation efficiency, it is crucial to have at the disposal of researchers organised sources of functional information on translation - plant translatomic maps similar to transcription maps. This would allow analyses of translatomic data, without extensive preprocessing of raw data available in public databases, and would provide ease of use. However, despite the large amount of experimental data illustrating the critical role of translational regulation in determining gene expression levels, only a small number of these data have been integrated into the only resource to date [9].
The lack of systematic and comprehensive studies on plant translatomics indicates significant gaps in the understanding of translation as an important regulatory step in the realisation of genetic information, and, in our opinion, this gap can be filled by developing a common experimental and computational methodology to the design and construction of translatomic maps.
We emphasise that the solution of this scientific problem requires an integrated approach in terms of methodology: obtaining plant material and mRNA fractions (resting and translationally active) corresponding to different physiological states of the plant; qualitative and quantitative assessment of different mRNA fractions through sequencing using modern technologies; and multifaceted analysis of sequencing results using bioinformatics methods: mapping, annotation of translatomes, quantitative analysis of translation profiles, assessment of differentiation profiles, and analysis of translation profiles.
It is quite justified, in our opinion, for the design of organised resources on translatomics (translatome maps) to partially rely on the experience of developing transcriptome maps, which have demonstrated their effectiveness and demand by the scientific community.
2. Experimental methodology for the creation of translatomic maps
In this section of the review, we will consider methods of separation and obtaining samples of actively translated mRNAs, additional experimental techniques for obtaining primary data, and sequencing methods. We will briefly describe the essence of each method, the potential of their combination, and discuss their applicability to translatomic projects (Figure 2).
2.1. Methods of separation and preparation of samples or fragments of actively translated mRNAs
According to the current view, the term "translatome" refers to the entire translated mRNA or RNC-mRNAs (RNC - ribosome nascent-chain complex) [4,6]. The main task of translatomics is to qualitatively and quantitatively assess such translatable mRNAs on a genome-wide scale. Thus, the primary experimental step is to divide the total mRNA pool into fractions with different translational status: translationally quiescent and actively translated mRNAs. Currently, most translatomics studies use several main methods: polysome profiling, full-length translating mRNA profiling (RNC), translational affinity purification of ribosomes (TRAP), ribosome footprinting (ribosome profiling) (Figure 2) [6,7].
2.1.1. Polysome profiling - to assess differential translation
The essence of the polysome profiling method is the separation of transcripts into mRNA fractions loaded with different numbers of ribosomes. For this method, a sucrose gradient with increasing concentration from top to bottom is used, on which a clarified cell extract of the tissue and/or whole plant of interest is layered [10,11]. The density of mRNA complexes with different numbers of ribosomes differs, which allows their separation during ultracentrifugation: molecules migrate within the gradient until they reach their equilibrium, with heavier molecules (polysomes) moving down the gradient to more concentrated layers than lighter complexes with single ribosomes (monosomes) or individual ribosomal subunits. And mRNAs not bound to ribosomes remain at the top of the gradient (Figure 2).
The polysome profiling method is well adapted to plant material, does not require any manipulation of growth or plant media, and provides a general picture of how ribosome load (and hence translation) varies in certain experimental samples or conditions. Nevertheless, it should be noted that polysome profiling is time and material intensive and requires obtaining and utilising more biological material compared to, for example, the method of obtaining total transcriptome samples.
2.1.2. Profiling of full-length translating mRNAs (RNC-seq) - to identify all actively translated mRNAs (overall quality composition of the translatome)
The essence of the method of obtaining all translating mRNAs is to separate the fraction of transcripts bound to ribosomes from free mRNA and other cellular components. For this method, a 30% sucrose cushion is used on which cell lysate is loaded and ultracentrifugation is performed. As a result, a pool of all translatable mRNAs (RNC-mRNA) can be collected, providing complete data on the qualitative composition of the translatome (Figure 2). It should be noted, however, that profiling of all translating mRNAs is somewhat easier to use compared to the polysome profiling method, but has its own technical complexity, which is related to the stability of mRNA-ribosome complexes. Namely, dissociation of ribosomes from mRNA and/or degradation of transcripts can occur, which can lead to systematic errors [12].
2.1.3. Translating Ribosome Affinity Purification (TRAP)
TRAP (translating ribosome affinity purification) – a method based on immunoprecipitation of labelled ribosomal protein and mRNA bound to the labelled ribosome. It allows the isolation of transcripts bound to at least one ribosome, i.e. those involved in the translation process. The first step of this method involves the creation of transgenic plants expressing a FLAG-tagged version of the ribosomal protein RPL18 in the tissue of interest. FLAG is a small epitope that does not affect ribosome function or polysome formation, but allows FLAG-tagged ribosomes to be isolated using antibodies. Biomaterial from transgenic plants is homogenised in buffer, the extract is purified by centrifugation and incubated with anti-FLAG agarose beads, then eluted and mRNA purified (Figure 2). The positive aspects of the TRAP method are the possibility of obtaining mRNA bound specifically to ribosomes but not to other RNA-binding proteins, which is difficult to do using other methods of separating translated mRNAs. In addition, the use of tissue-specific promoters that control the expression of labelled ribosomal protein allows the study of translation specific to cell type or developmental stage [6,10,13]. Nevertheless, this method has limitations: it requires the production of transgenic plants or the temporary expression of labelled ribosomal protein - this process requires time and skill, and the choice of the object may be limited to the range of organisms for which technologies have been developed to produce stable transformants. Overproduction of labelled ribosomal proteins, which can alter the structure and properties of ribosomes, should also be considered[12]
2.1.4. Ribosome profiling (Ribo-seq) - to search for regulatory sequences that determine the translation efficiency of individual transcripts
Ribosome footprinting (ribosome profiling) is a method based on enzymatic cleavage of mRNA not protected by ribosomes. It is known that ribosome covers a fragment of mRNA about 30 nucleotide residues long during translation. In this method, cell extract of plant tissue is treated with single-strand specific RNase, after which only mRNA fragments of 22-35 nucleotides, protected by ribosomes, remain in solution, samples of which can be obtained by separation by electrophoresis with subsequent separation and purification of fragments of a certain length from the gel (Figure 2). This method was originally developed for yeast [14], then adapted for several plant species [6,15]. The method provides information on the number and localisation of ribosomes on a particular mRNA, assuming that translated mRNAs are bound to ribosomes and that the number of ribosomes on an mRNA is an indicator of its translation efficiency. Nevertheless, this method has significant limitations, which are described in detail [see for an example review 6]. Some of them should be highlighted: (1) some RNA regions may form secondary structures or be protected by RNA-binding proteins rather than ribosomes and will be included for analysis; (2) it is impossible to distinguish the translation level of isoforms of a single gene or similar genes and a number of others. This should be taken into account when using primary data for subsequent qualitative and quantitative assessment of the translatome [7,16]. Additionally, ribosome profiling (compared to the other methods described above) requires a greater amount of starting material as well as material and time costs [6,17].
Thus, by applying the methods described above, samples of translated mRNAs can be obtained. It should be noted that both the number of mRNAs of each individual gene and the number of actively translated mRNAs can vary widely in the samples.
According to current data, mRNAs with a high level of representation (several thousand mRNA copies of an individual gene) can account for up to 20% of cellular mRNA (for 5-20 genes); mRNAs with a medium level of representation (several hundred copies of an individual gene) account for about 40-60% of cellular mRNA (for 500-2000 genes); the remaining 20-40% of mRNAs are represented by rare transcripts (from one to several tens of mRNA copies of an individual gene). Such huge differences in representation complicate the analysis of actively translated mRNAs at the whole genome level, as it leads to repeated sequencing of more represented transcripts
2.2. Additional Experimental Techniques for Obtaining Primary Data
To facilitate the identification of rare transcripts (mRNAs), researchers can use an additional methodological approach - cDNA normalisation using duplex-specific nuclease (DSN). This approach is highly efficient and is used to normalise a full-length enriched cDNA library [18]. The method is based on the kinetics of nucleic acid hybridisation [19] and unique properties of duplex-specific nuclease (SDS), strictly specific to double-stranded (ds) DNA [20]. The approach is based on the fact that after denaturation, ds cDNA flanked by known adaptors undergoes renaturation. During renaturation, frequently occurring transcripts are converted to the ds form more efficiently than those that occur less frequently. Thus, two fractions are formed, namely the frequently occurring ds- cDNA and the normalised single-stranded (ss) cDNA. The ds-cDNA fraction is then cleaved by DSN. The remaining normalised ss-DNA is amplified by PCR. In doing so, primers and reaction conditions are optimised to minimise the tendency for shorter fragments to amplify more efficiently than longer fragments. The normalised cDNA can then be used for sequencing. DSN-normalisation has been successfully applied to a variety of plant models [21]. Detailed protocols for DSN normalisation modifications are described in a number of publications [22,23]. Thus, cDNA normalisation reduces the prevalence of highly represented transcripts and equalises the number of unique transcripts in a cDNA sample, thereby dramatically increasing the probability of detecting rare transcripts. This approach is widely used for sequencing transcriptomes, but can also be useful for analysing actively translated mRNAs [24].
2.3. Sequencing methods
It should be noted that the methods of separation and obtaining samples or fragments of actively translated mRNA alone do not provide information on the translation efficiency of a specific transcript or the whole set of transcripts. For more detailed studies, it is necessary to combine these methods with others. Namely, with methods of quantifying the level of individual transcripts in the obtained samples using RNA sequencing, microarrays or quantitative PCR. This combination allows not only to see the differences in transcription and translation efficiency of a particular gene, but also to obtain general information about the differential translation of the transcripts of interest [7,25]. Currently, experimental data on expression profiles of samples at the whole genome level are usually obtained using two technologies: microarray or RNA-Seq. It should be emphasised that experimental data obtained by different technologies are similar to each other (Pearson correlation coefficient for different samples ranges from 0.70 to 0.83). Nevertheless, it has been convincingly proved that RNA-Seq allows detecting transcripts of a significantly larger number of genes (82.1% of all annotated genes versus 56.5% for microarrays), including tissue-specific genes, as well as better distinguishing the expression of paralogous genes [1]. Since all methods of separating and obtaining samples of actively translated mRNAs combine with sequencing methods, we next compare the two main sequencing platforms, and address the issues of additional experimental techniques in preparing samples for sequencing, and their applicability to constructing translatomic maps.
RNA-Seq is a widely used next-generation sequencing (NGS) methodology for transcriptome and translatome profiling, both for the identification of novel transcript sequences and for differential expression studies at the transcription and translation level [26].
The two main RNA-Seq technologies most popular with researchers are (1) short reads technology, and primarily the use of the Illumina platform, which allows accurate assessment of differential transcript representation in samples; (2) long reads technology (LRS) (such as Pacific Biosciences and Oxford Nanopore), which allows longer transcript sequences but with lower quality; nevertheless, LRS is highly sought after for the identification of spliced transcript isoforms.
Above we have briefly described the two most popular RNA-Seq technologies among researchers. Which sequencing platform is better for solving problems in translatomic mapping? For most users, next-generation sequencing is a choice between higher quality short reads, exemplified by the market-leading Illumina platform, or lower quality longer reads, exemplified by PacBio and Oxford Nanopore. Short-read sequencing approaches accurately quantify gene expression and work well in identifying alternative exon splicing, but often cannot identify which full-length alternative isoforms are expressed. Consequently, although short reads accurately quantify gene expression, they often cannot identify the correct isoform from which a read is derived because isoforms of the same gene are largely similar. Long reads technologies have a distinct advantage over short reads because they can reliably generate reads that span the entire isoform. This removes the difficult task of reconstructing possible transcript isoforms from fragmented read sequences and may improve our understanding of alternatively spliced isoforms of complex genes [27]. The growing popularity of third-generation sequencing methods that provide long RNA reads is due in part to their potential to rapidly and affordably combine the advantages of previous sequencing methods and full-length isoform profiling (Oxford Nanopore Technologies, 2021) [28].
Thus, polysome profiling combined with methods for quantifying the level of individual transcripts makes it possible to study the translational state of an entire plant or a specific tissue, i.e., to draw conclusions about translation efficiency at the global level or a specific mRNA of interest [25].
Like the polysome profiling method, the full-length translating mRNA profiling technique does not provide information on the pool of translated mRNAs, but complemented by sequencing of full-length transcripts of the obtained samples allows studying the complete sequences of all translated mRNAs. Based on the results of fractionation combined with sequencing (RNC-seq), it is possible to elucidate the specificity of transcript splicing variations in plant cells at certain stages of plant development and/or different organs. Additionally, information on the translational status of specific transcripts can also be obtained and new proteins, such as those encoded by "non-coding RNAs", can be searched for, thus expanding and/or updating the list of annotated transcripts (by directly proving their translational status), and indirectly identifying possible variations in their protein products. In this way, RNC-seq provides an accurate database of proteins that may exist in a sample. One example of the use of RNC-seq is the discovery of 1397 genes previously annotated as non-coding RNAs (ncRNAs) in the RefSeq database, some of which were later confirmed at the protein level [12].
Like the above methods used to analyse actively translated mRNAs, the TRAP method is complemented by sequencing. As a result, an insight into the translational state of a specific plant tissue can be obtained. TRAP was used to compare translatomes in response to various environmental factors and during photomorphogenesis and pollen growth.
The ribosome profiling method (Ribo-seq) identifies the location of the ribosome with codon accuracy, which can provide information about upstream open reading frames (uORFs) and non-canonical start codons.
It should be noted that combining several methods to separate and obtain samples or fragments of actively translated mRNAs and different sequencing technologies to study translation may allow overcoming the “pitfalls” of individual methods [7].
2.4. Theoretical and computational methodologies for the design and construction of translatomic maps
The volume and quality of raw sequencing data are the most important parameters affecting the success of their use for high-resolution translatomic mapping. Let us briefly review the most important of them - read length, sequencing depth, optimal RNA-Seq data volume, availability of a reference genome/transcriptome and its quality.
The length of reads is an important parameter affecting the success of mapping and, consequently, the amount of data obtained. Longer read lengths provide better mapping, which is not so important for species with well-assembled and annotated simple genomes, but is necessary for polyploids and other variants of complex genomes. In most current publications, read lengths range from 100 bp to a maximum of 150 bp [1]. The structure of the reads should also be mentioned: paired reads increase the resolution of mapping, so they are used in most of the works. It should be noted that single reads have also been successfully used for objects with good genome or transcriptome assembly [1]. This experimental design reduces the cost of sequencing compared to paired-end reads, allowing more samples to be analysed and/or sequencing depth to be increased.
Sequencing depth is another important parameter of RNA-Seq because of the trade-off between the cost of the experiment and the completeness of the resulting data. Some researchers believe that increasing sequencing depth allows for higher resolution analysis and, consequently, more reliable conclusions about gene expression at the genome level [1]. The current view is that greater sequencing depth is required to extract less abundant transcripts [26].
In addition to sequencing depth, the availability of a reference genome and its quality have a great influence on the results of RNA-Seq data analysis, in particular, for the construction of transcriptome maps. At the same time, however, read mapping on the transcriptome is now widely used for both model organisms and non-model objects with large genomes. For example, among the available transcriptome maps, nine have been mapped to the reference transcriptome specifically [1].
Potentially, when accumulating translatomic data, it is possible to generate plant translatomes using RNC-seq data, with accurate determination of isoforms, and use them for mapping and subsequent assessment of differential translation.
In our opinion, the analysis of differentially translated genes should be performed using RNA-Seq data from three samples: total RNA fraction, monosomal and polysomal fractions.
Two objectives should be addressed: (i) to map the reads obtained to a reference genome or transcript in order to calculate the number of transcripts of each gene in the samples (transcript abundance), and (ii) to identify differentially translated transcripts (i.e. DEG mRNAs at the translation level).
2.4.1. Mapping
To address the first task (mapping), a number of software tools have been developed using two approaches: the first is to construct an index based on the Burrows-Wheeler Transform (BWT), and allows mapping to the reference genome. This allows reads to be mapped when they do not fully match the reference, and also takes alternative splicing into account. Among the software tools that implement this approach, STAR is worth mentioning STAR [29], BWA [30] и Bowtie 2 [31].
Another recently developed approach is based on the use of k-measures: in this case, the reference transcriptome is represented as a de Bruijn graph, and reads are partitioned into fragments (k-measures), which are mapped according to the principle of exact correspondence. This algorithm is the basis for the programmes kallisto [32] and Salmon [33].
The use of indices in BWT allows you to determine the position of a read sequence coordinate in the genome and then compare it to a reference. The advantage of this method is that differences are allowed between the reference and the read, the extent of which can be specified when running the programme. However, due to the fact that the comparison of the read with the reference is done character by character, it takes a certain amount of time.
In the case of k-mers, the index is a set of short, about 30 bp, sequences. Each read is also partitioned into short sequences whose size corresponds to the size of the index, then the corresponding sequence in the index is searched. Due to the use of hash-table this process is much faster than sequence mapping in BWT, a read can only be mapped if the sequence k-means is the same in both cases. However, given that the reads are 100 or more nucleotides long, the probability of finding the corresponding k-mer is quite high.
Despite significant differences in approach, the above programmes show similar mapping efficiency results [34]. At the same time, the use of k-measures allows mapping reads an order of magnitude faster than in the case of BWT. In addition, it is worth noting that kallisto output can provide both information about the position of reads on the transcript in SAM format and the number of transcripts (transcript abundance), on the basis of which a search for differentially translated genes can be carried out subsequently.
2.4.2. Search and evaluation of differentially translated mRNAs
In pioneering work assessing translation efficiency, such as the study of regulation under the influence of mTORC [36] translation efficiency was evaluated using RPKM (reads per kilobase per million) [37] with certain modifications. However, the correctness of the results when using fold change can be affected by the experimental conditions and sequencing protocol, which leads to the need for additional statistical processing of the results [38,39].
Traditional means of searching for differentially expressed genes can be applied to solve this problem: EdgeR [40], DeSeq2 [41], limma [42,43].
In addition, software packages developed specifically to analyse the translation efficiency of ribosome profiling data are worth mentioning: RiboDiff [44] and Babel [45].
In the process of studying translation mechanisms, regulatory elements (non-translated regions, codon composition) are of particular interest, which, in turn, can be affected by alternative splicing. In this regard, the researcher may face the task of analysing the efficiency of translation not only at the gene level, but also at the level of isoforms. The sleuth programme can be recommended for this task [46], developed by the creators of the aforementioned programme kallisto.
The resulting analysis using the above resources - obtaining differential transcription or translation values for each gene and/or transcript (Log2FoldChange), accompanied by statistical significance values for the data (р-Value).
As mentioned above, we believe it is better to analyse differentially translated transcripts using RNA-Seq data from three samples: total mRNA fraction, mono- and polysomal fractions (Figure 3).
Below we will explain the basis of our considerations. The translation process is a dynamic process, and to correctly quantify the translation process for each transcript it is necessary to apply quantitative indicators. Such indicators can be calculated by normalisation, i.e. by bringing the data to a single form that will allow comparing them among themselves or using them to calculate the similarity of objects, as well as to reduce the potential inconsistency of information obtained in different scientific teams. Two indicators can be proposed as such (Figure 3):
(i) Translation Intensity (TI), i.e. a characteristic reflecting the potential of transcript (mRNA) involvement in the translation process, which can be calculated as a ratio of the level of translated mRNA (total level in poly- and monosomal fractions) to that for total mRNA in each transcript (e.g., in Log2FoldChange values). Thus, translation intensity will reflect the ability of a transcript (mRNA) to translate as a whole - the higher the value of this ratio, the more transcripts of an individual gene are involved in translation.
(ii) Translation efficiency (TE), i.e., a characteristic that reflects the potential of transcript performance in the translation process, which can be estimated by the ratio of transcript (mRNA) levels in the poly- and monosomal fractions of each transcript (e.g., in Log2FoldChange values). Thus, translation efficiency will reflect how successful translation of an individual transcript can be - the higher the value of such a ratio, the greater the probability of formation of more protein product of an individual gene transcript.
Thus, the TI and TE metrics may provide a basis for the development of a common computational methodology for the design and construction of translatomic maps in the future, and the use of these metrics will allow the correct comparison of data from different research teams and their integration into a common translatomics data pool (Figure 3).
2.4.3. Translatomic maps
First of all, we note that databases, including transcriptome maps, can be divided into two categories with different sets of tools: first, those designed for easy visualisation, and second, those that allow for additional analysis [1] (Figure 3). The choice of option for the design of translatomic maps is left to the researcher. Nevertheless, the option of translatomic maps with the possibility of additional data analysis seems to be the most effective, and will allow, in our opinion, to find out what precise molecular mechanisms underlie the formation of unique morphological and physiological properties of certain tissues and organs during ontogenesis, under the influence of environmental factors. In this case, a set of tools can be presented to the researcher, allowing, for example, to search for stably translated mRNAs; to determine tissue specificity of translation, as well as specificity of translation under the action of environmental stress factors on the plant; functional analysis of potential cis-regulatory elements important for the translation efficiency of the corresponding transcripts, evolutionary studies, etc.
3. Conclusions
Translation of mRNA into a protein product is an exquisitely regulated, and highly complex, process. The current view based on experimental evidence is that a systematic study of translation control in plants during growth, development, and stress events can clarify many fundamental questions and is needed to clarify the complex mechanisms of translation. The role of established translation rules as a cornerstone in biology for understanding plant gene expression is obvious. But at the same time, experimental evidence and theoretical predictions indicative of the complex, multi-level information encoded in the mRNA sequence are increasing at an accelerating pace. Given the key role of translation in the overall mechanism of genetic information realisation, and the fact that plants can exploit higher-order rules of mRNA regulation and decoding, it can be concluded that new knowledge of how critical each regulatory context in mRNA, as well as combinations of these contexts, are for translation efficiency is seen as crucial. In addition, compilation of lists of translated mRNAs (tissue-, organ-, stress-specific) has an important practical purpose: identification of target genes for genetic manipulations to increase economic productivity. All this taken together can bring the researcher closer to an important achievement - all stages of regulation of gene expression, including those introduced from outside, under his strict control, and thus will allow expanding the applied potential of translation mechanisms: laying the foundation for a new generation of transgenes obtained using genome editing technology, and, as a consequence, bringing great benefit to humanity
Author Contributions
A.A.T., O.N.M., I.V.D. and V.A.F. carried out the literature review and preparation of material for the review; A.A.T. and V.A.F. did preparation of original figures; and I.V.G.-P. wrote the manuscript. All authors were fully involved in preparing and revising the manuscript critically at its current state. All authors have read and agreed to the published version of the manuscript.
Funding
The authors would like to thank the Russian Science Foundation (RSF, funding number 22-14-00057) and the Ministry of Science and Higher Education of the Russian Federation (theme No. 122042700043-9).
Conflicts of Interest
The authors declare no conflict of interest”.
References
- Klepikova, A.V.; Penin, A.A. Gene Expression Maps in Plants: Current State and Prospects. Plants 2019, 8, 309. [Google Scholar] [CrossRef]
- Baerenfaller, K.; Grossmann, J.; Grobei, M.A.; Hull, R.; Hirsch-Hoffmann, M.; Yalovsky, S.; Zimmermann, P.; Grossniklaus, U.; Gruissem, W.; Baginsky, S. Genome-Scale Proteomics Reveals Arabidopsis Thaliana Gene Models and Proteome Dynamics. Science (1979) 2008, 320, 938–941. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on MRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef]
- Zhao, J.; Qin, B.; Nikolay, R.; Spahn, C.M.T.; Zhang, G. Translatomics: The Global View of Translation. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef]
- Urquidi Camacho, R.A.; Lokdarshi, A.; von Arnim, A.G. Translational Gene Regulation in Plants: A Green New Deal. Wiley Interdiscip Rev RNA 2020, 11, e1597. [Google Scholar] [CrossRef] [PubMed]
- Goldenkova-Pavlova, I.; Pavlenko, O.; Mustafaev, O.; Deyneko, I.; Kabardaeva, K.; Tyurin, A. Computational and Experimental Tools to Monitor the Changes in Translation Efficiency of Plant MRNA on a Genome-Wide Scale: Advantages, Limitations, and Solutions. Int J Mol Sci 2018, 20, 33. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni-Putman, S.M.; Stepanova, A.N. A Plant Biologist’s Toolbox to Study Translation. Front Plant Sci 2018, 9. [Google Scholar] [CrossRef]
- Sharma, V.; Salwan, R.; Sharma, P.N.; Gulati, A. Integrated Translatome and Proteome: Approach for Accurate Portraying of Widespread Multifunctional Aspects of Trichoderma. Front Microbiol 2017, 8, 275453. [Google Scholar] [CrossRef]
- Liu, W.; Xiang, L.; Zheng, T.; Jin, J.; Zhang, G. TranslatomeDB: A Comprehensive Database and Cloud-Based Analysis Platform for Translatome Sequencing Data. Nucleic Acids Res 2018, 46, D206–D212. [Google Scholar] [CrossRef]
- Mustroph, A.; Juntawong, P.; Bailey-Serres, J. Isolation of Plant Polysomal MRNA by Differential Centrifugation and Ribosome Immunopurification Methods. Methods Mol Biol 2009, 553, 109–126. [Google Scholar] [CrossRef] [PubMed]
- Lecampion, C.; Floris, M.; Fantino, J.R.; Robaglia, C.; Laloi, C. An Easy Method for Plant Polysome Profiling. Journal of Visualized Experiments 2016, 2016, 1–7. [Google Scholar] [CrossRef]
- Zhao, J.; Qin, B.; Nikolay, R.; Spahn, C.M.T.; Zhang, G. Translatomics: The Global View of Translation. International Journal of Molecular Sciences 2019, Vol. 20, Page 212 2019, 20, 212. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Meyerowitz, E.M. Cell-Type Specific Analysis of Translating RNAs in Developing Flowers Reveals New Levels of Control. Mol Syst Biol 2010, 6, 419. [Google Scholar] [CrossRef] [PubMed]
- Ingolia, N.T.; Ghaemmaghami, S.; Newman, J.R.S.; Weissman, J.S. Genome-Wide Analysis in Vivo of Translation with Nucleotide Resolution Using Ribosome Profiling. Science 2009, 324, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Chotewutmontri, P.; Stiffler, N.; Watkins, K.P.; Barkan, A. Ribosome Profiling in Maize. Methods Mol Biol 2018, 1676, 165–183. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.Y.; Calviello, L.; Wu, H.Y.L.; Li, F.W.; Rothfels, C.J.; Ohler, U.; Benfey, P.N. Super-Resolution Ribosome Profiling Reveals Unannotated Translation Events in Arabidopsis. Proc Natl Acad Sci USA 2016, 113, E7126–E7135. [Google Scholar] [CrossRef]
- Kabardaeva, K.V; Tyurin, A.A.; Pavlenko, O.S.; Gra, O.A.; Deyneko, I.V; Kouchoro, F.; Mustafaev, O.N.; Goldenkova-Pavlova, I. V Fine Tuning of Translation: A Complex Web of Mechanisms and Its Relevance to Plant Functional Genomics and Biotechnology. Russian Journal of Plant Physiology 2019, 66, 835–849. [Google Scholar] [CrossRef]
- Zhulidov, P.A.; Bogdanova, E.A.; Shcheglov, A.S.; Vagner, L.L.; Khaspekov, G.L.; Kozhemyako, V.B.; Matz, M. V.; Meleshkevitch, E.; Moroz, L.L.; Lukyanov, S.A.; et al. Simple CDNA Normalization Using Kamchatka Crab Duplex-Specific Nuclease. Nucleic Acids Res 2004, 32. [Google Scholar] [CrossRef]
- Young, B.D.; Anderson, M.L.M. Quantitative Analysis of Solution Hybridization. Nucleic Acid Hybridization: A Practical Approach. IRL Press, Washington DC 1985, 47–71.
- Shagin, D.A.; Rebrikov, D.V.; Kozhemyako, V.B.; Altshuler, I.M.; Shcheglov, A.S.; Zhulidov, P.A.; Bogdanova, E.A.; Staroverov, D.B.; Rasskazov, V.A.; Lukyanov, S. A Novel Method for SNP Detection Using a New Duplex-Specific Nuclease from Crab Hepatopancreas. Genome Res 2002, 12, 1935–1942. [Google Scholar] [CrossRef]
- Bogdanova, E.A.; Shagin, D.A.; Lukyanov, S.A. Normalization of Full-Length Enriched CDNA. Mol Biosyst 2008, 4, 205–212. [Google Scholar] [CrossRef]
- Bogdanova, E.A.; Shagina, I.; Barsova, E.V.; Kelmanson, I.; Shagin, D.A.; Lukyanov, S.A. Normalizing CDNA Libraries. Curr Protoc Mol Biol 2010, 90. [Google Scholar] [CrossRef]
- Shcheglov, A.S.; Zhulidov, P.A.; Bogdanova, E.A.; Shagin, D.A. Normalization of CDNA Libraries. Nucleic Acids Hybridization: Modern Applications 2007, 97–124. [Google Scholar] [CrossRef]
- Hoang, N.V.; Furtado, A.; Perlo, V.; Botha, F.C.; Henry, R.J. The Impact of CDNA Normalization on Long-Read Sequencing of a Complex Transcriptome. Front Genet 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Merchante, C.; Stepanova, A.N.; Alonso, J.M. Translation Regulation in Plants: An Interesting Past, an Exciting Present and a Promising Future. The Plant Journal 2017, 90, 628–653. [Google Scholar] [CrossRef]
- Patterson, J.; Carpenter, E.J.; Zhu, Z.; An, D.; Liang, X.; Geng, C.; Drmanac, R.; Wong, G.K.-S. Impact of Sequencing Depth and Technology on de Novo RNA-Seq Assembly. BMC Genomics 2019, 20, 604. [Google Scholar] [CrossRef] [PubMed]
- De Paoli-Iseppi, R.; Gleeson, J.; Clark, M.B. Isoform Age - Splice Isoform Profiling Using Long-Read Technologies. Front Mol Biosci 2021, 8. [Google Scholar] [CrossRef]
- Chen, Y.; Davidson, N.M.; Wan, Y.K.; Patel, H.; Yao, F.; Low, H.M.; Hendra, C.; Watten, L.; Sim, A.; Sawyer, C.; et al. A Systematic Benchmark of Nanopore Long Read RNA Sequencing for Transcript Level Analysis in Human Cell Lines. bioRxiv 2021. 1, 2021.04.21.440736. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-Optimal Probabilistic RNA-Seq Quantification. Nat Biotechnol 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nature Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Costa-Silva, J.; Domingues, D.; Lopes, F.M. RNA-Seq Differential Expression Analysis: An Extended Review and a Software Tool. PLoS One 2017, 12, e0190152. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Thoreen, C.C.; Chantranupong, L.; Keys, H.R.; Wang, T.; Gray, N.S.; Sabatini, D.M. A Unifying Model for MTORC1-Mediated Regulation of MRNA Translation. Nature 2012, 485, 109. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and Quantifying Mammalian Transcriptomes by RNA-Seq. Nature Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, L.M.; Lopiano, K.K.; Morse, A.M.; Amin, V.; Oberg, A.L.; Young, L.J.; Nuzhdin, S.V. RNA-Seq: Technical Variability and Sampling. BMC Genomics 2011, 12, 1–13. [Google Scholar] [CrossRef]
- Hansen, K.D.; Brenner, S.E.; Dudoit, S. Biases in Illumina Transcriptome Sequencing Caused by Random Hexamer Priming. Nucleic Acids Res 2010, 38, e131–e131. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol 2014, 15, 1–21. [Google Scholar] [CrossRef]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. Voom: Precision Weights Unlock Linear Model Analysis Tools for RNA-Seq Read Counts. Genome Biol 2014, 15, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Limma: Linear Models for Microarray Data. Bioinformatics and Computational Biology Solutions Using R and Bioconductor 2005, 397–420. [Google Scholar] [CrossRef]
- Zhong, Y.; Karaletsos, T.; Drewe, P.; Sreedharan, V.T.; Kuo, D.; Singh, K.; Wendel, H.-G.; Rätsch, G. RiboDiff: Detecting Changes of MRNA Translation Efficiency from Ribosome Footprints. Bioinformatics 2017, 33, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Olshen, A.B.; Hsieh, A.C.; Stumpf, C.R.; Olshen, R.A.; Ruggero, D.; Taylor, B.S. Assessing Gene-Level Translational Control from Ribosome Profiling. Bioinformatics 2013, 29, 2995–3002. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, H.; Bray, N.L.; Puente, S.; Melsted, P.; Pachter, L. Differential Analysis of RNA-Seq Incorporating Quantification Uncertainty. Nature Methods 2017, 14, 687–690. [Google Scholar] [CrossRef]
Figure 1.
The pathway of genetic information realisation and research methods for key biological molecules. 1. Transcription results in the formation of mRNA (transcriptome), which is quantitatively analysed by RNA-seq (RNA seq) or microarray (Chip seq) methods. The mRNA levels of individual genes vary widely (mRNAs of different genes are indicated in different colors). 2. Translation results in the formation of protein products, which are quantified mainly by mass spectrometry (MSM) methods (different color indicates proteins synthesised from different mRNAs. The same color of mRNA and protein products correspond to the same gene). An extremely weak quantitative correlation between mRNA level and protein abundance was experimentally confirmed. The main experimental approaches for the study of mRNA translational efficiency are outlined: polysome profiling (PP); profiling of full-length translating mRNAs (RNC); ribosome profiling (RP).
Figure 1.
The pathway of genetic information realisation and research methods for key biological molecules. 1. Transcription results in the formation of mRNA (transcriptome), which is quantitatively analysed by RNA-seq (RNA seq) or microarray (Chip seq) methods. The mRNA levels of individual genes vary widely (mRNAs of different genes are indicated in different colors). 2. Translation results in the formation of protein products, which are quantified mainly by mass spectrometry (MSM) methods (different color indicates proteins synthesised from different mRNAs. The same color of mRNA and protein products correspond to the same gene). An extremely weak quantitative correlation between mRNA level and protein abundance was experimentally confirmed. The main experimental approaches for the study of mRNA translational efficiency are outlined: polysome profiling (PP); profiling of full-length translating mRNAs (RNC); ribosome profiling (RP).
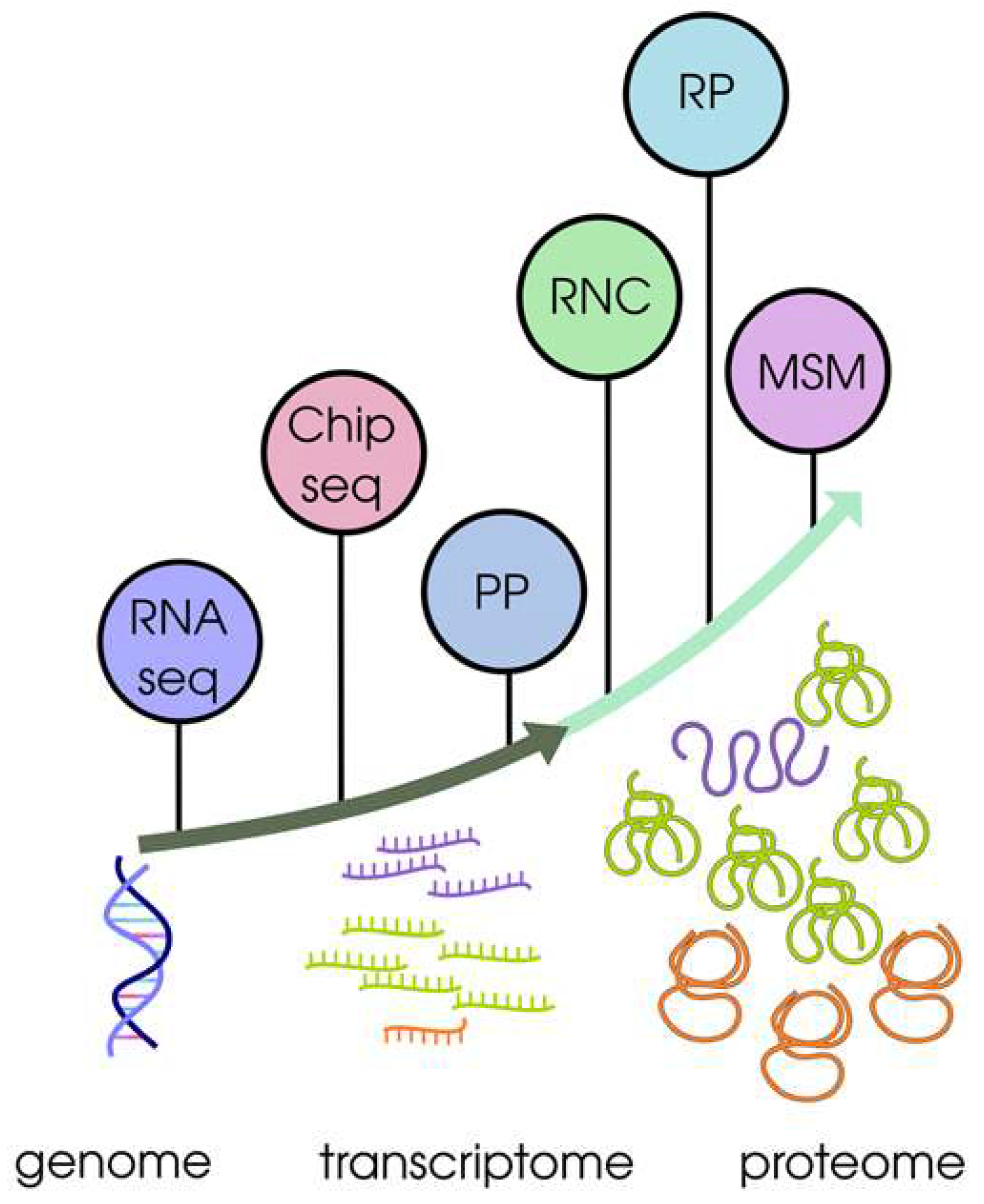
Figure 2.
Methods for separating and obtaining mRNA samples with different translational activities. From top to bottom: Polysome profiling, involving separation of mRNAs with different ribosome loadings in sucrose gradient, isolation and purification of mRNAs from different fractions (mono- and polysomal) and preparation of mRNAs for sequencing; ribosome profiling, involving obtaining ribosome-protected mRNA fragments after RNase treatment, purification and subsequent sequencing of mRNA fragments; full-length translating mRNA profiling (RNC), which includes obtaining all translating mRNAs on a sucrose cushion, isolating and purifying mRNA and preparing mRNA for sequencing; affinity purification of translating ribosomes (TRAP-seq), which includes incubating cell lysates with anti-FLAG agarose beads, isolating and purifying mRNA, and preparing mRNA for sequencing.
Figure 2.
Methods for separating and obtaining mRNA samples with different translational activities. From top to bottom: Polysome profiling, involving separation of mRNAs with different ribosome loadings in sucrose gradient, isolation and purification of mRNAs from different fractions (mono- and polysomal) and preparation of mRNAs for sequencing; ribosome profiling, involving obtaining ribosome-protected mRNA fragments after RNase treatment, purification and subsequent sequencing of mRNA fragments; full-length translating mRNA profiling (RNC), which includes obtaining all translating mRNAs on a sucrose cushion, isolating and purifying mRNA and preparing mRNA for sequencing; affinity purification of translating ribosomes (TRAP-seq), which includes incubating cell lysates with anti-FLAG agarose beads, isolating and purifying mRNA, and preparing mRNA for sequencing.
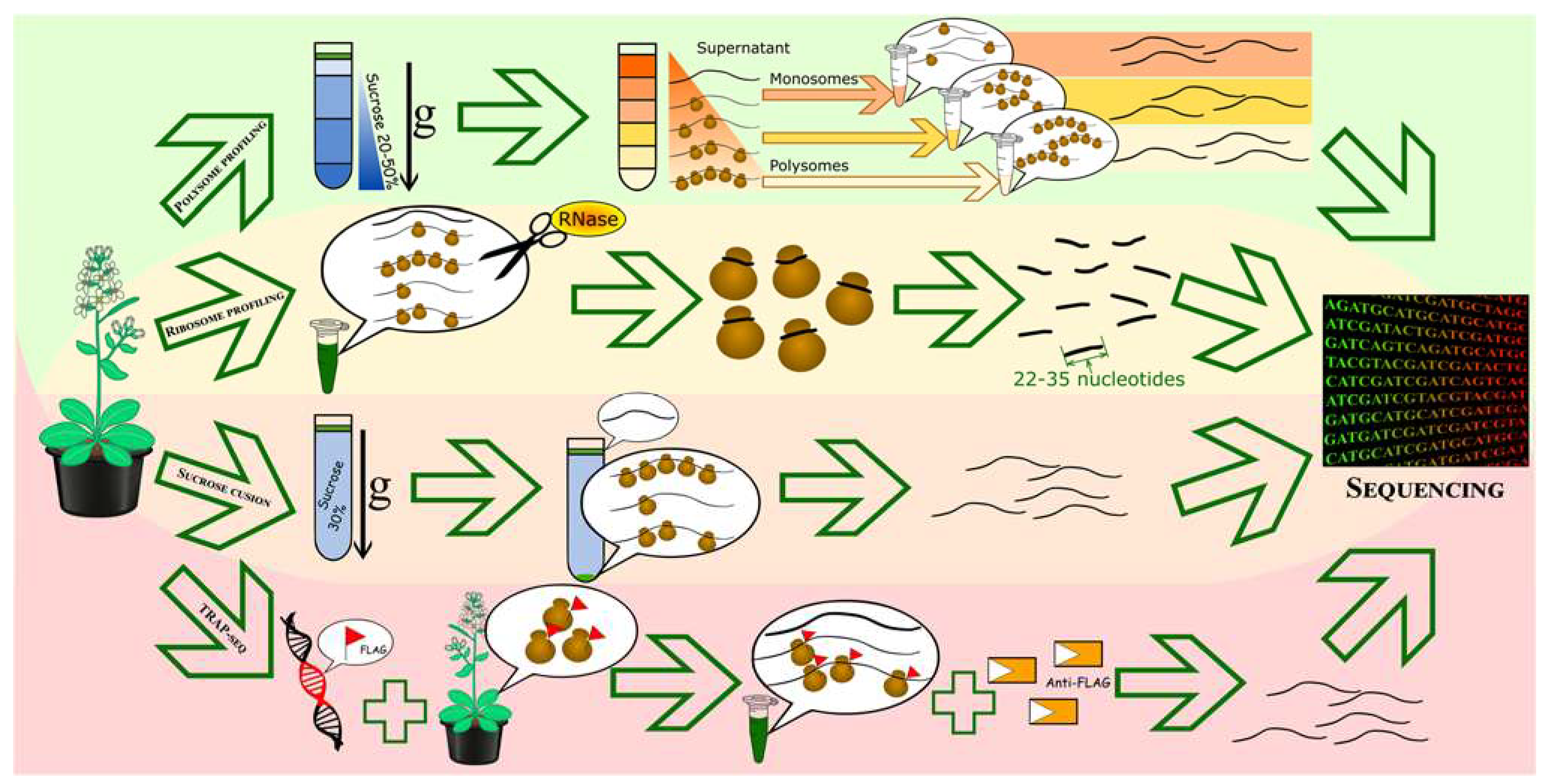
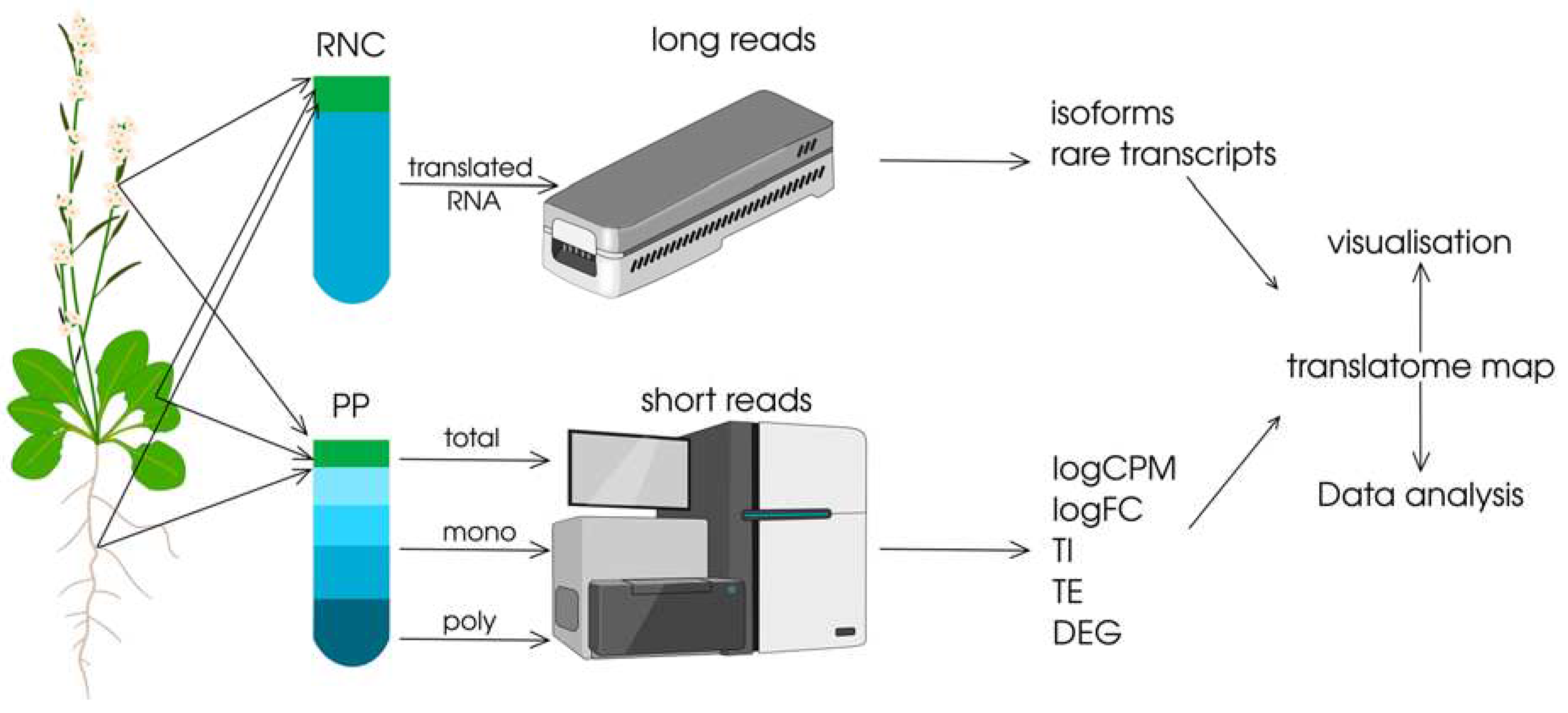
Figure 3.
Strategy of experimental and computational approaches towards the construction of translatomic maps of plants. 1. Collection of biological material for extensive sampling of plant tissues and/or organs, different developmental stages and/or environmental conditions; 2. Obtaining pool of all translatable mRNAs (RNC), pools of mRNAs with different translational activities (mono and poly) and total mRNA (PP); 3. Sequencing of RNC pools using long reads technology to obtain the qualitative composition of the translatome, including isoforms and rare transcripts, and sequencing of mRNA pools with different translational activity (mono and poly) and total mRNA (PP) using short reads technology to assess differential expression at the level of transcription and translation; 4. Use of computational methods to correctly quantify translation efficiency for each transcript (logCPM, logFC, TI, TE and DEG are quantitative metrics that can be used to assess differential translation of mRNAs, see description in text); 5. Design of translatome maps either with convenient visualisation or those that allow for additional analyses.
Figure 3.
Strategy of experimental and computational approaches towards the construction of translatomic maps of plants. 1. Collection of biological material for extensive sampling of plant tissues and/or organs, different developmental stages and/or environmental conditions; 2. Obtaining pool of all translatable mRNAs (RNC), pools of mRNAs with different translational activities (mono and poly) and total mRNA (PP); 3. Sequencing of RNC pools using long reads technology to obtain the qualitative composition of the translatome, including isoforms and rare transcripts, and sequencing of mRNA pools with different translational activity (mono and poly) and total mRNA (PP) using short reads technology to assess differential expression at the level of transcription and translation; 4. Use of computational methods to correctly quantify translation efficiency for each transcript (logCPM, logFC, TI, TE and DEG are quantitative metrics that can be used to assess differential translation of mRNAs, see description in text); 5. Design of translatome maps either with convenient visualisation or those that allow for additional analyses.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



