
Submitted:
01 September 2023
Posted:
05 September 2023
You are already at the latest version
Abstract
The pathogenesis of Cerebral Small Vessel Disease (CSVD) is largely unknown. Endothelial disfunction has been suggested as the turning point in CSVD development. In this study we tested the effect of plasma from CSVD patients on human cerebral microvascular endothelial cells with the aim of describing the pattern of endothelial activation. Plasma samples from 3 groups of young subjects have been tested: PTs (subjects affected by early stage CSVD); CTRLs (control subjects without abnormalities at MRI scanning); BDs (blood donors). Human Brain Endothelial Cells 5i (HBEC5i) were treated with plasma and total RNA was extracted. RNAs were pooled to reduce gene expression-based variability and NGS analysis was performed. Differentially expressed genes were highlighted comparing PTs, CTRLs and BDs with HBEC5i untreated cells. No significantly altered pathway was evaluated in BD related treatment. Regulation of p38 MAPK cascade (GO:1900744) was the solely pathway altered in CTRLs related treatment. Indeed, 36 different biological processes turned out to be deregulated after PTs treatment of HBEC5i, i.e., the cytokine-mediated signaling pathway (GO:0019221). Endothelial cells activate inflammatory pathways in response to stimuli from CSVD patients’ plasma, suggesting the pathogenetic role of neuroinflammation from the early asymptomatic phases of cerebrovascular disease.
Keywords:
neuroinflammation
; cytokines
; endothelial dysfunction
; cerebral small vessel disease
1. Introduction
Cerebral small vessel disease (CSVD) is one of the most common types of cerebrovascular diseases. With a prevalence that increases exponentially with aging, CSVD is one of the major risk factors for acute stroke and cognitive decline[1]. The pathogenesis of CSVD is largely unknown and consequently tailored preventive treatments are currently unavailable [1,2]. There is large evidence that classical vascular risk factors, above all hypertension, and micro-atherosclerosis of small arteries play a pivotal role, however, acquired or genetically determined structural abnormalities in the extracellular matrix have been demonstrated in several CSVD forms [3].
Endothelial disfunction has been suggested as the turning point in CSVD development. Conventional atherosclerosis, systemic inflammation, perivascular space abnormalities, as well as glymphatic system disruption, all seem to converge towards blood brain barrier dysfunction and finally, progressively, towards failure of vascular homeostasis[4,5,6,7]. However, CSVD includes several heterogenous diseases, and overlap with other disorders (Alzheimer’s Disease, Parkinson’s Disease, Multiple Sclerosis, rheumatologic diseases, etc.) and chronic conditions (such as chronic hypoxia, obesity, diabetes, congestive heart failure, metabolic syndrome etc.)[2,8,9,10,11].
It is not unexpected that, despite several studies explored this field, we still not know which pathways are chronically and firstly activated. That information would nevertheless be essential to indicate new drug treatments and prevention strategies in cerebrovascular disease [2].
Thus, in this study we aimed to investigate the possible effects of plasma from CSVD patients on human cerebral microvascular endothelial cell molecular expression with particular attention to pathways of endothelial activation and inflammation.
2. Materials and Methods
2.1. Patients’ samples selection process
This study was performed on a subgroup of plasma samples derived from the previous study[12], selecting 9 patients with CSVD (PT: 8 females and 1 male; mean age 50.6 ± 7.7 years) without history of cerebrovascular events and with less than 2 vascular risk factors and 7 control subjects from the same study without CSVD or other abnormalities at MRI scanning (CTRLs: 5 females and 2 males; mean age 51.0 ± 14.9 years). We also added 6 blood donors (BDs: 4 females and 2 males; mean age 33.3 ± 18.5 years) followed at our Transfusion Medicine Department.
Plasma of patients and controls were obtained by centrifugation from EDTA-treated blood samples and frozen at -80°C until use.
2.2. Human Brain Endothelial Cells Culture and Treatments
The Human Brain Endothelial Cells 5i (HBEC5i, ATCC® CRL3245 ™) were maintained in vessels coated with 0.1% Gelatin (ATCC® No. PCS999027) at 37 °C in 5% CO2 atmosphere in DMEM: F12 (D8437, Merk KGaA) supplemented with 40 μg/mL endothelial growth supplement (ECGS; E2759 Merk KGaA), Fetal Bovine Serum (FBS, Merk KGaA) to a final concentration of 10% and Penicillin– Streptomycin solution (P4333, Merk KGaA) to a final concentration of 1%.
HBEC5i cells were seeded at a density of 15,000 cells/cm2 and, the day after, the culture medium was replaced with fresh medium supplemented with Penicillin– Streptomycin solution to a final concentration of 1% and 15% v/v of plasma from CSVD patients (n=9), control (n=7) or blood donors (n=6).
Cells were treated for 24h. For untreated cells, culture medium was replaced with fresh medium supplemented with Penicillin– Streptomycin solution to a final concentration of 1% and FBS to a final concentration of 10%.
After treatments, HBEC5i were collected, and cell viability was determined using the trypan blue (T6146, Merk KGaA) exclusion method.
2.3. RNA isolation, library preparation and Next Generation Sequencing (NGS)
Total RNA was extracted from cells using the RNeasy Kits (QIAGEN), according to the manufacturer’s protocol. Extracted RNA was quantified using the Qubit RNA high-sensitivity assay kit (Q32852, Thermo Fisher Scientific) and stored at − 80 °C until use.
RNA from multiple biological replicates (9 PTs, 5 CTRLs, 4 BDs) were pooled prior sequencing to reduce gene expression-based variability as described in: [13].
TruSeq Stranded Total RNA with Ribo-Zero Human/Mouse/Rat kit (Illumina) was used for library preparation following the manufacturer’s instructions, starting with 200 ng of good quality RNA (R.I.N. >7) as input. Both RNA samples and final libraries were quantified by using the Qubit 2.0 Fluorometer (Thermo Fisher Scientific) and quality tested by Agilent 2100 Bioanalyzer RNA Nano assay (Agilent technologies). Libraries were then processed with Illumina cBot for cluster generation on the flowcell, following the manufacturer’s instructions and sequenced on 50 bp single-end mode at the on HiSeq2500 (Illumina). The CASAVA 1.8.2 version of the Illumina pipeline was used to processed raw data for both format conversion and de-multiplexing.
2.4. RNA-Seq bioinformatics analysis
Raw sequences were analyzed as previously described [14]. Briefly, sequencing reads quality was evaluated using the ShortRead (v1.44.3) R/Bioconductor package [15]. Quality, adapters and contamination filtering were performed using the Trimmomatic [16] command-line tool. Processed reads were aligned to the human reference genome GRCh37/hg19 and differentially expressed genes were identified using the DESeq2 (v1.26.0) R/Bioconductor package [17], considering as statistically significant the results having a log2 fold change (FC) ≥ 1.5 and a FDR adjusted p-value (i.e., q-value) ≤ 0.05. Extended gene annotations (including HGNC gene symbol, description and transcript type) were obtained using the biomaRt (v2.42.0) R/Bioconductor package [18]. Heatmaps were obtained by processing sequencing data with the online tool Morpheus (https:// software.broadinstitute.org/morpheus). Pathway analysis was performed with the web tool GOrilla (http://cbl-gorilla.cs.technion.ac.il/), setting a FDR adjusted p-value ≤ 0.05 for significance.
2.5. Target gene expression analysis
Complementary DNA (cDNA) was generated using the SuperScript™ IV VILO™ Master Mix (11756050, Thermo Fisher Scientific) using 2.5µg of total RNA, according to manufacturer’s protocols.
Quantitative PCR (qPCR) was performed using SsoAdvance Universal SYBR green super mix (Bio-Rad Laboratories) on a QuantStudio 3 System (Applied Biosystems). The QuantStudio Design and Analysis software v1.5.0 (Applied Biosystems), was used to calculate mRNA levels with the 2−∆∆Ct method, and GAPDH was used as reference. Oligonucleotide primers were purchased from Merk KGaA and their sequences are available upon request. All experiments were performed in triplicate.
2.6. Statistical analysis
Statistical analysis was performed using GraphPad Prism Software v.5 (GraphPad Software). Data were tested for normal distribution using the Kolmogorov–Smirnov test. After assessing the absence of a normal distribution, repeated measurements were analyzed by t-Student analysis followed by the Wilcoxon post-test for viability and one-way analysis of variance (ANOVA) followed by the Dunnett post-test for relative gene expression analysis. p values less than 0.05 were considered significant. Quantitative variables were expressed as mean ± standard deviation (SD).
3. Results
3.1. Treatments with plasma
To evaluate the hypothesis of the key role of endothelial dysfunction in CSVD, HBEC5i were treated for 24 hours with plasma from either CSVD patients (PTs), controls (CTRLs) or blood donors (BDs). We enrolled as controls age- and sex- matched subjects experiencing and undergoing brain MRI that displayed normal scans. Moreover, other neurological, vascular, and chronic inflammatory diseases were excluded during a clinical interview[12]. To set up the experiments and rule out any possible cellular activation mediated by plasma itself, plasma from blood donors was also used as a control.
To assess whether treatments with human plasma affected cell viability, HBEC5i cells were treated with plasma from blood donors, controls, and patients (final concentration 15% v/v) for 24 hours and viability was assessed by trypan blue assay (n=3). As shown in Figure 1, none of the treatments significantly affected cell viability.
3.2. Study of the effects of CSVD plasma incubation on molecular expression in HBEC5i cells
To evaluate the possible effects of plasma from CSVD patients at the molecular mechanism with particular attention to pathways related to endothelial dysfunction, RNA-sequencing was performed. HBEC5i cells were treated for 24 hours as previously described in the methods and total RNA was extracted. Replicates from the same treatment were pooled together and then underwent sequencing.
As represented in the heat map in Figure 2, HBEC5i showed a quite different response depending on the treatment performed.
After removing low quantity reads, 219, 175 and 174 differentially expressed genes were highlighted comparing treatment with plasma of PTs, CTRLs and BDs HBEC5i untreated cells, (at log2 fold change ≥ 1.5). In particular, 115 transcripts were up-regulated, and 104 transcripts were down-regulated in PTs-treated cells; 84 transcripts were up-regulated, and 91 transcripts were down-regulated in CTRLs-treated cells; 87 transcripts were up-regulated, and 87 transcripts were down-regulated in BD-treated cells.
Differentially expressed genes were then subjected to pathway analysis to outline which pathways were mostly affected by each treatment. Pathways commonly regulated were excluded from the analysis. No significantly altered pathway was evaluated in BD related treatment. Regulation of p38 MAPK cascade (GO:1900744) was the solely pathway altered in CTRLs-related treatment. Indeed, 36 different biological processes turned out to be deregulated after PTs plasma treatment of HBEC5i. Data are represented in Table 1.
Then, we focused on the cytokine-mediated signaling pathway (GO:0019221) From this pathway, we selected 3 representative cytokines involved in neuroinflammation and mRNA levels of CXCL8, LIF and IL12A were assessed by qPCR to confirm RNA-seq related up-regulation. Indeed, as shown in Figure 3, compared to untreated cells, all three transcripts turned out to be up regulated in treated cells.
4. Discussion
In this study, we herein showed that treating brain endothelial cells with plasma from patients with “non-lacunar, asymptomatic, CSVD” upregulates cytokine-mediated signalling pathways, possibly involved in neuroinflammation. This suggests that in those patients a low-grade inflammation at systemic level may be responsible of stressed endothelial cells at cerebral level.
Although this research does not clarify which molecule(s) specifically act on endothelial cells, it highlights the link between a pro-inflammatory phenotype and the early asymptomatic stages of cerebrovascular disease; moreover, it suggests that one or more specific biomarkers of endothelial activation and inflammation could be detected directly in the blood.
In the last decades the link between inflammation and CSVD has already been investigated and proposed as one of the key mainstays of its pathogenesis [19]. Beyond brain imaging, blood derived biomarkers have been investigated, both in the cerebrospinal fluid and in the plasma [20,21,22], but they are still poorly characterized. Indexes of overall immune status, such as “neutrophil-to-lymphocytes ratio” (NFL) and “systemic immune-inflammation” (SII), proved to be positively correlated with some CSVD features, although this evidence is only partially consistent all through the Literature [23]. Mid-regional pro-adrenomedulin (MR-proADM) has been proposed as biomarker of CSVD progression in a longitudinal observational study [24]. Systemic traditional inflammation runs parallel with immune cells trained by atherogenic substances exposure, perpetuating a proinflammatory phenotype: recent research showed that cytokine release is durably enhanced in established atherosclerosis and that cytokines such as CXCL8 and IL-17 are preferential biomarkers of trained immunity [25].
The cytokine-mediated signalling pathways and their consequent effects on cells, on diseases occurrence and eventually differential phenotyping, is still an evolving field, above all in neurological disorders. To the best of our knowledge, there is very few evidence of the role of the specific chemokines we found overexpressed in our CSVD sample.
CXCL8 is a widely studied chemokine, with several effects on cell interactions: it is chemo-attractive for neutrophil, chemotactic for endothelial cells, and stimulate stem cells, thus proving to be involved in acute inflammation and angiogenesis [26]. It showed several effects within the CNS as well, although often not completely understood, on proliferation, migration, survival of and interaction between neurons and glial cells. Some of these effects have been proposed as distinctive elements in selective inflammatory diseases of the CNS such as Multiple Sclerosis [27], but also in neuroinflammation associated to neurodegenerative diseases such as Alzheimer’s Disease [28]. Few studies also explored CXCL8 signalling in cerebrovascular disorders. Zheng et al. demonstrated in a microarray analysis, that CXCL8 overexpression (along with TNF, SOC3 and TNFAIP3) could serve as biomarker of both coronary artery disease (CAD) and Ischemic Stroke (IS) occurrence [29]. Moreover, in a rat model of acute stroke, inhibition of CXCL8 receptor with Reparixin improved neurological outcome along with a reduction in inflammatory response [30]. Inconsistent results in patients with sickle cells anaemia, in which poor outcome (i.e., number of recurrent VOC) seems to depend on high levels of IL-6, rather than CXCL8 [31], likely underline once more the heterogeneity of cerebrovascular diseases.
The Leukemia Inhibitory Factor (LIF) is an anti-inflammatory cytokine that acts in a signalling cascade that promotes cell survival and neuroprotection through MAPK, PI3K/Akt and JAK/STAT pathways. The activation of this cascade has been demonstrated both in rat models of stroke and in cultured neurons [32,33]. LIF receptor expression on plasma membrane occurring after a brain injury is also determinant in enhancing neuroprotective effect [33]. LIF overexpression in our treated endothelial cells suggests that those pathways could be activated also in CSVD, possibly trying to counteract endothelial damage.
A mouse and in vitro study recently explored the mechanism of microglia chemotaxis within ischemic brain tissue. IL12A is suggested to contribute to chemotactic signalling for neutrophils, together with other chemokines, soon after brain ischemia [34].
The availability of blood-based biomarkers could also pave the way for additional drug treatment of CSVD.
5. Conclusions
Consistently with the little evidence published in acute stroke animal and in vitro models, our study suggests that endothelial cells activate inflammatory pathways in response to CSVD patients’ plasma. Since previous studies indicate that the up-regulated patterns we found in cerebral endothelial cells are globally non-protective, similarly to acute ischemic stroke, we propose that neuroinflammatory pathways already activate in early and asymptomatic phases of cerebrovascular disease.
Biomarkers of neuroinflammation could be detected earlier, longitudinally, and directly in the blood of aging or at risk population, thus possibly allowing a more specific pharmacological treatment than traditional vascular preventive drugs.
Further research is needed to overcome limitations of this and other studies, above all trying to consider the high heterogeneity of CSVD and the lack of longitudinal assessment of inflammatory response.
Author Contributions
Conceptualization, A.C. and F.J.; methodology, A.C., C.M., and R.D.; patient’s selection and evaluation, blood sample collection, M.F, M.E.P. and R.G.; formal analysis, A.C. and C.M.; resources, M.R.V. and F.C.; data curation, A.C., C.M. and F.J.; writing—original draft preparation, A.C., C.M. and F.J.; writing—review and editing, M.F., M.R.V. and F.C.; supervision, M.R.V, M.F., and F.C.; All authors have read and agreed to the published version of the manuscript.”
Funding
This research received no external funding.
Institutional Review Board Statement
The study was performed in accordance with the principles of good clinical and laboratory practice. All patients and controls gave their informed consent to this retrospective study according to the Declaration of Helsinki and to the Italian legislation (Authorization of the Privacy Guarantor No. 9, 12th of December 2013). Our local ethic committee (Institutional Review Board of the Department of Medical Area, University of Udine) approved the study protocol (approval number 46/IRB/_gigli_16). All samples were anonymized.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jaime Garcia, D.; Chagnot, A.; Wardlaw, J.M.; Montagne, A. A Scoping Review on Biomarkers of Endothelial Dysfunction in Small Vessel Disease: Molecular Insights from Human Studies. International Journal of Molecular Sciences 2023, 24, 13114. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.E.; Taylor, J.L.; Smith, C.J.; Pritchard, H.A.T.; Greenstein, A.S.; Allan, S.M. Cardiovascular Comorbidities, Inflammation, and Cerebral Small Vessel Disease. Cardiovasc Res 2021, 117, 2575–2588. [Google Scholar] [CrossRef] [PubMed]
- Horsburgh, K.; Wardlaw, J.M.; van Agtmael, T.; Allan, S.M.; Ashford, M.L.J.; Bath, P.M.; Brown, R.; Berwick, J.; Cader, M.Z.; Carare, R.O.; et al. Small Vessels, Dementia and Chronic Diseases - Molecular Mechanisms and Pathophysiology. Clin Sci (Lond) 2018, 132, 851–868. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Norman, J.E.; Srinivasan, V.J.; Rutledge, J.C. Metabolic, Inflammatory, and Microvascular Determinants of White Matter Disease and Cognitive Decline. Am J Neurodegener Dis 2016, 5, 171–177. [Google Scholar] [PubMed]
- Walsh, J.; Tozer, D.J.; Sari, H.; Hong, Y.T.; Drazyk, A.; Williams, G.; Shah, N.J.; O’Brien, J.T.; Aigbirhio, F.I.; Rosenberg, G.; et al. Microglial Activation and Blood-Brain Barrier Permeability in Cerebral Small Vessel Disease. Brain 2021, 144, 1361–1371. [Google Scholar] [CrossRef]
- Tian, Y.; Zhao, M.; Chen, Y.; Yang, M.; Wang, Y. The Underlying Role of the Glymphatic System and Meningeal Lymphatic Vessels in Cerebral Small Vessel Disease. Biomolecules 2022, 12, 748. [Google Scholar] [CrossRef]
- Brown, R.; Benveniste, H.; Black, S.E.; Charpak, S.; Dichgans, M.; Joutel, A.; Nedergaard, M.; Smith, K.J.; Zlokovic, B.V.; Wardlaw, J.M. Understanding the Role of the Perivascular Space in Cerebral Small Vessel Disease. Cardiovasc Res 2018, 114, 1462–1473. [Google Scholar] [CrossRef]
- Jellinger, K.A. Alzheimer Disease and Cerebrovascular Pathology: An Update. J Neural Transm (Vienna) 2002, 109, 813–836. [Google Scholar] [CrossRef]
- Kang, P.; Ying, C.; Chen, Y.; Ford, A.L.; An, H.; Lee, J.-M. Oxygen Metabolic Stress and White Matter Injury in Patients With Cerebral Small Vessel Disease. Stroke 2022, 53, 1570–1579. [Google Scholar] [CrossRef]
- Maiuolo, J.; Muscoli, C.; Gliozzi, M.; Musolino, V.; Carresi, C.; Paone, S.; Ilari, S.; Mollace, R.; Palma, E.; Mollace, V. Endothelial Dysfunction and Extra-Articular Neurological Manifestations in Rheumatoid Arthritis. Biomolecules 2021, 11, 81. [Google Scholar] [CrossRef]
- Markus, H.S.; de Leeuw, F.E. Cerebral Small Vessel Disease: Recent Advances and Future Directions. Int J Stroke 2023, 18, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Janes, F.; Cifù, A.; Pessa, M.E.; Domenis, R.; Gigli, G.L.; Sanvilli, N.; Nilo, A.; Garbo, R.; Curcio, F.; Giacomello, R.; et al. ADMA as a Possible Marker of Endothelial Damage. A Study in Young Asymptomatic Patients with Cerebral Small Vessel Disease. Sci Rep 2019, 9, 14207. [Google Scholar] [CrossRef] [PubMed]
- Takele Assefa, A.; Vandesompele, J.; Thas, O. On the Utility of RNA Sample Pooling to Optimize Cost and Statistical Power in RNA Sequencing Experiments. BMC Genomics 2020, 21, 312. [Google Scholar] [CrossRef] [PubMed]
- Codrich, M.; Dalla, E.; Mio, C.; Antoniali, G.; Malfatti, M.C.; Marzinotto, S.; Pierobon, M.; Baldelli, E.; Di Loreto, C.; Damante, G.; et al. Integrated Multi-Omics Analyses on Patient-Derived CRC Organoids Highlight Altered Molecular Pathways in Colorectal Cancer Progression Involving PTEN. J Exp Clin Cancer Res 2021, 40, 198. [Google Scholar] [CrossRef]
- Morgan, M.; Anders, S.; Lawrence, M.; Aboyoun, P.; Pagès, H.; Gentleman, R. ShortRead: A Bioconductor Package for Input, Quality Assessment and Exploration of High-Throughput Sequence Data. Bioinformatics 2009, 25, 2607–2608. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol 2014, 15, 550. [Google Scholar] [CrossRef]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping Identifiers for the Integration of Genomic Datasets with the R/Bioconductor Package BiomaRt. Nat Protoc 2009, 4, 1184–1191. [Google Scholar] [CrossRef]
- Poggesi, A.; Pasi, M.; Pescini, F.; Pantoni, L.; Inzitari, D. Circulating Biologic Markers of Endothelial Dysfunction in Cerebral Small Vessel Disease: A Review. J Cereb Blood Flow Metab 2016, 36, 72–94. [Google Scholar] [CrossRef]
- Paolini Paoletti, F.; Simoni, S.; Parnetti, L.; Gaetani, L. The Contribution of Small Vessel Disease to Neurodegeneration: Focus on Alzheimer’s Disease, Parkinson’s Disease and Multiple Sclerosis. Int J Mol Sci 2021, 22, 4958. [Google Scholar] [CrossRef]
- Moretti, R.; Caruso, P. Small Vessel Disease: Ancient Description, Novel Biomarkers. Int J Mol Sci 2022, 23, 3508. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, G.A. Matrix Metalloproteinase-Mediated Neuroinflammation in Vascular Cognitive Impairment of the Binswanger Type. Cell Mol Neurobiol 2016, 36, 195–202. [Google Scholar] [CrossRef] [PubMed]
- L, J.; X, C.; D, Y.; J, J.; L, M.; Y, Y.; S, L.; A, J.; X, M.; H, L.; et al. Association of Inflammatory Markers with Cerebral Small Vessel Disease in Community-Based Population. Journal of neuroinflammation 2022, 19. [Google Scholar] [CrossRef]
- Kuriyama, N.; Ihara, M.; Mizuno, T.; Ozaki, E.; Matsui, D.; Watanabe, I.; Koyama, T.; Kondo, M.; Tokuda, T.; Tamura, A.; et al. Association between Mid-Regional Proadrenomedullin Levels and Progression of Deep White Matter Lesions in the Brain Accompanying Cognitive Decline. J Alzheimers Dis 2017, 56, 1253–1262. [Google Scholar] [CrossRef]
- Noz, M.P.; Ter Telgte, A.; Wiegertjes, K.; Joosten, L.A.B.; Netea, M.G.; de Leeuw, F.-E.; Riksen, N.P. Trained Immunity Characteristics Are Associated With Progressive Cerebral Small Vessel Disease. Stroke 2018, 49, 2910–2917. [Google Scholar] [CrossRef]
- Matsushima, K.; Yang, D.; Oppenheim, J.J. Interleukin-8: An Evolving Chemokine. Cytokine 2022, 153, 155828. [Google Scholar] [CrossRef]
- Watson, A.E.S.; Goodkey, K.; Footz, T.; Voronova, A. Regulation of CNS Precursor Function by Neuronal Chemokines. Neurosci Lett 2020, 715, 134533. [Google Scholar] [CrossRef]
- Shaik-Dasthagirisaheb, Y.B.; Conti, P. The Role of Mast Cells in Alzheimer’s Disease. Adv Clin Exp Med 2016, 25, 781–787. [Google Scholar] [CrossRef]
- Zheng, P.-F.; Liao, F.-J.; Yin, R.-X.; Chen, L.-Z.; Li, H.; Nie, R.-J.; Wang, Y.; Liao, P.-J. Genes Associated with Inflammation May Serve as Biomarkers for the Diagnosis of Coronary Artery Disease and Ischaemic Stroke. Lipids Health Dis 2020, 19, 37. [Google Scholar] [CrossRef]
- Villa, P.; Triulzi, S.; Cavalieri, B.; Di Bitondo, R.; Bertini, R.; Barbera, S.; Bigini, P.; Mennini, T.; Gelosa, P.; Tremoli, E.; et al. The Interleukin-8 (IL-8/CXCL8) Receptor Inhibitor Reparixin Improves Neurological Deficits and Reduces Long-Term Inflammation in Permanent and Transient Cerebral Ischemia in Rats. Mol Med 2007, 13, 125–133. [Google Scholar] [CrossRef]
- Domingos, I.F.; Pereira-Martins, D.A.; Sobreira, M.J.V.C.; Oliveira, R.T.D.; Alagbe, A.E.; Lanaro, C.; Albuquerque, D.M.; Blotta, M.H.S.L.; Araujo, A.S.; Costa, F.F.; et al. High Levels of Proinflammatory Cytokines IL-6 and IL-8 Are Associated with a Poor Clinical Outcome in Sickle Cell Anemia. Ann Hematol 2020, 99, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.M.; Pennypacker, K.R. The Role of the Leukemia Inhibitory Factor Receptor in Neuroprotective Signaling. Pharmacol Ther 2018, 183, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.M.; Collier, L.A.; Foran, E.A.; Leonardo, C.C.; Ajmo, C.T.; Pennypacker, K.R. Neuroprotective Activity of Leukemia Inhibitory Factor Is Relayed through Myeloid Zinc Finger-1 in a Rat Model of Stroke. Metab Brain Dis 2019, 34, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Pu, Z.; Bao, X.; Xia, S.; Shao, P.; Xu, Y. Serpine1 Regulates Peripheral Neutrophil Recruitment and Acts as Potential Target in Ischemic Stroke. J Inflamm Res 2022, 15, 2649–2663. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
(A) Cell viability was determined by trypan blue exclusion assay (n=3). Columns, mean; bars, SD. Treatment with plasma did not induce significant difference compared to untreated cells. (B) Representative phase contrast images (10x magnification) of HBEC5i untreated or treated with 15% v/v of human plasma for 24 hours.
Figure 1.
(A) Cell viability was determined by trypan blue exclusion assay (n=3). Columns, mean; bars, SD. Treatment with plasma did not induce significant difference compared to untreated cells. (B) Representative phase contrast images (10x magnification) of HBEC5i untreated or treated with 15% v/v of human plasma for 24 hours.
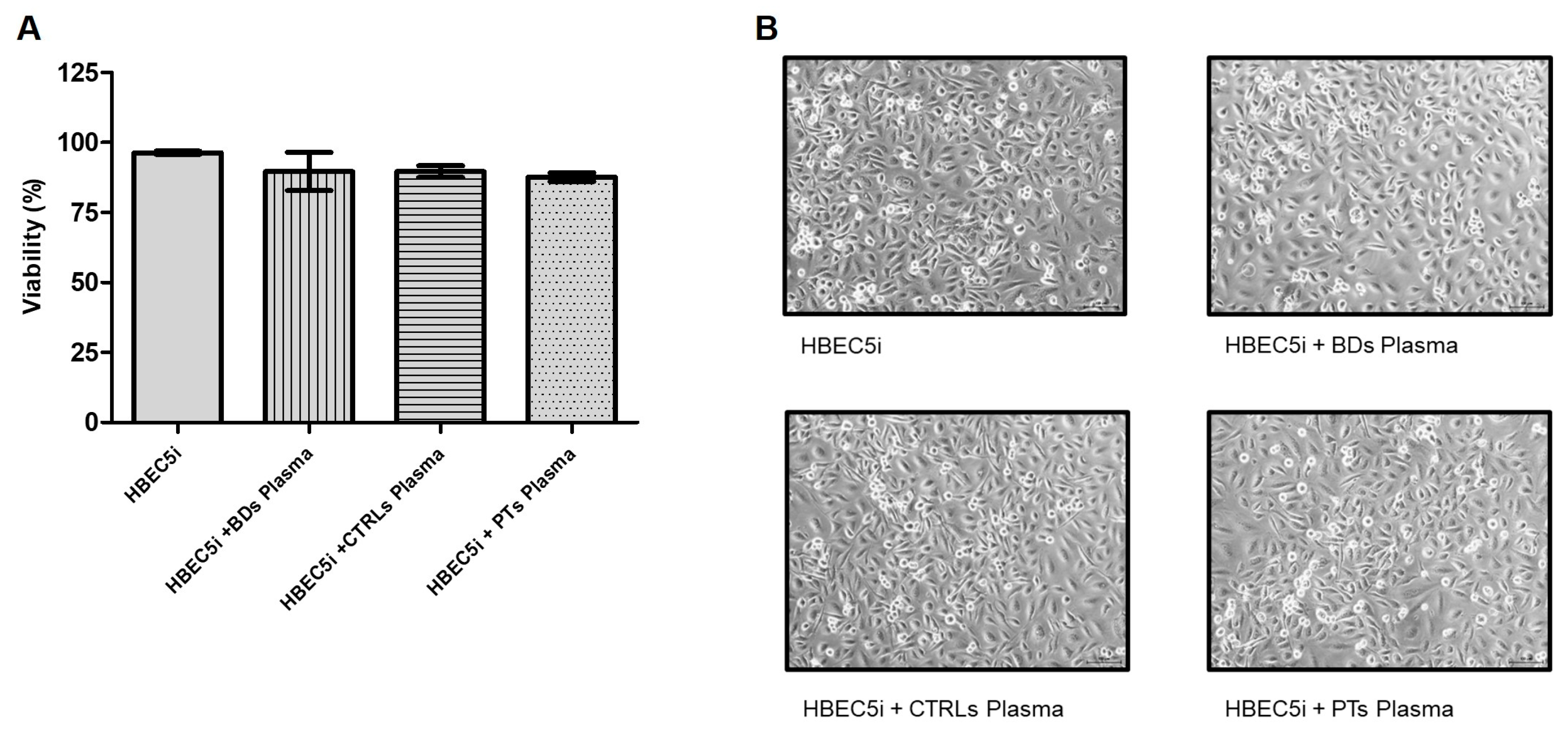
Figure 2.
Heatmap showing the differentially expressed genes comparing treatment with PTs, CTRLs and BDs plasma with HBEC5i untreated cells, respectively (at log2 fold change ≥ 1.5; see Methods for further details).
Figure 2.
Heatmap showing the differentially expressed genes comparing treatment with PTs, CTRLs and BDs plasma with HBEC5i untreated cells, respectively (at log2 fold change ≥ 1.5; see Methods for further details).
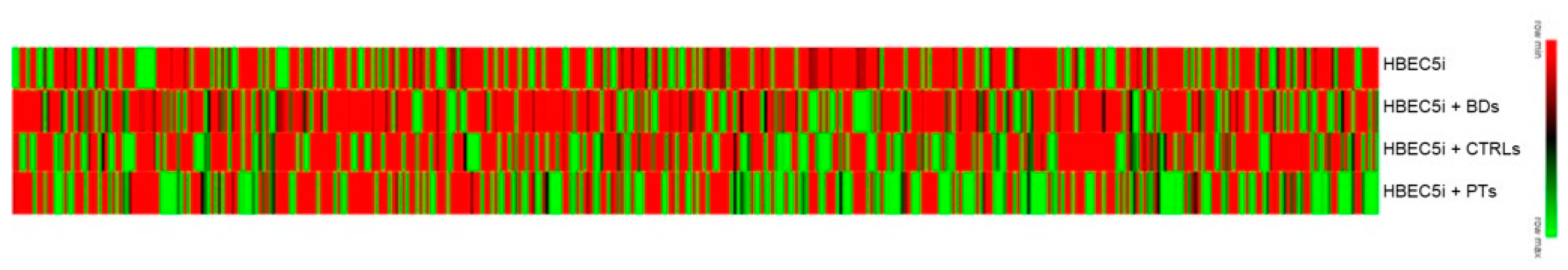
Figure 3.
HBEC5i were treated with 15% v/v of human plasma of BDs, CTRLs and PTs for 24 hours. Relative expression of CXCL8 (A), LIF (B) and IL12A (C) was analyzed by qPCR (n=4). Data were expressed as fold change over untreated cells. Columns, mean; bars, SD; * significant difference p<0.05. .
Figure 3.
HBEC5i were treated with 15% v/v of human plasma of BDs, CTRLs and PTs for 24 hours. Relative expression of CXCL8 (A), LIF (B) and IL12A (C) was analyzed by qPCR (n=4). Data were expressed as fold change over untreated cells. Columns, mean; bars, SD; * significant difference p<0.05. .
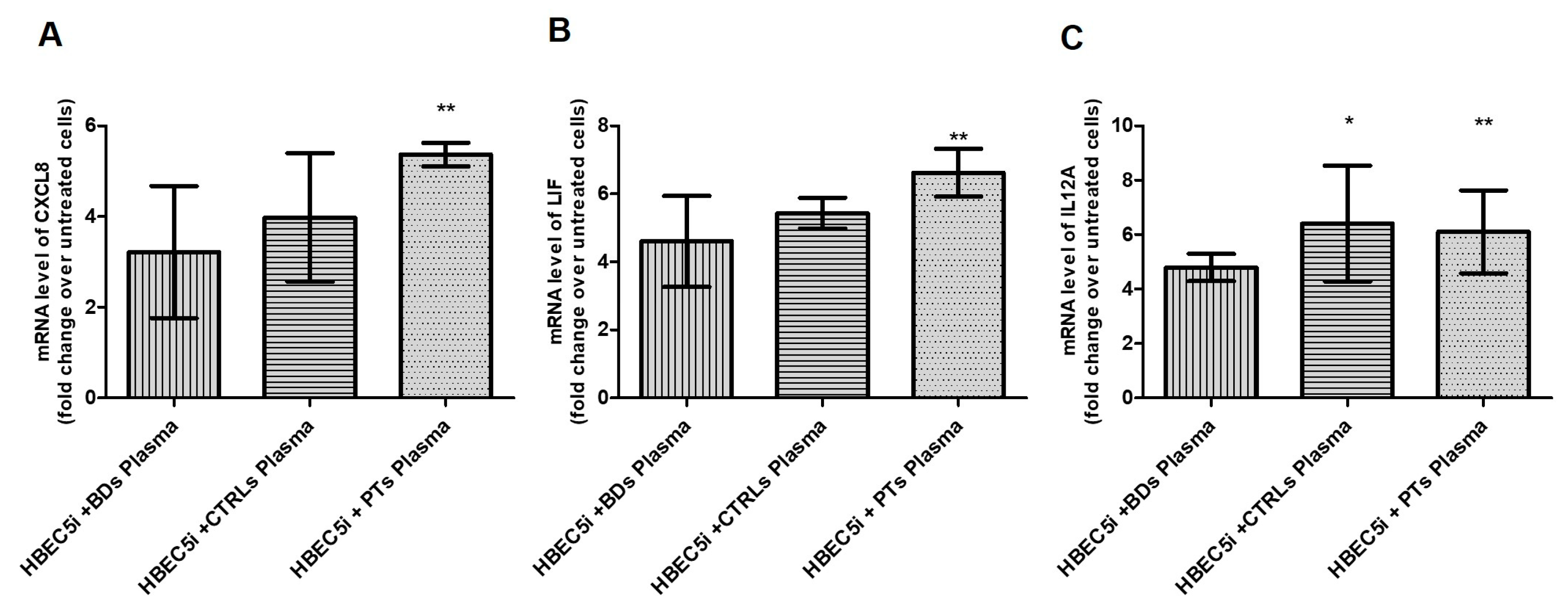

Table 1.
De-regulated pathways by CSVD treatment compared to untreated HBEC5i (only differentially regulated pathways in cells treated with plasma of patients are enlisted, p<0,05).
Table 1.
De-regulated pathways by CSVD treatment compared to untreated HBEC5i (only differentially regulated pathways in cells treated with plasma of patients are enlisted, p<0,05).
| GO Term | Description |
|---|---|
| GO:0036500 | ATF6-mediated unfolded protein response |
| GO:0007050 | cell cycle arrest |
| GO:0070887 | cellular response to chemical stimulus |
| GO:0071310 | cellular response to organic substance |
| GO:0051716 | cellular response to stimulus |
| GO:0033554 | cellular response to stress |
| GO:0019221 | cytokine-mediated signaling pathway |
| GO:0001892 | embryonic placenta development |
| GO:0030968 | endoplasmic reticulum unfolded protein response |
| GO:0043066 | negative regulation of apoptotic process |
| GO:0010648 | negative regulation of cell communication |
| GO:0060548 | negative regulation of cell death |
| GO:0043069 | negative regulation of programmed cell death |
| GO:0048585 | negative regulation of response to stimulus |
| GO:0023057 | negative regulation of signaling |
| GO:1901564 | organonitrogen compound metabolic process |
| GO:1990440 | positive regulation of transcription from RNA polymerase II promoter in response to endoplasmic reticulum stress |
| GO:0036003 | positive regulation of transcription from RNA polymerase II promoter in response to stress |
| GO:0042981 | regulation of apoptotic process |
| GO:0010646 | regulation of cell communication |
| GO:0051726 | regulation of cell cycle |
| GO:0031326 | regulation of cellular biosynthetic process |
| GO:0080135 | regulation of cellular response to stress |
| GO:0043067 | regulation of programmed cell death |
| GO:0031399 | regulation of protein modification process |
| GO:1905897 | regulation of response to endoplasmic reticulum stress |
| GO:0080134 | regulation of response to stress |
| GO:0023051 | regulation of signaling |
| GO:0042762 | regulation of sulfur metabolic process |
| GO:1901342 | regulation of vasculature development |
| GO:0042221 | response to chemical |
| GO:0034976 | response to endoplasmic reticulum stress |
| GO:0010033 | response to organic substance |
| GO:0006950 | response to stress |
| GO:0035966 | response to topologically incorrect protein |
| GO:0006986 | response to unfolded protein |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



