Submitted:
01 September 2023
Posted:
05 September 2023
You are already at the latest version
Abstract
The idealized rhombohedral unit cell of boron carbide is formed by a 12-atom icosahedron and a 3-atom linear chain. Phase transitions are second order and caused by the exchange of B and C sites or by vacancies in the structure. Nevertheless, the impact of such minimal structural changes on the properties can be significant. As the X-ray scattering cross sections of B and C atoms are very similar, the capability of X-ray fine structure investigation is restricted. Phonon spectroscopy helps closing this gap. Phase transitions known to date have been identified due to significant changes of properties: (1) The phase transition near the chemical composition B8C by clear change of the electronic structure; (2) The endothermic temperature-dependent phase transition at 712 K by the according change of specific heat; (3) The high-pressure phase transition at 33.2 GPa by the drastic change of optical appearance from opacity to transparency. These phase transitions affect IR- and Raman-active phonons and other solid-state properties. The phase transitions at B~8C and 712 K mean that a well-defined distorted structure is converted into another one each. In the high-pressure phase transition, an apparently well-defined distorted structure changes into a highly ordered one. In all these cases, the distribution of polar C atoms in the icosahedra plays a crucial role.
Keywords:
boron carbide
; phase transition
; structural disorder
; electronic properties
; phonons
1. Introduction
Boron carbide is one of the technically most important boron-rich solids. Outstanding properties like low density (2.52 g/cm³), high melting point (Tm ~ 2700 K) and extreme hardness (~40000 MPa) predestine boron carbide for application under conditions which are inaccessible for most other materials. Therefore, it is one of the best-investigated boron-rich solids, whose structures are based on B12 or related icosahedra. Complexity of structure and extraordinary electronic properties related have attracted attention. Regarding technical application, boron carbide is the most important boron-rich material at all. Since the sixties of last century hardness and chemical stability have been utilized in powders for grinding and polishing, in nozzles for sand blasting and in mortars (see A. Lipp [1]). Meanwhile, high-temperature ceramics and lightweight armoring of objects and persons have become important. High neutron absorption of the 10B isotope allows application for neutron shielding and control rods in nuclear devices. Domnich et al. [2] reviewed such application. In general, due to its extraordinary properties, boron carbide is highly promising for application under extreme conditions, like high temperatures, mechanical or chemical burdens. Mostly in such applications, details of the complex crystalline structure of boron carbide like the distribution of C atoms are of subordinate significance.
This does not apply in the case of the electronic properties. Optical appearance and electronic transport properties characterize boron carbide clearly as a p-type semiconductor (see [3,4]). However, untypical behavior indicates strong impact of structural defects. The high Seebeck coefficient (S ~ 300 mVK-1 up to at least 2000 K) is a promising potential for direct thermoelectric energy conversion (see [5]).
At least concerning details, the crystalline structure of boron carbide has not yet been finally determined. The formerly frequently and even currently occasionally assumed representative compound B4C (structure formula (B11C) CBC) does not exist in reality. B4.3C is the real compound achieved in commercially produced boron carbide [6,7] and in single crystals as well [8]. If B4C is obtained, excess carbon is precipitated in the structure, typically in the form of graphitic layers [9,10]. Unlike B4.3C at the carbon-rich limit, the boron-rich limit of the homogeneity range is less well defined. Few samples, whose C content was clearly below B11C, were unambiguously characterized as boron carbide, albeit with large error margin of lattice parameters (see [14] and references cited therein). Otherwise β-rhombohedral boron with clearly higher C content has been obtained (see [11] and references cited therein). Therefore, coexistence of both structures in a certain range cannot be excluded. Theoretical studies claim the structure model (B12) CBC to be the energetically most favorable, while various experiments have proved this compound to be the most distorted one in the whole homogeneity range (see ref. [3]).
Phase transitions, structural changes related and their effect on some other solid-state properties are object of this paper.
2. Structure
Figure 1 [12] shows the rhombohedral unit cells of α-rhombohedral (α-rh. B; idealized structure formula B12) boron and boron carbide (idealized structure formula (B12) CBC and (B11C) CBC) respectively. The actual structures differ more or less decisively. In α-rh. Boron, IR phonon spectra suggest the existence of additional single B atoms [13]. In boron carbide, whose homogeneity range extends from B4.3C at the carbon-rich to about B11C at the boron-rich limit, high-density structural defects have been identified and impact the electronic properties decisively [14,15].
The defects in boron carbide are mainly formed by the exchange of B and C atoms on regular sites or of minor concentrations of unoccupied sites. X-ray diffraction averaging larger volumes yields the basic parameters of boron carbide reliably, but it is largely unsuitable to analyze individual compositions of the components. Due to the similar X-ray scattering cross sections of 10B, 11B and 12C isotopes, the capability of X-ray fine structure investigation is significantly restricted. Phonon spectroscopy helps closing this gap.
Helpful in this respect is the comparison of the spectra of IR- and Raman-active phonons with those of α-rhombohedral boron having the same icosahedral structure, however without C atoms on polar sites of the icosahedron and without CBC or these replacing other structure elements. Figure 2 compares the IR-phonon spectra of single crystal natB4.3C boron carbide and α-rhombohedral boron (for according comparison of Raman spectra see Refs. [16,17,18]). The spectrum of α-rhombohedral B was discussed in ref. [19]. A detailed theoretical description was performed by Beckel et al. [20]. The spectra of boron carbide were analyzed by Werheit et al. (see Refs. [13,21] and references cited therein). The concentration of structure elements depending on the C content is shown in Figure 3. Changes of some icosahedral vibrations in boron carbide in connection with phase transitions are discussed below.
The existence of following defects in the structure in the form of deviations from the idealized structure formulas are generally confirmed:
C atom replacing B(1) in (B11C) CBC; that means one of the six B atoms on polar sites of the icosahedra is replaced by a C atom. The distribution of these C atoms is object of current investigation).
Carbon-rich limit of the homogeneity range: B4.3C (instead of B4C, formerly assumed to be the actual compound of boron carbide) [6,7,14]. Hence the real structure contains 5.7 at. % structural defects compared with the idealized (B11C) CBC structure. This applies also for high-quality single crystals, whose preparation is only possible in the chemical composition B4.3C [8] and for single crystals growing accidentally in industrial production.
Vacancies of the B(3) site (9 – 28 %, depending on composition) (see [14] and references cited therein).
CBB chains replacing CBC towards boron-rich boron carbides (Figure 3b, see [14] and references cited therein)
B4.3C is the least distorted compound at all [14,21]. It does not contain any free carbon, as for example systematically checked by Schwetz et al. [7,22] using a wet-chemical analysis for determining free and bound carbon in boron carbide separately. B4.3C is the only compound allowing to prepare high-quality single crystals [8].
Theoretical model calculations determined the idealized structure formula (B12) CBC of the compound B13C2 as the energetically most favorable [23,24]. In fact, B13C2 is the most distorted boron carbide at all. Its highly complex structure (B12)0.5(B11C)0.5 • (CBC)0.65(CBB)0.16(BB)0.19 shows that about 80% of the unit cells contain any kind of structural defect [4,25,26]. High concentrations of structural defects (with regard to idealized structures) are a generic property of boron carbide. structure.
The shares of structure elements in the unit cell depending on carbon content in Figure 3 [14] results preferably from the investigation of IR- and Raman-active phonons. The comparably low share of B☐B chains (☐, vacancy) has not been measured but is used to close the gap between the sum of atoms in the other structure elements determined and the chemical composition. TEM studies by Rasim et al. [27] suggest that a variety of further kinds of structural elements can replace the CBC chain. However, apart from this TEM study, there is no further experimental evidence. Therefore, it cannot be excluded, that such structural elements are provoked by the preparation process of the sample.
Many theoretical studies on structural details in boron carbide are based on Raman studies, which are affected by a severe systematic experimental error [16]. In cases of high Raman excitation energy, the spectra do not reflect bulk properties. Hence the results of accordingly performed calculations merely end in itself and do not yield scientific progress (see for example the controversial discussions in [28] and [29], [30] and [31], [32] and [33]).
3. Electronic structure of B4.3C at ambient conditions
Due to the availability of material, previous experimental studies of electronic properties are mainly restricted to polycrystalline boron carbide and especially to the compound B4.3C. This also applies when the compound is nominally designed as B4C, as this is in reality B4.3C with excess carbon typically distributed in the form of graphitic layers [10,22]. High quality B4.3C single crystals were used in references [8,34,56]. Figure 4 shows a collection of transition energies, consistently obtained from optical absorption like those shown in Figure 5, indicating interband transitions at 2.09 and 2.41 eV and from electrical transport measurements (see [3,4,35] and references cited therein).
Off the absorption edge in Figure 5 there is a peak absorption in polycrystalline boron carbide at 1.56 eV. On the first view surprising, the absorption of high-quality single crystal boron carbide in this range is considerably higher. XRS emission (Figure 5, Feng et al. [36]) and luminescence [35,37] (Figure 6) suggest a p-type exciton associated with the central B atom of the CBC chain. The luminescence peak consists of two components, whose intensity relation is 4:1. This corresponds quite well with the relation in natB4.3C 11B / 10B = 4.03, if random isotope distribution is assumed. The formation of excitons requires largely undistorted structures. In B4.3C single crystals this condition is better fulfilled than in polycrystals, and at low T better than at high T. This explains the peculiarities in Figure 5. Hence, the gap state “Exciton” in Figure 4 is not evoked by structural defects. However, effects of structural defects on the optical absorption cannot be excluded. For example, at high pressure (see below), structural distortion occurs by buckling-out of the central B atom in the CBC chain [15,38,39,56].
Former theoretical studies on boron carbide were not very helpful to understand the complex interactions between structural details and electronic properties [40,41,42,43,44,45,46], as they were based on idealized structure models deviating more or less considerably from the real structures. Thus, related conclusions were often in significant contrast to experimental results: For example, the theoretically claimed metallic character or even superconductivity [45] was opposite to the semiconducting behavior experimentally proved from the very beginning. Surprisingly, even in recent studies the real structure of boron carbide is frequently ignored.
4. Phase transition near the compound B8C
Reliable information on the electronic structure of more boron-rich boron carbide is obtained from electrical conductivity, measured between 5 and 2100 K by Werheit et al. [47], by Wood [48,49] and by Aselage et al. [50]. Results obtained by Amulele et al. [51] up to 9 GPa pressure are compatible.
The activation energies obtained from the electrical conductivity are displayed in Figure 7 showing that at ~B8C the homogeneity range is divided into two parts with significantly different electronic characteristics. The activation energy at about 80 meV found in both regions is uncertain as obtained in that range of temperature, where a continuous transition from thermal excitation to variable-range hopping takes place (see [47]).
It is expected that this phase transition is correlated with a structural change. This should be reflected in the phonon spectra. Figure 8 shows the IR- and Raman-active phonons of 11BxC boron carbide [34,52]. We analyze some phonons showing changes depending on the chemical composition.
The activation energy at about 80 meV found in both regions is uncertain as obtained in the range of temperature, where a continuous transition into the range of variable-range hopping takes place (see [47]).
4.1. IR-active phonon near 400 cm-1
The phonon near 400 cm-1 is composed of two components (see Figure 9a), whose frequencies (Figure 9b) and phonon strengths vary depending on the C content. Shirai et al. [53,54] and Vast et al. [46,55] attribute the IR phonon mode near 400 cm−1 to the central B atom in the chain vibrating perpendicularly to the chain axis [26]. Experimentally, this is confirmed for the component Ph 1 whose strength depending on the C content precisely matches with the share of CBC chains (Figure 9b). The considerably weaker component Ph 2 of this phonon belongs to the icosahedral structure, accordingly with counterpart in the spectrum of α-rhombohedral boron (see Figure 2). This phonon was already mentioned by Beckel et al. [20] but not described in detail (see [16]). Below, we perform an analysis of the temperature-dependence of this phonon.
In Figure 9, there is no change apparent suggesting a relation of this phonon to the phase transition at B8.1C. However, the mode Grüneisen parameter γ of the IR-phonon (-3.21 at low and +6.5 at high temperatures) deviating strikingly from all other phonons [56] suggest a peculiarity. The previously assumed involvement of this phonon in the T-dependent transition at 712 K [59] is checked below.
4.2. IR-active phonons near 700 cm-1
The spectra of isotope-enriched samples (Figure 10a) exhibit clear changes depending on C content. For interpretation, a denser sequence of spectra is required. Data on natBxC are at disposal ([57] Figure 10b). The results of the analysis (examples in (Figure 11a) are displayed in Figure 11b. Splitting of the component of this phonon near 760 cm-1 into three branches indicates a structural change at the phase transition. This phonon is correlated with the strong 705 cm-1 intra-icosahedral single-line A2u mode in α-rhombohedral boron, described by Beckel et al. [20], split in the boron carbide structure (see [16,31]).
4.3. IR-active phonons near 940 cm-1
The phonon near 940 cm-1 (Figure 12) represents an icosahedral vibration proved by the counterpart in the spectrum of α-rhombohedral boron (see Figure 2). According to Beckel et al. [20], this phonon consists of superimposing Eu and A2u modes. In boron carbide, the additional affect by polar C atoms in the icosahedra must be taken into account. This explains in principle the complexity of the phonon and of its change depending on C content.
The share of B11C icosahedra decreases from ~1 in B4.3C to ~0.1 in B10C. The main components (1) and (2) seem to vary continuously depending on the C content. There is no indication of a sudden change near B8C. Therefore, no involvement of this phonon in the phase transition at B~8C is discernible.
However, there is a significant change between B4.3C and B6.5C: two components of the phonon vanish completely. Unfortunately, there are no spectra in the intermediate range available, which could help explaining this behavior.
4.4. FIR-absorption
As indicated in the low-frequency region of Figure 8a, the FIR optical absorption increases towards very low wave numbers. Figure 13 shows this range extended down to 10 cm-1. This absorption is caused by the plasma-like absorption of free charge carriers, which is heavily damped in boron carbide (for details see [58]). With respect to the electric structure, Figure 13 shows that the concentration of free carriers increases with the C content decreasing. However, there is no effect of the phase transition at B~8C on the concentration of free holes discernable.
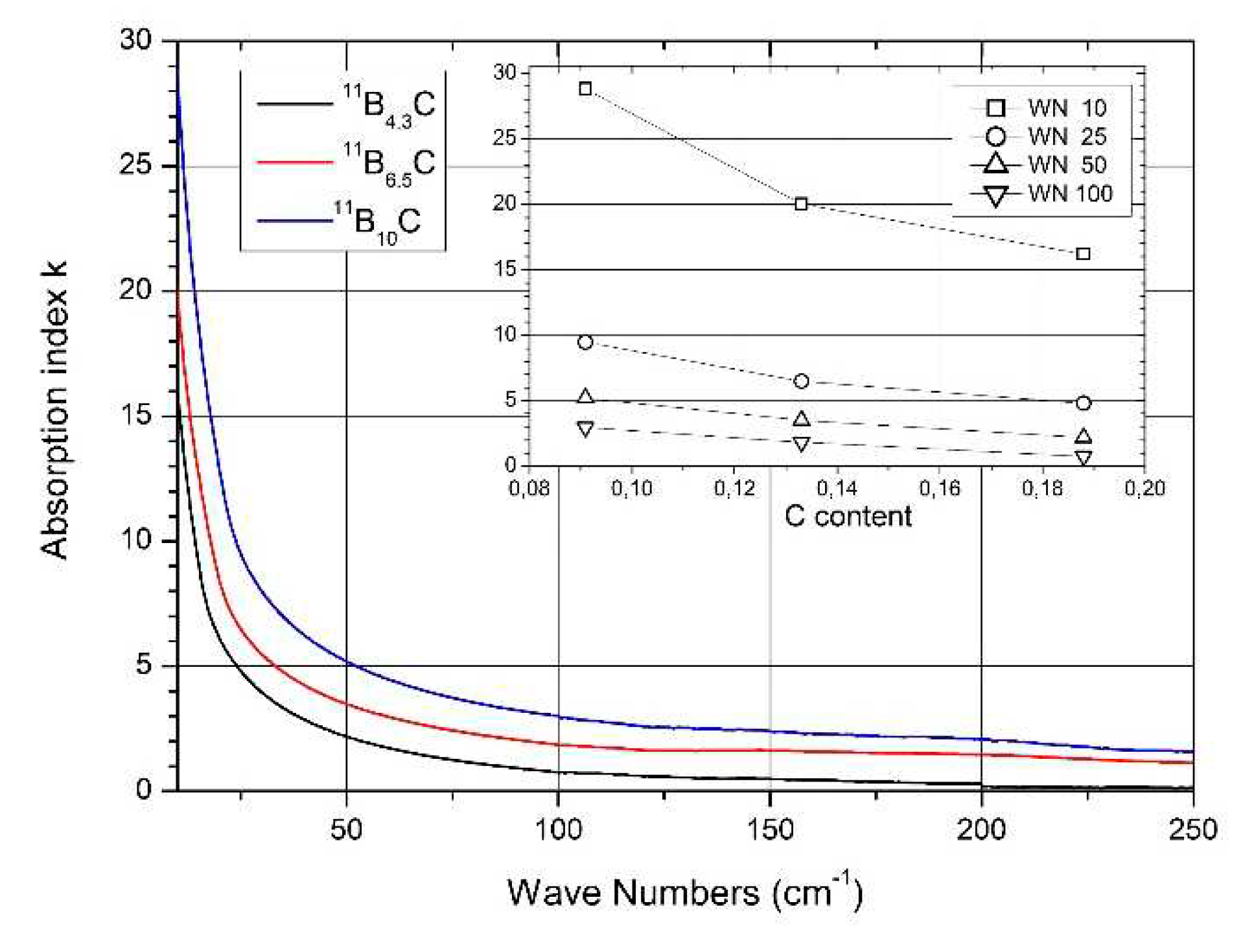
4.5. Raman-active phonons
The resolution of the Raman spectra in Figure 8b is restricted. Therefore, variations dependence on the C content cannot be doubtlessly identified, apart from the strong phonon doublet at 270/320 cm-1. This has no counterpart in the spectrum of α-rhombohedral boron. Therefore, it is to be attributed to the structural changes in boron carbide by C atoms: B11C icosahedra, CBC or CBB chains.
We attribute this doublet to the Eg phonon at 335 cm−1 described in the theoretical study of Shirai and Emura [54] as a rotation of the CBC chain associated with a wagging icosahedron, and assume that the splitting could result from the occupation of the atomic sites involved partially occupied by C or B atoms (see [12]).
The strengths of both components change monotonically depending on the carbon content. There is no indication that this phonon is involved in the phase transition at. B~8C.
5. Phase transition at 712 K
The T-dependent specific heat (Figure 15) shows an exothermic phase transition at 712 K, halfwidth 82 K [59]. The correlation of this phase transition with a structural change is proved by the electrical conductivity, whose activation energy 140 meV is correlated with content of B11C icosahedra and polar C atoms respectively (see above). At carbon contents below the phase transition B~8C it is no more detectable [47].
Yao et al. [60] described theoretically in this range a however endothermic phase transition that is related to the site exchange of carbon atoms within the icosahedra. Obviously, this process is not compatible.
Complete temperature-dependent phonon spectra of boron carbide are available below 450K only. Exception is the IR-active phonon mode near 400 cm-1 available between 100 and 800 K (Figure 15a) [59]. The spectra suggested a structural change by mode splitting near the phase transition. However, this splitting is feigned.
As shown above, the IR-active phonon mode near 400 cm-1 consists of two components. One of them is an icosahedral vibration identified by the equivalent vibration in α-rhombohedral boron (see Figure 2). As such it is mentioned by Beckel et al. [20] but not described in detail. Shirai et al. [54,61] and Vast et al. [46,55] attribute the second component to the central B atom in the CBC chain vibrating perpendicularly to the chain axis. We analyzed the IR absorption of this phonon accordingly. The parameters of both components are displayed in Figure 12b, showing that the before assumed split was feigned by the considerably different temperature dependence of the components.
Passing the phase transition, the frequency of the component representing the icosahedral vibration decreases clearly from 405 cm-1 at low T to 395 cm-1. Simultaneously, the phonon strength concerned increases marginally showing that the number of vibrating elements remains nearly unchanged. The atomic bonding force concerned falls slightly in the range of phase transition, but is nevertheless clearly measurable. It is surprising that the range of decrease is wider that the half width of the phase transition (Figure 14). Anyway. a certain effect of the phase transition on the electronic properties is clearly established.
The vibration of the central B atom in the chain behaves different. The frequency remains nearly constant showing that the bonds within the CBC chain are hardly affected by T. In contrast, the phonon strength representing the number of vibrating elements decreases considerably.
A certain difference of the structures below and above the phase transition is indicated by the frequency of the icosahedral phonon near 400 cm-1 (Figure 15b), which decreases from 405 cm-1 at low T to 392 cm-1 at high T. In contrast, the frequency of the bending mode of the central B atom in the CBC chain decreases at most marginally from ~414 to ~407 cm-1. The redistribution of polar C atoms in the phase transition affects the bonding force within the icosahedron and in consequence the electronic structure as well.
An important further characteristic of this phase transition can be obtained from the electrical conductivity at pressures up to 9 GPa [51]. The phase transition is reversible, however with considerable relaxation time.
6. Phase transition at 32 GPa
The drastic change from opacity to glasslike transparency is the visual appearance of the phase transition in boron carbide at 32 GPa. Hushur et al. [38] used the photos shown in Figure 17 for roughly estimating the band gap increasing at increasing pressure. This implies that the electronic structure of boron carbide known from ambient conditions [15] (see Figure 3) changes fundamentally at high pressure.
In Figure 17 [38], the estimated shift of the band gap is compared with theoretical model calculations on undistorted and distorted boron carbides [42,43,62,63,64,65] and experimental results [38,66,67]. Obviously, this phase transition converts boron carbide, as it is commercially available or prepared in laboratory scale and apparently basically highly distorted, into a largely undistorted state, which is at least close to the idealized models used in theoretical calculations.
At rising pressure, the band gap increases from values near 2 eV, experimentally determined [66] and theoretically attributed to a strongly distorted boron carbide [64] to gap widths between 3 and 4 eV, theoretically predicted for idealized meaning defect-free boron carbide.
Ektarawong et al. [64] studied how the distribution of icosahedral C atoms affects the electronic DOS. They used the idealized (B11C) CBC) structure and ignored changes in the CBC chain. In the icosahedra, 6 polar B(1) and 6 equatorial B(2) sites are occupied by B atoms, apart from one upper polar site p1 substituted by C. This C atom is slightly shifted towards the center of the icosahedron (see Ref. [25] and references cited therein). The energetically most favorable defect structures are generated by the transfer of the C atom to both neighboring p2 and p3 sites respectively, followed by transfers from p1 to the lower polar sites p4, p5 and p6 respectively. Ektarawong's RNG model is based on the assumption that the polar C atoms are randomly distributed over all six polar sites.
Rasim et al. [27] take the idealized (B12) CBC structure model for reference. As mentioned above, this model is not compatible with real boron carbide. Nevertheless, in this study it excludes interfering effects in icosahedra when calculating the effects on the DOS by exchanging the CBC chain by other structure elements of various chain structures.
Both approaches mark significant achievements compared with the previous theoretical studies based on idealized and therefore unrealistic structure models. In Figure 19 [15], we check to what extend the DOS of both model calculations are compatible with the electronic transitions in the empirical band scheme (Figure 4) based on experimental results. These fit excellently to Ektarawong’s RNG model, assuming random distribution of icosahedral C atoms over all polar sites [64].
Taking these results into account, the strong optically excited transitions between ~2 and ~3.5 eV, which are responsible for the opacity of boron carbide at ambient pressure, are evoked in any way by the polar C atoms in boron carbide. The strong optical absorption concerned fulfills the requirements of electronic interband transitions and has been interpreted accordingly. Compared with classical semiconductors, the polar C atoms in boron carbide differ decisively from dopants with one excess electron forming low density states in the gap. In boron carbide, the distribution of polar C atoms at room temperature induces high density electronic states. But a “simple” redistribution of the polar C atoms at high pressure apparently to a higher degree of order makes these high-density states completely disappear. This redistribution is reversible. That means, the largely defect-free structure at high pressure [38] is metastable only, while the distorted structure is the thermodynamically stable one, at least at ambient conditions.
The reversibility of the structural distortion induced by the C atoms randomly distributed over all six polar sites requires revising a sometimes-used model based on the assumption that the actual distorted structure of boron carbide is a non-equilibrium state only [60].
The DOS of boron carbide affected by structural elements replacing the CBC chain, calculated by Rasim et al. [27], is obviously not compatible with the high-pressure phase transition. Indeed, there are some experimentally determined narrow levels in the band scheme, which may be correlated with specific structural elements assumed to replace the CBC chain. However, the optical absorption related is obviously far too small for explaining the strong absorption in this range at ambient conditions.
The rearrangement of polar C atoms means a structural change and should therefore be verifiable in the phonon spectra. Actually, the whole structure is affected by the phase transition at 33 GPa. This is proved by the phonon mode Grüneisen parameters γ. In case of all IR- and Raman-active phonons, γ differs more or less considerably at pressure below and above the phase transition [12,29,56]. Unfortunately, in the pressure-dependent IR-phonon spectra of Chuvashova et al. [28], accordingly commented in [29] (some results are shown in Figure 19), the IR-active phonon near 400 cm-1 has been neglected. As shown above, a component of this phonon is sensitive regarding polar C atoms in B11C icosahedra.
The spectra of IR- and Raman-active phonons (Figure 8) exhibit strong, probably partially split peaks near 1100 cm-1. These are at least partially attributed to intra- and inter-icosahedral vibrations of polar atoms in the icosahedra. Hence, they should be affected by the rearrangement of polar C atoms.
In Figure 21, the Grüneisen coefficient of the 1093.3 cm-1 IR phonon above the phase transition exceeds that of the other phonons considerably. Moreover, the difference to the value below the phase transition is especially large. This suggests that the redistribution of polar C atoms increases the bonding force considerably.
The Grüneisen coefficients of concerning Raman-active phonons, determined in Figure 21, change significantly at the phase transition, obviously due to rearranging polar C atoms.
In the Raman spectra of boron carbide, the phonon doublet at 270/320 cm-1 is the most striking feature.
Werheit et al. [12] assigned this doublet to the 335 cm-1 Eg phonon that Shirai and Emura [54] attributed to a rotation of the 3-atom chain combined with a wagging icosahedron. Firstly, it is surprising, that this phonon splits, though there are only B11C icosahedra in B4.3C. This could be explained as follows: The translation vectors of atoms involved in this phonon (see Figure 1 in Ref. [54]) show that 8 of 12 atoms in the icosahedra and 4 of 6 polar sites respectively are involved. Assuming that the C atom is randomly distributed over all polar sites of the icosahedra, there are B11C icosahedra with C atom on sites, which are involved in this vibration, while others contain exclusively B atoms on such sites. Due to different mred, the phonon mode splits and the components Ra 1 (~ 270 cm-1; C on one of the relevant sites) and Ra 2 (~320 cm-1; B atoms only) could be attributed accordingly.
The negative Grüneisen coefficients of both components differ fundamentally from all other phonons. Normally, compression reduces the lattice parameters, leading to a positive Grüneisen parameter, at least in cases, when the force constant is not affected. However, exactly that seems to be the case in boron carbide in connection with the rearrangement of polar C atoms at the high-pressure phase transition.
In Figure 22, some parameters of both components of this doublet are displayed. For the evaluation had to be considered, that the spectra were obtained using a laser with problematic excitation energy (for details, see [17]). While the phonon frequencies are not affected this way, the phonon strengths A1 and A2 derived from the measured spectra depend uncontrollably on pressure. This influence is eliminated by determining the ratio A1/A2 of both components shown in Figure 20.
7. Discussion
The electronic structure of boron carbide is dominated by B11C icosahedra. In comparison, the influence of the 3-atom chain is of minor importance. This is obvious in the phase transition near the chemical composition B~8C, which is clearly related to the concentration of B11C icosahedra. The fact that this transition occurs at about 25% B11C instead of 50% shows the particular influence of polar C atom that is missing in the frequently used hypothetical structure model B12CBC. Whether the distribution of one C atom on 6 possible polar sites has any effect within this composition-dependent phase transition remains open.
However, the distribution of polar C atoms is doubtlessly important in B4.3C with ~100% B11C icosahedra. The phase transitions at 712 K and 33.2 GPa are directly related to the distribution of the polar C atoms. At ambient conditions, boron carbide is opaque. Its optical absorption in the visible range of the optical spectrum is high, due to optical transitions related to gap states shown in Figure 4. Hushur et al. [38,56] showed that high pressure changes the opacity drastically to transparency indicating a highly ordered structure. However, this is metastable only, as it retransforms into the disordered state reverting to normal conditions, albeit with considerable relaxation time. Accordingly, the distorted structure is energetically favored at ambient conditions. Applying this knowledge to the hypothetical structure B12CBC possibly explains, why this highly ordered structure is unstable compared with the distorted structure of natural boron carbide.
This leads to the conclusion that Ektarawong’s RNG model, assuming random distribution of polar C atoms over all polar sites at ambient conditions, is close to reality. The phase transitions at 712 K and 33.2 GPa both transform this distorted structure of boron carbide at ambient conditions (statistical distribution of 1 C atom per icosahedron to 6 polar sites) into a structure of higher order. This means occupation of anyway preferred polar sites; however, the actual distribution remains open. More detailed characterization of the phonon spectra might help.
In this study, the phonon spectra of boron carbides with natural isotope distribution and those highly enriched with 11B isotopes have been analyzed. Thus, isotopes could have a certain effect (for details, see [68]). The force constants of different isotopes are the same. Hence, the shift of the icosahedral phonon frequencies in B4.3C boron carbides, which are enriched with 12C and 13C respectively (see spectra in [52,69]), yields the relation of reduced masses involved.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Data are available on request.
Acknowledgments
The author thanks all coauthors of joint papers cited in this review for their contribution and particularly Murli Manghnani, University of Hawaii, Honolulu, USA, Karol Flachbarth, Institute of Experimental Physics, Slovak Academy of Sciences, Košice, Slovakia and Volodimir Filipov, I. N. Frantsevich Institute for Problems of Materials Science, Kiev, Ukraine, for their productive cooperation during the last years.
Conflicts of Interest
The author declares no conflict of interest.
References
- Lipp A., Technische Rundschau, 57(14), 5 (1965); 57(28), 19 (1965); 57(33), 5 (1966); 58(7), 3 (1966).
- Domnich V., Reynaud S., Haber R. A. and Chhowalla, M., J. Am. Ceram. Soc. 2011, 94, 3605.
- Werheit H., On Microstructure and Electronic Properties of Boron Carbide, in Ceramic Engineering and Science Proceedings (CESP; ICACC – S4 – 012 – 2014); Vol. 35, Issue 4, Adv. in Ceramic Armor X , Jerry C. LaSalvia (Editor )).
- Werheit H., Structural Defects: Essential elements of icosahedral boron-rich solids, in 2015 Sustainable Industrial Processing Summit, Vol. 8, Composite, Quasi-crystals and Nanomaterials, ed. By F. Kongoli, M. Pech-Canul, A. Kalemtas, H. Werheit, p. 159.
- Werheit H., Thermoelectric Properties of Boron-Rich Solids and their Possibilities of Technical Application, in: Proc. 25th International Conference on Thermoelectrics, Vienna, 2006, p. 159.
- Schwetz K.A. and Karduck P., J. Less Common. Met. 1991, 175, 1. [CrossRef]
- Schwetz K. A. and Karduck P., AIP Conf. Proc. 1990, 231, 405.
- Werheit H., Leithe-Jasper A., Tanaka T., Rotter H.W., Schwetz K.A., J. Solid State Chem. 2004, 177, 575.
- Werheit H. and Schwetz K.A., J. Phys. Chem. Solids. 1985, 46, 1331.
- Bandyopadhyay A. K., Beuneu F., Zuppiroli L. and Beauvy M., J. Phys. Chem. Solids 1984, 45, 207.
- Werheit H., Flachbart K., Pristáš G., Lotnyk D., Filipov V., Kuhlmann U., Shitsevalova N. and Lundström T., Semicond. Sci. Technol. 2017, 32, 095015.
- Werheit H., Manghnani M. H., Hushur A., Solid State Sciences 2019, 97, 105978.
- Werheit H., Kuhlmann U., J. Phys.: Condens. Matter 2012, 24, 305401.
- Werheit H., Solid State Sciences 2016, 60, 45.
- Werheit H., Solid State Sciences 2018, 86, 38.
- Werheit H., Rev. Sci. Instrum. 2019, 90, 043114.
- Werheit H., Phys. Rev. Mat. 2022, 6, 016601.
- Werheit H., Filipov V., Kuhlmann U., Schwarz U., Armbrüster M., Leithe-Jasper A., Tanaka T., Higashi I., Lundström T., Gurin V. N. and Korsukova M. M., Sci. Technol. Adv. Mater. 2010, 11, 023001.
- Werheit H., Kuhlmann U., J. Phys.: Condens. Matter 2012, 24, 305401.
- Beckel C. L., Youssaf M., Fuka M. Z., Raja S. Y. and Lu N. , Phys. Rev. B 1991, 44, 2535.
- Werheit H., Shalamberidze S., J. Phys.: Condens. Matter 2012, 24, 385406.
- Schwetz K.A., Hassler J., J. Less Common. Met. 1986,, 117, 7. [CrossRef]
- Bylander D.M., Kleinman L., Phys. Rev. B 1991, 43, 1487. [CrossRef]
- Kleinman L., AIP Conf. Proc. 1991, 231, 13.
- Werheit H., Boron carbide: on structural details and electronic properties, in: Jerry C. LaSalvia (Ed.), Ceramic Engineering and Science Proceedings 2014.
- Werheit H., Gerlach G., J. Phys. Condens. Matter, 2014, 26, 425801.
- Rasim K., Ramlau R., Leithe-Jasper A., Mori T., Burkhardt U., Borrmann H., Schnelle W., Carbogno C., Scheffler M. and Grin Y., Angew. Chem. 2018, 130, 6238.
- Chuvashova I., Gasharova B., Mathis Y.-L., Dubrovinsky L., Dubrovinskaia N., Z. Anorg. Allg. Chem. 2017, 643, 1357.
- Werheit H., Z. Anorg. Allg. Chem. 2018, 644, 353.
- Roma G., Gillet K., Jay A., Vast N., and Gutierrez G., Phys. Rev. Mater 2021. 5, 063601.
- Werheit H., Phys. Rev. Mat. 2022, 6, 016601.
- Sahu T., Bhattacharyya A., Gandi A.N., Physica B, 2022, 633, 413738.
- Werheit H., Physica B, 2023, 651, 414604.
- H. Werheit, T. Au, R. Schmechel, S.O. Shalamberidze, G.I. Kalandadze, A.M. Eristavi, J. Solid State Chem. 154, 79 (2000).
- Werheit H., J. Phys.: Condens. Matter, 2006, 18 10655.
- Feng Y, Seidler G T, Cross J O, Macrander A T and Rehr J J, 2004, Phys. Rev. B, 69, 125402.
- Schmechel R., Werheit H., Solid State Chem. 2000, 154, 68.
- Hushur A., Manghnani M.H., Werheit H., Dera P., Williams Q., J. Phys. Condens. Matter, 2016, 28, 045403.
- Korotaev P., Pokatashkin P., Yanilkin A., Comput. Mater. Sci., 2016, 12, 106.
- Armstrong D. R., Balland J., Perkins P. G., Will G. and Kirfel A., Acta Crystallogr. B 1983, 39, 324.
- Bullett D. W., J. Phys. C: Solid State Phys., 1982, 15, 415.
- Switendick A. C., The electronic structure of crystalline boron carbide I: B12 icosahedra and C–B–C chains in The Physics and Chemistry of Carbides, Nitrides and Borides, ed. R Freer (Dordrecht: Kluwer Academic) 1990, p 525.
- Bylander D. M., Kleinman L. and Lee S., Phys. Rev. B 1990, 42, 1394.
- Shirai K., Dekura H. and Masago A., J. Phys.: Conf. Ser. 2009, 176, 012001.
- Calandra M., Vast N. and Mauri F., Phys. Rev. B, 2004, 69, 224505.
- Vast N., Sjakste J. and Betranhandy E., J. Phys.: Conf. Ser.176 (2009) 012002.
- Werheit H., Herstell B., Winkelbauer W., Pristáš G., Gabáni S., Flachbart K., Shalamberidze S., Solid State Sciences 2022,132, 106987 and Solid State Sciences 2022, 134, 107055.
- Wood C., AIP Conf. Proc., 1986, 140, 206.
- Wood C., The transport properties of boron carbide prepared by different techniques, in: H. Werheit (Ed.), Proc. 9th Int. Symp. Boron Borides and Rel. Comp. Duisburg, Germany, 1987, p. 213.
- Aselage T. L., Emin D. and McCready S. S., Phys. Rev. B 2001, 64, 054302.
- Amulele G., Manghnani M. H., Werheit H., Lanati A. W., Clark S. M., Solid State Sciences 140 (2023) 107185.
- Werheit H., Rotter H.W., Meyer F.D., Hillebrecht H., Shalamberidze S.O., Abzianidze T.G. and Esadze G.G., J. Solid State Chem., 2004, 177, 569.
- Shirai K., Phys, Rev. B 1997, 55, 12235.
- Shirai K. and Emura S., J. Solid State Chem. 1997, 133, 93. [CrossRef]
- Lazzari R., Vast N., Besson J.M., Baroni S.and Dal Corso A., Phys. Rev. Lett. 83, 199, 3230.
- Werheit H., Manghnani M. H., Kuhlmann U., Hushur A., Shalamberidze S., Solid State Sciences, 2017, 72, 80.
- Kuhlmann U., PhD Thesis 1994, University of Duisburg, Germany.
- Werheit H. and Gerlach G., J. Phys.: Condens. Matter, 2014, 26, 425801.
- Werheit H., Hoffmann S., Gerlach G., Leithe-Jasper A., Tanaka T., Phase Transition in B4.3C Boron Carbide at moderate temperature 2015 Sustainable Industrial Processing Summit. Vol. 8: Composite, Quasi-crystals and Nanomaterials, Editors F. Kongoli, M. Pech-Canul, A. Kalemptas, H. Werheit, FLOGEN 2015 p. 183.
- Yao S., Huhn W. P., Widom M., Solid State Sciences, 2015, 47, 21.
- Shirai K., Phys. Rev. B, 1997, 55, 12235.
- Armstrong D. R., Balland J, Perkins P G, Will G and Kirfel A, Acta Crystallogr. B, 1983, 39 324.
- Bullett D. W., J. Phys. C: Solid State Phys., 1982, 15, 415.
- Ektarawong A., Simak S. I., Hultman L., Birch J. and Alling B, Phys. Rev. B, 2014, 90, 024204.
- Dekura H., Shirai K. and Yanase A., J. Phys.: Conf. Ser., 2010, 215, 012117.
- Werheit H 2015 Solid State Sci. 47 13.
- Werheit H, Janowitz C, Schmechel R, Tanaka T and Ishizawa Y, J. Solid State Chem. 1997, 133, 132.
- H. Werheit, U Kuhlmann, H W Rotter and S O Shalamberidze, J. Phys.: Condens. Matter, 2010, 22, 395401.
- Werheit H., Solid State Sciences, 2012, 14, 1559.
Figure 1.
Idealized rhombohedral unit cells of α-rhombohedral (structure formula B12) boron and boron carbide (structure formula B12 CBC) [12]. In (B11C) CBC at the carbon-rich limit, the icosahedral C atom occupies on of the B(1) polar sites.
Figure 1.
Idealized rhombohedral unit cells of α-rhombohedral (structure formula B12) boron and boron carbide (structure formula B12 CBC) [12]. In (B11C) CBC at the carbon-rich limit, the icosahedral C atom occupies on of the B(1) polar sites.
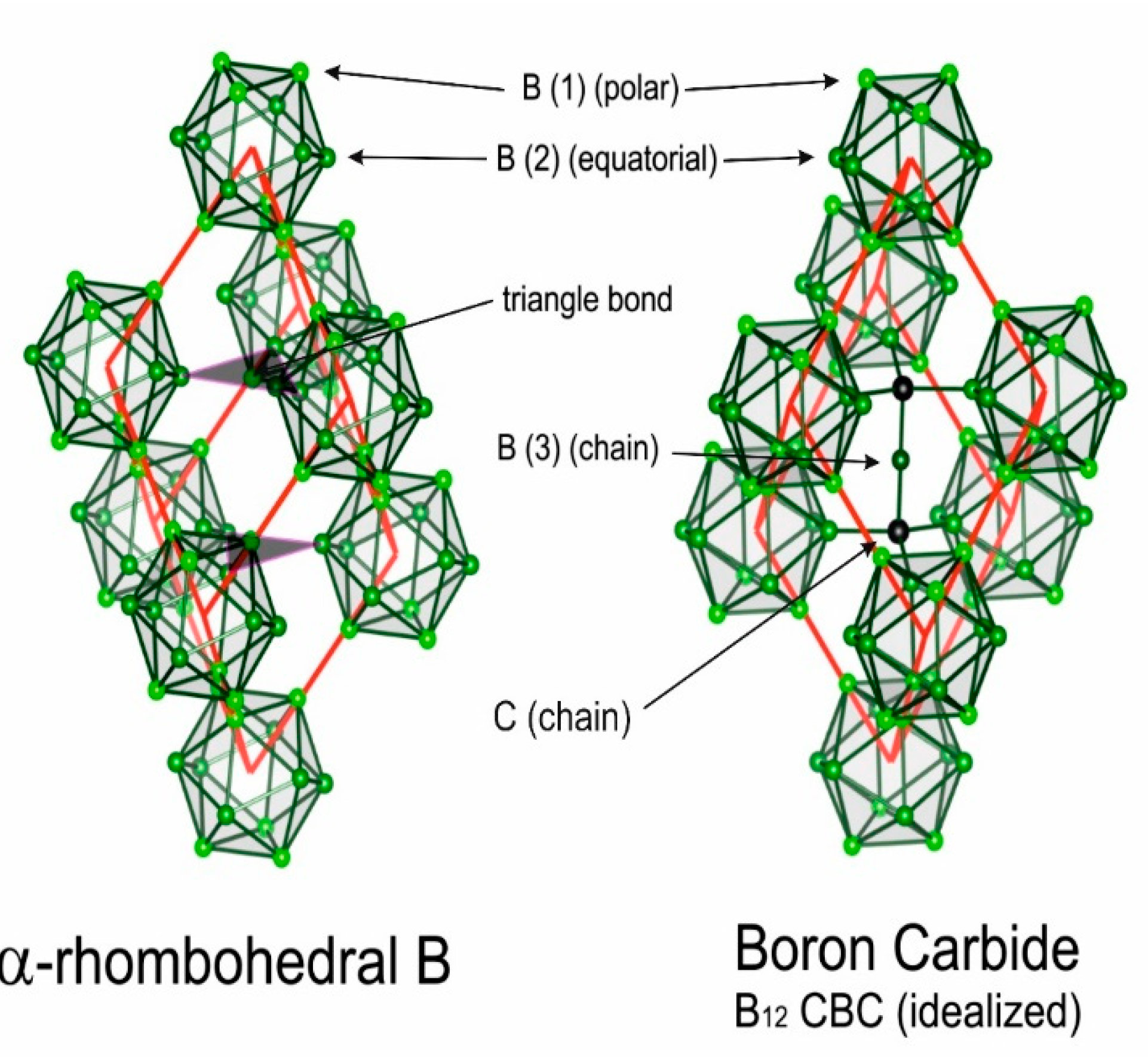
Figure 2.
IR-phonon spectra of B4.3C boron carbide and α-rhombohedral boron. The abscissas are shifted relative to one another to facilitate the correlation of icosahedral vibrations.
Figure 2.
IR-phonon spectra of B4.3C boron carbide and α-rhombohedral boron. The abscissas are shifted relative to one another to facilitate the correlation of icosahedral vibrations.
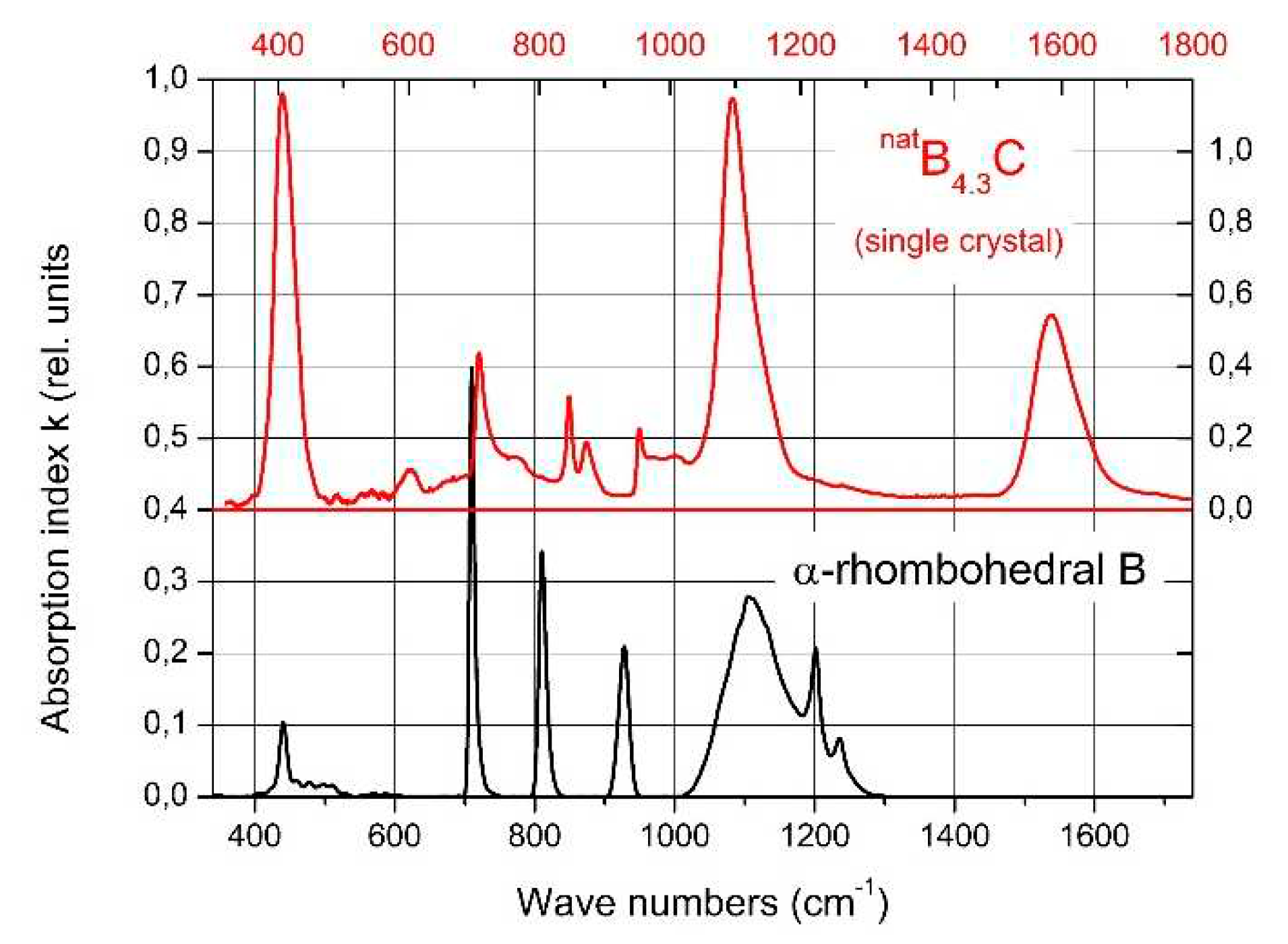
Figure 3.
Shares of structure elements in the unit cell of boron carbide [14]; a, B12 and B11C icosahedra, b, CBC, CBB and B☐B chains (☐, vacancy).
Figure 3.
Shares of structure elements in the unit cell of boron carbide [14]; a, B12 and B11C icosahedra, b, CBC, CBB and B☐B chains (☐, vacancy).
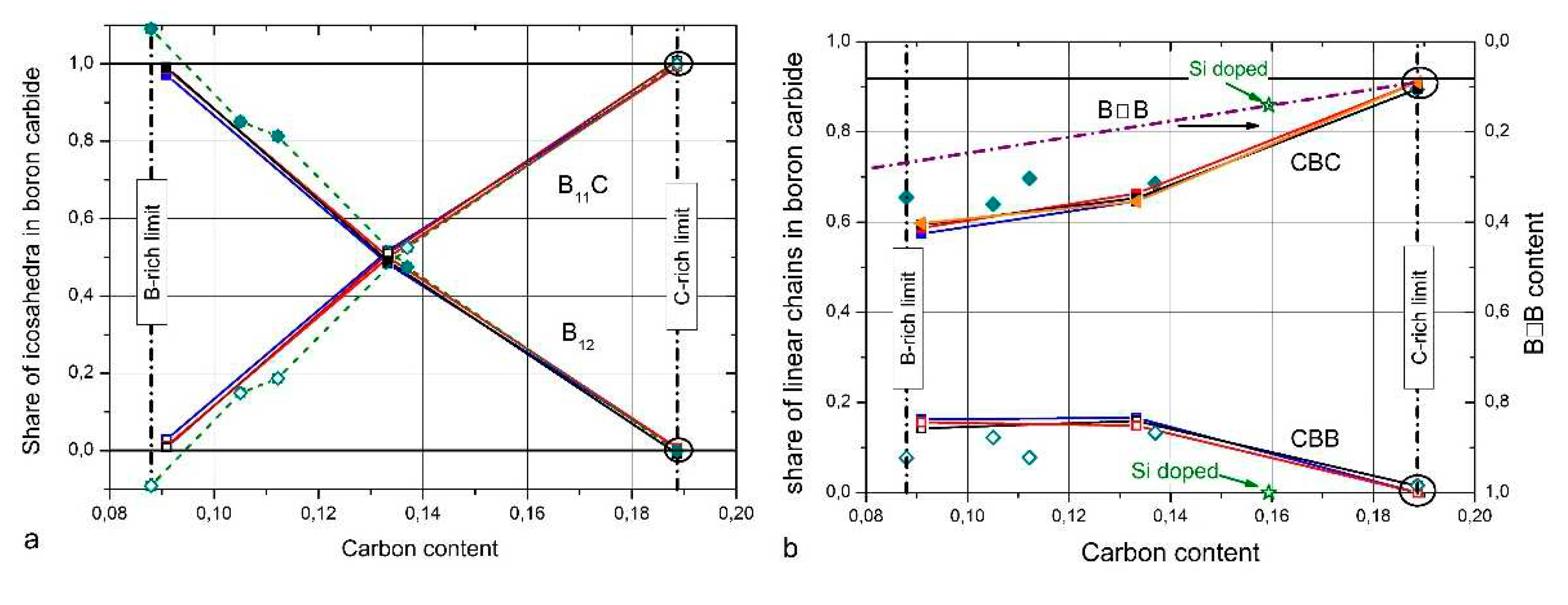
Figure 4.
Empirical band scheme of boron carbide based on experimental results (see [3,4] and references cited therein).
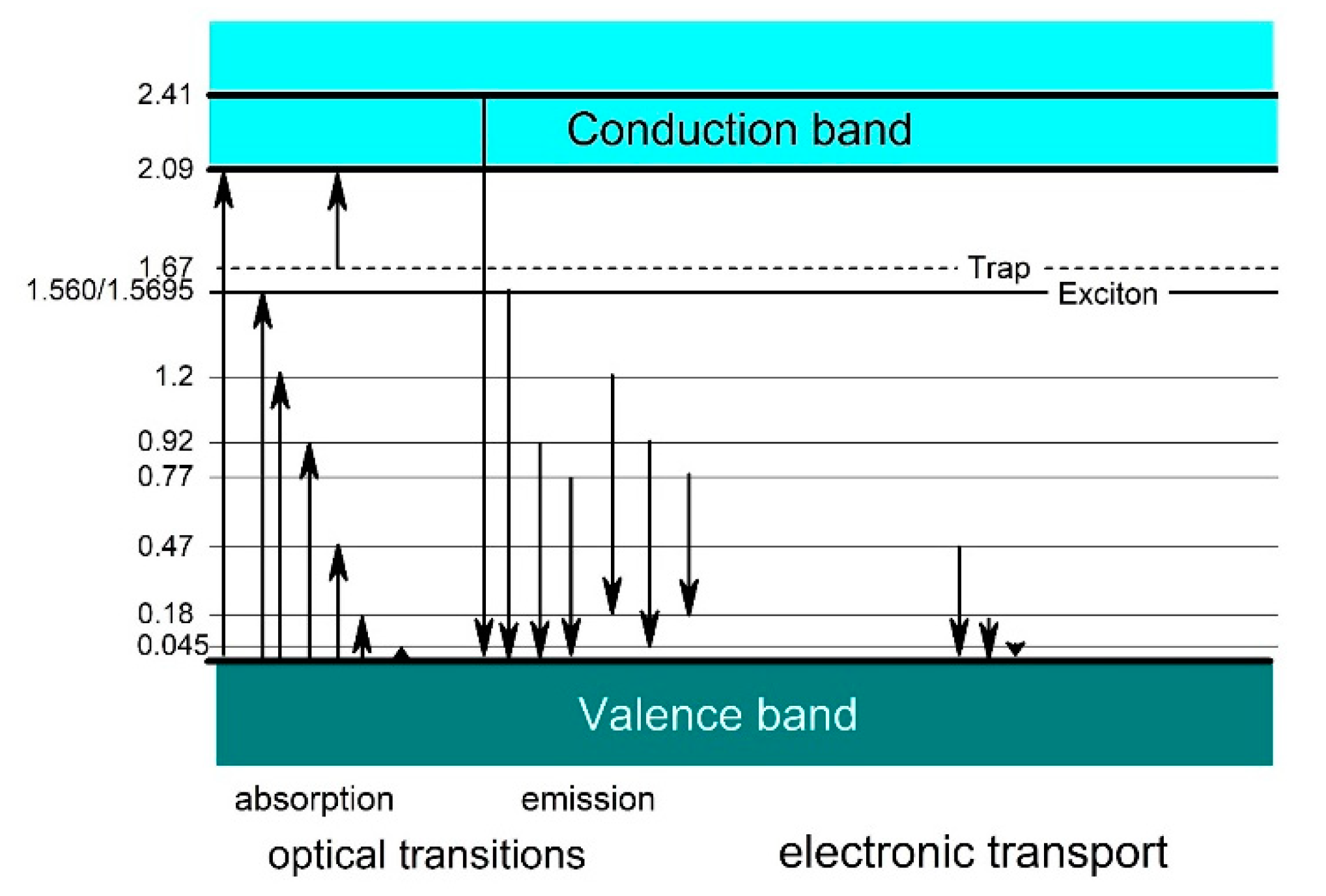
Figure 5.
B4.3C. (a) Optical absorption edge [3,4,35]. Symbols, polycrystalline material; line, single crystal. (b) X-ray Raman scattering (XRS) spectrum [36]. Symbols, selected experimental data for q = 1.05 °A-1; line, site-specific ab initio calculation for the background of icosahedral B atoms. We assume that the composition of this boron carbide corresponds to B4.3C, the carbon-rich limit of the homogeneity range; the composition B4C claimed by the authors does not exist (see above).
Figure 5.
B4.3C. (a) Optical absorption edge [3,4,35]. Symbols, polycrystalline material; line, single crystal. (b) X-ray Raman scattering (XRS) spectrum [36]. Symbols, selected experimental data for q = 1.05 °A-1; line, site-specific ab initio calculation for the background of icosahedral B atoms. We assume that the composition of this boron carbide corresponds to B4.3C, the carbon-rich limit of the homogeneity range; the composition B4C claimed by the authors does not exist (see above).
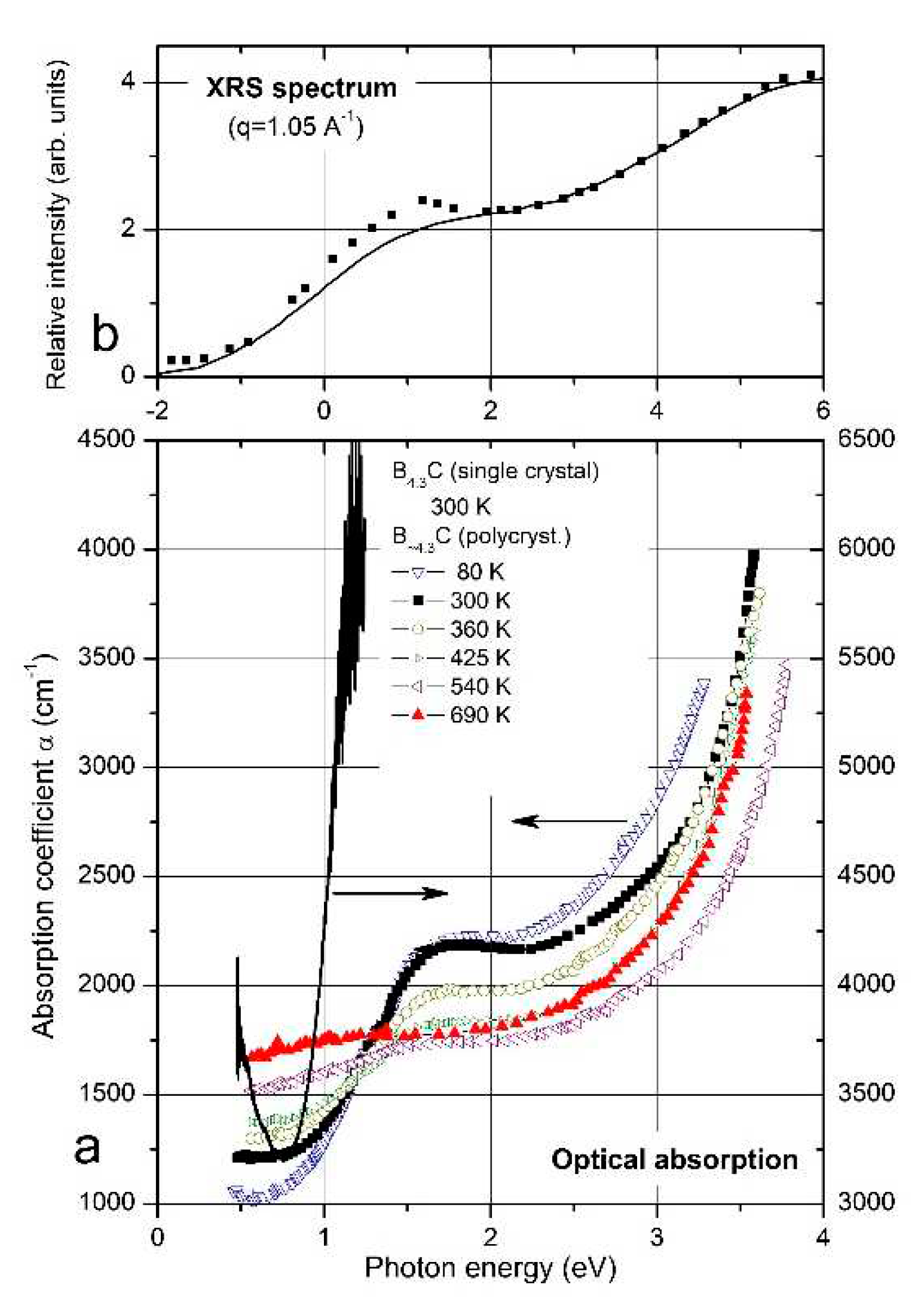
Figure 6.
B4.3C, photoluminescence spectrum at 290 K [35,37]. Excitation with the 514.5 nm (2.4 eV) line of an Ar laser; intensity 280 mW mm−2. Squares, experimental results; thin black lines, averaged experimental results, before and after subtracting the 1.56 eV model fit respectively; thick colored lines, recombination models of free excitons (1.560 and 1.5695 eV respectively).
Figure 6.
B4.3C, photoluminescence spectrum at 290 K [35,37]. Excitation with the 514.5 nm (2.4 eV) line of an Ar laser; intensity 280 mW mm−2. Squares, experimental results; thin black lines, averaged experimental results, before and after subtracting the 1.56 eV model fit respectively; thick colored lines, recombination models of free excitons (1.560 and 1.5695 eV respectively).
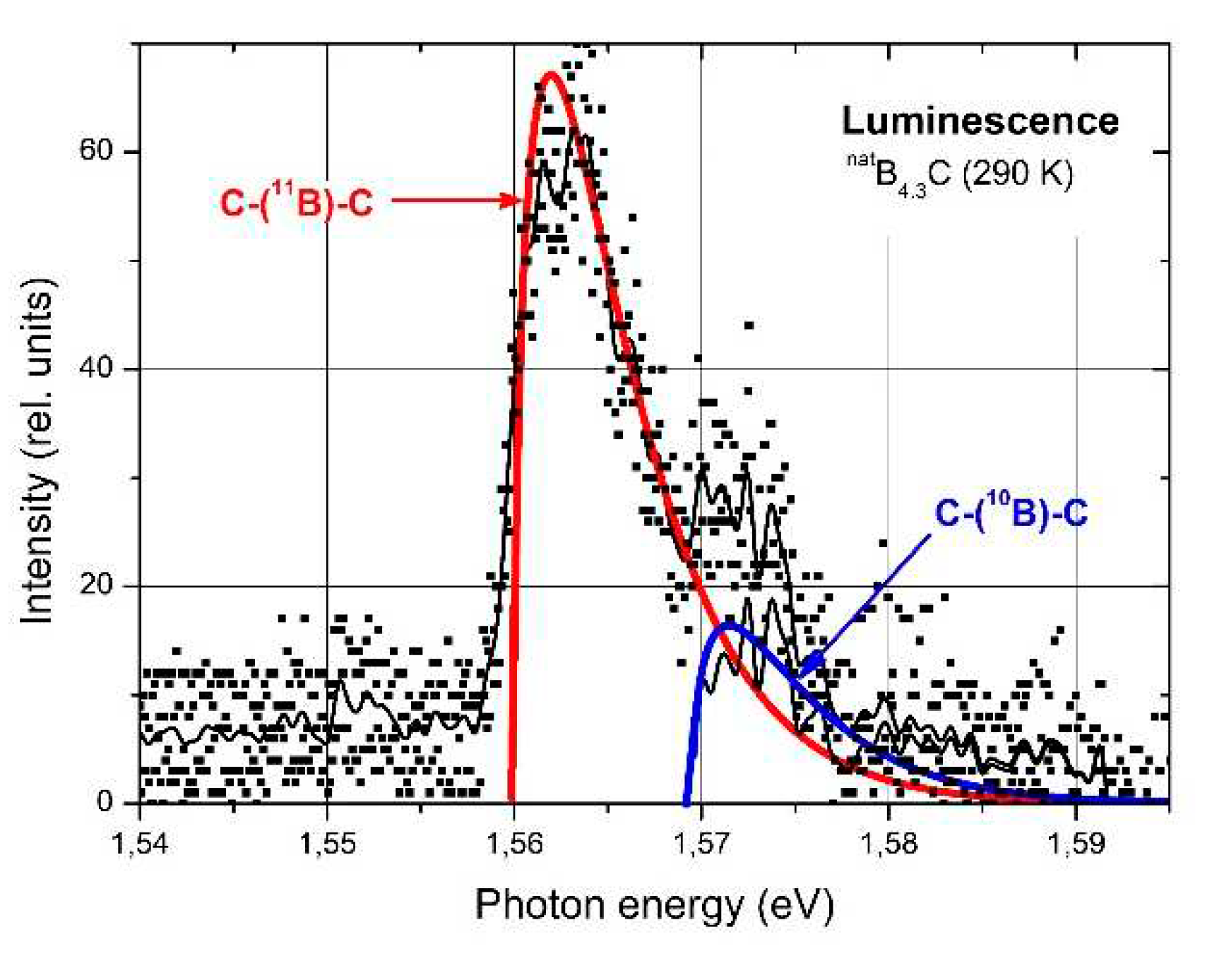
Figure 7.
Phase transition near the compound B8C.
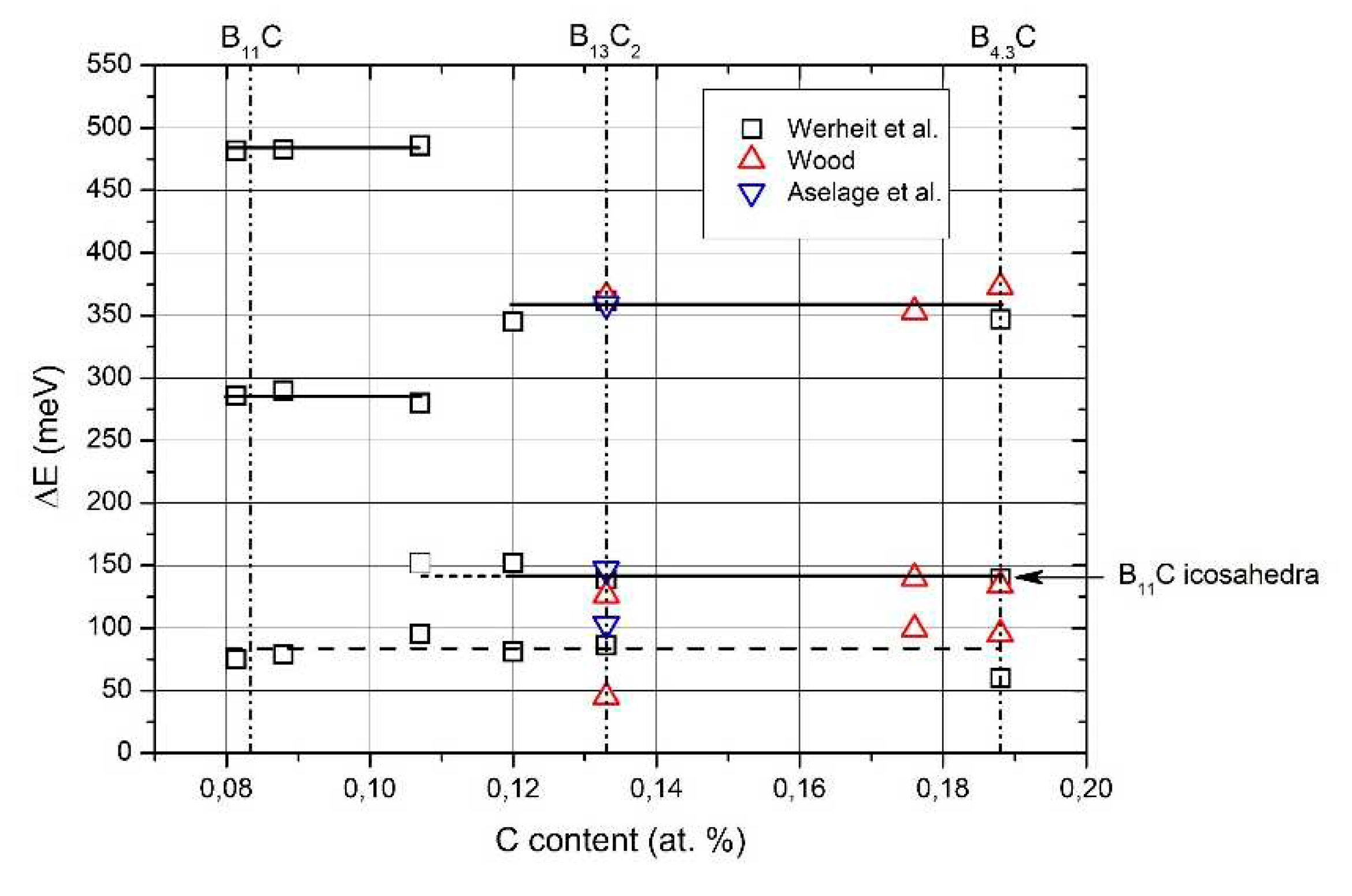
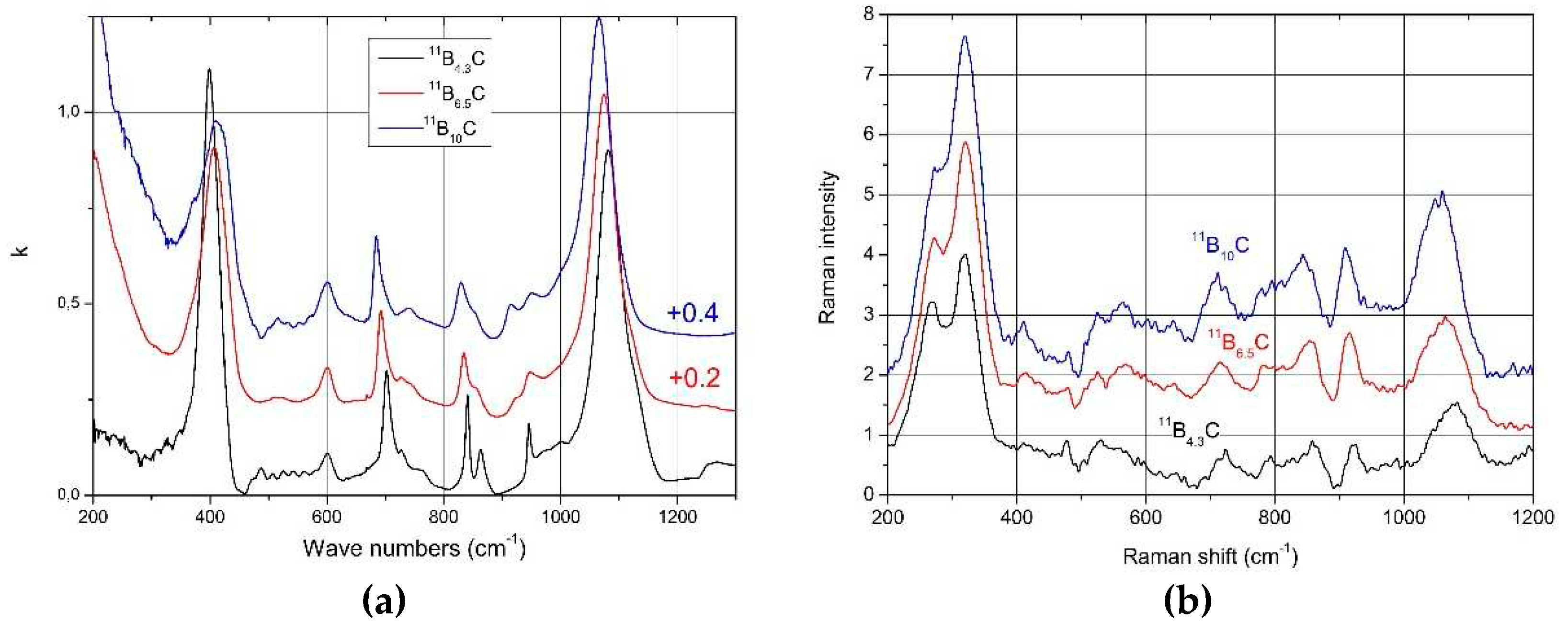
Figure 9.
a. IR-active phonon of 11BxC near 400 cm-1 (All fits were obtained using Origin Pro software); 9b. Phonon shift depending on C content and comparison between the reduction of phonon strength (components 1 and 2) and relation of the phonon strength of component #1 depending on C content compared with the relative share of CBC chains in the structure (obtained from the IR-active stretching vibration near 1600 cm-1 in Figure 2b).
Figure 9.
a. IR-active phonon of 11BxC near 400 cm-1 (All fits were obtained using Origin Pro software); 9b. Phonon shift depending on C content and comparison between the reduction of phonon strength (components 1 and 2) and relation of the phonon strength of component #1 depending on C content compared with the relative share of CBC chains in the structure (obtained from the IR-active stretching vibration near 1600 cm-1 in Figure 2b).
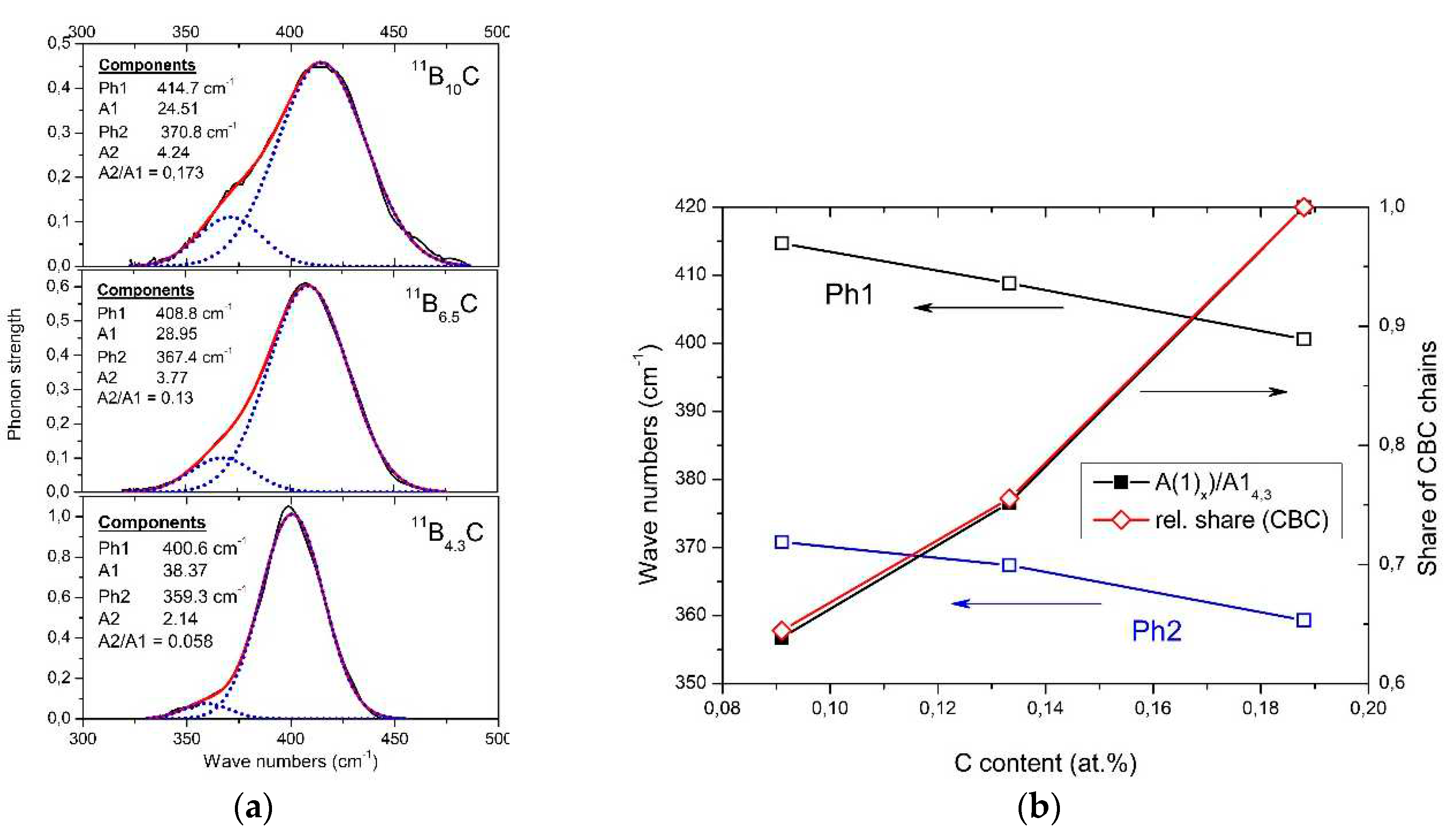
Figure 10.
IR-active phonons near 700 cm-1. a, 11BxC at 30 K; b, natBxC at 160 K, phonon-wavenumbers of natB4.3C and natB10.37C.
Figure 10.
IR-active phonons near 700 cm-1. a, 11BxC at 30 K; b, natBxC at 160 K, phonon-wavenumbers of natB4.3C and natB10.37C.
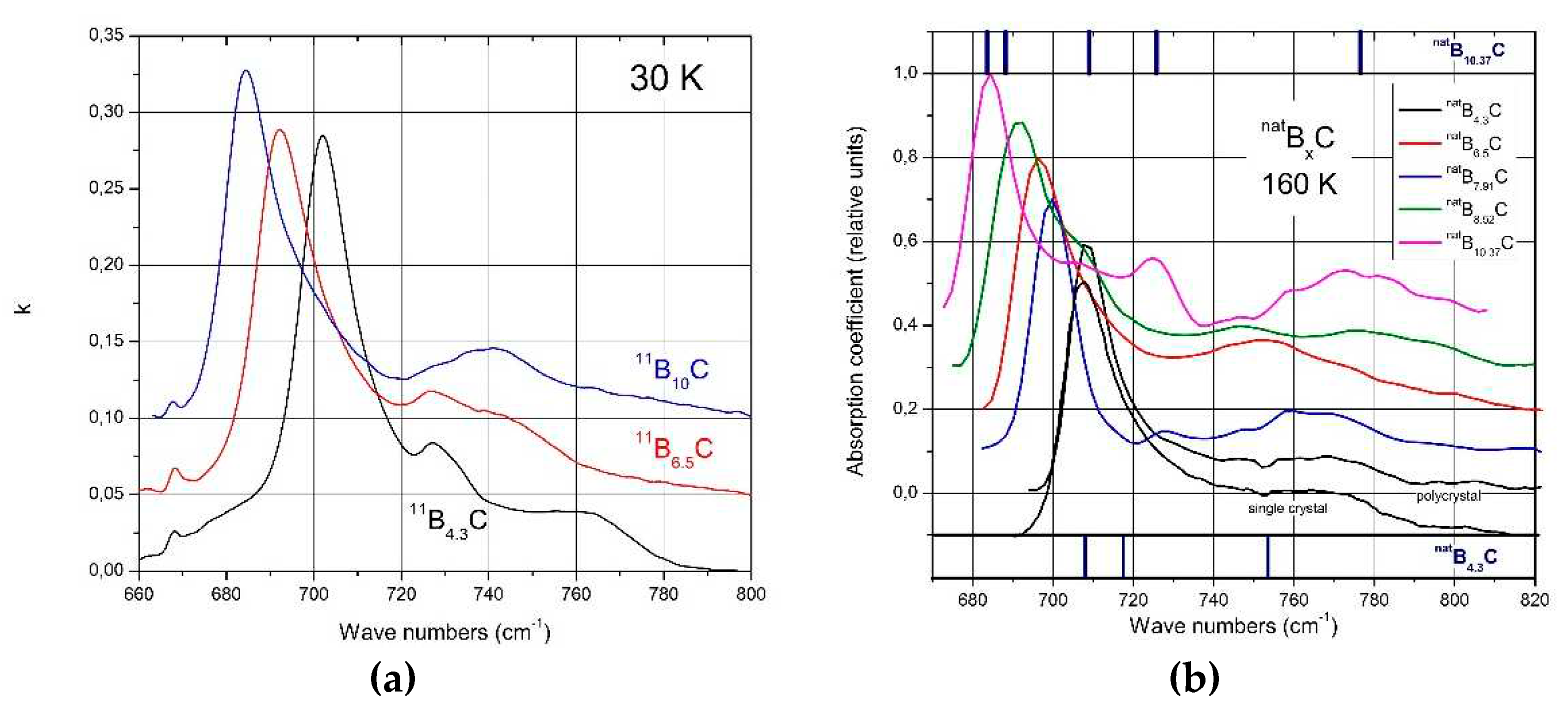
Figure 11.
IR-active phonons near 700 cm-1. a, fits obtained with OriginPro software. b., wave numbers of phonon components vs. C content. The results of the high-frequency components of B8.52C (C content 0,105) are rather uncertain.
Figure 11.
IR-active phonons near 700 cm-1. a, fits obtained with OriginPro software. b., wave numbers of phonon components vs. C content. The results of the high-frequency components of B8.52C (C content 0,105) are rather uncertain.
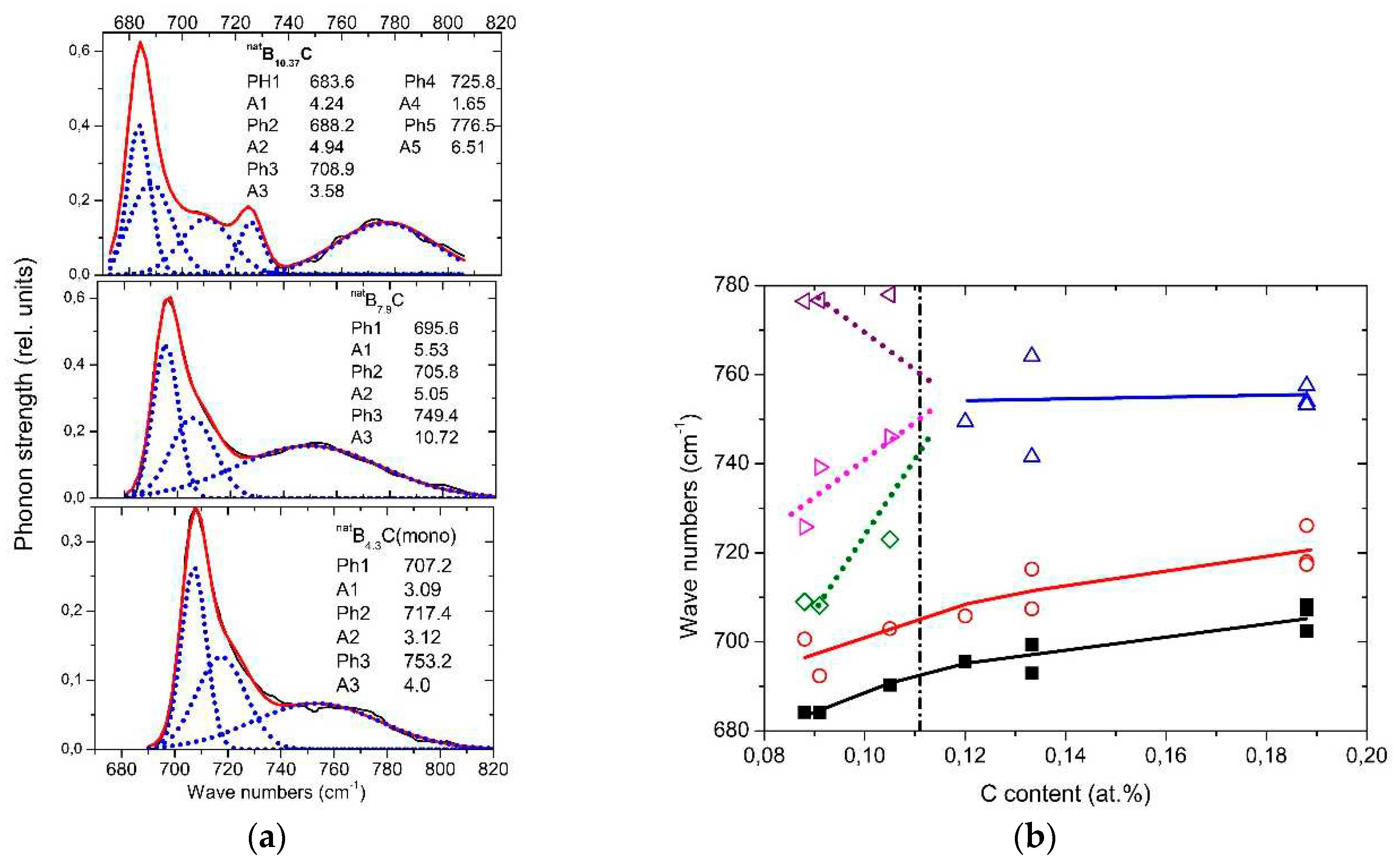
Figure 12.
IR-active phonons near 950 cm-1.
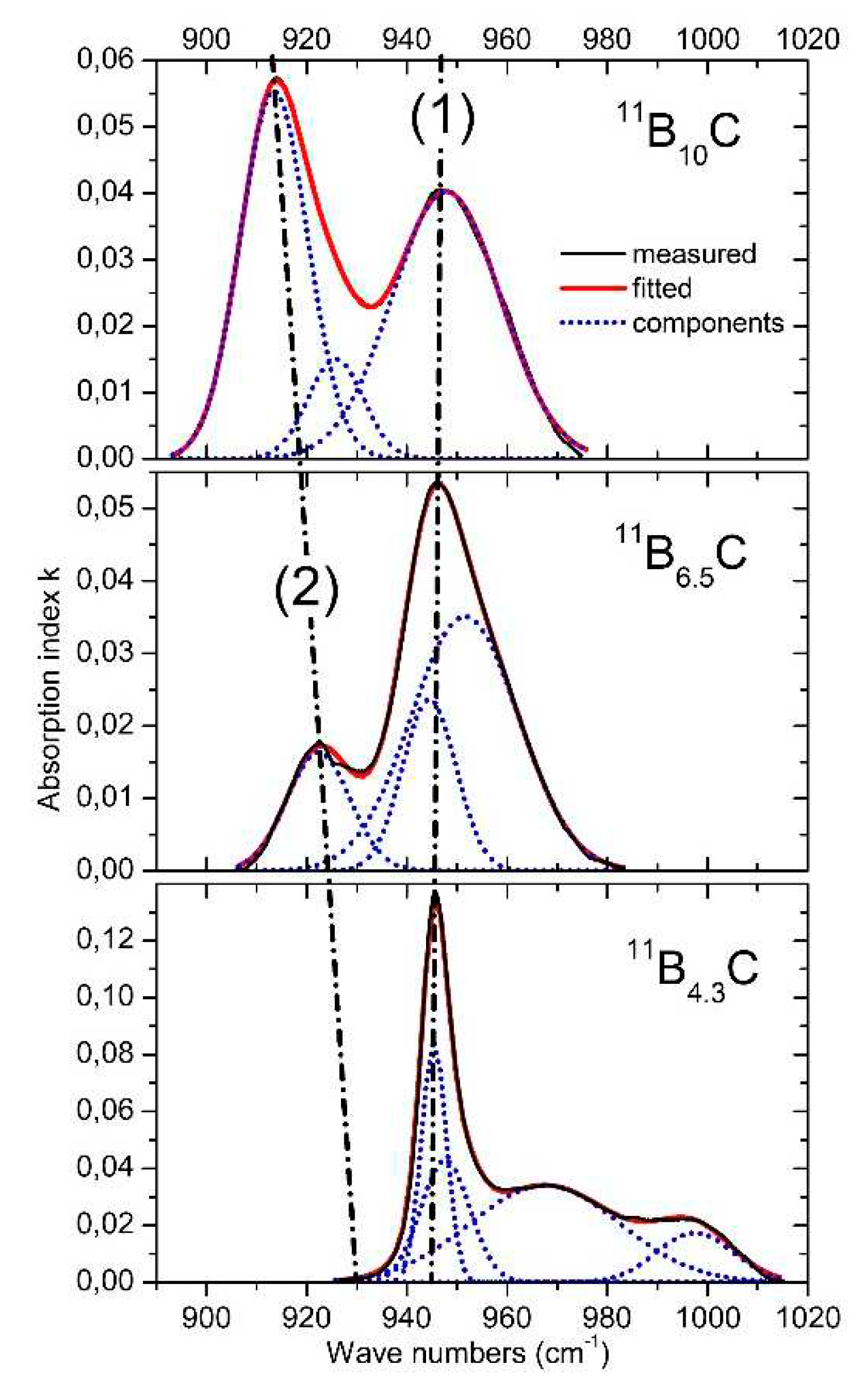
Figure 13.
IR-active phonons near 950 cm-1.
Figure 14.
Raman-active phonons of 11BxC at 270/320 cm-1.
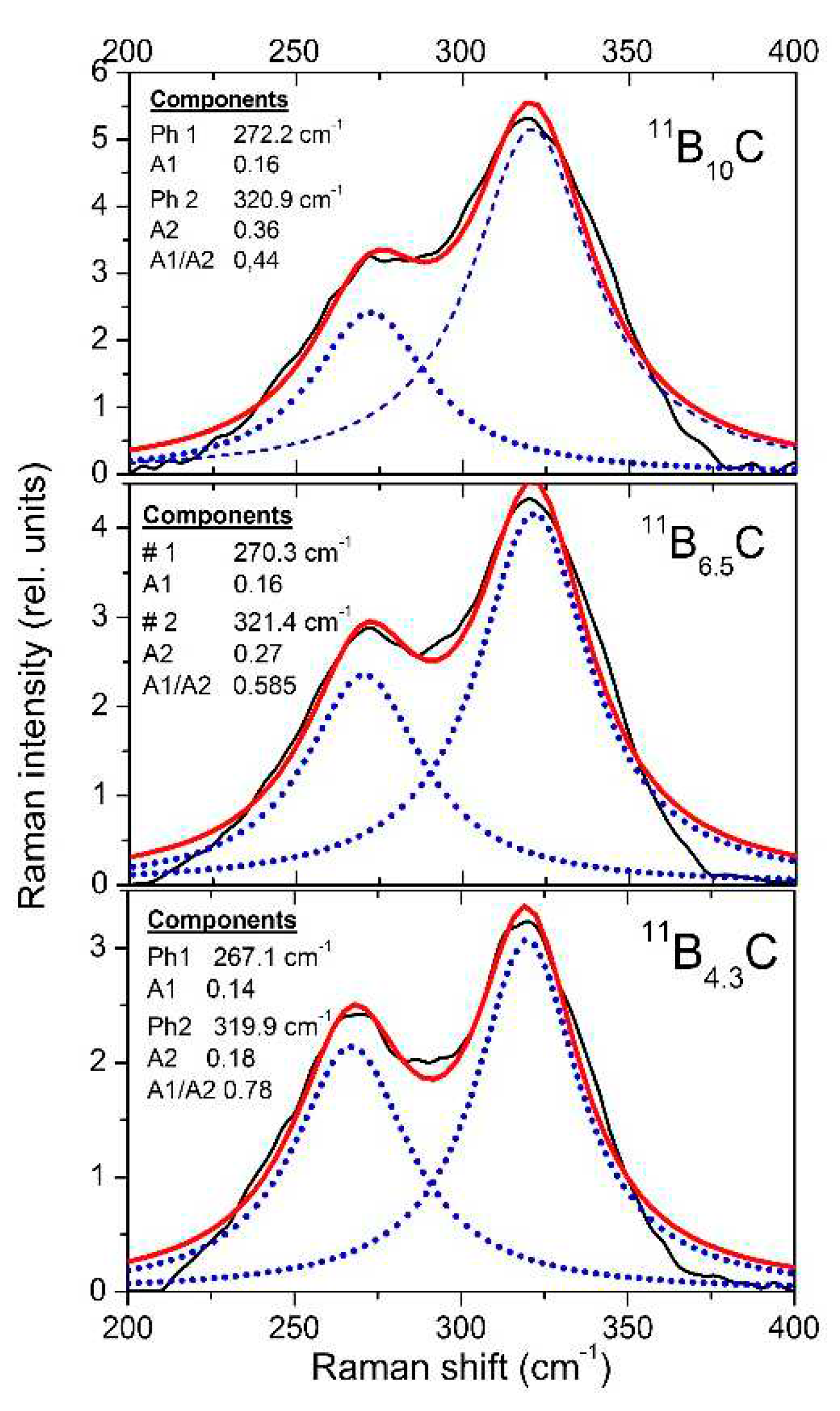
Figure 15.
Specific heat of B4.3C boron carbide vs. T. Black circles, measured; red dashed line, polynomial fit; blue, difference between measured data and fitted line; purple dotted line, 1st derivative.
Figure 15.
Specific heat of B4.3C boron carbide vs. T. Black circles, measured; red dashed line, polynomial fit; blue, difference between measured data and fitted line; purple dotted line, 1st derivative.
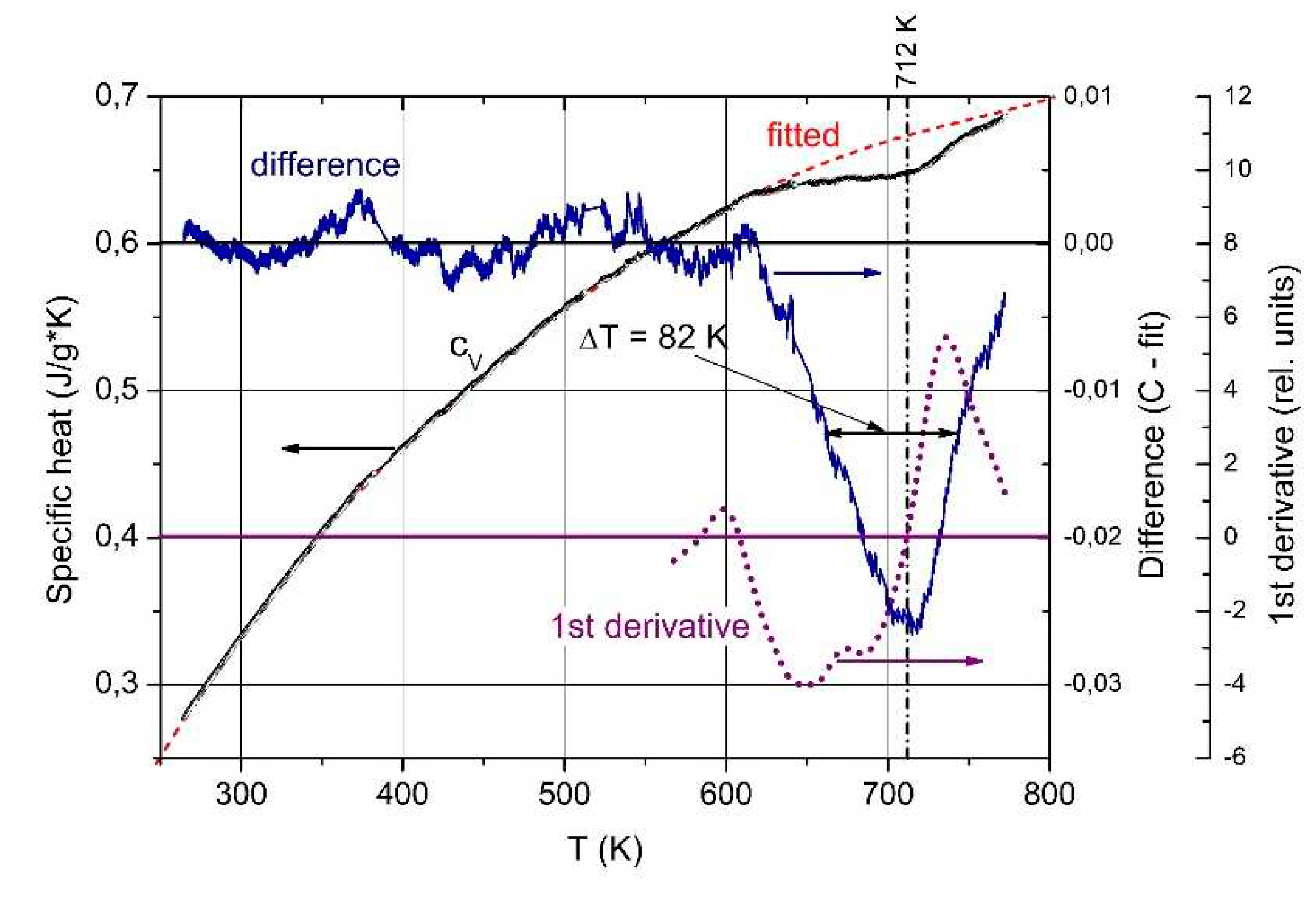
Figure 16.
IR-active phonon of natB4.3C boron carbide near 400 cm-1. a, absorption index k vs. wave number between 100 and 800 K; b, wave number and phonon strength of the components vs. T.
Figure 16.
IR-active phonon of natB4.3C boron carbide near 400 cm-1. a, absorption index k vs. wave number between 100 and 800 K; b, wave number and phonon strength of the components vs. T.
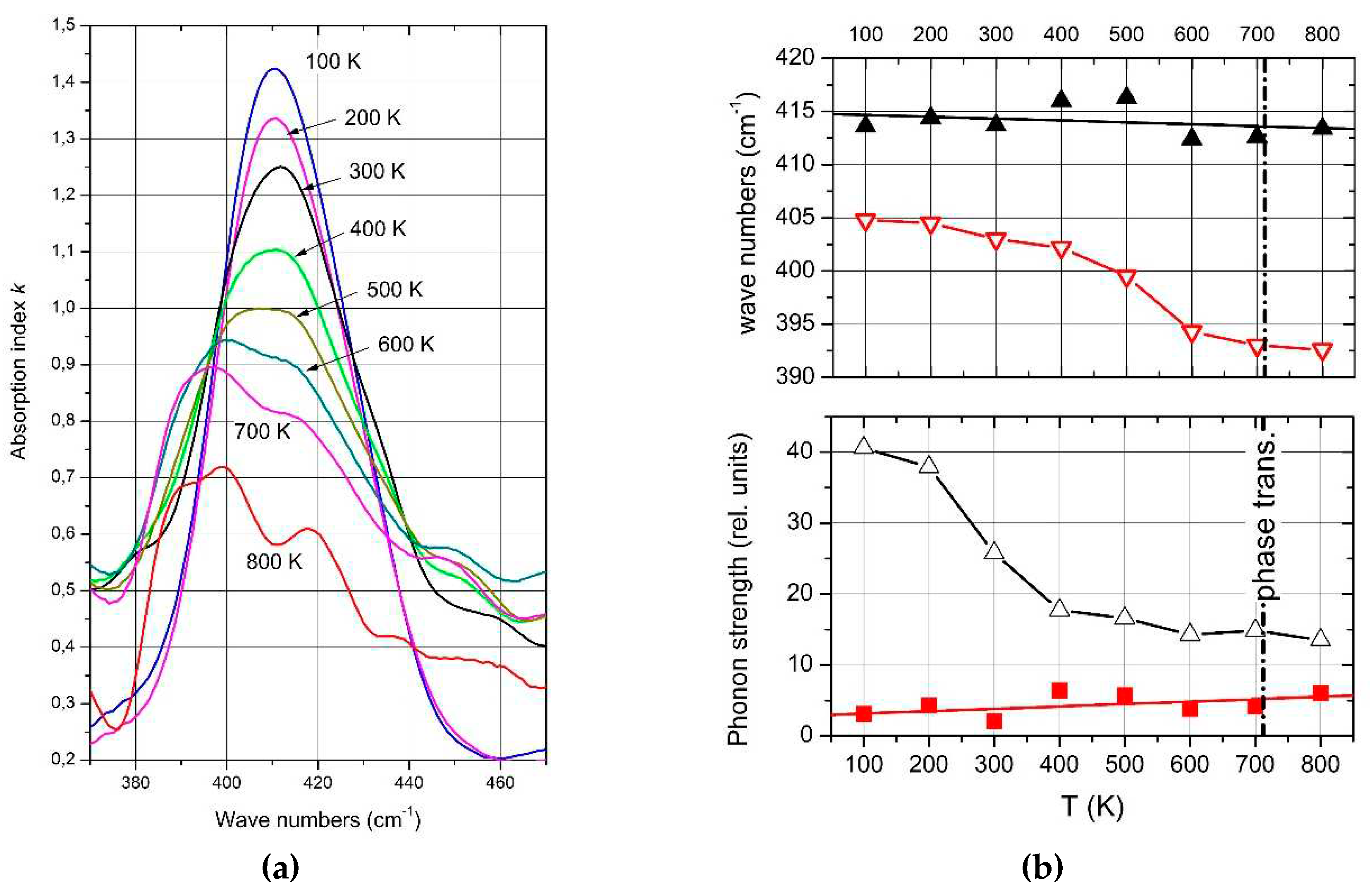
Figure 17.
Transient-light photos of a boron carbide B4.3C single crystal at various pressures [38].
Figure 17.
Transient-light photos of a boron carbide B4.3C single crystal at various pressures [38].

Figure 18.
Gap width of boron carbide depending on pressure. Black triangles, gap width, roughly estimated from the transient-light photos in Figure 13 [38]. Compared with experimental and theoretical results. Open diamonds [23,44,62,63,64]; pink filled diamond, ordered structure [64]; pink vertical bar, varying configurational disorder [65]; blue filled circles, pressure-dependence [38]. Experimental results: Blue open squares, ambient conditions [60,67]; dash-dotted lines, pressure-dependent decreasing gap [66].
Figure 19.
Electronic band scheme of B4.3C boron carbide [15] derived from optical and electrical measurements; DOS calculated by Ektarawong et al. (RNG model) [64] and Rasim et al. [27] for reference (data taken from diagram each). Vertical arrows show absorption (upward) and emission (downward) processes; the visible range is marked.
Figure 19.
Electronic band scheme of B4.3C boron carbide [15] derived from optical and electrical measurements; DOS calculated by Ektarawong et al. (RNG model) [64] and Rasim et al. [27] for reference (data taken from diagram each). Vertical arrows show absorption (upward) and emission (downward) processes; the visible range is marked.
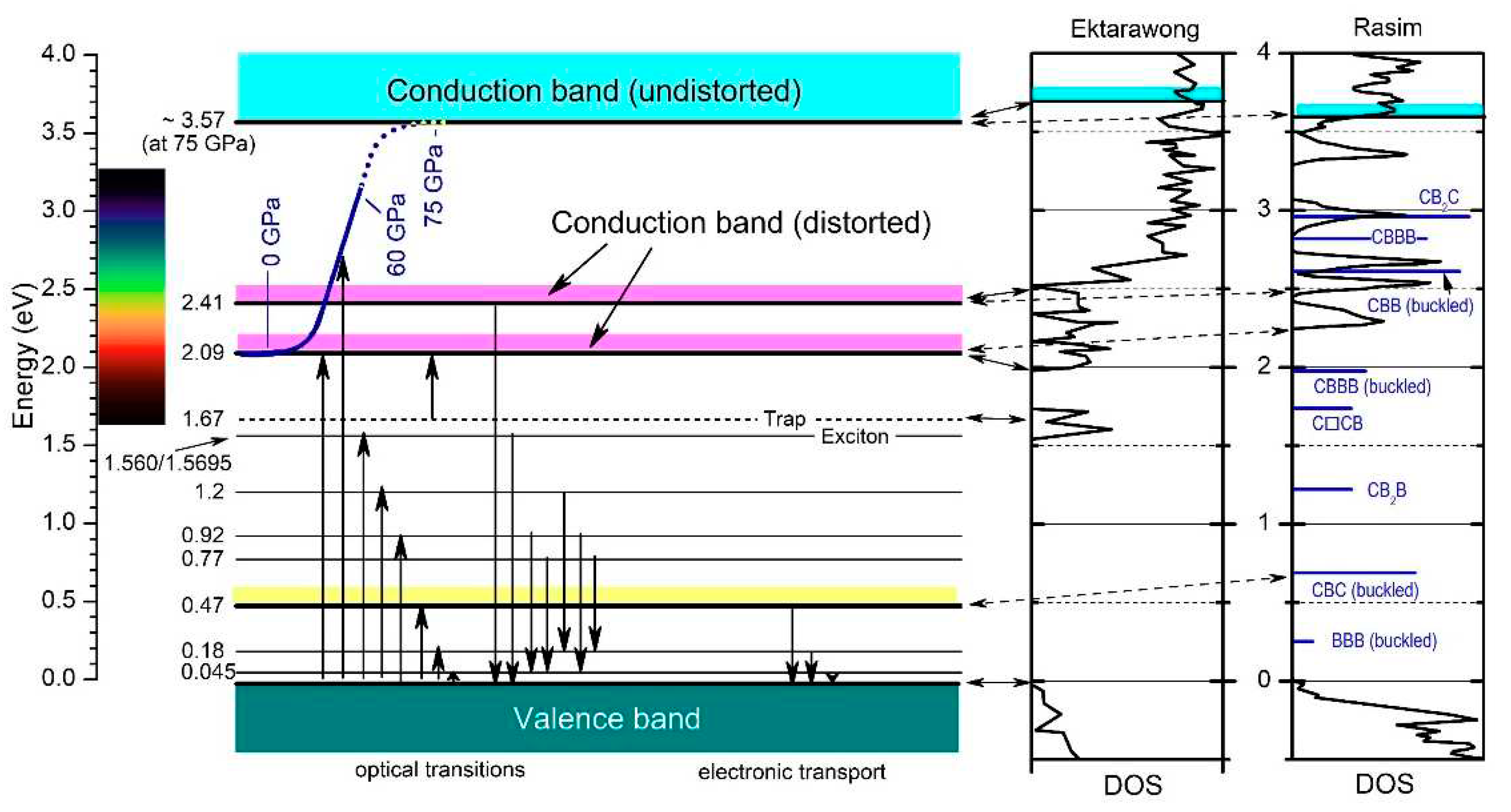
Figure 20.
IR phonon mode Grüneisen parameters below (▽) and above (△) the phase transition at 33.2 GPa. Insert, example of determining γ.
Figure 20.
IR phonon mode Grüneisen parameters below (▽) and above (△) the phase transition at 33.2 GPa. Insert, example of determining γ.
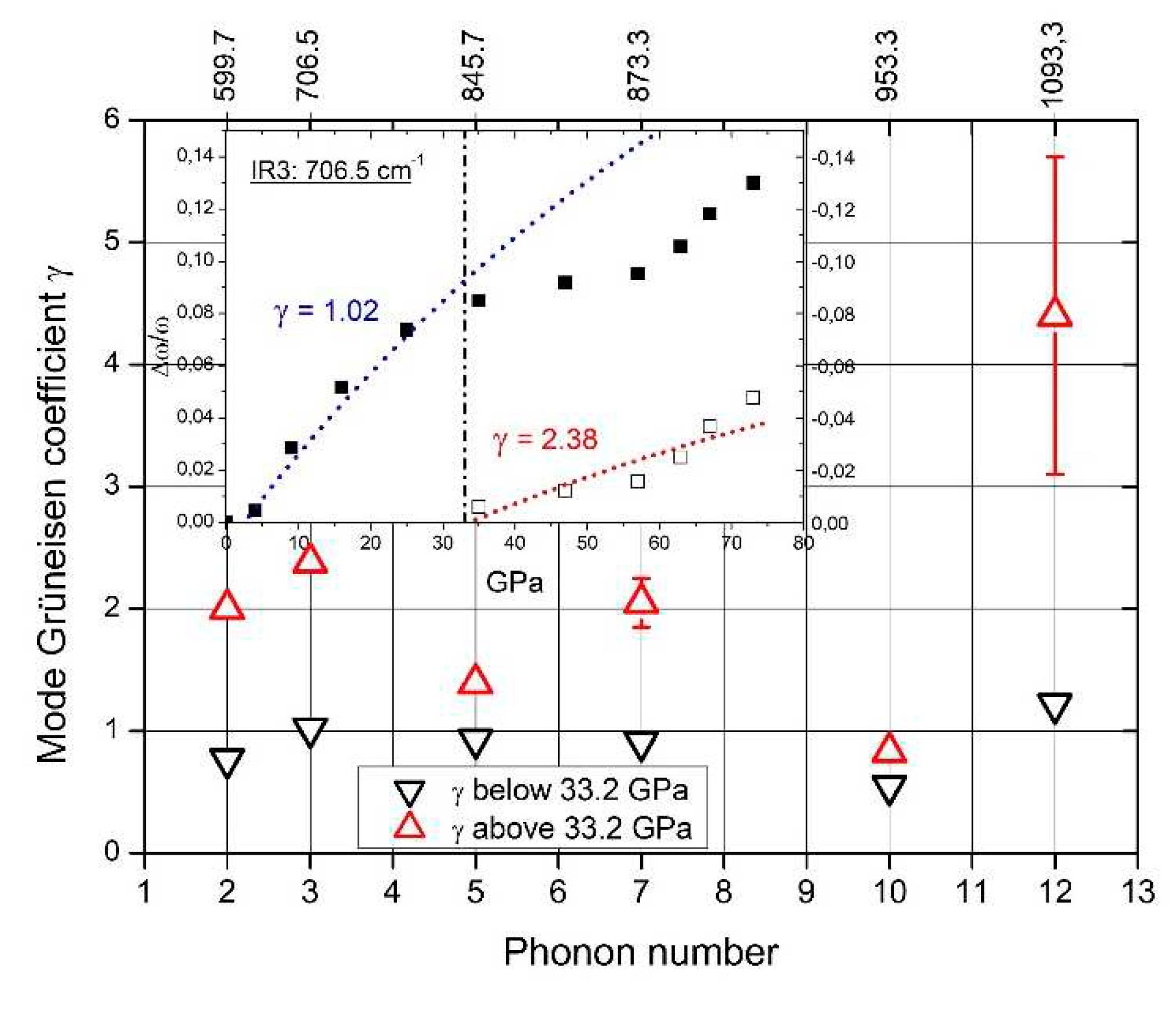
Figure 21.
Relative shift of phonon frequencies (left ordinate) and unit cell volume (right ordinate) vs. pressure, determining mode γ of typical IR-active phonons of B4.3C boron carbide [56]. Insert, FT-Raman spectrum of single-crystal B4.3C boron carbide.
Figure 21.
Relative shift of phonon frequencies (left ordinate) and unit cell volume (right ordinate) vs. pressure, determining mode γ of typical IR-active phonons of B4.3C boron carbide [56]. Insert, FT-Raman spectrum of single-crystal B4.3C boron carbide.
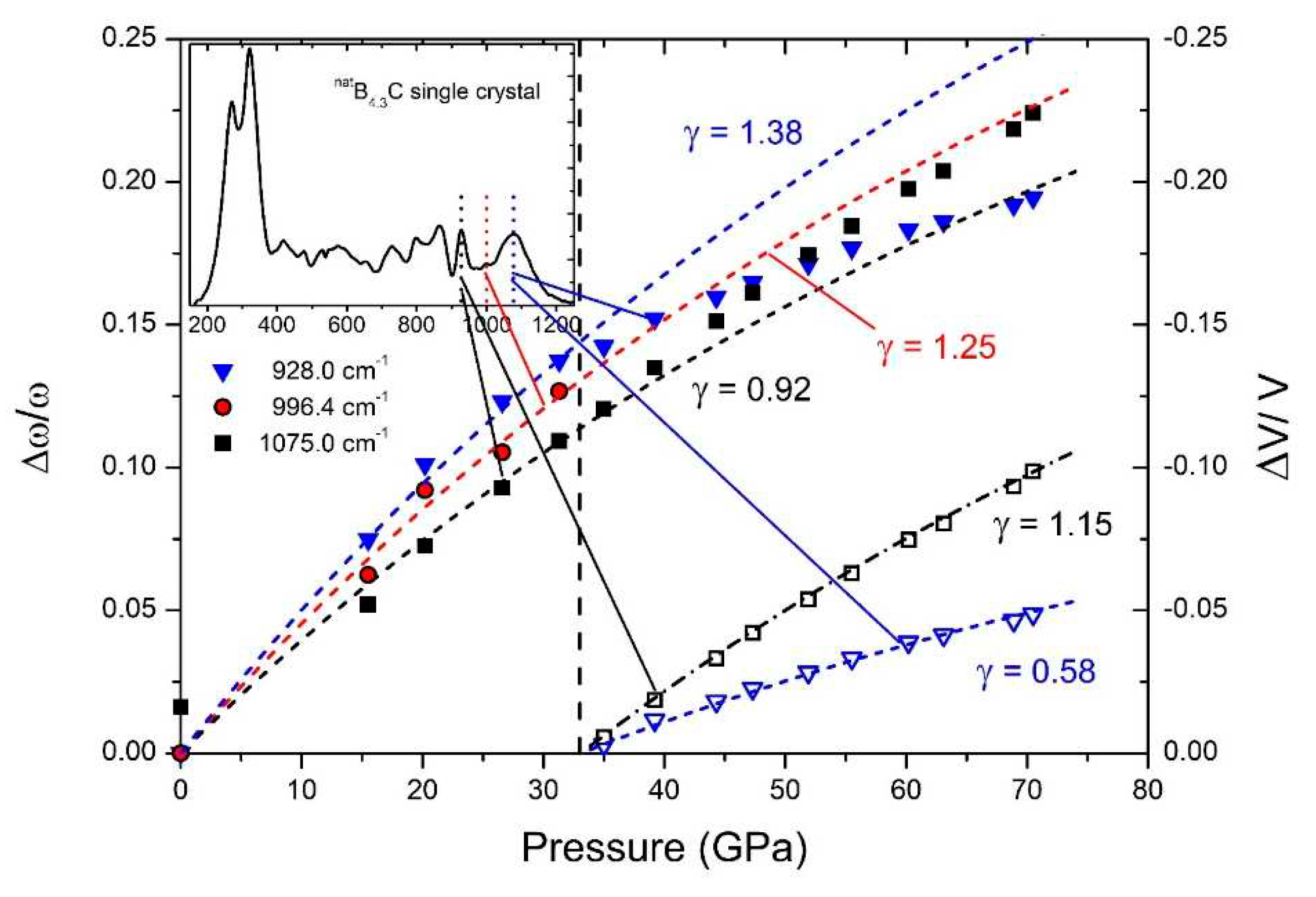
Figure 22.
Wave numbers of the Raman-active 270/320 cm-1 doublet and ratio of the phonon strengths of both components.
Figure 22.
Wave numbers of the Raman-active 270/320 cm-1 doublet and ratio of the phonon strengths of both components.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



