
Submitted:
01 September 2023
Posted:
04 September 2023
You are already at the latest version
Abstract
Retinitis Pigmentosa, defined more properly as rod-cone dystrophy, is the paradigm of diffuse inherited retinal dystrophies, one of the rare diseases with the highest prevalence in the worldwide population and one of the main causes of low vision in pediatric and elderly age groups. Advancement and understanding in molecular biology and gene editing technologies raised the interest in putting the basis for new therapeutic strategies in rare diseases. As a consequence, new possibilities for clinicians and patients are arising due to the feasibility of treating such a devastating disorder reducing its complications. The scope of this review is to focus on the pathomolecular mechanisms underlying RP better to understand the prospective for its treatment with innovative approaches.
Keywords:
Inherited Retinal Dystrophies
; Retinitis Pigmentosa
; therapeutic strategies
; cell therapies
1. Introduction
Retinitis pigmentosa (RP) is a relatively common inherited retinal disorder, with an estimated worldwide mean prevalence of 1:4000 people. It consists of progressive retinal degeneration due to the loss of photoreceptors leading to a severe visual impairment in the latest stages. RP may be inherited in an autosomal dominant, autosomal recessive or X-linked manner. Some digenic and mitochondrial forms have also been described. Actual RP prevalence is related to the geographic location of the reported study and, in consequence of this, it can be variable between 1:750 to 1:9000 individuals (1). Clinically, fundus examination usually shows diffuse dystrophy of the retinal pigment epithelium (RPE), waxy optic disc pallor, vessels narrowing and bone spicule pigmentation. Anyway, atypical forms with an unusual clinical picture may be found in clinical practice. The diagnosis may be easy in typical cases, but it may be challenging in atypical presentations as the disease may presents overlapping features with different retinal disorders. Additionally, RP is associated with an increased risk of other ocular complications, such as cataract and cystoid macular oedema (CME), which may cause additional visual disturbances (2) (3) (4). The pathophysiology of RP with such complications that may affect vision often leads to significant visual impairment at a young age, which can also have an impact on a patient’s physical and mental well-being. Researchers have identified mutations in over 100 genes that contribute to developing non-syndromic RP. Syndromic RP is even more complex due to molecular pathways acting in multiple organs. In the past, RP was thought to be untreatable, but recent medical advancements, such as genetic therapies, offer promising possibilities for slowing down or stopping, the degeneration of photoreceptors and potentially even restoring some level of visual function (5). Due to our enhanced comprehension of the cellular mechanisms and genetic factors associated with RP, as well as the eye’s immune-privileged nature, gene therapy has emerged as a highly promising treatment option for RP (6). Different studies on animal models have shown the potential benefit of gene therapy for RPE65-associated inherited retinal diseases. As a result, human clinical trials for gene therapy have been initiated, aiming to explore the potential benefits of this novel treatment approach (7) (8) (9) (10). The positive results in terms of both safety profiles and clinical endpoints in these trials have led to the approval of Voretigene Neparvovec as the first FDA-approved gene therapy for patients with RPE65-associated retinal dystrophies, now commercially available with the name of Luxturna® (11). In this review, we aim to discuss and highlight the different therapeutic approaches and the current clinical management of RP focusing on the alterations in the molecular pathways involved in the development of RP.
2. Pathomolecular Mechanisms of Retinitis Pigmentosa
Generally speaking, the pathomolecular mechanisms of RP involve primarily genetic mutations that disrupt the normal functioning of the retina and the retinal pigment epithelium (RPE) (12) through specific and common pathways.
These genes are involved in different cellular processes within the retina, including phototransduction, visual cycle, photoreceptor structure, and maintenance of retinal integrity. The specific genes and mutations define the precise mechanisms behind RP in individual cases (14). Nevertheless, since the genes activate different pathways and multiple mechanisms often converging to a more aspecific process, for better clarity we’ll describe firstly the main genes (summarized in Table 1), and their role, that are commonly involved in the pathogenesis of RP; this part will be followed by a discussion on the more general processes leading to retinal degeneration.
2.1.1. Main genes involved in RP
- Rhodopsin (RHO): mutations in the RHO gene are a common cause of autosomal dominant retinitis pigmentosa (adRP) (15) Rhodopsin is a protein found in the rod photoreceptor cells of the retina, and it plays a critical role in the phototransduction pathway, which converts light signals into electrical signals that can be interpreted by the brain. Many kinds of RHO gene mutations damage the structure or function of rhodopsin, leading to the disruption and degeneration of rod photoreceptor cells. Over 150 types of mutations of the RHO gene have been described (16) (17). Most cases present point mutations that determine the substitution of an amino acid with consequent alteration of the protein’s structure or function. In addition to these mutations others involving abnormalities in the protein’s folding or trafficking were also described. Mutations in the rhodopsin gene which lead to the development of autosomal dominant forms of retinitis pigmentosa are divided into three different classes (18), distinguished by the dysfunction of rhodopsin and the nature of its accumulation in cell culture. In class 1 mutations, the photopigment remains functional, and its bond with 11-cis-retinal remains intact, while the accumulation of protein in embryonic cell culture happens specifically on the cytoplasmic membrane. Class 2 mutations cause damage to the composition of the photopigment and the buildup of faulty protein in the endoplasmic reticulum. Class 3 encompasses mutations that result in the production of hyperphosphorylated rhodopsin, which exhibits a strong association with arrestin. The consequent rhodopsin-arrestin complex disturbs the structure of the endosomal compartment and impairs endocyte function. All these mutations have a strong impact on rhodopsin function as they can impair the light-sensitive ability of the protein or disrupt its signalling cascade within the photoreceptor cells (19) . These abnormalities can lead to defects in phototransduction, reduced sensitivity to light, and eventually, the death of rod photoreceptor cells.
- Peripherin/RDS (PRPH2): Mutations in the PRPH2 gene can cause autosomal dominant RP. The peripherin/RDS protein is involved in the structural integrity and function of photoreceptor outer segments. Specifically, PRPH2 is a transmembrane protein that is mainly expressed in rod and cone photoreceptor cells. It plays a crucial role in the structural integrity and organization of the photoreceptor outer segments, which are responsible for capturing and processing light signals. Mutations occurring in the PRPH2 gene can give rise to diverse structural anomalies in the peripherin 2 protein (20) . These abnormalities can impact crucial aspects such as protein folding, stability, and interactions with other proteins. The resultant disruption in the peripherin 2 function can hinder the formation of photoreceptor outer segments, consequently compromising the normal functionality of the photoreceptor cells. One common type of mutation in PRPH2 is the missense mutation which can affect the protein's folding, stability, and ability to interact with other proteins in the photoreceptor cells (21). Another type of common mutation is the frameshift mutation, mainly associated with anomalies in the protein's structure and function (20). Frameshift mutations often result in a truncated or non-functional Peripherin/RDS protein. Defective Peripherin/RDS protein can lead to mislocalization (22), aggregation, or degradation of the protein, affecting the integrity and function of the outer segments. This disruption ultimately results in the progressive degeneration of the photoreceptor cells and the characteristic symptoms of retinitis pigmentosa, such as night blindness, peripheral vision loss, and eventually central vision impairment.
- Cyclic Nucleotide-Gated (CNG) Channels. Mutations in genes encoding the CNG channels, such as CNGA1 and CNGB1, are associated with autosomal recessive RP (23). These channels are located in the outer segment of rod and cone photoreceptor cells and they are involved in the regulation of ion influx in response to light stimulation. They are responsible for the regulation of intracellular calcium and sodium ions, which are essential for phototransduction (the process by which light signals are converted into electrical signals in the retina thanks to hyperpolarization/depolarization phenomena). Mutations in genes encoding CNG channels can impair the normal function of these channels, disrupting the phototransduction process and leading to RP (24). Mutations in the CNGB1 and CNGA1 genes, which encode subunits of the CNG channels, have been associated with autosomal recessive RP (25). Mutations in the GNAT2 gene, which encodes the transducin alpha-subunit involved in CNG channel regulation, have also been linked to autosomal dominant RP. Impaired CNG channel function leads to abnormalities in the phototransduction process, where the conversion of light stimuli into electrical signals is disrupted (26). This alteration can result in reduced sensitivity to light, decreased visual acuity, and progressive vision loss, which are characteristic symptoms of RP. Additionally, dysfunctional CNG channels can lead to cellular stress, and oxidative damage, and ultimately trigger photoreceptor cell death (27). The loss of photoreceptor cells further contributes to the degeneration of the retina and the progression of RP.
- Retinal Pigment Epithelium-Specific 65 kDa Protein (RPE65). Mutations in the RPE65 gene are associated with autosomal recessive RP forms (28). The RPE65 gene encodes a protein called retinoid isomerohydrolase, which is primarily expressed in the retinal pigment epithelium (RPE) cells. RPE65 is involved in the visual cycle, a process that regenerates the visual pigment rhodopsin in photoreceptor cells. It plays a crucial role in converting all-trans-retinol to 11-cis-retinal, which is essential for the proper functioning of photoreceptor cells. Mutations in the RPE65 gene result in a loss or dysfunction of the RPE65 protein, disrupting the visual cycle and impairing the regeneration of 11-cis-retinal (29). As a consequence, there is a decreased availability of 11-cis-retinal, leading to compromised phototransduction and eventual degeneration of photoreceptor cells (30) (31). Mutations in the RPE65 gene are also associated with a severe form of retinitis pigmentosa known as Leber congenital amaurosis (LCA) or severe early childhood-onset retinal dystrophy (SECORD).
- Retinitis Pigmentosa GTPase Regulator (RPGR). Mutations in the RPGR gene are a major cause of X-linked RP (XLRP), which primarily affects males as the RPGR gene is located on the X chromosome and it is involved in the structure and function of the photoreceptor connecting cilium (32) (33). The RPGR protein is indeed predominantly localized in the connecting cilium and outer segment of photoreceptor cells in the retina. These cellular structures play critical roles in the phototransduction cascade and the maintenance of normal vision. Mutations in the RPGR gene can affect the normal function of the RPGR protein, leading to retinal degeneration in XLRP (34). The specific pathogenic mechanisms underlying RPGR-related retinal degeneration are not fully understood. However, it is believed that the mutations result in impaired ciliary transport, altered protein-protein interactions, or disrupted signalling pathways, ultimately leading to photoreceptor cell death and vision loss (32) (35).
- Cone-Rod Homeobox Protein (CRX): Mutations in the CRX gene are associated with autosomal dominant RP form (36). The Cone-Rod Homeobox Protein is a transcription factor that plays a crucial role in the development and function of photoreceptor cells in the retina. CRX is primarily expressed in cone and rod photoreceptor cells of the retina where it regulates the expression of genes involved in photoreceptor development, differentiation, and maintenance. CRX is essential for the proper formation and function of these specialized cells, which are responsible for capturing and processing light signals (37). CRX regulates the expression of genes encoding various photoreceptor-specific proteins, including opsins (light-sensitive pigments), transducin, and other important components of the phototransduction pathway. It helps establish the unique characteristics and functions of cone and rod photoreceptor cells, ensuring their proper light-sensitive abilities (38). Mutations in the CRX gene can disrupt the normal function of the CRX protein, leading to impaired development and function of photoreceptor cells (37). This can result in the degeneration of cones and rods with different specific effects depending on the kind of CRX mutation, resulting in isolated cone dysfunction to more generalized cone-rod dystrophy (39).
- Usher Syndrome Genes. It has been demonstrated that some forms of RP are associated with Usher syndrome (40), which involves both hearing loss and vision impairment. Genes associated with Usher syndrome, such as USH2A, MYO7A, CLRN1 and CDH23, can cause RP in addition to other symptoms. It is a heterogeneous condition with several genes implicated in its development. The most commonly associated genes with Usher syndrome and RP include: Mutations in the MYO7A gene account for the majority of Usher syndrome type 1(USH1) cases (41). MYO7A encodes the protein myosin VIIA, which is involved in the structure and function of hair cells in the inner ear and the development and maintenance of photoreceptor cells in the retina. It plays an important role in the renewal of the outer photoreceptor discs, in the distribution and migration of retinal pigment epithelium (RPE) melanosomes and phagosomes and the regulation of opsin transport in retinal photoreceptors. Mutations in the USH1C gene are also associated with Usher syndrome type 1. The USH1C gene encodes harmony, a scaffolding protein involved in the organization of hair cell stereocilia and the synaptic connections in the retina (41). Mutations in the USH2A gene are the most common cause of Usher syndrome type 2 (USH2). The USH2A gene encodes usherin, a protein involved in the maintenance of the structure and function of the photoreceptor cells and the hair cells of the inner ear (42) Mutations in the GPR98 gene, also known as ADGRV1 (Adhesion G Protein-Coupled Receptor V1) are associated with Usher syndrome type 2. The GPR98 gene encodes the protein G protein-coupled receptor 98, which is involved in the development and function of sensory cells in the inner ear and the retina. Mutations in the CLRN1 gene are associated with Usher syndrome type 3. The CLRN1 gene encodes clarin-1, a protein found in the hair cells of the inner ear and the photoreceptor cells of the retina (43) which seems to play an important role in the development and homeostasis as a regulatory element for the synapses within the retina.
2.1.2. Mechanisms involved in RP
As discussed above, the pathogenesis of RP involves a complex interplay of genetic, biochemical (44) and cellular mechanisms that ultimately lead to the degeneration of photoreceptor cells in the retina (45). Although the exact pathogenesis can change depending on the specific genetic mutations that act as initial triggers, in the next section we’ll describe an overview of more general and common processes that contribute to the development and progression of RP, also briefly illustrated in Figure 1.
Photoreceptor Cell Dysfunction
One of the first pathogenetic mechanism is the Photoreceptor Cell Dysfunction. RP primarily affects the photoreceptor cells in the retina, where mutations in genes involved in the visual cycle, phototransduction, and photoreceptor structure can lead to impaired functioning of these cells affecting the structure and function of photoreceptor proteins (13).
Protein misfolding and aggregation
Another mechanism that contributes to the development of retinitis pigmentosa is represented by protein misfolding and aggregation (46). In this case, mutations in specific genes can lead to the production of misfolded or unstable proteins. Abnormal proteins can accumulate within photoreceptor cells and trigger cellular stress responses, including endoplasmic reticulum (ER) stress and unfolded protein response (UPR). The accumulation of misfolded proteins and activation of stress pathways can contribute to photoreceptor cell dysfunction and degeneration (47).
Increase in Oxidative Stress levels
Another important pathomolecular mechanism is the increase of Oxidative Stress levels, encouraged by the damage to the normal cellular processes in the retina.
Accumulation of reactive oxygen species (ROS) can damage cellular components, including DNA, proteins, and lipids. This oxidative stress can further contribute to the degeneration of photoreceptor cells in RP (48). In RP, oxidative stress occurs due to multiple factors related to the degenerative processes in the retina. For example, in RP, there is evidence of reduced antioxidant capacity in the retina, leading to an imbalance between ROS production and antioxidant defence (Impaired Antioxidant Defense). This imbalance results in the accumulation of ROS, which can damage cellular components, including photoreceptor cells (49). The consequences of oxidative stress in RP contribute to the ongoing degeneration of photoreceptor cells, exacerbating the visual impairment experienced by individuals with the condition (50). Strategies aimed at reducing oxidative stress and enhancing the antioxidant defence system have been investigated as potential therapeutic approaches for RP. These include the use of antioxidants, such as vitamins C and E, and other compounds that can mitigate ROS-induced damage (51) (52).
Mitochondrial dysfunction
In the context of retinitis pigmentosa another important pathogenetic mechanism is mitochondrial dysfunction as it contributes to the disease progression and degeneration of photoreceptor cells in the retina (53).
Mutations in genes associated with mitochondrial function, such as those encoding mitochondrial proteins or involved in mitochondrial DNA maintenance, can lead to mitochondrial dysfunction in RP. Impaired energy production and increased oxidative stress associated with dysfunctional mitochondria can contribute to photoreceptor cell death (54). Mitochondria are responsible for generating energy in the form of adenosine triphosphate (ATP) through oxidative phosphorylation and photoreceptor cells have high energy demands due to their role in capturing and processing light signals. Mitochondrial dysfunction in RP can lead to impaired ATP production, resulting in energy deficits that can compromise the survival and function of photoreceptor cells (55). Moreover, mitochondrial dysfunction can contribute to an imbalance between the production and scavenging of reactive oxygen species, as ROS are byproducts of mitochondrial metabolism and excessive ROS production due to mitochondrial dysfunction can lead to oxidative stress, causing damage to cellular components, including photoreceptor cells (56). Mitochondria are also involved in regulating calcium homeostasis in cells. Disruption of calcium signalling due to mitochondrial dysfunction can impact various cellular processes, including phototransduction and cell survival (57). Dysregulated calcium levels can trigger apoptotic pathways and contribute to the degeneration of photoreceptor cells in RP (58).
Mitochondria play a critical role in apoptosis and their dysfunction can trigger the release of pro-apoptotic factors, such as cytochrome c, leading to the activation of apoptotic pathways and the subsequent death of photoreceptor cells (53). Finally, mitochondrial dysfunction can result in the accumulation of toxic reactive metabolites, such as reactive aldehydes and lipid peroxidation products. These metabolites can damage cellular components, including lipids, proteins, and DNA, further contributing to the degeneration of photoreceptor cells in RP (53). The exact mechanisms underlying mitochondrial dysfunction in RP can vary depending on the specific genetic mutations involved. Several genes associated with RP are known to affect mitochondrial function directly or indirectly (13). Understanding the specific mitochondrial defects and their impact on retinal cells is essential for developing targeted therapeutic strategies to mitigate mitochondrial dysfunction and potentially slow down the progression of RP (59).
Apoptosis
Apoptosis represents another pathogenetic mechanism underlying RP (60). Mutations in genes such as Fas ligand, c-Fos, and NRL have been implicated in regulating apoptosis in photoreceptor cells. Dysregulation of these genes can lead to increased cell death and accelerate the progression of RP (61). The principal mechanism underlying the activation of the apoptosis process in retinitis pigmentosa is the photoreceptor cell Stress. As described before photoreceptor cell stress can arise from factors such as oxidative stress, mitochondrial dysfunction, calcium dysregulation, and protein misfolding underlying molecular defects, which can activate apoptotic pathways (62). Classic apoptotic pathways are activated by cellular stress in RP involving the activation of pro-apoptotic proteins and the release of cytochrome c from the mitochondria leading to caspases finally activation (60). The apoptotic process in RP progresses over time, leading to the progressive loss of photoreceptor cells in the retina. Initially, the rod photoreceptor cells are more severely affected, leading to night blindness and peripheral vision loss. Later, cone photoreceptor cells may also undergo apoptosis, resulting in further visual impairment, including central vision loss and colour vision defects.
Retinal Remodeling
Another remarkable mechanism, characteristic of retinitis pigmentosa, is called retinal remodelling. Retinal remodelling is a phenomenon that refers to the structural and functional changes that take place in the retina in response to the progressive degeneration of photoreceptor cells (63). These alterations include the synaptic connections and gene expression profiles of various retinal cell types, contributing to the overall dysfunction of the retina. The pathogenetic mechanism underlying retinal remodelling can be classified into 4 different kinds of alterations, involving different kinds of retinal cells (64):
- Neuronal Rearrangement. As photoreceptor cells degenerate, there is a reorganization of the remaining retinal neurons, including bipolar cells, horizontal cells, and amacrine cells. These neurons undergo structural changes and establish new connections with each other to compensate for the loss of photoreceptor input (65).
- Bipolar Cell Dystrophy: Bipolar cells, the second-order neurons in the visual pathway, also undergo structural and functional changes in RP. They may exhibit abnormal dendritic sprouting or retraction, leading to the formation of ectopic synapses. These changes can result in altered signal processing and contribute to visual abnormalities in RP (66).
- Müller Cell Gliosis: Müller cells are the major glial cells in the retina and play a crucial role in maintaining retinal homeostasis. In response to photoreceptor cell degeneration, Müller cells undergo gliotic changes, becoming activated and hypertrophic (67). This gliosis involves changes in gene expression, increased production of glial fibrillary acidic protein (GFAP), and alterations in their structural morphology. Müller cell gliosis can have both protective and detrimental effects on retinal function and can influence the survival and function of remaining retinal neurons (68).
- Synaptic Remodeling: In RP, there is a reorganization of synaptic connections in the retina. As photoreceptor cells degenerate, the synaptic contacts between photoreceptor cells and downstream neurons, such as bipolar cells and horizontal cells, change. New synaptic connections may form between bipolar cells and surviving cones or between bipolar cells and other retinal neurons. This synaptic remodelling can lead to altered signal processing and contribute to the rewiring of the retinal circuitry (69).
Retinal remodelling in RP has severe functional consequences. The rewiring of the retinal circuitry can enable surviving neurons to receive input from a broader area of the retina, potentially enhancing their sensitivity to light. However, the remodelling processes can also disrupt normal signal processing and contribute to visual dysfunction, including changes in visual acuity, contrast sensitivity, and colour vision. Understanding retinal remodelling in RP is important for the development of potential therapeutic interventions. Strategies aimed at modulating or harnessing the adaptive aspects of retinal remodelling while minimizing the maladaptive changes are being investigated as potential approaches to slow down the progression of the disease and restore visual function in RP patients (70).
Inflammation And Immune Responses
Finally, the last but not least important pathogenetic mechanism that significantly contributes to the onset and progression of retinitis pigmentosa is represented by inflammation and Immune Responses (71). While the primary cause of RP is genetic mutations, inflammation and immune responses can exacerbate the degenerative processes in the retina and can be classified into 5 different mechanisms:
- 5.
- Microglial Activation: Microglia, the resident immune cells of the retina, become activated in response to photoreceptor cell death and degeneration. Activated microglia release pro-inflammatory cytokines, chemokines, and reactive oxygen species. While microglial activation initially aims to clear debris and promote tissue repair, chronic or excessive activation can lead to neuroinflammation and further damage to the retina (72).
- 6.
- Infiltration of Immune Cells In some cases of RP, immune cells from the bloodstream can infiltrate the retina, further contributing to the inflammatory response. These immune cells, including macrophages and T cells, release inflammatory mediators that can exacerbate retinal damage (73).
- 7.
- Cytokine Imbalance In RP, there is evidence of an imbalance in cytokine signalling in the retina. Pro-inflammatory cytokines, such as tumour necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6), are upregulated, while anti-inflammatory cytokines, such as interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β), are downregulated. This imbalance can perpetuate the inflammatory response and contribute to the degeneration of photoreceptor cells (74) (75).
- 8.
- Complement System Activation: Activation of the complement system can lead to the deposition of complement proteins on photoreceptor cells and subsequent immune-mediated damage (73).
- 9.
- Oxidative Stress and Inflammation: Oxidative stress, resulting from the imbalance between reactive oxygen species (ROS) production and antioxidant defence mechanisms, can further contribute to inflammation in RP. ROS can activate various intracellular signalling pathways involved in inflammatory responses, amplifying the inflammatory cascade and exacerbating retinal damage (76).
Modulating immune responses through anti-inflammatory strategies, immunomodulatory therapies, or targeted interventions to regulate specific immune cell functions are areas of active research for potential therapeutic approaches in RP (77).
3. Cell-Based Therapies for Retinitis Pigmentosa
Understanding the interplay between inflammation, immune responses, and the degenerative processes in RP is crucial for developing targeted therapies aimed at modulating the immune system, reducing inflammation and preserving retinal function. In the next section, the development of cell-based therapies for retinitis pigmentosa is going to be discussed with the latest updates.
As emphasized previously, understanding all these molecular mechanisms underlying RP is crucial for the development of targeted therapies. Current research efforts focus on strategies such as gene therapy, stem cell transplantation, neuroprotective agents, and optogenetics to address specific molecular defects and slow down the progression of RP (44).
In this context particularly cell-based therapies hold great promise for the treatment of RP (78), aiming to replace or restore the function of degenerated photoreceptor cells in the retina or to release some growth factors to enhance cell survival, growth and function of retinal cells. The main approaches of cell-based therapy for the treatment of RP will be discussed below and summarized in Figure 2.
Retinal Cells Transplantation
This approach involves transplanting healthy retinal cells, such as photoreceptor cells or RPE cells, into the degenerated retina. These transplanted cells can integrate into the existing retinal tissue and potentially restore visual function (79). Various sources of donor cells are being investigated, including stem cell-derived photoreceptor cells and RPE cells. The goal of Photoreceptor Cell Transplantation is to replace the lost or damaged photoreceptor cells in the retina and several approaches are being explored including:
- Fetal Retinal Tissue Transplantation. Fetal retinal tissue, obtained from donor fetuses, can be transplanted into the subretinal space of RP patients. The transplanted cells can integrate into the host retina and potentially improve visual function. However, the availability of fetal tissue is limited, and immunological compatibility needs to be considered (80).
- Stem Cell-Derived Photoreceptor Cell Transplantation. Pluripotent stem cells, such as embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), can be differentiated into photoreceptor-like cells in vitro (81) (82). These cells can then be transplanted into the retina to replace the degenerated photoreceptor cells.
RPE Cell Transplantation
On the other hand, the RPE Cell Transplantation is described as the most innovative and easiest-to-apply therapeutic approach (83). The RPE plays a critical role in supporting and maintaining the health of photoreceptor cells it is established that in RP, RPE dysfunction contributes to photoreceptor degeneration. RPE cell transplantation aims to replace the damaged RPE cells and provide a supportive environment for photoreceptor survival. RPE cells can be derived from different sources and different therapeutic approaches have been described (84) .
Supportive Cells Transplantation
In addition to photoreceptor and RPE cells, other supportive cell types can be transplanted to enhance the survival and function of existing retinal cells. For example, Müller glial cells, Schwann cells, central nervous system stem cells or olfactory ensheathing cells (OECs) can be transplanted to provide neurotrophic support, promote retinal regeneration, and modulate the retinal microenvironment (85) (86) (87).
Stem Cell Therapy
Stem cell therapy holds significant promise for the treatment of retinitis pigmentosa as stem cells have the potential to differentiate into various cell types, including photoreceptor cells and RPE cells, aiming to replace the lost or damaged photoreceptor cells and restore visual function (88) (89) (90). Several therapeutic approaches have been reported using different kinds of stem cells like pluripotent stem cells, such as embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), bone marrow-derived stem cells including multipotent cells such as MSCs (Mesenchymal Stromal Cells).
ESCs and iPSCs have the remarkable ability to differentiate into various cell types, including retinal cells like photoreceptors and other retinal neurons with direct retinal-like cell transplantation. As it is known, RPE cells play a crucial role in supporting and maintaining the health of photoreceptor cells. In RP, dysfunctional RPE cells contribute to photoreceptor degeneration and pluripotent stem cells can be differentiated into stem cell-derived RPE cells for subretinal space transplantation to restore RPE function. These transplanted cells can provide nutritional support, phagocytosis of photoreceptor outer segments, and maintenance of the blood-retinal barrier (91) (92).On the other hand, photoreceptor cell replacement can represent a remarkable therapeutic alternative. In this approach stem cell-derived photoreceptor cells can be transplanted into the subretinal space of RP patients to replace the lost or damaged photoreceptor cells. Differentiation protocols are used to generate photoreceptor-like cells from pluripotent stem cells, aiming to recapitulate the development and function of native photoreceptor cells. Transplanted photoreceptor cells should integrate into the host retina and establish functional connections with the remaining retinal circuitry (93) (94).
A very promising type of adult stem cells in this field are the Mesenchymal Stromal Cells that allow to overcome the limits represented by the use of pluripotent stem cells such as ethical, and immune rejection issues and the risk of viral integrations and oncogene expression. MSCs are a type of adult stem cell that can be obtained from various sources, including bone marrow, adipose tissue, and umbilical cord tissue (95). The idea behind using MSCs for RP treatment is to harness their regenerative and immunomodulatory properties to protect or replace damaged retinal cells and preserve or restore vision, according to different therapeutical mechanisms such as the release of paracrine factors and the immunomodulatory effects (96) (97). The paracrine effects of MSC are well described in the literature as MSCs can secrete various growth factors, cytokines, and neurotrophic factors that create a supportive environment for retinal cell survival and function. The paracrine effects of MSCs may help protect and promote the survival of existing retinal cells, including photoreceptors, and potentially slow down the progression of RP (98) (99). Moreover, MSCs have anti-inflammatory properties resulting in immunomodulatory effects which can help modulate immune responses in the retina. Thanks to the release of specific cytokines and growth factors, MSCs suppress the activation and proliferation of immune cells, such as T cells and macrophages, and reduce the production of pro-inflammatory cytokines (100). By modulating the immune response, MSCs may help alleviate the chronic inflammation associated with RP and create a more favourable environment for retinal cell survival. Moreover, MSCs have been shown to have anti-apoptotic (cell survival-promoting) and anti-oxidative effects (101). These properties can help protect retinal cells from cell death caused by oxidative stress and apoptosis, which are key features of RP pathogenesis. By reducing oxidative stress and promoting cell survival, MSCs may contribute to the preservation of retinal function in RP (102). Finally, it has been demonstrated that MSCs have a strong angiogenic potential as they can promote the formation of new blood vessels by enhancing angiogenesis (97) (103). This is a crucial aspect because in some cases of RP, vascular abnormalities and compromised blood flow in the retina contribute to disease progression. Thanks to their angiogenic potential MSCs may help improve retinal blood supply and support the survival and function of retinal cells.
Although MSCs show promise for RP therapy, further research is needed to optimize the therapeutic strategies, including determining the optimal route and timing of MSC administration, understanding the mechanisms of action, and addressing concerns such as cell survival, migration, and long-term effects (98). Clinical trials are ongoing to assess the safety and efficacy of MSC-based therapies in RP, and their outcomes will provide valuable insights into the potential of MSCs for treating this retinal disorder (104).
Finally, other types of cells that are being studied for cellular therapy approaches include Olfactory Ensheathing Cells (OECs), a type of glial cells capable of continuous growth and regeneration of olfactory axons into the CNS and Human Neural Progenitors (105). Approaches with these kinds of cells are aimed at providing an intrinsic continuous supply of neurotrophic factors, reducing the gliotic injury response of Muller cells and rescuing long-term vision function and associated morphologic substrates by protecting dying host neurons (106).
Despite stem cell therapies appearing extremely promising, there are still some crucial aspects to consider for their in vivo application. For example, therapy for RP requires careful consideration of immunological factors to ensure cell survival and minimize the risk of immune rejection (88) (107). Autologous transplantation could help to minimize immune responses Immune modulation strategies, such as immunosuppressive medications or genetic engineering of cells to evade immune recognition, are also being explored to enhance graft survival.
In addition, there is still a need for optimization of transplantation techniques as the success of stem cell therapy for RP depends on several factors, including the efficient generation of desired cell types, transplantation techniques, and the survival, integration, and functionality of transplanted cells (108). Current research is indeed moving towards refining the differentiation protocols, optimizing cell delivery methods, and promoting the long-term survival and integration of transplanted cells (109) (110).
For all these reasons stem cell therapy for RP is still in the experimental stage, and clinical trials are ongoing to evaluate its safety and efficacy. Challenges remain, such as the development of standardized protocols, ensuring long-term functionality and stability of transplanted cells, and addressing ethical considerations. However, the potential of stem cell therapy to restore visual function and halt the progression of RP provides hope for future treatments.
Optogenetic Therapy
Another kind of absolutely innovative cell-based therapy for RP is Optogenetic Therapy, whose application is proposed in cases where the photoreceptor cells are severely degenerated (111). While it is not a traditional stem cell therapy where stem cells are transplanted directly into the body, optogenetic therapy does involve the introduction of genetically modified proteins into cells to alter their behaviour and function. This approach involves the introduction of light-sensitive proteins into other retinal cells, such as remaining retinal ganglion cells or bipolar cells, to restore light sensitivity (112). The modified cells can then respond to light and transmit signals to the visual processing centres in the brain. Briefly, the optogenic therapy in RP utilizes light-sensitive proteins, such as microbial opsins (e.g., channelrhodopsin and halorhodopsin), that can be introduced into non-photosensitive cells in the retina. These proteins respond to specific wavelengths of light, allowing them to activate or inhibit the cells in response to light stimulation (113).
On the other hand, non-photosensitive cells, such as RGCs or bipolar cells, are genetically modified to express light-sensitive proteins. This can be achieved by delivering viral vectors carrying the genes encoding the light-sensitive proteins into the cells (113). The viral vectors integrate the genes into the cell's genome, enabling the cells to produce light-sensitive proteins. Once the target cells are genetically modified to express the light-sensitive proteins, they can respond to light stimulation. External light sources, such as specialized goggles or glasses, are used to deliver specific wavelengths of light to the retina. When the light-sensitive proteins are activated or inhibited by the light, they initiate electrical signals that can be transmitted to the visual processing centres in the brain. By introducing light sensitivity to non-photosensitive cells in the retina, optogenetic therapy aims to restore visual function in individuals with RP (114). Although the restored vision may not fully replicate natural vision, it can provide the ability to perceive light and distinguish basic shapes and objects, potentially improving the quality of life for patients with very advanced stages of RP (115). Optogenetic therapy is still in the early stages of development and research, and clinical trials are underway to evaluate its safety and efficacy in human subjects with RP. Several factors need to be considered for its successful implementation, including optimizing the targeting and expression of light-sensitive proteins, ensuring long-term functionality of the modified cells, and developing reliable and non-invasive methods of light delivery. Additionally, the therapy's long-term effects, potential side effects, and compatibility with individual patient characteristics need to be thoroughly investigated. Nonetheless, optogenetic therapy holds promise as a potential treatment strategy for RP and represents an exciting avenue for future research and clinical applications. In the next section, a few examples of optogenetic therapies being explored for the treatment of retinitis pigmentosa are briefly described:
- Channelrhodopsin-based therapy: Channelrhodopsin-2 (ChR2) is a light-sensitive protein derived from algae. In optogenetic therapy for RP, ChR2 is introduced into retinal ganglion cells (RGCs) or bipolar cells. When activated by light of specific wavelengths, ChR2 can depolarize the cells and initiate electrical signals, mimicking the function of photoreceptor cells. This approach aims to restore light sensitivity and enable visual information to be transmitted to the brain (116).
- Halorhodopsin-based therapy: Halorhodopsin (NpHR) is a light-sensitive protein that responds to yellow or amber light. In optogenetic therapy, NpHR can be introduced into bipolar cells or RGCs to allow the cells to be inhibited in response to light stimulation. By selectively inhibiting specific cell types, such as ON or OFF bipolar cells, the retinal circuitry can be modulated to enhance visual processing and restore functional vision (117) (118).
- Red-shifted opsin-based therapy: In addition to ChR2 and NpHR, other light-sensitive proteins with red-shifted absorption spectra are being explored for optogenetic therapy in RP. These proteins, such as ReaChR or ChrimsonR, can be activated by longer wavelengths of light, including red or near-infrared light. By utilizing these red-shifted opsins, optogenetic therapy can potentially penetrate deeper into the retina and improve light sensitivity in RP patients (119).
Strategies for Promoting the Survival and Function of Existing Retinal Cells
Another aspect of cell-based therapies for RP involves promoting the survival and function of existing retinal cells. Various approaches, including the transplantation of supportive cells or the delivery of neurotrophic factors, are being explored to enhance survival and protect the remaining retinal cells from further degeneration (120). The neuroprotection and cell survival enhancement in RP could be achieved through the administration of neurotrophic factors like brain-derived neurotrophic factor (BDNF), ciliary neurotrophic factor (CNTF), and glial cell line-derived neurotrophic factor (GDNF) delivered to the retina with intravitreal injections where they promote the survival of retinal cells (121) (122). Besides neurotrophic factors, anti-apoptotic agents such as caspase inhibitors and Bcl-2 family proteins can be used to prevent or reduce the apoptosis of retinal cells and promote their survival (123). As discussed before, apoptosis represent one of the most important pathomolecular mechanism of RP along with the chronic inflammation process: For this reason, even anti-inflammatory strategies are being explored to reduce inflammation and its detrimental effects on retinal cells (124). This can be achieved through the use of anti-inflammatory drugs, immunomodulatory therapies, or other approaches that target inflammatory pathways and cytokines. Finally, the latest approach is represented by Oxidative Stress Management through the administration of Antioxidants and ROS scavengers, such as vitamin C, vitamin E, and coenzyme Q10 to reduce oxidative stress and protect retinal cells from damage (125) (126).
4. Conclusions
Based on what has been discussed so far, it’s clear that the comprehensive understanding of the pathogenetic mechanisms underlying retinitis pigmentosa represents the key to the development of new therapies and the enhancement of existing treatments. It is important to note that while cell-based therapies hold great potential, they are still in the experimental stages and further research is needed to refine their safety and efficacy. The proof of concept that a new era of treating rare diseases is about to start is the increasing number of ongoing clinical trials that are assessing the feasibility and effectiveness of these approaches in treating RP. A close collaboration between researchers, clinicians, and regulatory bodies is warranted for their successful translation into clinical practice (127) (128). Relevant clinical trials with cell-based therapies are listed in Table 2.
Author Contributions
Conceptualization, G.B and V.B.; methodology, V.B.; validation, F.B., R.C. and A.S.; formal analysis, V.B. and E.M.; investigation, V.B and G.B.; writing—original draft preparation, V.B., G.B. and E.M.; writing—review and editing, F.B., R.C.; visualization, F.B. and R.C.; supervision, R.C. and A.S.. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hamel, C. Retinitis pigmentosa. Orphanet J. Rare Dis. 2006, 1, 40–40. [Google Scholar] [CrossRef] [PubMed]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Fishman, G.A.; Anderson, R.J.; Lourenco, P. Prevalence of posterior subcapsular lens opacities in patients with retinitis pigmentosa. Br. J. Ophthalmol. 1985, 69, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, A.; Yamamoto, S.; Ogata, K.; Sugawara, T.; Hiramatsu, A.; Shibata, M.; Mitamura, Y. Macular abnormalities in patients with retinitis pigmentosa: prevalence on OCT examination and outcomes of vitreoretinal surgery. Acta Ophthalmol. 2011, 89, e122–e125. [Google Scholar] [CrossRef]
- Kumaran, N.; Moore, A.T.; Weleber, R.G.; Michaelides, M. Leber congenital amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions. Br. J. Ophthalmol. 2017, 101, 1147–1154. [Google Scholar] [CrossRef]
- Wu, K.Y.; Kulbay, M.; Toameh, D.; Xu, A.Q.; Kalevar, A.; Tran, S.D. Retinitis Pigmentosa: Novel Therapeutic Targets and Drug Development. Pharmaceutics 2023, 15, 685. [Google Scholar] [CrossRef]
- Maguire, A.M.; High, K.A.; Auricchio, A.; Wright, J.F.; Pierce, E.A.; Testa, F.; Mingozzi, F.; Bennicelli, J.L.; Ying, G.-S.; Rossi, S.; et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: a phase 1 dose-escalation trial. Lancet 2009, 374, 1597–1605. [Google Scholar] [CrossRef]
- Bainbridge, J.W.; Smith, A.J.; Barker, S.S.; Robbie, S.; Henderson, R.; Balaggan, K.; Viswanathan, A.; Holder, G.E.; Stockman, A.; Tyler, N.; et al. Effect of Gene Therapy on Visual Function in Leber's Congenital Amaurosis. N. Engl. J. Med. 2008, 358, 2231–2239. [Google Scholar] [CrossRef]
- Sodi, A.; Banfi, S.; Testa, F.; Della Corte, M.; Passerini, I.; Pelo, E.; Rossi, S.; Simonelli, F. ; Italian IRD Working Group RPE65-associated inherited retinal diseases: consensus recommendations for eligibility to gene therapy. Orphanet J. Rare Dis. 2021, 16, 1–11. [Google Scholar] [CrossRef]
- Acland, G.M.; Aguirre, G.D.; Ray, J.; Zhang, Q.; Aleman, T.S.; Cideciyan, A.V.; Pearce-Kelling, S.E.; Anand, V.; Zeng, Y.; Maguire, A.M.; et al. Gene therapy restores vision in a canine model of childhood blindness. Nat. Genet. 2001, 28, 92–95. [Google Scholar] [CrossRef]
- Pierce, E.A.; Bennett, J. The Status ofRPE65Gene Therapy Trials: Safety and Efficacy. Cold Spring Harb. Perspect. Med. 2015, 5, a017285. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Pang, J.-J.; Zhang, H.; Mansfield, D. Retinitis Pigmentosa: Disease Mechanisms, Diagnosis, and Therapies. J. Ophthalmol. 2015, 2015, 1–1. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Di Iorio, E.; Barbaro, V.; Ponzin, D.; Sorrentino, F.S.; Parmeggiani, F. Retinitis pigmentosa: Genes and disease mechanisms. Curr. Genom. 2011, 12, 238–249. [Google Scholar] [CrossRef]
- Daiger, S.P.; Sullivan, L.S.; Bowne, S.J. Genes and mutations causing retinitis pigmentosa. Clin. Genet. 2013, 84, 132–141. [Google Scholar] [CrossRef]
- Athanasiou, D.; Aguila, M.; Bellingham, J.; Li, W.; McCulley, C.; Reeves, P.J.; Cheetham, M.E. The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog. Retin. Eye Res. 2018, 62, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Athanasiou, D.; Aguila, M.; Bellingham, J.; Li, W.; McCulley, C.; Reeves, P.J.; Cheetham, M.E. The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog. Retin. Eye Res. 2018, 62, 1–23. [Google Scholar] [CrossRef]
- Lewin, A.S.; Rossmiller, B.; Mao, H. Gene Augmentation for adRP Mutations in RHO. Cold Spring Harb. Perspect. Med. 2014, 4, a017400–a017400. [Google Scholar] [CrossRef]
- Chuang, J.-Z.; Vega, C.; Jun, W.; Sung, C.-H. Structural and functional impairment of endocytic pathways by retinitis pigmentosa mutant rhodopsin-arrestin complexes. J. Clin. Investig. 2004, 114, 131–140. [Google Scholar] [CrossRef]
- Liu, X.; Feng, B.; Vats, A.; Tang, H.; Seibel, W.; Swaroop, M.; Tawa, G.; Zheng, W.; Byrne, L.; Schurdak, M.; et al. Pharmacological clearance of misfolded rhodopsin for the treatment of RHO -associated retinitis pigmentosa. FASEB J. 2020, 34, 10146–10167. [Google Scholar] [CrossRef]
- Peeters, M.H.; Khan, M.; Rooijakkers, A.A.; Mulders, T.; Haer-Wigman, L.; Boon, C.J.; Klaver, C.C.; van den Born, L.I.; Hoyng, C.B.; Cremers, F.P.; et al. PRPH2 mutation update: In silico assessment of 245 reported and 7 novel variants in patients with retinal disease. Hum. Mutat. 2021. [Google Scholar] [CrossRef]
- Coco-Martin, R.M.; Sanchez-Tocino, H.T.; Desco, C.; Usategui-Martín, R.; Tellería, J.J. PRPH2-Related Retinal Diseases: Broadening the Clinical Spectrum and Describing a New Mutation. Genes 2020, 11, 773. [Google Scholar] [CrossRef] [PubMed]
- Stuck, M.W.; Conley, S.M.; Naash, M.I. PRPH2/RDS and ROM-1: Historical context, current views and future considerations. Prog. Retin. Eye Res. 2016, 52, 47–63. [Google Scholar] [CrossRef]
- Gerhardt, M.J.; Priglinger, S.G.; Biel, M.; Michalakis, S. Biology, Pathobiology and Gene Therapy of CNG Channel-Related Retinopathies. Biomedicines 2023, 11, 269. [Google Scholar] [CrossRef]
- Michalakis, S.; Becirovic, E.; Biel, M. Retinal Cyclic Nucleotide-Gated Channels: From Pathophysiology to Therapy. Int. J. Mol. Sci. 2018, 19, 749. [Google Scholar] [CrossRef]
- Gerhardt, M.J.; Petersen-Jones, S.M.; Michalakis, S. CNG channel-related retinitis pigmentosa. Vis. Res. 2023, 208, 108232. [Google Scholar] [CrossRef]
- Gerhardt, M.J.; Priglinger, S.G.; Biel, M.; Michalakis, S. Biology, Pathobiology and Gene Therapy of CNG Channel-Related Retinopathies. Biomedicines 2023, 11, 269. [Google Scholar] [CrossRef] [PubMed]
- Duricka, D.L.; Brown, R.L.; Varnum, M.D. Defective trafficking of cone photoreceptor CNG channels induces the unfolded protein response and ER-stress-associated cell death. Biochem. J. 2011, 441, 685–696. [Google Scholar] [CrossRef]
- Wagg, C.; Bender, S.F.; Widmer, F.; van der Heijden, MGA. Soil biodiversity and soil community composition determine ecosystem multifunctionality. PNAS 2014, 111, 5266–5270. [Google Scholar] [CrossRef] [PubMed]
- Sallum, J.M.F.; Kaur, V.P.; Shaikh, J.; Banhazi, J.; Spera, C.; Aouadj, C.; Viriato, D.; Fischer, M.D. Epidemiology of Mutations in the 65-kDa Retinal Pigment Epithelium (RPE65) Gene-Mediated Inherited Retinal Dystrophies: A Systematic Literature Review. Adv. Ther. 2022, 39, 1179–1198. [Google Scholar] [CrossRef]
- Sallum, J.M.F.; Kaur, V.P.; Shaikh, J.; Banhazi, J.; Spera, C.; Aouadj, C.; Viriato, D.; Fischer, M.D. Epidemiology of Mutations in the 65-kDa Retinal Pigment Epithelium (RPE65) Gene-Mediated Inherited Retinal Dystrophies: A Systematic Literature Review. Adv. Ther. 2022, 39, 1179–1198. [Google Scholar] [CrossRef]
- Thompson, D.A.; Gyürüs, P.; Fleischer, L.L.; Bingham, E.L.; McHenry, C.L.; Apfelstedt-Sylla, E.; Zrenner, E.; Lorenz, B.; Richards, J.E.; Jacobson, S.G.; et al. Genetics and phenotypes of RPE65 mutations in inherited retinal degeneration. Investig. Ophthalmol. Vis. Sci. 2000, 41, 4293–4299. [Google Scholar]
- He, S.; Parapuram, S.K.; Hurd, T.W.; Behnam, B.; Margolis, B.; Swaroop, A.; Khanna, H. Retinitis Pigmentosa GTPase Regulator (RPGR) protein isoforms in mammalian retina: Insights into X-linked Retinitis Pigmentosa and associated ciliopathies. Vis. Res. 2008, 48, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Vinikoor-Imler, L.C.; Simpson, C.; Narayanan, D.; Abbasi, S.; Lally, C. Prevalence of RPGR-mutated X-linked retinitis pigmentosa among males. Ophthalmic Genet. 2022, 43, 581–588. [Google Scholar] [CrossRef]
- Murga-Zamalloa, C.A.; Atkins, S.J.; Peranen, J.; Swaroop, A.; Khanna, H. Interaction of retinitis pigmentosa GTPase regulator (RPGR) with RAB8A GTPase: implications for cilia dysfunction and photoreceptor degeneration. Hum. Mol. Genet. 2010, 19, 3591–3598. [Google Scholar] [CrossRef]
- Murga-Zamalloa, C.; Swaroop, A.; Khanna, H. Multiprotein Complexes of Retinitis Pigmentosa GTPase Regulator (RPGR), a Ciliary Protein Mutated in X-Linked Retinitis Pigmentosa (XLRP). 664. [CrossRef]
- Sun, C.; Chen, S. Gene Augmentation for Autosomal Dominant CRX-Associated Retinopathies. 1415. [CrossRef]
- Swain, P.K.; Chen, S.; Wang, Q.-L.; Affatigato, L.M.; Coats, C.L.; Brady, K.D.; A Fishman, G.; Jacobson, S.G.; Swaroop, A.; Stone, E.; et al. Mutations in the Cone-Rod Homeobox Gene Are Associated with the Cone-Rod Dystrophy Photoreceptor Degeneration. Neuron 1997, 19, 1329–1336. [Google Scholar] [CrossRef] [PubMed]
- Freund, C.L.; Gregory-Evans, C.Y.; Furukawa, T.; Papaioannou, M.; Looser, J.; Ploder, L.; Bellingham, J.; Ng, D.; Herbrick, J.-A.S.; Duncan, A.; et al. Cone-Rod Dystrophy Due to Mutations in a Novel Photoreceptor-Specific Homeobox Gene () Essential for Maintenance of the Photoreceptor. Cell 1997, 91, 543–553. [Google Scholar] [CrossRef]
- Chen, S.; Wang, Q.-L.; Xu, S.; Liu, I.; Li, L.Y.; Wang, Y.; Zack, D.J. Functional analysis of cone-rod homeobox (CRX) mutations associated with retinal dystrophy. Hum. Mol. Genet. 2002, 11, 873–884. [Google Scholar] [CrossRef]
- Fuster-García, C.; García-Bohórquez, B.; Rodríguez-Muñoz, A.; Aller, E.; Jaijo, T.; Millán, J.M.; García-García, G. Usher Syndrome: Genetics of a Human Ciliopathy. Int. J. Mol. Sci. 2021, 22, 6723. [Google Scholar] [CrossRef]
- Lenassi, E.; Saihan, Z.; Cipriani, V.; Stabej, P.L.Q.; Moore, A.T.; Luxon, L.M.; Bitner-Glindzicz, M.; Webster, A.R. Natural History and Retinal Structure in Patients with Usher Syndrome Type 1 Owing to MYO7A Mutation. Ophthalmology 2013, 121, 580–587. [Google Scholar] [CrossRef]
- Nagel-Wolfrum, K.; Fadl, B.R.; Becker, M.M.; A Wunderlich, K.; Schäfer, J.; Sturm, D.; Fritze, J.; Gür, B.; Kaplan, L.; Andreani, T.; et al. Expression and subcellular localization ofUSH1C/harmonin in human retina provides insights into pathomechanisms and therapy. Hum. Mol. Genet. 2022, 32, 431–449. [Google Scholar] [CrossRef]
- Ratnam, K.; Västinsalo, H.; Roorda, A.; Sankila, E.-M.K.; Duncan, J.L. Cone Structure in Patients With Usher Syndrome Type III and Mutations in the Clarin 1 Gene. JAMA Ophthalmol 2013, 131, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liu, S.; Li, P.; Yao, K. Retinitis Pigmentosa: Progress in Molecular Pathology and Biotherapeutical Strategies. Int. J. Mol. Sci. 2022, 23, 4883. [Google Scholar] [CrossRef] [PubMed]
- Manley, A.; Meshkat, B.I.; Jablonski, M.M.; Hollingsworth, T. Cellular and Molecular Mechanisms of Pathogenesis Underlying Inherited Retinal Dystrophies. Biomolecules 2023, 13, 271. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; LaVail, M.M. Misfolded Proteins and Retinal Dystrophies. 664. [CrossRef]
- Tzekov, R.; Stein, L.; Kaushal, S. Protein Misfolding and Retinal Degeneration. Cold Spring Harb. Perspect. Biol. 2011, 3, a007492–a007492. [Google Scholar] [CrossRef]
- Vingolo, E.M.; Casillo, L.; Contento, L.; Toja, F.; Florido, A. Retinitis Pigmentosa (RP): The Role of Oxidative Stress in the Degenerative Process Progression. Biomedicines 2022, 10, 582. [Google Scholar] [CrossRef]
- Gallenga, C.E.; Lonardi, M.; Pacetti, S.; Violanti, S.S.; Tassinari, P.; Di Virgilio, F.; Tognon, M.; Perri, P. Molecular Mechanisms Related to Oxidative Stress in Retinitis Pigmentosa. Antioxidants 2021, 10, 848. [Google Scholar] [CrossRef]
- Domènech, E.B.; Marfany, G. The Relevance of Oxidative Stress in the Pathogenesis and Therapy of Retinal Dystrophies. Antioxidants 2020, 9, 347. [Google Scholar] [CrossRef]
- Komeima, K.; Rogers, B.S.; Lu, L.; Campochiaro, P.A. Antioxidants reduce cone cell death in a model of retinitis pigmentosa. Proc. Natl. Acad. Sci. 2006, 103, 11300–11305. [Google Scholar] [CrossRef]
- Ren, X.; Léveillard, T. Modulating antioxidant systems as a therapeutic approach to retinal degeneration. Redox Biol. 2022, 57, 102510. [Google Scholar] [CrossRef]
- Lefevere, E.; Toft-Kehler, A.K.; Vohra, R.; Kolko, M.; Moons, L.; Van Hove, I. Mitochondrial dysfunction underlying outer retinal diseases. Mitochondrion 2017, 36, 66–76. [Google Scholar] [CrossRef]
- Barot, M.; Gokulgandhi, M.R.; Mitra, A.K. Mitochondrial Dysfunction in Retinal Diseases. Curr. Eye Res. 2011, 36, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, X.; A, L.; Gao, H.; Zhao, M.; Ge, L.; Li, M.; Yang, C.; Gong, Y.; Gu, Z.; et al. Alleviation of Photoreceptor Degeneration Based on Fullerenols in rd1 Mice by Reversing Mitochondrial Dysfunction via Modulation of Mitochondrial DNA Transcription and Leakage. Small 2023, e2205998. [Google Scholar] [CrossRef] [PubMed]
- others, Dafni Vlachantoni and. Evidence of severe mitochondrial oxidative stress and a protective effect of low oxygen in mouse models of inherited photoreceptor degeneration, Human Molecular Genetics, Volume 20, Issue 2, , Pages 322–335, ht. 15 January.
- Valeria Marigo, Meltem Kutluer, Li Huang, Antonella Comitato, Davide Schiroli, Frank Schwede, Andreas Rentsch, Per A R Ekstrom, Francois Paquet-Durand. Decrease of intracellular calcium to restrain rod cell death in retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 2019;60(9):4866.
- Das, S.; Chen, Y.; Yan, J.; Christensen, G.; Belhadj, S.; Tolone, A.; Paquet-Durand, F. The role of cGMP-signalling and calcium-signalling in photoreceptor cell death: perspectives for therapy development. 473, 1411. [Google Scholar] [CrossRef]
- Wang, J.; Li, M.; Geng, Z.; Khattak, S.; Ji, X.; Wu, D.; Dang, Y. Role of Oxidative Stress in Retinal Disease and the Early Intervention Strategies: A Review. Oxidative Med. Cell. Longev. 2022, 2022, 1–13. [Google Scholar] [CrossRef]
- Li, Z.-Y.; Milam, A.H. Apoptosis in Retinitis Pigmentosa. 1995; 8. [Google Scholar] [CrossRef]
- Wong, P. Apoptosis, retinitis pigmentosa, and degeneration. Biochem. Cell Biol. 1994, 72, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Newton, F.; Megaw, R. Mechanisms of Photoreceptor Death in Retinitis Pigmentosa. Genes 2020, 11, 1120. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.W.; Kondo, M.; Terasaki, H.; Lin, Y.; McCall, M.; Marc, R.E. Retinal remodeling. Jpn. J. Ophthalmol. 2012, 56, 289–306. [Google Scholar] [CrossRef]
- Jones, B.W.; Pfeiffer, R.L.; Ferrell, W.D.; Watt, C.B.; Marmor, M.; Marc, R.E. Retinal remodeling in human retinitis pigmentosa. Exp. Eye Res. 2016, 150, 149–165. [Google Scholar] [CrossRef]
- Marc, R.E.; Jones, B.W.; Watt, C.B.; Strettoi, E. Neural remodeling in retinal degeneration. Prog. Retin. Eye Res. 2003, 22, 607–655. [Google Scholar] [CrossRef]
- Martínez-Gil, N.; Maneu, V.; Kutsyr, O.; Fernández-Sánchez, L.; Sánchez-Sáez, X.; Sánchez-Castillo, C.; Campello, L.; Lax, P.; Pinilla, I.; Cuenca, N. Cellular and molecular alterations in neurons and glial cells in inherited retinal degeneration. Front. Neuroanat. 2022, 16, 984052. [Google Scholar] [CrossRef]
- Gao, H.; Huang, X.; He, J.; Zou, T.; Chen, X.; Xu, H. The roles of microglia in neural remodeling during retinal degeneration. . 2021, 37, 1–10. [Google Scholar] [CrossRef]
- William, C. Gordon, Eric J. Knott, Kristopher G. Sheets, Cornelius E. Regan, Jr., Nicolas G. Bazan. Müller Cell Reactive Gliosis Contributes To Retinal Degeneration In Ccl2-/-/Cx3cr1-/- Mice. Invest. Ophthalmol. Vis. Sci. 2011;52(14):1375.
- Soto, F.; Kerschensteiner, D. Synaptic remodeling of neuronal circuits in early retinal degeneration. Front. Cell. Neurosci. 2015, 9, 395. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, R.L.; Jones, B.W. Current perspective on retinal remodeling: Implications for therapeutics. Front. Neuroanat. 2022, 16, 1099348. [Google Scholar] [CrossRef] [PubMed]
- Noailles, A.; Maneu, V.; Campello, L.; Gómez-Vicente, V.; Lax, P.; Cuenca, N. Persistent inflammatory state after photoreceptor loss in an animal model of retinal degeneration. Sci. Rep. 2016, 6, 33356. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Xiao, J.; Wang, K.; So, K.-F.; Tipoe, G.L.; Lin, B. Suppression of Microglial Activation Is Neuroprotective in a Mouse Model of Human Retinitis Pigmentosa. J. Neurosci. 2014, 34, 8139–8150. [Google Scholar] [CrossRef]
- Mohan, K.V.; Mishra, A.; Muniyasamy, A.; Sinha, P.; Sahu, P.; Kesarwani, A.; Jain, K.; Nagarajan, P.; Scaria, V.; Agarwal, M.; et al. Immunological consequences of compromised ocular immune privilege accelerate retinal degeneration in retinitis pigmentosa. Orphanet J. Rare Dis. 2022, 17, 1–12. [Google Scholar] [CrossRef]
- Olivares-González, L.; Velasco, S.; Campillo, I.; Rodrigo, R. Retinal Inflammation, Cell Death and Inherited Retinal Dystrophies. Int. J. Mol. Sci. 2021, 22, 2096. [Google Scholar] [CrossRef] [PubMed]
- Okita, A.; Murakami, Y.; Shimokawa, S.; Funatsu, J.; Fujiwara, K.; Nakatake, S.; Koyanagi, Y.; Akiyama, M.; Takeda, A.; Hisatomi, T.; et al. Changes of Serum Inflammatory Molecules and Their Relationships with Visual Function in Retinitis Pigmentosa. Investig. Opthalmology Vis. Sci. 2020, 61, 30–30. [Google Scholar] [CrossRef]
- Murakami, Y.; Nakabeppu, Y.; Sonoda, K.-H. Oxidative Stress and Microglial Response in Retinitis Pigmentosa. Int. J. Mol. Sci. 2020, 21, 7170. [Google Scholar] [CrossRef]
- Zhao, L.; Hou, C.; Yan, N. Neuroinflammation in retinitis pigmentosa: Therapies targeting the innate immune system. Front. Immunol. 2022, 13, 1059947. [Google Scholar] [CrossRef]
- Hinkle, J.W.; Mahmoudzadeh, R.; Kuriyan, A.E. Cell-based therapies for retinal diseases: a review of clinical trials and direct to consumer “cell therapy” clinics. Stem Cell Res. Ther. 2021, 12, 1–9. [Google Scholar] [CrossRef]
- Tezel, T.; Ruff, A. Retinal cell transplantation in retinitis pigmentosa. Taiwan J. Ophthalmol. 2021, 11, 336–347. [Google Scholar] [CrossRef]
- MacLaren, R.; Pearson, R.; MacNeil, A.; Douglas, R.H.; Salt, T.E.; Akimoto, M.; Swaroop, A.; Sowden, J.; Ali, R. Retinal repair by transplantation of photoreceptor precursors. Nature 2006, 444, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Yanai, A.; Laver, C.; Joe, A.W.; Gregory-Evans, K. Efficient Production of Photoreceptor Precursor Cells from Human Embryonic Stem Cells. 1307. [CrossRef]
- Li, Y.; Tsai, Y.-T.; Hsu, C.-W.; Erol, D.; Yang, J.; Wu, W.-H.; Davis, R.J.; Egli, D.; Tsang, S.H. Long-term Safety and Efficacy of Human-Induced Pluripotent Stem Cell (iPS) Grafts in a Preclinical Model of Retinitis Pigmentosa. Mol. Med. 2012, 18, 1312–1319. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.Z.; Utheim, T.P.; Eidet, J.R. Retinal Pigment Epithelium Transplantation: Past, Present, and Future. J. Ophthalmic Vis. Res. 2022, 17, 574–580. [Google Scholar] [CrossRef]
- Alexander, P.; Thomson, H.A.J.; Luff, A.J.; Lotery, A.J. Retinal pigment epithelium transplantation: concepts, challenges, and future prospects. Eye 2015, 29, 992–1002. [Google Scholar] [CrossRef] [PubMed]
- Heravi, M.; Rasoulinejad, S.A. Potential of Müller Glial Cells in Regeneration of Retina; Clinical and Molecular Approach., 13, 50–59.
- Eastlake, K.; Lamb, W.; Luis, J.; Khaw, P.; Jayaram, H.; Limb, G. Prospects for the application of Müller glia and their derivatives in retinal regenerative therapies. Prog. Retin. Eye Res. 2021, 85, 100970. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, Z.; Gu, P. Stem/progenitor cell-based transplantation for retinal degeneration: a review of clinical trials. Cell Death Dis. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, R.C. Stem cell therapy for retinal diseases: update. Stem Cell Res. Ther. 2011, 2, 1–10. [Google Scholar] [CrossRef]
- Wong, I.Y.-H.; Poon, M.-W.; Pang, R.T.-W.; Lian, Q.; Wong, D. Promises of stem cell therapy for retinal degenerative diseases. Graefe's Arch. Clin. Exp. Ophthalmol. 2011, 249, 1439–1448. [Google Scholar] [CrossRef]
- He, Y.; Zhang, Y.; Liu, X.; Ghazaryan, E.; Li, Y.; Xie, J.; Su, G. Recent Advances of Stem Cell Therapy for Retinitis Pigmentosa. Int. J. Mol. Sci. 2014, 15, 14456–14474. [Google Scholar] [CrossRef]
- Jin, Z.-B.; Okamoto, S.; Xiang, P.; Takahashi, M. Integration-Free Induced Pluripotent Stem Cells Derived from Retinitis Pigmentosa Patient for Disease Modeling. STEM CELLS Transl. Med. 2012, 1, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Idelson, M.; Alper, R.; Obolensky, A.; Ben-Shushan, E.; Hemo, I.; Yachimovich-Cohen, N.; Khaner, H.; Smith, Y.; Wiser, O.; Gropp, M.; et al. Directed Differentiation of Human Embryonic Stem Cells into Functional Retinal Pigment Epithelium Cells. Cell Stem Cell 2009, 5, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Vugler, A.; Lawrence, J.; Walsh, J.; Carr, A.; Gias, C.; Semo, M.; Ahmado, A.; da Cruz, L.; Andrews, P.; Coffey, P. Embryonic stem cells and retinal repair. Mech. Dev. 2007, 124, 807–829. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.-K.; Tosi, J.; Kasanuki, J.M.; Chou, C.L.; Kong, J.; Parmalee, N.; Wert, K.J.; Allikmets, R.; Lai, C.-C.; Chien, C.-L.; et al. Transplantation of Reprogrammed Embryonic Stem Cells Improves Visual Function in a Mouse Model for Retinitis Pigmentosa. Transplantation 2010, 89, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Bianco, P.; Robey, P.G.; Simmons, P.J. Mesenchymal Stem Cells: Revisiting History, Concepts, and Assays. Cell Stem Cell 2008, 2, 313–319. [Google Scholar] [CrossRef]
- Mannino, G.; Russo, C.; Longo, A.; Anfuso, C.D.; Lupo, G.; Furno, D.L.; Giuffrida, R.; Giurdanella, G. Potential therapeutic applications of mesenchymal stem cells for the treatment of eye diseases. World J. Stem Cells 2021, 13, 632–644. [Google Scholar] [CrossRef]
- Adak, S.; Magdalene, D.; Deshmukh, S.; Das, D.; Jaganathan, B.G. A Review on Mesenchymal Stem Cells for Treatment of Retinal Diseases. Stem Cell Rev. Rep. 2021, 17, 1154–1173. [Google Scholar] [CrossRef]
- Chen, X.; Jiang, Y.; Duan, Y.; Zhang, X.; Li, X. Mesenchymal-Stem-Cell-Based Strategies for Retinal Diseases. Genes 2022, 13, 1901. [Google Scholar] [CrossRef]
- Holan, V.; Palacka, K.; Hermankova, B. Mesenchymal Stem Cell-Based Therapy for Retinal Degenerative Diseases: Experimental Models and Clinical Trials. Cells 2021, 10, 588. [Google Scholar] [CrossRef]
- Reboussin. ; Buffault, J.; Brignole-Baudouin, F.; Goazigo, A.R.-L.; Riancho, L.; Olmiere, C.; Sahel, J.-A.; Parsadaniantz, S.M.; Baudouin, C. Evaluation of neuroprotective and immunomodulatory properties of mesenchymal stem cells in an ex vivo retinal explant model. J. Neuroinflammation 2022, 19, 1–17. [Google Scholar] [CrossRef]
- Stavely, R.; Nurgali, K. The emerging antioxidant paradigm of mesenchymal stem cell therapy. STEM CELLS Transl. Med. 2020, 9, 985–1006. [Google Scholar] [CrossRef]
- Angeloni, C.; Gatti, M.; Prata, C.; Hrelia, S.; Maraldi, T. Role of Mesenchymal Stem Cells in Counteracting Oxidative Stress—Related Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 3299. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.L.S.; Kumar, S.; Mok, P.L. Cellular Reparative Mechanisms of Mesenchymal Stem Cells for Retinal Diseases. Int. J. Mol. Sci. 2017, 18, 1406. [Google Scholar] [CrossRef] [PubMed]
- Holan, V.; Palacka, K.; Hermankova, B. Mesenchymal Stem Cell-Based Therapy for Retinal Degenerative Diseases: Experimental Models and Clinical Trials. Cells 2021, 10, 588. [Google Scholar] [CrossRef] [PubMed]
- Ruitenberg, M.J.; Vukovic, J.; Sarich, J.; Busfield, S.J.; Plant, G.W.; Yang, H.; He, B.-R.; Hao, D.-J.; Su, Z.; He, C.; et al. Olfactory Ensheathing Cells: Characteristics, Genetic Engineering, and Therapeutic Potential. J. Neurotrauma 2006, 23, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Huo, S.J.; Li, Y.C.; Xie, J.; Li, Y.; Raisman, G.; Zeng, Y.X.; He, J.R.; Weng, C.H.; Yin, Z.Q. Transplanted Olfactory Ensheathing Cells Reduce Retinal Degeneration in Royal College of Surgeons Rats. Curr. Eye Res. 2012, 37, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Bianco, P.; Robey, P.G.; Simmons, P.J. Mesenchymal Stem Cells: Revisiting History, Concepts, and Assays. Cell Stem Cell 2008, 2, 313–319. [Google Scholar] [CrossRef]
- Mannino, G.; Russo, C.; Longo, A.; Anfuso, C.D.; Lupo, G.; Furno, D.L.; Giuffrida, R.; Giurdanella, G. Potential therapeutic applications of mesenchymal stem cells for the treatment of eye diseases. World J. Stem Cells 2021, 13, 632–644. [Google Scholar] [CrossRef]
- Adak, S.; Magdalene, D.; Deshmukh, S.; Das, D.; Jaganathan, B.G. A Review on Mesenchymal Stem Cells for Treatment of Retinal Diseases. Stem Cell Rev. Rep. 2021, 17, 1154–1173. [Google Scholar] [CrossRef]
- Chen, X.; Jiang, Y.; Duan, Y.; Zhang, X.; Li, X. Mesenchymal-Stem-Cell-Based Strategies for Retinal Diseases. Genes 2022, 13, 1901. [Google Scholar] [CrossRef]
- Holan, V.; Palacka, K.; Hermankova, B. Mesenchymal Stem Cell-Based Therapy for Retinal Degenerative Diseases: Experimental Models and Clinical Trials. Cells 2021, 10, 588. [Google Scholar] [CrossRef] [PubMed]
- Reboussin. ; Buffault, J.; Brignole-Baudouin, F.; Goazigo, A.R.-L.; Riancho, L.; Olmiere, C.; Sahel, J.-A.; Parsadaniantz, S.M.; Baudouin, C. Evaluation of neuroprotective and immunomodulatory properties of mesenchymal stem cells in an ex vivo retinal explant model. J. Neuroinflammation 2022, 19, 1–17. [Google Scholar] [CrossRef]
- Stavely, R.; Nurgali, K. The emerging antioxidant paradigm of mesenchymal stem cell therapy. STEM CELLS Transl. Med. 2020, 9, 985–1006. [Google Scholar] [CrossRef]
- Angeloni, C.; Gatti, M.; Prata, C.; Hrelia, S.; Maraldi, T. Role of Mesenchymal Stem Cells in Counteracting Oxidative Stress—Related Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 3299. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.L.S.; Kumar, S.; Mok, P.L. Cellular Reparative Mechanisms of Mesenchymal Stem Cells for Retinal Diseases. Int. J. Mol. Sci. 2017, 18, 1406. [Google Scholar] [CrossRef]
- Holan, V.; Palacka, K.; Hermankova, B. Mesenchymal Stem Cell-Based Therapy for Retinal Degenerative Diseases: Experimental Models and Clinical Trials. Cells 2021, 10, 588. [Google Scholar] [CrossRef] [PubMed]
- Ruitenberg, M.J.; Vukovic, J.; Sarich, J.; Busfield, S.J.; Plant, G.W.; Yang, H.; He, B.-R.; Hao, D.-J.; Su, Z.; He, C.; et al. Olfactory Ensheathing Cells: Characteristics, Genetic Engineering, and Therapeutic Potential. J. Neurotrauma 2006, 23, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Huo, S.J.; Li, Y.C.; Xie, J.; Li, Y.; Raisman, G.; Zeng, Y.X.; He, J.R.; Weng, C.H.; Yin, Z.Q. Transplanted Olfactory Ensheathing Cells Reduce Retinal Degeneration in Royal College of Surgeons Rats. Curr. Eye Res. 2012, 37, 749–758. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zhang, Y.; Liu, X.; Ghazaryan, E.; Li, Y.; Xie, J.; Su, G. Recent Advances of Stem Cell Therapy for Retinitis Pigmentosa. Int. J. Mol. Sci. 2014, 15, 14456–14474. [Google Scholar] [CrossRef]
- Klassen, H.J.; Ng, T.F.; Kurimoto, Y.; Kirov, I.; Shatos, M.; Coffey, P.; Young, M.J. Multipotent Retinal Progenitors Express Developmental Markers, Differentiate into Retinal Neurons, and Preserve Light-Mediated Behavior. Investig. Opthalmology Vis. Sci. 2004, 45, 4167–4173. [Google Scholar] [CrossRef]
- Jiang, C.; Klassen, H.; Zhang, X.; Young, M. Laser injury promotes migration and integration of retinal progenitor cells into host retina. Mol. Vis. 2010, 16, 983–990. [Google Scholar] [PubMed]
- West, E.; Pearson, R.; Tschernutter, M.; Sowden, J.; MacLaren, R.; Ali, R. Pharmacological disruption of the outer limiting membrane leads to increased retinal integration of transplanted photoreceptor precursors. Exp. Eye Res. 2008, 86, 601–611. [Google Scholar] [CrossRef]
- Sahel, J.-A.; Boulanger-Scemama, E.; Pagot, C.; Arleo, A.; Galluppi, F.; Martel, J.N.; Degli Esposti, S.; Delaux, A.; de Saint Aubert, J.-B.; de Montleau, C.; et al. Partial recovery of visual function in a blind patient after optogenetic therapy. Nat. Med. 2021, 27, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Busskamp, V.; Picaud, S.; Sahel, J.A.; Roska, B. Optogenetic therapy for retinitis pigmentosa. Gene Ther. 2012, 19, 169–175. [Google Scholar] [CrossRef]
- De Silva, S.R.; Moore, A.T. Optogenetic approaches to therapy for inherited retinal degenerations. J. Physiol. 2022, 600, 4623–4632. [Google Scholar] [CrossRef] [PubMed]
- Busskamp, V.; Duebel, J.; Balya, D.; Fradot, M.; Viney, T.J.; Siegert, S.; Groner, A.C.; Cabuy, E.; Forster, V.; Seeliger, M.; et al. Genetic Reactivation of Cone Photoreceptors Restores Visual Responses in Retinitis Pigmentosa. Science 2010, 329, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Sakai, D.; Tomita, H.; Maeda, A. Optogenetic Therapy for Visual Restoration. Int. J. Mol. Sci. 2022, 23, 15041. [Google Scholar] [CrossRef] [PubMed]
- Alekseev, A.; Gordeliy, V.; Bamberg, E. Rhodopsin-Based Optogenetics: Basics and Applications. 2501. [CrossRef]
- Chow, B.Y.; Han, X.; Dobry, A.S.; Qian, X.; Chuong, A.S.; Li, M.; Henninger, M.A.; Belfort, G.M.; Lin, Y.; Monahan, P.E.; et al. High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature 2010, 463, 98–102. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, S.; Flossmann, T.; Gao, S.; Witte, O.W.; Nagel, G.; Holthoff, K.; Kirmse, K. Optimized photo-stimulation of halorhodopsin for long-term neuronal inhibition. BMC Biol. 2019, 17, 1–17. [Google Scholar] [CrossRef]
- Shen, Y.; Campbell, R.E.; Côté, D.C.; Paquet, M.-E. Challenges for Therapeutic Applications of Opsin-Based Optogenetic Tools in Humans. Front. Neural Circuits 2020, 14, 41. [Google Scholar] [CrossRef]
- Yin, X.; He, T.; Chen, R.; Cui, H.; Li, G. Impact of neurotrophic factors combination therapy on retinitis pigmentosa. J. Int. Med Res. 2020, 48. [Google Scholar] [CrossRef]
- Falsini, B.; Iarossi, G.; Chiaretti, A.; Ruggiero, A.; Manni, L.; Galli-Resta, L.; Corbo, G.; Abed, E. NGF eye-drops topical administration in patients with retinitis pigmentosa, a pilot study. J. Transl. Med. 2016, 14, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sieving, P.A.; Caruso, R.C.; Tao, W.; Coleman, H.R.; Thompson, D.J.S.; Fullmer, K.R.; Bush, R.A. Ciliary neurotrophic factor (CNTF) for human retinal degeneration: Phase I trial of CNTF delivered by encapsulated cell intraocular implants. Proc. Natl. Acad. Sci. USA 2006, 103, 3896–3901. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.Y.; Kulbay, M.; Toameh, D.; Xu, A.Q.; Kalevar, A.; Tran, S.D. Retinitis Pigmentosa: Novel Therapeutic Targets and Drug Development. Pharmaceutics 2023, 15, 685. [Google Scholar] [CrossRef]
- Ortega, J.T.; Jastrzebska, B. Neuroinflammation as a Therapeutic Target in Retinitis Pigmentosa and Quercetin as Its Potential Modulator. Pharmaceutics 2021, 13, 1935. [Google Scholar] [CrossRef]
- Komeima, K.; Rogers, B.S.; Campochiaro, P.A. Antioxidants slow photoreceptor cell death in mouse models of retinitis pigmentosa. J. Cell. Physiol. 2007, 213, 809–815. [Google Scholar] [CrossRef]
- Lee, S.Y.; Usui, S.; Zafar, A.-B.; Oveson, B.C.; Jo, Y.-J.; Lu, L.; Masoudi, S.; Campochiaro, P.A. N-acetylcysteine promotes long-term survival of cones in a model of retinitis pigmentosa. J. Cell. Physiol. 2010, 226, 1843–1849. [Google Scholar] [CrossRef]
- Sharma, A.; Jaganathan, B.G. Stem Cell Therapy for Retinal Degeneration: The Evidence to Date. Biol. Targets Ther. 2021, ume 15, 299–306. [Google Scholar] [CrossRef]
- Hinkle, J.W.; Mahmoudzadeh, R.; Kuriyan, A.E. Cell-based therapies for retinal diseases: a review of clinical trials and direct to consumer “cell therapy” clinics. Stem Cell Res. Ther. 2021, 12, 1–9. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of general processes underlying Retinitis Pigmentosa.
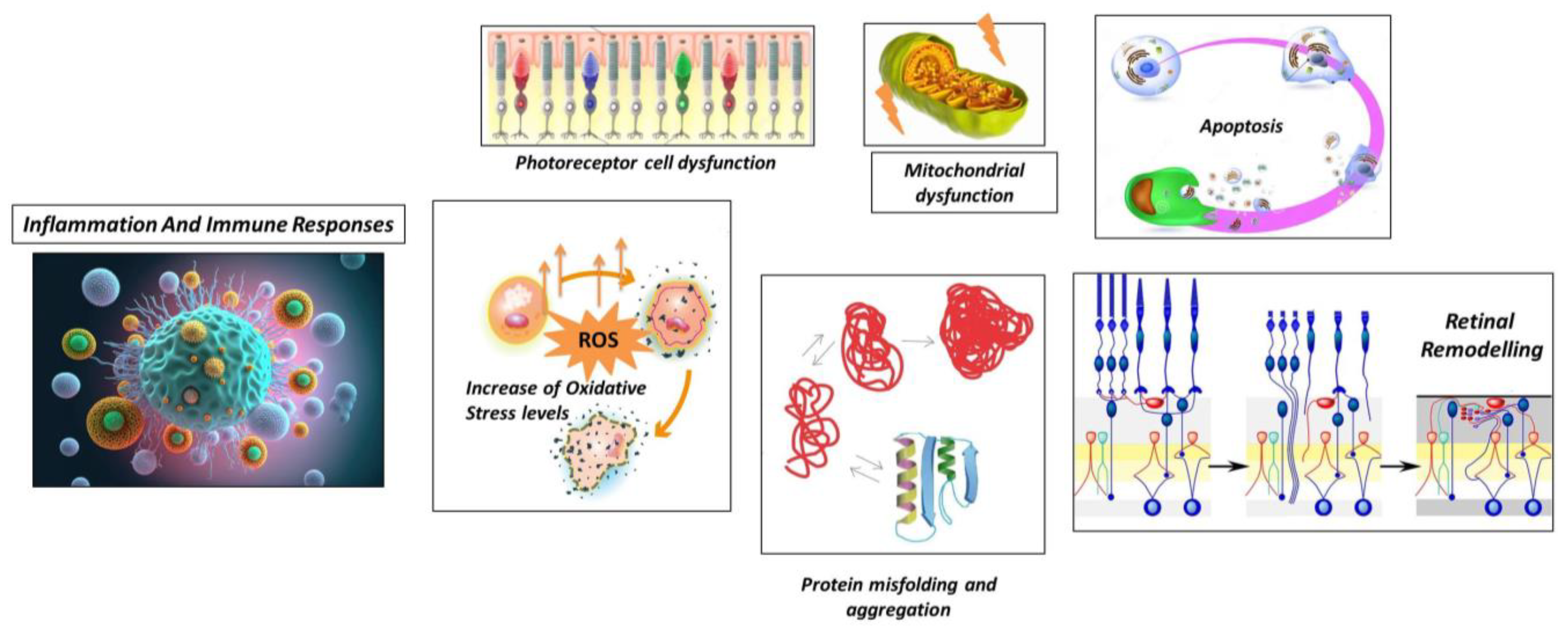
Figure 2.
Schematic representation of various therapeutic strategies for treating Retinitis Pigmentosa.
Figure 2.
Schematic representation of various therapeutic strategies for treating Retinitis Pigmentosa.
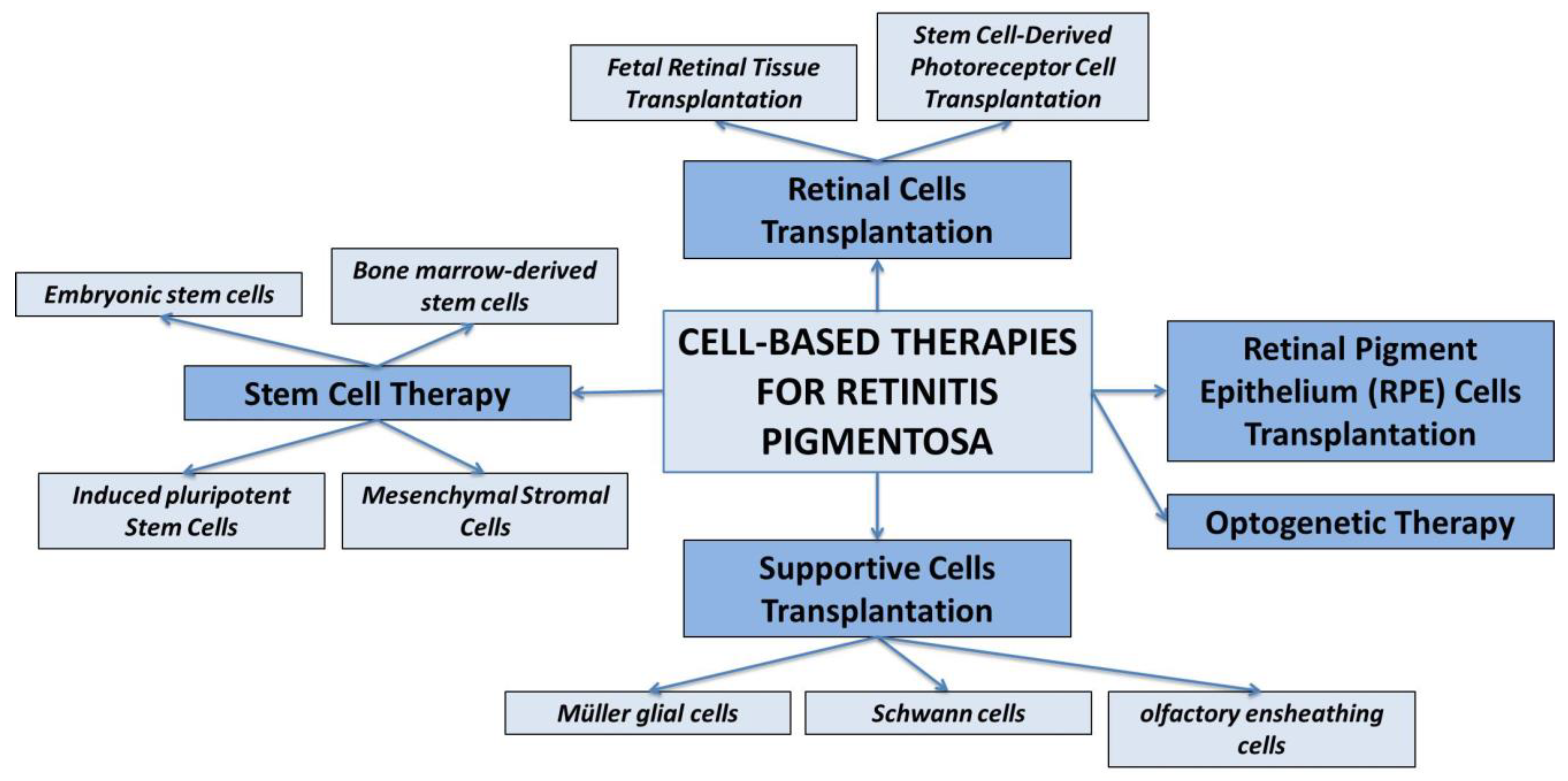

Table 1.
Function, effects of mutations and effects on retina’s structure of the main genes commonly involved in the pathogenesis of retinitis pigmentosa.
Table 1.
Function, effects of mutations and effects on retina’s structure of the main genes commonly involved in the pathogenesis of retinitis pigmentosa.
| GENE/PROTEIN | FUNCTION | EFFECTS OF MUTATIONS | EFFECTS ON RETINA’S STRUCTURE |
| Rhodopsin (RHO) | found in rod cells, plays a central role in phototransduction and rod photoreceptor cell health | alteration of the protein’s structure/function, abnormalities in the protein’s folding or trafficking | disruption and degeneration of rod photoreceptor cells, defects in phototransduction, reduced sensitivity to light |
| Peripherin/RDS (PRPH2) | found in the rod and cone cells plays a crucial role in the structural integrity and organization of the photoreceptor outer segments (essential for disk morphogenesis) | alteration of protein folding, stability, and interactions with other proteins. | Alteration of integrity and function of the outer segments, progressive degeneration of the photoreceptor associated with peripheral vision loss, and central vision impairment. |
| Cyclic Nucleotide-Gated (CNG) Channels | Non selective cation channels located in the outer segment of rod and cone photoreceptor cells are involved in the regulation of ion influx in response to light stimulation | impair the normal function of these channels | abnormalities in the phototransduction process, reduced sensitivity to light, decreased visual acuity, and progressive vision loss. Dysfunctional CNG channels can lead to cellular stress, oxidative damage |
| Retinal Pigment Epithelium-Specific 65 kDa Protein (RPE65) | component of the vitamin A visual cycle of the retina which supplies the 11-cis retinal chromophore of the photoreceptors opsin visual pigments | loss or dysfunction of the RPE65 protein, disrupting the visual cycle and impairing the regeneration of 11-cis-retinal | progressive loss of photoreceptors reduced sensitivity to light, decreased visual acuity |
| Retinitis Pigmentosa GTPase Regulator (RPGR) | predominantly localized in the connecting cilium and outer segment of photoreceptor cells in the retina, plays critical roles in the phototransduction cascade | impair the normal function | impaired ciliary transport, altered protein-protein interactions, or disrupted signaling pathways, leading to photoreceptor cell death and vision loss |
| Cone-Rod Homeobox Protein (CRX) | photoreceptor-specific transcription factor which plays a role in the differentiation of photoreceptor cells. This homeodomain protein is necessary for the maintenance of normal cone and rod function. | disrupt the normal function | impaired development and function of photoreceptor cells associated with degeneration of cones and rods |
|
Usher Syndrome Genes (MYO7A, USH1C, USH2A, GPR98, CLRN1) |
MYO7A encodes the protein myosin VIIA, involved in the development and maintenance of photoreceptor cells | impair the normal function | impaired development and function of photoreceptor cells associated with degeneration |
| USH1C encodes a scaffold protein involved in the organization of hair cell stereocilia and the synaptic connections in the retina | |||
| USH2A encodes a protein that contains laminin EGF motifs involved in the maintenance of the structure and function of the photoreceptor cells (maintenance of periciliary membrane complex) | |||
| GPR98 encodes the protein ADGRV1 involved into the development of photoreceptors ( maintenance of periciliary membrane complex) | |||
| CLRN1 encodes the protein Clarin 1 which play important role in development and homeostasis of the photoreceptor cells ( regulatory element for the synapses within the retina)an |
Table 2.
Relevant Clinical Trials with cell-based therapies for the treatment of retinitis pigmentosa www.clinicaltrials.gov last update 02.08.23.
Table 2.
Relevant Clinical Trials with cell-based therapies for the treatment of retinitis pigmentosa www.clinicaltrials.gov last update 02.08.23.
| ID | NAME | PHASE | AIM | METHODS |
| NCT02320812 | A Prospective, Multicenter, Open-Label, Single-Arm Study of the Safety and Tolerability of a Single, Intravitreal Injection of Human Retinal Progenitor Cells (jCell) in Adult Subjects With Retinitis Pigmentosa (RP) | 1/2 | Test the safety,tolerability and efficacy (impact on visual status) of the administration of a single dose of jCell | single intravitreal injection of 0.5 - 3.0x106 human retinal progenitor cells (hRPC- jCell) |
| NCT04925687 | Phase 1 Study of Intravitreal Autologous CD34+ Stem Cell Therapy for Retinitis Pigmentosa (BMSCRP1) | 1 | Determine safety and feasibility of injection of autologous CD34+ stem cells harvested from bone marrow. | Intravitreal injection of autologous CD34+ cells harvested from bone marrow under GMP conditions |
| NCT04763369 | Investigation of Therapeutic Efficacy and Safety of UMSCs for the Management of Retinitis Pigmentosa (RP) | 1/2 | Investigate the safety and therapeutic efficacy of UMSC (umbilical cord derived mesenchymal stem cells) injection employing two different routes (sub-tenon injection versus suprachoroidal injection) | sub-tenon and suprachoroidal injection of UMSCs |
| NCT05909488 | Role of UC-MSC and CM to Inhibit Vision Loss in Retinitis Pigmentosa Phase I/II | 2/3 | Investigate the safety and therapeutic efficacy of peribulbar injection of Umbilical Cord-derived Mesenchymal Stem Cell (UC-MSC) with Conditioned Medium (CM) | Peribulbar injection of 1.5- 5 x 106UC-MSC + CM |
| NCT03944239 | Safety and Efficacy of Subretinal Transplantation of Clinical Human Embryonic Stem Cell Derived Retinal Pigment Epitheliums in Treatment of Retinitis Pigmentosa | 1/2 | Test the safety and therapeutic efficacy of clinical level human embryonic stem cells derived retinal pigment epithelium transplantation | Subretinal transplantation of clinical human embryonic stem cell derived retinal pigment epitheliums |
| NCT01531348 | Intravitreal Injection of MSCs in Retinitis Pigmentosa | 1 | Determine feasibility and safety of Adult Human Bone Marrow-Derived Mesenchymal Stem Cells (BM-MSC) by Intravitreal Injection | Intravitreal Injection of 1x 106 BM-MSC in balanced salt solution |
| NCT03073733 | Safety and Efficacy of Intravitreal Injection of Human Retinal Progenitor Cells in Adults With Retinitis Pigmentosa | 2 | Evaluation of safety and efficacy of intravitreal injection of human retinal progenitor (hRPC) | Intravitreal injection of 3.0-6.0x 106 of human retinal progenitor cells (hRPC) suspended in clinical grade medium |
| NCT04284293 | CNS10-NPC for the Treatment of RP | 1 | Assess the safety and tolerability of two escalating doses of clinical grade human fetal cortical-derived neural progenitor cells (CNS10-NPC).Determine if CNS10-NPC can engraft and survive long-term in the retina of transiently immunosuppressed subjects.Obtain evidence that subretinal injection of CNS10-NPC can favorably impact the progression of vision loss in subjects with moderate RP. | human neural progenitor cells (CNS10-NPC) sub retinal space implantation |
| NCT02709876 | Autologous Bone Marrow-Derived CD34+, CD133+, and CD271+ Stem Cell Transplantation for Retinitis Pigmentosa | 1/2 | Assess the safety and efficacy of purified adult autologous bone marrow derived CD34+, CD133+, and CD271+ stem cells through a 48 month follow-up period. The combination of these three cell types was based on their diverse potentialities to differentiate into specific functional cell types to regenerate damaged retinal tissue | Intravitreal injection of Bone marrow-derived CD34+, CD133+, CD271+ stem cells in 1.0 ml normal saline |
| NCT03963154 | Interventional Study of Implantation of hESC-derived RPE in Patients With RP Due to Monogenic Mutation | 1/2 | Study the safety, tolerability and preliminary efficacy of implantation into one eye of hESC-derived RPE (Human Embryonic Stem Cell Derived Retinal Pigment Epithelium (RPE)) | Implantation into one eye of hESC-derived RPE (Human Embryonic Stem Cell Derived Retinal Pigment Epithelium (RPE)) |
| NCT03566147 | Treatment of RP and LCA by Primary RPE Transplantation | Early 1 | Study the Safety and Preliminary Efficacy of Human primary Retinal Pigment Epithelial (HuRPE) Cells Subretinal Transplantation | subretinal space transplantation of 0.3- 1 x106 HuRPE cells through a standard surgical approach. |
| NCT03772938 | Stem Cells Therapy in Degenerative Diseases of the Retina | 1 | Investigation of the safety and efficacy of intravitreal injection of autologous bone marrow-isolated stem/progenitor cells with different selected phenotypes. Especially, this clinical trial is designated to test the therapeutic (pro-regenerative and neuro-protective) functions of different stem/progenitor cell populations able to secrete bioactive neurotrophic factors. | intravitreal injection of Human autologous bone marrow-derived stem/progenitor cell |
| NCT05147701 | Safety of Cultured Allogeneic Adult Umbilical Cord Derived Mesenchymal Stem Cells for Eye Diseases | 1 | Study the safety and efficacy of intravenous and sub-tenon delivery of cultured allogeneic adult umbilical cord derived mesenchymal stem cells (UC-MSCs) | intravenous and sub-tenon injection of 1 x106 allogeneic adult umbilical cord derived mesenchymal stem cells |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



